Submitted:
30 January 2024
Posted:
31 January 2024
Read the latest preprint version here
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by resting tremor, bradykinesia, rigidity, postural instability, that also includes non-motor symptoms such as mood dysregulation. Dopamine (DA) is the primary neurotransmitter involved in this disease, but cholinergic imbalance has also been implicated. Current intervention in PD is focused on replenishing central DA, which provides remarkable temporary symptomatic relief but does not address the neuronal losss and the progression of the disease. It has been well established that neuronal nicotinic cholinergic receptors (nAChRs) can regulate DA release and that nicotine itself may have neuroprotective effects. Recent studies have identified nAChRs in nonneuronal cell types including glial cells, where they may regulate inflammatory responses. Given the crucial role of neuroinflammation in dopaminergic degeneration, and involvement of microglia and astrocytes in this response, glial nAChRs may provide a novel therapeutic target in prevention and/or treatment of PD. In this review, following a brief discussion of PD, we focus on the role of glial cells and specifically their nAChRs in PD pathology and/or treatment.
Keywords:
Parkinson’s disease
; Dopamine
; Acetylcholine
; Nicotine
; Nicotinic receptors
; Microglia
; Astroglia
; Oligodendrocyte
; NG2 Cells
; Alpha-Synuclein
; Toll-Like receptors
; Neuroinflammation
; Neuroprotection
Introduction
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disorder, where global epidemiological data show over 8.5 million individuals afflicted with it [1]. Though many of the motor features of PD are dopamine (DA) responsive, symptoms such as balance problems, do not respond well to such treatments. Non-motor symptoms including fatigue, depression, sleep problems, cognitive difficulties, variations in blood pressure, urinary problems, and constipation, also do not respond to what might be termed as “DA-replacement therapy.” Thus, it is concluded that deficiencies in other neurotransmitter systems including nicotinic cholinergic system, may underlie these features [2,3,4]. As such, there is interest in targeting these other neurotransmitters functions to treat specific dopamine-resistant aspects of PD.
One of the challenges in PD is underscored by the fact that its pathogenesis is not totally clear. Epidemiological data show approximately 15% of PD patients have a hereditary form, and 5-10% have a monogenic mendelian inheritance that, so far, has 23 loci and 19 causative genes [5,6]. Moreover, mutations in autosomal dominant genes such as SNCA, LRRK2, VPS35, or autosomal recessive genes such as PRKN, PINK1, DJ-1 may lead to PD. On the other hand, recessive DNAJC6 mutations can present as atypical parkinsonism [7]. Additionally, potential contribution of environmental toxins such as herbicides (e.g., paraquat), insecticides (e.g., rotenone), excess accumulation of iron or manganese [3,8,9,10], as well as exposure to endogenous toxins such as salsolinol or aminochrome to PD pathology have been verified [3,11,12].
Studies involving the hereditary form of PD, imaging, post-mortem, and extensive pre-clinical models have implicated a set of molecular and cellular alterations. These include loss of dopaminergic neurons in the nigrostriatal pathway [13], mitochondrial damage [14], dysfunction in autophagy/ mitophagy [15], accumulation of alpha-synuclein aggregates [16], and neuroinflammation, underscored by gliosis [17]. Regarding the latter, it is important to note that in the brain four main subsets of glial cells (microglia, astrocytes, oligodendrocytes, and synantocytes or NG2 cells), have been identified. For each subset, the functional heterogeneity, beneficial or detrimental roles in maintaining homeostasis are under intense investigation.
Thus, given the important role of glial cells in the pathogenesis of PD [17,18,19,20], and implicated nAChRs regulation of these cells [17,18,21], our aim in this review is to provide not only an update on glial nAChRs role in glial function but also potential exploitation of this knowledge in identifying novel targets for this devastating disease.
PD pathophysiology and current treatments
The main pathological alterations in PD are the accumulation of Lewy bodies, composed mainly of alpha-synuclein (α-Syn), and loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) that leads to striatal DA deficiency [22,23,24]. This dopaminergic loss results in motor deficits characterized by akinesia, rigidity, resting tremor, and postural instability as well as non-motor symptoms that also involve other neurotransmitter systems [3,24,25]. The non-motor symptoms may include cognitive deficits (e.g., mild to severe memory impairment), emotional changes (e.g., depression, apathy, and anxiety), sleep perturbations (e.g., insomnia/hypersomnia), autonomic dysfunction (e.g., bladder disturbances, orthostatic hypotension, sweating), sensory symptoms (e.g., pain, visual and olfactory disturbances) and gastrointestinal (GI) symptoms (e.g., constipation, nausea) [24,26,27]. The most common treatment is focused on DA replacement via L-dopa (L-3,4-dihydroxyphenylalanine), which is DA precursor. However, not only the efficacy of this drug is invariably reduced in a few years, but long-term treatment may cause severe dyskinesia or involuntary movements [3,25,28,29]. Hence, extensive efforts to find novel therapies are ongoing.
The usual clinical treatment for PD aims to replace DA and inhibit the motor symptoms by using L-dopa or inhibitors of enzymes that metabolize DA, such as monoamine oxidase B (MAO-B) or catechol-O-methyltransferase (COMT) [30]. L-Dopa is usually combined with carbidopa to inhibit its peripheral metabolism. L-dopa appears to be the best option for stiffness and akinesia, improving the patient’s quality of life [24,31], however, its chronic usage induces side effects, such as motor fluctuations, dyskinesia, and psychosis [24,32]. More recently, deep brain stimulation has been employed for inhibiting motor symptoms with a relatively high effectiveness, although surgery-related complications remain a possibility [24,33]. Due to limited efficacy and/or complications of current interventions, extensive effort, particularly towards the prevention of neurodegeneration, has been expended. In this regard, neuronal nAChRs have emerged as a viable target [4,34,35]. Interestingly, mutation in RIC3, a chaperone of neuronal nicotinic acetylcholine receptor subunit α-7 (CHRNA7), has been implicated in PD [7]. However, recent elucidation of functional presence of nAChRs in non-neuronal cells (i.e., glia) may offer additional novel targets in preventing and/or slowing down the disease progression. The following delves into this possibility.
Glial Cells
The glia represents the biggest population of cells in the human brain, with 10 times more cells than the neurons [36,37]. These cells perform several pivotal functions such as energetic support for neurons [38,39,40], formation of the blood-brain barrier (BBB) [41,42], regulation of neurotransmitters [43,44,45], development and remodeling of synapses [46,47,48], detoxification [49,50,51], control of the fluid/electrolyte homeostasis [52], control of metabolism [53,54], neuroendocrine function [55] innate immunity response [56,57], and myelination [58,59]. These functions confer on them key role in maintaining homeostasis, disruption of which can lead to neuropsychiatric and neurodegenerative diseases [59,60,61,62,63,64].
As mentioned above, four main subsets of glial cells (microglia, astrocytes, oligodendrocytes and synantocytes or NG2 cells) in the central nervous system (CNS) have been identified (Figure 1). Here, following description of each, specific roles of nAChRs in these cells in relevance to PD is discussed.
Microglia
Microglia, constituting 10%–15% of all CNS cells, are an important subset of glial cells acting as the innate immune response, and play a key role in neuroinflammation, responsible for degeneration of dopaminergic neurons in PD [65,66,67,68]. Indeed, microglia are considered the resident macrophages with a vital role in maintaining CNS homeostasis as they eliminate pathogens and cell residue through phagocytic activity (Figure 1) They are also crucial regulators of neurogenesis, participate in the formation and elimination of neuronal synapses, and control the number of neuronal precursor cells. Furthermore, microglia serve as the main antigen-presenting cells and mediate autoimmune effector T-cell infiltration into the brain [69]. Three states of microglia consisting of resting, activated and phagocytic have been identified. At the resting stage, they are highly ramified, but when activated in response to injury or insult, they contract, assume an enlarged cell body, and proliferate. Finally, they transform into full-blown phagocytic microglia. However, if overactivated, neuroinflammatory and neurodegenerative disorders may ensue [65,67,68,70]. In general, depending on their activation phase, they are categorized as M1 microglia, which lead to neuroinflammation and neurotoxicity, and M2 microglia, which stimulate anti-inflammatory and neuroprotective effects [69,71,72].
Microglial polarization into M1 or M2 phenotype occurs due to perturbation in the microglial micro-environment. Under physiological conditions that are in the resting state, microglial morphology is characterized by a small cell body and very fine, highly ramified processes, which allow these cells to screen their local environment for signs of pathogens or cellular damage. Because of this continuous activity, it has been proposed that this stage of microglia be designated as “surveilling” microglia rather than resting state [73,74,75]. When microglia are activated, their cell body enlarges, their processes become shorter or withdrawn, and they assume a round or amoebic shape, which allows for their migration to sites of injury and gain phagocytic abilities. Thus, amoeboid microglia, characterized by completely retracted processes and a swollen cell soma migrate to the site of injury to phagocytose harmful debris and invaders. This morphological transformation may reflect disease-specific functional cell states. Interestingly, a fourth morphology of microglia in mice, so-called rod-like microglial cells, which do not exhibit planar processes and show fewer secondary branches as well as narrowing of cell and soma have also been reported [75,76,77]. Activated microglia express microglial receptors, including the triggering receptor expressed on myeloid cells-2 (TREM2), low-density lipoprotein receptor-related protein 1 (LRP1), calcium-sensing receptor (CASR), toll-like receptors 2 and 4 (TLR2 and TLR4). TLRs are a well-characterized family of pattern recognition receptors (PRRs), expressed by microglia and astrocytes (discussed below), that sense pathogens or endogenous debris released by damaged cells and initiate the innate immune response. The contribution of microglial TLR signaling to CNS pathology has been extensively investigated, and potential therapeutic targets have been suggested [69,78]. In addition, all glial cells including microglia express nAChR, which is the emphasis of this review.
Microglia- Synucleinopathies
As the name implies, synucleinopathies refers to neurodegenerative diseases that share a pathologic accumulation of the protein α-Syn, which, as alluded to earlier, can cause neuronal death as in PD. Although the etiology of synucleinopathies remains unknown, microglia have been directly implicated in its pathogenesis. It is worth reiterating that microglia offer neuroprotection in the early stages of α-Syn accumulation, however, in chronic disease conditions, structural varieties of inherently disordered α-Syn result in varied microglial receptor-mediated interactions. Thus, whereas initially microglial receptors enable cellular recognition and uptake of α-Syn, in chronic disease states, physiological functions of microglia are overwhelmed. At this stage, α-Syn processing within microglia results in neuroinflammation and neurodegeneration, which can also spread to other brain regions [79,80]. Under pathological states or when the physiological functions of microglia are overwhelmed, they take on aberrant phenotypes, which helps the spread of α-Syn and promote disease progression [66,79,80]. Microglial activation in neurodegenerative diseases involves mitochondrial and cellular metabolism dysregulation, leading to the build-up of reactive oxygen species (ROS), amino acids, iron, and eventual inflammation and cell death [66,79,80]. Recently, it was revealed that circadian rhythm also plays an essential role in microglia activation and function and that disruption of this rhythm can lead to neurodegenerative diseases [81].
Synucleinopathies are also considered a hallmark of dementia, including PD dementia (PDD) and Lewy body dementia (LBD), where the latter, comprising roughly 20–30% of all dementia cases, is the third most common form of dementia in the world. Even in Alzheimer’s disease (AD), up to 50% of cases reveal co-morbid α-Syn pathology. Cell-to-cell transmission of α-Syn aligns well with the clinical course of the disease and is now an accepted mechanism underlying neurodegenerative diseases. What may cause α-Syn misfolding in the first place is believed to involve a combination of factors including aging, environmental factors, and exposure to toxins, which ultimately overwhelms the microglia’s ability to maintain homeostasis and leads to neurodegeneration [81,82]. Aberrant circadian rhythmicity may also lead to extensive α-Syn aggregation [83]. Interestingly, microglial gene and protein expression also follow circadian patterns, and hence, microglia’s activity (e.g. synaptic pruning) and response to an immune challenge may vary depending on the time of the day [84]. Thus, circadian rhythm dysfunction may precipitate neurodegenerative consequences via microglial activation and synucleinopathies.
Overall, it may be concluded that microglia play a pivotal role in the spread of α-Syn and eventual pathophysiology of synucleinopathies, including those related to circadian rhythm dysfunction. Therefore, extensive effort is expended in elucidating the molecular mechanism(s) of microglial interactions with α-Syn in the hope of identifying novel therapeutic targets for prominent neurodegenerative diseases including PD [79,80]. It is also worth mentioning that TLR 4 is required for α-synuclein dependent activation of microglia [85,86]
Very recently, however, it was discovered that when α-Syn is phosphorylated, its structure changes in a way that promotes interactions with other proteins, and lets it act as a brake to keep activity in certain neuronal circuits in check, suggesting that it might have a role in healthy brains [87]. But why relatively low-frequency events in a healthy brain, when accumulated over a lifetime, can trigger the pathological accumulation of α-Syn into Lewy bodies, leading to PD and Lewy body dementias is still unknown [88].
In summary, it may be suggested that α-Syn activation of microglia via TLRs results in microglial inflammatory response and eventual destruction of dopaminergic neurons leading to PD.
Astroglia
Astroglia or astrocytes are star-shaped cells that make up between 17% and 61% of the cells in the human brain, depending on the area. Like microglia, they exhibit heterogeneous phenotypes in response to various insults, a process known as astrocyte reactivity [89]. Astrocytes perform myriad essential functions including maintenance and accuracy of brain signaling, recycling of neurotransmitters, modulation of ionic environment and providing metabolic support for the neurons, regulation of cholesterol and sphingolipid metabolism, and maintenance of the blood–brain barrier [89,90] (Figure 1).
Several subclasses of astrocytes have been identified. The most numerous ones are protoplasmic astrocytes that have a stellate morphology and are prominent in layers II–VI of the cortical gray matter and are found in all mammals. Two other distinct subtypes are found only in primates and humans and reside in either layer I or layer VI of cortical layers. These interlaminar astrocytes have high expression of glial fibrillary astrocytic protein (GFAP) that are commonly used as markers in their identification. Moreover, astrocytes express several neurotransmitter receptors including both α4- and α7-containing nAChRs [91,92,93,94].
Although astrocytes are pivotal for brain functioning, homeostasis, and detoxification [38,39,95,96], their role in PD pathogenesis is not completely known. Reports show an increase in α-Syn-immunoreactive astrocytes in postmortem human brains [97]. Moreover, in-vitro studies suggest astrocytes are susceptible to dysfunction induced by α-Syn [98] or aminochrome [99,100,101,102,103]. As mentioned, α-Syn is an unfolded protein that accumulates in Lewy bodies, a hallmark of PD [104]. Aminochrome, on the other hand, is an endogenous DA neurotoxin [11]. Astrocytes are not well equipped with receptors recognizing pathogens compared to microglia, however, when activated by polarized microglia, they become reactive, release inflammatory mediators, and modulate inflammation [69]. Thus, astrocytes can amplify proinflammatory signals released by microglia and contribute to neuronal degeneration, suggesting synergistic collusion [105,106,107] (Figure 1). Interestingly, like microglia, α-Syn activation of astrocytes is dependent on the presence of TLR4 on these cells [85,86]. Moreover, an intimate interaction between astrocytes and neurons, as well as between astrocytes and microglia, referred to as crosstalk, has recently been highlighted [89,90]. As our knowledge of such cross talks expands, novel intervention in neurodegenerative diseases, including PD is anticipated [89,90,108]. Furthermore, because of nAChRs presence on these cells and their known functional role (discussed below) specific targets may be suggested [91,92,94,109].
Oligodendrocytes
Oligodendrocytes (OLs), once considered static glue, are not only the myelinating cells of CNS, but are also plastic and adaptive to changes in CNS [110] (Figure 1). Four phases in life cycle of OLs have been identified: (1) OLs precursor cells (OPCs) giving rise to birth, migration, and proliferation of OLS. Remarkably, OPCs by themselves constitute a subclass (4th subset) of glial cells that are described in detail below; (2) morphological differentiation, characterized by an expansive network of processes; (3) ensheathment and generation of compact myelin around target axons; and (4) metabolic and trophic support of the encased axon. It is of relevance to note that in the peripheral nervous system, neuroglia that are equivalent to OLs are called Schwann cells.
The importance of OLs in the pathogenesis of PD has also been verified (Figure 1). A single-core human transcriptomics atlas for the substantia nigra (SN) revealed that distinct neuropsychiatric disorders associated with neuron-specific genes converge on shared loci within OLs and OPCs [111]. These cells represent 75% of all glial cells in the adult CNS. In addition to axonal myelination, OLs control extracellular potassium concentration, and as mentioned above, provide metabolic and trophic supply to myelin, secrete glial and brain-derived neurotrophic factors (GDNF and BDNF), and modulate the axonal growth [110,113], all of which highlight their importance in functioning of CNS. Like microglia and astrocytes, OLs also express TLRs, which are considered of significant importance in myelin formation [58,114,115]. Importantly, and of direct relevance to our discussion, dysregulation of these glial cells, which contain nAChRs, can contribute to the pathogenesis of PD [116].
Synantocytes (NG2 cells)
The fourth subset of glial cells in CNS are synantocytes or neuron glial 2, or nerve/glial antigen 2 (NG2) cells. These cells, also referred to as OPCs, are primarily identified by the expression of two key markers: the chondroitin sulfate proteoglycan NG2 and the platelet-derived growth factor receptor alpha (PDGFRα) [110,117]. These cells display a combination of features including (i) an almost uniform distribution in both grey and white matter areas; (ii) a complex stellate morphology; (iii) a tendency to intimately associate with neuronal cell bodies and dendrites; (iv) the ability to keep proliferating in the adult brain; and (v) a latent ability to give rise to astrocytes and neurons that may be recruited to areas of lesion [110,117,118].
While originally the main role ascribed to NG2 cells was that of progenitors for OLs, later it became more obvious that they have further functions in the brain including potential roles in demyelinating and neurodegenerative diseases such as multiple sclerosis and AD, as well as traumatic brain injury, glioma, epilepsy, and electroconvulsive therapy for depression [119,120]. Additionally, as an early marker of pericyte activation in pathological conditions, NG2 cells were suspected of playing a role in experimental autoimmune encephalomyelitis (EAE), a disease associated with increased BBB permeability, and neuroinflammation [121]. It was suggested that NG2 cells exert their effects via stimulation of reactive T cells and controlling IL-12 expression [121].
More recently, it was suggested that NG2 cells play a critical role in modulation of neuroinflammation [122] and neurovascular unit formation during development [123] (Figure 1). Although yet no TLRs have yet been identified in these cells, their importance in angiogenesis and oligodendrogenesis following acute ischemic stroke has been reported [123]. Importantly, their ability to receive synapses from neurons and affect neuronal plasticity and behavior, and their containment of nAChRs [124,125] suggest their potential use in specific therapeutic interventions.
Nicotine
Nicotine, the primary psychoactive agent in tobacco leaves, is highly addictive and has therefore led to the widespread use of tobacco, with over one billion smokers globally. Severe consequences of smoking on almost every organ of the body and manifestation of numerous diseases, which result in nearly 500,000 deaths in US alone and nearly 8 million people worldwide are staggering statistics. Diseases associated with smoking include cancers, especially that of the lung, and cancers of the mouth and throat, voice box, esophagus, stomach, kidney, pancreas, liver, bladder, cervix, colon and rectum, and acute myeloid leukemia. Additionally, the risks of heart disease, stroke, diabetes, chronic obstructive pulmonary disease (COPD), including emphysema and chronic bronchitis, certain eye diseases, tuberculosis, immune dysfunction including rheumatoid arthritis, are well established (WHO and CDC websites). However, nicotine itself may have many therapeutic potentials, including neuroprotection (discussed below). Specifically, as our understanding of the mechanism of action of nicotine expands, more selective therapeutic targets become available. The following provides an up-to-date summary of the current knowledge on nAChRs, with specific implications in PD.
nAChRs
The action of nicotine is mediated via nAChRs, which act by directly opening a channel, allowing for the influx of sodium (Na+) and calcium (Ca²+) [126,127,128,129]. The nAChRs are pentamers composed of different subunits such as alpha (α), beta (β), or delta (δ). To date, an overall, 16 homologous mammalian nAChR subunits have been identified [126,128]. Many different nAChR subtypes due to subunit combination form in different areas such as the neuromuscular junction, autonomic ganglia and in CNS. However, the subunit structures of these receptors are different depending on the area. For example, only the neuromuscular receptors contain the delta subunit, whereas the autonomic ganglia and CNS nAChRs contain only alpha and beta subunits, albeit in different combinations. In the brain, the predominant subtypes consist of alpha4 and beta2 or the homomeric alpha7 subtypes [126,128,129,130].
The α7 subtype of nicotinic receptors (α7nAChRs) is one of the most abundant nicotinic receptor subtypes in CNS and both neurons and nonneuronal cells express it. When activated, α7nAChRs become permeable to cations and promote cellular responses including modulation of PI3K/Akt signaling cascade. This results in stimulation of anti-apoptotic molecules of the Bcl-2 family, Bcl-2 and Bcl-xl, and reduction of proapoptotic molecules, and hence promoting cell survival [131]. α7nAChR is also a key protein in the cholinergic anti-inflammatory pathway (discussed in detail below) that links the nervous and the immune systems [132].
More recently, there has been the suggestion of potential metabotropic signaling responses by α7 nAChRs through heterotrimeric G proteins in both neuronal and immune cells [133]. Furthermore, and of relevance to our discussion below, not only non-ionic signaling mechanisms via nAChRs have been demonstrated in immune cells, but also some nAChRs may be activated by endogenous ligands other than ACh [134].
Non-neuronal nAChRs also exist and are expressed in lung epithelial, endothelial, fibroblast cells, and in muscles. Chronic nicotine exposure differentially affects the expression of different receptor subtypes. For example, there is an increase in α5 subunit of nAChRs in epithelial cells, a decrease in α3 subunit airway fibroblast cells [135]. Further distinction between nicotinic receptor subtypes is evident in their central distribution as well as their physiological roles. Thus, alpha7 or low affinity subtype, are most abundant in the hippocampus and are believed to have a prominent role in neuronal growth and survival and be involved in cognitive functions, particularly attentional processes [35] These receptors are also implicated in pain modulation [136]. On the other hand, high affinity α4β2 subtype, are more prominent in mesolimbic or nigrostriatal pathways and are involved in rewarding or addictive behavior, locomotor activity and antinociception [137].
It is now evident that nAChRs not only play important roles in neuronal functions and addiction to nicotine [138,139], but may also offer therapeutic targets for various neuropsychiatric and neurodegenerative disorders including PD [25,140,141], depression [140,142,143], mild cognitive impairment or Alzheimer’s disease [144,145,146,147], ischemia [148], catalepsy [149], schizophrenia [4,147,150], pain [134,151], energy balance [152,153], autoimmune disorders [154], and even in sleep-wake cycle dysregulation [155]. These receptors are also abundantly expressed in variety of immune cells including B cells, T cells, macrophages and microglia, and are believed to contribute to the anti-inflammatory effects of nicotine [156,157,158,159]. Indeed, nicotine has been shown to inhibit pro-inflammatory cytokines such as tumor necrosis factor- α (TNF-α), IL-1, and IL-6, without affecting the anti-inflammatory cytokines such as IL-10 [158,159,160,161]. This effect of nicotine, in addition to its interaction with ACE2 via nicotinic receptors has led to the suggestion of a potential role of selective nicotinic receptors in interfering with SARS-Cov2 virus entry and hence improving COVID-19 condition [135,162]. In relation to cancers such as lung cancer, there is an increase in expression of nAChRs, which has been associated with cell proliferation, epithelial-to-mesenchymal cell transition, angiogenesis, and apoptosis prevention [163]. In relation to diabetes, a role for nAChRs in the release of glucoregulatory hormones, as well as glucose tolerance and insulin sensitivity was recently suggested [153]. Therefore, modulation of the nicotinic receptors by appropriate concentrations of nicotine may be exploited for a plethora of therapeutic purposes.
Nicotine for PD
It is now evident that the normal function of the basal ganglia is dependent on the equilibrium between the midbrain dopaminergic and striatal cholinergic systems. Acetylcholine can regulate striatal DA release via interaction with nicotinic receptors [25,141]. Moreover, in animal models of PD (e.g., 6-OHDA lesioned rodents) the impairments in DA release appear to be exacerbated by a loss of nAChRs, which suggests that nicotinic agonists may ameliorate the dopaminergic imbalance and may be useful therapeutic targets for PD. Indeed, several in-vitro and in-vivo studies in rodents and primates including genetically modified mice, have verified the protective effects of nicotine against neuronal damage induced by 6-OHDA, MPTP, rotenone, paraquat, methamphetamine, glutamate, and β-amyloid [3]. Nicotine also protects against salsolinol-induced toxicity in SH-SY5Y cells [164]. Salsolinol, an endogenous product of aldehyde and DA condensation is frequently used to induce selective toxicity to dopaminergic neurons. SH-SY5Y cells, derived from human neuroblastoma are commonly used as a cellular model of dopaminergic neurons to investigate novel treatments for PD [165,166]. It is now evident that both alpha4-beta2 and alpha7 nicotinic receptors are involved in the protective effects of nicotine [3,164,167]. Similarly, the damage inflicted by aminochrome, a neurotoxic molecule derived from DA oxidation, RCSN-3 cells (derived from substantia nigra of adult rat), could also be protected by nicotine [168]. More recently, protective effects of nicotine against the toxicity induced by manganese and iron in SH-SY5Y cells with implications for PD were reported [165,166]. Additionally, it was shown in-vitro that nicotine protects PC12 neural cells against 1-methyl-4-phenylpyridinium ion (MPP+) toxicity via activation of alpha7 nAChR/PI3K/Trx-1 and suppression of endoplasmic reticulum stress [169,170]. This finding was recently complemented by the findings that nicotine also alleviates MPTP-induced nigrostriatal damage through modulation of JNK and ERK signaling pathways in a mouse model of PD [171]. Moreover, using D-line α-Syn transgenic mice, and a humanized neuronal model of synucleinopathies, it was revealed that nicotine attenuates α-Syn-provoked neuropathology visa activation of α4β2 nicotinic receptors [172]. In a similar in-vivo study, it was shown that nicotine attenuates motor deficits in an α-Syn PD model [173]. Thus, manipulating both α4β2 and α7nAChRs may effectively mitigate synucleinopathies [159,172,174].
The anti-fibrillogenic and fibril-destabilizing activity of nicotine in addition to its ability to promote the clearance of α-Syn, may be of critical importance in its inhibition of Lewy bodies [175,176,177,178,179,180,181]. As alluded to earlier, the accumulation of Lewy bodies, composed primarily of α-Syn, is a hallmark of PD pathology. Indeed, it is believed that synucleinopathies not only contribute to movement disorders but also to cognitive and social impairment associated with PD [181,182,183]. In further support of the contention that targeting nAChRs may be a novel therapeutic strategy for PD treatment [92,184], it was recently reported that nicotine’s prevention of synucleinopathies or α-Syn toxicity may be due to its interaction with α7nAChRs and inhibition of apoptosis as well as interaction with synaptic vesicle glycoprotein [179,185].
Mode of nicotine administration as a critical factor in PD
The well-established inverse relationship between smoking and PD and documented neuroprotective effects of nicotine prompted several clinical trials with nicotine patch in PD [186]. However, no apparent benefit was noted with such mode of nicotine administration [186]. This lack of response to nicotine patch was likely due to steady release of nicotine and prolonged nicotinic receptor desensitization [3,186]. Although, nicotine patch, by maintaining a steady plasma concentration of nicotine may be an effective intervention for smoking cessation as desensitization of the central nicotinic receptors in critical brain reward circuitry may help ameliorate the withdrawal effects of nicotine, it is unlikely to be of therapeutic efficacy in PD. This contention is underscored by the fact that a pulsatile stimulation of nicotinic receptors, like that experienced by smokers, is necessary re-stimulate the nicotinic receptors and hence provide neuroprotection and be of benefit in PD [3,186]. It is also of utmost importance to distinguish between pure nicotine and nicotine derived from burning tobacco leaves, where the latter contains xenobiotics, including carcinogens and other toxins. Moreover, nicotine itself may not only be effective in ameliorating PD symptoms and retard the progression of the disease but may also counter L-dopa-induced dyskinesia [3,186].
It is worth mentioning that current efforts in developing selective nicotinic receptor subtype agonists or nicotinic receptor modulators that would be of similar or better potency and/or efficacy than nicotine but without its addictive properties, could be of significant therapeutic triumph [3,187,188,189].
nAChRs - Microglia and Gut-Brain Axis
Microglia, as discussed in detail above, are resident macrophages of CNS. Macrophages refer to cells that serve as vital defenders in response to invading pathogens and various stimuli. During the peripheral immune challenge, inflammatory cytokines can signal the brain for activation of the immunomodulatory mechanism via the vagus nerve (Figure 2). This involves activation of the cholinergic anti-inflammatory pathway, release of ACh from the vagus nerve, activation of α7nAChRs on peripheral macrophages to restore homeostasis [190,191,192,193] (Figure 2).
Moreover, the cholinergic anti-inflammatory pathway functions as an interface between the brain and the immune system [192]. It is noteworthy that the vagus nerve has also been implicated in brain-immune interaction through manipulation of the gut-brain axis (GBA) (Figure 2). GBA, considered to play a central role in many diseases including neurodegenerative/neuropsychiatric disorders, is influenced by the gut microbiota (GM) via a variety of molecules and neurotransmitters including short-chain fatty acids (SCFAs) [192,194,195]. GBA communication occurs through several pathways, including the immune system, the enteric nervous system (ENS, a complex of neuronal and glial network controlling the function of the gut), microbial metabolites, and the vagus nerve [196]. GM may also influence neural function via its interaction with microglia and astroglia [196]. Thus, neuroinflammation and its devastating consequences can arise from an imbalance in GM referred to as dysbiosis, or disruption in any component of the GBA.
Regarding ENS, it is important to note that the status of the gut is communicated to the brain via sensory neurons arising from the dorsal root, nodose ganglia, neurons of the autonomic nervous system, and immune cells of the gut. A critical component in GBA is the vagus nerve, which is essential for coordinating the immune system’s response to bacteria, pathogens, or toxins. In this regard, the afferent vagus nerve is considered the main retrograde signaling system, bringing information from the gut to the brain. On the other hand, the efferent vagus nerve, containing the cholinergic anti-inflammatory activity, helps control TNF-α and other cytokines secreted by macrophages in the gut [197]. One element common to all these gut-brain systems is the expression of nAChRs that serve myriad roles in GBA (Figure 2). These roles include mediating fast synaptic transmission between autonomic pre- and postganglionic neurons, modulating neurotransmitter release from peripheral sensory and enteric neurons, and controlling cytokine release from immune cells [194].
Involvement of GM in pathogenesis of PD via immunological, neuroendocrine, and direct neural mechanisms is well acknowledged. The sequence of events is believed to start with GM dysbiosis which triggers the loss of dopaminergic neurons via mitochondrial dysfunction and systemic inflammation (Figure 2). Moreover, GM dysbiosis results in activation of enteric neurons and enteric glial cells leading to aggregation of α-Syn. Curiously, α-Syn is also synthesized by the sensory cells of the gut mucosa and can be transported to CNS via vagus nerve [198,199] (Figure 2). Indeed, according to Braak hypothesis, α-synuclein misfolding begins in the gut and spreads "prion-like" via the vagus nerve into the lower brainstem and ultimately to the midbrain leading to PD [197]. Because of its enteroendocrine cell-mediated interaction with GM, the vagus nerve may also provide a potential pathway through which gut microorganisms influence host feeding behavior and metabolic control of physiological and pathological conditions, also suggesting a role for vagus nerve in appetite control, development of obesity, and diabetes [200].
Because nAChRs play a critical role in the function of GBA their manipulation along this axis and in microglia may provide novel interventions in neuropsychiatric/neurodegenerative diseases (Figure 3). As mentioned above, sufficient data is now available to justify the use of pulsatile nicotine in PD. This contention is further strengthened by the findings that nicotine may modulate the release of inflammatory cytokines and expression of TLRs [201,202]. Indeed, recent findings indicate inhibition of TLRs by cotinine, a metabolite of nicotine via α7nAChR [35].
As our understanding of the intricate interactions between GM, microglia and specifically the role of nAChRs in these scenarios expands, more therapeutic targets may become available. In this regard, recent discovery of partial duplication of α7nAChR, known as dup α7, is emerging as a significant factor in macrophage polarization and neuro-inflammation [192]. Additionally, nAChRs may not only provide a therapeutic target but may also function as a diagnostic tool. For example, it has been proposed that imaging of microglia and nAChRs in the living brain may be able to identify the stage of AD [203].
nAChRs - Astroglia
Astroglia as mentioned above also express nAChRs. Indeed, the role and manipulation of astrocytes nAChRs in neurodegenerative diseases such as AD and PD are well documented (Figure 3). Thus, stimulation of these receptors by nicotine or other agonists may suppress synaptotoxicity, amyloidosis, neuroinflammation, and oxidative stress, all of which are strongly implicated in AD and PD [92,204]. Specifically, it was shown that manipulation of α7nAChRs suppressed reactive astrogliosis, release of cytokines such as, IL-6, IL-1β and TNF- α, gliotransmitters such as ATP and glutamate, and potentially, Aβ plaque deposition. Cholinergic projection arising from the nucleus basalis of Meynert (NBM), and projecting to the entire cortical layer, the olfactory tubercle, hippocampus, and the amygdala, has been functionally associated with the control of attention and arousal, both key functions for learning and memory. A role for NBM in pain modulation was also recently reported [205]. Interestingly, astrocytic α7nAChRs, by maintaining Ca2+ homeostasis are considered essential for synaptic plasticity across cortical and hippocampal regions [206,207]. Moreover, stimulation of these receptors on astroglia leads to inhibition of the NF-κB and activation of the Nrf2 pathways, conferring anti-inflammatory effects on astroglia [208,209]. Additionally, potential diagnostic use of α7nAChRs as markers of reactive astrogliosis, at least in AD, has been proposed [207]. Using astrocyte-specific manipulations, it was shown that potentiating astrocyte Ca2+ signaling in the hippocampal CA1 region enhances temporal association, deemed essential for memory formation, whereas attenuation of astrocyte Ca2+ signaling had the opposite effect [94]. Curiously, these effects were mediated primarily by astrocytic α4 containing nAChR subunit. The same subunit was implicated in cognitive enhancement effects of nicotine [94]. Recently, presence of other subunit containing nAChR, including α4 in ventral tegmental area (VTA), an area implicated in reward circuitry was verified, suggesting a potential role for these astrocytic nAChRs in addictive behavior [93]. Thus, the significance of both α4 and α7 containing nAChRs in astrocyte functions are evident (Figure 3). Additionally, like in microglia, astrocytic TRLs also play a pivotal role in sterile inflammation and pathogenesis of neurodegenerative diseases [86]. Thus, further elucidation of specific role of nAChR subtypes in astroglial TRLs may provide more selective and targeted intervention in PD.
In summary, although more studies are required to investigate potential role of specific astroglial nAChRs in neuropsychiatric/neurodegenerative disorders, at this juncture, it may be suggested that activating α7nAChRs in these cells may confer neuroprotection by decreasing inflammation and oxidative stress, whereas stimulation of both α7 and α4 containing nAChRs in astrocytes may be of therapeutic potential in cognitive improvements (Figure 3).
nAChRs – Oligodendrocytes (OLs)
OLs play a substantial role in the central nervous system by forming myelin sheath which is critical in accelerating nerve conduction and maintaining neuronal signaling process. Demyelination, due to death of OLs and loss of myelin sheaths may lead to clinical disorders such as multiple sclerosis (MS), chronic cerebral hypoperfusion, stroke, dementia, and schizophrenia [113]. Increased myelin formation may be brought about by enhancing the signaling between neurons and OLs, which can lead to OPCs proliferation, differentiation, and maturation, as well as integrations of neural pathways [210,211]. Enhanced neuronal activity may be induced directly or indirectly by various methods such as optogenetics, chemogenetics, transcranial stimulation, sensory stimulation [212,213], or potentially via pharmacological manipulation. Regarding the latter, and of relevance to the current topic, the involvement of ACh in myelin formation and the presence of both muscarinic and nAChRs in OLs has been verified. In fact, demyelination disorders are associated with defects in cholinergic anti-inflammatory signaling pathways mediated by α7nAChRs (Figure 3). Interestingly, whereas nicotinic stimulation boosts OPC maturation and myelin regeneration, muscarinic stimulation has the opposite effect in that OPC differentiation and myelin regeneration are retarded [21,214]. Nonetheless, specific impact of nAChRs signaling in relation to OLs function in general, and to PD in particular, has yet to be fully revealed [113]. Moreover, since OLs also express TLRs, elucidation of nAChRs interaction with TLRs in these cells may offer novel therapeutic targets not only in demyelination diseases but also in PD [116].
nAChRs - NG2 cells
NG2-glia are heterogeneous glial cells with distinct properties whose dysfunction can affect neuronal plasticity, leading to neurological and behavioral consequences [125]. Regarding the latter, the role of NG2 cells in stress-related mental disorders was recently reviewed [215]. It was concluded that dissecting the complex biology of NG2 glial cells and further understanding of their causal role in stress response and stress-related psychopathologies, may provide novel interventions in such behavioral disorders [215].
NG2 cells alter their function in response to insults including viral encephalopathy, making them potential targets to prevent the development of epilepsy following viral infection [216]. Although it has been well established that neurons synapse on NG2 cells and have a modulatory role in their development and regeneration, very recently it was revealed that NG2 cells may themselves act as neural progenitor cells. This finding, which was reported in the cortex of adult mouse [217], if verified in human neuronal system, can have a wide implication in providing regenerative interventions in neurodegenerative diseases [217].
As mentioned earlier, NG2 cells may play a critical role in modulation of neuroinflammation [204]. NG2-positive cells co-expressing ionized calcium-binding adaptor molecule 1 (Iba1), were identified in SNpc and striatum of a rat model of PD [218]. This finding together with the observation that ablation of NG2 cells exacerbates dopaminergic neuronal cell loss in a mouse model of PD, suggest that NG2 cells may act as negative regulators of neuroinflammation [204]. Moreover, a critical role of NG2 cells in protecting neurovascular unit and angiogenesis after acute ischemic stroke was recently suggested [123] (Figure 3). It was proposed that exosomes derived from dental pulp stem cells may promote NG2-glia proliferation and differentiation, and hence reduce tissue damage due to acute ischemic stroke [123]. Exosomes are small extracellular vesicles secreted by various stem cells and are potent mediators of intercellular communication and tissue repair [147,219]. Recently, clinical applications of exosomes in acute skin wound healing, orthopedic surgery, neurosurgery, plastic surgery, general surgery, cardiothoracic surgery, urology, head and neck surgery, ophthalmology, and obstetrics and gynecology, and other diseases induced by cancer, ischemia, or inflammation were suggested [220,221].
Regarding neuroinflammation, distinct roles for TLRs as well as nAChRs in neuroinflammatory diseases including PD is well documented (see above). Although yet no TLRs have been identified in NG2 cells, these cell’s possession of α7nAChRs is confirmed [124,125] (Figure 3). Moreover, a recent report implicates striatal NG2-glia in L-dopa induced dyskinesia [222]. Curiously, in a mouse model of preeclampsia, it was reported that nicotine has favorable modifications of the trophoblast-derived exosomes [223]. Exosomes by carrying genetic material such as microRNAs, can regulate cell function and may not only serve as a biomarker of disease state, but may also be of therapeutic potential [219–221,2024]. Therefore, it would be of significant interest to determine whether nAChRs may have a role in exosome production and/or effect, particularly in relation to neurodegenerative diseases.
Thus, further elucidation of potential role of nAChRs as well as interaction of these receptors with TLRRs in NG2 glial cells may provide novel intervention in neurodegenerative and/or neuropsychiatric diseases including PD.
Conclusions
In summary, based on pre-clinical and epidemiological data nicotine is a potential drug for PD. Although the significance of neuronal nAChRs in action of nicotine and other modulators of these receptors are well documented, only relatively recently the significance on non-neuronal nAChRs, particularly those expressed in glial cells have emerged. Given the significance of the glial cells in myriad of synaptic functions and eventual role in neuroinflammation, novel therapeutics targeting these receptors are envisioned. This contention is strengthened by emerging interactions between nicotinic and TLRs, where the latter plays a critical role in neuroinflammatory diseases. Furthermore, recent evidence of glial influence on GBA and potential manipulation of the axis by nAChRs warrant further investigation for their utility in neuropsychiatric/neurodegenerative diseases including PD.
Ethical Approval
Not applicable
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
ENS, SLC and VDAS were supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (n. processes 316590/2020-7, 307539/2018 and 303882/2022-0); RPU was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP: 2016/20796-2 and 2020/04709-8; VDAS was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (Visiting Research Fellowship at Howard University - EDITAL n.º 004/2021-PROPG CAPES-PRINT/UFBA); and YT was supported in part by the National Institutes of Health - NIH/NIAAA R03AA022479 and NIH/NIGMS (2 SO6 GM08016-39).
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization Parkinson Disease Available online:. Available online: https://www.who.int/news-room/fact-sheets/detail/parkinson-disease (accessed on 26 August 2022).
- Snijders, A.H.; Takakusaki, K.; Debu, B.; Lozano, A.M.; Krishna, V.; Fasano, A.; Aziz, T.Z.; Papa, S.M.; Factor, S.A.; Hallett, M. Physiology of Freezing of Gait. Ann Neurol 2016, 80, 644–659. [Google Scholar] [CrossRef]
- Tizabi, Y.; Getachew, B.; Aschner, M. Novel Pharmacotherapies in Parkinson’s Disease. Neurotox Res 2021, 39, 1381–1390. [Google Scholar] [CrossRef]
- Vallés, A.S.; Barrantes, F.J. Nicotinic Acetylcholine Receptor Dysfunction in Addiction and in Some Neurodegenerative and Neuropsychiatric Diseases. Cells 2023, Vol. 12, Page 2051 2023, 12, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The Genetic Background of Parkinson’s Disease: Current Progress and Future Prospects. Acta Neurol Scand 2016, 134, 314–326. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The Genetics of Parkinson Disease. Ageing Res Rev 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P.; Vijayaraghavan, A. Parkinson’s Disease - Genetic Cause. Curr Opin Neurol 2023, 36, 292–301. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sciences 2022, Vol. 12, Page 175 2022, 12, 175. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Pan-Montojo, F.; Reichmann, H. Considerations on the Role of Environmental Toxins in Idiopathic Parkinson’s Disease Pathophysiology. Translational Neurodegeneration 2014 3:1 2014, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Mannervik, B. A Preclinical Model for Parkinson’s Disease Based on Transcriptional Gene Activation via KEAP1/NRF2 to Develop New Antioxidant Therapies. Antioxidants 2023, 12, 673. [Google Scholar] [CrossRef]
- Voon, S.M.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Voon, K.G.L.; Yap, Y.J.; Koh, R.Y. The Mechanism of Action of Salsolinol in Brain: Implications in Parkinson’s Disease. CNS Neurol Disord Drug Targets 2021, 19, 725–740. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J. V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini-Reviews in Medicinal Chemistry 2020, 20, 754–767. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial Dysfunction in Parkinson’s Disease – a Key Disease Hallmark with Therapeutic Potential. Mol Neurodegener 2023, 18, 83. [Google Scholar] [CrossRef]
- Lizama, B.N.; Chu, C.T. Neuronal Autophagy and Mitophagy in Parkinson’s Disease. Mol Aspects Med 2021, 82, 100972. [Google Scholar] [CrossRef]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-First versus Body-First Parkinson’s Disease: A Multimodal Imaging Case-Control Study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef]
- Chen, K.; Wang, H.; Ilyas, I.; Mahmood, A.; Hou, L. Microglia and Astrocytes Dysfunction and Key Neuroinflammation-Based Biomarkers in Parkinson’s Disease. Brain Sci 2023, 13, 634. [Google Scholar] [CrossRef]
- Górska, A.; Markiewicz-Gospodarek, A.; Markiewicz, R.; Chilimoniuk, Z.; Borowski, B.; Trubalski, M.; Czarnek, K. Distribution of Iron, Copper, Zinc and Cadmium in Glia, Their Influence on Glial Cells and Relationship with Neurodegenerative Diseases. Brain Sci 2023, 13, 911. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Pajarillo, E.; Nyarko-Danquah, I.; Aschner, M.; Lee, E. Role of Astrocytes in Parkinson’s Disease Associated with Genetic Mutations and Neurotoxicants. Cells 2023, 12, 622. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, R.; Reid, A.J.; Tata, A.M. Emerging Roles of Cholinergic Receptors in Schwann Cell Development and Plasticity. Biomedicines 2022, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Hustad, E.; Aasly, J.O. Clinical and Imaging Markers of Prodromal Parkinson’s Disease. Front Neurol 2020, 11. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin Geriatr Med 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Prajjwal, P.; Flores Sanga, H.S.; Acharya, K.; Tango, T.; John, J.; Rodriguez, R.S.C.; Dheyaa Marsool Marsool, M.; Sulaimanov, M.; Ahmed, A.; Hussin, O.A. Parkinson’s Disease Updates: Addressing the Pathophysiology, Risk Factors, Genetics, Diagnosis, along with the Medical and Surgical Treatment. Annals of Medicine & Surgery 2023, 85, 4887–4902. [Google Scholar] [CrossRef]
- Quik, M.; Boyd, J.T.; Bordia, T.; Perez, X. Potential Therapeutic Application for Nicotinic Receptor Drugs in Movement Disorders. Nicotine Tob Res 2019, 21, 357–369. [Google Scholar] [CrossRef]
- Carroll, V.; Rossiter, R.; Blanchard, D. Non-Motor Symptoms of Parkinson’s Disease. Aust J Gen Pract 2021, 50, 812–817. [Google Scholar] [CrossRef]
- Onofrj, M.; Russo, M.; Carrarini, C.; Delli Pizzi, S.; Thomas, A.; Bonanni, L.; Espay, A.J.; Sensi, S.L. Functional Neurological Disorder and Somatic Symptom Disorder in Parkinson’s Disease. J Neurol Sci 2022, 433. [Google Scholar] [CrossRef]
- di Biase, L.; Pecoraro, P.M.; Carbone, S.P.; Caminiti, M.L.; Di Lazzaro, V. Levodopa-Induced Dyskinesias in Parkinson’s Disease: An Overview on Pathophysiology, Clinical Manifestations, Therapy Management Strategies and Future Directions. J Clin Med 2023, 12, 4427. [Google Scholar] [CrossRef] [PubMed]
- Bove, F.; Angeloni, B.; Sanginario, P.; Rossini, P.M.; Calabresi, P.; Di Iorio, R. Neuroplasticity in Levodopa-Induced Dyskinesias: An Overview on Pathophysiology and Therapeutic Targets. Prog Neurobiol 2024, 232, 102548. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Pal, G. Initial Management of Parkinson’s Disease. BMJ 2014, 349. [Google Scholar] [CrossRef]
- Tarakad, A.; Jankovic, J. Diagnosis and Management of Parkinson’s Disease. Semin Neurol 2017, 37, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F. Tratamento Da Doença de Parkinson. Arq Neuropsiquiatr 1995, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Malek, N. Deep Brain Stimulation in Parkinson’s Disease. Neurol India 2019, 67, 968. [Google Scholar] [CrossRef] [PubMed]
- Bye, L.J.; Finol-Urdaneta, R.K.; Tae, H.-S.; Adams, D.J. Nicotinic Acetylcholine Receptors: Key Targets for Attenuating Neurodegenerative Diseases. Int J Biochem Cell Biol 2023, 157, 106387. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Mendoza, C.; Iarkov, A. Nicotinic Acetylcholine Receptors and Learning and Memory Deficits in Neuroinflammatory Diseases. Front Neurosci 2023, 17. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front Hum Neurosci 2009, 3, 31. [Google Scholar] [CrossRef]
- Shi, J.; Huang, S. Comparative Insight into Microglia/Macrophages-Associated Pathways in Glioblastoma and Alzheimer’s Disease. Int J Mol Sci 2023, 25, 16. [Google Scholar] [CrossRef]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The Astrocyte Biochemistry. Semin Cell Dev Biol 2019, 95, 142–150. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-Neuron Metabolic Cooperation Shapes Brain Activity. Cell Metab 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Kim, J.D.; Copperi, F.; Diano, S. Microglia in Central Control of Metabolism. Physiology 2024, 39, 5–17. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int J Mol Sci 2023, 24, 17146. [Google Scholar] [CrossRef]
- Fernandes, V.M.; Auld, V.; Klämbt, C. Glia as Functional Barriers and Signaling Intermediaries. Cold Spring Harb Perspect Biol 2024, 16, a041423. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Novikov, N.I.; Brazhnik, E.S.; Kitchigina, V.F. Pathological Correlates of Cognitive Decline in Parkinson’s Disease: From Molecules to Neural Networks. Biochemistry (Moscow) 2023, 88, 1890–1904. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; Mccarthy, K.D.; Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. Journal of Neuroscience 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The Tripartite Glutamatergic Synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef] [PubMed]
- Rasia-Filho, A.A.; Calcagnotto, M.E.; von Bohlen und Halbach, O. Dendritic Spines; Rasia-Filho, A.A., Calcagnotto, M.E., von Bohlen und Halbach, O., Eds.; Springer International Publishing: Cham, 2023; Vol. 34, ISBN 978-3-031-36158-6. [Google Scholar]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide Detoxification by Brain Cells. J Neurosci Res 2005, 79, 157–165. [Google Scholar] [CrossRef]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochemical Research 2014 40:12 2014, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A. V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535. [Google Scholar] [CrossRef]
- Reed, M.M.; Blazer-Yost, B. Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cellular Physiology and Biochemistry 2022, 56, 12–30. [Google Scholar] [CrossRef]
- Ebling, F.J.P.; Lewis, J.E. Tanycytes and Hypothalamic Control of Energy Metabolism. Glia 2018, 66, 1176–1184. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Huang, N.; Xie, Y.; LiCausi, F.; Li, S.; Li, Y.; Sheng, Z.H. Oligodendrocytes Enhance Axonal Energy Metabolism by Deacetylation of Mitochondrial Proteins through Transcellular Delivery of SIRT2. Neuron 2021, 109, 3456–3472. [Google Scholar] [CrossRef]
- Clayton, R.W.; Lovell-Badge, R.; Galichet, C. The Properties and Functions of Glial Cell Types of the Hypothalamic Median Eminence. Front Endocrinol (Lausanne) 2022, 13. [Google Scholar] [CrossRef]
- Kofler, J.; Wiley, C.A. Microglia:Key Innate Immune Cells of the Brain. Toxicol Pathol 2011, 39, 103–114. [Google Scholar] [CrossRef]
- Chen, X.; Holtzman, D.M. Emerging Roles of Innate and Adaptive Immunity in Alzheimer’s Disease. Immunity 2022, 55, 2236–2254. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem Cell Res Ther 2022, 13, 117. [Google Scholar] [CrossRef]
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived Extracellular Vesicles in Homeostasis and Demyelination/Remyelination Processes. J Neurochem 2024, 168, 3–25. [Google Scholar] [CrossRef]
- Rahman, S.; Alzarea, S. Glial Mechanisms Underlying Major Depressive Disorder: Potential Therapeutic Opportunities. 2019; 159–178. [Google Scholar]
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. 2021; 3–19. [Google Scholar]
- Hanslik, K.L.; Marino, K.M.; Ulland, T.K. Modulation of Glial Function in Health, Aging, and Neurodegenerative Disease. Front Cell Neurosci 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhao G Shared and Disease-Specific Glial Gene Expression Changes in Neurodegenerative Diseases. Nat Aging 2023, 3, 246–247. [CrossRef] [PubMed]
- Zhu, H.; Guan, A.; Liu, J.; Peng, L.; Zhang, Z.; Wang, S. Noteworthy Perspectives on Microglia in Neuropsychiatric Disorders. J Neuroinflammation 2023, 20, 223. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Fernandez-Villalba, E.; Izura, V.; Lucas-Ochoa, A.; Menezes-Filho, N.; Santana, R.; de Oliveira, M.; Araújo, F.; Estrada, C.; Silva, V.; et al. Combined 1-Deoxynojirimycin and Ibuprofen Treatment Decreases Microglial Activation, Phagocytosis and Dopaminergic Degeneration in MPTP-Treated Mice. Journal of Neuroimmune Pharmacology 2021, 16, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, Z.; Lu, G.; Chan, W.; Zhang, Y.; Wong, G.C. Microglia Activation, Classification and Microglia-Mediated Neuroinflammatory Modulators in Subarachnoid Hemorrhage. Neural Regen Res 2022, 17, 1404. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct Target Ther 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sriram, K. Molecular Mechanisms Underlying Neuroinflammation Elicited by Occupational Injuries and Toxicants. Int J Mol Sci 2023, 24, 2272. [Google Scholar] [CrossRef] [PubMed]
- Saitgareeva, A.R.; Bulygin, K. V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The Role of Microglia in the Development of Neurodegeneration. Neurological Sciences 2020, 41, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The Dual Face of Microglia (M1/M2) as a Potential Target in the Protective Effect of Nutraceuticals against Neurodegenerative Diseases. Frontiers in Aging 2023, 4. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia Activation in Central Nervous System Disorders: A Review of Recent Mechanistic Investigations and Development Efforts. Front Neurol 2023, 14. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain: Figure 1. The Journal of Neuroscience 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Nimmerjahn, A. Two-Photon Imaging of Microglia in the Mouse Cortex In Vivo. Cold Spring Harb Protoc 2012, 2012, pdb–prot069294. [Google Scholar] [CrossRef]
- Leyh, J.; Paeschke, S.; Mages, B.; Michalski, D.; Nowicki, M.; Bechmann, I.; Winter, K. Classification of Microglial Morphological Phenotypes Using Machine Learning. Front Cell Neurosci 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod Microglia: Elongation, Alignment, and Coupling to Form Trains across the Somatosensory Cortex after Experimental Diffuse Brain Injury. J Neuroinflammation 2012, 9, 247. [Google Scholar] [CrossRef]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS One 2014, 9, e97096. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as Therapeutic Target in Central Nervous System Disorders. J Pharmacol Sci 2020, 144, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int J Mol Sci 2023, 24, 2477. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Meng, H. The Involvement of α-Synucleinopathy in the Disruption of Microglial Homeostasis Contributes to the Pathogenesis of Parkinson’s Disease. Cell Communication and Signaling 2024, 22, 31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, W.; Sun, Y.; Wu, M. New Insight on Microglia Activation in Neurodegenerative Diseases and Therapeutics. Front Neurosci 2023, 17. [Google Scholar] [CrossRef]
- Saramowicz, K.; Siwecka, N.; Galita, G.; Kucharska-Lusina, A.; Rozpędek-Kamińska, W.; Majsterek, I. Alpha-Synuclein Contribution to Neuronal and Glial Damage in Parkinson’s Disease. Int J Mol Sci 2023, 25, 360. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, S.; Kim, H.; Yoon, H.; Ha, S.; Kang, S.U. Molecular Crosstalk Between Circadian Rhythmicity and the Development of Neurodegenerative Disorders. Front Neurosci 2020, 14. [Google Scholar] [CrossRef]
- Guzmán-Ruiz, M.A.; Guerrero-Vargas, N.N.; Lagunes-Cruz, A.; González-González, S.; García-Aviles, J.E.; Hurtado-Alvarado, G.; Mendez-Hernández, R.; Chavarría-Krauser, A.; Morin, J.; Arriaga-Avila, V.; et al. Circadian Modulation of Microglial Physiological Processes and Immune Responses. Glia 2023, 71, 155–167. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like Receptor 4 Is Required for A-synuclein Dependent Activation of Microglia and Astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of Astroglial Toll-like Receptors (TLRs) in Central Nervous System Infections, Injury and Neurodegenerative Diseases. Brain Behav Immun 2021, 91, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Parra-Rivas, L.A.; Madhivanan, K.; Aulston, B.D.; Wang, L.; Prakashchand, D.D.; Boyer, N.P.; Saia-Cereda, V.M.; Branes-Guerrero, K.; Pizzo, D.P.; Bagchi, P.; et al. Serine-129 Phosphorylation of α-Synuclein Is an Activity-Dependent Trigger for Physiologic Protein-Protein Interactions and Synaptic Function. Neuron 2023, 111, 4006–4023. [Google Scholar] [CrossRef]
- Harvey, J.; Pishva, E.; Chouliaras, L.; Lunnon, K. Elucidating Distinct Molecular Signatures of Lewy Body Dementias. Neurobiol Dis 2023, 188, 106337. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-Astrocyte Omnidirectional Signaling in Neurological Health and Disease. Front Mol Neurosci 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front Neurosci 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yang, B.; Chen, Q.; Zhang, J.; Hu, J.; Fan, Y. Activation of A7 Nicotinic Acetylcholine Receptor Protects Against 1-Methyl-4-Phenylpyridinium-Induced Astroglial Apoptosis. Front Cell Neurosci 2019, 13. [Google Scholar] [CrossRef]
- Aryal, S.P.; Fu, X.; Sandin, J.N.; Neupane, K.R.; Lakes, J.E.; Grady, M.E.; Richards, C.I. Nicotine Induces Morphological and Functional Changes in Astrocytes via Nicotinic Receptor Activity. Glia 2021, 69, 2037–2053. [Google Scholar] [CrossRef]
- Gao, R.; Schneider, A.M.; Mulloy, S.M.; Lee, A.M. Expression Pattern of Nicotinic Acetylcholine Receptor Subunit Transcripts in Neurons and Astrocytes in the Ventral Tegmental Area and Locus Coeruleus. European Journal of Neuroscience 2023. [Google Scholar] [CrossRef]
- Ma, W.; Si, T.; Wang, Z.; Wen, P.; Zhu, Z.; Liu, Q.; Wang, J.; Xu, F.; Li, Q. Astrocytic A4-Containing NAChR Signaling in the Hippocampus Governs the Formation of Temporal Association Memory. Cell Rep 2023, 42, 112674. [Google Scholar] [CrossRef]
- Deitmer, J.W. Strategies for Metabolic Exchange between Glial Cells and Neurons. Respir Physiol 2001, 129, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nature Reviews Neuroscience 2018 19:4 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-Synuclein Immunoreactive Astrocytes in the Forebrain Parallels Stages of Intraneuronal Pathology in Sporadic Parkinson’s Disease. Acta Neuropathol 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y. Te; Ureshino, R.P.; et al. Overexpression of α-Synuclein in an Astrocyte Cell Line Promotes Autophagy Inhibition and Apoptosis. J Neurosci Res 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Huenchuguala, S.; Munõz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.F.; Couve, E.; Mannervik, B.; et al. Glutathione Transferase Mu 2 Protects Glioblastoma Cells against Aminochrome Toxicity by Preventing Autophagy and Lysosome Dysfunction. Autophagy 2014, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Huenchuguala, S.; Muñoz, P.; Graumann, R.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Protects Astrocytes from Aminochrome-Induced Toxicity. Neurotoxicology 2016, 55, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione Transferase-M2-2 Secreted from Glioblastoma Cell Protects SH-SY5Y Cells from Aminochrome Neurotoxicity. Neurotox Res 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Valdes, R.; Armijo, A.; Muñoz, P.; Hultenby, K.; Hagg, A.; Inzunza, J.; Nalvarte, I.; Varshney, M.; Mannervik, B.; Segura-Aguilar, J. Cellular Trafficking of Glutathione Transferase M2-2 Between U373MG and SHSY-S7 Cells Is Mediated by Exosomes. Neurotox Res 2021, 39, 182–190. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants 2022, 11, 296. [Google Scholar] [CrossRef]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis 2023, 14, 176. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat Rev Neurol 2023, 19, 395–409. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. Journal of Comparative Neurology 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Q.; Zhang, W.-J.; Su, D.-F.; Zhang, G.-Q.; Miao, C.-Y. Cellular Responses and Functions of A7 Nicotinic Acetylcholine Receptor Activation in the Brain: A Narrative Review. Ann Transl Med 2021, 9, 509–509. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.-P.; Kothary, R. Oligodendrocytes in a Nutshell. Front Cell Neurosci 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A Single-Cell Atlas of the Human Substantia Nigra Reveals Cell-Specific Pathways Associated with Neurological Disorders. Nat Commun 2020, 11, 4183–4183. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Majdan, M.; Awatramani, R.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Genetic and Physiological Evidence That Oligodendrocyte Gap Junctions Contribute to Spatial Buffering of Potassium Released during Neuronal Activity. The Journal of Neuroscience 2006, 26, 10984. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, J. Neuronal Activity and Remyelination: New Insights into the Molecular Mechanisms and Therapeutic Advancements. Front Cell Dev Biol 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Nomden, A.; van Noort, J.M.; Baron, W. Toll-like Receptors 2 and 3 Agonists Differentially Affect Oligodendrocyte Survival, Differentiation, and Myelin Membrane Formation. J Neurosci Res 2012, 90, 388–398. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like Receptors in the Pathogenesis of Neuroinflammation. J Neuroimmunol 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Bae, E.-J.; Pérez-Acuña, D.; Rhee, K.H.; Lee, S.-J. Changes in Oligodendroglial Subpopulations in Parkinson’s Disease. Mol Brain 2023, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Nishiyama, A. NG2 Cells (Polydendrocytes): Listeners to the Neural Network with Diverse Properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Kirdajova, D.; Anderova, M. NG2 Cells and Their Neurogenic Potential. Curr Opin Pharmacol 2020, 50, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-P.; Zhao, J.; Li, S. Roles of NG2 Glial Cells in Diseases of the Central Nervous System. Neurosci Bull 2011, 27, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Gallo, V. <scp>NG</Scp> 2-glia and Their Functions in the Central Nervous System. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood–Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol 2016, 132, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Q.; Yang, Q.; Gu, H.; Yin, Y.; Li, Y.; Hou, J.; Chen, R.; Sun, Q.; Sun, Y.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med 2019, 17, 204. [Google Scholar] [CrossRef]
- Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-Glia Is a Potential Target to Maintain the Integrity of Neurovascular Unit after Acute Ischemic Stroke. Neurobiol Dis 2023, 180, 106076. [Google Scholar] [CrossRef]
- Vélez-Fort, M.; Audinat, E.; Angulo, M.C. Functional A7-containing Nicotinic Receptors of NG2-expressing Cells in the Hippocampus. Glia 2009, 57, 1104–1114. [Google Scholar] [CrossRef]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of <scp>NG2</Scp> Glial Cells Affects Neuronal Plasticity and Behavior. Glia 2023, 71, 1481–1501. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int Rev Neurobiol 2015, 124, 3. [Google Scholar] [CrossRef]
- Papke, R.L.; Lindstrom, J.M. Nicotinic Acetylcholine Receptors: Conventional and Unconventional Ligands and Signaling. Neuropharmacology 2020, 168. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Changeux, J.P. Nicotinic Receptors: From Protein Allostery to Computational Neuropharmacology. Mol Aspects Med 2022, 84. [Google Scholar] [CrossRef]
- Nara, S.; Yamaguti, Y.; Tsuda, I. Review: Nicotinic Acetylcholine Receptors to Regulate Important Brain Activity—What Occurs at the Molecular Level? Cogn Neurodyn 2023. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.; Kudryavtsev, D.; Shelukhina, I.; Nikolaev, G.; Utkin, Y.; Tsetlin, V. Marine Origin Ligands of Nicotinic Receptors: Low Molecular Compounds, Peptides and Proteins for Fundamental Research and Practical Applications. Biomolecules 2022, Vol. 12, Page 189 2022, 12, 189. [Google Scholar] [CrossRef]
- Mugayar, A.A.; da Silva Guimarães, G.; de Oliveira, P.H.T.; Miranda, R.L.; dos Santos, A.A. Apoptosis in the Neuroprotective Effect of A7 Nicotinic Receptor in Neurodegenerative Models. J Neurosci Res 2023, 101, 1795–1802. [Google Scholar] [CrossRef]
- Keever, K.R.; Yakubenko, V.P.; Hoover, D.B. Neuroimmune Nexus in the Pathophysiology and Therapy of Inflammatory Disorders: Role of A7 Nicotinic Acetylcholine Receptors. Pharmacol Res 2023, 191, 106758. [Google Scholar] [CrossRef]
- Sinclair, P.; Kabbani, N. Ionotropic and Metabotropic Responses by Alpha 7 Nicotinic Acetylcholine Receptors. Pharmacol Res 2023, 197, 106975. [Google Scholar] [CrossRef]
- Hone, A.J.; McIntosh, J.M. Nicotinic Acetylcholine Receptors: Therapeutic Targets for Novel Ligands to Treat Pain and Inflammation. Pharmacol Res 2023, 190, 106715. [Google Scholar] [CrossRef]
- Salehi, Z.; Motlagh Ghoochani, B.F.N.; Hasani Nourian, Y.; Jamalkandi, S.A.; Ghanei, M. The Controversial Effect of Smoking and Nicotine in SARS-CoV-2 Infection. Allergy, Asthma & Clinical Immunology 2023, 19, 49. [Google Scholar] [CrossRef]
- Shelukhina, I.; Siniavin, A.; Kasheverov, I.; Ojomoko, L.; Tsetlin, V.; Utkin, Y. A7- and A9-Containing Nicotinic Acetylcholine Receptors in the Functioning of Immune System and in Pain. Int J Mol Sci 2023, 24, 6524. [Google Scholar] [CrossRef] [PubMed]
- Wills, L.; Ables, J.L.; Braunscheidel, K.M.; Caligiuri, S.P.B.; Elayouby, K.S.; Fillinger, C.; Ishikawa, M.; Moen, J.K.; Kenny, P.J. Neurobiological Mechanisms of Nicotine Reward and Aversion. Pharmacol Rev 2022, 74, 271–310. [Google Scholar] [CrossRef]
- Kim, K.; Picciotto, M.R. Nicotine Addiction: More than Just Dopamine. Curr Opin Neurobiol 2023, 83, 102797. [Google Scholar] [CrossRef]
- Sansone, L.; Milani, F.; Fabrizi, R.; Belli, M.; Cristina, M.; Zagà, V.; de Iure, A.; Cicconi, L.; Bonassi, S.; Russo, P. Nicotine: From Discovery to Biological Effects. Int J Mol Sci 2023, 24, 14570. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Getachew, B.; Csoka, A.B.; Manaye, K.F.; Copeland, R.L. Novel Targets for Parkinsonism-Depression Comorbidity. Prog Mol Biol Transl Sci 2019, 167, 1–24. [Google Scholar] [CrossRef]
- Liu, C. Targeting the Cholinergic System in Parkinson’s Disease. Acta Pharmacol Sin 2020, 41, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gandelman, J.A.; Newhouse, P.; Taylor, W.D. Nicotine and Networks: Potential for Enhancement of Mood and Cognition in Late-Life Depression. Neurosci Biobehav Rev 2018, 84, 289–298. [Google Scholar] [CrossRef]
- Conti, A.A.; Tolomeo, S.; Steele, J.D.; Baldacchino, A.M. Severity of Negative Mood and Anxiety Symptoms Occurring during Acute Abstinence from Tobacco: A Systematic Review and Meta-Analysis. Neurosci Biobehav Rev 2020, 115, 48–63. [Google Scholar] [CrossRef]
- Koukouli, F.; Changeux, J.P. Do Nicotinic Receptors Modulate High-Order Cognitive Processing? Trends Neurosci 2020, 43, 550–564. [Google Scholar] [CrossRef]
- Nop, O.; Senft Miller, A.; Culver, H.; Makarewicz, J.; Dumas, J.A. Nicotine and Cognition in Cognitively Normal Older Adults. Front Aging Neurosci 2021, 13. [Google Scholar] [CrossRef]
- Hahn, B.; Harvey, A.N.; Concheiro-Guisan, M.; Huestis, M.A.; Ross, T.J.; Stein, E.A. Nicotinic Receptor Modulation of the Default Mode Network. Psychopharmacology (Berl) 2021, 238, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, T.; Zhao, C.; Li, G. The Regulation of Exosome Generation and Function in Physiological and Pathological Processes. Int J Mol Sci 2023, 25, 255. [Google Scholar] [CrossRef]
- Han, T.; Wang, Q.; Lai, R.; Zhang, D.; Diao, Y.; Yin, Y. Nicotine Induced Neurocognitive Protection and Anti-Inflammation Effect by Activating α 4β 2 Nicotinic Acetylcholine Receptors in Ischemic Rats. Nicotine Tob Res 2020, 22, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Ionov, I.D.; Pushinskaya, I.I.; Gorev, N.P.; Frenkel, D.D.; Severtsev, N.N. Anticataleptic Activity of Nicotine in Rats: Involvement of the Lateral Entorhinal Cortex. Psychopharmacology (Berl) 2021, 238, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Terry, A. V.; Callahan, P.M. A7 Nicotinic Acetylcholine Receptors as Therapeutic Targets in Schizophrenia: Update on Animal and Clinical Studies and Strategies for the Future. Neuropharmacology 2020, 170, 108053. [Google Scholar] [CrossRef] [PubMed]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on A7 NAChRs. Curr Neuropharmacol 2018, 16, 415. [Google Scholar] [CrossRef] [PubMed]
- Seoane-Collazo, P.; Diéguez, C.; Nogueiras, R.; Rahmouni, K.; Fernández-Real, J.M.; López, M. Nicotine’ Actions on Energy Balance: Friend or Foe? Pharmacol Ther 2021, 219. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, X.; Kenny, P.J. Central and Peripheral Actions of Nicotine That Influence Blood Glucose Homeostasis and the Development of Diabetes. Pharmacol Res 2023, 194, 106860. [Google Scholar] [CrossRef]
- Pechlivanidou, M.; Ninou, E.; Karagiorgou, K.; Tsantila, A.; Mantegazza, R.; Francesca, A.; Furlan, R.; Dudeck, L.; Steiner, J.; Tzartos, J.; et al. Autoimmunity to Neuronal Nicotinic Acetylcholine Receptors. Pharmacol Res 2023, 192, 106790. [Google Scholar] [CrossRef]
- Arnold, E.C.; Soler-Llavina, G.; Kambara, K.; Bertrand, D. The Importance of Ligand Gated Ion Channels in Sleep and Sleep Disorders. Biochem Pharmacol 2023, 212, 115532. [Google Scholar] [CrossRef]
- Lunney, P.C.; Leong, R.W.L. Review Article: Ulcerative Colitis, Smoking and Nicotine Therapy. Aliment Pharmacol Ther 2012, 36, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Morioka, N.; Hisaoka-Nakashima, K.; Nakata, Y. Regulation by Nicotinic Acetylcholine Receptors of Microglial Glutamate Transporters: Role of Microglia in Neuroprotection. Nicotinic Acetylcholine Receptor Signaling in Neuroprotection 2018, 73–82. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, H.; Zou, M.; Yuan, Q.; Huang, Z.; Pan, X.; Zhang, W. Nicotine in Inflammatory Diseases: Anti-Inflammatory and Pro-Inflammatory Effects. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Matsuo, K. Nicotine Exerts a Stronger Immunosuppressive Effect Than Its Structural Analogs and Regulates Experimental Colitis in Rats. Biomedicines 2023, 11, 922. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Auyeung, K. Inflammatory Bowel Disease: Etiology, Pathogenesis and Current Therapy. Curr Pharm Des 2014, 20, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.P.; Watad, A.; Shoenfeld, Y. Nicotine and Autoimmunity: The Lotus’ Flower in Tobacco. Pharmacol Res 2018, 128, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Getachew, B.; Copeland, R.L.; Aschner, M. Nicotine and the Nicotinic Cholinergic System in COVID-19. FEBS J 2020, 287, 3656–3663. [Google Scholar] [CrossRef] [PubMed]
- Bele, T.; Turk, T.; Križaj, I. Nicotinic Acetylcholine Receptors in Cancer: Limitations and Prospects. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2024, 1870, 166875. [Google Scholar] [CrossRef]
- Copeland, R.L.; Leggett, Y.A.; Kanaan, Y.M.; Taylor, R.E.; Tizabi, Y. Neuroprotective Effects of Nicotine against Salsolinol-Induced Cytotoxicity: Implications for Parkinson’s Disease. Neurotox Res 2005, 8, 289–293. [Google Scholar] [CrossRef]
- Getachew, B.; Csoka, A.B.; Bhatti, A.; Copeland, R.L.; Tizabi, Y. Butyrate Protects Against Salsolinol-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox Res 2020, 38, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Getachew, B.; Aschner, M. Butyrate Protects and Synergizes with Nicotine against Iron- and Manganese-Induced Toxicities in Cell Culture. Neurotox Res 2024, 42, 3. [Google Scholar] [CrossRef] [PubMed]
- Getachew, B.; Csoka, A.B.; Aschner, M.; Tizabi, Y. Nicotine Protects against Manganese and Iron-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurochem Int 2019, 124, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Cuevas, C.; Villa, M.; Caviedes, P.; Segura-Aguilar, J.; Tizabi, Y. Protective Effects of Nicotine Against Aminochrome-Induced Toxicity in Substantia Nigra Derived Cells: Implications for Parkinson’s Disease. Neurotox Res 2012, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, X.; Zhou, X.; Wu, X.; Li, Y.; Yao, J.; Bai, J. Nicotine Suppresses the Neurotoxicity by MPP +/MPTP through Activating A7nAChR/PI3K/Trx-1 and Suppressing ER Stress. Neurotoxicology 2017, 59, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bi, W.; Zheng, K.; Zhu, E.; Wang, S.; Xiong, Y.; Chang, J.; Jiang, J.; Liu, B.; Lu, Z.; et al. Nicotine Prevents Oxidative Stress-Induced Hippocampal Neuronal Injury Through A7-NAChR/Erk1/2 Signaling Pathway. Front Mol Neurosci 2020, 13. [Google Scholar] [CrossRef]
- Ruan, S.; Xie, J.; Wang, L.; Guo, L.; Li, Y.; Fan, W.; Ji, R.; Gong, Z.; Xu, Y.; Mao, J.; et al. Nicotine Alleviates MPTP-Induced Nigrostriatal Damage through Modulation of JNK and ERK Signaling Pathways in the Mice Model of Parkinson’s Disease. Front Pharmacol 2023, 14. [Google Scholar] [CrossRef]
- Fares, M.B.; Alijevic, O.; Johne, S.; Overk, C.; Hashimoto, M.; Kondylis, A.; Adame, A.; Dulize, R.; Peric, D.; Nury, C.; et al. Nicotine-Mediated Effects in Neuronal and Mouse Models of Synucleinopathy. Front Neurosci 2023, 17. [Google Scholar] [CrossRef]
- Lai, J.Ic.; Porcu, A.; Romoli, B.; Keisler, M.; Manfredsson, F.P.; Powell, S.B.; Dulcis, D. Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model. Int J Mol Sci 2023, 24, 4204. [Google Scholar] [CrossRef]
- Liu, Q.; Emadi, S.; Shen, J.-X.; Sierks, M.R.; Wu, J. Human A4β2 Nicotinic Acetylcholine Receptor as a Novel Target of Oligomeric α-Synuclein. PLoS One 2013, 8, e55886. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Anti-Fibrillogenic and Fibril-Destabilizing Activity of Nicotine in Vitro: Implications for the Prevention and Therapeutics of Lewy Body Diseases. Exp Neurol 2007, 205, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hirohata, M.; Yamada, M. Alpha-Synuclein Assembly as a Therapeutic Target of Parkinson’s Disease and Related Disorders. Curr Pharm Des 2008, 14, 3247–3266. [Google Scholar] [CrossRef] [PubMed]
- Kardani, J.; Sethi, R.; Roy, I. Nicotine Slows down Oligomerisation of α-Synuclein and Ameliorates Cytotoxicity in a Yeast Model of Parkinson’s Disease. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 1454–1463. [Google Scholar] [CrossRef]
- Bono, F.; Mutti, V.; Savoia, P.; Barbon, A.; Bellucci, A.; Missale, C.; Fiorentini, C. Nicotine Prevents Alpha-Synuclein Accumulation in Mouse and Human IPSC-Derived Dopaminergic Neurons through Activation of the Dopamine D3- Acetylcholine Nicotinic Receptor Heteromer. Neurobiol Dis 2019, 129, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, Y.; Li, Y.; Xu, S.; Tao, T.; Hua, Y.; Zhang, J.; Fan, Y. Activation of A7-NAChRs Promotes the Clearance of α-Synuclein and Protects Against Apoptotic Cell Death Induced by Exogenous α-Synuclein Fibrils. Front Cell Dev Biol 2021, 9, 315. [Google Scholar] [CrossRef]
- Elgayar, S.A.M.; Hussein, O.A.; Mubarak, H.A.; Ismaiel, A.M.; Gomaa, A.M.S. Testing Efficacy of the Nicotine Protection of the Substantia Nigra Pars Compacta in a Rat Parkinson Disease Model. Ultrastructure Study. Ultrastruct Pathol 2022, 46, 37–53. [Google Scholar] [CrossRef]
- Kalsoom, I.; Wang, Y.; Li, B.; Wen, H. Research Progress of α-Synuclein Aggregation Inhibitors for Potential Parkinson’s Disease Treatment. Mini-Reviews in Medicinal Chemistry 2023, 23, 1959–1974. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Magen, I.; Bove, N.; Zhu, C.; Lemesre, V.; Dutta, G.; Elias, C.J.; Lester, H.A.; Chesselet, M.F. Chronic Nicotine Improves Cognitive and Social Impairment in Mice Overexpressing Wild Type α-Synuclein. Neurobiol Dis 2018, 117, 170–180. [Google Scholar] [CrossRef]
- Fan, T.-S.; Liu, S.C.-H.; Wu, R.-M. Alpha-Synuclein and Cognitive Decline in Parkinson Disease. Life 2021, 11, 1239. [Google Scholar] [CrossRef]
- Camilo Jurado-Coronel, J.; Avila-Rodriguez, M.; Capani, F.; Gonzalez, J.; Echeverria Moran, V.; E. Barreto, G. Targeting the Nicotinic Acetylcholine Receptors (NAChRs) in Astrocytes as a Potential Therapeutic Target in Parkinson’s Disease. Curr Pharm Des 2016, 22, 1305–1311. [Google Scholar] [CrossRef]
- Olsen, A.L.; Clemens, S.G.; Feany, M.B. Nicotine-Mediated Rescue of A-Synuclein Toxicity Requires Synaptic Vesicle Glycoprotein 2 in Drosophila. Movement Disorders 2023, 38, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Getachew, B. Nicotinic Receptor Intervention in Parkinson’s Disease: Future Directions. Clin Pharmacol Transl Med 2017, 1, 14. [Google Scholar] [PubMed]
- Mitra, S.; Khatri, S.N.; Maulik, M.; Bult-Ito, A.; Schulte, M. Allosterism of Nicotinic Acetylcholine Receptors: Therapeutic Potential for Neuroinflammation Underlying Brain Trauma and Degenerative Disorders. Int J Mol Sci 2020, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Manetti, D.; Dei, S.; Arias, H.R.; Braconi, L.; Gabellini, A.; Teodori, E.; Romanelli, M.N. Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators. Molecules 2023, 28, 1270. [Google Scholar] [CrossRef] [PubMed]
- Sanders, V.R.; Millar, N.S. Potentiation and Allosteric Agonist Activation of A7 Nicotinic Acetylcholine Receptors: Binding Sites and Hypotheses. Pharmacol Res 2023, 191, 106759. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The Cholinergic Anti-Inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol Med 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B. Cholinergic Modulation of the Immune System Presents New Approaches for Treating Inflammation. Pharmacol Ther 2017, 179, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Roa-Vidal, N.; Rodríguez-Aponte, A.S.; Lasalde-Dominicci, J.A.; Capó-Vélez, C.M.; Delgado-Vélez, M. Cholinergic Polarization of Human Macrophages. Int J Mol Sci 2023, 24, 15732. [Google Scholar] [CrossRef]
- Tracey, K.J. Fat Meets the Cholinergic Antiinflammatory Pathway. J Exp Med 2005, 202, 1017–1021. [Google Scholar] [CrossRef]
- Rueda Ruzafa, L.; Cedillo, J.L.; Hone, A.J. Nicotinic Acetylcholine Receptor Involvement in Inflammatory Bowel Disease and Interactions with Gut Microbiota. Int J Environ Res Public Health 2021, 18, 1189. [Google Scholar] [CrossRef]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10, 1838. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Traina, G. Neuroinflammation in the Brain and Role of Intestinal Microbiota: An Overview of the Players. J Integr Neurosci 2023, 22, 148. [Google Scholar] [CrossRef] [PubMed]
- Claudino dos Santos, J.C.; Lima, M.P.P.; Brito, G.A. de C.; Viana, G.S. de B. Role of Enteric Glia and Microbiota-Gut-Brain Axis in Parkinson Disease Pathogenesis. Ageing Res Rev 2023, 84, 101812. [Google Scholar] [CrossRef] [PubMed]
- Pan, I.; Issac, P.K.; Rahman, Md.M.; Guru, A.; Arockiaraj, J. Gut-Brain Axis a Key Player to Control Gut Dysbiosis in Neurological Diseases. Mol Neurobiol 2023. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Sokratian, A.; Chavez, K.R.; King, S.; Swain, S.M.; Snyder, J.C.; West, A.B.; Liddle, R.A. Gut Mucosal Cells Transfer α-Synuclein to the Vagus Nerve. JCI Insight 2023, 8. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.; Rizza, S.; Federici, M. Microbiota-Gut-Brain Axis: Relationships among the Vagus Nerve, Gut Microbiota, Obesity, and Diabetes. Acta Diabetol 2023, 60, 1007–1017. [Google Scholar] [CrossRef]
- Takbiri Osgoei, L.; Parivar, K.; Ebrahimi, M.; Mortaz, E. Nicotine Modulates the Release of Inflammatory Cytokines and Expression of TLR2, TLR4 of Cord Blood Mononuclear Cells. Iran J Allergy Asthma Immunol 2018. [Google Scholar] [CrossRef]
- Randall, C.A.; Sun, D.; Randall, P.A. Differential Effects of Nicotine, Alcohol, and Coexposure on Neuroimmune-Related Protein and Gene Expression in Corticolimbic Brain Regions of Rats. ACS Chem Neurosci 2023, 14, 628–644. [Google Scholar] [CrossRef]
- Takata, K.; Kimura, H.; Yanagisawa, D.; Harada, K.; Nishimura, K.; Kitamura, Y.; Shimohama, S.; Tooyama, I. Nicotinic Acetylcholine Receptors and Microglia as Therapeutic and Imaging Targets in Alzheimer’s Disease. Molecules 2022, 27, 2780. [Google Scholar] [CrossRef]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential Astrocytic Receptors and Transporters in the Pathogenesis of Alzheimer’s Disease. Journal of Alzheimer’s Disease 2019, 67, 1109–1122. [Google Scholar] [CrossRef]
- Oswald, M.J.; Han, Y.; Li, H.; Marashli, S.; Oglo, D.N.; Ojha, B.; Naser, P. V.; Gan, Z.; Kuner, R. Cholinergic Basal Forebrain Nucleus of Meynert Regulates Chronic Pain-like Behavior via Modulation of the Prelimbic Cortex. Nat Commun 2022, 13, 5014. [Google Scholar] [CrossRef]
- Koulousakis, P.; Andrade, P.; Visser-Vandewalle, V.; Sesia, T. The Nucleus Basalis of Meynert and Its Role in Deep Brain Stimulation for Cognitive Disorders: A Historical Perspective. Journal of Alzheimer’s Disease 2019, 69, 905–919. [Google Scholar] [CrossRef]
- Fontana, I.C.; Kumar, A.; Nordberg, A. The Role of Astrocytic A7 Nicotinic Acetylcholine Receptors in Alzheimer Disease. Nat Rev Neurol 2023, 19, 278–288. [Google Scholar] [CrossRef]
- Patel, H.; McIntire, J.; Ryan, S.; Dunah, A.; Loring, R. Anti-Inflammatory Effects of Astroglial A7 Nicotinic Acetylcholine Receptors Are Mediated by Inhibition of the NF-ΚB Pathway and Activation of the Nrf2 Pathway. J Neuroinflammation 2017, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on A7 Nicotinic Receptor. Int J Mol Sci 2021, 22, 4912. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.A.; Hughes, E.G. Neuron-Oligodendroglia Interactions: Activity-Dependent Regulation of Cellular Signaling. Neurosci Lett 2020, 727, 134916. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Shu, S.; Zhai, L.; Xia, S.; Cao, X.; Li, H.; Bao, X.; Liu, P.; Xu, Y. Optogenetic Stimulation of MPFC Alleviates White Matter Injury-Related Cognitive Decline after Chronic Ischemia through Adaptive Myelination. Advanced Science 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.-J.; Kukley, M. Different Patterns of Neuronal Activity Trigger Distinct Responses of Oligodendrocyte Precursor Cells in the Corpus Callosum. PLoS Biol 2017, 15, e2001993. [Google Scholar] [CrossRef] [PubMed]
- Maas, D.A.; Angulo, M.C. Can Enhancing Neuronal Activity Improve Myelin Repair in Multiple Sclerosis? Front Cell Neurosci 2021, 15. [Google Scholar] [CrossRef]
- Imamura, O.; Arai, M.; Dateki, M.; Oishi, K.; Takishima, K. Donepezil-induced Oligodendrocyte Differentiation Is Mediated through Estrogen Receptors. J Neurochem 2020, 155, 494–507. [Google Scholar] [CrossRef]
- Poggi, G.; Wennström, M.; Müller, M.B.; Treccani, G. NG2-Glia: Rising Stars in Stress-Related Mental Disorders? Mol Psychiatry 2023, 28, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Reactivity and Increased Proliferation of NG2 Cells Following Central Nervous System Infection with Theiler’s Murine Encephalomyelitis Virus. J Neuroinflammation 2020, 17, 369. [Google Scholar] [CrossRef] [PubMed]
- Janeckova, L.; Knotek, T.; Kriska, J.; Hermanova, Z.; Kirdajova, D.; Kubovciak, J.; Berkova, L.; Tureckova, J.; Camacho Garcia, S.; Galuskova, K.; et al. Astrocyte-like Subpopulation of <scp>NG2</Scp> Glia in the Adult Mouse Cortex Exhibits Characteristics of Neural Progenitor Cells. Glia 2024, 72, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Inden, M.; Minamino, H.; Abe, M.; Takata, K.; Taniguchi, T. The 6-Hydroxydopamine-Induced Nigrostriatal Neurodegeneration Produces Microglia-like NG2 Glial Cells in the Rat Substantia Nigra. Glia 2010, 58, 1686–1700. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology , Function , and Biomedical Applications of Exosomes. Science (1979) 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, M.; Liu, Q.; Yu, A.; Cheng, K.; Ma, J.; Murphy, S.; McNutt, P.M.; Zhang, Y. Insights into Optimizing Exosome Therapies for Acute Skin Wound Healing and Other Tissue Repair. Front Med 2024. [Google Scholar] [CrossRef]
- Tan, F.; Li, X.; Wang, Z.; Li, J.; Shahzad, K.; Zheng, J. Clinical Applications of Stem Cell-Derived Exosomes. Signal Transduct Target Ther 2024, 9, 17. [Google Scholar] [CrossRef]
- Nascimento, G.C.; Bortolanza, M.; Bribian, A.; Leal-Luiz, G.C.; Raisman-Vozari, R.; López-Mascaraque, L.; Del-Bel, E. Dynamic Involvement of Striatal NG2-Glia in L-DOPA Induced Dyskinesia in Parkinsonian Rats: Effects of Doxycycline. ASN Neuro 2023, 15, 175909142311559. [Google Scholar] [CrossRef]
- Kubo, A.; Matsubara, K.; Matsubara, Y.; Nakaoka, H.; Sugiyama, T. The Influence of Nicotine on Trophoblast-Derived Exosomes in a Mouse Model of Pathogenic Preeclampsia. Int J Mol Sci 2023, 24, 11126. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y. Emerging roles of O-GlcNAcylation in protein trafficking and secretion. J Biol Chem 2024, 23, 105677. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of glial-neuron interaction. Glial cells are essential for maintaining neuronal funtion and brain homeostasis. 4 types are depicted here. 1. Microglia act as innate immune cells in the brain and are critical in defense response. However, their disruption (overactivation) can lead to neuroinflammation and neurodegeneration. 2. Astrocytes perform several functions including providing energy support for neurons, angiogenesis, neurogenesis, and synaptic modulation, disruption of which results in blood-brain-barrier (BBB) damage. 3. Oligodendrocytes are pivotal for myelination and produce neurotrophic factors such as brain-derived neuroptrophic factor (BDNF) and glial-dervided neurotrophic factor (GDNF). Dysfunction in these cells results in demyelination. Synantocytes (NG2 cells), are oligodendrocyte progenitor cells and contribute to neurovascular maintenance and myelination. Their disruption leads to various damages including neuroinflammation and neuronal death. Figure created with BioRender.com.
Figure 1.
Schematic diagram of glial-neuron interaction. Glial cells are essential for maintaining neuronal funtion and brain homeostasis. 4 types are depicted here. 1. Microglia act as innate immune cells in the brain and are critical in defense response. However, their disruption (overactivation) can lead to neuroinflammation and neurodegeneration. 2. Astrocytes perform several functions including providing energy support for neurons, angiogenesis, neurogenesis, and synaptic modulation, disruption of which results in blood-brain-barrier (BBB) damage. 3. Oligodendrocytes are pivotal for myelination and produce neurotrophic factors such as brain-derived neuroptrophic factor (BDNF) and glial-dervided neurotrophic factor (GDNF). Dysfunction in these cells results in demyelination. Synantocytes (NG2 cells), are oligodendrocyte progenitor cells and contribute to neurovascular maintenance and myelination. Their disruption leads to various damages including neuroinflammation and neuronal death. Figure created with BioRender.com.
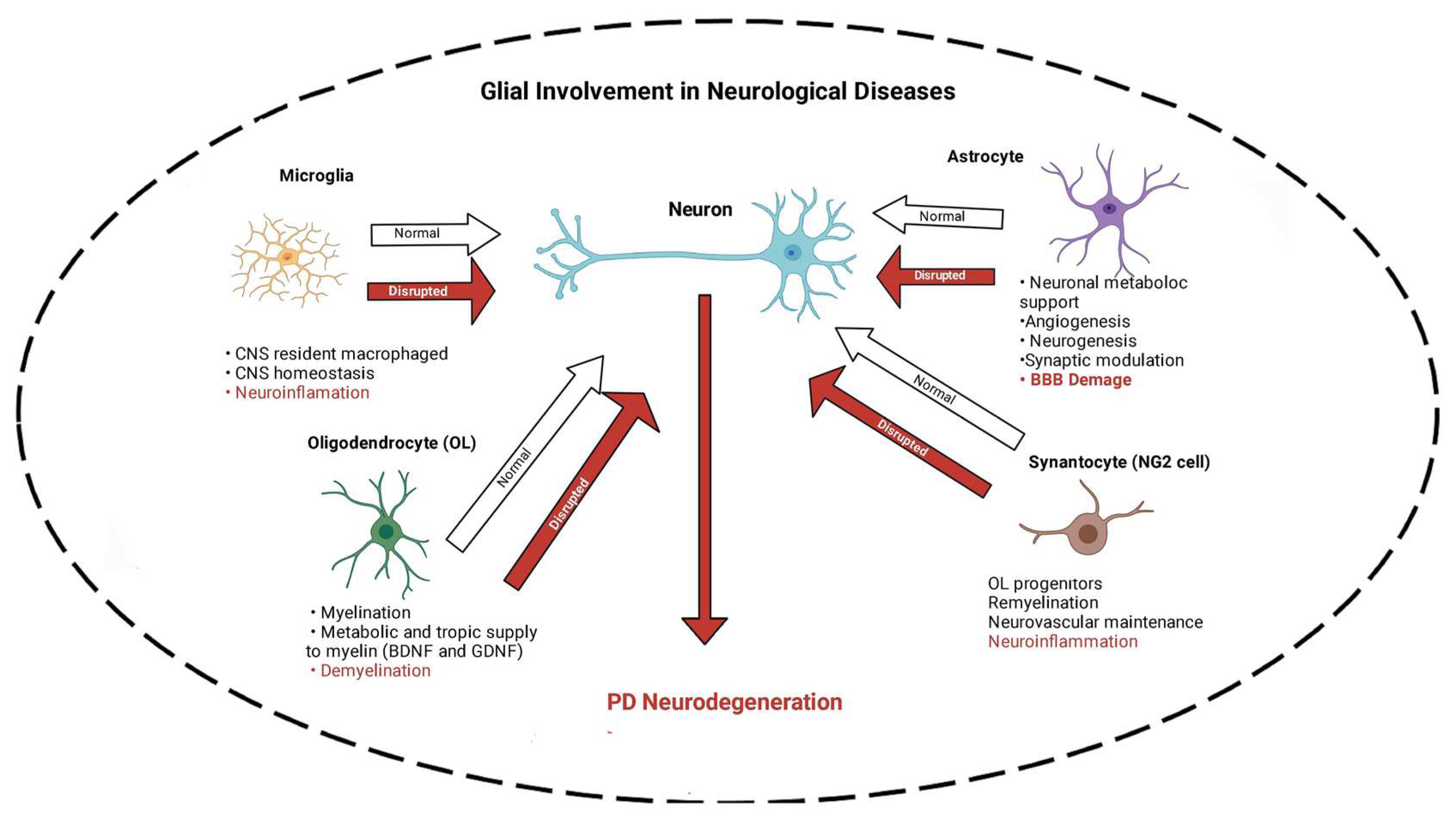
Figure 2.
Schematic diagram of gut-brain axis and cholinegic anti-inflmmatory pathway. Dysbiosis and increased intestinal permeability cause reactivity of enteric nervous system including enteric glial cells. This can result in Figure 7. nAChRs on peripheral macrophages. Figure created with BioRender.com.
Figure 2.
Schematic diagram of gut-brain axis and cholinegic anti-inflmmatory pathway. Dysbiosis and increased intestinal permeability cause reactivity of enteric nervous system including enteric glial cells. This can result in Figure 7. nAChRs on peripheral macrophages. Figure created with BioRender.com.
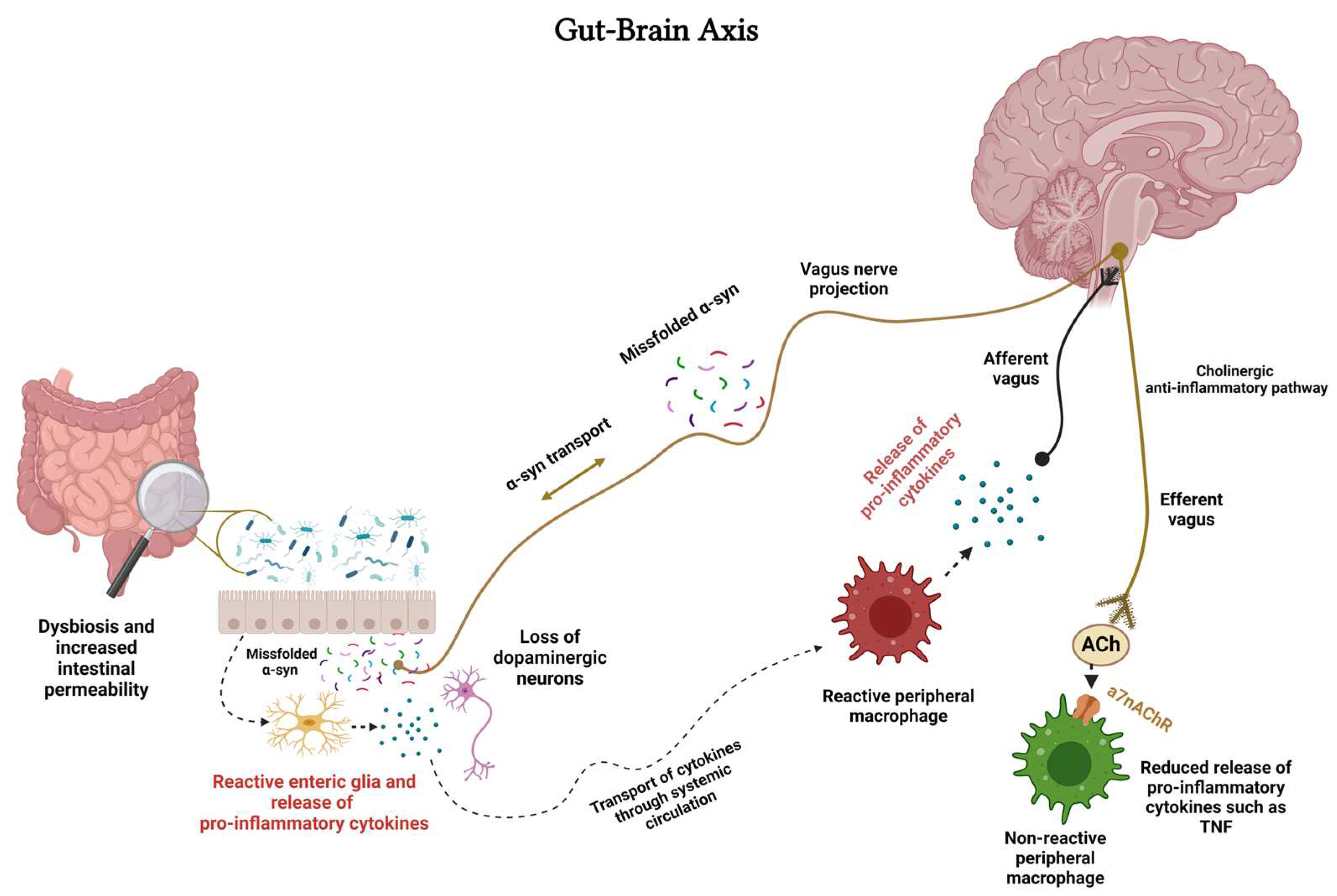
Figure 3.
Scematic diagram of nicotinic acetylcholine receptors (nAChRs) on glial and neuronal cells and consequence of activation by nicotine. Activation of selective nAChR in microglia and astrocytes results in an anti-inflammatory response. In oligodendrocytes (OLs), stimulation of nAChRs may result in remyelination and in NG2 cells, to anti-inflammatory response, although the latter has yet to be verified. Figure created with BioRender.com.
Figure 3.
Scematic diagram of nicotinic acetylcholine receptors (nAChRs) on glial and neuronal cells and consequence of activation by nicotine. Activation of selective nAChR in microglia and astrocytes results in an anti-inflammatory response. In oligodendrocytes (OLs), stimulation of nAChRs may result in remyelination and in NG2 cells, to anti-inflammatory response, although the latter has yet to be verified. Figure created with BioRender.com.
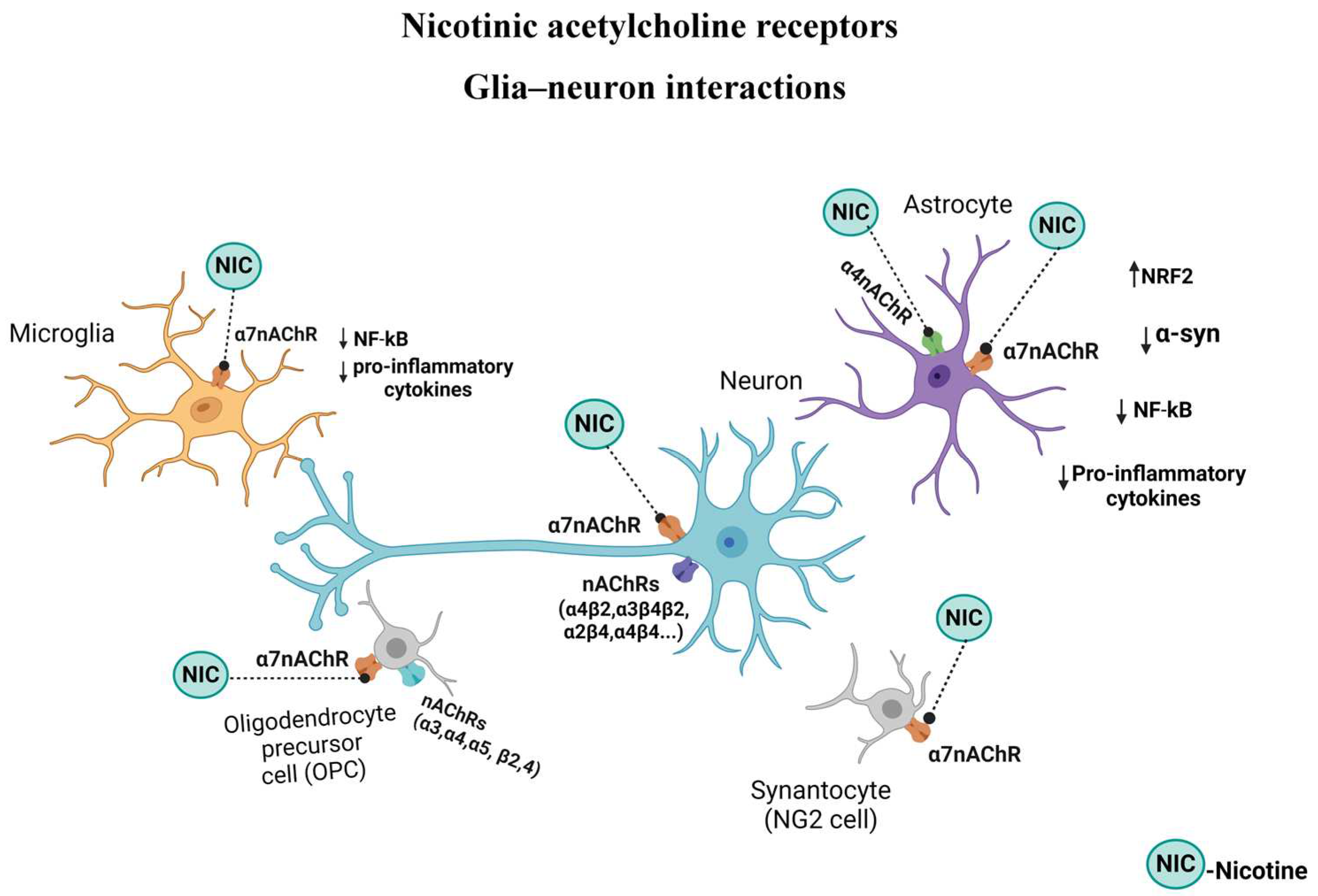
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



