
Submitted:
18 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Alveolar soft part sarcoma (ASPS) is a rare mesenchymal neoplasm. It affects young adults more and occurs more frequently in the extremities. ASPS is exceedingly rare in the nasal cavity. We report a case of ASPS in the nasal cavity of a 17-year-old female. Its most striking features included organoid and pseudoalveolar arrangement of tumor cells, which were large and polygonal, with vesicular nuclei and abundant eosinophilic cytoplasm rich in granules. There were PAS-positive, diastase-resistant crystalline rods within the cytoplasm. Our case displayed inapparent pseudoalveolar growth pattern and frank pleomorphism. The diagnosis of ASPS, which presents uncommon morphologic findings in unusual sites, is challenging. Recognition of ASPS is greatly assisted by attention to its characteristic cytological features, including intracytoplasmic crystals and immunohistochemistry showing absence of skeletal muscle, epithelial, and neuroendocrine markers.
Keywords:
alveolar soft part sarcoma
; nasal cavity
; pathology
; TFE3
1. Introduction
Alveolar soft part sarcoma (ASPS) is a rare malignant tumor of uncertain lineage. It has characteristic histological and genetic features [1,2]. ASPS mainly affects adolescents and young adults (15-35 years of age, median age 25 years), with a female predominance. ASPS predominantly occurs in extremities and trunk [3,4]. Although it is also known to occur in the head and neck [3,4,5,6,7,8,9,10,11,12], especially in children, sinonasal ASPS is exceedingly rare [5,6,7,8,9,10]. Only five cases in nasal cavity have been reported to date [6,7,8,9,10].
ASPS typically shows a nesting and pseudoalveolar growth pattern. Its epithelioid tumor cells have a large eosinophilic cytoplasm with periodic acid-Schiff (PAS)-positive, diastase-resistant crystals. In some cases, the peudoalveolar pattern as a result of loss of cell cohesion within tumor nests may be lost and the tumor is composed of sheets of epithelioid cells, which can result in diagnostic confusion, especially for tumors at unusual sites [5,10]. A portion of the tumor demonstrates histologic features atypical for ASPS [3,5]. It is characterized by nuclear pleomorphism, increased nuclear hyperchromatism, increased nuclear-cytoplasmic ratio, and decreased cytoplasmic eosinophilia.
We describe an extremely rare case of ASPS of nasal cavity lacking pseudoalveolar growth pattern and showing atypical cytomorphologic features in a 17-year-old female. It is important to perform an accurate diagnosis of ASPS based on its typical cytologic findings described above. ASPSCR1-TFE3 gene fusion is highly specific for ASPS. A anti-TFE3 antibody has proven to be of great value in confirming the diagnosis of ASPS [13]. It was also positive in our case.
2. Case Report
A 17-year-old female patient presented with a 2-year history of right nasal obstruction. She was currently taking medication for attention deficit hyperactivity disorder (ADHD) and depression. She had been treated for precocious puberty seven years ago. She had a history of traumatic cerebral hemorrhage 14 years ago. In addition, she had a family history of pharyngeal cancer in her grandfather. Nasopharyngoscopy revealed a polypoid mass in the right posterior nasal cavity (Figure 1). Paranasal sinus magnetic resonance imaging (PNS MRI) demonstrated a well-defined enhancing mass at the right nasal cavity, involving middle and inferior meatus, measuring 3x3x1.6 cm in dimensions. The mass exhibited medium signal intensity on T2 weighted image (Figure 2). These images were interpreted as juvenile nasopharyngeal angiofibroma.
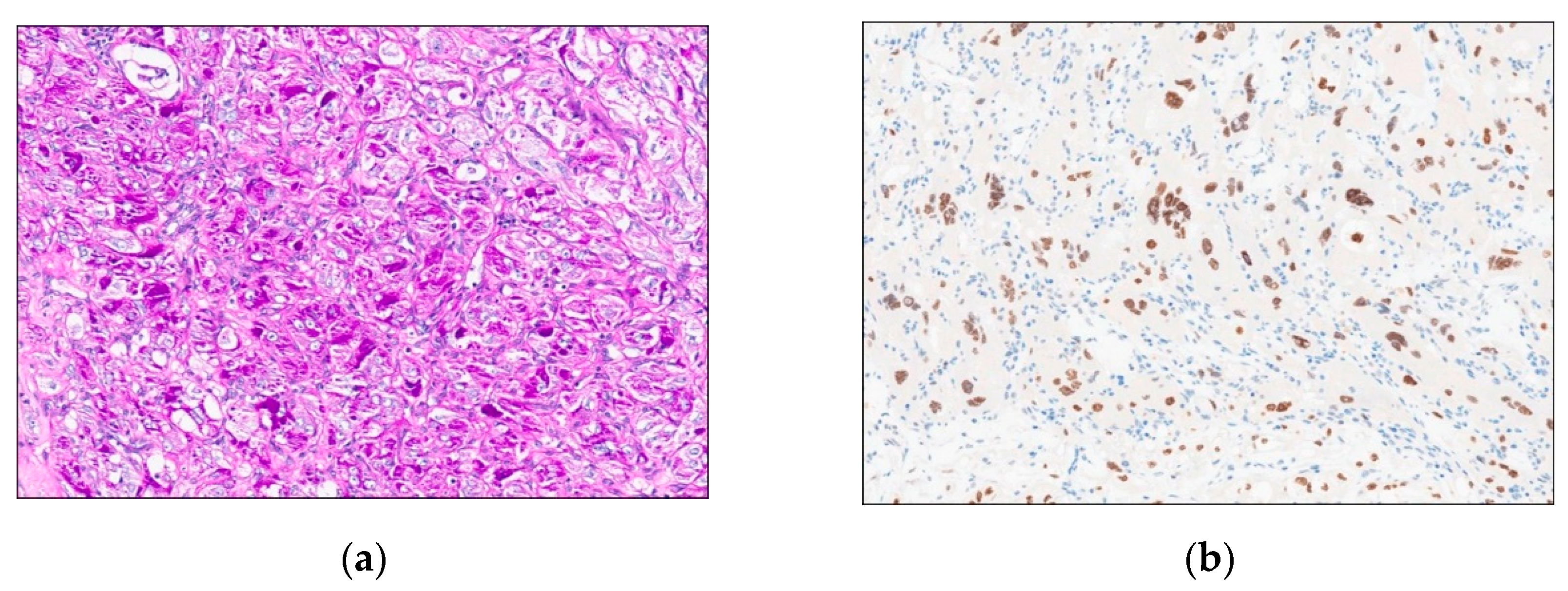
The patient underwent embolization and mass removal. On gross examination, the mass was 2.6cm in maximum diameter. The cut surface was rubbery in yellow to white tan color (Figure 3). A significant portion of the mass showed a sheet-like growth pattern consisting of pleomorphic tumor cells with hyperchromatic nuclei (Figure 4A). Nest formation surrounded by vascular channels of fine capillary size was focally observed, However, pseudoalveolar growth pattern as a result of loss of cell cohesion within tumor nests was not evident (Figure 4B). Epithelioid, polygonal tumor cells contained large, vesicular nuclei with prominent nucleoli and abundant, clear to eosinophilic cytoplasm. Mitoses were 0 to 1 per 10 high-power fields. Intracytoplasmic granules were frequently seen and PAS-positive, diastase-resistant (Figure 5A). Immunohistochemically, tumor cells were negative for panCK, EMA, SMA, desmin, S100 protein, CD56, CD31, CD34, HMB45, melan A, chromogranin A, synaptophysin, GFAP, and CD68. The tumor cells showed nuclear reactivity for TFE3 immunostaining (Figure 5B). The MIB-1 labeling index was approximately 5% in the highest proliferating areas. Based on these findings, the lesion was diagnosed as solid variant of ASPS. At the last follow-up, which was 6 months after the last surgery, the patient was clinically well without evidence of recurrence or metastasis.
3. Discussion
ASPS is a relatively rare soft tissue tumor of uncertain lineage with a characteristic morphology. ASPS usually presents as a deep-seated mass, located predominantly in the extremities preferentially in adolescents and young adults [1,2,3,4]. ASPS occurring in the sinonasal region was first reported in 1988 [8]. Since its first report, it has been very rarely reported [5,6,7,8,9,10]. Since there were only five cases of ASPS in the nasal cavity prior to the present case [6,7,8,9,10], it is not easy to perform its the differential diagnosis. In the case of sinonasal ASPS, it appears as a hypervascular polypoid mass at a young age. It is sometimes mistaken as a benign tumor in clinical practice. Our case was also clinically evaluated as a juvenile angiofibroma preoperatively.
Histologically, ASPS shows monotonous organoid and nest-like growth pattern consisting of large, round to polygonal cells usually displaying little variation in individual tumor cell size. Tumor cells have round, vesicular nuclei, prominent nucleoli, and abundant eosinophilic granular cytoplasm. Central discohesion within tumor nests results in characteristic pseudoalveolar-like structures. A portion of the tumor demonstrates histologic features atypical for ASPS [3,5], including nuclear hyperchromatism and pleomorphism, increased nuclear-cytoplasmic ratio, decreased cytoplasmic eosinophilia and granularity, mitotic figures necrosis, a less distinct nesting pattern and xantholomatous change. Diagnostic difficulties are encountered when the classical alveolar or pseudoalveolar pattern is absent and atypical cytologic features are seen. Our case displayed sheet-like to organoid growth pattern without typical discohesion, distinct areas of pleomorphic tumor cells with hyperchromatic nuclei and multinucleated giant cells. Our case has important diagnostic value as ASPS showing unusual histologic features can occur in the nasal cavity, an extremely rare area.
Tumor cells frequently show intracytoplasmic crystalline structures, which are PAS-positive and diastase-resistant. Diagnosis through ancillary testing used to confirm rhomboid crystals using an electron microscope in the past. However, gene fusion is now mainly confirmed through TFE3 immunostaining or molecular pathology techniques. ASPS has a characteristic der(170t(X;17)(p11.2;q25) translocation, which fuses the TFE3 transcription factor gene at Xp11 to a gene at 17q25, designated APSL [13,14]. Our case showed PAS-positive, diastase-resistant granules in the cytoplasm and TFE3 nuclear immunoreactivity even in areas with atypical histologic features.
Differential diagnosis of ASPS comprise tumors with large cells organized in nests with eosinophilic cytoplasm, such as malignant melanoma, renal cell carcinoma, adrenal cortical carcinoma, paragangioma, granular cell tumor, rhabdomyosarcoma, extrarenal rhabdoid tumor and perivascular epithelioid cell tumor (PEComa) [4,5]. All these differential diagnoses can be excluded with clinical correlation, adequate PAS and immunohistochemical analysis. ASPS typically lacks expression of S100 protein, chromogranin A, synaptophysin, myogenin, MyoD1, smooth muscle actin, HMB45, melan A, keratin, and EMA, allowing its distinction from the above-mentioned differential diagnosis.
Treatment of choice is surgical resection with sufficient margins. In the case of positive margins, adjuvant RT or chemotherapy is performed. However, the role of chemotherapy in ASPS is not proven [4,12]. ASPS behaves as a relatively indolent sarcoma, characterized by late metastases. A 5-year survival rate of 59~62% and a 10-year survival rate of 42~47% have been reported [1,4]. The prognosis is better when patients are younger, the size is less than 5 cm, and the stage is low [11]. Lingual and orbital tumors also have very high survival rates [12]. No specific survival data exist for tumor in the sinonasal region. In our case, at six months after surgery, there was no evidence of recurrence or metastasis.
Conclusively, this report highlights the difficulty in diagnosing ASPS when it lacks the classical pseudoalveolar pattern and shows atypical cytologic features, such as pleomorphism. When it occurs in the nasal cavity, a very unusual location, characteristic cytomorphology should be recognized and PAS, DPAS staining and TFE3 immunohistochemistry should be performed to accurately identify this tumor.
Author Contributions
Conceptualization, K.K. and K.U.C.; acquisition of clinical data, S.P. and S.W.K.; methodology, S.L. and Y.L.; investigation, K.S.C.; data curation, C.H.L. and G.Y.H.; writing—original draft preparation, K.K.; writing—review and editing, K.U.C.; supervision, A.K.; funding acquisition, K.U.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a 2-year research grant from Pusan National University.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of Pusan National University Hospital (protocol code 2309-038-019 and date of approval 5 November 2023).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Enzinger FM and Weiss SW. Alveolar soft part sarcoma. Soft Tissue Tumors. 6th edition. Saunders; 2013.
- Jambhekar NA, Ladanyi M. Alveolar soft part sarcoma. In: WHO Classification of Tumours Editorial Board, editor. WHO classification of tumours: soft tissue and bone tumours. 5th edition. Lyon: IARC; 2020. 297-299.
- Evans, HL. Alveolar soft part sarcoma. A study of 13 typical examples and one with a histologically atypical component. Cancer. 1985, 10, 912–917. [Google Scholar] [CrossRef]
- Shelke P, Sarode GS, Sarode SC, Anand R, Prajapati G, Patil S. Alveolar soft-part sarcoma of the oral cavity: A review of literature. Rare Tumors. 2018, 17, 10:2036361318810907.
- Fanburg-Smith JC, Miettinen M, Folpe AL, Weiss SW, Childers EL. Lingual alveolar soft part sarcoma; 14 cases: novel clinical and morphological observations. Histopathology. 2004, 45, 526–537. [CrossRef] [PubMed]
- P Chatterji, G N Purohit, I N Ramdev, N K Soni. Alveolar soft part sarcoma of the nasal cavity and paranasal sinuses. J Laryngol Otol. 1977, 91, 1003–1008. [CrossRef] [PubMed]
- Barbareschi M, Ferrero S, Ottaviani F. Alveolar soft part sarcoma of the nasal cavity. Pathologica. 1988, 80, 363–370.
- Rebinstein MI, Krake AF, McClatchey KD. Alveolar soft part sarcoma of the nasal cavity: report of a case and a review of literature. Laryngoscope. 1988, 98, 1246–1250. [CrossRef] [PubMed]
- Singh G, Sharma MC, Suri V, Sarkar C, Garg A, Singh M. Alveolar soft part sarcoma of the paranasal sinuses masquerading as a giant invasive pituitary adenoma. Ann Diagn Pathol. 2013, 17, 276–280. [CrossRef] [PubMed]
- Dutta R, Kakkar A, Sakthivel P, Kumar R, Seth R, Sharma MC. Alveolar Soft Part Sarcoma of the Oro-Maxillofacial Region in the Pediatric Age Group: Immunohistochemical and Ultrastructural Diagnosis of Two Cases. Head Neck Pathol. 2021, 15, 1303–1307. [CrossRef] [PubMed]
- Casanova M, Ferrari A, Bisogno G, Cecchetto G, Basso E, De Bernardi B, Indolfi P, Fossati Bellani F, Carli M. Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol. 2000, 11, 1445–1449. [CrossRef] [PubMed]
- A O Akinyamoju, O O Gbolahan2, B F Adeyemi. Characterization of alveolar soft part sarcoma of the tongue: A clinico-pathologic study and scoping review. Ann Ib Postgrad Med. 2020, 18, 122–134.
- Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, Healey JH, Ueda T, Yoshikawa H, Meloni-Ehrig A, Sorensen PH, Mertens F, Mandahl N, van den Berghe H, Sciot R, Dal Cin P, Bridge J. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001, 20, 48–57. [CrossRef]
- Pedram Argani1, Priti Lal, Brian Hutchinson, Man Yee Lui, Victor E Reuter, Marc Ladanyi. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol. 2003, 27, 750–761. [CrossRef] [PubMed]
Figure 1.
Nasopharyngoscopy demonstrated a protruding polypoid mass in nasal cavity.
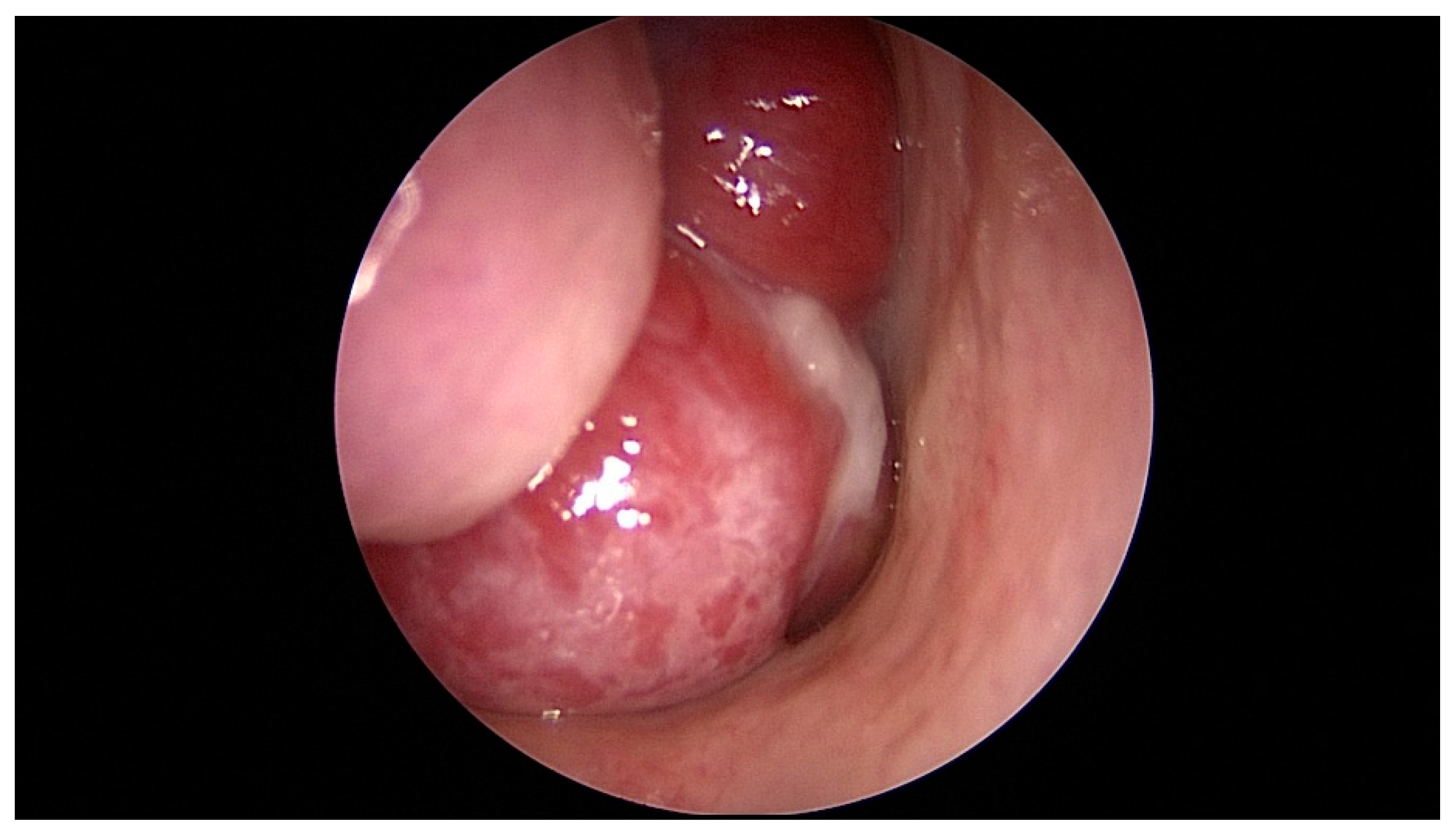
Figure 2.
PNS MRI showed a nasal cavity mass approximately 3 cm in size, well-circumscribed, and with medium signal intensity on T2-weighted image axial view.
Figure 2.
PNS MRI showed a nasal cavity mass approximately 3 cm in size, well-circumscribed, and with medium signal intensity on T2-weighted image axial view.
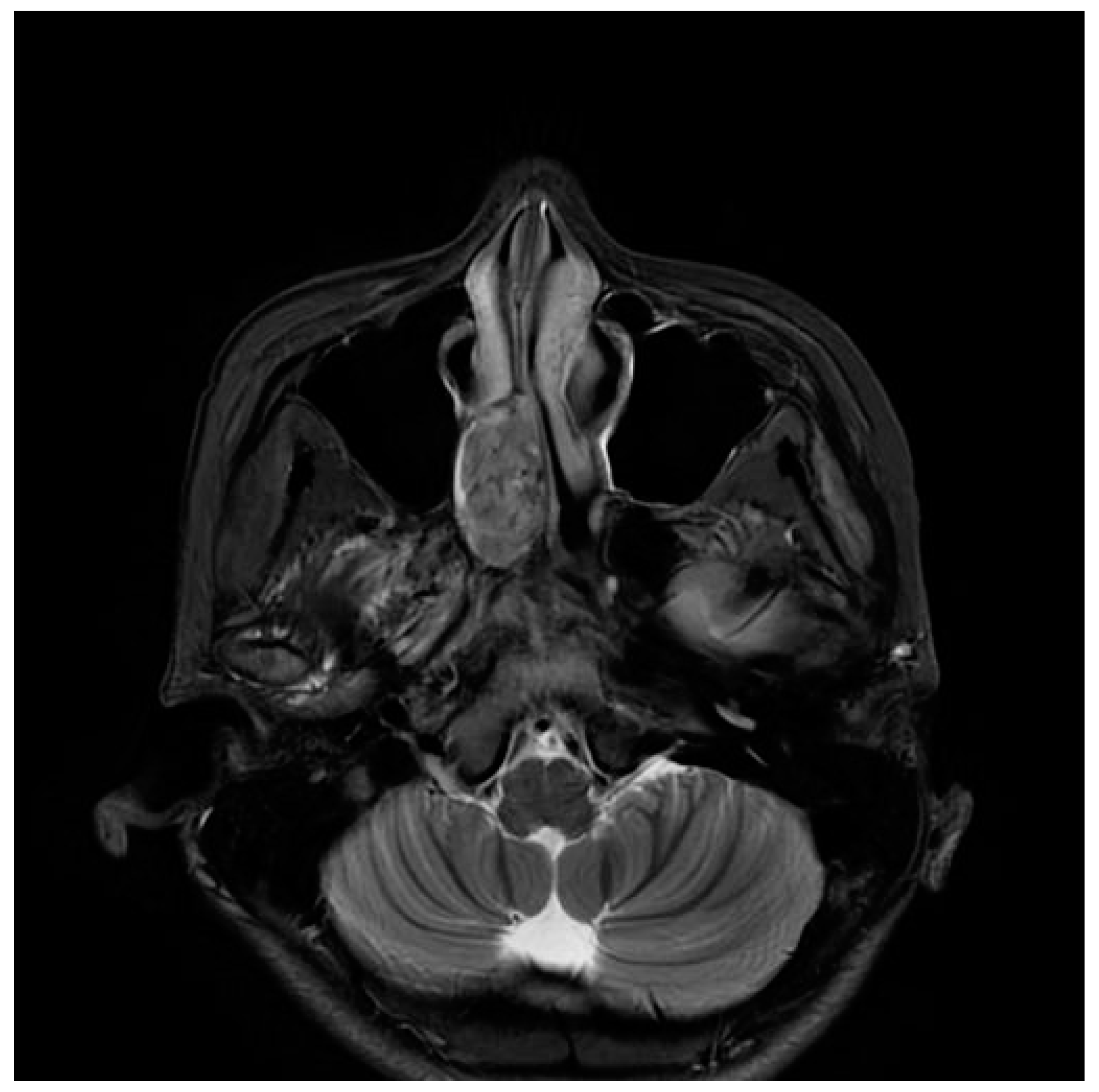
Figure 3.
Gross findings of a resected specimen. The mass was grossly 2.6 cm in maximum diameter and the cross-section was solid and yellowish-white.
Figure 3.
Gross findings of a resected specimen. The mass was grossly 2.6 cm in maximum diameter and the cross-section was solid and yellowish-white.
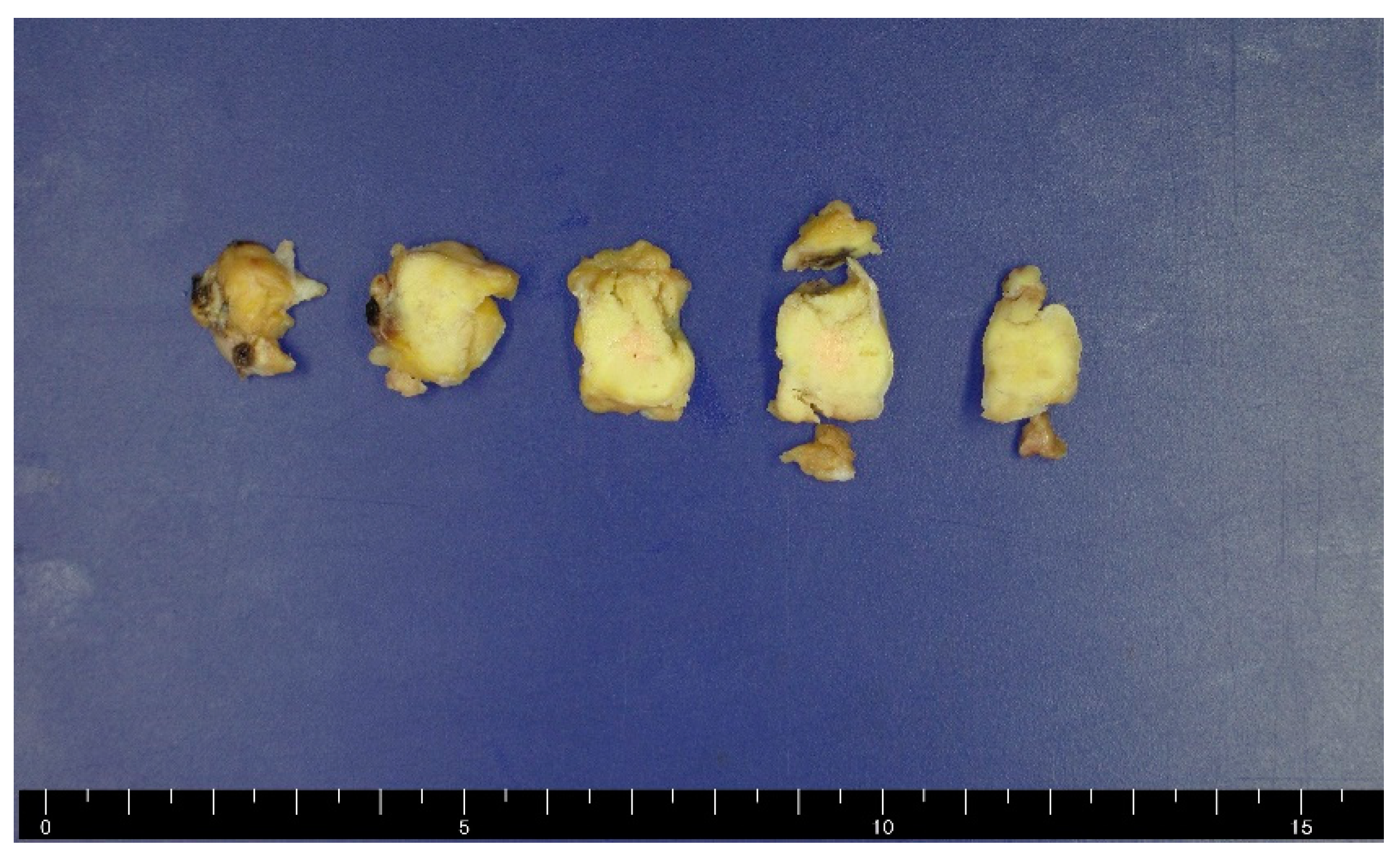
Figure 4.
Microscopic findings (H&E stain, x200). (a) The tumor exhibited sheet-like pattern of frankly pleomorophic tumor cells and (b) focal nest formation of relatively uniform tumor cells.
Figure 4.
Microscopic findings (H&E stain, x200). (a) The tumor exhibited sheet-like pattern of frankly pleomorophic tumor cells and (b) focal nest formation of relatively uniform tumor cells.
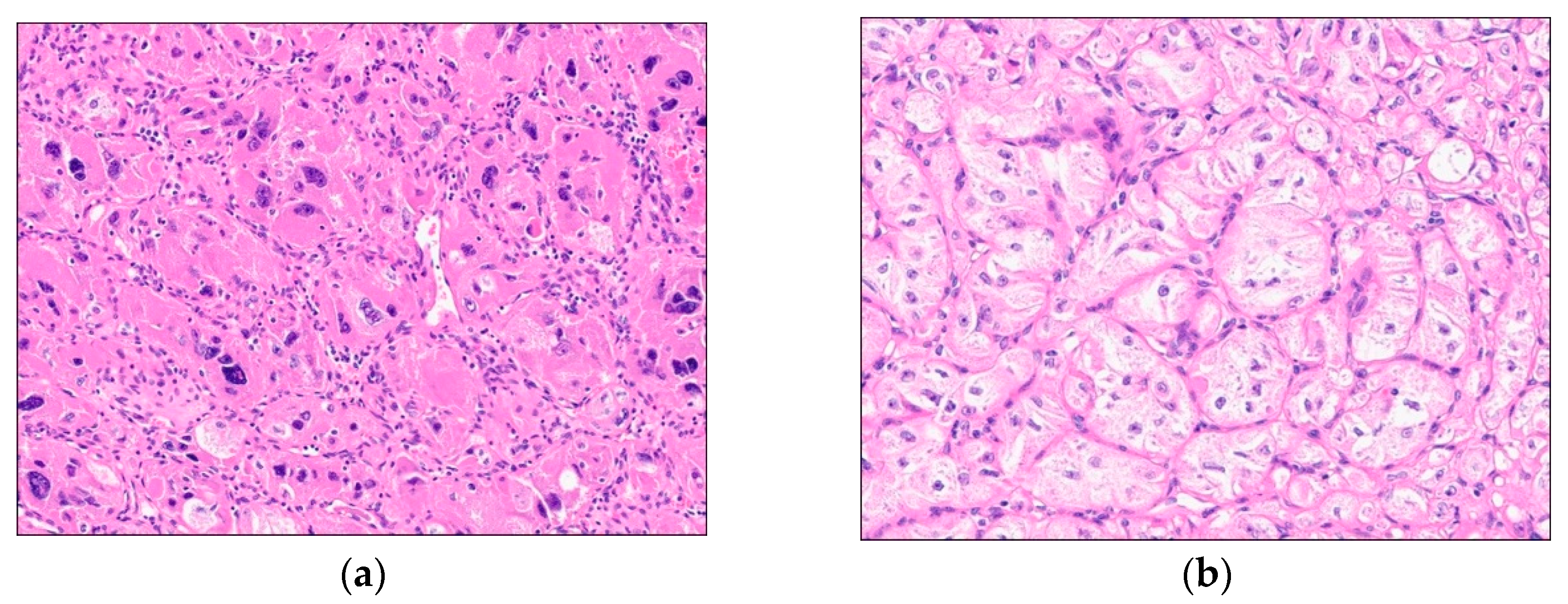
Figure 5.
Special and immunohistochemical staining. (a) PAS-positive, diastase-resistant cytoplasmic crystals were identified and (b) TFE3, a nuclear stain, was interpreted as positive throughout the tumor.
Figure 5.
Special and immunohistochemical staining. (a) PAS-positive, diastase-resistant cytoplasmic crystals were identified and (b) TFE3, a nuclear stain, was interpreted as positive throughout the tumor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



