Submitted:
02 January 2024
Posted:
03 January 2024
You are already at the latest version
Abstract
Neuromyelitis optica spectrum disorder (NMOSD), previously known as Devic disease or neuromyelitis optica (NMO), represents inflammatory conditions affecting the central nervous system. These disorders are characterized by severe immune-mediated demyelination and axonal damage, with a predilection for targeting optic nerves and the spinal cord. Key clinical features of NMOSD encompass acute episodes of bilateral or rapid sequential optic neuritis and transverse myelitis, typically exhibiting a relapsing course. These attacks endure for several days, and recovery varies, spanning from weeks to months. In this report, we present the case of a 35-year-old individual who initially complained of urinary retention, followed by decreasing vision in the left eye and subsequently in the right eye. These visual symptoms were succeeded by weakness in the right upper and lower limbs.
Keywords:
Neuromyelitis optica spectrum disorder (NMOSD)
; Aquaporin proteins
; AQP4 and AQP1
; longitudinally extensive transverse myelitis (LETM)
; Area Postrema Syndrome
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare autoimmune condition primarily affecting the central nervous system. It was first recognized in 1894 by Eugene Devic and Fernand Gault, but significant advancements in diagnosis and treatment only emerged in the twenty-first century [1].
Wingerchuk et al. (2015) have outlined the diagnostic criteria for NMOSD as established by the International Panel for Neuromyelitis Optica Diagnosis (IPND) in 2015. These criteria encompass clinical, imaging, and laboratory features, with the presence of AQP4 antibodies being a key diagnostic marker [5]. This article also explores the pathophysiology of NMOSD, shedding light on the immunological pathways involved and how AQP4 antibodies induce complement-dependent cytotoxicity and inflammation, leading to central nervous system damage. Additionally, the potential roles of other immune cells, such as T cells and B cells, in NMOSD development are considered [4].
Common symptoms of NMOSD include long-term inflammation of the spinal cord, severe optic neuritis, and persistent vomiting and hiccups [3]. NMOSD affects populations worldwide, with a higher prevalence among females and a greater incidence in Asian and African populations [6]. The treatment approach for NMOSD is multifaceted, encompassing acute management, prevention, and symptomatic relief through various classes of immunomodulator drugs [7].
Furthermore, the quality of life for NMOSD patients is influenced by factors such as the severity of symptoms, stigma, emotional distress, pain, fatigue, and occupational challenges. It is crucial for healthcare professionals to recognize these factors and provide appropriate interventions to enhance the overall quality of life for individuals living with NMOSD [9].
Patient Presentation
A 35-year-old male patient presented with several concerning symptoms over the past few days. His chief complaint included the inability to urinate for the last three days, decreasing vision that initially affected the left eye and progressed to the right eye within the same duration, and weakness in the right upper and lower limbs that began two days ago. There was no notable past medical history, personal history, or family history of similar conditions.
Upon initial evaluation, the patient's vital signs were measured as follows: blood pressure of 110/80 mm Hg, random blood sugar (RBS) of 121 mg/dl, partial pressure of oxygen in the blood at 98%, pulse rate of 80/min, and a respiratory rate of 16/min. The patient was alert, oriented, and conscious during the examination.
Ophthalmological assessment revealed bilaterally equal-sized pupils, which were mid-dilated and unresponsive to light stimulation. Neurological examination further confirmed weakness in the right upper and lower limbs, as well as weakness in the left lower limb. Detailed findings from the neurological examination are summarized in Table 1.
Routine blood tests showed generally normal results, with the exception of a mild increase in white blood cell count (leucocytosis). Serological tests for HIV, HbsAg (Hepatitis B surface antigen), HCV (Hepatitis C virus), and HSV (Herpes simplex virus) all returned negative results. Additional laboratory investigations are listed in Table 2.
Cerebrospinal fluid (CSF) analysis indicated elevated CSF protein levels and an increase in the number of CSF cells, with 100% of these cells being lymphocytes.
Other findings of CSF are mentioned in Table 3:
IMAGING STUDIES
1.5 T MRI OF BRAIN WITH ORBITS WITH CONTRAST + CERVICO-DORSAL SPINE + CONTRAST WITH SCREENING OF WHOLE SPINE:
MR imaging of the brain and orbits performed and high resolution T1 and T2-weighted serial sections obtained in the Sagittal and axial planes on a 1.5 Testa scanner with high strength gradients. Contiguous Fast Flair images were also obtained in the coronal plane.
Orbit:
T2W / STIR hyperintensity seen in both optic nerve and optic chiasma (left > right) which shows restricted diffusion and mild contrast enhancement---likely bilateral optic neuritis. Both eyeballs appear normal. No intraocular abnormality is seen. No evidence of retrobulbar mass lesion is seen. Extraocular muscles and lamina papyracia appear normal. Bony orbital wall appears normal.
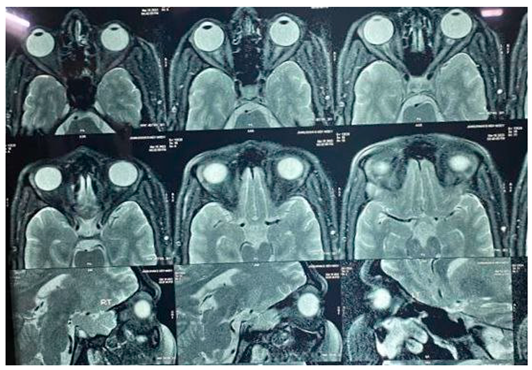
Brain:
No evident any periventricular white matter lesion seen. No evident changes of meningitis. Cerebral parenchyma appears normal. No definite focal or diffuse lesion is seen. The cerebellum, brainstem and pituitary gland are normal. The lateral, third and fourth ventricles are normal in size, shape, and position. The basal cisterns, fissures and sulci are unremarkable. Cervico-medullary junction does not reveal any abnormality.
Spine:
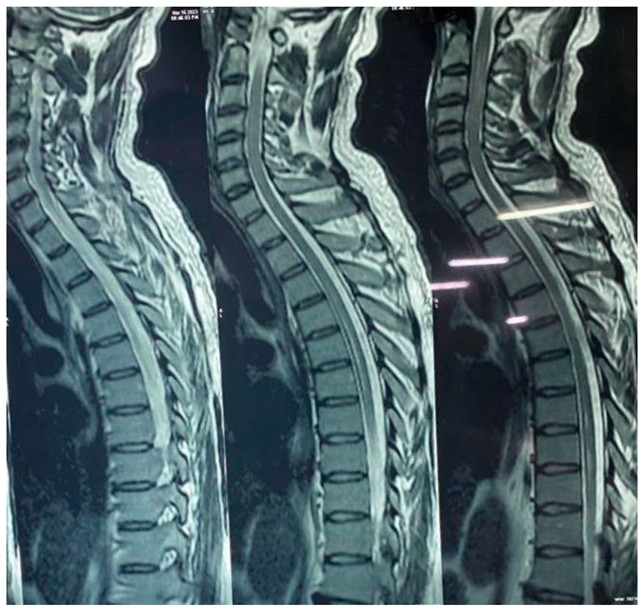
Long segment intramedullary T2W / STIR hyperintense signal changes seen involving cervical and dorsal cord extending from C2 to upto D11 level. It is seen involving entire circumferential of cord in cervical region and central cord in dorsal region.
Spondylotic changes are seen in the form multilevel disc desiccation and multilevel anterior and posterior osteophytes noted.
Ligamentum flavum, intervertebral foramina and facet joints do not reveal any abnormality.
No intraspinal mass or pre/paravertebral collection is seen.
IMPRESSION:
- Bilateral optic neuritis.
- Brain appears unremarkable.
- Abnormal signal /oedema involving spinal cord.
Imaging appearance may represent NEUROMYELITIS OPTICA (NMO).
Differential Diagnosis
Our patient presented with a complex clinical picture, and the initial differential diagnosis included several conditions, such as:
- Neuromyelitis Optica Spectrum Disorder (NMOSD)
- Multiple Sclerosis
- Myelin Oligodendrocyte Glycoprotein Antibody Associated Disease (MOGAD)
- Acute Disseminated Encephalomyelitis (ADEM)
- Idiopathic Transverse Myelitis
- Idiopathic Optic Neuritis
Following a comprehensive evaluation that encompassed clinical examination, laboratory investigations, and imaging findings, our patient was ultimately diagnosed with NMOSD.
Treatment
Upon diagnosis, the patient's treatment plan was initiated as follows:
- 1gm Methylprednisolone I.V Pulse Therapy: The patient received an initial course of intravenous Methylprednisolone pulse therapy, resulting in mild clinical improvement.
- Plasma Exchange Therapy (PLEX) and Physiotherapy: Subsequently, the patient underwent five cycles of Plasma Exchange Therapy (PLEX) alongside physiotherapy sessions to address the symptoms and promote recovery.
- IV Cyanocobalamin (1500 micrograms OD): As part of the treatment regimen, the patient was administered intravenous Cyanocobalamin at a daily dose of 1500 micrograms.
- Pain Management: Pain management was a crucial aspect of the patient's care. Initially, a Tramadol trial was administered, followed by a transition to Gabapentin (300mg TDS) to manage pain effectively.
Following ten days of therapy, the patient's condition improved, and they were subsequently discharged with the following ongoing medications:
- Prednisolone: To continue addressing the autoimmune component of NMOSD.
- Mycophenolate Mofetil: As part of the long-term immunosuppressive therapy plan.
Discussion
Neuromyelitis optica spectrum disorder (NMOSD) primarily affects central nervous system structures, including the optic nerve, dorsal medulla, brainstem, and the central gray matter of the spinal cord. This condition was first described by Eugène Devic (1858–1930) and Fernand Gault (1873–1936) back in 1894 [1,4].
Two key aquaporin proteins, AQP4 and AQP1, play a significant role in water flow regulation and are implicated in the development of brain edema in pathological conditions. Approximately 70% of NMO patients produce antibodies to AQP4, with the majority targeting an extracellular determinant in AQP4's C-loop. NMO-IgG primarily belongs to the IgG1 subclass, enabling complement activation [8]. In some AQP-4 negative cases, antibodies against the central nervous system's myelin oligodendrocyte glycoprotein (MOG) have been identified [7].
The prevalence of NMOSD among White populations is estimated at 1 per 100,000, with an annual incidence of 1 per million. AQP4-antibody disease shows a notably high female-to-male ratio (up to 9:1) and tends to manifest at around 40 years of age. Among East Asians, the frequency is higher, at 3.5 per 100,000, but it can go up to 10 per 100,000 among Black populations. MOG-antibody disease's yearly incidence stands at 1.6 per million individuals (adults: 1.3 per million, children: 3.1 per million) [5].
Clinical manifestations of NMOSD encompass optic neuritis, longitudinally extensive transverse myelitis (LETM), area postrema syndrome, acute brainstem syndrome, cerebral syndrome, and diencephalic syndrome [2]. Optic neuritis in NMO is marked by profound and enduring vision loss, with 80% of NMO-affected eyes experiencing significant visual acuity decline (20/200 or worse). This can result in unilateral or bilateral blindness. Some cases exhibit optic chiasm and optic tract involvement, leading to bitemporal or homonymous visual field abnormalities [10]. Myelitis can present as acute or subacute motor or sensory deficits in the limbs or trunk, along with bladder or bowel dysfunction. Acute brainstem syndrome can cause diplopia, facial palsy, and dysarthria, while area postrema syndrome manifests with intractable hiccups, nausea, and vomiting. Cerebral syndrome may bring about sudden confusion, seizures, or cognitive impairment, while diencephalic syndrome can lead to life-threatening hyponatremia [2].
LETM, characterized by a significant spinal cord lesion spanning three or more vertebral segments on spinal MRI, is often a prominent feature of neuromyelitis optica spectrum disorder (NMOSD). Symptoms typically include bladder dysfunction, paraparesis, quadriparesis, and visual impairment [8].
The International Panel for Neuromyelitis Optica Diagnosis (IPND) Diagnostic Criteria for NMOSD (2015)
Diagnostic Criteria for NMOSD with AQP-4 IgG:
- At least 1 Core Clinical Characteristic
- Positive Test for AQP4-IgG Using the Best Available Detection Method
- Exclusion of Alternative Diagnoses
Diagnostic Criteria for NMOSD without AQP-4 IgG or NMOSD with Unknown AQP-4 IgG Status:
- At least 2 Core Clinical Characteristics Resulting from One or More Clinical Attacks and Meeting All of the Following Requirements:
- At least 1 Core Clinical Characteristic Must Be One of the Following:
- Optic Neuritis
- Acute Myelitis with Longitudinally Extensive Transverse Myelitis (LETM)
- Area Postrema Syndrome (episode of otherwise unexplained hiccups or nausea and vomiting)
- Dissemination in Space (2 or More Different Core Clinical Characteristics)
- Fulfillment of Additional MRI Requirements, as Applicable
- Negative Tests for AQP-4 IgG Using the Best Available Detection Method, or Testing Unavailable
- Exclusion of Alternative Diagnosis
Core Clinical Characteristics:
- Optic Neuritis
- Acute Myelitis
- Area Postrema Syndrome (episode of otherwise unexplained hiccups or nausea and vomiting)
- Acute Brainstem Syndrome
- Symptomatic Narcolepsy or Acute Diencephalic Clinical Syndrome with NMOSD-Typical Diencephalic MRI Lesions
- Symptomatic Cerebral Syndrome with NMOSD-Typical Brain Lesions
Additional MRI Requirements for NMOSD without AQP-4 IgG and NMOSD with Unknown AQP-4 IgG Status:
- Acute Optic Neuritis: Requires brain MRI showing either (a) normal findings or only nonspecific white matter lesions, OR (b) optic nerve MRI with a T2-hyperintense lesion or T1-weighted gadolinium-enhancing lesion extending over >1/2 optic nerve length or involving the optic chiasm.
- Acute Myelitis: Requires associated intramedullary MRI lesion extending over >= 3 contiguous segments (LETM) OR >=3 contiguous segments of focal spinal cord atrophy in patients with a history compatible with acute myelitis.
- Area Postrema Syndrome: Requires associated dorsal medulla/area postrema lesions.
- Acute Brainstem Syndrome: Requires associated periependymal brainstem lesions.
Immunopathogenesis of NMOSD:
In the context of neuromyelitis optica spectrum disorder (NMOSD), B cells possessing AQP4-specific B cell receptors may assume the role of antigen-presenting cells, triggering the activation of autoreactive T cells that differentiate into the Th17 lineage. Th17 cells, in turn, can promote B cell maturation into plasma cells, leading to the production of antibodies targeting AQP4, known as NMO-IgG. These circulating antibodies, primarily originating outside the central nervous system (CNS), can penetrate the blood-brain barrier (BBB), including the endothelial basement membrane (BM), when the CNS vasculature's endothelium is inflamed, often instigated by Th17 cells [8].
AQP4-IgG antibodies interact with AQP4 located in the astrocyte feet, triggering complement activation and subsequent complement-dependent cytotoxicity. This cascade of events also attracts inflammatory cells such as eosinophils, neutrophils, and macrophages, leading to further disruption of the BBB and facilitating the entry of AQP4-IgG into the CNS. The degranulation of inflammatory cells and the damage to astrocytes contribute to oligodendrocyte injury, accelerating the loss of myelin sheaths, axonal damage, and resulting in neurological impairments [3].
Treatment Strategies for NMOSD:
In the management of acute NMOSD relapses, high-dose intravenous methylprednisolone is commonly employed as the initial treatment to mitigate inflammation. If, in the judgment of the attending physician, high-dose corticosteroids fail to provide substantial improvement, plasma exchange (PLEX) has proven effective in NMOSD. Preventive therapies for NMOSD include medications like Azathioprine, Mycophenolate Mofetil, and Rituximab. Additionally, two drugs, eculizumab and tocilizumab, are currently under investigation for their potential as preventive treatments for NMOSD [6].
Symptomatic management in NMOSD encompasses several approaches, including the use of antiepileptic medications, gabapentin, pregabalin, and levetiracetam, for pain control. Oral anti-spasticity agents, such as the GABA-B agonist baclofen and/or the α2 agonist tizanidine, are employed to manage spasticity. For urinary incontinence, anti-muscarinic medications like oxybutynin, darifenacin, or solifenacin, as well as selective beta-3 adrenergic receptor agonists like mirabegron, serve as the mainstay treatments [11].
References
- Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:8. Published 2013 Jan 15. [CrossRef]
- Huda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, Jacob A. Neuromyelitis optica spectrum disorders. Clin Med (Lond). 2019;19(2):169-176. [CrossRef]
- Wu Y, Zhong L, Geng J. Neuromyelitis optica spectrum disorder: Pathogenesis, treatment, and experimental models. Mult Scler Relat Disord. 2019;27:412-418. [CrossRef]
- Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189. [CrossRef]
- Hor JY, Asgari N, Nakashima I, et al. Epidemiology of Neuromyelitis Optica Spectrum Disorder and Its Prevalence and Incidence Worldwide. Front Neurol. 2020;11:501. Published 2020 Jun 26. [CrossRef]
- Kessler RA, Mealy MA, Levy M. Treatment of Neuromyelitis Optica Spectrum Disorder: Acute, Preventive, and Symptomatic. Curr Treat Options Neurol. 2016;18(1):2. [CrossRef]
- Page 3477, Chapter 445 : Neuromyelitis Optica, Harrison’s principles of Internal Medicine 21st edition Volume -2 , ISBN : 978-1-26-426851-1, MHID: 1-26-426851-3.
- Mitsdoerffer M, Kuchroo V, Korn T. Immunology of neuromyelitis optica: a T cell-B cell collaboration. Ann N Y Acad Sci. 2013;1283:57-66. [CrossRef]
- Meca-Lallana JE, Gómez-Ballesteros R, Pérez-Miralles F, et al. Impact of Neuromyelitis Optica Spectrum Disorder on Quality of Life from the Patients' Perspective: An Observational Cross-Sectional Study. Neurol Ther. 2022;11(3):1101-1116. [CrossRef]
- Levin MH, Bennett JL, Verkman AS. Optic neuritis in neuromyelitis optica. Prog Retin Eye Res. 2013;36:159-171. [CrossRef]
- Abboud H, Salazar-Camelo A, George N, et al. Symptomatic and restorative therapies in neuromyelitis optica spectrum disorders. J Neurol. 2022;269(4):1786-1801. [CrossRef]

Table 1.
| Higher mental function | Normal |
| Motor system | Muscle bulk and tone were normal in left upper limb. Reduced in Right upper and lower limb and left lower limb. Power : +5 in Left upper limb +1 in Left lower limb +1 in Right upper limb 0 in Right lower limb |
| Cranial Nerve examination | No light perception bilaterally Rest cranial nerves normal. |
| Sensory system | Normal |
| Reflexes | Biceps reflex : +2 Bilaterally Triceps reflex : +2 bilaterally Ankle reflex : +2 bilaterally Knee reflex : 0 bilaterally |
Table 2.
| Hb | 8.1 mg/dl |
| Platelets | 358 * 103 / cmm |
| RBC count | 3.21* 106 / cmm |
| Total WBC count | 12.11 * 103 / cmm |
| S. AQP-4 IgG | Negative |
Table 3.
| Physical examination | Normal |
| CSF Protein | 107.7 mg/dl |
| CSF Glucose | 60.38 mg/dl |
| CSF Chloride | 125.98 mmol/lit |
| CSF Total Cell Count | 30 (Lymphocytes – 100%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



