
Submitted:
25 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
Bulbophyllum is one of the largest genera and presents some of the most intricate taxonomic problems in the family Orchidaceae, including species of ornamental and medical importance. The lack of knowledge regarding the characterization of Bulbophyllum chloroplast (cp) genomes has imposed current limitations on our study. Here, we reported the complete cp genomes of seven Bulbophyllum species, including B. ambrosia, B. crassipes, B. farreri, B. hamatum, B. shanicum, B. triste and B. violaceolabellum, and compared with related taxa to provide a better understanding of their genomic information on taxonomy and phylogeny. A total of 28 Bulbophyllum cp genomes exhibit typical quadripartite structures with lengths ranging from 145,092 bp to 165,812 bp and GC content of 36.60% to 38.04%. Each genome contained 125–132 genes, encompassing 74–86 protein-coding genes, 38 tRNA genes, and eight rRNA genes. The genome arrangements, gene contents and length were similar with differences observed in ndh gene composition. A total of 18–49 long repeats and 38–80 simple sequence repeats (SSRs) were detected and the single-nucleotide (A/T) was dominant in Bulbophyllum cp genomes, with an obvious A/T preference. An analysis of relative synonymous codon usage (RSCU) revealed Leucine (Leu) was the most frequently used codon, while cysteine (Cys) was the least used. Six highly variable regions including ndhF-trnLUAG, trnTUGU-trnLUAA, trnFGAA-ndhJ, rps15-trnNGUU, rbcL-accD and psbI-trnSGCU were identified based on the ranking of the Pi values, had the potential to serve as DNA markers for species identification and phylogeny of the genus Bulbophyllum. Phylogenetic analysis based on the complete cp genome sequences and 68 protein-coding genes strongly supported 28 Bulbophyllum species can be divided into four branches and sects. Brachyantha, Cirrhopetalum, Leopardinae defined by morphology were non-monophyly. Our results enriched the genetic resources of Bulbophyllum species, providing valuable information to illustrate the complicated taxonomy, phylogeny and evolution process of the genus.
Keywords:
Bulbophyllum
; chloroplast genome
; molecular markers
; phylogenetics analysis
1. Introduction
Bulbophyllum, comprising approximately 2,200 species, is one of the largest genera of Orchidaceae and serves as an excellent model system for investigating orchid biodiversity [1,2,3]. Its distribution spans pantropical regions, including Africa, Madagascar, the Americas and the Asia-Pacific region [4]. Members of this genus exhibit epiphytic or lithophytic habits and typically possess one or two-leaved pseudobulbs with a labellum attaches to the base of the floral column via an elastic hinge [4,5]. Bulbophyllum demonstrates remarkable adaptability, flourishing in a variety of environments including subtropical dry forests and wet montane cloud forests [4,6]. Owing to its morphologically diverse lateral sepals that vary in size, shape, color and surface ornamentation, Bulbophyllum has economic importance attributable to ornamental uses [5]. Additionally, the aromatic compounds found in these orchids are significant for their medical benefits [7,8].
Bulbophyllum has a complex taxonomic history, with numerous proposals for generic delimitations and infrageneric classifications based on morphological characters since its establishment by Thouars in 1822 [9,10,11,12,13,14]. Two mainly perspectives exist on the morphological division of Bulbophyllum: either dividing the genus into multiple sections or categorizing the broad genus into several genera. Statistically, more than 50 genera have been merged into Bulbophyllum (e.g. Cirrhopetalum Lindl., Drymoda Lindl., Monomeria Lindl., Trias Lindl., and Sunipia Lindl.), and approximately 70–80 sections proposed alone in the Asia-Pacific region [4,15,16].
Phylogenetic analyses of Bulbophyllum using DNA sequence data have made significant progress recently. Most phylogenetic results supported the monophyly of a broadly defined Bulbophyllum and its continental taxa, such as Asian, African and Neotropical clades [1,4,17]. Hu et al. reconstructed the phylogenetic relationship in the Asian Cirrhopetalum alliance of Bulbophyllum based on combining four DNA sequence data (nrDNA: ITS, Xdh; cp DNA: matK and psbA-trnH, 117 taxa), supported an ameneded Cirrhopetalum alliance were monophyly [5]. Based on eight DNA sequence data (nrDNA: ITS, Xdh, OrcPI; cp DNA: atpI-atpH, ycf1, matK, trnD-trnE, psbA-trnH, 179 taxa), Gamisch et al. divided the Malagasy taxa into four clades [18]. These studies have clarified the phylogenetic relationships of different region in this group, but nodal support values of the main clades or lineages were moderate to low or lacking for some relationships. The number of accepted species continues to grow as new discoveries are reported [19,20,21], taxonomic work on Bulbophyllum became a major challenge that the further investigation into the relationships within the genus necessitates more detailed study.
With the continuous reduction in sequencing costs, the chloroplast (cp) genome has become a pivotal tool for investigating phylogenetic relationships within complex taxa. The cp genome offers several advantages, including unique mode of inheritance, highly conserved genome structure, and a moderate evolutionary rate [22,23]. Owing to these unique characteristics, cp genomes are widely used to explore the phylogenetic relationship among orchid clades. Liu et al. reconstructed the phylogenetic relationships of the Cleisostoma–Gastrochilus clades in Aeridinae based on the cp genomes robustly supporting this clade divided into six subclades with higher support rates and more stable topological structures than before [24]. Additionally, many studies conducted comprehensively compared differences analysis in orchid cp genomes to understand the structural characteristics and evolution patterns, such as Pholidota (13 species) and Paraphalaenopsis (3 species) [25,26]. Yang et al. compared and analysed cp genomes of 18 species from Asian and Netropical Bulbophyllum. The results show that the cp genome structure of Asian and Neotropical clades is different due to selection pressures under the condition of geographical isolation [27]. Furthermore, integrative analysis of multiple cp genomes can help to develop applicable molecular markers for species identification [28]. Five highly variable regions (ycf1, ndhA, ndhF, trnQ and trnK), the potential DNA markers, were found in four Liparis cp genomes [29]. Tang et al. analysed the cp genomes of sect. Macrocaulia in Bulbophyllum and proposed 20 intergenic regions and three coding genes of the most variable hot spot regions as candidate effective molecular markers [30].
To date, only a few cp genomes of Bulbophyllum have been sequenced and detailed cp genomic comparisons and phylogenetic analyses are lacking, which hindered our ability to further elucidate its interspecific relationships. In order to further clarify the phylogenetic relationships among species of the genus and to obtain useful genetic resource, we sequenced and assembled the cp genomes of seven Bulbophyllum species (B. ambrosia, B. crassipes, B. farreri, B. hamatum, B. shanicum, B. triste, B. violaceolabellum) and compared with other Bulbophyllum species published to investigate their relationships. Our results will provide valuable information for cp genome evolution, phylogenetic relationships and species identification of Orchidaceae.
2. Results
2.1. General Characteristics of the Chloroplast Genomes
The seven newly sequenced Bulbophyllum cp genomes were circular with the typical quadripartite structure, including a large single copy (LSC), a small single copy (SSC) and a pair of inverted repeats (IRs) (Figure 1). We combined the published cp genomes of 21 Bulbophyllum orchids with this study’s seven species to compare the basic cp genome features within the genus. The cp genomes lengths, number of genes, GC content, etc. of the 28 cp genomes are summarized in Table 1. As shown in Table 1, 28 Bulbophyllum cp genomes sizes were ranged from 145,092 bp (B. kwangtungense) to 165,812 bp (B. crassipes). The cp genomes were variable in LSC and SSC regions, with 77,088 to 87,177 bp and 11,089 to 18,632 bp, while conserved in IR regions, with sizes ranging from 25,465 to 26,919 bp. The GC content was relatively consistent, ranging from 36.60% to 38.04%, and the distribution of GC content across different regions was uneven, with about 43.18%, 34.93%, and 29.67% for the IR, LSC, and SSC regions, respectively (Table 1).
Each cp genome was annotated with a total of 113 unique genes, which included 79 protein-coding genes, 30 transfer RNAs (tRNAs), and four ribosomal RNAs (rRNAs) (Table 1). Most genes existed as single copies in either LSC or SSC regions. However, 19 genes were duplicated in IRs, encompassing seven protein-coding genes (ycf1, rpl2, rps7, ndhB, ycf2, rpl23, and rps12), eight tRNAs (trnNGUU, trnRACG, trnAUGC, trnLGAU, trnVGAC, trnLCAA, trnICAU, and trnHGUG) and four rRNAs (rrn23, rrn16, rrn5, and rrn4.5). Nine protein-coding genes and six tRNA genes contained one intron each, whereas genes ycf3 and clpP possessed two introns. The length of introns varied among different genes, with the longest intron found in the trnKUUU gene. Notably, the ndh genes were truncated or completely lost in more than half the species (Table 1, Supplementary Table S10). The highest degree of loss was the ndhF gene, which was observed in 11 species. The highest degree of pseudogenization was ndhD gene, which was pseudogenized in nine species. The species with simultaneous pseudogenization and loss of ndh gene are B. disciflorum, B. exaltatum, B. granulosum, B. hamatum, B. inconspicuum, B. kwangtungense, B. mentosum, B. ningboense, B. pingnanense, B. plumosum and B. tianguii. All functional genes could be categorized into three groups: those related to self-replication, photosynthesis, and others (Supplementary Table S1).
2.2. Repeat Sequence Characterization
We identified four types of long repeats: palindromic (P), forward (F), complementary (C), and reverse (R) elements (Figure 2A, Supplementary Table S2) in 28 Bulbophyllum cp genomes. Among these, all four categories were observed in 14 species, while 12 species contained three categories of repeats (C/R, F and P), two species (B. crassipes and B. farreri) exhibited two categories (P and F). The number of long repeat sequences ranging from 17 (B. kwangtungense) to 49 (B. disciflorum, B. gedangense, B. reptans and B. violaceolabellum). Across these 28 cp genomes, P were the most prevalent, ranging from 5 occurrences in B. hirtum to 25 occurrences in B. inconspicuum and B. pingnanense. Bulbophyllum cp genomes had fewer R and C repeats, and the highest counts of the two types were 25 R in B. reptans and 10 C in B. hirtum, respectively. Long repeat sequences in the range of 30–40 bp were the most frequently observed and ranged from 15 occurrences in B. shanicum to 47 occurrences in B. disciflorum. B. inconspicuum displayed the highest count of 40–50 bp repeats. The 50–60 bp repeat sequences were detected in 18 Bulbophyllum species, ranging from 1 to 6 occurrences. The 60–70 bp repeat sequences were only presented in B. crassipes, B. lingii, B. menghaiense, B. pentaneurum, B. pingnanense, B. shanicum and B. triste, ranging from 1 to 4 occurrences. The longest repeat sequences were 77 bp in B. ningboense (Figure 2B, Supplementary Table S3).
A total of 38 (B. leopardinum) to 80 (B. mentosum) SSRs were detected in the cp genome of the 28 Bulbophyllum species, and six categories of SSRs (mono-, di-, tri-, tetra-, penta- and hexanucleotide repeats) were identified (Figure 2C, 2D and Supplementary Table S4, S5). Mono-nucleotide repeats (SSR loci A/T) were the most abundant, accounting for 58.3% (B. lingii) to 81.7% (B. ambrosia), with counts varying from 47 to 58. This was followed by di-nucleotide repeats (6 to 13 occurrences, 8.8.% to 22.0%), tri-nucleotide repeats (0 to 4 occurrences, 5.9%), tetra-nucleotide repeats (2 to 13 occurrences, 2.6% to 17.6%), penta-nucleotide repeats (0 to 5 occurrences, 6.4%), and hexa-nucleotide, with the least number of SSRs (0 to 2 occurrences, 4.1%). All mononucleotide SSRs belonged to A or T type, and the majority of di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs were particularly rich in A or T (Figure 2D, Supplementary Table S5). In general, the distribution pattern of SSRs was unevenly across the 28 species. Mono-, di- and tetra- nucleotide repeats categories were observed in all species, while tri- and penta-nucleotide repeats were absent in 10 different species. Hexa-nucleotide repeats were only present in B. affine, B. farreri, B. gedangense, B. hamatum, B. ningboense, B. pentaneurum, B. pingnanense and B. plumosum.
2.3. Relative Synonymous Codon Usage Analysis
We analysed a total of 68 protein-coding genes among the 28 Bulbophyllum cp genomes, with the exception of the ndh genes due to incomplete gene loss and pseudogenization. These genes were encoded by a range of 17,226 codons in B. plumosum to 22,758 codons in B. shanicum (Figure 3, Supplementary Table S6). The codon usage patterns revealed a highly conserved codon usage bias (CUB). Leucine (Leu) was one of the most frequently occurring amino acids, appearing a total of 57,130 times across all 28 cp genomes. In contrast, Cysteine (Cys) was the least frequent, occurring only 6,515 times. Analysis of the relative synonymous codon usage (RSCU) indicated that UUA and AGA had the highest CUB, with average values of 1.934 and 1.894, respectively, while CGC and CUC had the lowest CUB, with average values of 0.374 and 0.397, respectively. Among the three stop codons, the frequency of UAA was the highest, accounting for 39.9%. The results also showed that 30 codons exhibited RSCU values greater than 1, and 32 codons exhibited values less than 1 (Figure 3, Supplementary Table S6). The RSCU values of AUG encoding for methionine (Met) and UGG encoding for tryptophan (Trp) were determined to be 1 in all seven species.
2.4. Expansion and Contraction of IRs, Sequence Divergence and Nucleotide Diversity
A comprehensive comparison of the boundaries between the LSC, IRs and SSC regions was conducted across the 28 Bulbophyllum species (Figure 4). The junctions between the IRs and SC regions exhibited a high degree of conservation. In the cp genomes of these 28 Bulbophyllum species, several key genes, namely rpl22, ndhF, ycf1, rps19, and psbA were found at the junction of LSC/ IRb, IRb/SSC, SSC/IRa, and IRa/LSC borders. The rpl22 gene, spanning from LSC to IRb, was primarily located in the LSC region ranged from 279–423 bp in length. B. hamatum and B. tianguii comprised 279 bp in the LSC region and 87 bp in the IRb region were the shortest. Furthermore, the IRb/SSC border of B. ambrosia, B. crassipes, B. farreri, B. gedangense, B. mentosum was located in the ndhF pseudogene, with just 70 bp located in the IRb region. The ndhF pseudogene was close to but did not overlap with the IRb/SSC junction in B. affine, B. andersonii, B. disciflorum, B. hirtum, B. kwangtungense, B. leopardimum, B. orientale, B. pectinatum, B. reptans. Within the SSC/IRa (JSA) region, the ycf1 gene spanned the SSC/IRa boundary, primarily residing in the SSC region, with lengths ranging from 4,308 bp (B. crassipes) to 5,469 bp (B. hamatum). In the case of B. hamatum and B. tianguii, the ycf1 gene was positioned to the left side of the JSA line, with a distance of 6 bp and 9 bp, respectively. In the IRa/LSC (JLA) region, the rps19 gene was situated on the left side of the JLA line, and the distance from the rps19 to the JLA line ranged from 229 bp (B. crassipes) to 290 bp (B. hamatum). The psbA gene was located on the right side of the JLA line, with distances ranging from 10 bp (B. kwangtungense, B. leopardinum and B. pectinatum) to 132 bp (B. ambrosia) (Figure 4).
The divergence of sequences in the cp genomes of 28 Bulbophyllum species was plotted using the mVISTA program with the annotated B. affine (LC556091) sequence as a reference (Figure 5). The results revealed sequences were significant conservation in Bulbophyllum cp genomes, particularly in the coding region. The highest variation was observed in the SSC region, followed by the LSC region and IR regions. Mauve visualization graphs also indicated that no significant gene rearrangement was detected among these cp genomes (Figure 6).
Nucleotide diversity value (Pi) for the coding regions and intergenic regions were calculated using DnaSP to further analyze the mutation hotpots in 28 Bulbophyllum species. The results showed that the Pi values ranged from 0 to 0.21413 (ndhF-trnLUAG) (Figure 7A, Supplementary Table S7). The IR regions exhibited the highest conservation with a value of 0.0035. The SSC region displayed the greatest nucleotide diversity (Pi = 0.0307), followed by the LSC region (0.0141). According to the ranking of the Pi values, six hypervariable regions were identified, including ndhF-trnLUAG (0.21413), trnTUGU-trnLUAA (0.09437), trnFGAA-ndhJ (0.09138), rps15-trnNGUU (0.08122), rbcL-accD (0.07534) and psbI-trnSGCU (0.06529). Additionally, the protein-coding genes displayed higher conservation (Figure 7B, Supplementary Table S8). Among these genes, ycf1 (0.02956), rps12 (0.02643), matK (0.02178), psbK (0.01599), and rps15 (0.01511) exhibited the highest Pi values.
2.5. Phylogenetic Analysis
The phylogenetic analysis of 28 Bulbophyllum species, based on two datasets comprising complete cp genomes and 68 protein-coding genes, revealed that the species formed four major clades (Figure 8, Supplementary Figure S1). The alignment matrix of complete cp genomes was 131,138 bp, with 12,031 variable sites and 6,201 parsimony informative sites. The matrix of 68 protein-coding genes was 59,417 bp and included 4,869 variable sites along with 2,404 parsimony informative sites. The topologies remained largely consistent within the two datasets, demonstrating strong support based on complete cp genomes (Bootstrap Support, BS ≥ 98, Posterior Probability, PP = 1.00) while the support was relatively moderate inferred by 68 protein-coding genes (BS ≥ 75, PP ≥ 0.70) (Figure 8, Supplementary Figure S1). Clade 1 (Neotropical clade) consisted of four species from different sections and clade 2 primarily consisted species from sect. Macrocaulia with robust support. In clade 3, sects Lemniscata (B. shanicum, B. triste and B. hirtum) and Racemosae (including B. crassipes and B. orientale) were sister groups with generally high support values in one subclade, while another subclade consisted of species from sects. Leopardinae, Trias, Stenochilus and Repantia. Clade 4 contained species assigned to sects Cirrhopetalum, Brachyantha, Leopardinae, Ephippium and Desmosanthes. A single species of sect. Brachyantha (B. farreri) appeared as a sister to two species from sect. Cirrhopetalum (including B. pingnanense and B. inconspicuum) with strong support (BP=100, PP = 1.00). Additionally, B. ambrosia and B. gedangense formed a separate and strongly supported clade. Notably, B. hamatum, a newly described species belonging to sect. Cirrhopetalum and recently published by Yan et al. [19], appeared as a sister to B. tianguii (sect. Brachyantha). Subsequently, B. violaceolabellum (sect. Brachyantha) was also a sister to these two species with strong support (BP=100, PP = 1.00).
3. Discussion
3.1. The Characteristics of Chloroplast Genomes
Owing to the highly conserved structure, uniparental inheritance and mutation rates were between those shown in the mitochondrial and nuclear genomes, cp genomes has been widely employed for investigating phylogenetic relationships [22,23]. Recently, orchids have become a focal point in phylogenetic studies due to their rich diversity, wide distributions and epiphytic habits. With the decreasing costs of sequencing, an increasing number of cp genomes evolution in Orchidaceae has been studied [31,32]. The genus Bulbophyllum, serves as one of the representative groups of orchid biodiversity [1,2,4], the cp genomes of their diversity patterns and evolutionary adaptations are attracting much attention [24,30,33].
This study sequenced complete cp genomes of seven orchid species in genus Bulbophyllum and compared with other 21 Bulbophyllum species in order to broaden the knowledge about the genome organization and molecular evolution of the Orchidaceae species. The obtained seven cp genomes of Bulbophyllum species in this study possessed typical quadripartite structure, with the genome sizes of these cp genomes varying from 145,092 bp (B. kwangtungense) to 165,812 bp (B. crassipes), and the GC content ranging from 36.60% (B. plumosum) to 38.04% (B. leopardinum), all of which fell within the normal range of cp genomes reported in previous studies [34,35]. The gene order and content were not different from those of its closely related Bulbophyllum species [27,30,33,36,37].
Although the general structure of Bulbophyllum cp genomes is conserved, differences in ndh gene composition were detected. The ndh genes encode the thylakoid NADH complex [38], which is frequently pseudogenized or lost in Orchidaceae [39,40]. Recently, studies of the orchid cp genomes have revealed that rampant independent loss of the ndh genes occurred in different orchid clades. The cp genome of E. pusilla contains truncated versions of ndhJ, C, D, B, G, and H, and lacks sequences for ndhK, F, E, A, and I [41], and the pseudogenization of ndh genes in the Cleisostoma-Gastrochilus clades is widespread [24]. In this study, all of the cp genomes showed evidence of gene pseudogenization or loss except B. affine, B. andersonii, B. crassipes, B. farreri, B. gedangense, B. hirtum, B. leopardinum, B. lingii, B. menghaiense, B. orientale, B. pectinatum, B. pentaneurum, B. shanicum, B. triste and B. violaceolabellum (Table 1, Supplementary Table S10). Some studies have suggested that inactivation of ndh genes may be associated with epiphytic habitats [42] and connected to the extreme water availability and use of CAM (Crassulacean acid metabolism) photosynthesis [24,43], such as the ndh genes were extensively pseudogenized in Cymbidium mannii, an epiphyte with constitutive CAM [44]. Although Bulbophyllum is primarily an epiphytic group and utilizes the CAM pathway [18,45], more research is needed to understand the relationship between the evolution of the CAM pathway or growth form and the cp genomes.
3.2. Repeat Sequence Analysis
As the inheritance variation, long repeat sequences with length greater than 30 bp are universal in angiosperms and considered to play an important role in genome stability and structural variation [46]. There were abundant long repeat sequences in the cp genomes of Bulbophyllum species in previous studies [33], and a total of 18–49 long repeats were detected in our study (Figure 3B). The palindromic (P) and forward (F) repeats were the most common long repeat sequences in our study (Figure 3A). Slight variation in the number of repeat units and their proportions occurred in different species. Additionally, the GC content of IR regions was much higher than compared to the LSC and SSC regions (Table 1), and these characteristics were also revealed in other plant species, primarily because of the presence of rRNA (rrn4.5, rrn5, rrn16, and rrn23) genes in this region [47].
Simple sequence repeats (SSRs) are highly abundant and randomly distributed throughout the genome, making them valuable genetic molecular markers for population genetic relationships and phylogenetic studies [48]. A number of SSR markers were discovered in several orchid genus such as Vanda [49] and Dendrobium [50]. The most abundant SSR type was the mononucleotide repeat, and the majority of SSRs in Bulbophyllum species was composed of A/T SSRs [27,30,33]. In this study, a total of 38–80 SSRs and six types of SSRs (mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats) were detected (Figure 3C, 3D). A/T SSRs were found to be more abundant than G/C SSRs (G/C was only detected in B. triste) and may be due to a bias towards A/T in cp genomes [51]. Of di- to hexa-nucleotide SSRs among Bulbophyllum species, most of SSRs were specific to each species (Figure 3D). These SSRs were distributed widely and randomly throughout the chloroplast genomes, and were usually located in the intergenic spacer (IGS) region, which is consistent with angiosperm cp genomes [30]. Most of the previous studies revealed that the richness of SSR types is various in different species, which may contribute to the genetic variations differently among species [52]. Notably, some of the SSRs repeats were highly specific, such as AC/GT and AAATCC/ATTTGG SSRs was only detected in the cp genomes of B. farreri, AGATAT/ATATCT SSRs was only detected in B. gedangense and ATCCCC/ATGGGG was only detected in B. hamatum, respectively. Furthermore, B. crassipes and B. orientale (the members of sect. Racemosae) possessed ACT/AGT SSRs, B. hirtum, B. shanicum, B. triste (the members of sect. Lemniscata) possessed AATCT/AGATT SSRs, which consistent with some results of phylogenetic analysis (Figure 3C, Figure 8). Thus, these SSRs have the potential to be specific molecular markers for establishing molecular evolutionary history and demographic diversity of Bulbophyllum species. These results are significant for identifying and analyzing genetic diversity in Bulbophyllum.
3.3. Codon Usage Analysis
Codons are involved in protein translation, vital for the genetic information transfer process of an organism. Codon usage bias is a significant factor in cp genomes evolution, influencing gene functions expression. Organisms with close genetic relationships exhibit similar codon usage bias [53]. These studies can help to clarify evolutionary relationships and improve the efficiency of gene expression in research utilizing genetic transformation [54]. More recently, a variety of orchids cp genomes have been sequenced, allowing for the comprehensive analysis of codon preferences [55,56]. The codon usage bias in Bulbophyllum cp genomes showed similar patterns, as indicated by the comparative analysis of RSCU values (Figure 4, Supplementary Table S6). According to the RSCU analysis, it was found that the most of the frequently used codons (RSCU > 1) ended in A or U, while the less frequently used codons (RSCU < 1) ended in C or G. Among all codons, leucine (Leu) had the highest occurrence, while cysteine (Cys) had the lowest frequency. This trend is consistent with observations in most angiosperm cp genomes [57] and further demonstrates the high conservation in 28 Bulbophyllum species.
3.4. Expansion and Contraction of IRs, Sequence Divergence and Nucleotide Diversity
Boundary shifts between the IRs and SC regions are a common occurrence in the evolution of angiosperms and are the main factors contributing to the differences in the length and gene content of cp genomes [55]. For instance, the IR region of the cp genome of Pelargonium × hortorum was expanded extensively, its length was increased to 76 kb [58]. In general, the gene arrangement of the IR/SC boundary was highly conserved (Figure 5), with some differences in the IR/SSC junction were detected. In B. hamatum and B. tianguii, the ycf1 gene was completely located within the SSC region, while in the other species, the ycf1 gene crossed over JSA. At the junction between JSB, some species lost the ndhF gene. This result indicated that there was no significant expansion or contraction in the IR regions of Bulbophyllum. This may be one of the primary factors contributing to the high conservation of the cp genome structure.
The divergent regions as molecular markers could provide abundant valuable information for DNA barcoding and phylogenetic studies, and numerous phylogenetic reconstructions researches using divergent hotspots [59]. Recently, various plastid markers have been proposed for Orchidaceae. Dong et al. suggested that eleven mutational hotspot regions could be used as potential DNA barcodes, including five non-coding regions (ndhB intron, ccsA-ndhD, rpl33-rps18, ndhE-ndhG, and ndhF-rpl32) and six coding regions (rps16, ndhC, rpl32, ndhI, ndhK, and ndhF) [60]. We identified several prominent divergent regions in this study, including ndhF-trnLUAG, trnTUGU-trnLUAA, trnFGAA-ndhJ, rps15-trnNGUU, rbcL-accD and psbI-trnSGCU (Figure 7A). These regions exhibited a nucleotide diversity greater than 0.065. The psbI-trnS, ndhF-trnL, trnF-ndhJ and trnT-trnL regions have been identified or utilized in previous studies of Bulbophyllum [27,30,61,62], further supports previous results. Although four protein-coding genes (ycf1, matK, psbK and rps12) also showed high Pi values, they are still highly conserved, with nucleotide exceeding 0.015 (Figure 7B). Futhermore, IR regions were highly conserved and had more mutation sites compared to the coding region, which is consistent with previous studies in Orchidaceae [40,55] (Figure 5). Our results suggest that intergenic regions may be more suitable for DNA barcode investigation in Bulbophyllum.
3.5. Phylogenetic Analysis
Complete cp genomes are valuable resources for analyzing phylogenetic relationships, they have been extensively used for phylogenetic analysis across different plant groups [24,32]. Our phylogenetic analysis of Bulbophyllum, based on complete cp genome and 68 CDS (Figure 8, Supplementary Figure S1), provided strong support for the monophyly of Neotropical clade, sects. Lemniscata, Racemosae and Macrostylida (BS ≥ 98, PP =1.00), in agreement with previous studies [5,27,30,33]. The branch topology and node support rates compared to the phylogenetic relationships constructed using traditional molecular markers also improved [4,5] (Figure 8, Supplementary Figure S1). In addition, B. ambrosia, previously assigned to sect. Leopardinae, was distantly related to other two species (B. leopardimum and B. pectinatum) [5], a result corroborated here. It was noteworthy that B. hamatum, being a member of sect. Cirrhopetalum was closely related to B. omerandrum based on morphological comparison [19], was close to two species from sect. Brachyantha (B. tianguii and B. violaceolabellum) with high support (Figure 8, Supplementary Figure S1). Two species, i.e., B. ningboense and B. gedangense, were identified as unplaced along the spine of Bulbophyllum by Lin et al. and Luo et al. [63,64]. B. ningboense, a species similar to B. chrondriophorum morphologically, was sister to B. pingnanense and B. inconspicuum (Figure 8), with lateral sepals connated partly and sub-umbellate raceme [19], basically in accordance with the characteristics of sect. Cirrhopetalum. The phylogenetic analysis further strongly supported that B. ningboense is closely related to B. pingnanense within the sect. Cirrhopetalum. B. gedangense, morphologically similar to B. psychoon and B. levinei, was close to the single species B. ambrosia. It appears that more sampling and more evidence are required to understand the evolutionary history of B. gedangense. Our results generally indicated that there was an overlap of species from different sections, especially sects. Brachyantha, Cirrhopetalum and Leopardinae. The conclusions of previous studies that the boundaries between these sections should be reevaluated [5,27,65]. However, our phylogenetic analysis showed that species from sect. Ephippium and sect. Desmosanthes, sect. Stenochilus and sect. Reptantia respectively were sister groups, might due to sampling limited. Therefore, additional cp genomes from Bulbophyllum individuals are necessary to further investigate phylogeny, especially at lower taxonomic levels.
4. Materials and Methods
4.1. Taxon Sampling and DNA Sequencing
In this study, we sequenced seven Bulbophyllum species (B. ambrosia, B. crassipes, B. farreri, B. hamatum, B. shanicum, B. triste and B. violaceolabellum), and their voucher specimens were stored at the herbarium of the College of Forestry, Fujian Agriculture and Forestry University (FJFC). Total genome DNA was extracted using a modified cetyltrimethylammonium bromide (CTAB) method [66]. Sequencing was carried out at Berry Genomics (Beijing, China) using the Illumina HiSeq 2500 platform, with a read length of 150 bp. Approximately 10 Gb of raw data were obtained for each species. In addition to our newly sequenced data, we downloaded available chloroplast genomes of 21 other Bulbophyllum species from GenBank (Table 1).
4.2. Chloroplast Genome Assembly and Annotation
We employed the GetOrganelle pipeline v1.7.5 for de novo cp genome assembly with the default parameters [67]. Subsequently, the “fastg” file was manually examined, and lower-quality fragments were removed using Bandage v.0.8.1 to obtain circular cp genomes [68]. Gene annotation was carried using PGA (Plastid Genome Annotator) software [69] with Bulbophyllum lingii (MW161052) as the reference genome. Manual checking and adjustments of the annotation results, including the determination of initiation and termination codon positions and identification of gene pseudogenization or loss were performed using the Dual Organellar GenoMe Annotator (DOGMA) [70] and Geneious v11.0.11 [71]. Further, the circular genome map was generated online using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw, accessed on 1 November, 2023) [72]. The annotated cp genome sequences have been submitted to NCBI (Table 1). All cp genomes obtained from NCBI underwent reannotation using PGA tool. Geneious v11.0.11 was employed to analyze the length and guanine-cytosine (GC) content of the entire chloroplast genome, including the Large Single Copy (LSC), Small Single Copy (SSC), and Inverted Repeat (IR) regions. Additionally, we examined the number of genes and categories.
4.3. Repeat sequence Characterization
We identified four types of long repeats within the chloroplast genomes of 28 Bulbophyllum species using the REPuter program (https://bibiserv.cebitec.uni-bielefeld.de/reputer, accessed on 1 November, 2023) [73]. The parameters for repeat identification were set as follows: (1) hamming distance = 3; (2) minimum repeat size≥30 bp; and (3) maximum computed repeats of 50 bp. To determine the positions and types of microsatellites (SSRs), we employed the microsatellite identification tool MISA, available online at https://webblast.ipk-gatersleben.de/misa/ [74]. We used the following thresholds: 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively [30]. The characteristics of repeat sequences were visualized using the R package ggplot2 [75].
4.4. Relative Synonymous Codon Usage Analysis
Codon usage and relative synonymous codon usage (RSCU) values were estimated using Codon W, accessible at http://codonw.sourceforge.net/ (accessed on 1 November, 2023) [76]. To minimize sampling errors, we excluded repeat sequences and protein-coding regions (CDSs) shorter than 300 bp from the codon usage calculations. This step was necessary since short CDS can lead to estimation errors in codon usage. TBtools v1.1047 was employed to create the heat map for the RSCU analysis [77].
4.5. Genome Structure Comparisons and Sequence Divergence Analysis
To investigate variations in the boundaries of the LSC/IR/SSC regions in 28 Bulbophyllum chloroplast genomes, we conducted the SC/IR boundary analyses using Perl script CPJSdraw.pl (https://github.com/xul962464/CPJSdraw, accessed on 1 November, 2023). For visualizing identity across the 28 cp genomes, we employed the shuffle-LAGAN mode of the mVISTA program, with B. affine (LC556091) as the reference genome (http://genome.lbl. gov/vista/mvista/submit.shtml, accessed on 1 November, 2023) [78]. Mauve was utilized to perform analyses of cp genome rearrangement using default “seed families” and default values. In all sequences, one of the IR regions was consistently removed [79]. Nucleotide variability (Pi) for the 28 Bulbophyllum cp genomes and the 68 protein-coding genes was calculated using DnaSP v6.0 with a window length of 100 bp and a step size of 25 bp [80].
4.6. Phylogenetic Analysis
In accordance with previous molecular systematic studies [27,33,37], we selected a total of 33 chloroplast genomes from 33 species for this study. The selection includes 28 species from Bulbophyllum and five species from Dendrobium (D. chrysanthum, D. findlayanum, D. hercoglossum, D. longicornu and D. moschatum) serve as the outgroups (Supplementary Table S9). A total of 68 protein-coding genes (excluding ndh genes due to their widespread loss or truncation in Bulbophyllum) were extracted using PhyloSuite v1.2.2 [81] and aligned them using MAFFT v.7 [82]. The complete chloroplast genomes were aligned by MAFFT and trimmed using TrimAl v1.2 to remove poorly aligned positions [83]. For phylogenetic analysis, we utilized the CIPRES Science Gateway, specifically RaxML-HPC2 on XSEDE 8.2.12, PAUP on XSEDE 4.a168 and MrBayes on XSEDE 3.2.7, applying three methods including maximum likelihood (ML), maximum parsimony (MP) and Bayesian inference (BI) [84]. In ML analysis, we specified the GTRGAMMA model for all datasets and calculated bootstrap values on 1000 bootstrap replicates using heuristic searches [85,86]. In MP analysis, we conducted a heuristic search involving 1000 random addition sequence repeats, employing TBR branch switching. All characters were treated as equally weighted and unordered. In BI analysis, we utilized the GTR + I + Γ substitution model with MrBayes v. 3.2.7 [87]. The Markov chain Monte Carlo (MCMC) algorithm was run for 10,000,000 generations, with one tree sampled every 100 generations. We discarded the first 25% of trees as burn-in to construct majority-rule consensus trees and estimate posterior probabilities (PP).
5. Conclusions
In this study, we obtained the cp genomes of seven Bulbophyllum species (B. ambrosia, B. crassipes, B. farreri, B. hamatum, B. shanicum, B. triste, and B. violaceolabellum) and compared them with 21 related species to investigate cp genome differences. We found that these cp genomes exhibited high similarity in terms of the genome size, gene content, gene order and differences observed in ndh gene composition. Additionally, long repeat sequences in the cp genomes of Bulbophyllum species were abundant with an obvious A/T preference. A number of exclusive SSRs presented in some species are useful molecular markers for species identification and detecting genetic diversity. RSCU analysis revealed that the codon usage bias in Bulbophyllum cp genomes showed similar patterns. Six highly variable regions (ndhF-trnLUAG, trnTUGU-trnLUAA, trnFGAA-ndhJ, rps15-trnNGUU, rbcL-accD and psbI-trnSGCU) were identified as potential specific DNA barcodes, serving as mutational hotspots for further phylogenetic studies. Based on cp genomes sequences, 28 Bulbophyllum species can be divided into four clades and sects. Brachyantha, Cirrhopetalum, Leopardinae defined by morphology were non-monophyly. This study further supports the significance of cp genomes in elucidating the phylogeny of Bulbophyllum.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
M.-H.L. and Y.G.: Conceptualization. Y.W. and M.-Y.Z.: Methodology, Software; Y.W.; M.-Y.Z.; S.L.; Z.-J.L. and S.Z.: Data curation, Writing-Original draft preparation, Writing Reviewing and Editing. Y.W. and H.-X.W.: Validation; Resources. All authors read and approved the final manuscript.
Funding
This research was supported by the National Key Research and Development Program of China (2023YFD1600504).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are provided within this manuscript and supplementary materials.
Acknowledgments
We acknowledge the technical support by lab staff during the conduction of lab experiments, Ding-Kun Liu, Xiong-De Tu, Cheng-Yuan Zhou.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gravendeel, B.; Smithson, A.; Slik, F.J.W.; Schuiteman, A. Epiphytism and pollinator specialization: Drivers for orchid diversity? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2004, 359, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Gamisch, A.; Comes, H.P. Clade-age-dependent diversification under high species turnover shapes species richness disparities among tropical rainforest lineages of Bulbophyllum (Orchidaceae). BMC Evol. Biol. 2019, 19, 1–16. [Google Scholar] [CrossRef]
- POWO. Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. Published on the Internet. Available online: http://www.plantsoftheworldonline.org/ (accessed on 15 December 2023).
- Pridgeon, A.M.; Cribb, P.J.; Chase, M.W.; Rasmussen, F.N. Genera Orchidacearum Vol. 6 Epidendroideae; Oxford University Press: New York, NY, USA, 2014; pp. 1–687. [Google Scholar]
- Hu, A.-Q.; Gale, S.W.; Liu, Z.-J.; Suddee, S.; Hsu, T.-C.; Fischer, G.A.; Saunders, R.M.K. Molecular phylogenetics and floral evolution of the Cirrhopetalum alliance (Bulbophyllum, Orchidaceae): Evolutionary transitions and phylogenetic signal variation. Mol. Phylogenetics Evol. 2020, 143, 106689. [Google Scholar] [CrossRef] [PubMed]
- Dressler, R.L. Phylogeny and Classification of the Orchid Family; Dioscorides Press: Portland, OR, USA, 1993; pp. 1–311. [Google Scholar]
- Chen, Y.-G.; Xu, J.-J.; Yu, H.; Qing, C.; Zhang, Y.-L.; Wang, L.-Q.; Liu, Y.; Wang, J.-H. Cytotoxic phenolics from Bulbophyllum odoratissimum. Food Chem. 2008, 107, 169–173. [Google Scholar] [CrossRef]
- Lalitharani, S.; Mohan, V.R.; Maruthupandian, A. Pharmacognostic investigations on Bulbophyllum albidum (Wight) Hook. F. Int. J. PharmTech Res. 2011, 3, 556–562. [Google Scholar]
- Reichenbach, H.G. Catalog der Orchideen-Sammlung von G.W. Schiller zu Overlgönne an der Elbe. Annal, Botan. System. 1861, 6, 264. [Google Scholar]
- Bentham, G. Notes on Orchideæ. Biol. J. Linn. Soc. Lond. 1881, 18, 281–360. [Google Scholar] [CrossRef]
- Bentham, G.; Hooker, J.D. Orchideae. In Genera Plantarum: Ad Exemplaria Imprimis in Herberiis Kewensibus Servata Definite, 1st ed.; Lovell Reeve & Co., Limited: London, UK, 1883; Volume 3, pp. 466–636. Available online: https://www.biodiversitylibrary.org/item/186440#page/768/mode/1up (accessed on 15 December 2023).
- Seidenfaden, G.; Wood, J.J.; Holttum, R.E. The Orchids of Peninsular Malaysia and Singapore; Olsen: Tokyo, Japan, 1992; pp. 1–779. [Google Scholar]
- Garay, L.A.; Hamer, F.; Siegerist, E.S. The genus Cirrhopetalum and the genera of the Bulbophyllum alliance. Nord J Bot. 1994, 14, 609–646. [Google Scholar] [CrossRef]
- Clements, M.A.; Jones, D.L. Nomenclatural changes in the Australian and New Zealand Bulbophyllinae and Eriinae (Orchidaceae). The orchadian 2002, 13, 498–501. [Google Scholar]
- Vermeulen, J.J.; Schuiteman, A.; De Vogel, E.F. Nomenclatural changes in Bulbophyllum (Orchidaceae; Epidendroideae). Phytotaxa 2014, 166, 101–113. [Google Scholar] [CrossRef]
- Seidenfaden, G. Orchid Genera in Thailand VIII: Bulbophyllum Thou. Dansk Bot. Ark. 1979, 33, 1–228. [Google Scholar]
- Chase, M.W.; Cameron, K.M.; Freudenstein, J.V.; Pridgeon, A.M.; Salazar, G.; Van den Berg, C.; Schuiteman, A. An updated classification of Orchidaceae. Bot. J. Linn. Soc. 2015, 177, 151–174. [Google Scholar] [CrossRef]
- Gamisch, A.; Winter, K.; Fischer, G.A. Evolution of Crassulacean Acid Metabolism (CAM) as an escape from ecological niche conservatism in Malagasy Bulbophyllum (Orchidaceae). New Phytol. 2021, 231, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Li, X.-W.; Wu, J. Bulbophyllum hamatum (Orchidaceae), a new species from Hubei, central China. Phytotaxa 2021, 523, 269–272. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, P.-Y.; Xu, X.-W.; Zhao, Z.; Lin, Y.-J.; Liu, Z.-J. Bulbophyllum contortum (Orchidaceae, Malaxideae), a new species from Yunnan, China. Phytotaxa 2022, 560, 295–300. [Google Scholar] [CrossRef]
- Miao, J.-L.; Zhang, H.-Y.; Zhu, W.-T.; Liu, Z.; Ji, H.-Y.; Liu, Z.-J.; Zhai, J.-W. Bulbophyllum deergongense (Dendrobiinae; Malaxideae; Epidendroideae; Orchidaceae), a new species from Tibet, China. Phytotaxa 2023, 619, 197–202. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S. Sequencing Angiosperm Plastid Genomes Made Easy: A Complete Set of Universal Primers and a Case Study on the Phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 2019, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-K.; Tu, X.-D.; Zhao, Z.; Zeng, M.-Y.; Zhang, S.; Ma, L.; Zhang, G.-Q.; Wang, M.-M.; Liu, Z.-J.; Lan, S.R.; et al. Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma-Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 2020, 145, 106729. [Google Scholar] [CrossRef]
- Li, L.; Wang, W.; Zhang, G.; Wu, K.; Fang, L.; Li, M.; Liu, Z.; Zeng, S. Comparative analyses and phylogenetic relationships of thirteen Pholidota species (Orchidaceae) inferred from complete chloroplast genomes. BMC Plant Biol. 2023, 23, 269. [Google Scholar] [CrossRef]
- Chen, J.; Wang, F.; Zhao, Z.; Li, M.; Liu, Z.; Peng, D. Complete Chloroplast Genomes and Comparative Analyses of Three Paraphalaenopsis (Aeridinae, Orchidaceae) Species. Int. J. Mol. Sci. 2023, 24, 11167. [Google Scholar] [CrossRef]
- Yang, J.-P.; Zhang, F.-W.; Ge, Y.-J.; Yu, W.-H.; Xue, Q.-Q.; Wang, M.-T.; Wang, H.-M.; Xue, Q.-Y.; Liu, W.; Niu, Z.-T.; et al. Effects of geographic isolation on the Bulbophyllum chloroplast genomes. BMC Plant Biol. 2022, 22, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-N.; Lao, J.; Liu, H.; Zhang, W.-X.; He, W.; Zhong, C.; Xie, J.; Zhang, S.-H.; Jin, J. Characterization of the chloroplast genome of medicinal herb Polygonatum cyrtonema and identification of molecular markers by comparative analysis. Genome 2023, 66, 80–90. [Google Scholar] [CrossRef]
- Chen, X.-H.; Ding, L.-N.; Zong, X.-Y.; Xu, H.; Wang, W.-B.; Ding, R.; Qu, B. The complete chloroplast genome sequences of four Liparis species (Orchidaceae) and phylogenetic implications. Gene 2023, 888, 147760. [Google Scholar] [CrossRef]
- Tang, H.; Tang, L.; Shao, S.; Peng, Y.; Li, L.; Luo, Y. Chloroplast genomic diversity in Bulbophyllum section Macrocaulia (Orchidaceae, Epidendroideae, Malaxideae): Insights into species divergence and adaptive evolution. Plant Divers. 2021, 43, 350–361. [Google Scholar] [CrossRef]
- Lallemand, F.; Logacheva, M.; Le Clainche, I.; Berard, A.; Zheleznaia, E.; May, M.; Jakalski, M.; Delannoy, E.; Le Paslier, M.C.; Selosse, M.A. Thirteen new plastid genomes from mixotrophic and autotrophic species provide insights into heterotrophy evolution in Neottieae orchids. Genome Biol. Evol. 2019, 11, 2457–2467. [Google Scholar] [CrossRef]
- Tu, X.-D.; Liu, D.-K.; Xu, S.-W.; Zhou, C.-Y.; Gao, X.-Y.; Zeng, M.-Y.; Zhang, S.; Chen, J.-L.; Ma, L.; Zhou, Z.; et al. Plastid phylogenomics improves resolution of phylogenetic relationship in the Cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, Orchidaceae). Mol. Phylogenet. Evol. 2021, 164, 107269. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Páez, M.; Vieira, L.D.N.; Baura, V.A.D.; Balsanelli, E.; Souza, E.M.D.; Cevallos, M.C.; Smidt, E.D.C. Comparative plastid genomics of neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front. Plant Sci. 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Vickrey, T.L. Structural diversity among plastid genomes of land plants. In Advances in Botanical Research; Chaw, S.M., Jansen, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 58, pp. 263–292. [Google Scholar]
- Wen, Y.; Qin, Y.; Shao, B.; Li, J.; Ma, C.; Liu, Y.; Yang, B.; Jin, X. The extremely reduced, diverged and reconfigured plastomes of the largest mycoheterotrophic orchid lineage. BMC Plant Biol. 2022, 22, 448. [Google Scholar] [CrossRef]
- Li, M.-K.; Tang, L.; Deng, J.-P.; Tang, H.-Q.; Shao, S.-C.; Xing, Z.; Luo, Y. Comparative chloroplast genomics of three species of Bulbophyllum section Cirrhopetalum (Orchidaceae), with an emphasis on the description of a new species from Eastern Himalaya. PeerJ 2023, 11, e14721. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, Z.; Fan, Y.; Zhu, F.; Chen, Y.; Ding, X. Comparative plastomic analysis of three Bulbophyllum medicinal plants and its significance in species identification. Acta Pharm. Sin. 2020, 55, 2736–2745. [Google Scholar]
- Sazanov, L.A.; Burrows, P.A.; Nixon, P.J. The plastid ndh genes code for an NADH-specific dehydrogenase: isolation of a complex I analogue from pea thylakoid membranes. Proc. Natl. Acad. Sci. 1998, 95, 1319–1324. [Google Scholar] [CrossRef]
- Feng, Y.L.; Wicke, S.; Li, J.W.; Han, Y.; Lin, C.S.; Li, D.Z.; Zhou, T.T.; Huang, W.C.; Huang, L.Q.; Jin, X.H. Lineage-specific reductions of plastid genomes in an orchid tribe with partially and fully mycoheterotrophic species. Genome Biol. Evol. 2016, 8, 2164–2175. [Google Scholar] [CrossRef]
- Niu, Z.; Zhu, S.; Pan, J.; Li, L.; Sun, J.; Ding, X. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 2017, 7, 2073. [Google Scholar]
- Pan, I.C.; Liao, D.C.; Wu, F.H.; Daniell, H.; Singh, N.D.; Chang, C.; Shih, M.C.; Chan, M.T.; Lin, C.S. Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PloS one 2012, 7, e34738. [Google Scholar] [CrossRef]
- Martín, M.; Sabater, B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010, 48, 636–645. [Google Scholar] [CrossRef]
- Strand, D.D.; D'Andrea, L.; Bock, R. The plastid NAD (P) H dehydrogenase-like complex: structure, function and evolutionary dynamics. Biochem. J. 2019, 476, 2743–2756. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Chen, F.; Zhang, G.-Q.; Zhang, S.-N.; Xiong, J.-S.; Lin, Z.G.; Cheng, Z.-M.; Liu, Z.-J. Origin and mechanism of crassulacean acid metabolism in orchids as implied by comparative transcriptomics and genomics of the carbon fixation pathway. The Plant J. 2016, 86, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.-Q.; Gale, S.W.; Liu, Z.-J.; Fisher, G.A.; Saunders, R.M.K. Diversification slowdown in the Cirrhopetalum alliance (Bulbophyllum, Orchidaceae): Insights from the evolutionary dynamics of crassulacean acid metabolism. Front. Plant Sci. 2022, 13, 794171. [Google Scholar] [CrossRef]
- Nanjala, C.; Wanga, V.O.; Odago, W.; Mutinda, E.S.; Waswa, E.N.; Oulo, M.A.; Mkala, E.M.; Kuja, J.; Yang, J.X.; Dong, X.; et al. Plastome structure of 8 Calanthe s.l. species (Orchidaceae): comparative genomics, phylogenetic analysis. BMC Plant Biol. 2022, 22, 1–22. [Google Scholar] [CrossRef]
- Alzahrani, D.A.; Yaradua, S.S.; Albokhari, E.J.; Abba, A. Complete chloroplast genome sequence of Barleria prionitis, comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genom. 2020, 21, 1–19. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Teh, S.L.; Chan, W.S.; Janna, O.A.; Parameswari, N. Development of expressed sequence tag resources for Vanda Mimi Palmer and data mining for EST-SSR. Mol. Biol. Rep. 2011, 38, 3903–3909. [Google Scholar] [CrossRef]
- Kanga, J.Y.; Lua, J.J.; Qiua, S.; Chen, Z.; Liu, J.J.; Wang, H.Z. Dendrobium SSR markers play a good role in genetic diversity and phylogenetic analysis of Orchidaceae species. Sci. Hortic. 2015, 183, 160–166. [Google Scholar] [CrossRef]
- Sablok, G.; Mudunuri, S.B.; Patnana, S.; Popova, M.; Fares, M.A.; Porta, N.L. ChloroMitoSSRDB: open source repository of perfect and imperfect repeats in organelle genomes for evolutionary genomics. DNA Res. 2013, 20, 127–133. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome Biol. 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Zhang, W.J.; Zhou, J.; Li, Z.F.; Wang, L.; Gu, X.; Zhong, Y. Comparative Analysis of Codon Usage Patterns Among Mitochondrion, Chloroplast and Nuclear Genes in Triticum aestivum L. J. Integr. Plant Biol. 2007, 49, 246–254. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Duan, H.; Tao, K.; Luo, Y.; Li, Q.; Li, L. Complete chloroplast genome structural characterization of two Phalaenopsis (Orchidaceae) species and comparative analysis with their alliance. BMC Genom. 2023, 24, 359. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Ul Rehman, W.; Kaur, A.; Das, S.; Joseph, J.; Singh, A.; Bhardwaj, P. Comprehensive Codon Usage Analysis Across Diverse Plant Lineages. bioRxiv 2023, 567812. [Google Scholar]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef]
- Menezes, A.; Resende-Moreira, L.C.; Buzatti, R.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef]
- Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef]
- Fischer, G.A.; Sieder, A.; Cribb, P.J.; Kiehn, M. Description of two new species and one new section of Bulbophyllum (Orchidaceae) from Madagascar. Adansonia sér 2007, 3, 1. [Google Scholar]
- Hosseini, S.; Dadkhah, K.; Go, R. Molecular systematics of genus Bulbophyllum (Orchidaceae) in Peninsular Malaysia based on combined nuclear and plastid DNA sequences. Biochem. Syst. Ecol. 2016, 65, 40–48. [Google Scholar] [CrossRef]
- Lin, H.-L.; Li, X.-P.; Zhang, J.-H.; Shen, B. Bulbophyllum ningboense, a new species of Orchidaceae from Zhejiang, China. Journal of Zhejiang A&F University 2014, 31, 847–849. [Google Scholar]
- Luo, Y.; Deng, J.-P.; Peng, Y.-L.; Li, J.-W. Bulbophyllum gedangense (Orchidaceae, Epidendroideae, Malaxideae), a new species from Tibet, China. Phytotaxa, 2020, 453, 145–150. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Liu, Z.-J.; Wu, X.-Y.; Huang, J.-X. Bulbophyllum lipingtaoi, a new orchid species from China: evidence from morphological and DNA analyses. Phytotaxa 2017, 295, 218–226. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A Modified CTAB Protocol for Plant DNA Extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis (2nd Ed.). Measurement 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 1, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Bio. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gatew. Comput. Environ. Workshop. 2010, 3, 1–8. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits of phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
Figure 1.
Chloroplast genome maps for seven Bulbophyllum species. Genes on the inside of the large circle are transcribed clockwise, and those on the outside are transcribed counter clockwise. The color-coding of the genes is determined according to their annotation functions. The GC content of the chloroplast genomes is represented by the dashed area.
Figure 1.
Chloroplast genome maps for seven Bulbophyllum species. Genes on the inside of the large circle are transcribed clockwise, and those on the outside are transcribed counter clockwise. The color-coding of the genes is determined according to their annotation functions. The GC content of the chloroplast genomes is represented by the dashed area.
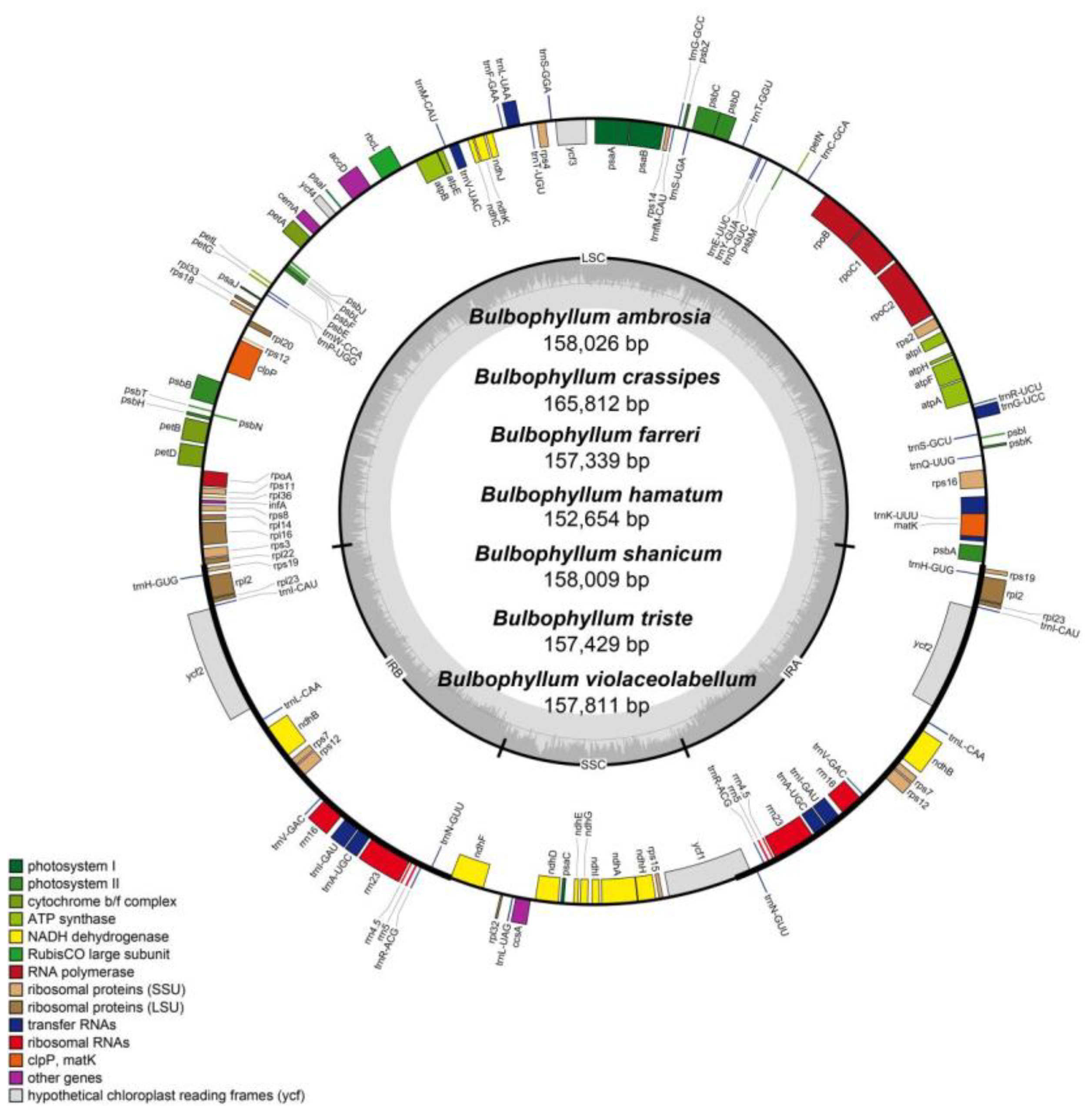
Figure 2.
Summary of sequence repeats across the 28 Bulbophyllum cp genomes. (A) Variation in repeat abundance and type; (B) Number of long repeats by sequence length; (C) Frequency of identified SSR motifs (mono-, di-, tri-, tetra-, penta- and hexa-); (D) Frequency of classified repeat types (considering sequence complementary).
Figure 2.
Summary of sequence repeats across the 28 Bulbophyllum cp genomes. (A) Variation in repeat abundance and type; (B) Number of long repeats by sequence length; (C) Frequency of identified SSR motifs (mono-, di-, tri-, tetra-, penta- and hexa-); (D) Frequency of classified repeat types (considering sequence complementary).

Figure 3.
RSCU value of the codons in the 28 Bulbophyllum cp genomes.

Figure 4.
Comparison of junctions between the LSC, SSC, and IR regions among 28 Bulbophyllum cp genomes.
Figure 4.
Comparison of junctions between the LSC, SSC, and IR regions among 28 Bulbophyllum cp genomes.

Figure 5.
Global alignment of 28 Bulbophyllum cp genomes using mVISTA with B. affine as reference. Thick, gray arrows above the alignment indicate the orientation and position of each gene. The Y-axis represents the identity percentage, ranging from 50 to 100%.
Figure 5.
Global alignment of 28 Bulbophyllum cp genomes using mVISTA with B. affine as reference. Thick, gray arrows above the alignment indicate the orientation and position of each gene. The Y-axis represents the identity percentage, ranging from 50 to 100%.
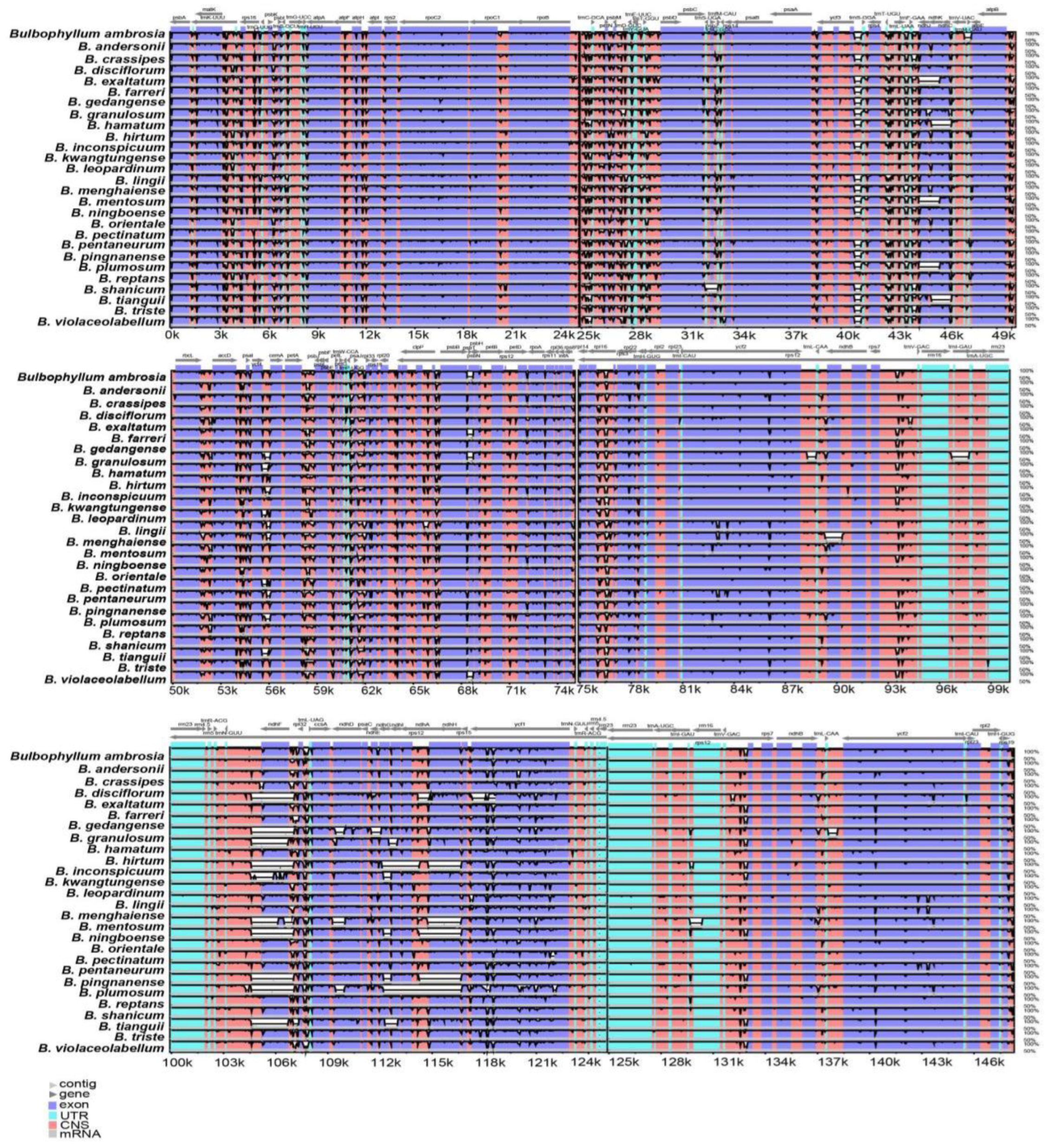
Figure 6.
Alignment of the 28 Bulbophyllum cp genomes (mauve graphs). Local collinear blocks within each alignment are represented by blocks of the same color connected with lines.
Figure 6.
Alignment of the 28 Bulbophyllum cp genomes (mauve graphs). Local collinear blocks within each alignment are represented by blocks of the same color connected with lines.

Figure 7.
The nucleotide diversity (Pi) of 28 Bulbophyllum cp genomes and 68 protein-coding sequences. (A) For the nucleotide diversity of the complete cp genomes using a sliding window test, four mutation hotspot regions were annotated. The window size was set to 100 bp and the sliding windows size was 25 bp. X-axis, the position of the midpoint of a window; Y-axis, Pi values of each window. (B) The nucleotide diversity of 68 protein-coding sequences. X-axis, gene; Y-axis, Pi values of each gene.
Figure 7.
The nucleotide diversity (Pi) of 28 Bulbophyllum cp genomes and 68 protein-coding sequences. (A) For the nucleotide diversity of the complete cp genomes using a sliding window test, four mutation hotspot regions were annotated. The window size was set to 100 bp and the sliding windows size was 25 bp. X-axis, the position of the midpoint of a window; Y-axis, Pi values of each window. (B) The nucleotide diversity of 68 protein-coding sequences. X-axis, gene; Y-axis, Pi values of each gene.
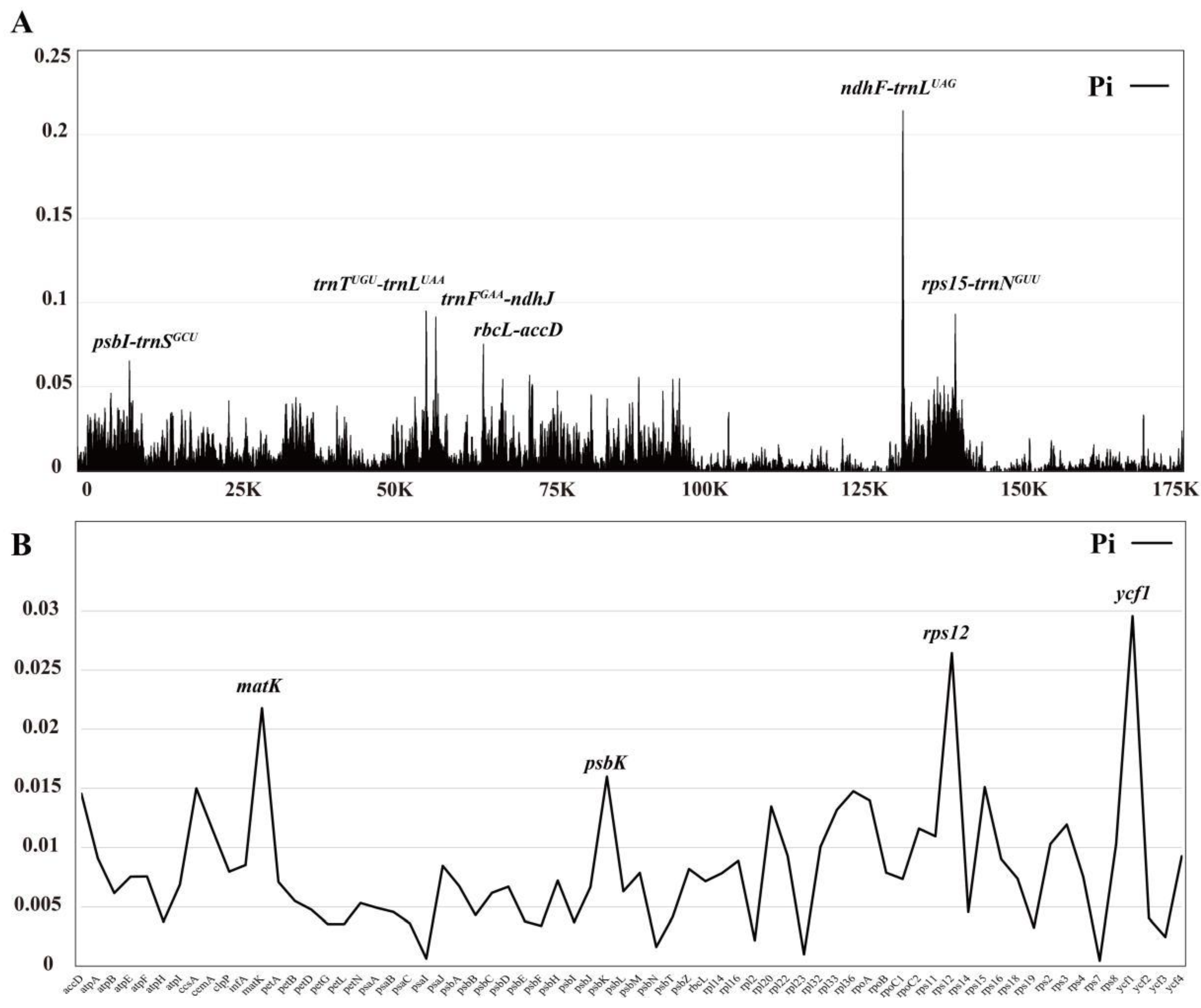
Figure 8.
Phylogenetic tree obtained by maximum-likelihood analysis based on complete cp genomes. The numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BPML, BPMP, PP). *Node is 100 bootstrap percentage or 1.00 posterior probability. The previous recognized sections from Pridgeon et al. [4] and Hu et al. [5] are highlighted by the color of branches.
Figure 8.
Phylogenetic tree obtained by maximum-likelihood analysis based on complete cp genomes. The numbers near the nodes are bootstrap percentages and Bayesian posterior probabilities (BPML, BPMP, PP). *Node is 100 bootstrap percentage or 1.00 posterior probability. The previous recognized sections from Pridgeon et al. [4] and Hu et al. [5] are highlighted by the color of branches.
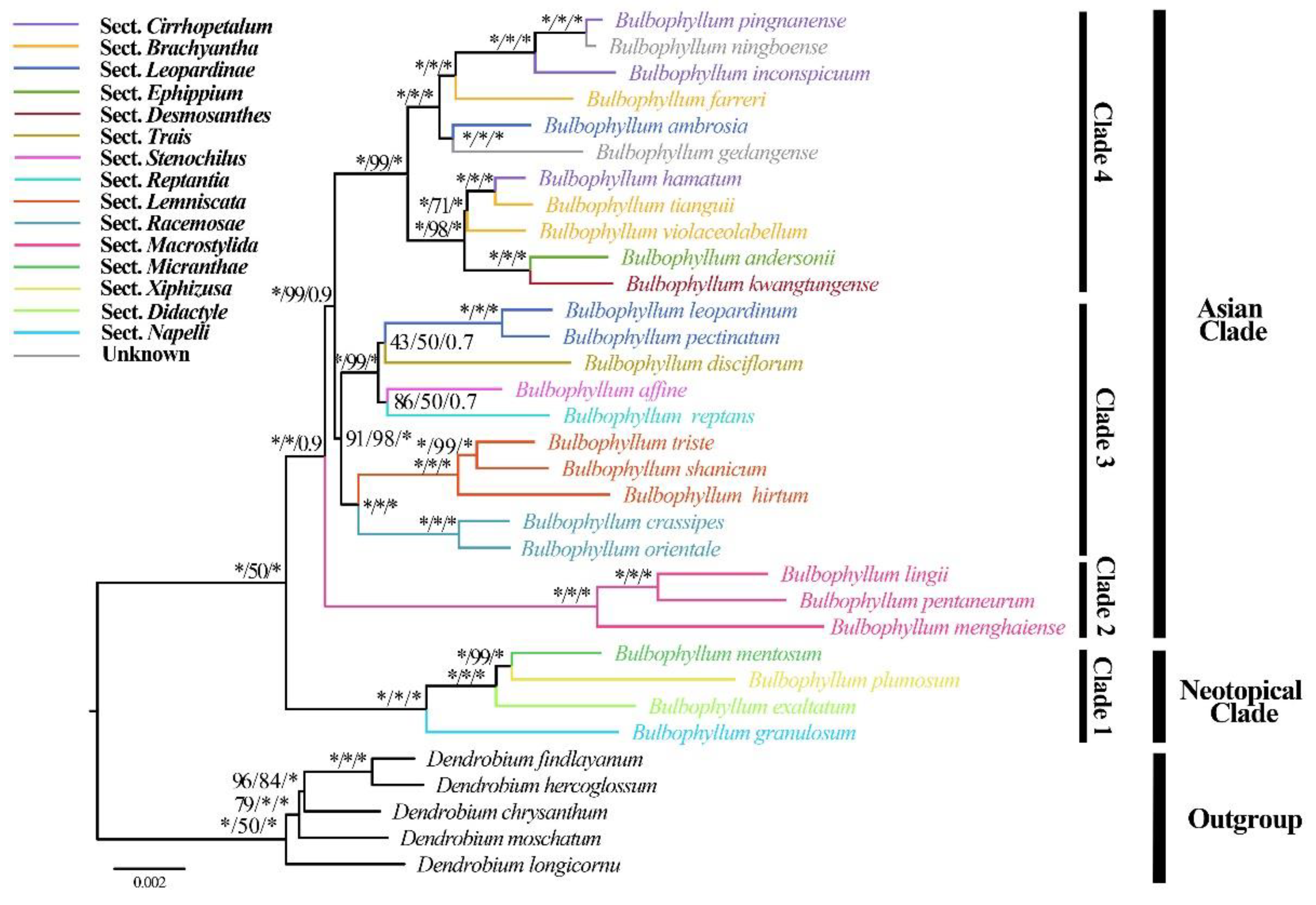

Table 1.
Features of the complete chloroplast genomes of 28 Bulbophyllum species.
| Species | Specimen voucher | Accession No. |
Size (bp) | Number of genes (unique) |
Protein-coding genes (unique) |
tRNA genes (unique) | rRNA genes (unique) | ndh genes loss/pseudogenization | GC% (Total) |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | LSC | SSC | IR | |||||||||
| B. affine | - | LC556091 | 148,230 | 78,178 | 17,280 | 26,386 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.86 |
| B. ambrosia | * | * | 158,026 | 85,821 | 18,622 | 26,804 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 36.95 |
| B. andersonii | Yang202201 | LC703293 | 148,255 | 78,074 | 17,449 | 26,366 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.83 |
| B. crassipes | * | * | 165,812 | 85,690 | 18,293 | 30,927 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.29 |
| B. disciflorum | - | LC498826 | 148,554 | 79,001 | 16,797 | 26,378 | 131 (112) | 77 (70) | 38 (30) | 8 (4) | 1/8 | 37.94 |
| B. exaltatum | Fiorini 218 (HBCB) | NC_048480 | 150,410 | 83,335 | 15,380 | 25,847 | 129 (110) | 76 (70) | 38 (30) | 8 (4) | 3/7 | 36.80 |
| B. farreri | * | * | 157,339 | 85,560 | 18,228 | 26,788 | 132 (113) | 86(79) | 38(30) | 8 (4) | -/- | 36.96 |
| B. gedangense | Y. Luo et al., 1239 | MW161053 | 158,524 | 86,200 | 18,632 | 26,846 | 132 (113) | 86 (79) | 38(30) | 8 (4) | -/- | 36.80 |
| B. granulosum | Mancinelli 1059 (UPCB) | NC_048481 | 151,112 | 84,492 | 15,690 | 25,465 | 128 (110) | 76 (69) | 38 (30) | 8 (4) | 7/2 | 36.70 |
| B. hamatum | * | * | 152,654 | 84,132 | 16,881 | 25,822 | 128 (113) | 80 (79) | 38 (30) | 8 (4) | 4/2 | 36.95 |
| B. hirtum | Yang202105 | LC642724 | 147,382 | 77,587 | 17,129 | 26,333 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.96 |
| B. inconspicuum | PDBK2012-0213 | MN200377 | 149,548 | 85,760 | 12,136 | 25,826 | 127 (108) | 78 (71) | 38 (30) | 8 (4) | 5/3 | 37.00 |
| B. kwangtungense | Yang202107 | LC642722 | 145,092 | 77,192 | 15,376 | 26,262 | 129 (110) | 82 (75) | 38 (30) | 8 (4) | 3/1 | 37.98 |
| B. leopardinum | Yang202102 | LC642723 | 147,514 | 77,762 | 16,996 | 26,378 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 38.04 |
| B. lingii | Y. Luo et al., 2247 | MW161052 | 156,689 | 84,607 | 18,244 | 26,919 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 36.80 |
| B. menghaiense | XY Wang & ZF Xu 202,003 | MW161050 | 156,550 | 84,663 | 18,105 | 26,891 | 131 (112) | 85 (78) | 38 (30) | 8 (4) | -/- | 36.70 |
| B. mentosum | Fiorini 323 (HBCB) | NC_048482 | 150,217 | 83,640 | 13,895 | 26,341 | 125 (106) | 74 (68) | 38 (30) | 8 (4) | 7/5 | 36.70 |
| B. ningboense | - | MW683325 | 151,052 | 86,020 | 13,348 | 25,842 | 128 (109) | 80 (73) | 38 (30) | 8 (4) | 4/2 | 37.00 |
| B. orientale | Yang202104 | LC642725 | 147,388 | 77,392 | 17,206 | 26,395 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 38.01 |
| B. pectinatum | - | LC556092 | 147,169 | 77,478 | 17,529 | 26,081 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 38.01 |
| B. pentaneurum | Y. Luo et al., 2252 | MW161051 | 156,182 | 84,240 | 18,266 | 26,838 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 36.80 |
| B. pingnanense | J.F. Liu 201312 | MW822749 | 151,224 | 86,017 | 13,497 | 25,855 | 128 (109) | 80 (73) | 38 (30) | 8(4) | 4/2 | 37.00 |
| B. plumosum | Imig 606 (HAC) | NC_048479 | 146,401 | 83,260 | 11,089 | 26,026 | 125 (106) | 74 (68) | 38 (30) | 8(4) | 7/5 | 36.60 |
| B. reptans | Yang202106 | LC642726 | 146,928 | 77,088 | 17,038 | 26,401 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.98 |
| B. shanicum | * | * | 158,009 | 85,657 | 18,253 | 27,062 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 36.99 |
| B. tianguii | - | MZ983368 | 151953 | 83,780 | 16,683 | 25746 | 127 (108) | 77 (70) | 38 (30) | 8 (4) | 5/4 | 37.00 |
| B. triste | * | * | 157,429 | 87,177 | 18,199 | 27,039 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 37.04 |
| B. violaceolabellum | * | * | 157,811 | 85,751 | 18,445 | 26,820 | 132 (113) | 86 (79) | 38 (30) | 8 (4) | -/- | 36.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



