
Submitted:
04 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadien-3,5-dione], a component of Curcuma longa L. rhizomes, displays various biological and pharmacological activities. However, it is poorly bioavailable and unstable in physiological pH, which has led more stable and effective curcumin analogs (e.g., monoketone curcuminoids, or MKCs) to be synthesized, and their biological activities to be described. In this review, we cover papers published between 2019 and 2023 on the antimicrobial, anticancer, antioxidant, and antiparasitic actions as well as other less common MKC biological and pharmacological activities. We also address the Claisen-Schmidt condensation as a standard procedure to synthesize MKCs.
Keywords:
Claisen-Schmidt condensation
; deketene curcuminoid
; diarylpentanoids
; monocarbonyl curcuminoids
; monoketone curcuminoids
1. Introduction
Curcumin (or 1,7- (4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) (I, Figure 1), also known as diferuloylmethane, is one of the main hydrophobic phenolic compounds identified in Curcuma longa L. (Zingiberaceae) rhizomes (Hani et al., 2023). Curcumin and its analogs demethoxycurcumin (II) and bisdemethoxycurcumin (III), also present in C. longa rhizomes, are commonly referred to as curcuminoids [1].
Besides being used as a supplement, seasoning, food preservative, flavoring, and coloring in the food industry [2], curcumin has various biological and pharmacological activities, including antioxidant [3], anti-inflammatory [4], antimicrobial [5], anticancer [6], and antiparasitic [7] actions. However, limitations have prevented it from being approved as a therapeutic agent. For example, curcumin is poorly bioavailable because it is little absorbed and rapidly metabolized [8]. Moreover, it has low chemical stability under physiological conditions because its β-diketone moiety is prone to hydrolysis [9]. Furthermore, it undergoes rapid light-induced decomposition [10]
The diverse biological activities of curcumin and its low bioavailability and stability have motivated the synthesis of curcumin analogs bearing a modified β-diketone portion or new substituents in the aromatic moiety [9]. Replacing the β-diketone portion with a monocarbonyl portion increases the chemical stability of the resulting monoketone curcuminoid (MKC) under physiological conditions [11] and provides compounds with interesting biological actions [12]. However, MKCs (IV, Figure 3) have controversial and confusing nomenclature. In the literature, the terms “diphenylpentanoids”, “dibenzylidene ketones”, “1,5-diaryl-penta-1,4-diene-3-ones”, “diarylpentanoids”, “monoketone curcuminoids”, “C5-curcuminoids”, “monocarbonyl curcuminoids”, “deketene curcumin”, and “monocarbonyl curcumin” are used to refer to them. Nevertheless, the general term “curcuminoids”, which includes the dicarbonyl compounds, has been the most used. The lack of a uniform nomenclature for MKCs not only makes searching them in the literature challenging, but also causes their importance to be underestimated.
According to Moreira and co-workers, symmetric (Ar = Ar’) and asymmetric (Ar ≠ Ar’) MKCs are classified on the basis of the carbon atoms of the C5 unit, and the classification depends on whether these carbon atoms are part of an acyclic skeleton or belong to a cyclic fraction. When MKCs bear a cyclic C5 unit, this unit can be part of a cyclopentanone (V), cyclohexanone (VI), piperidin-4-one (VII), tetrahydro-4H-pyran-4-one (VIII), or a tetrahydro-4H-thiopiran-4-one (IX) (Figure 2) [13].
Although extensive reviews on the MKC biological activities have been published over the last decade [11,14,15], a review spanning a more recent period would be welcome. Here, we provide an overview of the recent literature (2019–2023) on the biological activities of MKCs and address some aspects of their synthesis and biological activities (e.g., their insecticidal activity) that have not been discussed yet.
2. Synthesis of Monoketone Curcuminoids (MKCs)
2.1. The Claisen-Schmidt Condensation (CSC) as a Source of MKCs
2.1.1. General Aspects and Stereoselectivity
Unlike curcumin, which can be obtained from natural sources, MKCs can be only produced by synthetic chemical methods. Claisen-Schmidt condensation (CSC) between an aromatic aldehyde and a cyclic (e.g., cyclopentanone, cyclohexanone, piperidin-4-one, tetrahydro-4H-pyran-4-one, or a tetrahydro-4H-thiopiran-4-one) or aliphatic (e.g., propanone) ketone is the standard procedure to synthesize MKCs (Scheme 1). Various aromatic aldehydes can be used because they do not undergo enolization and hence cannot function as the nucleophilic component of the reaction. CSC can be base- or acid-catalyzed; in both cases, excess aromatic aldehyde is employed to ensure that MKC is the final product (Scheme 1a) [16]. Alternatively, excess ketone instead of excess aromatic aldehyde can be used, so that the ketone condenses with one aldehyde only. The resulting product can then react with a different aromatic aldehyde, to produce an asymmetric MKC (Scheme 1b) [17]. Recently, Yadav and Wagh published an extensive review of CSC [18], so we will only address some selected aspects of this reaction.
Enone formation from the aromatic aldol is thermodynamically favored, driven by extended conjugation between the carbonyl and the aromatic ring. A trans-double bond (E configuration) is preferentially formed in acid or base-catalyzed CSC (Scheme 2). This stereoselectivity arises during the elimination step and stems from both steric and stereoelectronic factors. In the case of base-catalyzed CSC, the hydroxyl group is expelled as a hydroxide anion (OH-) through an E1cb mechanism that requires the carbanion (enolate ion) filled p orbital to be in anti with the C‒OH bond σ* orbital [19,20]. Among the two possible conformers (A and B, Scheme 2a) that allow these orbitals to be in anti-arrangement, conformer B is the most stable—the bulkiest phenyl and acetyl groups are located on opposite sides. Consequently, conformer B reacts faster (through a lower-energy transition state) than conformer A, to give the double bond with E configuration As for acid-catalyzed CSC (Scheme 2b), the stereoselectivity originates from an E1-like transition state, where the C-O bond cleavage is considerable [18].
2.1.2. Base-Catalyzed Claisen-Schmidt Condensation
In the standard CSC procedure, acetone, cyclopentanone, cyclohexanone, piperidin-4-one, tetrahydro-4H-pyran-4-one, or tetrahydro-4H-thiopiran-4-one reacts with aromatic aldehyde in a sodium hydroxide (NaOH) alcoholic (methanolic or ethanolic) solution. Initially, aqueous NaOH solution is added to ethanol (EtOH), and the resulting solution is then added to the reaction vessel containing the ketone at 0 °C and stirred for a few minutes. Next, the aromatic aldehyde is added to the vessel under stirring at room temperature. In some methodologies, at the end of the reaction, the reaction mixture is neutralized by adding a hydrochloric acid (HCl) solution to the vessel [21] In most cases, the resulting solid is separated from the reaction mixture by vacuum filtration and washed with ice water, to remove excess base. Finally, the solid product is dried and recrystallized from hexane/ethyl acetate, EtOH, or EtOH/ to obtain pure crystals [16,21,22,23,24].
Besides NaOH, calcium hydroxide (Ca(OH)2) has been used as a basic catalyst in CSC to obtain MKCs. Homogeneous basic catalysts allow the reaction to be conducted at lower temperature, in a shorter time. Protocols based on catalysts like NaOH/EtOH, potassium hydroxide/EtOH, piperidine/HCl, piperidine, and L-Proline/EtOH have been reported as well. Nevertheless, these methods have certain limitations: excess or stoichiometric amounts of reactants are employed, and the reactants can be corrosive and may not be recoverable [18].
Zhang and co-workers synthesized a series of MKCs by CSC between (E)-4-phenylbut-3-en-2-one and benzaldehyde catalyzed by Ca(OH)2 in diluted EtOH medium (20% v/v) at 60 oC. Reaction for 48 h gave dibenzylideneacetone 1 (Figure 3) in 47% yield. Then, the authors tested different experimental conditions of temperature, catalyst amount, and reaction scale. A temperature of 80 oC, Ca(OH)2 at 10%, and 10 or 100 mmol of each reagent afforded compound 1 in 81% and 80% yield, respectively, after column chromatography and filtration. After the authors optimized the reaction conditions, they tested other aromatic aldehydes and demonstrated that electron-deficient aldehydes are preferable because they give higher MKC yield after the product is purified [25].
2.1.3. Acid-Catalyzed CSCs
Although some experimental aspects have led base-catalyzed CSC to be more often used to synthesize MKCs, acid-catalyzed CSC is preferred when the aromatic aldehyde structure contains acid sites other than the α-carbonyl hydrogen (e.g., the hydrogen of phenol hydroxyl groups). Acetic acid/HCl gas [26], acetic acid/H2SO4 [27]or Lewis acids like zinc chloride, niobium pentachloride, or cesium carbonate have recently been employed to obtain MKCs [18].
2.2. Synthesis of MKCs through Oxidative Catalysis
Waldron and co-workers designed a method to synthesize benzylacetone and 4-(4-methoxyphenyl)-2-butanone from benzyl alcohol and 4-methoxybenzyl alcohol in a flow system composed of three micropacked bed reactors operating at 115, 130, and 120 oC, respectively. The methodology employs gold-palladium (Au-Pd) nanoparticles supported on titanium dioxide (TiO2) and affords MKCs as secondary products. In this system, the compounds are synthesized via oxidation, aldolic condensation, and reduction pathways; AuPd/TiO2, TiO2 anastase, and 1 wt% Pt/TiO2 are used for oxidation, C-C coupling, and reduction, respectively. During coupling between the alcohol and the ketone, two aldehyde molecules couple to the ketone, to produce MKCs 1 and 11 (Figure 3). Lower temperatures (80–100 oC) promote selectivity for the desired product (> 80%), while higher temperatures favor benzaldehyde production for the coupling reaction between benzaldehyde and the ketone [28].
2.3. Using Ionic Liquids and Apolar Solvents in the Synthesis of MKCs
Interest in using ionic liquids (ILs) in aldol condensation reactions has increased because ILs are a safer and cleaner reaction medium than organic solvents [19]. Thus, some authors have used ILs to obtain MKCs. For instance, Subhedar and co-workers designed a method to synthesize compounds 174-185 (Figure xx) by using the IL [Et3NH+][HSO4−] as a medium/catalyst via a one-pot multicomponent approach. The authors proposed that the reaction starts with aldehyde carbonyl protonation by [Et3NH+][HSO4−], which is followed by piperidone enolization and further nucleophilic attack to the carbonyl of the aromatic aldehyde, with the consequent formation of a C-C bond. Subsequent protonation and water elimination generates compound 160. The IL increases the electrophilicity of the carbon atom of the C-Cl bond in 2-chloro-N-phenylacetamide (Scheme 3). Then, nucleophilic substitution of 2-chloro-N-phenylacetamides for- intermediate 159 accelerates C-N bond formation, to produce compounds 214-218 [19].
3. Biological Activities of MKCs
3.1. An Overview of the Biological Activities of MKCs
Over the last decade, diverse biological activities, such as antimicrobial, anticancer, anti-inflammatory, antiangiogenic, antioxidant, anticoagulant, antidiabetic, and antiparasitic actions, have been reported for MKCs. Some of these biological activities (e.g., anticancer [13,29], antibacterial [14], and anti-inflammatory [30] actions) have been the subject of recent reviews. This section will focus on an update of these activities and on studies that have not been discussed yet. In addition, other activities that have not been reviewed (for example, antidiabetic, antiangiogenic, anticoagulant, antiparasitic, and insecticidal actions) will be addressed (Table 1). The structures of compounds listed in Table 1 are shown in Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9 and Figure 10.
3.2. Anticancer Activity
Cancer is one of the main causes of death worldwide. The disease consists of abnormal cell proliferation, with later invasion of different body parts. Current cancer treatment requires that chemotherapy, radiation therapy, surgery, and even hormonal therapy be combined [60] The MKC anticancer activity has been extensively investigated and was the subject of two excellent reviews, one by Rodrigues and co-workers in 2019 [29] and the other by Moreira and co-workers in 2020 [13]. Here, we will only update the literature on the MKC anticancer activity with papers published between 2020 and 2023, which were not included in the abovementioned reviews.
Recent literature on the MKC anticancer activity has focused on the MKC antiproliferative activity (i.e., tumor cell growth suppression) and cytotoxicity (i.e., toxicity to cells). The mechanisms underlying such activities have also been addressed. For instance, when it comes to antiproliferative action, cancer cell lysis, inhibition of tumor cell growth and division, or triggering of genetic apoptosis pathways have been investigated; as for cytotoxicity, cell cycle arrest or delay and DNA degradation up to a certain level have been studied.
The cytokine tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a promising experimental cancer therapeutic drug that is currently being tested in clinical trials [32] TRAIL induces apoptosis by binding to its specific receptors called “death receptors” to kill tumor cells selectively. Because the development of TRAIL to treat many human tumor cells has been reported, compounds that can sensitize cancer cell lines to TRAIL are needed. Prasad and co-workers reported that compound 1 potentiatesTRAIL-induced apoptosis in colon cancer lines and converts TRAIL-resistant cells to TRAIL-sensitive. The authors also found that compound 1 decreases the antiapoptotic protein expression and increases the apoptotic protein expression via activation of the ROS and CHOP (C/EBP homologous transcription factor) pathways [32].
Compound 7 inhibits human lung cancer cells by impairing ER stress-mediated apoptosis [61]. This compound is being evaluated as an anticancer agent at the pre-clinical stage. Chen and co-workers assessed the inhibitory effects of compound 7 against three gastric cancer cell lines (SGC-7901, BGC-823, and SNU-2016), to find that, at concentrations ranging from 9 to 12 µM, this compound reduces the viability of the three cancer cell lines by 50%, whereas curcumin only elicits the same effect at concentrations higher than 30 µM. This reduction in cancer cell viability is due to increased levels of reactive oxygen species (ROS) in the cells. The authors also identified the protein thioredoxin/thioredoxin reductase (TrxR) as a potential target of compound 7. This flavoenzyme is overexpressed in human lung cancers where increased tumor growth and drug resistance have been observed [34].
Because compound 57 is being evaluated in pre-clinical trials, Zhang and co-workers investigated the mechanism and target of curcumin A in colon cancer cells. This compound targets TrxR1 and increases ROS levels, to activate the JNK signaling pathway in human colon cancer cells. Moreover, in combination with cisplatin, compound 57 enhances the growth inhibition in colon cancer cells and increases the ROS accumulation. In combination with cisplatin in vivo. Besides that, curcumin A was shown to inhibit tumor growth in a colon cancer xenograft model and to attenuate the body weight loss caused by treatment with cisplatin [33].
Compounds 227 and 171 display anticancer activity. Compound 171 kills lung cancer lines by producing ROS and activating apoptotic mechanisms [62]. Compound 171 kills ovarian carcinoma cell lines via apoptotic mechanisms, but the role played by ROS production in compound 171-mediated cell death is unclear [63]. Monroe and co-workers combined compound 171 or 227 with cisplatin (cis-diaminedichloroplatinum(II)) and investigated the inhibitory effects of these combinations on the viability of the A549 lung cancer cell line. The authors reported 24-h IC50 of 1.74 ± 0.28, 13.82 ± 0.63, and 10.91 ± 0.19 µM for compound 227, compound 171, and cisplatin, respectively. Moreover, they reported that cisplatin and compounds 227 and 171 affect the apoptosis-induced factor (AIF), caspase-12, c-Jun N-terminal kinase (JNK), mitogen-activated protein kinase (MAPK), and Src expression similarly, but they do not impact the caspase-3/7, -8, and -9 activities. They found that the auditory threshold shifts induced by cisplatin decrease when this drug is combined with compound 171 or 227, so these combinations might prevent cisplatin ototoxicity (i.e., the cisplatin side effect of damaging the inner ear or causing balance issues). On the other hand, combination with compound 171 or 227 may counteract the cisplatin effect by increasing ROS production [38].
More recently, Ghosh and co-workers synthesized 20 analogs of compound 171 (Table 1) and assessed their inhibitory effects on pancreatic cancer cells. The authors found that compounds 206 and 207 act against MiaPaCa-2 (IC50 = 0.29 ± 0.12 µM and 0.31 ± 0.05 µM, respectively) and Panc-2 (IC50 = 0.51 ± 0.15 µM and 0.53 ± 0.20 µM, respectively) cancer cells more effectively than irinotecan, the positive control (IC50 = 1.29 ± 0.36 µM and 49 ± 0.58 µM against MiaPaCa-2 and Panc-2, respectively). The authors also found that compounds 213 and 211 are moderately active against these pancreatic carcinoma cancer cells (compound 213: IC50 = 0.37 ± 0.14 µM and 0.64 ± 0.20 µM against MiaPaCa-2 and Panc-2, respectively; compound 211: IC50 = 0.32 ± 0.12 µM and 0.77 ± 0.18 µM against MiaPaCa-2 and Panc-2, respectively, and that they display antiproliferative activity after 24 h and up to 72 h. The authors observed treatment with 182 and compound 207 for 72 h is not toxic to THP1 cells (a monocyte cell line) at a concentration of 10 µM. Additionally, the authors found that the pro-apoptotic activity of compound 207 in pancreatic cancer cells surpasses the apoptosis induction properties of compound 171, and that compound 207 activates caspase-3, suppresses the antiapoptotic BCL2 and BCL-XL expression, and increase PARP cleavage in pancreatic cancer cells. They concluded that the antipancreatic cancer activity is favored by the N-acryloyl-piperidin-4-one moiety combined with 3,4-difluoro- or 3-fluoro-4-methoxy-substituted phenyl rings [22].
Curcumin A (10) inhibits human gastric cancer cells with no observable toxicity to normal cells [64]. This MKC also inhibits prostate [65] and cervical [66] cancer cell lines and induces cancer cell death by activating the apoptosis pathway in gastric [67] and colorectal [68] cancer cells. Lee and co-workers reported that curcumin A induces potent cytotoxicity against glioblastoma U-87 MG (EC50 = 6.78±1.04 µM) and SH-SY5Y neuroblastoma (EC50 = 4.72 ± 1.05 µM) cells and displays antiproliferative effects on both types of cells. The authors also verified that the caspase-3 activity increases and Bcl-2 concentration decreases in a dose- and time-dependent increase upon treatment with curcumin A, indicating that this MKC induces apoptosis in these cells [35]. Also in 2021, Wahab and co-workers reported higher cytotoxicity of curcumin A-treated androgen-independent prostate cancer (AIPC) cell lines DU 145 (EC50 = 7.57 ± 0.2 µM) and PC-3 (EC50 = 7.80 ± 0.7 µM) compared to curcumin (EC50 = 34.25 ± 2.7 µM and 27.77 ± 6.4 µM against DU 145; and PC-3 cell lines, respectively). The authors also verified that compound 10 displays higher dose- and time-dependent antiproliferative activity against AIPC cells than curcumin. On the basis of morphological observations, increased caspase-3 activity, and reduced Bcl-2 protein levels in these cells, the authors concluded that curcumin A induces apoptosis and inhibits the DU 145 and PC-3 cell migration. They suggested that the curcumin A anti-cancer activity is due to modulation of differentially expressed genes (DEGs) associated with the cell cycle-apoptosis and PI3K pathways [36]. Additionally, Tajuddin and co-workers reported that curcumin A (10) exhibits higher inhibitory effect on two types of non-small cell lung cancer (NSCLC) cells – squamous cell carcinoma (NCI-H520, EC50 = 4.7±0.1 µM) and adenocarcinoma (NCI-H23, EC50 = 3.7±0.4 µM) – compared to curcumin (NCI-H520, EC50 = 25.2 ± 1.7 µM; and NCI-H23, EC50 = 18.5 ± 0.7 µM). The authors also found that curcumin A promotes apoptosis, increases the caspase-3 activity, and decreases the Bcl-2 protein concentration in both cell lines in a time- and dose-dependent manner [37].
Huber and co-workers evaluated the in vitro antiproliferative activity of curcumin (I) and 31 MKCs against human cancer cell lines, namey A2780 (ovarian), C33A (cervix), and MDA-MB-231 (breast) (Table 1). They found that compound 159, which is a 4-hydroxy-cyclohexanone-derived MKC, displays the lowest IC50 (0.68 μM, 0.69 μM, and 0.92 μM, respectively), which are lower than the cisplatin IC50 (1.30 μM, 3.69 μM, and 19.13 μM, respectively). A comparison of the IC50 values of a series of 4-hydroxy-cyclohexanone-derived MKCs revealed that the IC50 is influenced by the lipophilicity and electronic and steric properties of the aryl substituents. The relative potency of the most active 4-hydroxy-cyclohexanone-derived MKCs decreases in the order meta-NO2 (compound 159) > 4′-pyridyl (compound 167) > meta-Cl (compound 155) > ortho-NO2 (compound 158) > para-NO2 (compound 157) > 3′-pyridyl (compound 166) > hydrogen (compound 152). Physicochemical parameters, estimated for other 4-hydroxy-cyclohexanone-type MKCs synthesized by the authors, indicate that compound 167 [IC50 =0.76 μM (A2780), 2.69 μM (C33A), 1.28 μM (MDA-MB-231)] is expected to be more bioavailable than of curcumin (I). Based on circular dichroism measurements, the authors concluded that these MKCs do not bind to DNA in vitro [27].
Recently, Yu and co-workers synthesized seven derivatives (compounds 39-45) of curcumin A (10) and assessed their cytotoxicity to human hepatocellular carcinoma cells (HepG2). According to the authors, compound 43 exhibits higher antiproliferative activity (IC50 = 11.33 µM) than curcumin (IC50 = 32.83 µM), the positive control. Mechanistic studies revealed that compound 43 inhibits the ability of HepG2 cells to form clones and suppresses HepG2 cell migration, thereby inhibiting liver cancer progression. Furthermore, the results of these studies indicates that compound 43 dissipated the mitochondrial transmembrane potential, an early event in apoptosis, downregulates the pp-Bcl/Bcl-2 ratio, and upregulates c-caspase 3/caspase 3 ratio [23].
Protein p53 is a powerful tumor suppressor that plays a central role in cell cycle regulation, apoptosis, and DNA repair, among other mechanisms. The p53 pathway is activated under cellular stress signals, compromising tumor development and growth and preventing damaged cells from proliferating. However, p53 is inactivated by interacting with endogenous negative regulators (e.g., murine double minute (MDM)2 and MDMX) in cells with oncogenic potential. Moreira and co-workers carried out an in silico study to evaluate the potential of an MKC library to disrupt the interaction between p53 and MDM2/X. Thereafter, the authors synthesized the compounds that scored the highest during the docking studies on drug-likeness and ADMET prediction properties and evaluated their antiproliferative activity in colon cancer HCT116 and fibroblasts HFF-1. Among the 27 MKCs synthesized by the authors (228-254), compounds 237 and 254 were shown to display the highest in vitro antiproliferative activity in HCT116 cells and low toxicity in normal cells. Thereafter, the authors evaluated the potential of compounds 237 and 254 these compounds to interact with p53-MDM2/X was evaluated through yeast-based assays, to find that compound 237 is a potential p53-MDM2/X dual inhibitor, and that the antiproliferative effect of this compound in HCT116 cells is associated with the inducted cell cycle arrest, apoptosis, PARP cleavage, and increased expression of p53 and its transcriptional targets, p21 and PUMA [39]. In the same year, Novais and co-workers investigated the mechanism of action of compound 254 in cancer cells and verified that the compound inhibits the growth of a potent tumor with high selectivity index (SI). The authors reported that the antiproliferative activity of compound 254 stems from mitosis inhibited by perturbed microtubules. This causes irreversible defects in chromosome congression during mitosis, as well as prolonged spindle assembly checkpoint-dependent mitotic arrest with subsequent massive apoptosis [40].
Taleb and co-workers used to modified techniques to prepare poly-nano micelles by using poly-caprolactone polyurethane β-cyclodextrin (PCL-PU-βCD) amphiphilic copolymer containing compound 1 or 35. The authors reported that, compared to pure curcumin, the resultingnanocomposites have controlled sustained release, promising physicochemical properties, low cytotoxicity, and a satisfactory IC50 in a breast tumor cell culture (MCF-7) compared to pure curcumin [41].
Razali and co-workers synthesized compounds 179 and 192 and assessed their cytotoxicity in LN-18 human glioblastoma cells as compared to curcumin. The authors found that compounds 179 and 192 (IC50 = 2.4 µM and 4 µM, respectively) are more effective than curcumin (IC50 = 31 µM) and kill LN-18 cells in a concentration-dependent manner after treatment for 14 h. In addition, the authors found that compounds 179 and 192 are selective to LN-18 cells compared to the non-cancerous HBEC-5i cell line, with SI (selective index) of 2.33 and 2.25, respectively. Moreover, the authors reported that compounbds 179 and 192 significantly increases the superoxide anion and hydrogen peroxide levels after treatment for 2 h and 6 h, respectively, confirming that oxidative stress is involved in cell death induced by these compounds. Finally, the authors described higher antimigratory effects of compounds 179 and 192 through inhibition of LN-18 cell’s migration and invasion were also reported to be higher as compared to curcumin (I) [42].
More recently, Clariano and co-workers evaluated the potential of 17 MKCs (Table 1) to treat colorectal cancer and found that these compounds are 1.3 to 13 times more effective than curcumin in HCT116 cells. Besides that, all the tested compounds proved to be more stable in PBS (phosphate buffer saline) with 20% acetonitrile at 37oC for 48 h than curcumin and obeyed the ‘drug-likeness properties. The authors identified MKC 26 as the most promising antitumoral compound, with an IC50 of 1.95 µM [21].
3.3. Antimicrobial Activity
3.3.1. Antibacterial Activity
MKCs have been extensively reported for their antimicrobial activity against bacteria, fungi, and viruses. In recent years, some excellent reviews on the MKC antibacterial activity of have been published [11,14]. Therefore, this review will only cover the literature on the MKC antibacterial activity of s published since 2021.
Polaquini and co-workers synthesized and evaluated eight assymmetric MKCs (compounds 49, 51-56) against Mycobacterium tuberculosis and a panel of Gram-positive and Gram-negative bacteria based on Minimum Inhibitory Concentration (MIC) values. According to these authors, compounds 52, 54, and 56 showed broad spectrum and potent antibacterial activity, mainly against M. tuberculosis (MIC = 0.9 µg/mL), Acinetobacter baumannii (MIC = 3.9, 7.8, and 3.9 µg/mL, respectively), and methicillin-resistant Staphylococcus aureus (MIC = 15.6, 15.6, and 7.8 µg/mL, respectively. The authors reported that compound 51 is more selective than compounds 54 and 56 in cytotoxicity assays in human lung cells, with a S ranging from 5.4 to 15.6. Moreover, compound 52 is not genotoxic to A549 cells and is more stable than curcumin in phosphate buffer (pH 7.4) for 24 at 37 oC. Finally, the authors demonstrated that compound 52 can disrupt the B. sutilis divisome without damaging its cytoplasmic membrane. The authors also suggested that the presence of a p-OH substituent is more relevant to the antibacterial activity than the presence of m-OMe group [49].
Gagandeep and co-workers investigated the antibacterial activity of 17 water-soluble MKCs (74-90) against M. tuberculosis (H37Rv), to find that all the compounds exhibit good to moderate antibacterial activity, with MIC99 ranging from 3.12 to 25.0 µM. Compounds 76 and 89 were shown to be the most potent, with MIC99 ranging from 3.12 to 6.25 µM. The authors reported that these compounds are nonhemolytic, nontoxic, and stable under physiological and reducing conditions. Apart from that, compound 89 has moderate in vitro metabolism by liver human microsomes (half-life of 1.2 h; intrinsic clearance of 1.12 mL/h/mg) [69].
Nivedha and co-workers investigated the antimicrobial activity of compound 188 against Escherichia coli and Staphylococcus aureus was by using the inhibition zone method. The authors reported that 188 at 40 µg/mL displays the largest inhibition zone against both bacteria [16]. More recently, Jonathan and co-workers investigated the antimicrobial activity of compounds 190 and 191 against Bacillus subtilis, S. aureus, E. coli, and Pseudomonas aeruginosa by using the disc diffusion method, to find that both the compounds control the E. coli growth, even at low concentrations. In fact, compound 190 has even higher antibacterial activity than ampicillin, the positive control [50].
3.3.2. Antifungal Activity
Nagargoje and co-workers synthesized 16 chlorocarcinoline-based MKCs (Table 1) and assessed their antifungal activity against Candida albicans (CA), Fusarium oxysporum (FO), Aspergillus flavus (AF), Aspergillus niger (AN), and Cryptococcus neoformans (CN). The authors compared the MIC of these compounds and miconazole (positive control). They found that compound 100 and 197 are the most active against all the fungal strains (MIC= 12.5-25 μg/mL), and that compounds 47, 48, 99, 114, 116, 117, 194, 195 and 196 exhibit the same antifungal potency as the positive control (miconazole). The authors reported that the cyclopentanone-derived MKCs (compounds 99 and 100) and acetone-derived MKCs (compounds 46, 47, and48) have higher activity than the positive control (miconazole). However, the greater antifungal activity of compound 197 compared to compound 196 may be related to the presence of the methyl group. This result agrees with the authors’ hypothesis that the presence of electron-donating groups in the quinoline moiety increases the antifungal potential. In addition, piperidone (in compounds 194 and 195) and the N-methylpiperidone spacer (in compounds 196 and 197) also increase the antifungal activity, which may also be due to the presence of electron-releasing groups in the aromatic rings. Comparison between the fungicidal potential of these MKCs and the positive control indicates that most of the tested compounds are more active than miconazole [12].
In the same year, the same authors reported the antifungal activity of nine propargyl MKCs (Table 1) and tested their antifungal activities against C. albicans, F. oxysporum, A. flavus, A. niger, and C. neoformans, to find that some compounds are more effective against the selected fungi than the positive control (miconazole): compounds 120 and 147 against C. albicans (MIC = 12.5 µg/mL); compounds 120 and 199 against F. oxysporum (MIC = = 12.5 µg/mL), compound 183 against A. niger (MIC = = 12.5 µg/mL), and compound 120 against C. neoformans (MIC = = 12.5 µg/mL) [48].
2.3.3. Antiviral Activity
Two recent reviews have highlighted the antiviral potential of curcumin [70,71]. Although Kumari and co-workers have reported that curcumin A (10) displays more potent anti-HIV activity (IC50 = 2 µM) against peripheral blood mononuclear cells (PBMCs) than curcumin (IC50 = 12 µM) [72], the antiviral activity of MKCs has been little exploited.
The dengue virus has prevalence in tropical and subtropical regions of the world, nevertheless, it is considered a human pathogen of global importance. When crcumin (I) was tested as an inhibitor of the dengue virus (DENV2 NS2B/NS3 protease), it was found to inhibit viral protease weakly. On the other hand, curcumin A (10) and compound 95 and 104 inhibit the dengue virus more potently and with higher selectivity index (SI > 10) than curcumin [47]. In vitro tests of compounds 10, 95, and 107 against the dengue virus showed that they inhibit viral replication and infectivity (plaque) better than they inhibit protease activity. The IC50 of these compounds are 39.17 ± 6.69 µM (compound 10), 43.88 ± 10.14 µM (compound 95) and 60.98 ± 8.7 µM (compound 107). The authors also assessed whether these compounds inhibit DENV2 infectivity during formation of infectious particles. The authors obtained EC50 (PFU) of 2.68 ± 0.51 µM (compound 10), 5.37 ± 0.49 µM (compound 95), and 2.34 ± 0.22 µM (compound 107), which demonstrated that these MKCs analogs inhibit formation of infectious particles more effectively than curcumin. Only compound 95 (a cyclopentanone-derived MKC) proved to be less toxic than native curcumin (I). The authors showed that the three analogs have selectivity indices (SI) greater than 10 [47].
3.3. Anti-Inflammatory Activity
The MKC anti-inflammatory activity has been extensively investigated. However, this MKC activity has been recently reviewed by Cuainoglou and Hadjipavlou-Litina [30], so we will only cover the literature from 2019 to 2023.
Leong and co-workers investigated the anti-inflammatory activity of 13 asymmetric MKCs (58-70). First, the authors tested all the compounds at 50 µM and found that nine of them (59, 60, 61, 62, 63, 65, 68, 69, and 70) suppress the NO production significantly, by over 50%. According to the authors, compound 65 (IC50 = 17.5 µM) exhibits the strongest NO inhibitory activity, whereas compounds 59-63, and 68-70 display IC50 ranging from 19.7 to 31.5 µM. On the basis of the greater NO inhibitory activity of hydroxylated compounds 63 and 65 compared to their methoxylated (compounds 60 and 666) and halogenated (compounds 58 and 69) analogs, the authors concluded that the hydroxyl group plays a key role in the MKC anti-inflammatory activity. Similarly, the authors found that the a meta-substituted MKCs (compounds 64 and 66) tend to exhibit higher NO inhibitory effects than the para-substituted MKCs (compounds 63 and 67), regardless of the nature of the para-substituent. On the other hand, the higher NO inhibitory activity of the multi-methoxylated MKCs (compounds 61, 62, and 68) compared to the mono-methoxylated MKCs (compounds 60 and 67) led the authors to conclude that the anti-inflammatory activity of MKCs is enhanced by increased electron density in the aromatic ring [44].
Tham and co-workers reported the anti-inflammatory properties of 107 in cellular modes of inflammation and improved mouse survival inlethal sepsis. The authors also used a mouse model for allergic asthma to investigate the therapeutic effect of BHMC on acute airway inflammation. To this end, the authors sensitized and challenged mice with ovalbumin (OVA) to increase airway hyper-responsiveness (AHR) and pulmonary inflammation. Next, compound 107 was intraperitoneally administred at 0.1, 1, and 10 mg/kg doses. This showed that compound 107 significantly reduces the number of eosinophils, lymphocytes, macrophages, and neutrophils at all the tested doses. Moreover, this MKC decreases the Th2 cytokine (IL-4, IL-5 and IL-13) levels in bronchoalveolar lavage fluid (BALF) as compared to OVA-challenged mice. Additionally, the authors described that the three 107 doses (0.1, 1, and 10 mg/kg) suppress inflammatory cell infiltration in the peribronchial and perivascular regions similarly to dexamethasone at 1 mg/kg. At 1 or 10 mg/kg, compound 107 also inhibits goblet cell hyperplasia. These findings demonstrate that compound 107 attenuates acute airway inflammation associated with allergic asthma [46].
3.4. Antioxidant Activity
Antioxidants can prevent some cells from being damaged by harmful reactive species called free radicals. Free radicals have been associated with diseases like cancer, diabetes, liver damage, autoimmune diseases, and heart disease, among others. Thus, antioxidants with the potential to eliminate free radicals play an important role in curing and preventing these diseases [73,74].
The search for new antioxidants led Nagargoje and co-workers to evaluate 16 MKCs (Table 1) based on synthesized 2-chloroquinoline for their in vitro radical scavenging activity; the authors used butylated hydroxytoluene (BHT) as the positive control. The IC50 values revealed that most of the tested compounds display higher radical scavenging activity than BHT. Compounds 117 (IC50=5.39±0.14 µg/mL) and 47 (IC50=6.37±0.55 µg/mL) were found to be three times more active than BHT, followed by compounds 99 (IC50=7.44±0.16 µg/mL), 46 (IC50=8.92±0.18 µg/mL), 48 (IC50=9.62±0.76 µg/mL), and 195 (IC50=9.66±0.53 µg/mL), which are about twice more potent than BHT (IC50=16.47±0.18). These results suggested that the monocarbonyl nucleus present in MKCs contribute to the effect of eliminating radicals by the resonance phenomenon. In addition, structure-activity data indicated that chlorine substitution in the quinoline structure markedly increases the antioxidant activity, as seen in compound 117 compared to compound 114 with less chlorine substitution. Besides that, the authors found that the MKCs derived from acetone are more active than the MKCs derived from cyclopentanone and cyclohexanone. Finally, substitution at position 7 of the quinoline nucleus was found to boost the antioxidant activity as compared to other regioisomers [12].
In the same year, Nagargoje and co-workers assessed the in vitro antioxidant activity of the nine propargylated MKCs (Table 1) in terms of their radical scavenging activity. The authors found that the acetone-derived MKCs display increased antioxidant activity compared to the cyclopentanone-, cyclohexanone-, and 4-piperidone-derived MKCs. According to these authors, compound 38 (IC50 = 12.78 ± 0.71 µg/mL), 101 (IC50 = 13.18 ± 0.34 µg/mL), 198 (IC50 = 15.78 ± 0.47 µg/mL), and 199 (IC50 = 16.17 ± 0.11 µg/mL) displayed greater or equal antioxidant potential as compared with BHT (IC50 = 16.47 ± 0.18 µg/mL), which was used as positive control. The authors noticed a trend in the antioxidant and antifungal activities of these compounds, except in the case of compound 38, which was inactive against fungal strains of C. albicans, F. oxysporum, A. flavus, A. niger, and C. neoformans and exhibited the greatest antioxidant activity [48].
Recently, the antioxidant activity of compound 188 has been investigated by using nine different assays (2,2’-azino-bis-3-ethyl benzothiazoline-6-sulfonic acid (ABTS•+),1,1-diphenyl-2-picryl hydroxyl (DPPH•) radical scavenging assay, nitric oxide scavenging assay, ferric reducing antioxidant power (FRAP), hydrogen peroxide scavenging, superoxide anion radical scavenging assay (O2•−), reducing ability assay, and phosphomolybdenum assay metal ion chelating assay), to find that compound 188 at 40 µg/mL displays antioxidant activity in all of them [16].
3.5. Antiparasitic Activity
Many MKCs are active against parasites that cause malaria [75], and Chagas’ disease [76]. Over the last five years, most of the studies regarding the MKC antiparasitic activity have focused on the antiparasitic activity against Leishmania species, as discussed, below.
Leishmania donovani is the intracellular protozoa parasite that causes visceral leishmaniasis (also known as kala-azar), a neglected tropical disease. Chauhan and co-workers investigated the antileishmanial effects of compound 1 against L. donovani. In 2018, the authors reported the in vitro effects of compound 1 against L. donovani promastigotes and intracellular amastigotes. They found that compound 1 inhibits intracellular amastigotes more effectively than promastigotes (IC50 = 7.43 ± 1.88 μg/mL and 17.80 ± 1.42 μg/mL, respectively), at IC50 close to those measured for miltefosine (0.01–10.9 μg/mL). Regarding the cytotoxicity in vitro, compound 1 kills amastigote forms selectively compared to J774A BALB/c mouse cells, with a SI of 15.34. The authors showed that 1 significantly reduces the trypanothione/trypanothione reductase (TR) system of Leishmania cells [51]. In another study, published in the same year, the authors tracked putative autophagosomes by using the GFP-ATG8 gene as marker to prove that the autophagic cell death promoted by compound 1 in L. donovani is due to autophagic vacuolization [52]. In 2019, the same authors found that the Ldrab6 gene provides L. donovani with resistance via a mechanism of drug-thiol conjugation and sequestration by ABC transporter multidrug resistance-protein A (MRPA) [53]. Further mechanistic studies carried out by the same group indicated that compound 1 can inhibit the GTPase activity of L. donovani Rab6 protein (LdRab6), thereby suggesting that this protein can be a potential target of compound 1 [54].
Silva and co-workers investigated the antiparasitic activity of 18 MKCs (Table 1) against Trichomonas vaginalis. The authors showed that of these 18 curcumin analogs, compounds 1, 23, and 97 are the most effective against T. vaginalis after exposure to MKC for 24 h. For compounds 1 and 23 at 100 µM, the authors observed that parasite viability is reduced by a 98.8%, while compound 97 reduces the parasite viability by 70% as compared to the negative control. Compounds 1, 23, and 97 gave MIC of 80, 90, and 200 μM, respectively, and IC50 of 50, 50, and 70 μM, respectively. The authors also observed that all the propanone-derived-MKCs induce some percentage of inhibition of 100 μM trophozoites, indicating that the substituents present in the aromatic rings and the chain length between the aromatic rings play an essential part in the MKC antiparasitic activity. Thus, compounds that lack substituents (1) or electron-withdrawing substituents such as halogens (2-Cl in compound 23, 4-F in compound 22, and 4-Cl in compound 24) are more active than compounds 20 and 11, which contained electron-donating substituents (i.e., methyl and methoxy, respectively) in the aromatic rings. As for cyclohexanone-derived MKCs derivatives, only compounds containing both electron-releasing and electron-withdrawing substituents (–Cl for 97 and –CH3 for 92) inhibit trophozoite growth. The analogs derived from cyclopentanone and cinnamaldehyde do not reduce trophozoite viability at the tested concentrations. However, the length of the hydrocarbon binder between the aromatic rings is the main factor underlying the antiparasitic activity of these compounds. Compound 1 (5-carbon ligand) was shown to be the most potent inhibitor among the evaluated molecules, while its 9-carbon ligand analog, without substituent in the aromatic rings, does not inhibit trophozoites growth. In other words, the shorter length of the hydrocarbon binder, the greater the antiparasitic activity [24]. Nevertheless, the α-β-unsaturated system is crucial for the MKC antiparasitic activity, as in the case of the MKC antimicrobial activity [77,78].
The in vitro effects of five acetone-derived MKCs (compounds 1, 11, 12, 14, and 24) were evaluated against trypomastigotes and amastigotes of four strains (Brazil, CA-I/72, Sylvio X10/4, and Sylvio X10/7) of Trypanosoma cruzi, the parasite that causes Chagas’ disease (or American trypanosomiasis). Compound 12 (or diveratralacetone) was described to be the most active MKC, with higher anti-Trypanosoma cruzi than curcumin. Its IC50 is lower than 10 μM (1.51–9.63 μM), and its SI is higher than 10 in non-infected C2C12mammalian cells. When tested in female BALB/ mice at oral dose of 300 or 1,000 mg/kg, compound 12 and curcumin do not produce toxic effects or mortality along 14 days. The presence of oxygenated groups such as methoxy at the para-position of the aromatic ring plays a key role in the MKC activity against T. cruzi [55].
More recently, Francisco and co-workers screened 27 MKCs (Table 1)) against Trypanosoma brucei, the parasite that causes Human African Trypanosomiasis (HAT). The authors identified compound 130 (IC50 = 0.20 ± 0.03 µM) as the most active against T. b. brucei, with a CC50 of 2.88 ± 0.86 µM in Humam Embrionic Kidney (HEK293) cells (SI > 14). Time-kill experiments revealed that compound 132 (EC99 = 0.90 ± 0.16 µM) at 1, 2, or 4 µM killed all the parasites at all concentrations tested, whereas pentamidine (positive control) require more than 30 h to kill all the parasites at 0.5, 1, and 2 µM. The antitrypanosomal activity of compound 132 has been tentatively attributed to two main structural features: 1) the enone moiety, which can deplete the essential thiols by Michael addition during reaction with trypanothione, thereby killing the parasite, and 2) the nitro group, which can be involved in ROS production [56].
3.6. Other Activities
3.6.1. Anti-Angiogenic Activity
Angiogenesis is the generation of new blood vessels in tissues from the pre-existing vasculature. Although angiogenesis is crucial for several physiological and pathological processes, such as wound healing, it can also aggravate tumor progression, some ophthalmic conditions, rheumatoid arthritis, and obesity [31].
The vascular endothelial growth factor (VEGF) is one of the potential anti-angiogenic targets, and its receptors (VEGFR1, VEGFR2, and VEGFR3) are characterized by tyrosine kinase activity [31]. Sun and co-workers found that compound 8 can decrease the phosphorylated forms of serine/threonine kinase Akt, extracellular signal-regulated kinase, and p-38 nitrogen-activated protein, thereby suppressing the downstream protein kinase activation of VEGF. This results in migration inhibition and tube formation in human umbilical vein endothelial cells, arrests microvessel outgrowth from rat aortic rings, and suppresses neovascularization in chicken chorioallantoic membrane. Compound 8 has greater antiangiogenic activity and higher bioavailability than curcumin [31,79]
3.6.2. Anticoagulant Activity
Ahmed and co-workers (2018) evaluated the anticoagulant activity of compounds 103 and 72 by plasma recalcification time (PRT) and bleeding time (BT) experiments and found that these compounds inhibit arachidonic acid (AA)-induced platelet aggregation (IC50 of 65.2 µM and 37.7 µM, respectively) and adenosine diphosphate (ADP)-induced platelet aggregation (IC50 of 750.4 µM and 422 µM, respectively). Aspirin, used as the positive control in the experiments, provided IC50 of 10.01 µM and 308.4 µM, respectively. On the other hand, at concentrations of 30, 100, 300, or 100 µM, compounds 103 and 72 increase the coagulation time 137±2.12, 182.8±5.59, 224.6±8.37 and 284±9.46 s and 128±2.16, 150.6±2.29, 186±3.25 and 223±4.47 s, respectively. At a concentration of 440 µM, heparin increases the coagulation time to 379.40±9.17 s compared to the saline group. On the other hand, at the same concentration, 103 and 72 increase the plasma recalcification time compared to the saline group) respectively. Finally, treatment with 103 and 72 at doses of 100, 300, or 1000 µg/kg prolong the bleeding time [43].
3.6.3. Antidiabetic Activity
α-Glicosidase inhibitors (AGIs) reduce incident type-2 diabetes. AGIs lower blood glucose by delaying carbohydrate digestion and intestinal absorption of compounds 139, 135, and 140 were found to display greater α-glucosidase inhibitory activity compared to curcumin (IC50 = 30.9 µM), with IC50 between 19.4 and 24.9 µM. Compound 139 displays the strongest activity (IC50 = 19.4 µM). Compounds 138, 136, and 137 exhibit moderate α-glucosidase inhibitory activity, with IC50 values ranging from 25 to 35 µM). Given that all the brominated analogs (compounds 134, 139, 135, and 140) inhibit α-glucosidase strongly, the authors concluded that the bromo group is important for α-glucosidase inhibitory activities. However, the position of the substituent in the aromatic ring does not affect the anti-α-glucosidase potential of targeted compounds [44].
Aldose Reductase (AR) enzyme has recently been reported to induce diabetic complications [45]. Khondare and co-workers investigated the aldose reductase inhibition (ARI) effects of a series of 11 MKCs (Table 1) on the basis of their capabilities to reduce dl-glyceraldehyde in the presence of NADPH as a reductant. The authors showed that compounds 8 and 11 showed promising ARI activities, with IC50 values of 5.73 ± 0.28 and 5.95 ± 0.27 µM, respectively. The authors observed that methyl and methoxy substituents in the aromatic rings provide the compound with more significant ARI activity than those compounds with other substituents at the aromatic rings, indicating that the substitution pattern in the phenyl ring affect the MKC ARI activity. Given that most of the MKCs proved to be more active than the corresponding chalcones, the authors concluded that the AR inhibition increases with increasing conjugation [45].
3.6.4. Insecticidal Activity
There are few studies on the MKC insecticidal potential. To mention, Rain and co-workers tested the insecticidal activity of MKCs 1, 2, 5, and 34 against the “cotton mealy bug” (Phenacoccus solenopsis Tinsley (Hemiptera: Pseudococcidae). Alone, compound 2 was shown to display slight activity against P. solenopsis third-instar nymphs of after 24 h, causing about 30% mortality. In combination with neem oil, this compound was demonstrated to have almost 1.5 times higher activity (about 52% mortality) [57].
Anstrom and co-workers reported the mosquitocidal effects of six MKCs (compounds 3, 16, 50, 71, 108, and 148). According to these authors, compounds 50, 16, 108, 148, and 3 inhibit cholesterol binding to Aedes aegypti sterol carrier protein-2, with EC50 values of 12.11, 62.87, 2.38, 2.02, and 0.65 µM, respectively. On the other hand, compounds 71 displays larvicidal activity against fourth instar Ae. aegypti (LC50 = 17.29 µM) [58].
More recently, Matiadis and co-workers analyzed the larvicidal effects of nine MKCs (4, 6, 10, 11, 12, 16, 93, 148, and 149) against Aedes albopictus and Culex pipiens were reported by. Among the tested MKCs, the tetramethoxylated compound 12 was shown to be the most effective against third- to fourth-instar larvae of Ae. albopictus (LC50 = 23.6 ppm) and Cx. pipiens (LC50 = 32.5 ppm) after treatment for 24 h [59].
4. Concluding Remarks
The synthetic methodologies available for obtaining MKCs are attractive because they are mostly accessible, simple, fast, and low-cost. Moreover, MKCs are easy to isolate and can be obtained in good yields, which allows their synthesis to be scaled up. Besides, most of the methodologies are “green” and do not use toxic solvents. In addition, MKCs are more stable (and, in some cases, more active) than curcumin because they are less prone to retro-aldol decomposition.
MKCs display diverse biological and pharmacological activities, being their anticancer and antimicrobial actions noteworthy. The higher selectivity of MKCs for bacterial cells compared to mammalian cells and their low toxicity at antibacterial concentrations make them a promising class of antimicrobial compounds. Regarding the anticancer activity, the nature of the substituents in the aromatic rings strongly influences MKC cytotoxicity and selectivity.
In conclusion, this review has discussed the attractive aspects of the methodologies available for MKC synthesis and the promising activities of these compounds, to show that, notwithstanding the biological and pharmacological potential of MKCs, they are still less exploited than curcumin.
Author Contributions
Conceptualization, A.E.M.C. and L.G.M.; investigation, T.M.V., L.S.T., and V.C.G.H.; writing—original draft preparation, T.M.V., L.S.T, and V.C.G.H.; writing—review and editing, A.E.M.C.; funding acquisition, L.G.M. and A.E.M.C. All the authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the São Paulo Research Foundation (FAPESP), grant numbers 16/24456-1, and 16/19272-9.
Acknowledgments
A.E.M.C. is grateful to the National Council for Scientific and Technological Development of Research and Development (CNPq) for fellowships (proc. 303821/2016 7).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, C.; Lu, H.-F.; Chen, Y.-H.; Chen, J.-C.; Chou, W.-H.; Huang, H.-C. Curcumin, demethoxycurcumin, and bisdemethoxycurcumin induced caspase-dependent and –independent apoptosis via Smad or Akt signaling pathways in HOS cells. BMC Complement. Med. Therap. 2020, 20, 68. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, M.; Yuan, Y. The potential of curcumin-based co-delivery systems for applications in the food industry: Food preservation, freshness monitoring, and functional food. Food Res. Int. 2023, 171, 113070. [Google Scholar] [CrossRef] [PubMed]
- Bērziņa, L.; Mieriņa, I. Antiradical and antioxidant activity of compounds containing 1,3-dicarbonyl moiety: an overview. Molecules 2023, 28, 6203. [Google Scholar] [CrossRef]
- Shi, L.; Qu, Y.; Li, Z.; Fan, B.; Xu, H.; Tang, J. In vitro permeability and bioavailability enhancement of curcumin by nanoemulsion via pulmonary administration. Curr. Drug Deliv. 2019, 16, 751–758. [Google Scholar] [CrossRef]
- Denison, H.J.; Schwikkard, S.L.; Khoder, M.; Kelly, A.F. The chemistry, toxicity and antibacterial activity of curcumin and its analogues. Planta Med. 2023. early access. [Google Scholar] [CrossRef]
- Alam, S.; Verma, S.; Fátima, K.; Luqman, S.; Srivastava, S.K.; Khan, F. Pharmacophore & QSAR guided design, synthesis, pharmacokinetics and in vitro evaluation of curcumin analogs for anticancer activity. Curr. Med. Chem. 2024, 31, 620–639. [Google Scholar] [CrossRef]
- Rai, M.; Ingle, A.P.; Pandit, R.; Paralikar, P.; Anasane, N.; Dos Santos, C.A. Curcumin and curcumin-loaded nanoparticles: antipathogenic and antiparasitic activities. Exp. Rev. Anti Infect. Ther. 2020, 18, 367–379. [Google Scholar] [CrossRef]
- Omidian, H.; Wilson, R.L.; Chowdhury, S.D. Enhancing therapeutic efficacy of curcumin: advances in delivery systems and clinical applications. Gels 2023, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Gagandeep; Kumar, P. ; Kandi, S.K.; Mukhopadhyay, K.; Rawat, D.S. Synthesis of novel monocarbonyl curcuminoids, evaluation of their efficacy against MRSA, including ex vivo infection model and their mechanistic studies. Eur. J. Med. Chem. 2020, 195, 112276. [Google Scholar] [CrossRef]
- Chakraborti, S.; Dhar, G.; Dwivedi, V.; Das, A.; Poddar, A.; Chakraborti, G.; Basu, G.; Chakrabarti, P.; Surolia, A.; Bhattacharyya, B. Stable and potent analogues derived from the modification of the dicarbonyl moiety of curcumin. Biochemistry 2013, 52, 7449–7460. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019, 182, 111631. [Google Scholar] [CrossRef]
- Nagargoje, A.A.; Akolkar, S.V.; Siddiqui, M.M.; Subhedar, D.D.; Sangshetti, J.N.; Khedkar, V.M.; Shingate, B.B. Quinoline based monocarbonyl curcumin analogs as potential antifungal and antioxidant agents: synthesis, bioevaluation and molecular docking study. Chem. Biodiv. 2020, 17, e1900624. [Google Scholar] [CrossRef]
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Diarylpentanoids with antitumor activity: a critical review of structure-activity relationship studies. Eur. J. Med. Chem. 2020, 192, 112177. [Google Scholar] [CrossRef]
- Abdullah, M.A.; Faudzi, S.M.M.; Nasir, N.M. A review on biological properties and synthetic methodologies of diarylpentadienones. Mini-Rev. Med. Chem. 2021, 21, 1058–1070. [Google Scholar] [CrossRef]
- Kumar, B.; Singh, V.; Shankar, R.; Kumar, K.; Rawal, R.K. Synthetic and medicinal prospective of structurally modified curcumins. Curr. Top. Med. Chem. 2017, 17, 148–161. [Google Scholar] [CrossRef]
- Nivedha, J.; Kanimozhi, K.; Olikkavi, S.; Vidhyasagar, T.; Vijayakumar, N.; Uma, C.; Rajeswari, K.; Vennila, L. In vitro screening for antioxidant and antimicrobial properties of 3,5-bis(e-thienylmethylene) piperidin-4-one, a curcumin analogue. Pharmacogn. Res. 2022, 14, 276–283. [Google Scholar] [CrossRef]
- Hawbecker, B.L.; Kurtz, D.W.; Putnam, T.D.; Ahlers, P.A.; Gerber, G.D. The aldol condensation: a simple teaching model for organic laboratory. J. Chem. Ed. 1978, 55, 540–541. [Google Scholar] [CrossRef]
- Yadav, G.D.; Wagh, D.P. Claisen-Schmidt condensation using green catalytic processes: a critical review. ChemistrySelect 2020, 5, 9059–9085. [Google Scholar] [CrossRef]
- Subhedar, D.D.; Shaikh, M.H.; Nagargoje, A.A.; Akolkar, S.V.; Bhansali, S.G.; Sarkar, D.; Shingate, B.B. Amide-linked monocarbonyl curcumin analogues: efficient synthesis, antitubercular activity and molecular docking study. Polycycl. Aromat. Compd. 2020, 42, 2655–2671. [Google Scholar] [CrossRef]
- Mohrig, J.R.; Beyer, B.G.; Fleischhacker, A.S.; Ruthenburg, A.J.; John, S.G.; Snyder, D.A.; Nyffeler, P.T.; Noll, R.J.; Penner, N.D.; Phillips, L.A.; et al. Does activation of the anti proton, rather than concertedness, determine the stereochemistry of base-catalyzed 1,2-elimination reactions? Anti stereospecificity in E1cb eliminations of β-3-trifluoromethylphenoxy esters, thioesters, and ketones. J. Org. Chem. 2012, 77, 2819–2828. [Google Scholar] [CrossRef] [PubMed]
- Clariano, M.; Marques, V.; Vaz, J.; Awam, S.; Afonso, M.B.; Perry, M.J.; Rodrigues, C.M.P. Monocarbonyl analogs of curcumin with potential to treat colorectal cancer. Chem. Biodiv. 2023, 20, e2023002. [Google Scholar] [CrossRef]
- Ghosh, H.; Bhattacharyya, S.; Schobert, R.; Dandawate, P.; Biersack, B. Fluorinated and N-acryloyl-modified 3,5-di[(E)-benzylidene]piperidin-4-one curcuminoids for the treatment of pancreatic carcinoma. Pharmaceutics 2023, 15, 1921. [Google Scholar] [CrossRef]
- Yu, P.; Cao, W.; Zhao, L.; Han, Q.; Yang, S.; Yang, K.; Pan, X.; Wang, Q.; Wang, Y. Design, synthesis, and antitumor evaluation of novel mono-carbonyl curcumin analogs in hepatocellular carcinoma cell. Pharmaceuticals 2022, 15, 950. [Google Scholar] [CrossRef]
- Silva, C.C.; Pacheco, B.S.; Nascimentos das Neves, R.; Dié Alves, M.S.; Sena-Lopes, Â.; Moura, S.; Borsuk, S.; Pereira, C.M. Antiparasitic activity of synthetic curcumin monocarbonyl analogues against Trichomonas vaginalis. Biomed. Pharmacother. 2019, 111, 367–377. [Google Scholar] [CrossRef]
- Zhang, H.; Han, M.; Yang, C.; Yu, L.; Xu, Q. Gram-scale preparation of dialkylideneacetones through Ca(OH)2 - catalyzed Claisen-Schmidt condensation in dilute aqueous EtOH. Chin. Chem. Lett. 2019, 30, 263–265. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Synthesis of new symmetric cyclic and acyclic halocurcumin analogues typical precursors for hybridization. Rev. Chem. Intermed. 2020, 46, 5307–5323. [Google Scholar] [CrossRef]
- Huber, I.; Zupkó, I.; Gyovai, A.; Horváth, P.; Kiss, E.; Gulyás-Fekete, G.; Schmidt, J.; Perjési, P. A novel cluster of C5-curcuminoids: design, synthesis, in vitro antiproliferative activity and DNA binding of bis(arylidene)-4-cyclanone derivatives based on 4-hydroxycyclohexanone scaffold. Res. Chem. Intermed. 2019, 45, 4711–4735. [Google Scholar] [CrossRef]
- Waldron, C.; Cao, E.; Cattaneo, S.; Brett, G.L.; Miedziak, P.J.; Wu, G.; Sankar, M.; Hutchings, G.J.; Gavriilidis, A. Three step synthesis of benzylacetone and 4-(4-methoxyphenyl)butan-2-one in flow using micropacked bed reactors. Chem. Eng. J. 2019, 377, 119976. [Google Scholar] [CrossRef]
- Rodrigues, F.C.; Kumar, N.V.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: an up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef] [PubMed]
- Chauinoglou, E.; Hadjipavlou-Litina, D. Curcumin analogues and derivatives with anti-proliferative and anti-inflammatory activity: Structural characteristics and molecular targets. Ex. Opin. Drug Discov. 2019, 14, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Ward, N.; Panahi, Y.; Sahebkar, A. Anti-angiogenic activity of curcumin in cancer therapy: a narrative review. Curr. Vasc. Pharmacol. 2019, 17, 262–269. [Google Scholar] [CrossRef]
- Prasad, S.; Yadav, V.R.; Ravindran, J.; Aggarwal, B.B. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2019, 71, 538–549. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, P.; Shen, X.; Shao, R.; Wang, B.; Shen, H.; Zhang, J.; Xia, Y.; Zou, P. Curcuminoid WZ26, a TrxR1 inhibitor, effectively inhibits colon cancer cell growth and enhances cisplatin-induced cell death through the induction of ROS. Free Radic. Biol. Med. 2019, 14, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, X.; Zhang, X.; Wang, L.; Cao, P.; Rajamanickam, V.; Wu, C.; Zhou, H.; Cai, Y.; Liang, G.; et al. Curcuminoid B63 induces ROS-mediated paraptosis-like cell death by targeting TrxR1 in gastric cells. Redox Biol. 2019, 21, 101061. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Q.; Rajadurai, P.; Abas, F.; Othman, I.; Naidu, R. Proteomic analysis on anti-proliferative and apoptosis effects of curcumin analog, 1,5-bis(4-hydroxy-3-methyoxyphenyl)-1,4-pentadiene-3-one-treated human glioblastoma and neuroblastoma cells. Front. Mol. Biosci. 2021, 8, 645856. [Google Scholar] [CrossRef] [PubMed]
- Wahab, N.A.A.; Abas, F.; Othman, I.; Naidu, R. Diarylpentanoid (1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadiene-3-one) (MS13) exhibits anti-proliferative, apoptosis induction and anti-migration properties on androgen-independent human prostate cancer by targeting cell cycle–apoptosis and PI3K signalling pathways. Front. Pharmacol. 2021, 12, 707335. [Google Scholar] [CrossRef]
- Tajuddin, W.N.B.W.M.; Abas, F.; Othman, I.; Naidu, R. Molecular mechanisms of antiproliferative and apoptosis activity by 1,5-bis(4-hydroxy-3-methoxyphenyl)1,4-pentadiene-3-one (MS13) on human non-small cell lung cancer cells. Int. J. Mol. Sci. 2021, 22, 7424. [Google Scholar] [CrossRef] [PubMed]
- Monroe, J.D.; Hodzic, D.; Millay, M.H.; G., P.B.; Smith, M.E. Anti-cancer and ototoxicity characteristics of the curcuminoids, CLEFMA and EF24, in combination with cisplatin. Molecules 2019, 24, 3889. [CrossRef] [PubMed]
- Moreira, J.; Almeida, J.; Loureiro, J.B.; Ramos, H.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. A diarylpentanoid with potential activation of the p53 pathway: combination of in silico screening studies, synthesis, and biological activity evaluation. ChemMedChem 2021, 16, 2969–2981. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Moreira, J.; Palmeira, A.; Amorim, I.; Pinto, M.; Cidade, H.; Bousbaa, H. BP-M345, a new diarylpentanoid with promising antimitotic activity. Molecules 2021, 26, 7139. [Google Scholar] [CrossRef]
- Taleb, S.A.A.; Sobh, R.A.; Mourad, R.M. Investigating the Effect of loading curcuminoids using PCL-PU-βCD nano-composites on physico-chemical properties, in-vitro release, and ex-vivo breast cancer cell-line. Biointerface Res. Appl. Chem. 2022, 12, 4074–4102. [Google Scholar] [CrossRef]
- Razali, N.S.C.; Lam, K.W.; Rajab, N.F.; Jamal, A.R.A.; Kamaluddin, N.F.; Chan, K.M. Curcumin piperidone derivatives induce anti-proliferative and anti-migratory effects in LN-18 human glioblastoma cells. Sci. Rep. 2022, 12, 13131. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Khan, A.; Abbass, M.; Filho, E.R.; Din, Z.U.; Khan, A. Synthesis, characterization, molecular docking, analgesic, antiplatelet and anticoagulant effects of dibenzylidene ketone derivatives. Chem. Cent. J. 2018, 12, 134. [Google Scholar] [CrossRef]
- Leong, S.W.; Awin, T.; Faudzi, S.M.M.; Maulidiani, M.; Shaari, K.; Abas, F. Synthesis and biological evaluation of asymmetrical diarylpentanoids as antiinflammatory, anti-α-glucosidase, and antioxidant agents. Med. Chem. Res. 2019, 28, 2002–2009. [Google Scholar] [CrossRef]
- Kondhare, D.; Deshmukh, S.; Lade, H. Curcumin Analogues with aldose reductase inhibitory activity: synthesis, biological evaluation, and molecular docking. Processes 2019, 7, 417. [Google Scholar] [CrossRef]
- Tham, C.L.; Yeoh, S.Y.; Harith, H.H.; Israf, D.A. A synthetic curcuminoid analogue, 2,6-bis-4-(hydroxyl-3-methoxybenzylidine)-cyclohexanone (BHMC) ameliorates acute airway inflammation of allergic asthma in ovalbuminsensitized mice. Mediators Inflamm. 2021, 2021, 9725903. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Pilankatta, R.; Teramoto, T.; Sajith, A.M.; Nwulia, E.; Kulkarni, A.; Padmanabhan, R. Inhibition of dengue virus by curcuminoids. Antivir. Res. 2019, 162, 71–78. [Google Scholar] [CrossRef]
- Nagargoje, A.A.; Akolkar, S.V.; Subhedar, D.D.; Shaikh, M.H.; Sangshetti, J.N.; khedari, V.M.; Shingate, B.B. Propargylated monocarbonyl curcumin analogues: synthesis, bioevaluation and molecular docking study. Med. Chem. Res. 2020, 29, 1902–1913. [Google Scholar] [CrossRef]
- Polaquini, C.R.; Marques, B.C.; Ayusso, G.M.; Morão, L.G.; Sardi, J.C.O.; Campos, D.L.; Silva, I.C.; Cavalca, L.B.; Scheffers, D.-J.; Rosalen, P.L.; et al. Antibacterial activity of a new monocarbonyl analog of curcumin MAC 4 is associated with divisome disruption. Bioorg. Chem. 2021, 109, 104668. [Google Scholar] [CrossRef]
- Jonathan, D.R.; Thendral, E.D.; Priya, M.K.; Shirmila, D.A.; Fathima, A.A.; Yuvashri, R.; Usha, G. Investigations on 3D-structure, properties and antibacterial activity of two new curcumin derivatives. J. Mol. Struct. 2023, 1292, 136063. [Google Scholar] [CrossRef]
- Chauhan, I.S.; Rao, G.S.; Shankar, J.; Chauhan, L.K.S.; Kapadia, G.J.; Singh, N. Chemoprevention of leishmaniasis: in-vitro antiparasitic activity of dibenzalacetone, a synthetic curcumin analog leads to apoptotic cell death in Leishmania donovani. Parasitol. Int. 2018, 67, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Chauhan, I.S. MicroRNA expression profiling of dibenzalacetone (DBA) treated intracellular amastigotes of Leishmania donovani. Exp. Parasitol. 2018, 193, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, I.S.; Rao, G.S.; Subba, G.; Singh, N. Enhancing the copy number of Ldrab6 gene in Leishmania donovani parasites mediates drug resistance through drug-thiol conjugate dependent multidrug resistance protein A (MRPA). Acta Trop. 2019, 199, 105158. [Google Scholar] [CrossRef]
- Chauhan, I.S.; Marwa, S.; Rao, G.S.; Singh, N. Antiparasitic dibenzalacetone inhibits the GTPase activity of Rab6 protein of Leishmania donovani (LdRab6), a potential target for its antileishmanial effect. Parasitol. Res. 2020, 119, 2991–3003. [Google Scholar] [CrossRef]
- Souza, J.M.; Vieira, T.M.; Cândido, A.C.B.B.; Tezuka, D.Y.; Rao, G.S.; Albuquerque, S.; Crotti, A.E.M.; Siqueira-Neto, J.L.; Magalhães, L.G. In vitro anti-Trypanosoma cruzi activity enhancement of curcumin by its monoketone tetramethoxy analog diveratralacetone. Curr. Res. Parasitol. Vector Borne Dis. 2021, 1, 100031. [Google Scholar] [CrossRef] [PubMed]
- Francisco, K.R.; Monti, L.; Yang, W.; Park, H.; Liu, L.J.; Watkins, K.; Amarasinghe, D.K.; Nalli, M.; Polaquini, C.R.; Regasini, L.O.; et al. Structure-activity relationship of dibenzylideneacetone analogs against the neglected disease pathogen, Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2023, 81, 129123. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Jain, S.; Gautam, R.D. Investigation of insecticidal activity of some α,β-unsaturated carbonyl compounds and their synergistic combination with natural products against Phenacoccus solenopsis tinsley. J. Plant Prot. Res. 2012, 52, 146–155. [Google Scholar] [CrossRef]
- Anstrom, D.M.; Zhou, X.; Kalk, C.N.; Song, B.; Lan, Q. Mosquitocidal properties of natural product compounds isolated from chinese herbs and synthetic analogs of curcumin. J. Med. Entomol. 2012, 49, 350–355. [Google Scholar] [CrossRef]
- Matiadis, D.; Liggri, P.G.V.; Kritsi, E.; Tzioumaki, N.; Zoumpoulakis, P.; Papachristos, D.P.; Balatsos, G.; Sagnou, M.; Michaelakis, A. Curcumin derivatives as potential mosquito larvicidal agents against two mosquito vectors, Culex pipiens and Aedes albopictus. Int. J. Mol. Sci. 2021, 22, 8915. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, Y.; Peng, J.; guo, L.; Hu, J.; Cao, M.; zhang, X.; Zhang, H.; Wang, Z.; Li, X.; et al. A synthetic compound, 1,5-bis(2-methoxyphenyl)penta- 1,4-dien-3-one (B63), induces apoptosis and activates endoplasmic reticulum stress in non-small cell lung cancer cells. Int. J. Cancer 2011, 131, 1455–1465. [Google Scholar] [CrossRef]
- Sahoo, K.; Dozmorov, M.G.; Anant, S.; Awasthi, V. The curcuminoid CLEFMA selectively induces cell death in H441 lung adenocarcinoma cells via oxidative stress. Invest. New Drugs 2012, 30, 558–567. [Google Scholar] [CrossRef]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.-P.; Wang, S.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple anticancer activities of EF24, a novel curcumin analog, on human ovarian carcinoma cells. Reprod. Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef]
- Zou, P.; Xia, Y.; Chen, T.; Zhang, J.; Wang, Z.; Chen, W.; Chen, M.; Kanchana, K.; Yang, S.; Liang, G. Selective killing of gastric cancer cells by a small molecule targeting ROS-mediated ER stress activation. Mol. Carcinog. 2016, 55, 1073–1086. [Google Scholar] [CrossRef]
- Citalingam, K.; Abas, F.; Lajis, N.H.; Othman, I.; Naidu, R. Antiproliferative effect and induction of apoptosis in androgen-independent human prostate cancer cells by 1,5-bis(2-hydroxyphenyl)-1,4-pentadiene-3-one. Molecules 2015, 20, 3406–3430. [Google Scholar] [CrossRef] [PubMed]
- Paulraj, F.; Abas, F.; Lajis, N.H.; Othman, I.; Hassan, S.S.; Naidu, R. The curcumin analogue 1, 5-bis(2-hydroxyphenyl)-1,4-pentadiene-3-one induces apoptosis and downregulates E6 and E7 oncogene expression in HPV16 and HPV18-infected cervical cancer cells. Molecules 2015, 20, 11830–11860. [Google Scholar] [CrossRef]
- Yoshida, T.; Maruyama, T.; Miura, M.; Inoue, M.; Fukuda, K.; Shimazu, K.; Taguchi, D.; Kanda, H.; Oshima, M.; Iwabuchi, Y.; et al. Dietary intake of pyrolyzed deketene curcumin inhibits gastric carcinogenesis. J. Funct. Foods 2018, 50, 192–200. [Google Scholar] [CrossRef]
- Cen, L.; Hutzen, B.; Ball, S.; DeAngelis, S.; chen, C.L.; Fuchs, J.R.; Li, C.; Li, P.-K.; Lin, J. New structural analogues of curcumin exhibit potent growth suppressive activity in human colorectal carcinoma cells. BMC Cancer 2009, 9, 99. [Google Scholar] [CrossRef]
- Gagandeep; Singh, M.; Kidawi, S.; Das, U.S.; Velpandian, T.; Singh, R.; Rawat, D.S. Monocarbonyl curcuminoids as antituberculosis agents with their moderate in-vitro metabolic stability on human liver microsomes. J. Biochem. Mol. Toxicol. 2021, 2021, e22754. [CrossRef]
- Jennings, M.R.; Parks, R.J. Curcumin as an antiviral agent. Viruses 2020, 12, 1242. [Google Scholar] [CrossRef]
- Ardebili, A.; Pouriayevali, M.H.; Aleshikh, S.; Zahani, M.; Ajorloo, M.; Izanloo, A.; Siyadapanah, A.; Nikoo, H.R.; Wilairatana, P.; Coutinho, H.D.M. Antiviral therapeutic potential of curcumin: an update. Molecules 2021, 26, 6994. [Google Scholar] [CrossRef]
- Kumari, N.; Kulkami, A.A.; Lin, X.; McLean, C.; Ammosova, T.; Ivanov, A.; Hipolito, M.; Nekhai, S.; Nwilia, E. Inhibition of hiV-1 by curcumin a, a novel curcumin analog. Drug Des. Devel. Ther. 2015, 9, 5051–5060. [Google Scholar] [CrossRef]
- Amran, N.; Rani, A.N.A.; Mahmud, R.; Yin, K.B. Antioxidant and cytotoxic effect of Barringtonia racemosa and Hibiscus sabdariffa fruit extracts in MCF-7 human breast cancer cell line. Pharmacogn. Res. 2020, 8, 66–70. [Google Scholar] [CrossRef]
- Shah, S.S.; Shah, D.; Khan, I.; Ahmad, S.; Ali, U.; Rahman, A.U. Synthesis and antioxidant activities of schiff bases and their complexes: an updated review. Biointerface Res. Appl. Chem. 2020, 10, 6936–6963. [Google Scholar] [CrossRef]
- Dohutia, C.; Chetia, D.; Gogoi, K.; Sarma, K. Design, in silico and in vitro evaluation of curcumin analogues against Plasmodium falciparum. Exp. Parasitol. 2017, 175, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; Bilbao, N.V.; Aguirre-López, B.; Cabrera, N.; Mazariegos, S.D.; et al. Potent and selective inhibitors of Trypanosoma cruzi triosephosphate isomerase with concomitant inhibition of cruzipain: inhibition of parasite growth through multitarget activity. ChemMedChem 2016, 11, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.M.; Santos, I.A.; Silva, T.S.; Martins, C.H.G.; Crotti, A.E.M. Antimicrobial activity of monoketone curcuminoids against cariogenic bacteria. Chem. Biodiv. 2018, 15, 1–6. [Google Scholar] [CrossRef]
- Vieira, T.M.; Ambrosio, M.A.L.V.; Martins, C.H.G.; Crotti, A.E.M. Structure-antimicrobial activity relationships of monoketone curcuminoids. Int. J. Complement. Alt. Med. 2018, 11, 347–350. [Google Scholar] [CrossRef]
- Sun, Y.; Du, L.; Liu, Y.; Li, X.; Li, M.; Jin, Y. Transdermal delivery of the in situ hydrogels of curcumin and its inclusion complexes of hydroxypropyl-β-cyclodextrin for melanoma treatment. Int. J. Pharm. 2014, 469, 31–39. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of curcumin (I), demethoxycurcumin (II), and bisdemethoxycurcumin (III).
Figure 1.
Chemical structures of curcumin (I), demethoxycurcumin (II), and bisdemethoxycurcumin (III).
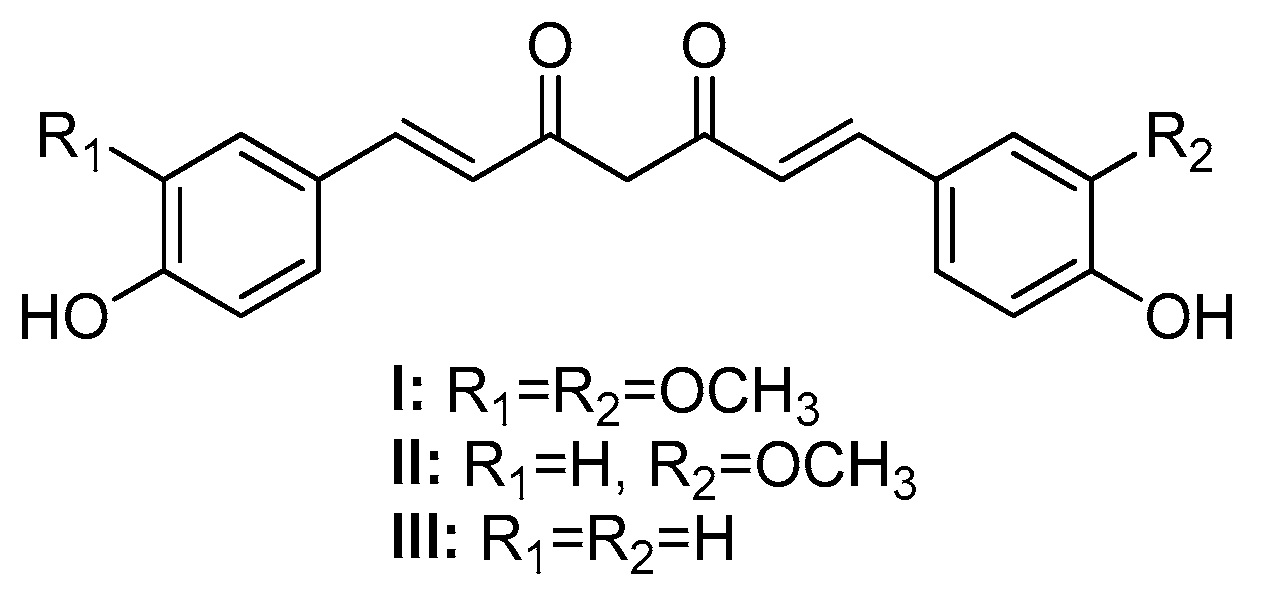
Figure 2.
Structure of MKCs displaying an acyclic (IV) and a cyclic C5 bridge (V-IX) [13].
Figure 2.
Structure of MKCs displaying an acyclic (IV) and a cyclic C5 bridge (V-IX) [13].
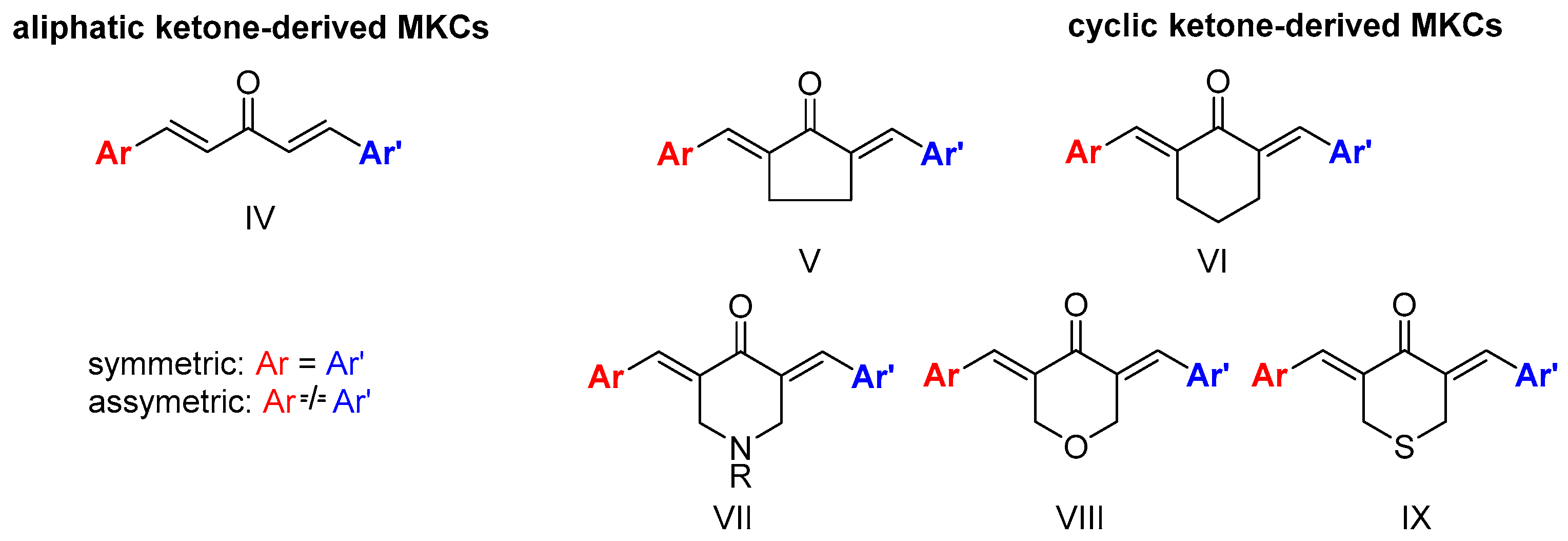
Scheme 1.
MKC synthesis by the Claisen-Schmidt condensation [17].
Scheme 1.
MKC synthesis by the Claisen-Schmidt condensation [17].
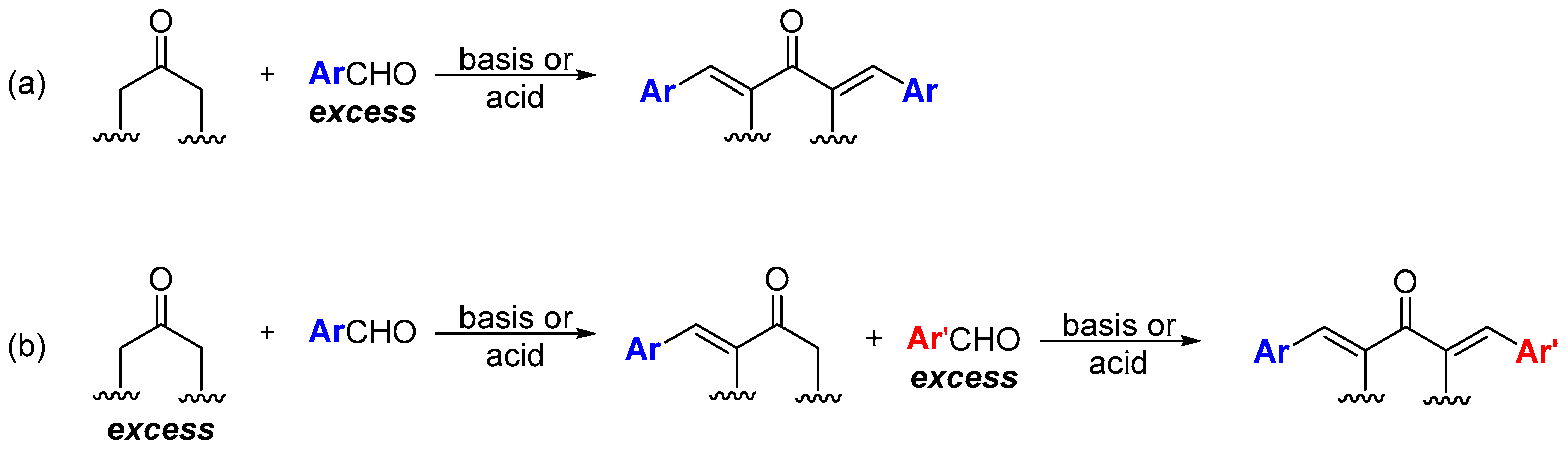
Scheme 2.
Possible conformers involved in double bond formation through the E1cb mechanism (base-catalyzed CSC) and like-E1 mechanism (acid-catalyzed CSC).
Scheme 2.
Possible conformers involved in double bond formation through the E1cb mechanism (base-catalyzed CSC) and like-E1 mechanism (acid-catalyzed CSC).
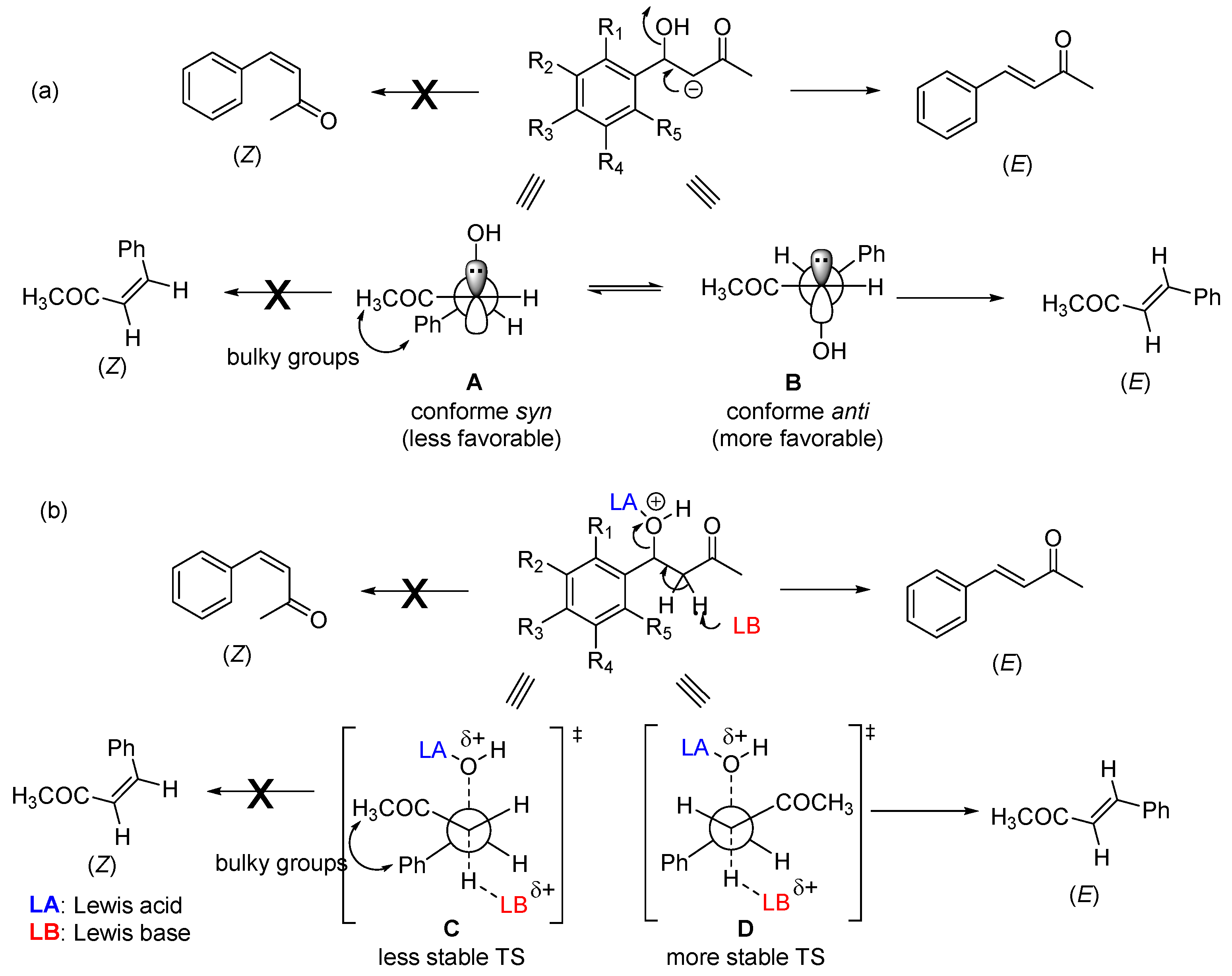
Scheme 3.
Mechanism of the IL-assisted formation of MKCs 214-218 from 159 [19].
Scheme 3.
Mechanism of the IL-assisted formation of MKCs 214-218 from 159 [19].
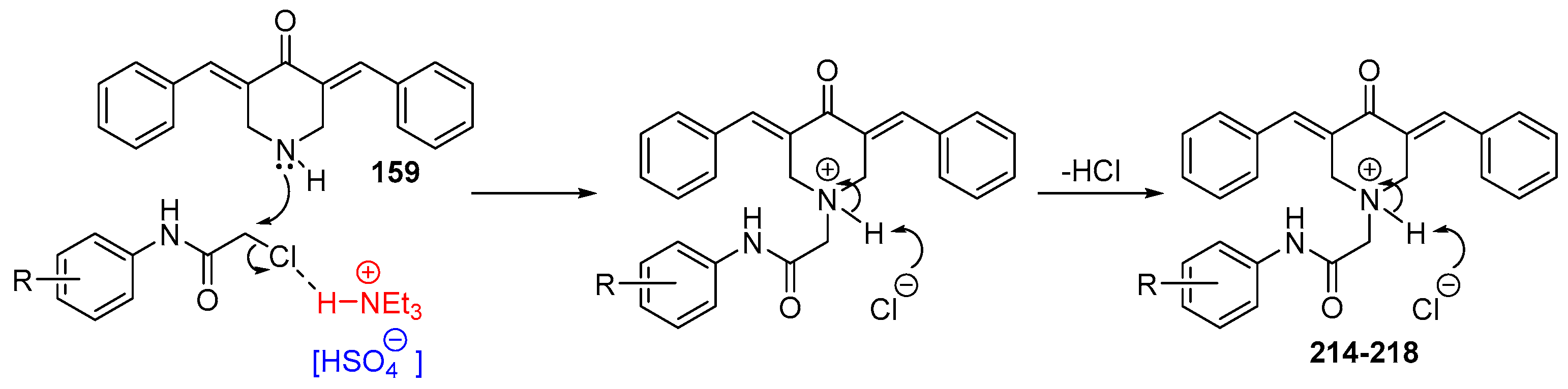
Figure 3.
Chemical structures of symmetric acetone-derived MKCs 1-48.
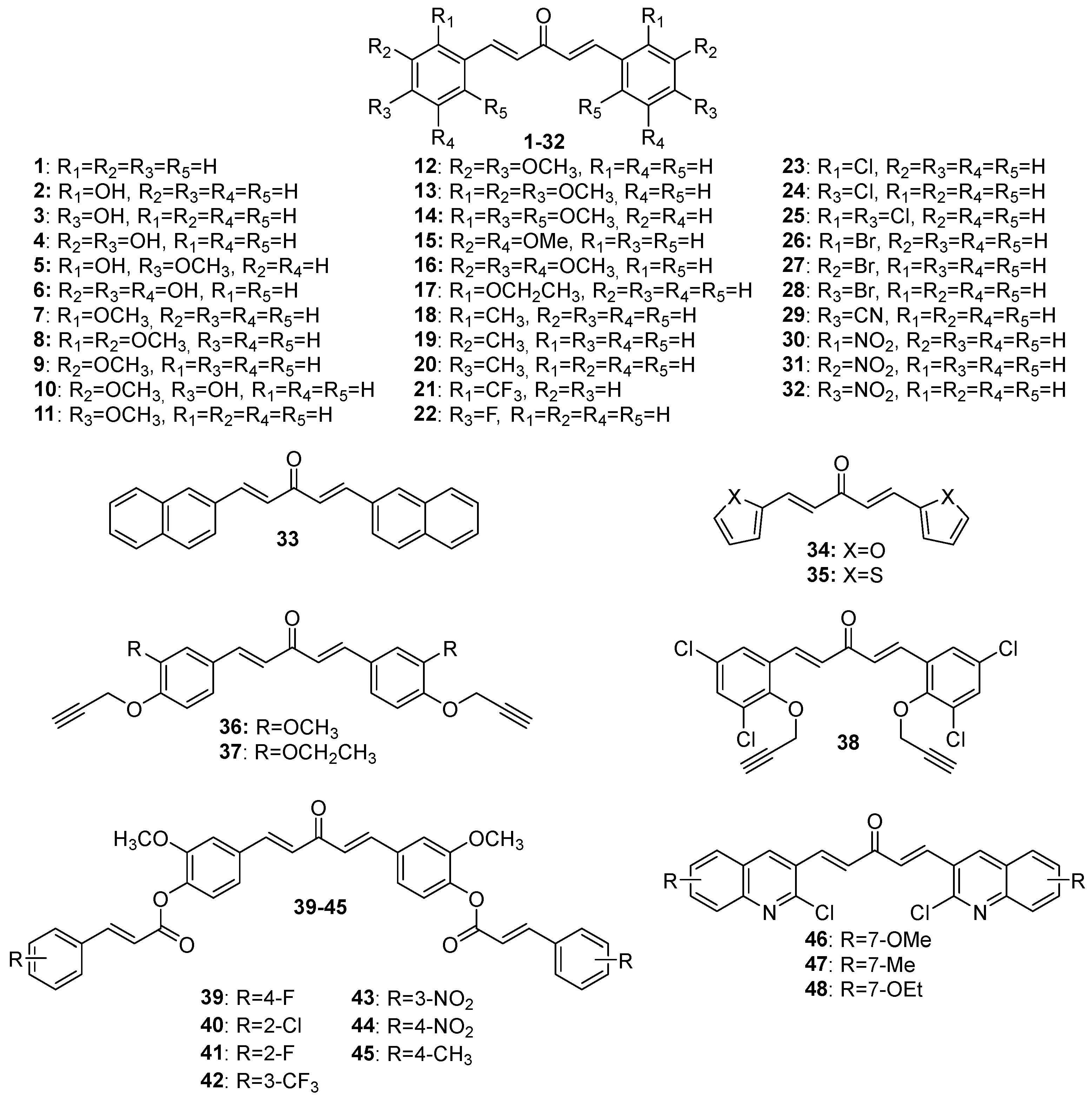
Figure 4.
Chemical structures of asymmetric acetone-derived MKCs 49-73.
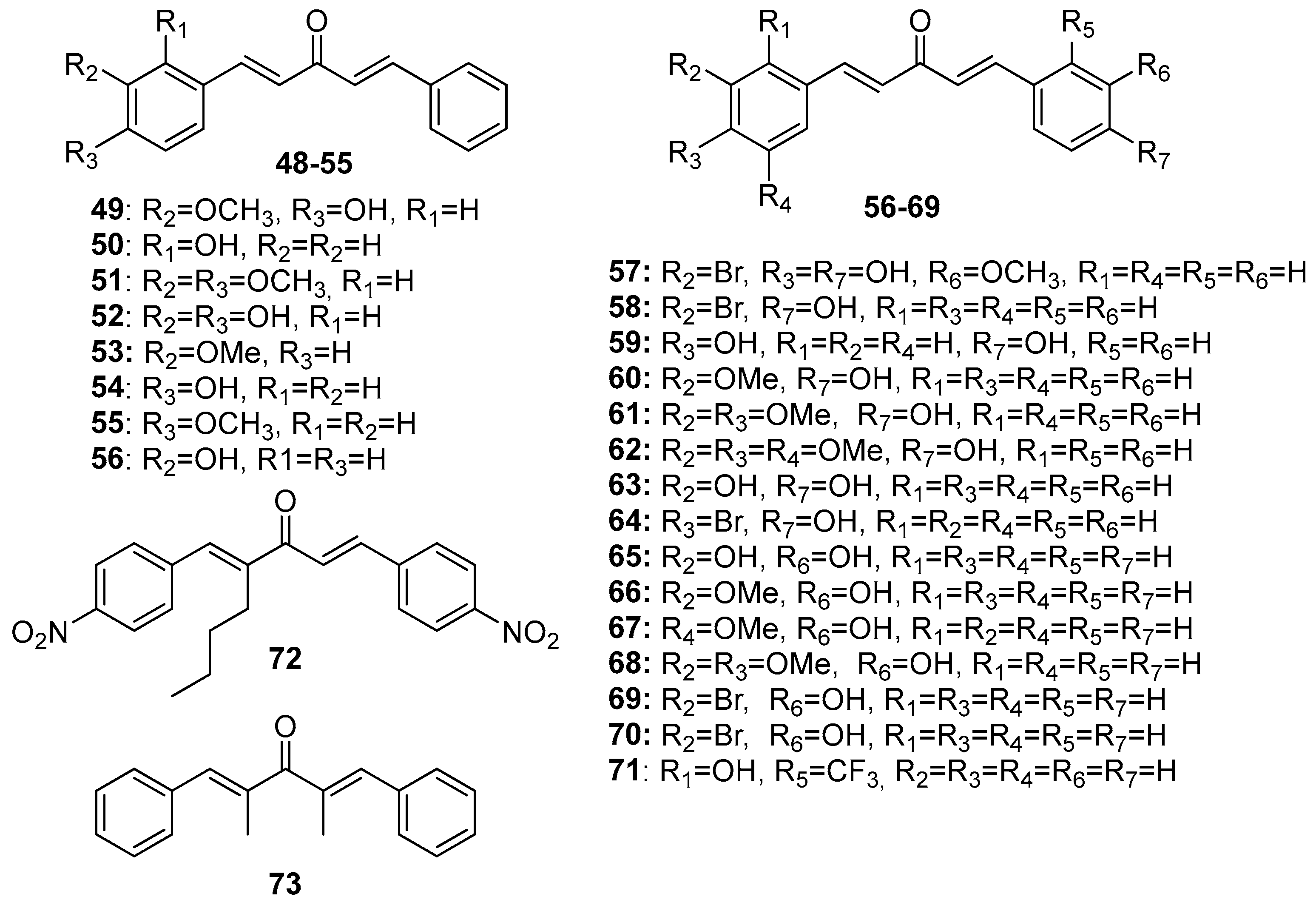
Figure 5.
Chemical structures of cyclopentanone-derived MKCs 74-103.
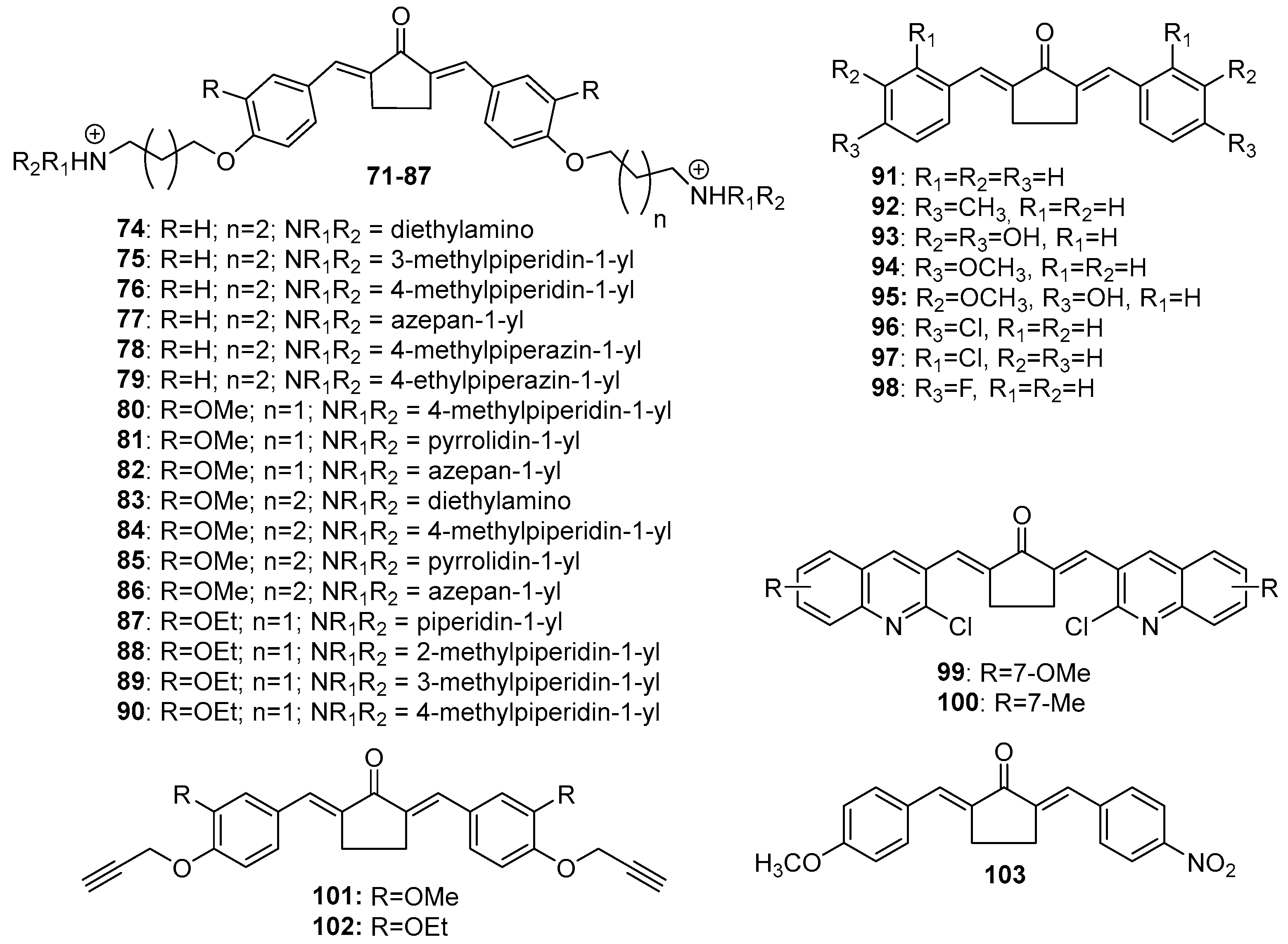
Figure 6.
Chemical structures of cyclohexanone-derived MKCs 104-151.
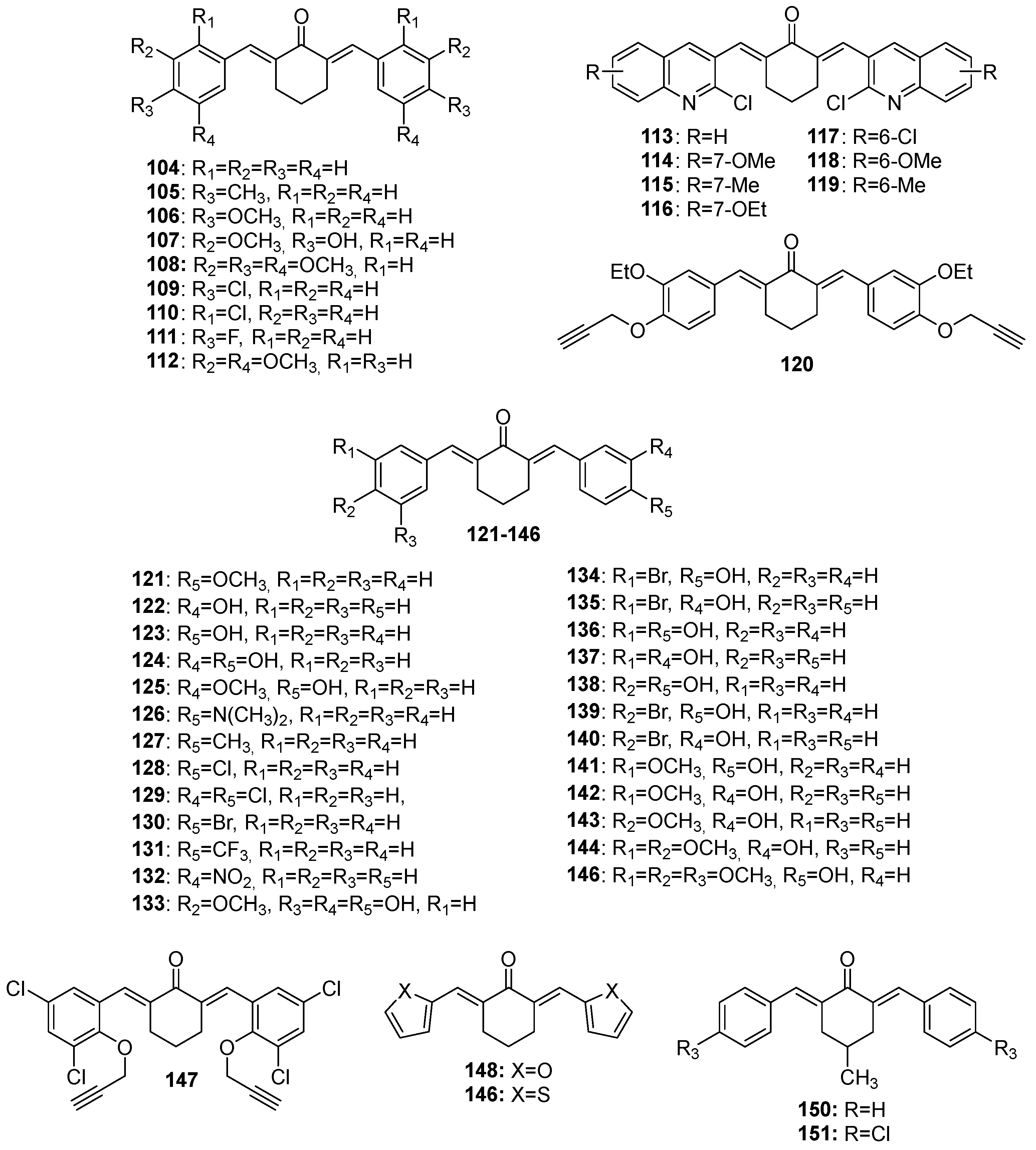
Figure 7.
Chemical structures of 4-hydroxy-cyclohexanone-derived MKCs 152-169.
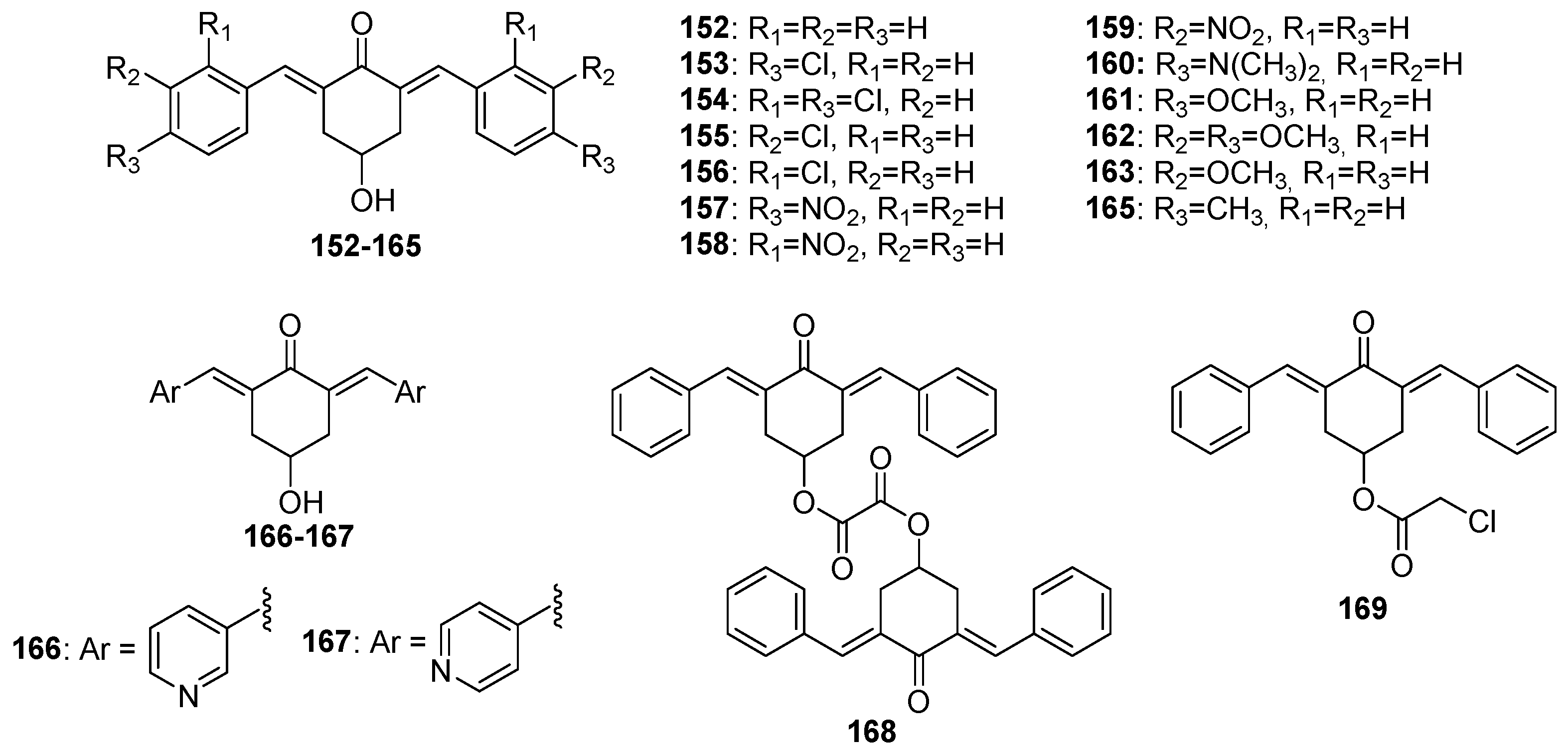
Figure 8.
Chemical structures of 4-piperidone-derived MKCs 170-227.
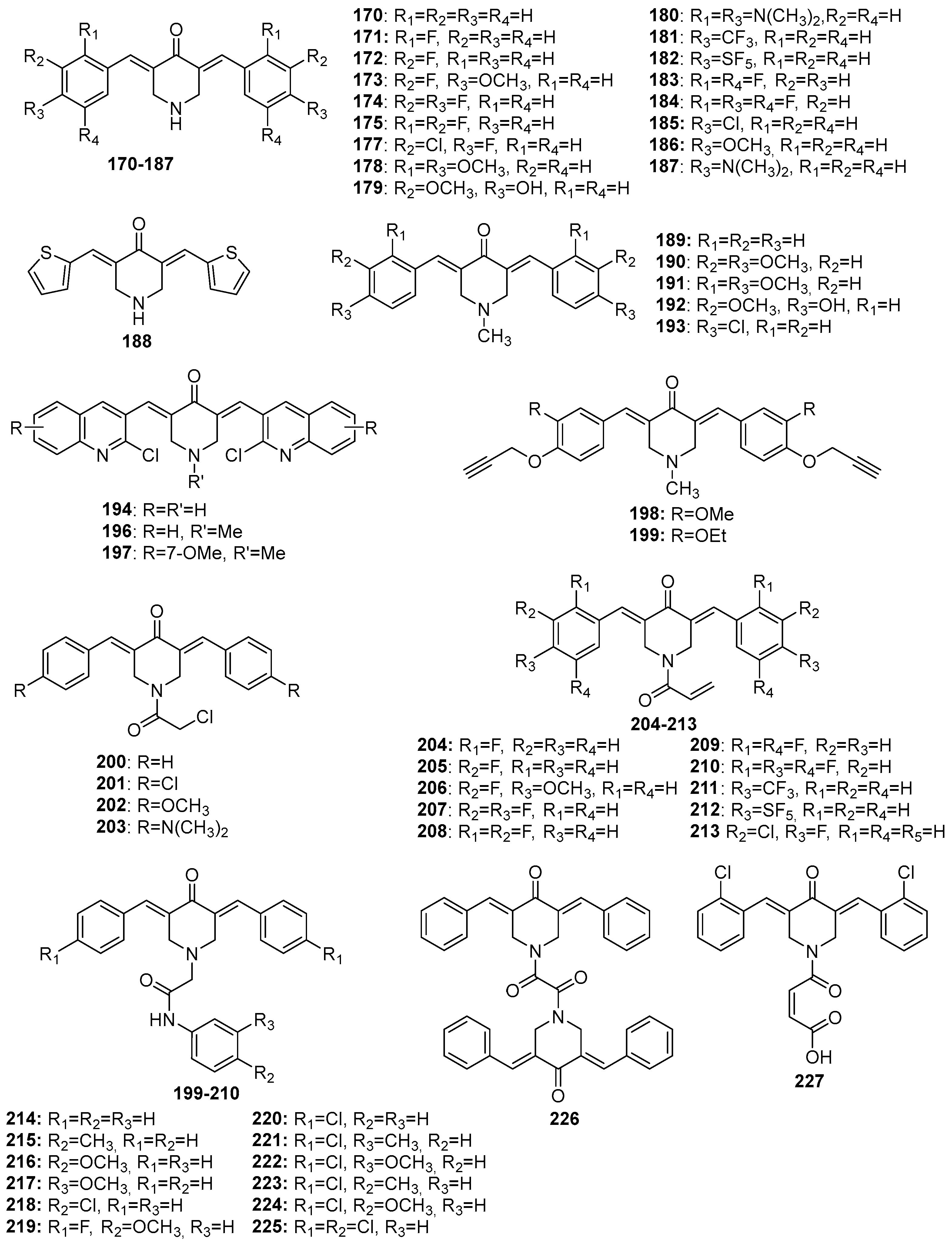
Figure 9.
Chemical structures of pyran-4-one-derived MKCs 228-254.

Table 1.
Biological activities of MKCs addressed in this review article.
| Activity | Compounds | Ref. |
|---|---|---|
| Antiangiogenic | 8 | [31] |
| Anticancer | 1 | [32] |
| 57 | [33] | |
| 7 | [34] | |
| 10 | [35,36,37] | |
| 171, 227 | [38] | |
| 10, 73, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 185, 186, 187, 189, 193, 200, 201, 202, 203, 226 | [27] | |
| 228, 229, 230, 231, 232, 233, 234, 235, 236, 237*, 238, 239, 240, 241, 242, 243, 244, 245, 246, 247, 2487, 249, 250, 251, 252, 253, 254* | [39] | |
| 254 | [40] | |
| 1, 35 | [41] | |
| 39, 40, 41, 42, 43, 44, 45 | [23] | |
| 179, 192 | [42] | |
| 171, 172*, 173, 174, 175, 177, 181, 182, 183, 184, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213 | [22] | |
| 1, 7, 9, 11, 13, 17, 18, 21, 22, 24, 26, 27, 28, 29, 30, 31, 32 | [21] | |
| Anti-coagulant | 72, 103 | [43] |
| Anti-diabetic | 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146 | [44] |
| 1, 8*, 11*, 16, 19, 20, 24, 25, 28, 31, 33 | [45] | |
| Anti-inflammatory | 58, 59, 60, 61, 62, 63, 64, 65*, 66, 67, 68, 69, 70 | [44] |
| 107 | [46] | |
| Antimicrobial | 10, 95*, 107 | [47] |
| 46, 47, 48, 99, 100*, 113, 114, 115, 116, 117, 118, 119, 194, 195, 196, 197* | [12] | |
| 36, 37, 38, 101, 102, 120*, 147, 198, 199 | [48] | |
| 1, 49, 50, 51, 52*, 53, 54, 55 | [49] | |
| 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89*, 90 | [9] | |
| 188 | [16] | |
| 190*, 191 | [50] | |
| Antioxidant | 46, 47, 48, 99, 100*, 113, 114, 115, 116, 117, 118, 119, 194, 195, 196, 197* | [12] |
| 36, 37, 38, 101, 102, 120*, 147, 198, 199 | [48] | |
| 188 | [16] | |
| Antiparasitic | 1 | [51,52,53,54] |
| 1, 11, 20, 22, 23, 24, 91, 92, 94, 96, 97, 98, 104, 105, 106, 109, 110, 111 | [24] | |
| 1, 11, 12*, 14, 24 | [55] | |
| 1, 9, 10, 11, 12, 15, 16, 20, 22, 23, 27, 112, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 171 | [56] | |
| Insecticidal | 1, 2, 5, 34 | [57] |
| 3, 16, 50, 71, 108, 148 | [58] | |
| 4, 6, 10, 11, 12*, 16, 93, 148, 149 | [59] |
* Most active compound among the tested MKCs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



