
Submitted:
24 November 2023
Posted:
30 November 2023
You are already at the latest version
Abstract
Metal complexes of mesoionic carbenes (MICs) of the triazolylidene-type and their derivatives have gained increasing attention in the fields of electrocatalysis and photochemistry. The redox-activity of these metal complexes is critical for their applications in both the aforementioned fields. Easy accessibility and modular synthesis open a wide field for the design of ligands, such as bidentate ligands. The combination of a MIC with a pyridyl unit in a bidentate ligand setup increases the π-acceptor properties of the ligands while retaining their strong σ-donor properties. The analogy with the well-established 2,2'-bipyridine ligand allows conclusions to be drawn about the influence of the mesoionic carbene (MIC) moiety in tetracarbonyl group 6 complexes in cyclic voltammetry and (spectro-)electrochemistry (SEC). However, the effects of the different connectivity in pyridyl-MIC ligands remains underexplored. Based on our previous studies, we present here a thorough investigation of the influence of the two different pyridyl-MIC constitutional isomers on the electrochemical and the UV-vis-NIR/IR/EPR spectroelectrochemical properties of group 6 carbonyl complexes. Moreover, the presented complexes were investigated for the electrochemical conversion of CO2 using two different working electrodes, providing a fundamental understanding of the influence of the electrode material in the precatalytic activation.
Keywords:
mesoionic carbenes
; (spectro)electrochemistry
; carbonyl ligands
; group 6 carbonyls
; EPR spectroscopy
1. Introduction
In 2001, Sharpless and co-workers coined the term "click" chemistry, to describe modular reactions with a wide scope, and high yields, producing only mild inoffensive byproducts [1]. The azide-alkyne cycloaddition reaction is arguably one of the best examples of a click reaction. The thermally induced 1,3-dipolar cycloaddition between alkynes and azides results in a mixture of two regioisomers [2]. In 2002, two groups independently discovered the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC), generating exclusively the 1,4-regioisomer of 1,2,3-triazole [3,4].
Methylation of 1,2,3-triazoles leads to the formation of so-called triazolium salts in near quantitatively yields [5,6,7]. They represent one of the most important precursors for triazolylidenes, a class of carbenes that are better known as abnormal N-heterocyclic carbenes (aNHC) or mesoionic carbenes (MIC). This classification arises from the fact that while following octet rules, no resonance structures can be drawn for MICs without charge separation, unlike their well-established N-heterocyclic carbenes (NHC) counterparts [5,7,8,9]. Therefore, not surprisingly, the synthetic scope of MICs has expanded rapidly, opening up the possibility of introducing additional donor substituents, such as pyridine, to generate bidentate ligands [10,11,12,13,14,15,16] or post-modifications to N-heterocyclic olefins (NHO) [17,18] and mesoionic imines (MII), [19,20] which are promising candidates for small molecule activation [5].
Suntrup et al., in 2017 showed, that the insertion of a pyridyl moiety into 1,2,3-triazole- and 1,4-triazolylidene-based Re(I) carbonyl complexes drastically improves the overall acceptor character of the ligand, while the incorporation of a MIC unit results in a greater donor strength compared to the well-established bpy ligand [21]. The robustness towards reductive electrochemistry provided the basis for the investigation of a series of pyridyl-MIC Re(I) complexes in the electrochemical reduction of CO2 to generate CO with high selectivity and to study their photophysical properties [16].
However, many of the most promising electrocatalysts explored contain expensive and rare metals, which preclude their large-scale applications.[22,23,24,25,26,27]. In recent years, great efforts have been made to develop more earth-abundant photo- and electrocatalysts for the activation of small molecules based on carbenes [28,29,30,31,32,33,34,35,36,37,38,39].
Group 6 metal complexes are attractive candidates because of their natural occurrence, such as molybdenum in the active site of enzymes that convert CO2 to formate [40].
Recent reports have shown that the isoelectronic and isostructural group 6 metal complexes of [M(bpy)(CO)4] (M = Cr, Mo, W), as well [M(L)(CO)4] (L = "non-innocent" ligands) with Mo and W are capable of electrocatalytic conversion of CO2 [28,29,33,36,37,39].
Tory et al. and Clark et al. reported the (spectro-)electrochemical properties of group 6 complexes [M(bpy-R)(CO)4] (R = 5,5' H, 5,5' tBu) and demonstrated their activity in CO2 reduction on a gold (Au WE) and glassy carbon working electrode (GC WE), respectively [28,39]. The results indicate two important facts: first, the substitution of the bpy moiety results in a shift of the reduction potential for the precatalytic activation, and second, the change of the working electrode from a platinum working electrode (Pt WE) to an Au WE shifts the onset potential for electrocatalytic CO2 reduction by +0.6 V, similar to what was reported for the group 7 electrocatalysts [25]. Based on these results, Neri et al. investigated the role of the electrode-catalyst interaction using vibrational sum frequency generation spectroscopy (VSFG) providing an insight into the mechanism at the electrode surface [36]. Cyclic voltammetric measurements with an Au WE show an equilibrium between the one-electron reduced species [Mo(bpy)(CO)4]− and [Mo(bpy)(CO)3]− after CO dissociation. In contrast, using a Pt WE two-electron reduction is required to generate the precatalytically active species.
Recently, we have presented a series of two 1,4-pyidyl-MIC group 6 carbonyl complexes [M(L)(CO)4] (M = Cr, Mo, W) with two different constitutional isomers (L: C−C = pyridyl-4-triazolylidene [41] and C−N = pyridyl-1-triazolylidene [42,43,44]) that exhibit excellent photophysical and photochemical properties, making them suitable candidates in photo-induced small molecule activation [43,44,45,46]. For the first time, details of the influence of the two constitutional isomers were reported in the chemically and electrochemically oxidized [Cr(L)CO)4] complexes, providing detailed insights into the extraordinary donor properties [41]. In addition, a comprehensive study of precatalytic activation in [Rh(Cp*)] complexes for electrochemical H+ reduction was reported, demonstrating the capability in small molecule activation with both ligands (Scheme 1) [47].
Scheme 1.
Relative donor/acceptor strength of constitution isomers C−C (left) and C−N (right).
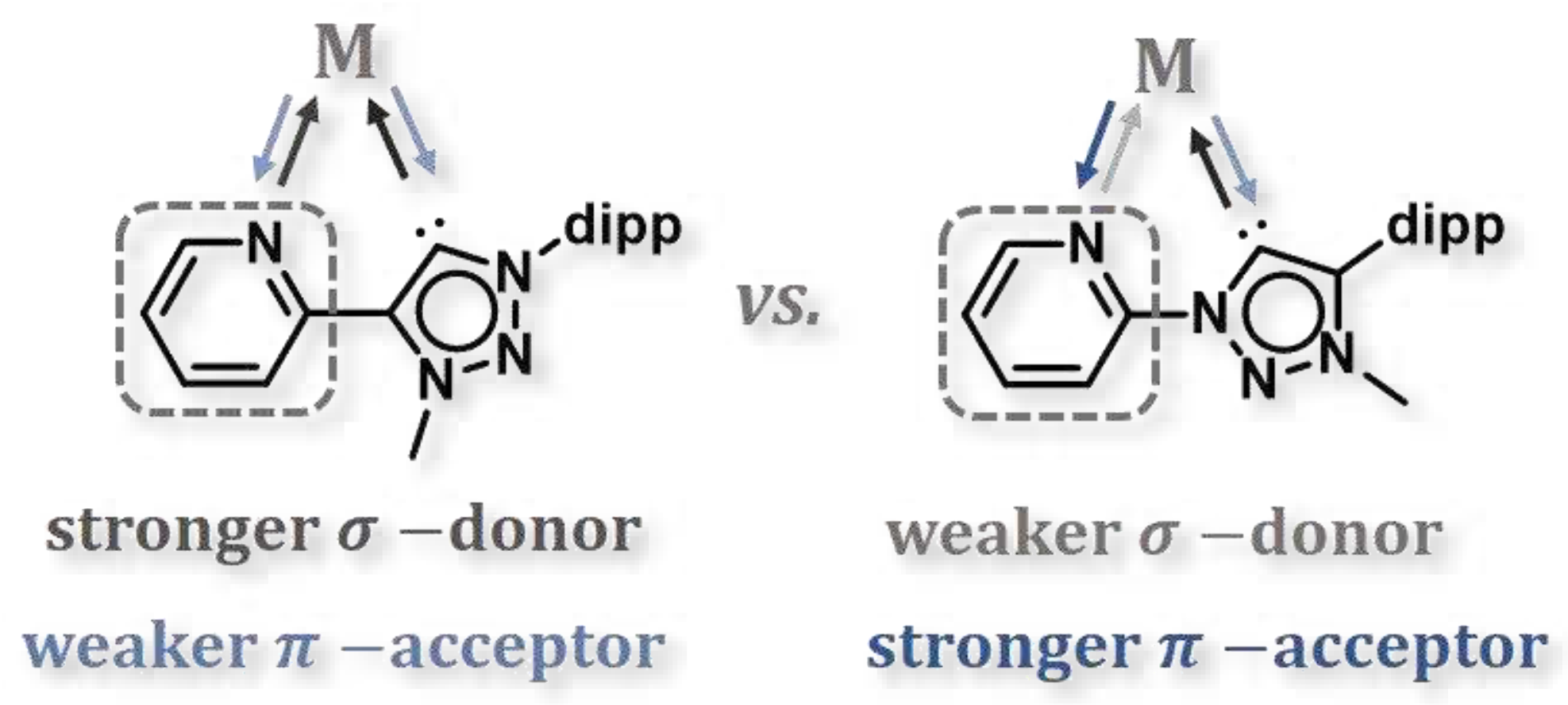
Based on our previous studies, we report here on a comprehensive electrochemical and spectroelectrochemical investigation of [M(C−N)(CO)4] and [M(C−N)(CO)4] [42] (M = Cr, Mo, W) to gain a fundamental understanding of the effects of the two constitutional isomers on their electronic structures and to perform reactivity of the complexes in electrochemical CO2 reduction as a function of the electrode material.
2. Results and Discussion
The triazolium salts [H(C−C)](BF4), [21] [H(C−N)](OTf) [42] and the complexes [M(C−C)(CO)4] [41] and [M(C−N)(CO)4] [42,43] were synthesized according to a previously reported protocol (Scheme 2).
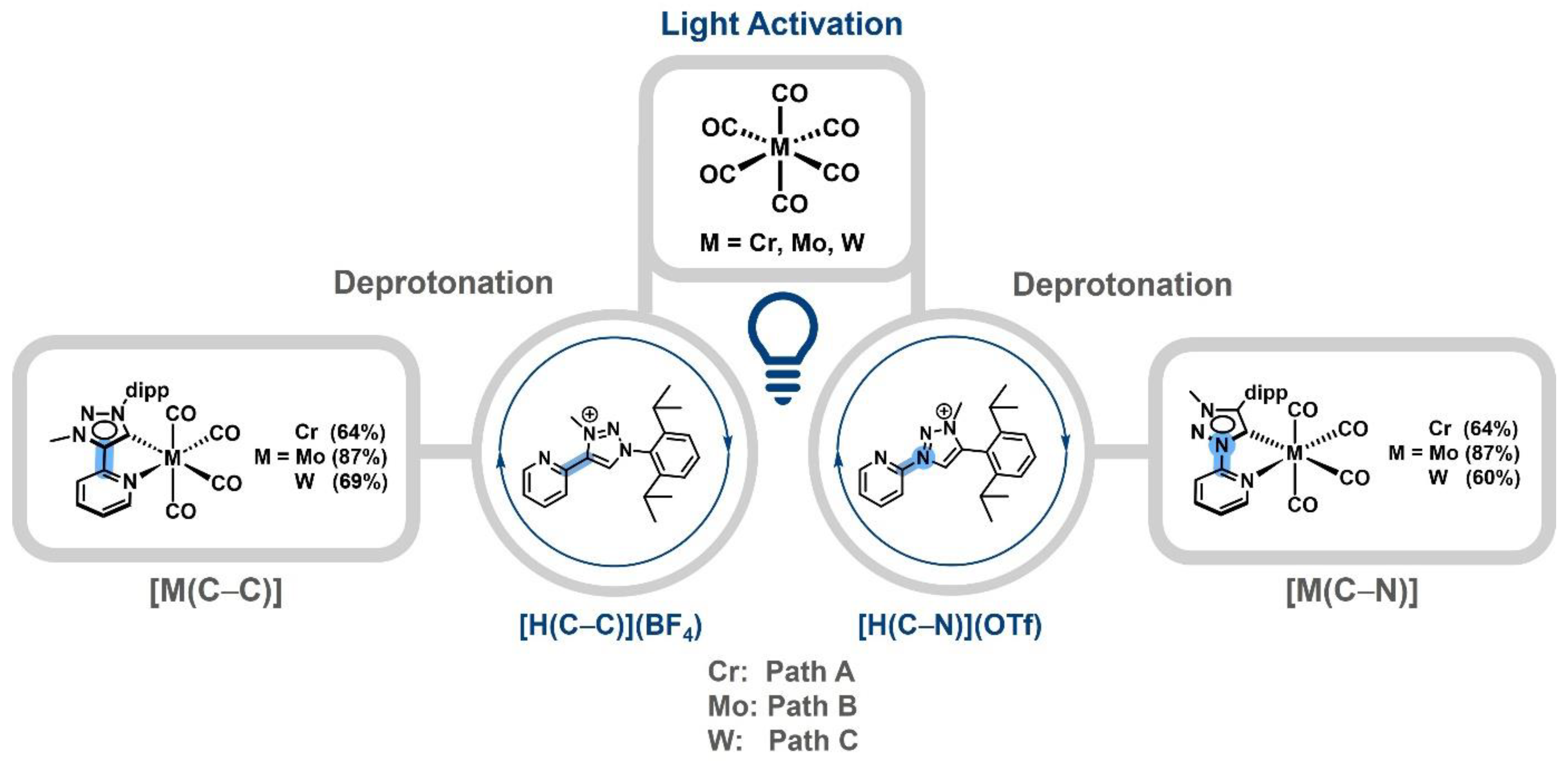
The light-induced activation of the corresponding [M(CO)6] followed by the addition of [H(C−C)](BF4) or [H(C−N)](OTf) and subsequent deprotonation with NEt3 leads to the chromium and tungsten complexes [M(C−C)(CO)4] and [M(C−N)(CO)4] after chromatographic workup and recrystallization, while in the case of molybdenum, the precursor [Mo(nbd)(CO)4] (nbd = norbornadiene) was synthesized and further converted in the presence of a base to isolate [Mo(C−C)(CO)4] or [Mo(C−N)(CO)4], respectively.
2.1. Cyclic Voltammetry with a GC WE and EPR-SEC
The redox potentials measured from cyclic voltammetry are often, but not always, used for gauging the donor/acceptor properties of the ligands in metal complexes. A reversible metal-centered oxidation, as observed for [Cr(C−C)(CO)4] [41] and [Cr(C−N)(CO)4], [42] allows to estimate the overall donor strength of the ligand, while a reversible ligand-centered reduction can be used to determine indirectly the acceptor capacity of the ligand.
Previous reports from our group already established a stronger donor strength of the ligand in [Cr(C−C)(CO)4] compared to [Cr(C−N)(CO)4] [41,42]. The same trend in this regard is observed for the higher homologs [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Mo, W). However, the oxidations of the respective complexes are irreversible as a consequence of the kinetic lability of the CO ligands and the possibility to form complexes with higher coordination numbers in the oxidized complexes (Figure 1, see SI, S6) [48,49]. The oxidation potentials follow the trend according to the ionization energy of the central metal atom (Cr > Mo > W) [50].
Figure 1.
Cyclic voltammograms of [W(C−C)(CO)4] (top) and [W(C−N)(CO)4] (bottom) in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode.
Figure 1.
Cyclic voltammograms of [W(C−C)(CO)4] (top) and [W(C−N)(CO)4] (bottom) in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode.
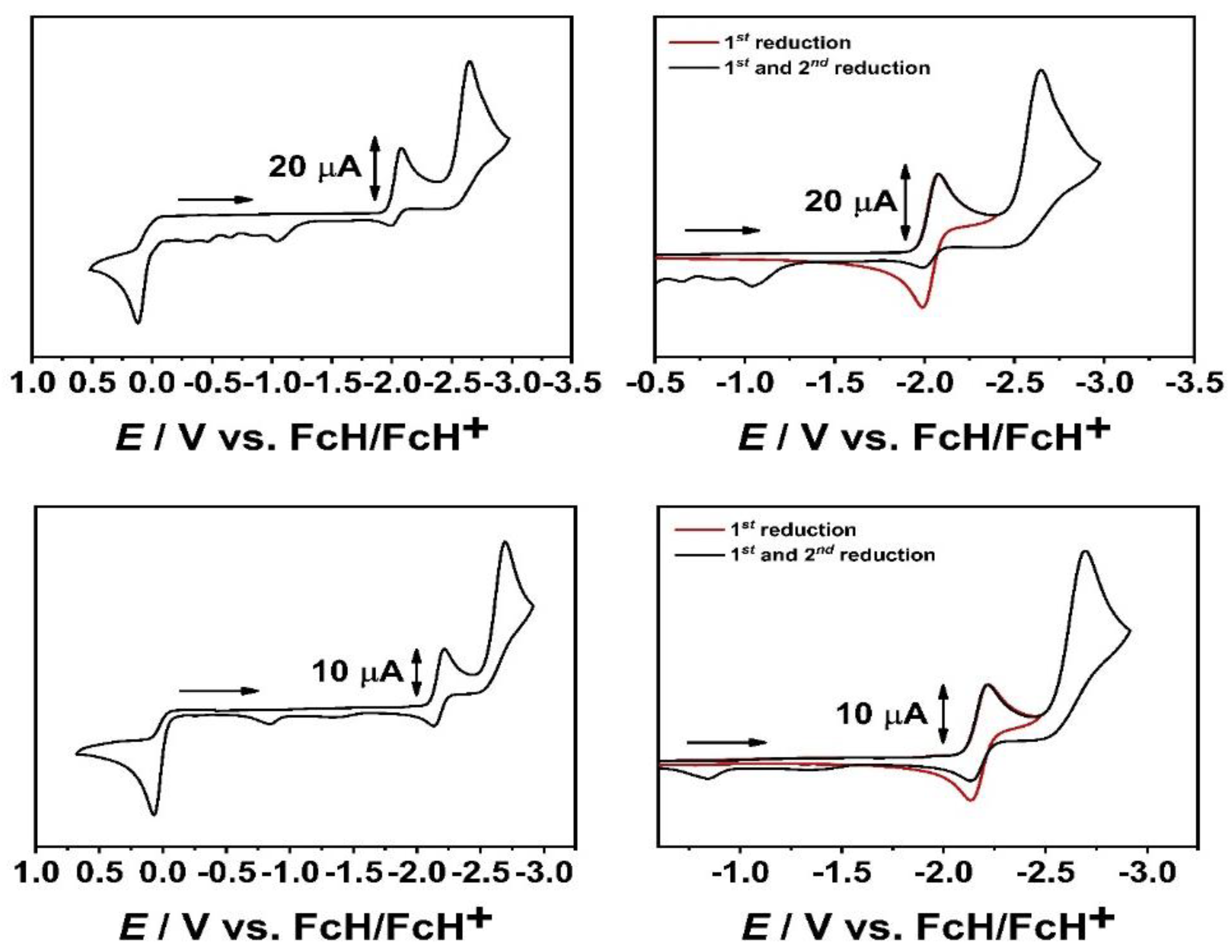
All presented complexes on the other hand show a reversible first reduction, followed by a second irreversible reduction, whereas in the series of [M(C−N)(CO)4] a third reduction process is observed at lower scan rates (Table 1, see SI, 2.) [42]. The reduction potentials presented in Table 1 are in good agreement with the aforementioned acceptor properties of the constitutional isomers. In the case of [M(C−N)(CO)4] (M = Cr, Mo, W), the first reduction is shifted to more anodic potential compared to [M(C−C)(CO)4] (M = Cr, Mo, W), indicating the greater acceptor ability of the C−N linked constitutional isomer in the complexes.
To get detailed insights into the electronic structure of the first reduction, electron paramagnetic resonance (spectro-)electrochemistry (EPR-SEC) was performed with an Au WE in 0.1 M Bu4NPF6/CH3CN (Figure 2 and Table 2, see SI, 3.).
Upon reduction at room temperature, line-rich EPR spectra at g = 2.003 are observed for all complexes, showing hyperfine coupling to all four 14N nuclei within the central pyridyl-MIC ligand framework (Table 2). The hyperfine-coupling constants of the 14N nuclei and the spin density plots of the respective complexes reveal a strong interaction of the electron spin with the N2 and N3 nuclei of the reduced 1,2,3-triazolylidene (MIC) moiety and to a smaller extent with the 14N nuclei of the pyridyl-N and the N1 nuclei of the MIC unit. Only in the case of [W(C−C)]−, a strong coupling to only one 14N nucleus is observed. A plausible explanation might be the stronger delocalization of the electron spin within the C−C isomer. The [W(C−C)]− complex shows 1H hyperfine coupling to overall ten 1H nuclei. In contrast, the analogue [Cr(C−C)(CO)4]− and [Mo(C−C)(CO)4]− complexes display 1H hyperfine coupling to four 1H nuclei, which can be assigned to the pyridyl-H. The strong interaction of the electron spin within the pyridyl moiety is also present in [W(C−C)(CO)4]−. The complex shows a strong 1H coupling constant to four 1H nuclei, indicating a predominant localization within the central pyridyl-MIC framework. However, small hyperfine couplings with six additional 1H nuclei are observed. Even though the spin density plot of [W(C−C)]− does not directly indicate the localization of the electron spin at the different ligand fragments, the coupling to three 1H nuclei of the methyl group at the MIC moiety and three 1H nuclei of the 2,6-diisopropylphenyl (= dipp) substituent are reasonable.
Figure 2.
EPR spectrum and spin density plot (B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006) of [Cr(C−C)] (top left), [Mo(C−C)] (top right), [W(C−C)] (bottom left) and [W(C−N)] (bottom right) in 0.1 M NBu4PF6/CH3CN with an Au working electrode during the first reduction (black: experimental, red: simulation).
Figure 2.
EPR spectrum and spin density plot (B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006) of [Cr(C−C)] (top left), [Mo(C−C)] (top right), [W(C−C)] (bottom left) and [W(C−N)] (bottom right) in 0.1 M NBu4PF6/CH3CN with an Au working electrode during the first reduction (black: experimental, red: simulation).
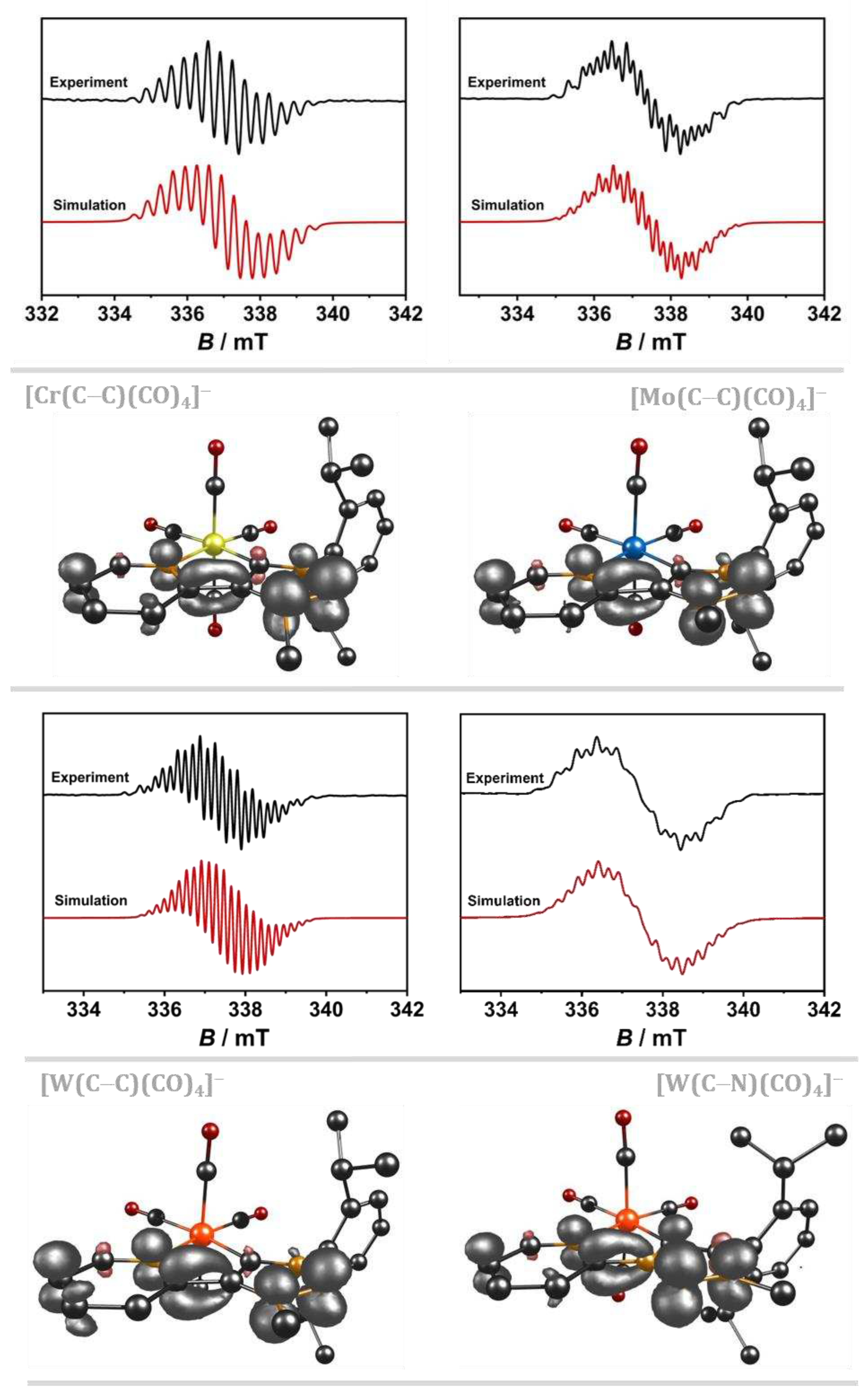
The influence of the constitutional isomers is shown in the EPR spectrum of [W(C−C)(CO)4]− and [W(C−N)(CO)4] −. The analogue tungsten C−N complex displays a stronger coupling of the electronic spin with the four 14N nuclei in the pyridyl-MIC moiety. Consequently, the line-rich spectrum shows an increased line-broadening of the isotropic signal. In contrast to its C−C counterpart, only six 1H hyperfine couplings are observed in [W(C−N)(CO)4]−. This observation could indicate an increased localization of the electron spin at the MIC moiety and consequently leading to a decreased contribution of the dipp-substituent. The stronger localization at the MIC moiety of the C−N linked isomer is further affirmed by the stronger 1H hyperfine coupling to the methyl group. The EPR spectrum shows 1H coupling constants of up to 7.19 MHz, while the C−C linked analogue shows only weak couplings of up to 1.81 MHz. Additionally, three strong 1H hyperfine couplings to three pyridyl-H are observed, confirming the significant localization at the pyridyl-MIC framework within the C−N isomer.
Unfortunately, no clear trend regarding the influence of the central metal atom in the series of [M(C−C)(CO)4]− and [M(C−N)(CO)4]− (M = Cr, Mo, W) could be observed, despite all metal ions showing a coupling with the ligand-centered radical [42].
To further shine light on the influence of the constitutional isomers in [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W), IR-SEC with an Au WE in 0.1 M NBu4PF6/CH3CN was conducted.
2.2. IR-Spectroelectrochemistry
In contrast to cyclic voltammetry, IR spectroscopy of the complexes [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) under investigation is a common method for the characterization of the electronic structure due to the characteristic CO stretching frequencies.
The IR spectra of [M(C−C)(CO)4] and [M(C−N)(CO)4] in CH2Cl2 show four CO stretching frequencies, as a consequence of the lowering of symmetry around the metal center (Table 3, Figure 3). Even though the number of bands observed in the IR spectra are identical, their positions are shifted depending on the electronic nature of the ligands and the central metal atoms.
Figure 3.
IR spectra of [M(C−C)(CO)4] [41] (left) and [M(C−N)(CO)4] [42] (right) in CH2Cl2 (M = Cr: black, Mo: grey, W: light grey).
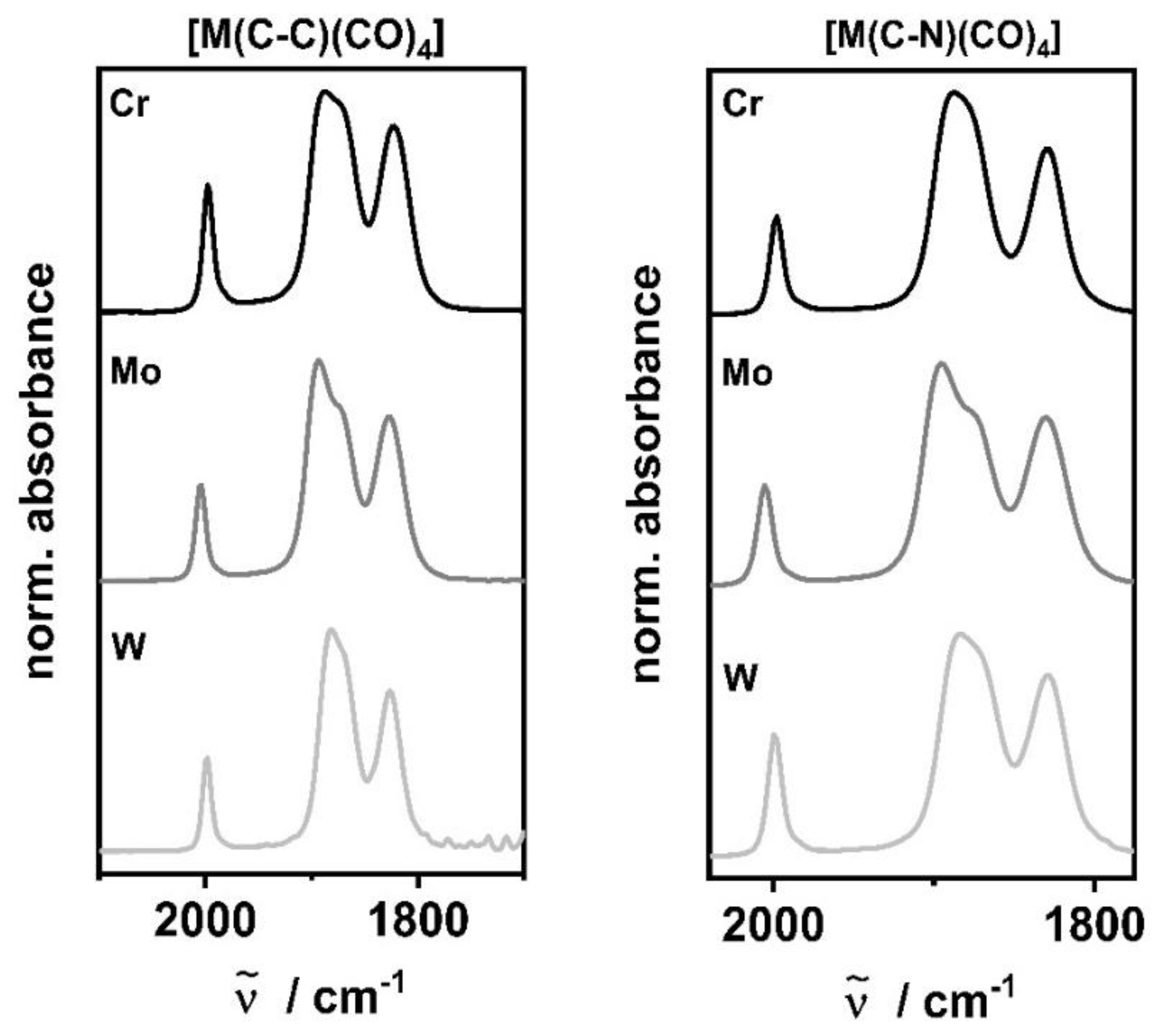
Concerning the net-electron density of the [M(CO)4] fragment with the incorporated pyridyl-MIC ligands, the averaged CO stretching frequencies presented in Table 3 further confirm the greater donor strength of the chelating ligand observed in [M(C−C)(CO)4] compared to [M(C−N)(CO)4].
The influence of the constitutional isomers becomes in particular evident upon one-electron reduction of [M(C−C)(CO)4] and [M(C−N)(CO)4] during IR-SEC (Figure 4, see SI, 4.). Within the series of [M(C−N)(CO)4], a clean conversion of the native species into the reduced [M(C−N)(CO)4]− complexes is observed, as indicated by the isosbestic points during the IR-SEC measurements. The shift of the frequencies by about 20 cm-1 to lower wavenumbers confirms the predominantly ligand-centered reduction and are in good agreement with our calculated changes in the CO stretching frequencies of [M(C−N)(CO)4]− (see SI, 4.). [41,42].
Figure 4.
Changes in the IR spectra of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) in CH3CN/0.1 M Bu4NPF6 with an Au working electrode during the first reduction.
Figure 4.
Changes in the IR spectra of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) in CH3CN/0.1 M Bu4NPF6 with an Au working electrode during the first reduction.
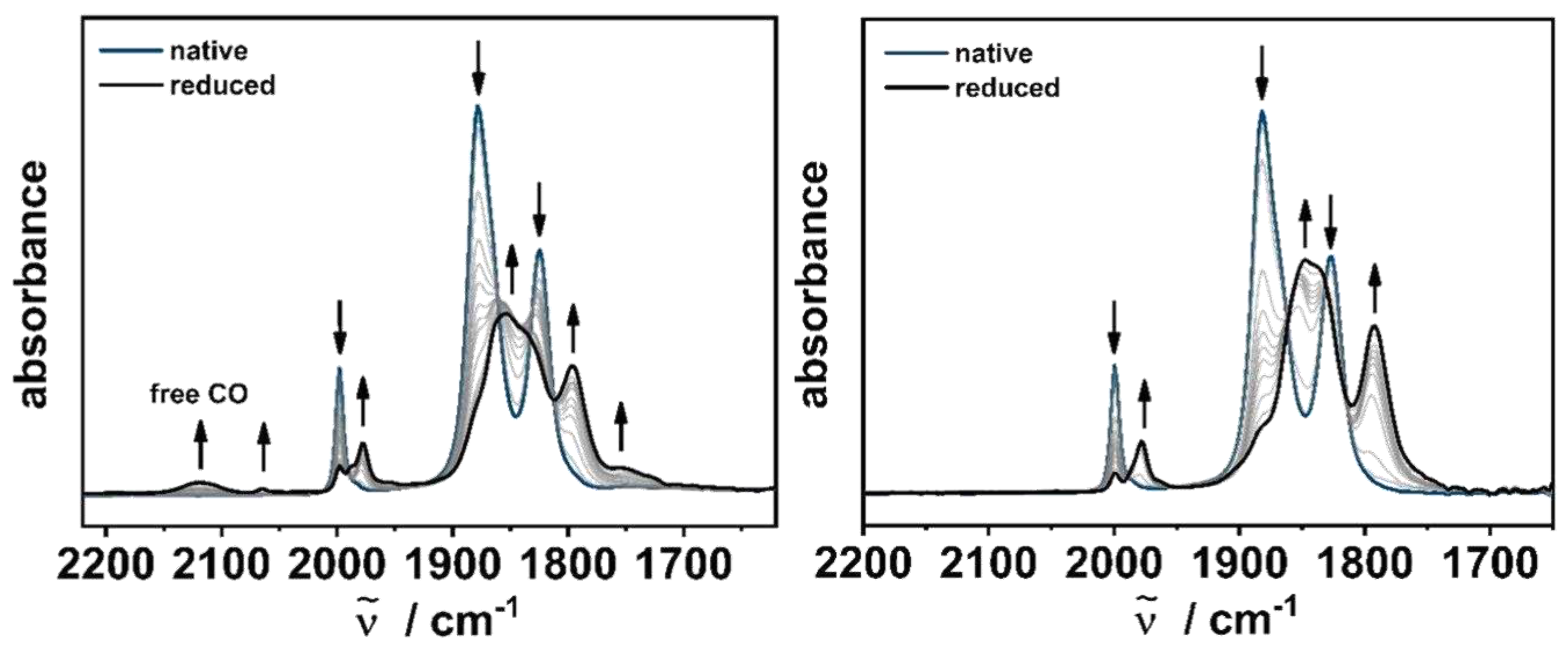
However, the picture changes upon reduction of the other isomer. All complexes within the series show at least two new species in the IR-SEC measurements, as indicated by the formation of several new IR bands.
Probably the most significant change can be assigned to the newly formed band at 2119 cm-1. Torey et al. described a similar observation after the reduction of [Mo(bpy)(CO)4] [39]. The IR band at 2130 cm-1 could be assigned to adsorbed CO at the Au electrode surface. Furthermore, in-depth investigations by VSFG by Neri et al. confirmed the dissociative EC-mechanism of CO upon reduction at an Au WE [36]. Based on these results and our theoretical calculations (see SI, 4.), the reduced species could likely be a mixture of the one-electron reduced [M(C−C)(CO)4]− species, the coordinatively unsaturated complex [M(C−C)(CO)3]− and/or the solvent adduct [M(C−C)(CH3CN)(CO)3]−, formed after subsequent CO dissociation.
In addition, comparison of the IR spectra before and after electrolysis in the OTTLE cell clearly indicate the partial decomposition of [M(C−C)(CO)4] after reduction, whereas only minor decomposition products are observed in the series of [M(C−N)(CO)4], after prolonged electrolysis [42]. These results provide useful information on the stabilization of the ligand-centered radical based on the different linkage in the two constitutional isomers, as the CO cleavage observed in [M(C−C)(CO)4] gives access to an open coordination site for potential electrocatalytic applications, such as the electrochemical CO2 reduction [28,29,33,36,37,39].
To confirm the reversibility in the series of [M(C−N)(CO)4] and the EC mechanism observed for [M(C−C)(CO)4] upon reduction, UV/vis/NIR-SEC measurements were performed.
2.3. UV/vis/NIR-Spectroelectrochemistry
UV/vis/NIR-SEC is a commonly employed technique to test either pure electrochemical reversibility or reversibility following an EC mechanism [51].
All presented complexes display electronic transitions in the visible to near UV region (300−550 nm), which can be assigned to metal-to-ligand charge transfers (MLCT) with an additional contribution of the axial CO ligands in the ground state and excited state (see SI, 6.20−6.40) [41,42]. Within the series of [M(C−C)(CO)4], the MLCT transitions are blue-shifted compared to [M(C−N)(CO)4], which is in good agreement with the previously described acceptor properties of the C−N linked constitutional isomer. However, a significant contribution of the aromatic substituent is observed in [M(C−N)(CO)4] (see SI, 6.40) [42].
The electrochemical reduction of [M(C−C)(CO)4] leads to broad transitions in the visible and NIR region (650−2100 nm, Figure 5 and S30−S31). According to TD-DFT calculations, these bands can be assigned to an intra-ligand charge transfer (ILCT) from the reduced C−C linked ligand to the 2,6-diisopropylphenyl substituent and a ligand-to-ligand charge transfer (LLCT) from the reduced ligand to the axial CO ligands. The absorption bands in the 380−400 nm range, are best described as metal-ligand-to-ligand charge transfer (MLLCT) from the [M(CO)4] fragment to the pyridyl-MIC ligand and all four CO ligands.
Upon reduction of [M(C−N)(CO)4], weak bands are observed in the visible and NIR region (700−2100 nm), which can be assigned to ILCTs and LLCTs from the reduced ligand to the axial CO ligands, the pyridyl-MIC moiety and the aromatic substituent (see SI, 6.40). Additionally, more discrete transitions are observed in the 550−700 nm region, indicating a more localized ligand-centered radical, which is in good agreement with the aforementioned EPR-SEC results. The electronic transitions in this range are best described as a mixture of ILCTs, MLLCTs, and LLCTs.
Figure 5.
Changes in the UV/VIS spectra of [W(C−C)(CO)4] (left, Inset: 650−2050 nm) and [W(C−N)(CO)4] (right, Inset: 750−2090 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with an Au working electrode.
Figure 5.
Changes in the UV/VIS spectra of [W(C−C)(CO)4] (left, Inset: 650−2050 nm) and [W(C−N)(CO)4] (right, Inset: 750−2090 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with an Au working electrode.
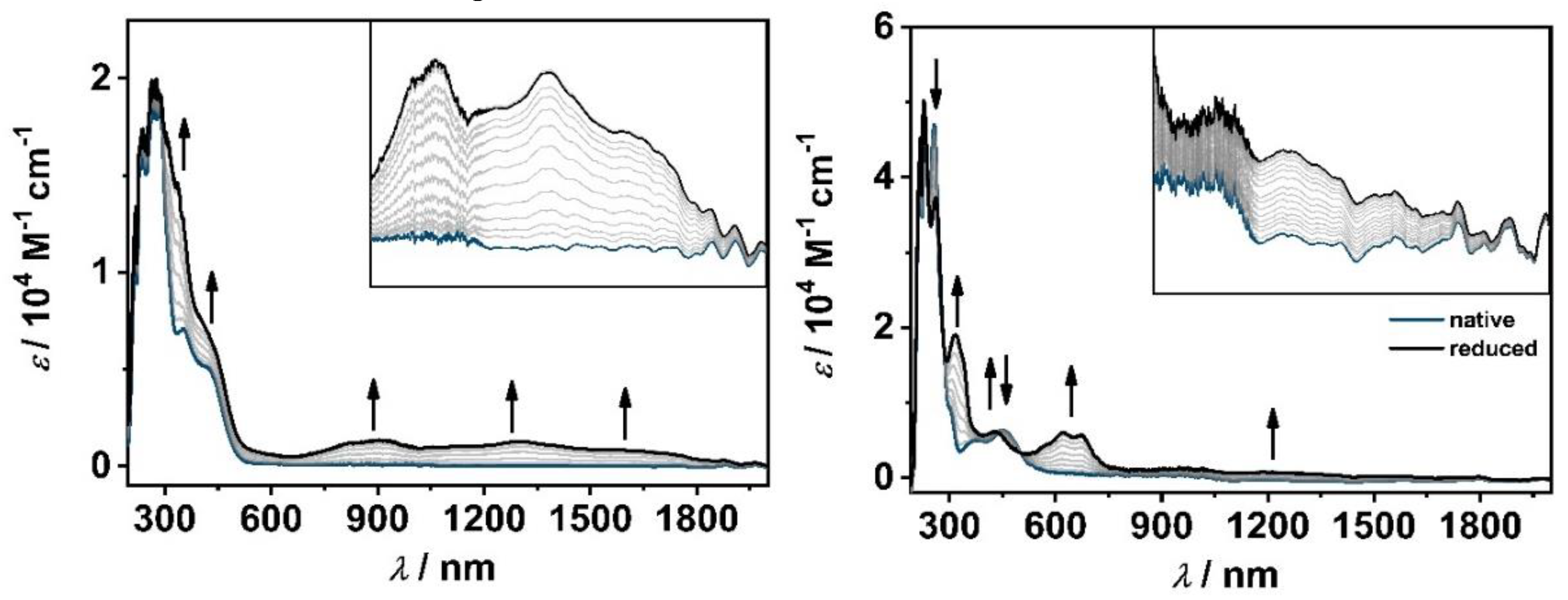
The partial degradation of [M(C−C)(CO)4]− by a EC mechanism is confirmed by the decrease in absorption maxima during electrolysis in the OTTLE cell in the visible and NIR region and further supported by comparing the UV/vis/NIR spectra before and after UV/vis/NIR-SEC (see SI, 5.10−5.30). Only in the case of [Mo(C−C)(CO)4], the UV/vis/NIR spectrum of the starting complex could be recovered completely. A similar observation was already described by Tory et al. proposing the recoordination of the CO ligand to the metal center within the experimental setup [39]. In contrast, no degradation within the series of [M(C−N)(CO)4] is detected, confirming the complete reversibility of the first ligand-centered reduction (see SI, 5.40).
Based on our UV/vis/NIR- and IR-SEC measurements we can conclude, that the C−N linkage in [M(C−N)(CO)4] results in an increased stabilization of the ligand-centered radical, while the reduction of the C−C pyridyl-MIC ligand shows an EC mechanism, leading to CO dissociation (Scheme 3).
Scheme 3.
Simplified mechanism of the one-electron reduction in [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) at the Au WE surface (grey: alternative reaction pathways).
Scheme 3.
Simplified mechanism of the one-electron reduction in [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) at the Au WE surface (grey: alternative reaction pathways).
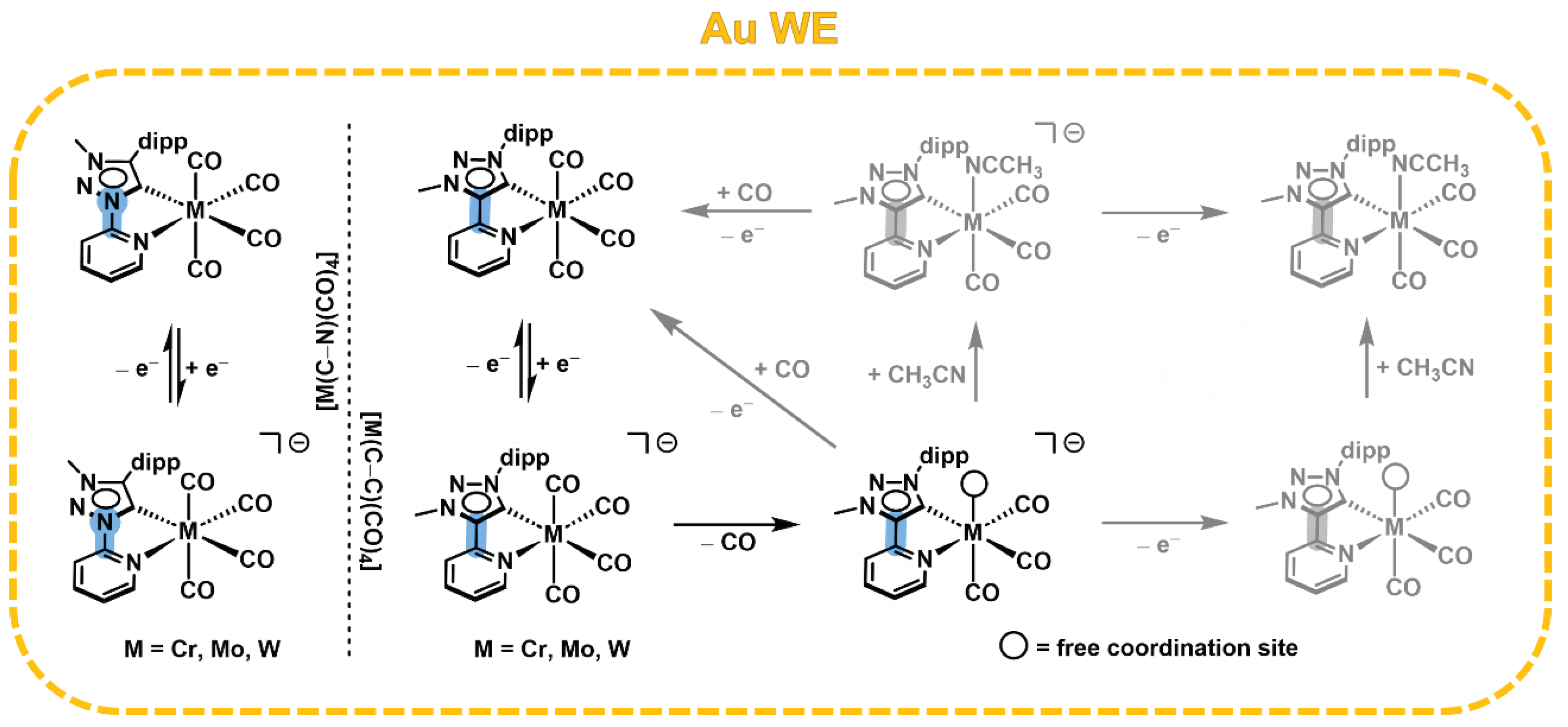
An associative mechanism for the CO dissociation is unlikely, as it would generate a 21 VE species of the already electron-rich [M(C−C)(CO)4] complex. Therefore, we propose a dissociative mechanism after the first reduction, leading to the 17 VE species [M(C−C)(CO)3]−. However, the intermediate is coordinatively unsaturated and thus accessible to solvent coordination, generating the complex [M(C−C)(CH3CN)(CO)3]−.
Based on the previously described reversibility of [Mo(C−C)(CO)4] in UV/vis/NIR-SEC, a stepwise mechanism following the oxidation of the proposed intermediates could lead to the regeneration of the parent complex [39].
Furthermore, the irreversibility of [M(C−C)(CO)4] suggests the formation of multiple species after UV/vis/NIR- and IR-SEC (see SI, 4.20−4.40). The newly generated IR bands after IR-SEC at 1888 cm-1 and 1776 cm-1 (M = W), as well as the IR bands at 1907 cm-1 and 1781 cm-1 (M = Mo), are in good accordance with the previously reported photo-induced formation of the axial solvent adduct [M(C−C)(CH3CN)ax(CO)3] after CO dissociation, supporting the proposed EC mechanism [45].
Interestingly, the IR bands of the decomposition products at 1938 cm-1 and 1799 cm-1 in [W(C−C)(CO)4] and 1946 cm-1 and 1810 cm-1 in [Mo(C−C)(CO)4], respectively, are well-described as the trans-positioned pyridyl [M(C−C)(CH3CN)trans-N(CO)3] and MIC [M(C−C)(CH3CN)trans-C(CO)3] solvato complexes, indicating a fluxional reorganization of the CO ligands after electrochemically induced CO dissociation [45].
Inspired by these results, we reinvestigated all the presented complexes by cyclic voltammetry using an Au WE and in the electrochemical reduction of CO2 with a GC and Au WE, respectively.
2.4. Cyclic Voltammetry with an Au WE and Electrochemical CO2 Reduction
The cyclic voltammograms of [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) with an Au WE show the same electrochemical redox processes as observed with a GC WE (Figure 6, see SI, 2.). The second reduction shifts to more cathodic potential, while in the potential range from −1.5 V to 0.0 V, only minor changes are observed.
The separation of the first reduction from the second reduction leads to a reversible first reduction in all presented complexes, as indicated by the current ratio of 1, the peak-to-peak separation of 0.07 V and the absence of oxidative processes between −1.5 V to 0.0 V (Figure 6, see SI, 2.). The reversibility of the first reduction in the series of [M(C−C)(CO)4] (M = Cr, Mo, W) is a direct consequence of the applied scan rate of 100 mV/s leading to a fast electron transfer processes instead of an EC mechanism, accompanied by CO dissociation, which is observed during electrolysis in the OTTLE cell in the IR- and UV/vis/NIR-SEC measurements.
The second reduction appears completely irreversible in the series of [M(C−C)(CO)4] and [M(C−N)(CO)4] and is further confirmed by the appearance of additional oxidation processes in the range from −1.5 V to 0.0 V.
Earlier reports by Hartl and co-workers on [Mo(CO)4(bpy)] showed a reversible first reduction, generating the monoanionic [Mo(CO)4(bpy)]− species using an Au WE [39]. The second irreversible reduction results in the formation of the coordinatively unsaturated [Mo(CO)3(bpy)]2− complex after CO dissociation. On sweeping back to cathodic potentials, rapid recoordination of the CO ligand is proposed, as indicated by the near recovery of the first reversible reduction in [Mo(CO)4(bpy)]−.
As judged by the cyclic voltammetry for [M(C−C)(CO)4] and [M(C−N)(CO)4], no such intermediate could be detected after the second reduction with an Au WE, even at lower scan rates of 25 mV/s (see SI, 2.). Notably, lowering the scan rate leads to the complete disappearance of the oxidative processes between −1.5 V and 0.0 V. Reversible coordination of one of the pyridyl-MIC moieties after the second reduction, can therefore not be ruled out, due to its electron-rich nature [52,53,54,55].
In presence of CO2 under non-protic conditions, the influence of the metal center, the electrode material, and the constitutional isomers reveal their full potential in the electrochemical activation of CO2 (Figure 7, see SI, 7.).
In the series of [M(C−C)(CO)4], only the chromium complex shows a catalytic current with a GC WE after the first catalytic cycle, while no catalytic current is observed for the higher homologues. Instead, an overpotential ( 280 mV) [56] is observed after the second reduction, which could be a the consequence of adduct formation with CO2, leading to deactivation of the catalysts, as previously reported by Kubiak and co-workers [37].
The second catalytic cycle in [Cr(C−C)(CO)4] shows similar reactivity as described for its higher homologues. To verify whether the catalyst is a real homogenous catalyst or deposited on the electrode surface, a rinse test was performed (see SI, S60) [57]. As judged by the experimental data, no heterogenous reactivity can be detected, which supports the CO2 adduct formation within the series of [M(C−C)(CO)4].
In contrast, in the series of [M(C−N)(CO)4], a catalytic current at high potentials > 3.0 V is detected (see SI, 7.), which underlines that the fine-tuning of the ligand can have a major impact on the catalytic performance. Unfortunately, high applied potentials for the electrocatalytic transformation of CO2 prevented us from further product analysis. Hence, we focused on the influence of the electrode material to shift the onset potential for the electrochemical conversion of CO2 with an Au WE (Figure 7, see SI, 7.).
According to our IR-SEC measurements, the first reduction of [M(C−C)(CO)4] with an Au WE leads to CO dissociation creating an open coordination site for binding CO2. However, the weaker acceptor properties of the C−C linked pyridyl-MIC ligand compared to its bpy counterpart shifts the onset potential to higher cathodic potential, preventing us to investigate the catalytic conversion under the experimental conditions, giving only access to the precatalytic activation (see SI, 7.).
To our surprise, the electrochemical conversion of CO2 with the greater acceptor ligand in [M(C−N)(CO)4], results in a catalytic current close to the potential window of a saturated CO2/CH3CN solution, which is in conflict with our previously described IR-SEC measurements. A plausible explanation could be the formation of only traces of [M(C−N)(CO)3]− at the electrode surface, capable of electrocatalytically reducing CO2, as previously described by Cowan and co-workers [36].
Analysis of the results in the electrochemical conversion of CO2 with [M(C−C)(CO)4] and [M(C−N)(CO)4] using an Au WE show that the onset potential can be shifted drastically up to +730 mV vs. a GC WE by the right choice of the ligand and the electrode material, as shown in the case of [W(C−N)(CO)4].
3. Conclusions
The influence of the constitutional isomers on the redox and the spectroscopic properties of group 6 carbonyl complexes was investigated by cyclic voltammetry, EPR-, IR- and UV/vis/NIR-SEC. According to cyclic voltammetry, the different linkage of the constitutional isomers results in a greater donor strength of the C−C linked pyridyl-MIC ligand and to a lower acceptor ability compared to its C−N counterpart, which could be further confirmed by IR spectroscopy. The changes in the electronic structure have a tremendous influence on the redox-properties of [M(C−C)(CO)4] and [M(C−N)(CO)4]. Based on our EPR-SEC measurements, the first ligand-centered reduction leads to an increased delocalization of the electron spin within the C−C linked isomer. This observation was further supported by UV/vis/NIR-SEC measurements and TD-DFT calculations of the singly-reduced species, indicating an enhanced localization of the charge distribution in [M(C−N)(CO)4]−. Upon reduction, IR-SEC measurements of [M(C−C)(CO)4] show a EC mechanism leading to CO dissociation using an Au WE, while in the case of [M(C−N)(CO)4] a complete electrochemically reversible one-electron reduction was observed. Additionally, UV/vis/NIR- SEC measurements were performed to confirm the pure reversibility of the first ligand-centered reduction or reversibility following an EC mechanism. In the case of [Mo(C−C)(CO)4], the initial spectra could be fully recovered, indicating reversible binding of CO following an EC mechanism. Based on these results, all presented complexes were further tested for the electrochemical conversion of CO2 using a GC and an Au WE. Performing the electrochemical CO2 reduction with a GC WE indicates that all complexes of the series [M(C−N)(CO)4] are capable to electrochemically convert CO2 at high potentials, while the [M(C−C)(CO)4] complexes tend to generate CO2 adducts after the second reduction. The change of electrode material leads to a shift of the onset potential of about +730 mV. However, the catalytic performance close to the potential window of the CO2 saturated 0.1 M CH3CN/Bu4NPF6 precluded further analysis of the product formation. Qualitatively, all presented complexes are capable to activate CO2 by changing the working electrode from GC to Au. Through this study we were able to demonstrate that minor changes in the ligand framework, metal center and experimental setup can have a tremendous influence on the electrochemical, spectroelectrochemical and electrocatalytic performance in such systems.
4. Experimental Section
The synthesis of the complexes [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) was performed according to the previously reported literature procedures [41,42,43,45].
General Procedures, Materials and Instrumentation
Caution! Compounds containing azides are potentially explosive. Although we never experienced any problems during synthesis or analysis, all compounds should be synthesized only in small quantities and handled with great care!
Unless otherwise noted, all reactions were carried out using standard Schlenk-line-techniques under an inert atmosphere of argon (Linde Argon 4.8, purity 99.998%) or in a glovebox (Glovebox Systemtechnik, GS095218).
Commercially available chemicals were used without further purification. The solvents used for metal complex synthesis and catalysis were available from MBRAUN MB-SPS-800 solvent System and degassed by standard techniques prior to use. The identity and purity of compounds were established via 1H and 13C NMR spectroscopy, elemental analysis and mass spectrometry.
Solvents for cyclic voltammetry and UV/vis- and EPR-spectroelectrochemical measurements were dried and distilled under argon and degassed by common techniques prior to use. Column chromatography was performed over silica 60 M (0.04 − 0.063 mm).
1H and 13C{1H} NMR spectra were recorded on a Bruker Advance 400 spectrometer at 19 – 22 °C. Chemical shifts are reported in ppm referenced to the residual solvent peaks [58].
The following abbreviations are used to represent the multiplicity of the signals: s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), sept (septet).
Mass spectrometry was performed on an Agilent 6210 ESI-TOF.
Elemental analyses were performed with an Elementar Micro Cube elemental analyser.
The light-induced syntheses were performed with a LOT-QuantumDesign Arc Lamp (150 W, Xe OF).
Electrochemistry
Cyclic voltammograms were recorded with a PalmSens4 potentiostat or PAR VersaStat (Ametek), respectively, with a conventional three-electrode configuration consisting of a glassy carbon working electrode or gold working electrode, a platinum auxiliary electrode, and a coiled silver wire as a pseudo reference electrode. The (decamethyl)ferrocene/(decamethyl)ferrocenium couple was used as internal reference. All measurements were performed at room temperature with a scan rate between 25 and 1000 mV. The experiments were carried out in absolute Acetonitrile containing 0.1 M Bu4NPF6 (Sigma Aldrich, ≥ 99.0%, electrochemical grade) as the supporting electrolyte.
Spectroelectrochemistry
UV/vis spectra were recorded with a Avantes spectrometer consisting of a light source (AvaLight-DH-S-Bal), a UV/vis detector (AvaSpec-ULS2048), and an NIR detector (AvaSpec-NIR256-TEC). IR spectra were recorded with a BRUKER Vertex 70 FT-IR or Nicolet 6700 FT-IR spectrometer, respectively. UV/vis-spectroelectrochemical measurements were carried out in an optically transparent thin-layer electrochemical (OTTLE) [59,60] cell (CaF2 windows) with a gold-mesh working electrode, a platinum-mesh counter electrode, and a silver-foil pseudo reference. EPR spectra at the X-band frequency (ca. 9.5 GHz) were obtained with a Magnettech MS-5000 benchtop EPR spectrometer equipped with a rectangular TE 102 cavity and a TC HO4 temperature controller. The measurements were carried out in synthetic quartz glass tubes. For EPR spectroelectrochemistry, a three-electrode setup was employed using two Teflon-coated platinum wires (0.005 in. bare and 0.008 in. coated) as the working and counter electrodes and a Teflon-coated silver wire (0.005 in. bare and 0.007 in coated) as the pseudo reference electrode. The experiments were carried out in absolute Acetonitrile containing 0.1 M Bu4NPF6 as the supporting electrolyte. The same solvents as for the CV measurements were used for each compound.
Calculations
The program package ORCA 4.1. was used for all DFT calculations [61]. Starting from the molecular structure obtained from X-ray diffraction geometry optimizations were carried out using the B3LYP [62,63] functional and no symmetry restrictions were imposed during the optimization. For tungsten relativistic effects in zero-order regular approximation (ZORA) were included [64]. All calculations were performed with empirical Van der Waals correction (D3) [65,66,67,68] The restricted and unrestricted DFT methods were employed for closed and open shell molecules respectively unless stated otherwise. Convergence criteria were set to default for geometry-optimization (OPT), and tight for SCF calculations (TIGHTSCF). Triple-ζ–valence basis sets (def2-TZVP) [69] were employed for all atoms. Calculations were performed using resolution of the identity approximation [70,71,72,73,74,75,76] with matching auxiliary basis sets [77,78] for geometry optimizations and numerical frequency calculations and the RIJCOSX (combination of the resolution of the identity and chain of spheres algorithms) approximation for single point calculations using the B3LYP functional. Low-lying excitation energies were calculated with time-dependent DFT (TD-DFT). Solvent effects were taken into account with the conductor-like polarizable continuum model, CPCM [79]. Spin densities were calculated according to the Mulliken population analysis [80]. The absence of imaginary frequency Spin densities, molecular orbitals and difference densities were visualized with the modified Chemcraft 1.8 program [81]. All molecular orbitals are illustrated with an iso value of 0.052. All calculated TD-DFT spectra are Gaussian broadened with a band width of 25 at half height unless otherwise noted.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Cyclic voltammograms of [Cr(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode; Figure S2: Cyclic voltammograms of [Cr(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a gold working electrode; Figure S3: Scan rate dependency in cyclic voltammetry of complex [Cr(C−C)(CO)4] in CH3CN and 0.1 M NBu4PF6 (GC: left; Au: right); Figure S4: Cyclic voltammograms of [Cr(C−N)(CO)4] in CH3CN and 0.1 M Bu4NPF6 with a scan rate of 100 mV/s and a gold working electrode; Figure S5: Scan rate dependency in cyclic voltammetry of complex [Cr(C−N)(CO)4] in CH3CN and 0.1 M NBu4PF6 and a gold working electrode; Figure S6: Cyclic voltammograms of [Mo(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode; Figure S7: Cyclic voltammograms of [Mo(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a gold working electrode; Figure S8: Scan rate dependency in cyclic voltammetry of complex [Mo(C−C)(CO)4] in CH3CN and 0.1 M NBu4PF6 (GC: left; Au: right); Figure S9: Cyclic voltammograms of [Mo(C−N)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a gold working electrode; Figure S10: Scan rate dependency in cyclic voltammetry of complex [Mo(C−N)(CO)4] in CH3CN and 0.1 M NBu4PF6 with a gold working electrode; Figure S11: Cyclic voltammograms of [W(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode; Figure S12: Cyclic voltammograms of [W(C−C)(CO)4] in CH3CN and 0.1 M Bu4NPF6 with a scan rate of 100 mV/s and a gold working electrode; Figure S13: Scan rate dependency in cyclic voltammetry of complex [W(C−C)(CO)4] in CH3CN and 0.1 M NBu4PF6 (GC: left; Au: right); Figure S14: Cyclic voltammograms of [W(C−N)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a glassy carbon working electrode; Figure S15: Cyclic voltammograms of [W(C−N)(CO)4] in CH3CN and 0.1 M Bu4NPF6 at a scan rate of 100 mV/s and a gold working electrode; Figure S16: Scan rate dependency in cyclic voltammetry of complex [W(C−N)(CO)4] in CH3CN and 0.1 M NBu4PF6 (GC: left; Au: right); Table S1: Redox potentials of [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) in CH3CN and 0.1 M NBu4PF6 at 100 mV/s with a glassy carbon working electrode; Table S2. Redox potentials of [M(C−C)(CO)4]and [M(C−N)(CO)4] (M = Cr, Mo, W) in CH3CN and 0.1 M NBu4PF6 at 100 mV/s with a gold working electrode; Figure S17: EPR spectrum of [Cr(C−C)(CO)4]− (left, room temperature) in 0.1 M NBu4PF6/CH3CN with a gold working electrode and spin density plot (right, B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006); Table S3: EPR simulation data of complex [Cr(C−C)(CO)4]-; Figure S18: EPR spectrum of [Mo(C−C)(CO)4]− (left, room temperature) in 0.1 M NBu4PF6/CH3CN with a gold working electrode and spin density plot (right, B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006); Table S4. EPR simulation data of complex [Mo(C−C)(CO)4]-; Figure S19: EPR spectrum of [W(C−C)(CO)4]− (left, room temperature) in 0.1 M NBu4PF6/CH3CN with a gold working electrode and spin density plot (right, B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006); Table S4: EPR simulation data of complex [W(C−C)(CO)4]-; Figure S20: EPR spectrum of [W(C−N)(CO)4]− (left, room temperature) in 0.1 M NBu4PF6/CH3CN with a gold working electrode and spin density plot (right, B3LYP/RIJCOSX/D3BJ/def2-TZVP, iso value = 0.006); Table S5: EPR simulation data of complex [W(C−N)(CO)4]-; Table S6: Composition of [M(C−C)(CO)4]− and [M(C−N)(CO)4]− (M = Cr, Mo, W) in Loewdin spin density; Figure S21: IR spectra of [M(C−C)(CO)4] (left) and [M(C−N)(CO)4] (right) in CH2Cl2 (M = Cr: black, Mo: grey, W: light grey); Table S7: CO frequencies of [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) in CH2Cl2; Figure S22: Changes in the IR spectra of [Cr(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a Au working electrode during the first reduction (left) and after IR-SEC (right); Table S8: Experimental and calculated changes (B3LYP/RIJCOSX/D3BJ/def2-TZVP) in the CO frequencies of [Cr(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a Au working electrode during the first reduction; Figure S23: Top left: Calculated (red) and experimental (grey) IR spectra of [Cr(C−C)(CO)4], Top right: Calculated (red) and experimental (grey) IR spectra of [Cr(C−C)(CO)4]− and bottom centered: Overlay of calculated [Cr(C−C)(CO)4] (blue) and [Cr(C−C)(CO)4]− (black) (B3LYP/RIJCOSX/D3BJ/def2-TZVP, FWHM = 32); Figure S24: Changes in the IR spectra of [Mo(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction (left) and after IR-SEC (right); Table S9: Experimental and calculated changes (B3LYP/RIJCOSX/D3BJ/def2-TZVP) in the CO frequencies of [Mo(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction; Figure S25: Top left: Calculated (red) and experimental (grey) IR spectra of [Mo(C−C)(CO)4], Top right: Calculated (red) and experimental (grey) IR spectra of [Mo(C−C)(CO)4]− and bottom centered: Overlay of calculated [Mo(C−C)(CO)4] (blue) and [Mo(C−C)(CO)4]− (black) (B3LYP/RIJCOSX/D3BJ/def2-TZVP, FWHM = 32); Figure S26: Changes in the IR spectra of [W(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction (left) and after IR-SEC (right); Table S10: Experimental and calculated changes (B3LYP/RIJCOSX/D3BJ/def2-TZVP) in the CO frequencies of [W(C−C)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction; Figure S27: Top left: Calculated (red) and experimental (grey) IR spectra of [W(C−C)(CO)4], Top right: Calculated (red) and experimental (grey) IR spectra of [W(C−C)(CO)4]− and bottom centered: Overlay of calculated [W(C−C)] (blue) and [W(C−C)(CO)4]− (black) (B3LYP/RIJCOSX/D3BJ/ZORA/def2-TZVP, FWHM = 32); Figure S28: Changes in the IR spectra of [W(C−N)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction (left) and after IR-SEC (right); Table S11: Experimental and calculated changes (B3LYP/RIJCOSX/D3BJ/def2-TZVP) in the CO frequencies of [W(C−N)(CO)4] in CH3CN/0.1 M Bu4NPF6 with a gold working electrode during the first reduction; Figure S29. Top left: Calculated (red) and experimental (grey) IR spectra of [W(C−N)(CO)4], Top right: Calculated (red) and experimental (grey) IR spectra of [W(C−N)(CO)4]− and bottom centered: Overlay of calculated [W(C−N)(CO)4] (blue) and [W(C−N)(CO)4]− (black) (B3LYP/RIJCOSX/D3BJ/ZORA/def2-TZVP, FWHM = 32); Figure S30: Changes in the UV/VIS spectra of [Cr(C−C)(CO)4] (left, Inset: 600−1800 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with a gold working electrode and after UV/vis/NIR-SEC (right); Figure S31: Changes in the UV/VIS spectra of [Mo(C−C)(CO)4] (left, Inset: 600−2000 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with a gold working electrode and after UV/vis/NIR-SEC (right); Figure S32: Changes in the UV/VIS spectra of [W(C−C)(CO)4] (left, Inset: 650−2050 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with a gold working electrode and after UV/vis/NIR-SEC (right); Figure S33: Changes in the UV/VIS spectra of [W(C−N)(CO)4] (left, Inset: 750−2090 nm) in CH3CN/0.1 M Bu4NPF6 during the first reduction with a gold working electrode and after UV/vis/NIR-SEC (right); Figure S34: α-HOMO (left) and α-LUMO (right) of complex [Cr(C−C)(CO)4]−; Figure S35: β-HOMO (left) and β-LUMO (right) of complex [Cr(C−C)(CO)4]−; Table S12: Energies of selected orbitals; Table S13: Selected experimental UV/vis data of [Cr(C−C)(CO)4]− together with selected TD-DFT calculations; Figure S36: Involved TD-DFT orbitals of complex [Cr(C−C)(CO)4]−; Figure S37: Calculated TD-DFT spectrum with discrete transitions of [Cr(C−C)(CO)4]−; Table S14: Selected MO energies of [Cr(C−C)(CO)4]−; Table S15: XYZ coordinates of optimized [Cr(C−C)(CO)4]−; Figure S38: HOMO (left) and LUMO (right) of complex [Mo(C−C)(CO)4]; Figure S39: α-HOMO (left) and α-LUMO (right) of complex [Mo(C−C)(CO)4]-; Figure S40: β-HOMO (left) and β-LUMO (right) of complex [Mo(C−C)(CO)4]−; Table S16: Energies and compositions of selected orbitals; Table S17: Selected experimental UV/vis data of [Mo(C−C)] together with selected TD-DFT calculations; Figure S41: Involved TD-DFT orbitals of complex [Mo(C−C)(CO)4]; Figure S42: Calculated TD-DFT spectrum with discrete transitions of [Mo(C−C)(CO)4]; Table S18: Selected experimental UV/vis data of [Mo(C−C)(CO)4]− together with selected TD-DFT calculations; Figure S43: Involved TD-DFT orbitals of complex [Mo(C−C)(CO)4]−; Figure S44: Calculated TD-DFT spectrum with discrete transitions of [Mo(C−C)(CO)4]−; Table S19: Selected MO energies of [Mo(C−C)(CO)4] and [Mo(C−C)(CO)4]-; Table S20: XYZ coordinates of optimized [Mo(C−C)(CO)4]; Table S21: XYZ coordinates of optimized [Mo(C−C)(CO)4]−; Figure S45: HOMO (left) and LUMO (right) of complex [W(C−C)(CO)4]; Figure S46. α-HOMO (left) and α-LUMO (right) of complex [W(C−C)(CO)4]−; Figure S47: β-HOMO (left) and β-LUMO (right) of complex [W(C−C)(CO)4]−; Table S22: Energies and compositions of selected orbitals; Table S23: Selected experimental UV/vis data of [W(C−C)(CO)4] together with selected TD-DFT calculations; Figure 48: Involved TD-DFT orbitals of complex [W(C−C)(CO)4]; Figure S49: Calculated TD-DFT spectrum with discrete transitions of [W(C−C)(CO)4]; Table S24: Selected experimental UV/vis data of [W(C−C)(CO)4]− together with selected TD-DFT calculations; Figure S50: Involved TD-DFT orbitals of complex [W(C−C)(CO)4]−; Figure S51: Calculated TD-DFT spectrum with discrete transitions of [W(C−C)(CO)4]−; Table S25: Selected MO energies of [W(C−C)(CO)4] and [W(C−C)(CO)4]−; Table S26: XYZ coordinates of optimized [W(C−C)(CO)4]; Table S27: XYZ coordinates of optimized [W(C−C)(CO)4]−; Figure S52: HOMO (left) and LUMO (right) of complex [W(C−N)(CO)4]; Figure S53. α-HOMO (left) and α-LUMO (right) of complex [W(C−N)(CO)4]−; Figure S54: β-HOMO (left) and β-LUMO (right) of complex [W(C−N)(CO)4]−; Table S28: Energies and compositions of selected orbitals; Table S29: Selected experimental UV/vis data of [W(C−N)(CO)4] together with selected TD-DFT calculations; Figure S55: Involved TD-DFT orbitals of complex [W(C−N)(CO)4]; Figure S56: Calculated TD-DFT spectrum with discrete transitions of [W(C−N)(CO)4]; Table S30: Selected experimental UV/vis data of [W(C−N)(CO)4]− together with selected TD-DFT calculations; Figure 57: Involved TD-DFT orbitals of complex [W(C−N)(CO)4]−; Figure S58: Calculated TD-DFT spectrum with discrete transitions of [W(C−N)(CO)4]−; Table S31: Selected MO energies of [W(C−N)(CO)4] and [W(C−N)(CO)4]−; Table S32: XYZ coordinates of optimized [W(C−N)(CO)4]; Table S33: XYZ coordinates of optimized [W(C−N)(CO)4]−; Figure S59: Cyclic voltammograms of [Cr(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working electrode (left: first cycle, right: second cycle); Figure S60: Rinse test of [Cr(C−C)(CO)4] (1 mM, black, second cycle) and freshly prepared saturated CO2 solution (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working electrode; Figure S61: Cyclic voltammograms of [Cr(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode; Figure S62: Cyclic voltammograms of [Cr(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working; Figure S63: Cyclic voltammograms of [Cr(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode; Figure S64: Cyclic voltammograms of [Mo(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working; Figure S65: Cyclic voltammograms of [Mo(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode; Figure S66: Cyclic voltammograms of [Mo(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working; Figure S67: Cyclic voltammograms of [Mo(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode; Figure S68: Cyclic voltammograms of [W(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working; Figure S69: Cyclic voltammograms of [W(C−C)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode; Figure S70. Cyclic voltammograms of [W(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC working; Figure S71: Cyclic voltammograms of [W(C−N)(CO)4] (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a gold working electrode.
Author Contributions
Conceptualization, T.B. and B.S..; Methodology, T.B.; Software, T.B.; Validation, T.B. and B.S.; Formal Analysis, T.B. and B.S.; Investigation, T.B.; Resources, B.S.; Data Curation, T.B.; Writing – Original Draft Preparation, T.B..; Writing – Review & Editing, B.S..; Visualization, T.B..; Supervision, B.S..; Project Administration, B.S..; Funding Acquisition, B.S.
Funding
This research was funded by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster).
Data Availability Statement
The data that support the findings of this study are available in the supporting material of this article.
Conflicts of Interest
There are no conflicts of interest to declare.
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. https://doi.org/10.1002/1521-3773(20010601)40:11<2004:AID-ANIE2004>3.0.CO;2-5.
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. 2002, 114, 2708–2711. https://doi.org/10.1002/1521-3757(20020715)114:14%3C2708:AID-ANGE2708%3E3.0.CO;2-0.
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [CrossRef]
- Maity, R.; Sarkar, B. Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes. JACS Au 2022, 2, 22–57. [CrossRef]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as Versatile Abnormal Carbene Ligands for Late Transition Metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [CrossRef]
- Schweinfurth, D.; Hettmanczyk, L.; Suntrup, L.; Sarkar, B. Metal Complexes of Click-Derived Triazoles and Mesoionic Carbenes: Electron Transfer, Photochemistry, Magnetic Bistability, and Catalysis. Z. Anorg. Allg. Chem. 2017, 643, 554–584. [CrossRef]
- Crabtree, R.H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. [CrossRef]
- Donnelly, K.F.; Petronilho, A.; Albrecht, M. Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: Synthesis, properties, and application in catalysis and beyond. Chem. Commun. 2013, 49, 1145–1159. [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [CrossRef]
- Bolje, A.; Košmrlj, J. A Selective Approach to Pyridine Appended 1,2,3-Triazolium Salts. Org. Lett. 2013, 15, 5084–5087. [CrossRef]
- Hohloch, S.; Suntrup, L.; Sarkar, B. Arene–Ruthenium(II) and −Iridium(III) Complexes with “Click”-Based Pyridyl-triazoles, Bis-triazoles, and Chelating Abnormal Carbenes: Applications in Catalytic Transfer Hydrogenation of Nitrobenzene. Organometallics 2013, 32, 7376–7385. [CrossRef]
- Kralj, J.; Bolje, A.; Polančec, D.S.; Steiner, I.; Gržan, T.; Tupek, A.; Stojanović, N.; Hohloch, S.; Urankar, D.; Osmak, M.; et al. Half-Sandwich Ir(III) and Os(II) Complexes of Pyridyl-Mesoionic Carbenes as Potential Anticancer Agents. Organometallics 2019, 38, 4082–4092. [CrossRef]
- Sabater, S.; Müller-Bunz, H.; Albrecht, M. Carboxylate-Functionalized Mesoionic Carbene Precursors: Decarboxylation, Ruthenium Bonding, and Catalytic Activity in Hydrogen Transfer Reactions. Organometallics 2016, 35, 2256–2266. [CrossRef]
- Saha, S.; Yadav, S.; Reshi, N.U.D.; Dutta, I.; Kunnikuruvan, S.; Bera, J.K. Electronic Asymmetry of an Annelated Pyridyl–Mesoionic Carbene Scaffold: Application in Pd(II)-Catalyzed Wacker-Type Oxidation of Olefins. ACS Catal. 2020, 10, 11385–11393. [CrossRef]
- Suntrup, L.; Stein, F.; Klein, J.; Wilting, A.; Parlane, F.G.L.; Brown, C.M.; Fiedler, J.; Berlinguette, C.P.; Siewert, I.; Sarkar, B. Rhenium Complexes of Pyridyl-Mesoionic Carbenes: Photochemical Properties and Electrocatalytic CO2 Reduction. Inorg. Chem. 2020, 59, 4215–4227. [CrossRef]
- Hansmann, M.M.; Antoni, P.W.; Pesch, H. Stable Mesoionic N-Heterocyclic Olefins (mNHOs). Angew. Chem. 2020, 132, 5831–5836. [CrossRef]
- Liang, Q.; Song, D. Recent advances of mesoionic N-heterocyclic olefins. Dalton Trans. 2022, 51, 9191–9198. [CrossRef]
- Huang, S.; Wu, Y.; Huang, L.; Hu, C.; Yan, X. Synthesis, Characterization and Photophysical Properties of Mesoionic N-Heterocyclic Imines. Chem. Asian J. 2022, 17, e202200281. [CrossRef]
- Rudolf, R.; Neuman, N.I.; Walter, R.R.M.; Ringenberg, M.R.; Sarkar, B. Mesoionic Imines (MIIs): Strong Donors and Versatile Ligands for Transition Metals and Main Group Substrates. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200653. [CrossRef]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [CrossRef]
- Collin, J. Electrochemical Reduction of Carbon Dioxide Mediated by Molecular Catalysts. Coord. Chem. Rev. 1989, 93, 245–268. [CrossRef]
- Gonell, S.; Massey, M.D.; Moseley, I.P.; Schauer, C.K.; Muckerman, J.T.; Miller, A.J.M. The Trans Effect in Electrocatalytic CO2 Reduction: Mechanistic Studies of Asymmetric Ruthenium Pyridyl-Carbene Catalysts. J. Am. Chem. Soc. 2019, 141, 6658–6671. [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun. 1984, 328–330. [CrossRef]
- Smieja, J.M.; Kubiak, C.P. Re(bipy-tBu)(CO)3Cl-improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49, 9283–9289. [CrossRef]
- Todorova, T.K.; Huan, T.N.; Wang, X.; Agarwala, H.; Fontecave, M. Controlling Hydrogen Evolution during Photoreduction of CO2 to Formic Acid Using Rh(R-bpy)(Cp*)Cl+ Catalysts: A Structure-Activity Study. Inorg. Chem. 2019, 58, 6893–6903. [CrossRef]
- Windle, C.D.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [CrossRef]
- Clark, M.L.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W.; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts. Chem. Sci. 2014, 5, 1894–1900. [CrossRef]
- Franco, F.; Cometto, C.; Sordello, F.; Minero, C.; Nencini, L.; Fiedler, J.; Gobetto, R.; Nervi, C. Electrochemical Reduction of CO2 by M(CO)4 (diimine) Complexes (M = Mo, W): Catalytic Activity Improved by 2,2′-Dipyridylamine. ChemElectroChem 2015, 2, 1372–1379. [CrossRef]
- Franco, F.; Pinto, M.F.; Royo, B.; Lloret-Fillol, J. A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem. Int. Ed. 2018, 57, 4603–4606. [CrossRef]
- Friães, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Royo, B. Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules 2021, 26. [CrossRef]
- Gonell, S.; Lloret-Fillol, J.; Miller, A.J.M. An Iron Pyridyl-Carbene Electrocatalyst for Low Overpotential CO2 Reduction to CO. ACS Catal. 2021, 11, 615–626. [CrossRef]
- Grice, K.A.; Saucedo, C. Electrocatalytic Reduction of CO2 by Group 6 M(CO)6 Species without "Non-Innocent" Ligands. Inorg. Chem. 2016, 55, 6240–6246. [CrossRef]
- Huang, C.; Liu, J.; Huang, H.-H.; Ke, Z. Recent progress in electro- and photo-catalytic CO2 reduction using N-heterocyclic carbene transition metal complexes. Polyhedron 2021, 203, 115147. [CrossRef]
- Machan, C.W.; Stanton, C.J.; Vandezande, J.E.; Majetich, G.F.; Schaefer, H.F.; Kubiak, C.P.; Agarwal, J. Electrocatalytic Reduction of Carbon Dioxide by Mn(CN)(2,2'-bipyridine)(CO)3: CN Coordination Alters Mechanism. Inorg. Chem. 2015, 54, 8849–8856. [CrossRef]
- Neri, G.; Donaldson, P.M.; Cowan, A.J. The Role of Electrode-Catalyst Interactions in Enabling Efficient CO2 Reduction with Mo(bpy)(CO)4 As Revealed by Vibrational Sum-Frequency Generation Spectroscopy. J. Am. Chem. Soc. 2017, 139, 13791–13797. [CrossRef]
- Sieh, D.; Lacy, D.C.; Peters, J.C.; Kubiak, C.P. Reduction of CO2 by Pyridine Monoimine Molybdenum Carbonyl Complexes: Cooperative Metal-Ligand Binding of CO2. Chem. Eur. J. 2015, 21, 8497–8503. [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [CrossRef]
- Tory, J.; Setterfield-Price, B.; Dryfe, R.A.W.; Hartl, F. [M(CO)]4 (2,2′-bipyridine)] (M = Cr, Mo, W) Complexes as Efficient Catalysts for Electrochemical Reduction of CO2 at a Gold Electrode. ChemElectroChem 2015, 2, 213–217. [CrossRef]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. PNAS USA 2008, 105, 10654–10658. [CrossRef]
- Bens, T.; Walter, R.R.M.; Beerhues, J.; Schmitt, M.; Krossing, I.; Sarkar, B. The Best of Both Worlds: Combining the Power of MICs and WCAs to generate Stable and Crystalline CrI-tetracarbonyl Complexes with π-Accepting Ligands. Chem. Eur. J. 2023, e202301205. [CrossRef]
- Bens, T.; Boden, P.; Di Martino-Fumo, P.; Beerhues, J.; Albold, U.; Sobottka, S.; Neuman, N.I.; Gerhards, M.; Sarkar, B. Chromium(0) and Molydenum(0) Complexes with a Pyridyl-Mesoionic Carbene Ligand: Structural, (Spectro)electrochemical, Photochemical, and Theoretical Investigations. Inorg. Chem. 2020, 59, 15504–15513. [CrossRef]
- Boden, P.; Di Martino-Fumo, P.; Bens, T.; Steiger, S.; Albold, U.; Niedner-Schatteburg, G.; Gerhards, M.; Sarkar, B. NIR-Emissive Chromium(0), Molybdenum(0), and Tungsten(0) Complexes in the Solid State at Room Temperature. Chem. Eur. J. 2021, 27, 12959–12964. [CrossRef]
- Boden, P.J.; Di Martino-Fumo, P.; Bens, T.; Steiger, S.T.; Marhöfer, D.; Niedner-Schatteburg, G.; Sarkar, B. Mechanistic and Kinetic Investigations of ON/OFF (Photo)Switchable Binding of Carbon Monoxide by Chromium(0), Molybdenum(0) and Tungsten(0) Carbonyl Complexes with a Pyridyl-Mesoionic Carbene Ligand. Chem. Eur. J. 2022, 28, e202201038. [CrossRef]
- Bens, T.; Marhöfer, D.; Boden, P.; Steiger, S.T.; Suntrup, L.; Niedner-Schatteburg, G.; Sarkar, B. A Different Perspective on Tuning the Photophysical and Photochemical Properties: The Influence of Constitutional Isomers in Group 6 Carbonyl Complexes with Pyridyl-Mesoionic Carbenes. Inorg. Chem. 2023, 62, 16182–16195. [CrossRef]
- Tang, M.; Cameron, L.; Poland, E.M.; Yu, L.-J.; Moggach, S.A.; Fuller, R.O.; Huang, H.; Sun, J.; Thickett, S.C.; Massi, M.; et al. Photoactive Metal Carbonyl Complexes Bearing N-Heterocyclic Carbene Ligands: Synthesis, Characterization, and Viability as Photoredox Catalysts. Inorg. Chem. 2022, 61, 1888–1898. [CrossRef]
- Bens, T.; Walter, R.R.M.; Beerhues, J.; Lücke, C.; Gabler, J.; Sarkar, B.. Isolation and Reactivity of Chlorido, Solvato, Reduced and Hydridic Cp*Rh Complexes with Pyridyl-MIC (MIC = Mesoionic Carbene) Ligands: A Combined Crystallographic, (Spectro)electrochemical and Theoretical Investigation 2023, under revision.
- Bohnenberger, J.; Schmitt, M.; Feuerstein, W.; Krummenacher, I.; Butschke, B.; Czajka, J.; Malinowski, P.J.; Breher, F.; Krossing, I. Completing the triad: Synthesis and full characterization of homoleptic and heteroleptic carbonyl and nitrosyl complexes of the group VI metals. Chem. Sci. 2020, 11, 3592–3603. [CrossRef]
- Vlček Jr., A. Highlights of the spectroscopy, photochemistry and electrochemistry of [M(CO)4(α-diimine)] complexes, M=Cr, Mo, W. Coord. Chem. Rev. 2002, 230, 225–242. [CrossRef]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Huheey - Anorganische Chemie; Walter de Gruyter: Berlin/Boston, 2008, ISBN 978-3-11-030433-6.
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [CrossRef]
- Crawley, M.R.; Kadassery, K.J.; Oldacre, A.N.; Friedman, A.E.; Lacy, D.C.; Cook, T.R. Rhenium(I) Phosphazane Complexes for Electrocatalytic CO2 Reduction. Organometallics 2019, 38, 1664–1676. [CrossRef]
- Knebel, W.J.; Angelici, R.J. Kinetic and Equilibrium Studies of Bi- and Tridentate Chelate Ring-Opening Reactions of Metal Carbonyl Complexes. Inorg. Chem. 1974, 13, 632–637. [CrossRef]
- Knebel, W.J.; Angelici, R.J. Mechanism of Chelate Ring-Opening in Metal Carbonyl Complexes. Inorg. Chem. 1974, 13, 627–631. [CrossRef]
- Lee, C.-C.; Ke, W.-C.; Chan, K.-T.; Lai, C.-L.; Hu, C.-H.; Lee, H.M. Nickel(II) Complexes of Bidentate N-Heterocyclic Carbene/Phosphine Ligands: Efficient Catalysts for Suzuki Coupling of Aryl Chlorides. Chem. Eur. J. 2007, 13, 582–591. [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [CrossRef]
- Lee, K.J.; McCarthy, B.D.; Dempsey, J.L. On decomposition, degradation, and voltammetric deviation: The electrochemist's field guide to identifying precatalyst transformation. Chem. Soc. Rev. 2019, 48, 2927–2945. [CrossRef]
- The Merck-index, 1991.
- Krejčik, M., Daněk, M., Hartl, F.J. Electroanal. Chem. Interfacial 1991, 317, 179.
- Klein, J.; Stuckmann, A.; Sobottka, S.; Suntrup, L.; van der Meer, M.; Hommes, P.; Reissig, H.-U.; Sarkar, B. Ruthenium Complexes with Strongly Electron-Donating Terpyridine Ligands: Effect of the Working Electrode on Electrochemical and Spectroelectrochemical Properties. Chem. Eur. J. 2017, 23, 12314–12325. [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput Mol Sci 2012, 2, 73–78. [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phy. 1993, 98, 5648–5652. [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phy. 1998, 109, 392–399. [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phy. 2010, 132, 154104–1-154104-19. [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [CrossRef]
- Weigend, Florian, Ahlrichs, Reinhart. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phy. 2011, 135, 144105–1-144105-11. [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [CrossRef]
- Neese, F.; Olbrich, G. Efficient use of the resolution of the identity approximation in time-dependent density functional calculations with hybrid density functionals. Chem. Phy. Lett. 2002, 362, 170–178. [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [CrossRef]
- Petrenko, T.; Kossmann, S.; Neese, F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J. Chem. Phy. 2011, 134, 054116–1-054116-14. [CrossRef]
- Vahtras, O.; Almlöf, J.; Feyereisen, M.W. Integral approximations for LCAO-SCF calculations. Chem. Phy. Lett. 1993, 213, 514–518. [CrossRef]
- Whitten, J.L. Coulombic potential energy integrals and approximations. J. Chem. Phy. 1973, 58, 4496–4501. [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials (Chem. Phys. Letters 240 (1995) 283-290). Chem. Phy. Lett. 1995, 242, 652–660. [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phy. 1955, 23, 1833–1840. [CrossRef]
- Zhurko, G.A. Chemcraft-Graphical Program for Visualization of Quantum Chemistry Computations; Ivanovo (Russia), 2023.
Figure 6.
Cyclic voltammograms of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) in CH3CN and 0.1 M Bu4NPF6 with a scan rate of 100 mV/s and an Au electrode.
Figure 6.
Cyclic voltammograms of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) in CH3CN and 0.1 M Bu4NPF6 with a scan rate of 100 mV/s and an Au electrode.
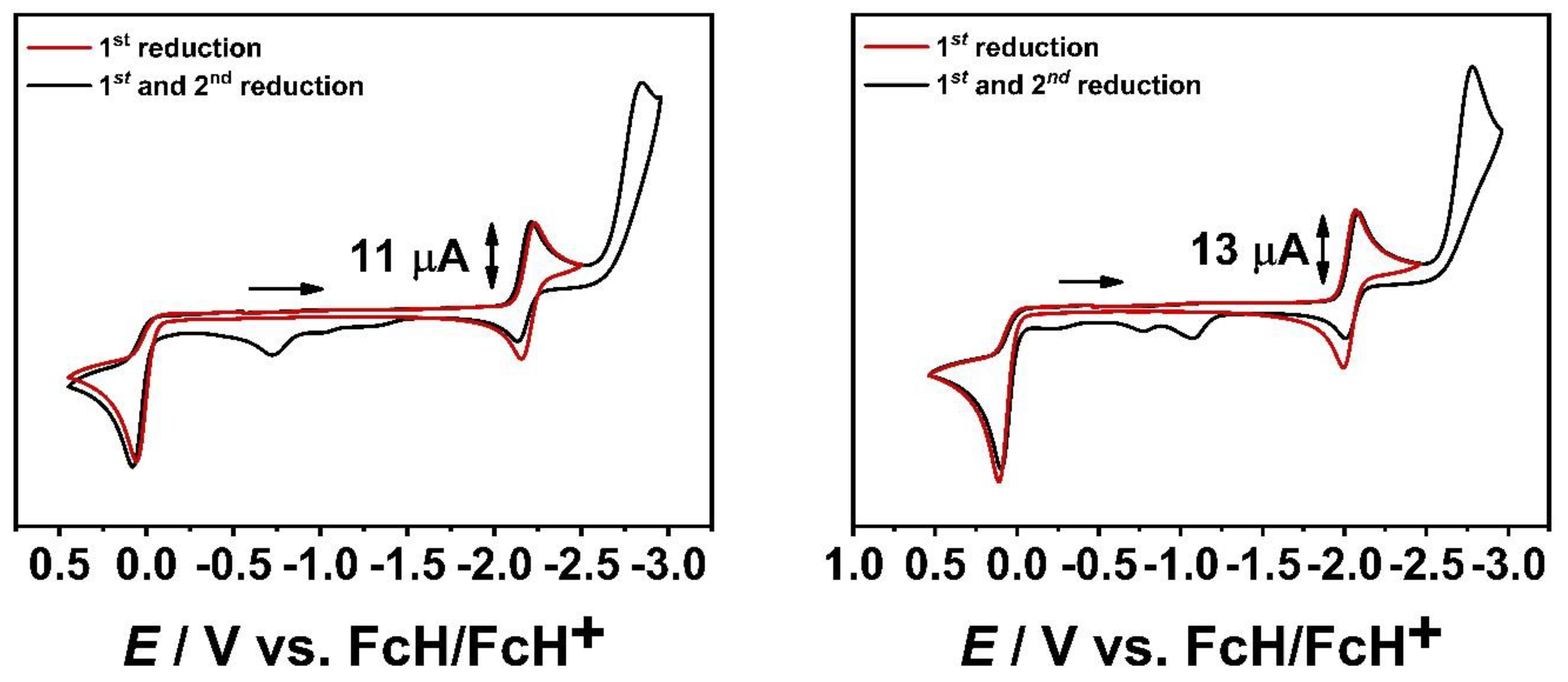
Figure 7.
Cyclic voltammograms of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC WE (top) and an Au WE (bottom).
Figure 7.
Cyclic voltammograms of [W(C−C)(CO)4] (left) and [W(C−N)(CO)4] (right) (1 mM, black) and in the presence of CO2 (red) at 100 mV/s in CH3CN/0.1 M Bu4NPF6 with a GC WE (top) and an Au WE (bottom).
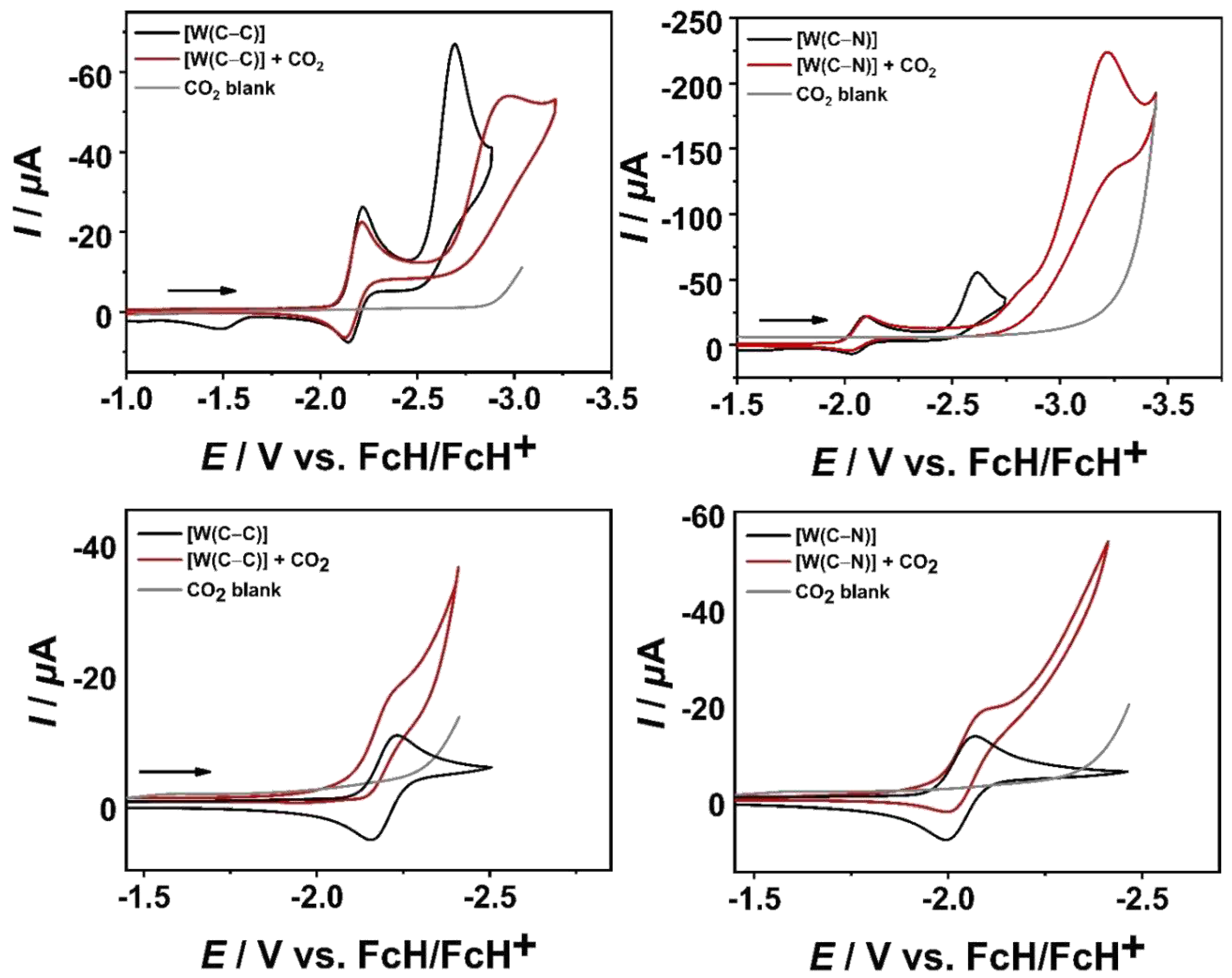

Table 1.
Redox potentials of [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) in CH3CN and 0.1 M NBu4PF6 at 100 mV/s with a glassy carbon working electrode.
Table 1.
Redox potentials of [M(C−C)(CO)4] and [M(C−N)(CO)4] (M = Cr, Mo, W) in CH3CN and 0.1 M NBu4PF6 at 100 mV/s with a glassy carbon working electrode.
| . | 1. Red. / V ΔEp |
2. Red. / V |
1. Ox. / V ΔEp |
||
| [Cr(C−C)(CO)4] [41] | 2.26 | 0.07 | 2.80 | 0.21 | 0.07 |
| [Cr(C−N)(CO)4] [42] | 2.16 | 0.07 | 2.79 | 0.17 | 0.07 |
| [Mo(C−C)(CO)4] | 2.21 | 0.07 | 2.70 | 0.07a | |
| [Mo(C−N)(CO)4] [42] | 2.10 | 0.08 | 2.68 | 0.08a | |
| [W(C−C)(CO)4] | 2.19 | 0.08 | 2.69 | 0.07a | |
| [W(C−N)(CO)4] | 2.05 | 0.06 | 2.65 | 0.12a | |
a.
Table 2.
EPR simulation data of [M(C−N)(CO)4]− (M = Cr, Mo, W) and [W(C−N)(CO)4]−.
| [Cr(C−C)(CO)4]− | [Mo(C−C)(CO)4]− | [W(C−C)(CO)4]− | [W(C−N)(CO)4]− | |
|---|---|---|---|---|
| g | 2.0030 | 2.0033 | 2.0028 | 2.0032 |
| AM | 5.90 | 1.97 | 13.10 | 2.65 |
| AN1 | 17.90 | 10.37 | 10.39 | 16.92 |
| AN2 | 17.80 | 4.95 | 5.60 | 14.22 |
| AN3 | 11.00 | 16.10 | 5.60 | 6.47 |
| AN4 | 9.60 | 9.60 | 4.53 | 5.82 |
| AH1 | 11.70 | 12.00 | 4.16 | 16.20 |
| AH2 | 9.50 | 11.90 | 20.27 | 13.77 |
| AH3 | 3.00 | 10.50 | 20.27 | 13.22 |
| AH4 | 2.00 | 14.45 | 15.36 | 7.19 |
| AH5 | - | - | 1.01 | 3.19 |
| AH6 | - | - | 1.01 | 3.19 |
| AH7 | - | - | 0.63 | - |
| AH8 | - | - | 1.81 | - |
| AH9 | - | - | 0.49 | - |
| AH10 | - | - | 0.50 | - |
| lwppa / mT | [0 0.123] | [0 0.121] | [0 0.054] | [0 0.161] |
a The first value corresponds to Gaussian and the second to Lorentzian shape.
Table 3.
CO stretching frequencies of [M(C−C)] and [M(C−N)] (M = Cr, Mo, W) in CH2Cl2.
| [Cr(C−C)(CO)4][41] | 1998 | 1890 | 1875 (sh) | 1822 | 1896 |
| [Cr(C−N)(CO)4][42] | 1998 | 1890 | 1878 (sh) | 1830 | 1899 |
| [Mo(C−C)(CO)4] | 2004 | 1894 | 1876 (sh) | 1827 | 1900 |
| [Mo(C−N)(CO)4][42] | 2006 | 1896 | 1876 (sh) | 1830 | 1902 |
| [W(C−C)(CO)4] | 1998 | 1882 | 1870 (sh) | 1826 | 1894 |
| [W(C−N)(CO)4] | 2000 | 1884 | 1873 (sh) | 1830 | 1897 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



