
Submitted:
25 December 2023
Posted:
28 December 2023
You are already at the latest version
Abstract
In this work, Density Functional Theory (DFT) calculations were employed to study the photocatalytic CO2 to CO reduction by a series of Pt(II) square planar complexes with general formula [Pt(5-R-dpb)Cl] (dpb = 1,3-di(2-pyridyl)benzene anion, R = H, N,N-dimethylaniline,T thiophene, diazaborinine). The CO2 to CO conversion process is thought to proceed via two main steps, namely the photocatalytic/reduction step and the main catalytic step. The simulated absorption spectra exhibit strong bands in the 200 – 400 nm UV-Vis region. The calculated excited state reduction potentials are in the range 0.32 — -0.36 V revealing that the reductive quenching of the T1 state of the complexes could be modulated with suitable changes of the N^C^N pincer ligands. The CO2 fixation and activation by the ‘real’ three coordinated Pt(II) catalyst/intermediate is predicted to be favorable with Pt-CO2 bond dissociation energies, D0 in the range -36.9—-10.3 kcal/mol. The nature of the Pt-CO2 bond is complex with covalent, hyperconjugative and H-bonding interactions prevailing the repulsive electrostatic interactions. The main catalytic cycle is estimated to be a favorable exergonic process.
Keywords:
Photocatalytic CO2 reduction
; DFT/TDDFT
; Pt(II) pincer complexes
; T1 excited state electrochemistry
; CO2 fixation/activation by Pt
1. Introduction
The problem of global overheating dates back long before the industrial revolution. The excessive use of fossil fuels and the massive release of greenhouse gases into the atmosphere have turned the term "global warming" into an uncomfortable reality of modern life. CO2, although not a primary greenhouse gas, nevertheless has a significant contribution to global warming. In 2012, global CO2 emissions, which come mainly from the burning of fossil fuels, reached a total of 34.5 billion tons [1].
The steadily increasing concentration of CO2 in the atmosphere is now a real problem. CO2 emissions from the massive consumption of fossil fuels are largely responsible for global climate change: an increase in the concentration of CO2 in the atmosphere causes global warming as well as ocean acidification from the uptake of atmospheric CO2 [2].
Man's growing needs for energy lead to the ominous conclusion that CO2 emissions will not remain stagnant, but will increase even more. This results in the scientific community focusing on how the abundance of CO2 will be used to produce materials of commercial interest. Only 1‰ of the total abundance of CO2 is currently used for chemical synthesis, which is mainly due to its chemical inertness (it is highly oxidized in nature) but also to the fact that capturing and storing CO2 is an expensive process [3].
Many studies have been focused on how to deal with the problem of the continuous increase in atmospheric CO2 while at the same time there are ongoing efforts and studies not only to capture CO2 but at the same time to catalytically convert it into "fine chemicals" such as CO, CH3OH and HCOOH [3].
Among various strategies to achieve this goal, the biggest and most attractive challenge is the efficient conversion of CO2 into useful compounds using sunlight as an energy source [4]. Studies on this issue have shown the photochemical and electrochemical reduction of CO2 to CO, to CH3OH, but also to HCOOH, using transition metal electrodes, metal complexes, semiconductors as well as some organic molecules [5,6,7,8].
Much research has focused on how to sequester CO2 from coal plant emissions, which account for 76% of all global emissions [9]. Recent advances in catalyst design combined with the reduced cost of clean energy has made the use of CO2 more attractive as a feedstock for chemical production. Particular interest has focused on the conversion of CO2 to CO as a step in the petrochemical production process [10].
Mainly there are two approaches for the CO2 reduction namely either via heterogeneous or homogeneous catalysis. Both approaches could be achieved either electrocatalytically or photocatalytically. The heterogeneous electrocatalytic CO2 reduction is done with electrolysis with the aid for example of nanostructured materials [11] while heterogeneous photocatalytic CO2 reduction utilizes mainly various types of semiconductors as photocatalysts [12]. On the other hand, the homogeneous electrochemical/photochemical CO2 reduction is mainly based on transition metal complexes [13]. So far, coordination complexes of numerous metals have been studied as catalysts such as the first row Mn, Fe, Co, Ni and Cu, also the second row Ru, Rh, Pd, as well as the third row W, Re, Os, Ir [13].
Up to now, the Ru and Re complexes have received the major focus and are the most widely studied. Probably, the interest for these complexes stems from the inspiring, seminal works of Lehn et al. [14,15,16], who studied the electro/photocatalytic CO2 reduction by Re(I) and Ru(II) complexes. Since then, many other Re(I) and Ru(II) complexes have been studied for the electro/photocatalytic CO2 reduction [2,17,18].
The reduction of CO2 to either CO or HCOOH proceeds via the following chemical equations:
CO2 + 2e− + 2H+ → CO + H2O
CO2 + 2e− + 2H+ → HCOOH
The reduction potentials of the above 2e-/2H+ reductions are -0.52 and -0.61 V vs SHE [19] and the e- are provided either from the electrodes in the case of electrocatalysis or by a ‘sacrificial donor’ e.g. TEOA in the case of photocatalysis. At least for Re(I) and Ru(II) complexes, the proposed mechanism involves, in almost all cases, the formation of an coordinatively unsaturated, 17e- very reactive intermediate [2]. Thus, for example, in the reduction of CO2 with cis-[Ru(bpy)2(CO)X]n+ a mixture of CO/HCOO- is obtained [19]. Two mechanisms are proposed to operate in conjunction, having in common the very reactive, five-coordinated, cis-[Ru(bpy)2(CO)]0 intermediate. The product selectivity depends upon the reactivity of this intermediate towards CO2 or H+ [2]. The so called η1-CO2 complex mechanism operates upon CO2 capture by the cis-[Ru(bpy)2(CO)]0 intermediate, forming the cis-[Ru(bpy)2(CO)(η1-CO2)] adduct which upon protonations yields CO. On the other hand, in the so called hydride mechanism, operating simultaneously with η1-CO2 complex mechanism, the cis-[Ru(bpy)2(CO)]0 intermediate captures H+ forming a hydrido intermediate which upon reduction and CO2 insertion into the Ru-H bond yields HCOO-. However, it should be noticed that the unsaturated five-coordinated [Ru(bpy)2(CO)]0 intermediate has not yet been detected [2]. Accordingly, there have been proposed two other mechanisms for the reduction of CO2 [18].
Ishitani et al. [20], using various spectroscopic techniques, demonstrated the existence of the fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] formed upon coordination of TEOA- anion to the metal centre of the 17e- five-coordinated fac-[ReI(bpy)(CO)3]+ intermediate. They also demonstrated the existence of the fac-[ReI(bpy)(CO)3(O(CO)OCH2CH2NR2)] produced upon insertion of CO2 into the Re-O bond of the fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] intermediate. A similar, CO2 insertion product has been observed also for the [Ru(dmb)2(CO)2]2+ complex in DFM-TEOA solution [21]. The CO2 to CO reduction upon coordination of TEOA- anion and subsequent CO2 insertion has also been scrutinized for Re(I) complexes with the aid of DFT calculations [22].
Finally, based upon both experimental [23,24] as well as theoretical [25] studies there is also another proposed mechanism for the CO2 reduction by transition metal complexes thought to operate via dimerization of the five-coordinated, very reactive intermediate. Hence, Muckerman et al. [25] employing DFT electronic structure calculations, showed that CO2 is inserted between two five-coordinated [Re(dmb)(CO)3]· (dmb = 4,4′-dimethyl-bpy), radical intermediates bridging their metal centers. Through second CO2 addition to this dimeric species, and via a transition state, CO is produced.
Although, so far complexes of a large number of transition metals have been studied for the electro/photocatalytic CO2 reduction, Pt complexes have attracted very little attention. Ceballos et al. [26], demonstrated the electrocatalytic CO2 reduction to formate using the [Pt(dmpe)2](PF6)2 complex with high Faradaic efficiency and low overpotential. The vast majority of studies on CO2 reduction, involving Pt metal have been devoted to heterogeneous catalysis. For example, Zhang et al. [27], dealt with the photocatalytic reduction of CO2 to CH4, under the influence of ultraviolet radiation, with water as solvent on a TiO2 catalyst with Pt. Quite numerous other studies have been also appeared in the literature [28,29,30,31,32,33,34] concerning the heterogeneous CO2 transformation.
To the best of our knowledge, neither experimental nor theoretical studies have been done so far, for the photocatalytic CO2 to CO reduction employing Pt(II) square planar complexes as catalysts. Accordingly, we instigated to scrutinize, by means of electronic structure calculations, the photocatalytic reduction of CO2 to CO using square planar Pt(II) cyclometalated complexes bearing N^C^N coordinating substituted dipyridylbenzene ligands of the general formula [Pt(5-R-dpb)Cl] (dpb = 1,3-di(2-pyridyl)benzene anion, R = H, N,N-dimethylaniline, thiophene, bodipy). Our targets are: 1) To study the photophysical properties of these complexes in order to investigate if they could be used as both photosensitizers as well as catalysts, 2) to explore the mechanism of the catalytic reaction and 3) to probe how the nature of the N^C^N substituted bipyridyl ligand affects both the photophysical properties and mechanism of action of the Pt(II) complexes under study.
2. Results and Discussion
The molecular structure of the square planar Pt(II) complexes employed for the CO2 to CO photocatalytic reduction in this study is depicted in Scheme 1.
The CO2 to CO photocatalytic reduction is thought to proceed via two steps: 1) The photoexcitation/reductive quenching step and 2) the actual catalytic cycle. The two steps are depicted in Figure 1 along with the molecular structures of all species participating in the overall process. Let us start examining first the initial step which henceforth we shall call it the Photoexcitation/Reductive Quenching step and next we will deal with the second step which we shall call it the Catalytic Cycle step.
2.1. Photoexcitation/Reductive Quenching step
In the first step, the Pt(II) catalyst, upon irradiation, is excited to its first singlet excited state, S1. For a third row transition metal complex like those under study, it is expected the transition to occur from the S1 state to the first triplet excited state, T1 via Intersystem Crossing, ISC due to Spin-Orbit Coupling, SOC. Then, if the lifetime, τ of the T1 state of the complex is large enough, a reductive quenching occurs where the complex upon accepting an e- from a donor, D e.g. TEOA or TEA, yields the One Electron Reduced, OER complex (Figure 1). Notice that, the experimentally determined τ of the T1 state of 1 has been found to be 7.2 μs, in CH2Cl2 solvent at 298ºC [35] which is large enough to permit reductive quenching. The optimized geometries of 1 – 4 in their S0 and T1 states as well the geometries of the respective OER species, along with selected structural parameters calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent, are given in Figure 2 as well as in Figures S1–S3 of the Supplementary Material. The calculated structural parameters of 1 in its S0 ground state are in excellent agreement with the respective structural parameters derived from the X-ray structural analysis of this complex [36]. Thus, for example, the difference between the calculated and X-ray derived bond lengths around the coordination sphere is in the range of only 0.001 – 0.016 Å while the calculated bond angles differ by only 0.3 – 0.5º from the respective X-ray experimental values. Upon excitation to the T1 excited state, no significant differences in the structural parameters could be observed (Figure 1b and Figures S1–S3). Accordingly, the changes of the Pt-Cl and Pt-C coordination bonds are in the ranges 0.03 – 0.001 Å and 0.009 – 0.019 Å respectively while the changes of the Pt-N coordination bond lie within the range 0.0 – 0.015 Å. On the other hand, the bond angles around the coordination sphere change slightly by about 1º in all cases, 1 - 4. Finally, the same holds also for the organic framework of the N^C^N pincer ligand, where the structural changes upon the S0 → T1 excitation are negligible. Next, the one electron reduction of the T1 state causes more significant structural changes. Thus, there are more obvious structural differences between the optimized geometries of the T1 state and the OER species (Figure 1c and Figures S1–S3). The most striking structural change is observed in the Pt-Cl coordination bond which is elongated by 0.007 - 0.032 Å in the OER species as compared to the T1 state species. The same holds also, if we compare the OER species with the S0 state species Also the Pt-C coordination bond is elongated by 0.035 Å in contrast to the two Pt-N coordination bonds which are shortened.
Finally, the bond angles around the coordination sphere do not change significantly upon one electron reduction.
2.1.1. Absorption Spectra
Since excitation of the precursor complexes, 1 - 4 is a prerequisite in the photocatalytic CO2 conversion, we set out to study their absorption spectra in DCM by means of TDDFT calculations. The simulated absorption spectra of 1 – 4 in DCM at the TDDFT/CAM-B3LYP/Def2-TZVP level are depicted in Figure 3. Inspection of Figure 3 reveals that the simulated absorption spectra of 1 – 4 exhibit two bands while 1, exhibits also a shoulder. The strongest band, of highest energy for 1, appears around 225 nm accompanied by a shoulder at 243 nm. Finally, the lowest energy band, of medium intensity, appears at 344 nm. In terms of Natural Transition Orbitals (NTOs), the band peaking at 225 nm, arises mainly from an electronic transition at 225 nm which is due to two electron excitations between the NTO pairs depicted in Figure 4. Based upon the shape of these NTOs the band could be assigned as MLCT/IL/LL´CT. Next, the shoulder around 243 nm, arises mainly from an electronic transition at 253 nm due to two electronic excitations (Figure 4) and is assigned as MC/LC/MLCT. Finally, the lowest energy band at 344 nm, arise from an electronic transition which due to one electronic excitation involving an NTO pair depicted in Figure 4. This band could be assigned as MLCT/LC. Upon substitution of one H of the N^C^N pincer ligand with strong donor groups such as PhNMe2 in complex 2 or thiophene in complex 3, the simulated absorption spectra change significantly. Accordingly, for complexes 2 and 3 we observe a weak band, of highest energy, peaking at 229 and 224 nm respectively. Next, there is the strongest band, of lower energy, at 276 nm for both 2 and 3. Finally, there is a shoulder, of lowest energy, in the region 370 – 380 nm. The NTO pairs, relevant to the most significant bands appearing in the absorption spectra of 2 and 3, are depicted in Figures S4 and S5 respectively (See Supplementary Materials). Thus, the bands in the absorption spectra of 2 at 229 nm and of 3 at 224 nm arise mainly from electronic transitions at 227 and 224 nm respectively. Each of these two electronic transitions are due to two electronic excitations between the NTO pairs depicted in Figures S4 and S5. Based upon the shapes of these NTO pairs, the highest energy bands in the absorption spectra of 2 and 3 are assigned as MLCT/IL. Next, the strongest bands in the absorption spectra of 2 and 3, peaking at 276nm, are due to mainly an electronic transition at 263 and 280 nm respectively. The latter, are due to two electronic excitations each and based upon the shape of the participating NTO pairs, we could assign the strongest bands as MLCT/IL. Finally, the shoulder, of lowest energy, appearing in the spectra of 2 and 3 in the region 370 – 380 nm is due to mainly an electronic transition at 376 and 366 nm respectively and based upon the shape of the participating NTOs are assigned as MLCT/IL.
Upon introduction of the diazaborinine group in the N^C^N pincer ligand, the simulated absorption spectrum of 4 changes dramatically with respect to the other complexes under study. Accordingly, the spectrum is red shifted, exhibiting two main bands, one in the UV region, peaking at 260 nm, and the other in the visible region, peaking around 400 nm. The former, arises mainly from an electronic transition at 257 nm while the latter arises from an electronic transition at 422 nm. The electronic transition at 257 nm is due to two electronic excitations and based upon the shapes of the participating NTO pairs (Figure S6) could be assigned as MLCT/IL. On the other hand, the electronic transition at 422 nm is almost solely due to one electronic excitation between NTOs located at the diazaborinine substituent (Figure S6) and therefore the band in the visible region is assigned as IL.
2.1.2. Excited State Electrochemistry
One of the most important steps in the photocatalytic CO2 conversion by transition metal catalysts is the reductive quenching of their T1 state upon receiving an e from a sacrificial donor e.g. TEOA. Therefore, we instigated to study the T1 excited state reduction potentials by means of DFT calculations. The ground state redox potentials could be calculated employing the Born – Haber cycle [37] depicted schematically in Scheme 2.
The standard absolute ground-state reduction potential, E0red is calculated by the following equation:
where F is the Faraday constant (23.061 kcal per volt gram equivalent) and Z is unity for one-electron redox processes. ΔG°(soln., redox) is obtained from the following equation:
E0red = −ΔG°(soln. redox)/ZF,
ΔG°(soln., redox) = ΔG°(gas, redox) + ΔG°(solv.,[Pt(5-R-dpb)Cl]•-) - ΔG°(solv.,[Pt(5-R-dpb)Cl], S0)
In Table 1 are tabulated the values of the reduction potentials calculated for complexes 1 – 4.
2.2. Catalytic Cycle Step
The initial photoexcitation/reductive quenching step ends with the formation of the OER species. The latter, upon losing a chloride ligand, yields a three coordinated intermediate, ImA which starts the actual catalytic cycle (Figure 1). ImA could be considered as the ‘true’ catalyst while the initial Pt(II) complex could be considered as a ‘precatalyst’. Based upon earlier studies [2], the Catalytic Cycle Step could proceed upon one electron reduction of ImA with the aid of an electron donor, D like for example TEOA yielding ImB. Next, there is one of the most important steps i.e. the CO2 addition to the metal center of ImB. After two subsequent protonations (e.g. with TEOA which can act as proton source as well [2]) we arrive, through transition state TS, to the carbonyl intermediate, ImF. A second electron reduction of the latter yields ImG from which we obtain CO and the initial ‘true’ catalyst, ImA.
2.2.1. The η1-CO2 complex
Since the CO2 capture/activation step, upon coordination to the metal center of the catalyst, is considered to be of paramount importance in the electro/photocatalytic CO2 conversion [38,39,40], we set out to study in depth the structural, bonding and electronic properties of ImC.
Structural Properties
The optimized geometry of 1_ImC calculated at the PBE0-D3/Def2-TZVP level, in DCM solvent, is depicted in Figure 5a while those of 2_ImC, 3_ImC, and 4_ImC are given in Figures S5–S7 of the Supporting Information.
Perusal of Figure 5a and Figures S5–S7 reveals that, in all complexes, CO2 is coordinated to the Pt metal center via the C atom in a η1 bonding mode. The complex formed via the η1-CO2 bonding mode is considered to be of pivotal importance for the majority of the electron transfer reactions for the photo/electrocatalytic CO2 conversion [2,40]. The Pt-CO2 bond length is estimated to be 2.132 - 2.135 Å indicative of a bonding interaction. Upon coordination to the Pt metal center, the CO2 molecule is activated and from linear becomes bended with a <O-C-O bond angle equal to 120.8º while the C-O bonds are equal to 1.270 Å, elongated by about 0.110 Å as compared to the ‘free’ CO2 molecule (1.160 Å).
Bonding and Electronic Properties
The covalent nature of the Pt-CO2 bond is reflected in the shape of HOMO calculated for 1_ImC and depicted schematically in Figure 5b (the HOMOs of 2_ImC, 3_ImC and 4_ImC). The HOMO is a bonding MO mainly constructed by the in-phase combination of the Pt dz2 AO with the π* MO of CO2 in line with previous studies [38]. The bond dissociation energy, D0 of the Pt-CO2 bond was found to be -36.8, -36.9, -35.1 and -10.3 kcal/mol for 1_ImC, 2_ImC, 3_ImC and 4_ImC respectively. Obviously, substitution in position 5 of the N^C^N pincer ligand has no significant impact on D0(Pt-CO2). Exception is complex 4, where the introduction of the diazaborinine group reduces D0(Pt-CO2) to almost more than a third as compared to the rest of the complexes. Nevertheless, based upon the magnitude of D0(Pt-CO2), the interaction between ImB and CO2 is expected to be a relatively favorable interaction.
To further analyze the Pt-CO2 bond in ImC, we employed the Atoms in Molecules, AIM method. In Figure 5c are depicted the Bond Critical Points, BCPs found with AIM method performed for 1_ImC (similar BCPs are detected also for intermediate ImC formed by complexes 2 – 4). According to Bader’s [41,42] theory, the presence of a BCP between two atoms indicates bond formation. Inspection of Figure 5c reveals the existence of a BCP between the Pt metal center and C atom of the CO2 ligand, indicating a Pt-CO2 bonding interaction. It has been proposed that [43,44] that based upon the values of certain properties of BCPs, the bonding interactions could be classified into three categories: (a) Pure closed-shell interactions (e.g., ionic bonds, hydrogen bonds and van der Waals interactions) characterized by |VBCP|/GBCP < 1 (∇2ρBCP > 0 and HBCP > 0); (b) pure open-shell (covalent) interactions characterized by |VBCP|/GBCP > 2 (∇2ρBCP < 0 and HBCP < 0); and (c) intermediate bonds with 1 < |VBCP|/GBCP < 2 (i.e., ∇2ρBCP > 0 and HBCP < 0) were VBCP is the potential energy density at BCP, GBCP is the kinetic energy density at BCP, ∇2ρBCP is the Laplacian of electron density, ρ at BCP and finally HBCP is the energy density at BCP. Accordingly, the calculated values of these properties for the BCP found between Pt and CO2 in intermediates 1_ImC – 4_ImC are given in Table 2.
Inspection of Table 1 reveals that the AIM parameters calculated for the Pt-CO2 BCP, do not vary significantly between the ImC intermediates of 1 – 4. Based upon the values of AIM parameters given in Table 1, we can assume that Pt-CO2 interaction falls into the third category i.e. intermediate nature arising from an interplay of covalent, electrostatic, charge transfer and probably weak dispersion forces components. However, the latter should be excluded for the Pt-CO2 bond, in terms of the Reduced Density Gradient, RDG defined by the following equation [45]:
where, ρ(r) is the electron density ρ at point r. The 3D isosurface map of RDG, depicted in Figure 5d, reveals absence of any weak interactions between the Pt metal centre and CO2 (notice that the green isosurface represents weak interactions regions). Similar RDG maps are observed for complexes 2 – 4 as well. In addition, inspection of Figure 5c reveals existence of two BCPs located between the O atoms of CO2 and the H atoms of the N^C^N pincer ligand. Also, the RDG function (Figure 5d) reveals non covalent interactions between those atoms. Therefore, these H bond interactions, further contribute to the overall interaction of CO2 with Im_B intermediate.
Another, useful method for analysing the chemical bond, is the NBO method, which is a partitioning scheme for the electronic charge distribution in a molecule. Thus, we employed NBO calculations to analyse the Pt-CO2 bond from this point of view. The results of the NBO analysis are given in Table 3.
The Natural Charges obtained from NBO analysis, are found to be 0.235 – 0.260 for Pt and 0.494 – 0.497 for C atom of the CO2 ligand. This, results in a repulsive electrostatic interaction between Pt and CO2 in line with previous findings [38,46]. NBO analysis reveals also the existence of a bonding, BD, σ NBO, which is depicted in Figure 6a for 1_ImC (similar BD NBOs are observed also for the rest respective intermediates).
The linear combinations of the BD[σ(Pt-CO2)] NBOs found for 1_ImC – 4_ImC are given in Table 2. Thus, for example, the σ(Pt-C) bonding NBO of 1_ImC has an occupation number equal to 1.857 |e|, arising from the interaction of the sp2.23d1.08 hybrid orbital of Pt (23.15% s, 51.66% p and 25.03% d character) with the sp1.88 hybrid orbital of the C donor atom of CO2 (34.72% s and 65.25% p character), and is described as σ(Pt–C) = 0.558hPt + 0.830hC. In Figure 6b are also depicted the shapes of the NBOs participating in donor – acceptor, hyperconjugative interactions related to the Pt-CO2 bond in 1_ImC. According to NBO analysis [47], there is a stabilization of a system due to charge transfer interactions between specific donor-acceptor NBOs. The stalization energy, ΔE(2) arising from these hyperconjugative (stereoelectronic) interactions is given by the following equation:
where qi is the occupancy of the donor NBO, Fij are the off-diagonal Fock-matrix elements, εi and εj is the energy of the donor and acceptor NBO respectively. The two hyperconjugative interactions, related to Pt-CO2 bond, given in Figure 6b involve σ(Pt–C) and σ*(Pt–C) NBOs and stabilizing the system by overall 86.1 kcal/mol. The overall stabilization due to hyperconjugative interactions for 2_ImC, 3_ImC and 4_ImC is 84.5, 85.5and 80.5 kcal/mol respectively.
ΔE(2) = qiFij/(εi – εj)
Another, method for analyzing a chemical bond is the Charge Decomposition Analysis, CDA [48]. We employed the latter to study the Pt-CO2 interaction for 1_ImC representative case. The net charge transfer from the Pt fragment towards CO2 is calculated to be marginal, amounting to only 0.048 |e| (the donation, d and backdonation, b terms were found to be -0.023 and -0.071 |e| respectively and are somewhat balanced). Finally, the charge polarization term, r has a negative value equal to -0.350 indicating electronic charge removal from the overlapping into the non-overlapping regions upon formation of 1_ImC. The orbital interaction diagram for the formation of 1_ImC from fragments [Pt(dpb)]- (Fragm. 1) and CO2 (Fragm. 2) is depicted in Scheme 3.
Perusal of Scheme 3 reveals that the bonding HOMO of the complex 1_ImC, reflecting the covalent Pt-CO2 interaction, is composed by 34% of LUFO+1 of Fragm. 1 and 55% of LUFO of Fragm, 2. In other words, the HOMO of 1_ImC, is constructed by the in-phase interaction of the, mainly Pt dz2 character, LUFO+1 of the [Pt(dpb)]- fragment with a π* MO of the CO2 fragment.
2.2.2. Energetic Reaction Profiles
The free energy reaction profiles calculated for 1 - 4, corresponding to the respective catalytic cycle step (Figure 1) are given in Figure 7. Inspection of the energetic profiles reveals that the CO2 to CO conversion by the ‘real’ catalyst, ImA is a strongly exergonic reaction. Upon reduction, ImA yields the reactive intermediate ImB which subsequently captures a CO2 molecule to yield the η1-CO2 complex, ImC. Successive protonations of the latter result in the formation of the, immensely stabilized, intermediate ImE. Next, ImE is converted to the carbonyl intermediate, ImF through a concerted mechanism and via a transition state, TS, surmounting an activation barrier of around 38 kcal/mol. Finally, upon second electron reduction we obtain CO while regenerating the ‘real’ catalyst, ImA.
The optimized geometries along with selected structural parameters of all intermediates and TSs involved in the Catalytic Cycle are given in Figures S7–S10. Perusal of the later reveals that the substitution of one H atom of 1 by an R substituent (Scheme 1) has no significant impact on the structural parameters of the [Pt(5-R-dpb)Cl] complexes.
3. Computational Methods
Full geometry optimization has been performed for all species under study, without symmetry constraints, using the 1997 hybrid functional of Perdew, Burke and Ernzerhof [49,50,51,52,53,54] as implemented in the program Gaussian16W [55]. This functional uses 25% exchange and 75% weighting correlation and is denoted as PBE0. Dispersion interactions were accounted by using the D3 version of Grimme dispersion with Becke−Johnson damping [56]. The Def2-TZVP basis set for all atoms was used for the geometry optimizations. The computational protocol will be hereafter denoted as PBE0-GD3BJ/Def2-TZVP. All stationary points have been identified as minima (number of imaginary frequencies, NImag = 0). Natural bond orbital (NBO) population analysis was done employing the methodology by Weinhold [47] as implemented in G16W software. The atoms in molecules (AIM) of Bader [41], the reduced gradient density (RDG) method [57] and the CDA method [48] were used as implemented in Multiwfn software [58]. The Gibbs free energy was calculated to be 298.15 K and 1 atm pressure. Solvent effects were calculated via the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEF-PCM), which is the default method of G16W (self-consistent reaction field (SCRF)) [59] while Dichloromethane, DCM was used as the solvent. Time-dependent density functional theory (TD-DFT) calculations [60,61,62] were performed on the ground-state, S0 equilibrium geometries in DCM solvent using the CAM-B3LYP/Def2-TZVP/PCM computational protocol, taking account the first 30 excited states.
4. Conclusions
The photocatalytic reduction of CO2 to CO by [Pt(5-R-dpb)Cl] (dpb = 1,3-di(2-pyridyl)benzene anion, R = H, N,N-dimethylaniline, thiophene, bodipy), Pt(II) square planar complexes, has been studied by means of DFT electronic structure calculations. The overall process is thought to proceed via two main steps: (i) Photoexcitation/Reduction of the initial complexes and (ii) the main catalytic cycle, producing CO. The TDDFT simulated absorption spectra showed that these complexes absorb mainly in the UV region. However, a small change in the N^C^N pincer ligand i.e. substitution in position 5 with diazaborinine shifts the absorption spectrum to the red, exhibiting a strong band at 400 nm in the visible region. Thus, it is anticipated that changes in the pincer ligand could provide complexes that absorb in the visible, making them ideal for use as photocatalysts for CO2 to CO conversion. Next, the excited state reduction potentials, E0*red, dictating the reductive quenching of the Pt(II) complexes in their T1 state, have been calculated by DFT. Based upon E0*red, values we conclude that the oxidizing ability of the Pt(II) complexes in their T1 state follows the series 1 > 3 > 2 meaning that 1 is expected to be more easily reduced amongst all complexes under study. The second step of the photocatalytic CO2 to CO conversion starts with a three coordinated Pt(II), ImA intermediate which upon one e- reduction captures CO2 and after successive protonations and reductio yields CO. The Pt-CO2 bond of ImC intermediate, being crucial for the hole process, has been scrutinized. It is revealed that this bond is relatively strong with D0 in the range -36.9—-10.3 kcal/mol, exhibiting a complex nature comprising mainly covalent and hyperconjugative interaction which compensate the repulsive electrostatic interactions. The Pt-CO2 bond involves also H bonding with the N^C^N pincer ligand. Overall, the catalytic cycle is estimated to be strongly exergonic process. Taking into account that 1 exhibits a very high T1 excited state lifetime, τ, a whole new series of Pt(II) complexes could be synthesized bearing suitable pincer ligands in order to absorb in the visible, making them ideal for the CO2 to CO conversion using sunlight.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Optimized geometries of 2 in S0 ground state, 2_S0 in T1 excited state, 2_T1 and of One Electron Reduced form, 2_OER, calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S2: Optimized geometries of 3 in S0 ground state, 3_S0 in T1 excited state, 3_T1 and of One Electron Reduced form, 3_OER, calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S3: Optimized geometries of 4 in S0 ground state, 4_S0 in T1 excited state, 4_T1 and of One Electron Reduced form, 4_OER, calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S4: NTO pairs for the most significant electronic transitions of the simulated absorption spectra of 2 (hole, h+ at the left, electron, e- at the right). Figure S5: NTO pairs for the most significant electronic transitions of the simulated absorption spectra of 3 (hole, h+ at the left, electron, e- at the right). Figure S6: NTO pairs for the most significant electronic transitions of the simulated absorption spectra of 4 (hole, h+ at the left, electron, e- at the right). Figure S7: Optimized geometries of all species involved in the catalytic cycle of CO2 to CO conversion by 1 calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S8: Optimized geometries of all species involved in the catalytic cycle of CO2 to CO conversion by 2 calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S9: Optimized geometries of all species involved in the catalytic cycle of CO2 to CO conversion by 3 calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Figure S10: Optimized geometries of all species involved in the catalytic cycle of CO2 to CO conversion by 4 calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent. Table S1: Cartesian Coordinates and Energetic Data of the optimized geometries of all species calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent.
Author Contributions
Conceptualization, A.T.; validation, A.T.; formal analysis, A.T. and A.S.; investigation, A.T. and A.S.; data curation, A.T. and A.S.; writing—original draft preparation, A.T. and A.S.; writing—review and editing, A.T.; visualization, A.T. and A.S.; supervision, A.T.; project administration, A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lahijani, P.; Zainal, Z. A.; Mohammadi M.; Mohamed, A. R. Conversion of the greenhouse gas CO2 to the fuel gas CO via the Boudouard reaction: A review Renew. Sustain. Energy Rev. 2015, 41, 615–632. https://doi.org/10.1016/j.rser.2014.08.034. [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 2018, 373, 333–356. https://doi.org/10.1016/j.ccr.2017.11.023. [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W. A.; Kühn, F. E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chemie - Int. Ed. 2011, 50, 8510–8537. https://doi.org/10.1002/anie.201102010. [CrossRef]
- Morris, A. J.; Meyer, G. J.; Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 2009, 42, 1983–1994. https://doi.org/10.1021/ar9001679. [CrossRef]
- Benson, E. E.; Kubiak, C. P.; Sathrum, A. J.; Smieja, J. M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. https://doi.org/10.1039/b804323j. [CrossRef]
- Takeda, H.; Koike, K.; Morimoto, T.; Inumaru, H.; Ishitani, O. Photochemistry and photocatalysis of rhenium(I) diimine complexes, Adv. Inorg. Chem. 2011, 63, 137-186. https://doi.org/10.1016/B978-0-12-385904-4.00007-X. [CrossRef]
- Costentin, C.; Robert M.; Savéant, J. M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. https://doi.org/10.1039/c2cs35360a. [CrossRef]
- Das, S.; Wan Daud, W. M. A. Photocatalytic CO2 transformation into fuel: A review on advances in photocatalyst and photoreactor. Renew. Sustain. Energy Rev. 2014, 39, 765–805. https://doi.org/10.1016/j.rser.2014.07.046. [CrossRef]
- Vandezande, J. E.; Schaefer, H. F. CO2 Reduction Pathways on MnBr(N-C)(CO)3 Electrocatalysts. Organometallics 2018, 37, 337–342. https://doi.org/10.1021/acs.organomet.7b00743. [CrossRef]
- Paquin, F.; Rivnay, J.; Salleo, A.; Stingelin, N.; Silva, C. Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors. J. Mater. Chem. C, 2015, 3, 10715–10722. https://doi.org/10.48550/arXiv.1310.8002. [CrossRef]
- Yang, C.; Wang, Y.; Qian, L.; Al-Enizi, A. M.; Zhang, L.; Zheng, G. Heterogeneous Electrocatalysts for CO2 Reduction. Appl. Energy Mater. 2021, 4, 1034−1044. https://doi.org/10.1021/acsaem.0c02648. [CrossRef]
- Li, K.; Peng, B.; Peng, T. Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels. ACS Catal. 2016, 6, 7485−7527. https://doi.org/10.1021/acscatal.6b02089. [CrossRef]
- Ishida H., Electrochemical/Photochemical CO2 Reduction Catalyzed by Transition Metal Complexes. In Carbon Dioxide Chemistry, Capture and Oil Recovery. Iyad, K., Shaya, J., Srour, H., Eds.; InTechOpen Ltd. London, UK, 2018; pp. 17 – 40.
- Lehn, J.-M.; Ziessel, R. Photochemical generation of carbon monoxide and hydrogen by reduction of carbon dioxide and water under visible light irradiation. Proc. Natl. Acad. Sci. 1982, 79, 701–704. https://doi.org/10.1073/pnas.79.2.701. [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–Co2+ combinations as homogeneous catalysts. Chem. Commun., 1983, 536–538. https://doi.org/10.1039/C39830000536. [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2′-Bipyridine)tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69, 1990–2012. https://doi.org/10.1002/hlca.19860690824. [CrossRef]
- Elgrishi, N.; Chambers, M. B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761-796. https://doi.org/10.1039/c5cs00391a. [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. & Photobiol. C: Photochem. Rev. 2015, 25, 106–137. https://doi.org/10.1016/j.jphotochemrev.2015.09.001. [CrossRef]
- Voyame, P.; Toghill, K. E.; Méndez, M. A.; Girault, H.H. Photoreduction of CO2 Using [Ru(bpy)2(CO)L]n+ Catalysts in Biphasic Solution/Supercritical CO2 Systems. Inorg. Chem. 2013, 52, 10949–10957. https://doi.org/10.1021/ic401031j. [CrossRef]
- Morimoto, T.; Nakajima, T.; Sawa, S.; Nakanishi, R.; Imori, D.; Ishitani, O. CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine. J. Am. Chem. Soc. 2013, 135, 16825−16828. https://doi.org/10.1021/ja409271s. [CrossRef]
- Tamaki, Y.; Morimoto, T.; Koike, K.; Ishitani, O. Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc. Natl. Acad. Sci. 2012, 109, 15673–15678. https://doi.org/10.1073/pnas.1118336109. [CrossRef]
- Tsipis,A. C.; Sarantou, A. A. Dalton Trans. DFT insights into the photocatalytic reduction of CO2 to CO by Re(I) complexes: the crucial role of the triethanolamine “magic” sacrificial electron donor. 2021, 50, 14797–14809. https://doi.org/10.1039/d1dt02188e. [CrossRef]
- Hayashi, Y.; Kita, S.; Brunschwig, B. S.; Fujita E. Involvement of a Binuclear Species with the Re-C(O)O-Re Moiety in CO2Reduction Catalyzed by Tricarbonyl Rhenium(I)Complexes with Diimine Ligands: Strikingly Slow Formation of the Re-Re and Re-C(O)O-Re Species from Re(dmb)(CO)3S (dmb = 4,4’-Dimethyl-2,2’-bipyridine, S = Solvent). J. Am. Chem. Soc. 2003, 125, 11976-11987. https://doi.org/10.1021/ja035960a. [CrossRef]
- Sullivan, B. P.; Bolinger, C. M.; Conrad, D.; Vining, W. J.; Meyer, T. J. One- and Two-electron Pathways in the Electrocatalytic Reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2’-bipyridine). J. Chem. Soc., Chem. Commun. 1985, 1414–1416. https://doi.org/10.1039/C39850001414. [CrossRef]
- Agarwal, J.; Fujita, E.; Schaefer, III, H. F.; Muckerman, J. T. Mechanisms for CO Production from CO2 Using Reduced Rhenium Tricarbonyl Catalysts. J. Am. Chem. Soc. 2012, 134, 5180-5186. https://doi.org/10.1021/ja2105834. [CrossRef]
- Ceballos B. M.; Yang, J. Y. Directing the reactivity of metal hydrides for selective CO2 reduction. Proc. Natl. Acad. Sci. 2018, 115, 12686–12691. https://doi.org/10.1073/pnas.1811396115. [CrossRef]
- Zhang, Q. H.; Han, W. D.; Hong Y. J.; Yu, J. G. Photocatalytic reduction of CO2 with H2O on Pt-loaded TiO2 catalyst. Catal. Today 2009, 148, 335–340. https://doi.org/10.1016/j.cattod.2009.07.081. [CrossRef]
- Wang, W.; An, W.; Ramalingam, B.; Mukherjee, S.; Niedzwiedzki, D. M.; Gangopadhyay, S.; Biswas, P. Size and Structure Matter: Enhanced CO2 Photoreduction Efficiency by Size-Resolved Ultrafine Pt Nanoparticles on TiO2 Single Crystals. J. Am. Chem. Soc. 2012, 134, 11276−11281. https://doi.org/10.1021/ja304075b. [CrossRef]
- Katsumata, K. I.; Sakai, K.; Ikeda, K.; Carja, G.; Matsushita, N.; Okada, K. Preparation and photocatalytic reduction of CO2 on noble metal (Pt, Pd, Au) loaded Zn-Cr layered double hydroxides. Mater. Lett. 2013, 107, 138–140. https://doi.org/10.1016/j.matlet.2013.05.132. [CrossRef]
- Xie, S.; Wang, Y.; Zhang, Q.; Fan, W.; Denga, W.; Wang, Y. Photocatalytic reduction of CO2 with H2O: Significant enhancement of the activity of Pt–TiO2 in CH4 formation by addition of MgO. Chem. Commun. 2013, 49, 2451–2453. https://doi.org/10.1039/C3CC00107E. [CrossRef]
- Xiong, Z.; Lei, Z.; Kuang, C. C.; Chen, X.; Gong, B.; Zhao, Y.; Zhang, J.; Zheng, C.; Wu, J. C. S. Selective photocatalytic reduction of CO2 into CH4 over Pt-Cu2O TiO2 nanocrystals: The interaction between Pt and Cu2O cocatalysts. Appl. Catal. B Environ. 2017, 202, 695–703. https://doi.org/10.1016/j.apcatb.2016.10.001. [CrossRef]
- Kočí, K.; Dang Van, H.; Edelmannová, M.; Reli, M.; Wu, J. C. S. Photocatalytic reduction of CO2 using Pt/C3N4 photocatalyts. Appl. Surf. Sci. 2020, 503, 144426. https://doi.org/10.1016/j.apsusc.2019.144426. [CrossRef]
- Tasbihi, M.; Kočí, K.; Edelmannová, M.; Troppová, I.; Reli, M.; Schomäcker, R. Pt/TiO2 photocatalysts deposited on commercial support for photocatalytic reduction of CO2. J. Photochem. Photobiol. A Chem. 2018, 366, 72–80. https://doi.org/10.1016/j.jphotochem.2018.04.012. [CrossRef]
- Xu, J.; Liu, X.; Zhou, Z.; Deng, L.; Liu, L.; Xu, M. Platinum Nanoparticles with Low Content and High Dispersion over Exfoliated Layered Double Hydroxide for Photocatalytic CO2 Reduction. Energy and Fuels 2021, 35, 10820–10831. https://doi.org/10.1021/acs.energyfuels.1c00820. [CrossRef]
- Williams, J. A. G.; Beeby, A.; Davies, S.; Weinstein, J. A.; Wilson, C. An Alternative Route to Highly Luminescent Platinum(II) Complexes: Cyclometalation with N^C^N-Coordinating Dipyridylbenzene Ligands. Inorg. Chem. 2003, 42, 8609–8611. https://doi.org/10.1021/ic035083+. [CrossRef]
- Cárdenas, D. J.; Echavarren, A. M.; Ramírez de Arellano, M. C. Divergent Behavior of Palladium(II) and Platinum(II) in the Metalation of 1,3-Di(2-pyridyl)benzene. Organometallics 1999, 18, 3337–3341. https://doi.org/10.1021/om990125g. [CrossRef]
- Demissie, T. B.; Ruud, K.; Hansen, J. H. DFT as a Powerful Predictive Tool in Photoredox Catalysis: Redox Potentials and Mechanistic Analysis. Organometallics, 2015, 34, 4218–4228. https://doi.org/10.1021/acs.organomet.5b00582. [CrossRef]
- Nandal, N.; Jain, S. L. A review on progress and perspective of molecular catalysis in photoelectrochemical reduction of CO2. Coord. Chem. Rev. 2022, 451, 214271. https://doi.org/10.1016/j.ccr.2021.214271. [CrossRef]
- Grice, K. A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78-95. https://doi.org/10.1016/j.ccr.2017.01.007. [CrossRef]
- Kinzel, N. W.; Werl, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. https://doi.org/10.1002/anie.202006988. [CrossRef]
- Bader, R. F. W. Atoms in molecules—a quantum theory. Oxford University Press, Oxford, 1990. [CrossRef]
- Bader, R. F. W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. https://doi.org/10.1021/jp981794v. [CrossRef]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 383, 238–239. https://doi.org/10.1016/S0010-8545(02)00252-7. [CrossRef]
- Espinosa, E; Alkorta, I; Elguero, J; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529−5542. https://doi.org/10.1063/1.1501133. [CrossRef]
- Johnson, E. R.; Shahar Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A. J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498−6506. https://doi.org/10.1021/ja100936w. [CrossRef]
- Yin, X.; Moss, J. R. Recent developments in the activation of carbon dioxide by metal complexes. Coord. Chem. Rev. 1999, 181, 27-59. https://doi.org/10.1016/s0010-8545(98)00171-4. [CrossRef]
- Reed, A. E.; Curtiss, L. A.; F. Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899-926. https://doi.org/10.1021/cr00088a005. [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem.1995, 99, 9352-9362. https://doi.org/10.1021/j100023a009. [CrossRef]
- Vetere, V.; Adamo, C.; Maldivi, P. Performance of the `parameter free' PBE0 functional for the modeling of molecular properties of heavy metals. Chem. Phys. Lett., 2000, 325, 99–105. https://doi.org/10.1016/s0009-2614(00)00657-6. [CrossRef]
- Adamo, C.; Barone, V. Inexpensive and accurate predictions of optical excitations in transition-metal complexes: the TDDFT/PBE0 route. Theor. Chem. Acc., 2000, 105, 169–172. https://doi.org/10.1007/s002140000202. [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys., 1999, 110, 6158–6170. https://doi.org/10.1063/1.478522. [CrossRef]
- Ernzerhof, M.; Scuseria, G. E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys., 1999, 110, 5029–5036. https://doi.org/10.1063/1.478401. [CrossRef]
- Adamo, C.; Scuseria, G. E.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model. J. Chem. Phys., 1999, 111, 2889–2899. https://doi.org/10.1063/1.479571. [CrossRef]
- Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett., 1997, 274, 242–250. https://doi.org/10.1016/s0009-2614(97)00651-9. [CrossRef]
- Gaussian 16W, Revision C.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2016.
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem. 2011, 32, 1456–1465. https://doi.org/10.1002/jcc.21759. [CrossRef]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent Interaction Analysis in Fluctuating Environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. https://doi.org/10.1021/ct4001087. [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyser. J. Comput. Chem. 2012, 33, 580–592. https://doi.org/10.1002/jcc.22885. [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. https://doi.org/10.1021/cr9904009. [CrossRef]
- Van Gisbergen, S. J. A.; Kootstra, F.; Schipper, P. R. T.; Gritsenko, O. V.; Snijders, J. G. Baerends, E. J. Density-functional-theory response-property calculations with accurate exchange-correlation potentials Phys. Rev. A - At. Mol. Opt. Phys., 1998, 57, 2556–2571. https://doi.org/10.1103/PhysRevA.57.2556. [CrossRef]
- Jamorski, C.; Casida, M. E.; Salahub, D. R. Dynamic polarizabilities and excitation spectra from a molecular implementation of time-dependent density-functional response theory: N2 as a case study. J. Chem. Phys., 1996, 104, 5134–5147. https://doi.org/10.1063/1.471140. [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett., 1996, 256, 454–464. https://doi.org/10.1016/0009-2614(96)00440-x. [CrossRef]
Scheme 1.
The molecular structures of Pt(II) complexes under study.
Figure 1.
Proposed calculated mechanism for the CO2 to CO photocatalytic reduction by the [Pt(5-R-dpb)Cl], 1 - 5 Pt(II) complexes.
Figure 1.
Proposed calculated mechanism for the CO2 to CO photocatalytic reduction by the [Pt(5-R-dpb)Cl], 1 - 5 Pt(II) complexes.
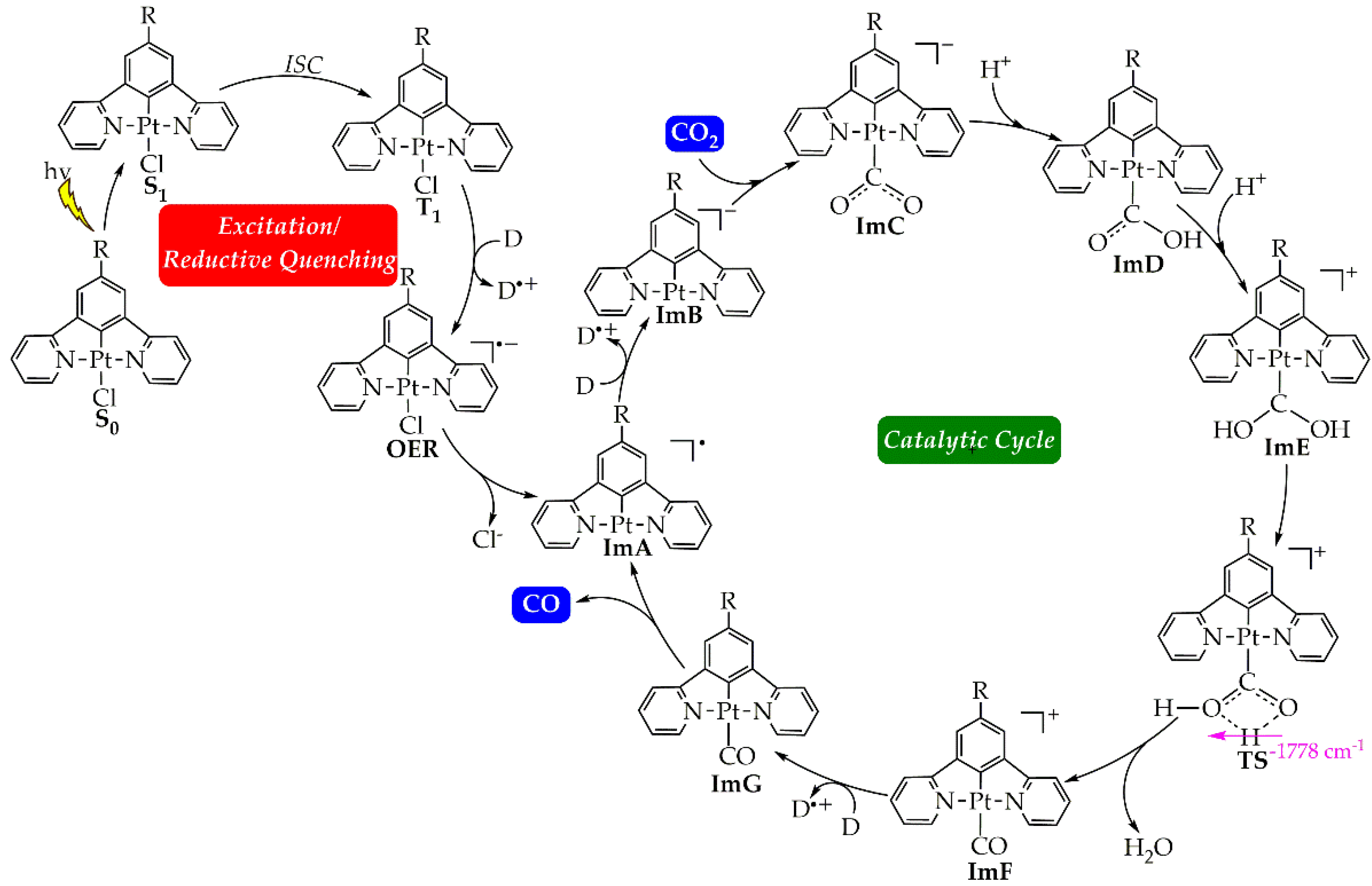
Figure 2.
Optimized geometries of 1 in (a) S0 ground state, (b) in T1 excited state and (c) of One Electron Reduced form, 1_OER, calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent.
Figure 2.
Optimized geometries of 1 in (a) S0 ground state, (b) in T1 excited state and (c) of One Electron Reduced form, 1_OER, calculated at the PBE0-GD3BJ/Def2-TZVP level, in DCM solvent.
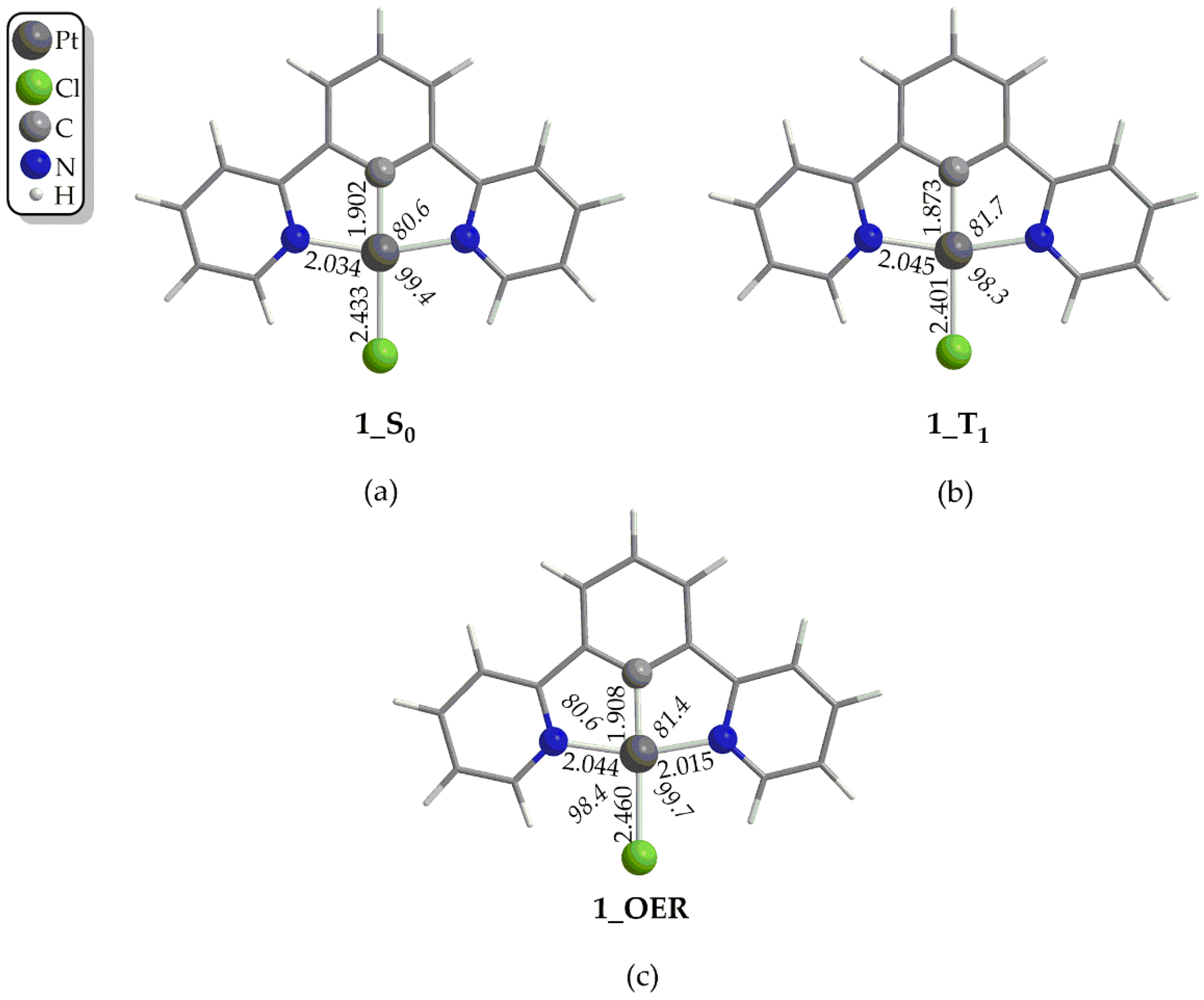
Figure 3.
Simulated absorption spectra of 1 – 4 in DCM at the TDDFT/CAM-B3LYP/Def2-TZVP level.
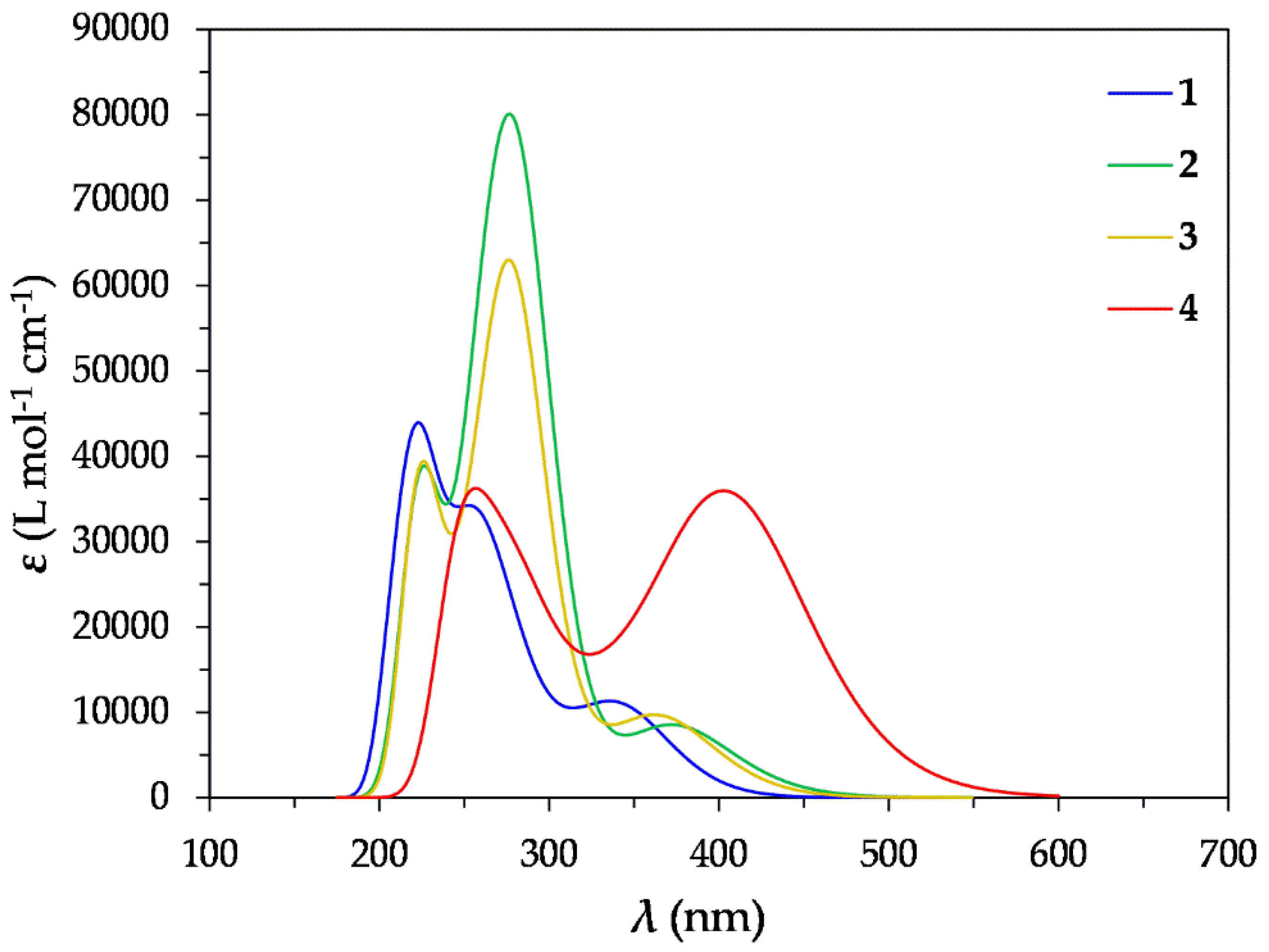
Figure 4.
NTO pairs for the most significant electronic transitions of the simulated absorption spectra of 1 (hole, h+ at the left, electron, e- at the right).
Figure 4.
NTO pairs for the most significant electronic transitions of the simulated absorption spectra of 1 (hole, h+ at the left, electron, e- at the right).
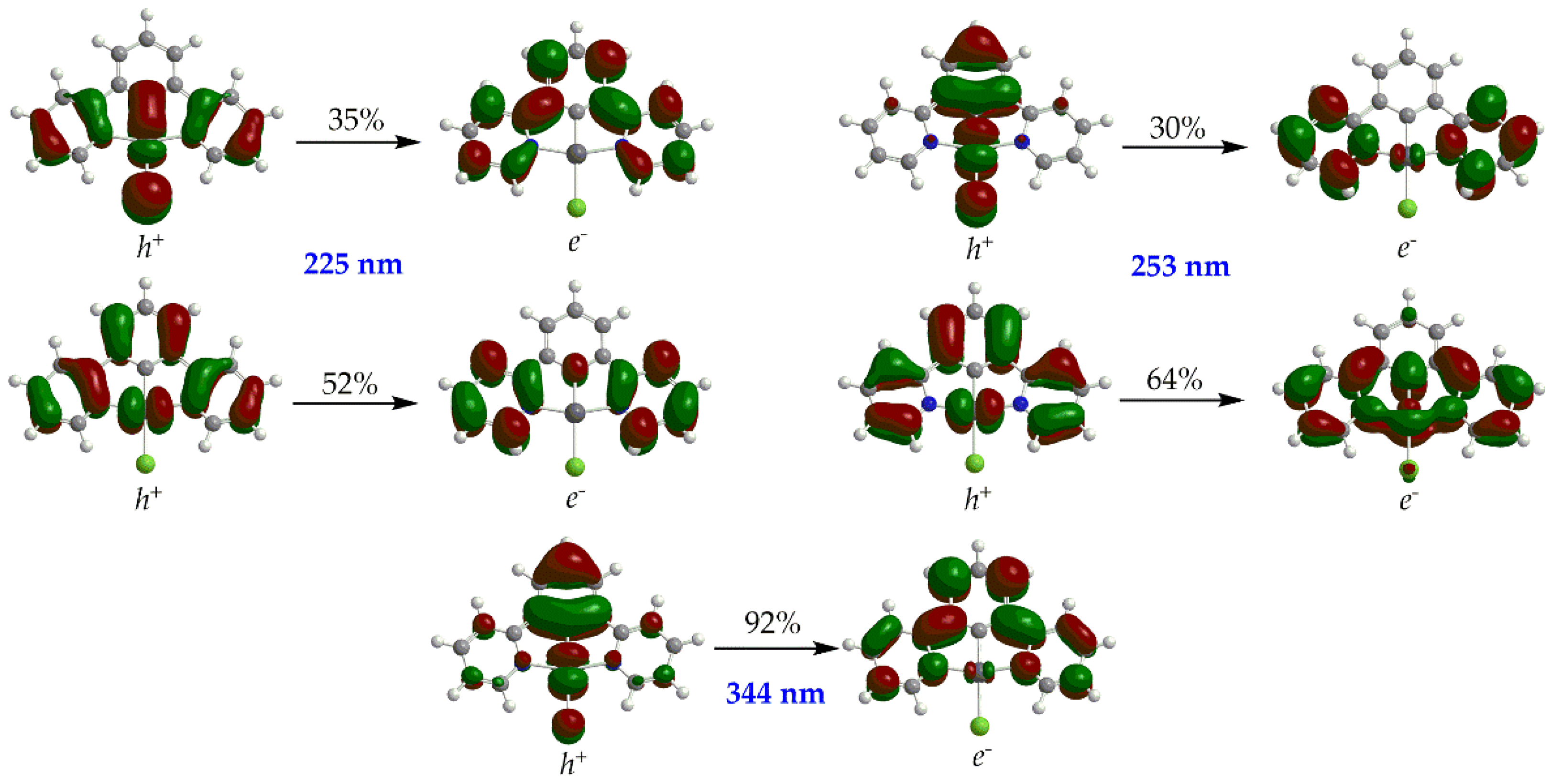
Scheme 2.
Born – Haber cycle for calculation of ground state reduction potentials, E0red (above) and Latimer diagram for the calculation of T1 excited state reduction potentials, E0*red (below).
Scheme 2.
Born – Haber cycle for calculation of ground state reduction potentials, E0red (above) and Latimer diagram for the calculation of T1 excited state reduction potentials, E0*red (below).
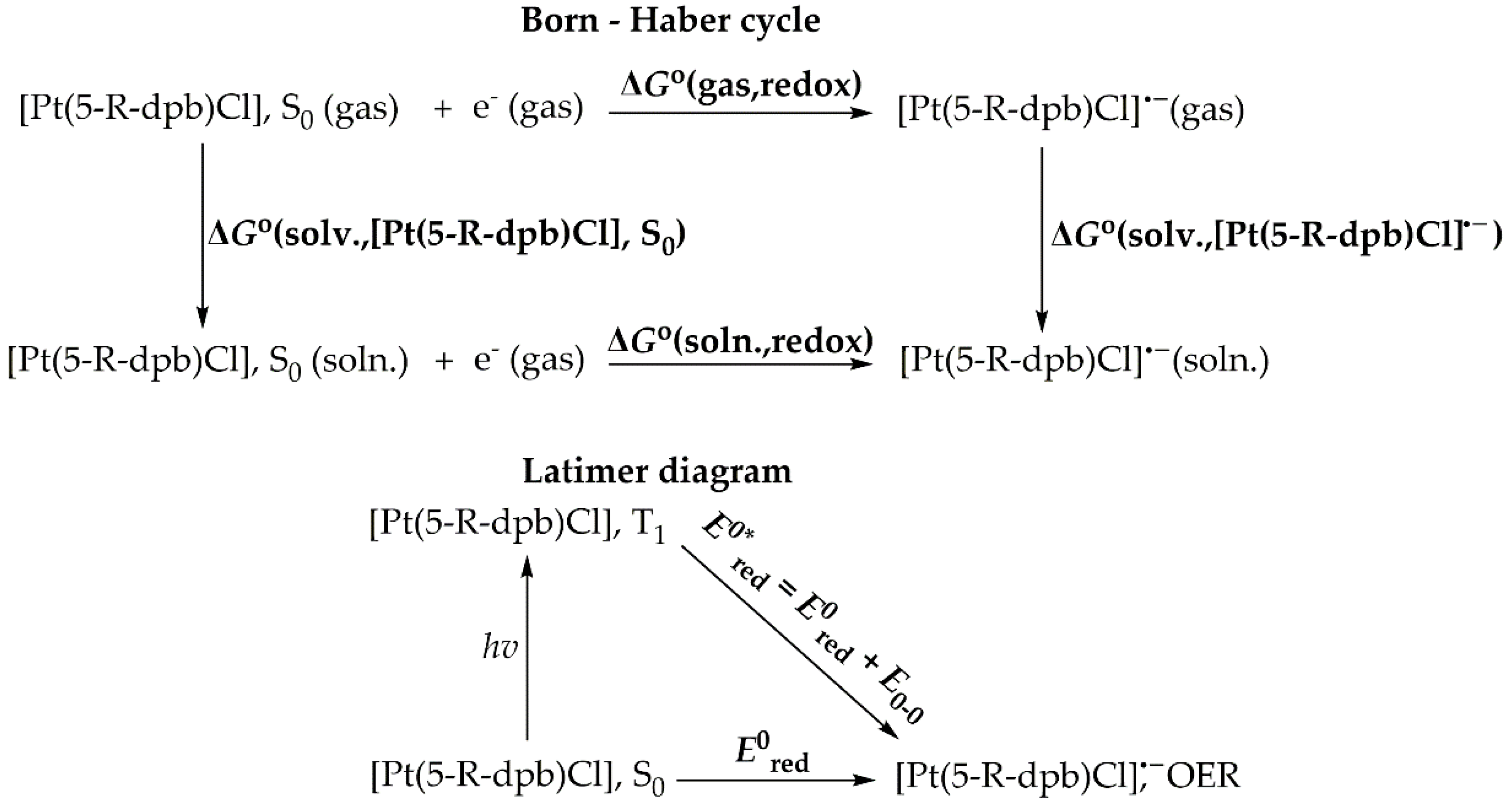
Figure 5.
(a) Optimized geometry of 1_ImC, (b) 3D surface plot of HOMO, (c) BCPs (orange spheres) and (d) 3D surface of RDG function.
Figure 5.
(a) Optimized geometry of 1_ImC, (b) 3D surface plot of HOMO, (c) BCPs (orange spheres) and (d) 3D surface of RDG function.
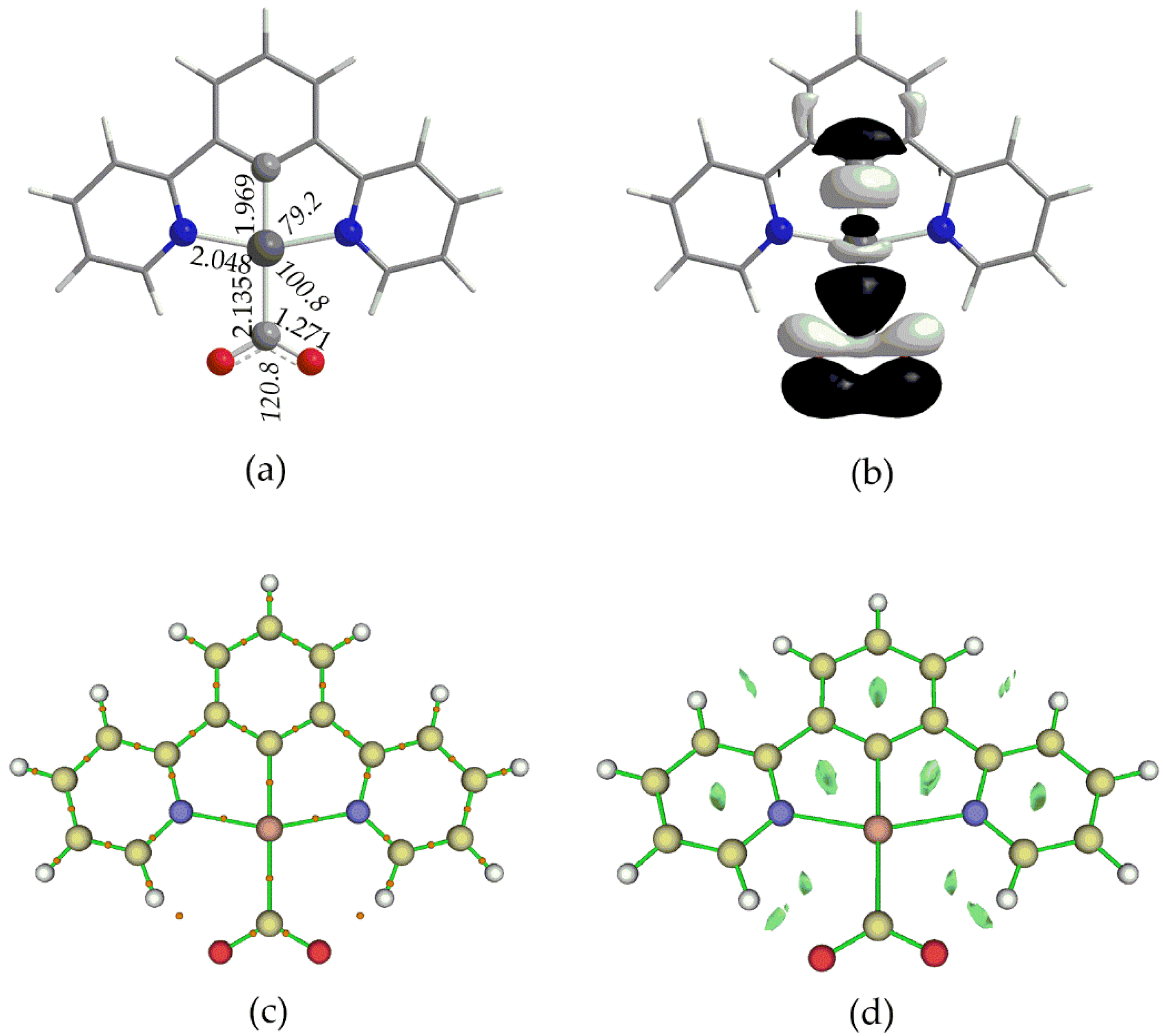
Figure 6.
3D isosurface plots of (a) σ(Pt-C) BD NBO and (b) of donor and acceptor NBOs participating in the Pt-C hyperconjugative interactions.
Figure 6.
3D isosurface plots of (a) σ(Pt-C) BD NBO and (b) of donor and acceptor NBOs participating in the Pt-C hyperconjugative interactions.
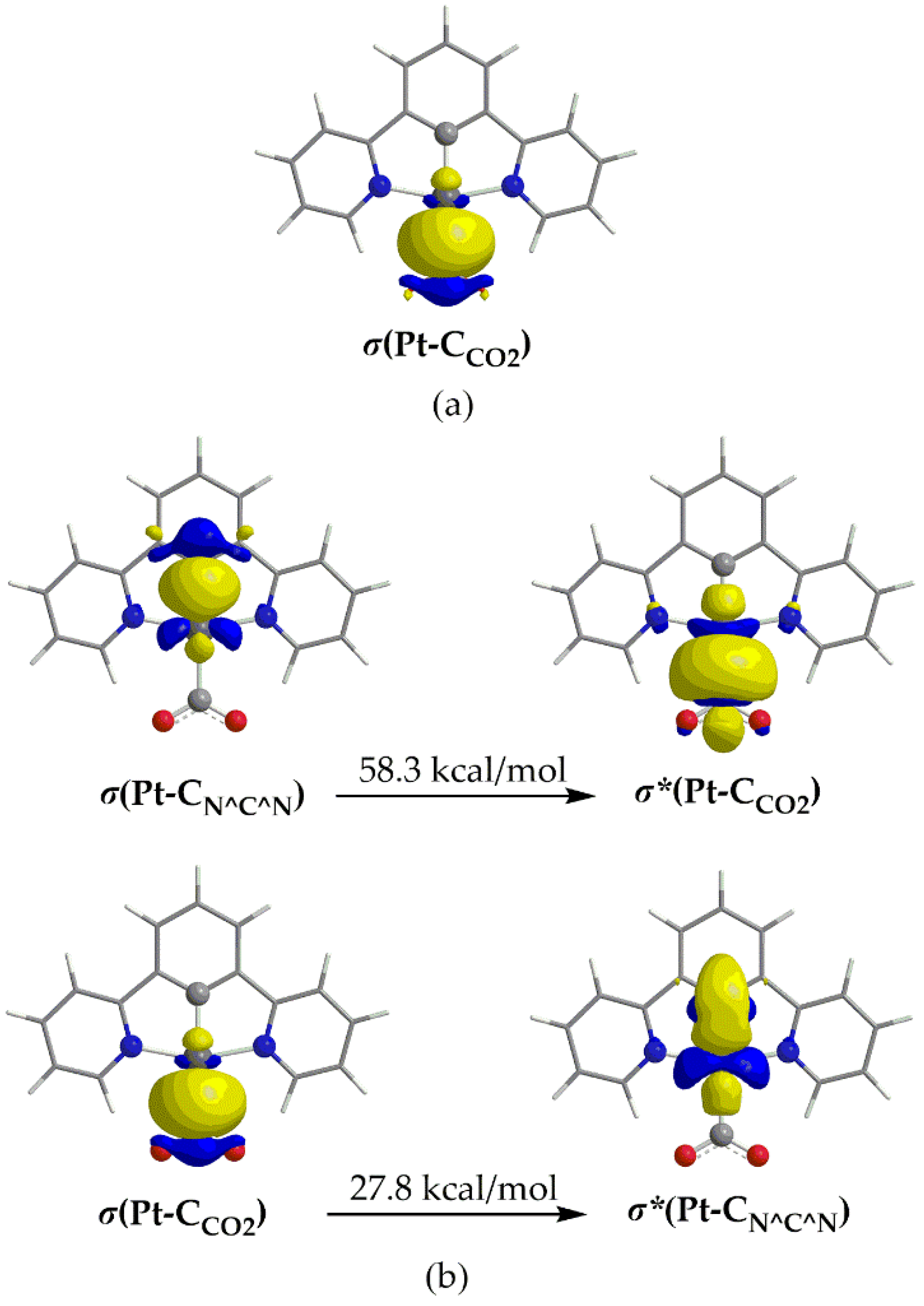
Scheme 3.
Orbital Interaction Diagram for the formation of 1_ImC (Complex) from fragments [Pt(dpb)]- (Fragm. 1) and CO2 (Fragm. 2).
Scheme 3.
Orbital Interaction Diagram for the formation of 1_ImC (Complex) from fragments [Pt(dpb)]- (Fragm. 1) and CO2 (Fragm. 2).
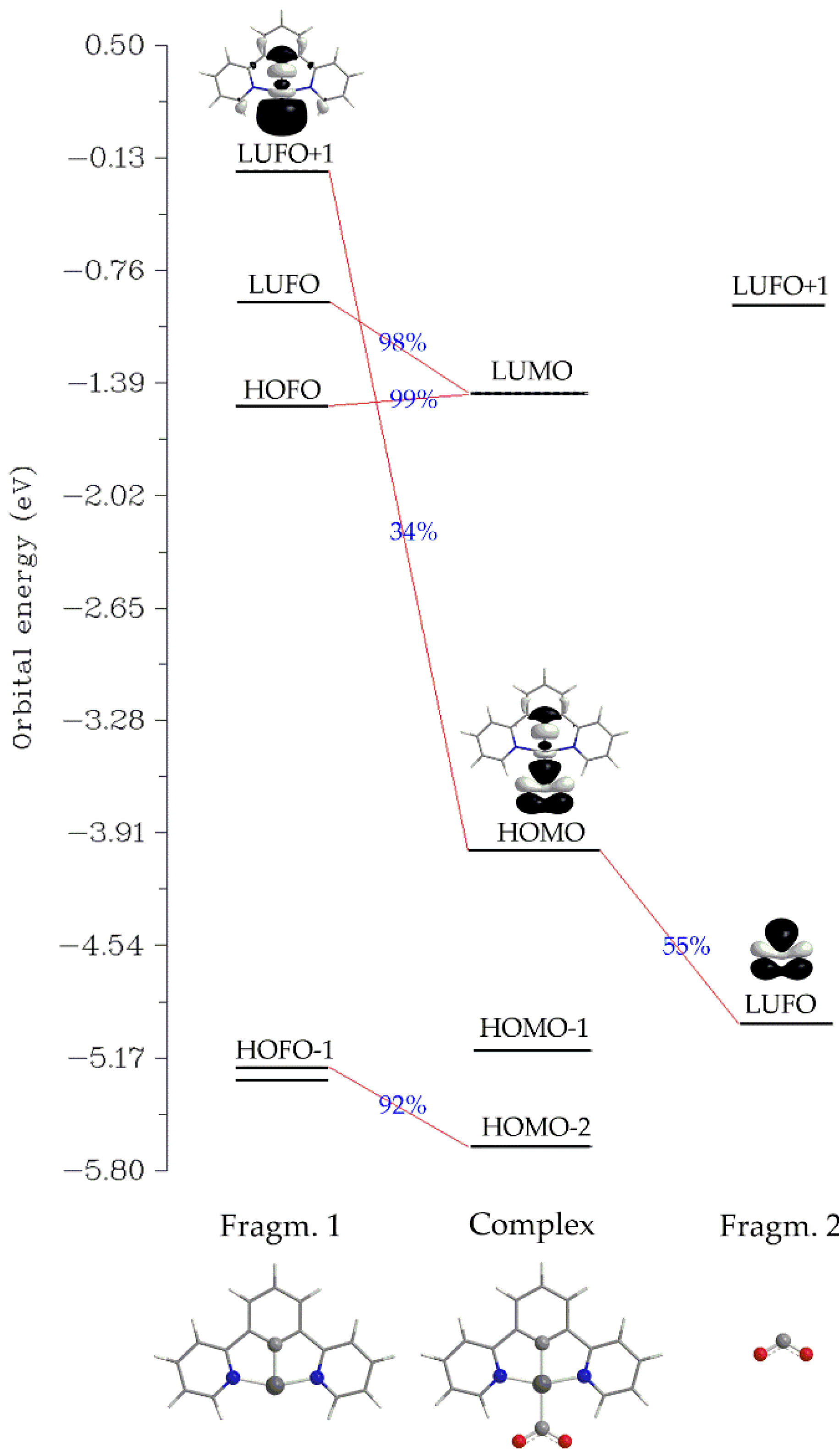
Figure 7.
Free energy, ΔG (in kcal/mol), reaction profiles of 1 – 4 in DCM solvent calculated at the PBE0-GD3BJ/Def2-TZVP level (numbers in blue for 1, in green for 2, in orange for 3 and in red for 4).
Figure 7.
Free energy, ΔG (in kcal/mol), reaction profiles of 1 – 4 in DCM solvent calculated at the PBE0-GD3BJ/Def2-TZVP level (numbers in blue for 1, in green for 2, in orange for 3 and in red for 4).
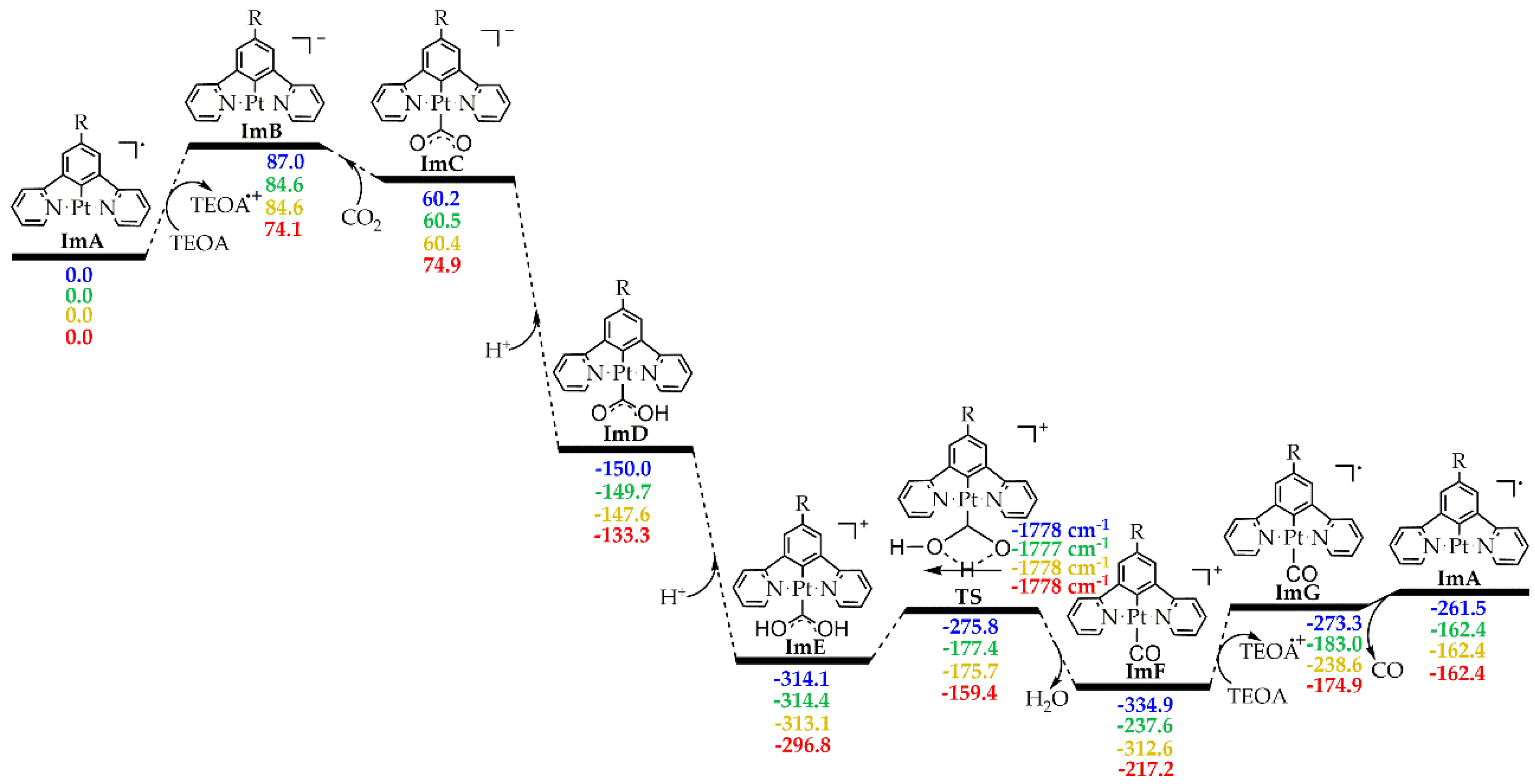

Table 1.
Reduction potentials (in Volts) of 1 – 4 in S0 state, E0red, in T1 state, E0*red, along with the respective values vs SHE.
Table 1.
Reduction potentials (in Volts) of 1 – 4 in S0 state, E0red, in T1 state, E0*red, along with the respective values vs SHE.
| Complex | E0red | E0*red | E0red vs SHE | E0*red vs SHE c |
| 1 | -2.48 | 1.80 | 0.32 | 4.60 |
| 2 | -2.46 | 1.82 | -0.36 | 3.92 |
| 3 | -2.54 | 1.74 | -0.34 | 3.94 |
| 4 | -3.48 | 0.80 | -1.88 | 2.40 |
Inspection of Table 1 reveals that the oxidizing ability of the Pt(II) complexes in their T1 state follows the series 1 > 4 > 3 > 2 i.e. 1 is anticipated to be more easily reduced amongst all complexes under study. In contrast, the oxidizing ability of the Pt(II) complexes in their S0 ground state, somewhat differs from that observed for the T1 state, following the order 2 > 1 > 3 > 4.
Table 2.
Topological and energetic properties of ρ(r) calculated at the (3,–1) bond critical point (BCP) located at the Pt-CO2 bond of intermediates 1_ImC – 4_ImC.
Table 2.
Topological and energetic properties of ρ(r) calculated at the (3,–1) bond critical point (BCP) located at the Pt-CO2 bond of intermediates 1_ImC – 4_ImC.
| Species | ρBCPa | ρBCPb | GBCPc | VBCPc | |VBCP|/GBCP | HBCPc | GBCP/ρBCP |
| 1_ImC | 0.124 | 0.137 | 0.089 | -0.145 | 1.629 | -0.056 | 0.718 |
| 2_ImC | 0.125 | 0.138 | 0.090 | -0.147 | 1.633 | -0.057 | 0.720 |
| 3_ImC | 0.125 | 0.135 | 0.090 | -0.147 | 1.633 | -0.057 | 0.720 |
| 4_ImC | 0.125 | 0.130 | 0.088 | -0.145 | 1.648 | -0.057 | 0.704 |
1 in eÅ-3. 2 in eÅ-5. 3 in kJ mol−1 (atomic unit volume)−1. 4 in kJ mol−1 electron−1. The bond degree parameter HBCP/ρBCP represents either the covalence (HBCP < 0) or the softening (HBCP > 0) degree of the interaction.
Table 3.
Natural Charges, Q, Bonding NBOs, BD and hyperconjugative interactions stabilization energy, ΔE(2) relevant to the Pt-CO2 in 1_ImC – 4_ImC.
Table 3.
Natural Charges, Q, Bonding NBOs, BD and hyperconjugative interactions stabilization energy, ΔE(2) relevant to the Pt-CO2 in 1_ImC – 4_ImC.
| Species | QPt | QC1 | BD[σ(Pt-CO2)] | ΔE(2) | |
| σ(Pt-CN^C^N)→σ*(Pt-CO2) | σ(Pt-CO2)→σ*(Pt-CN^C^N) | ||||
| 1_ImC | 0.235 | 0.495 | 0.558hPt + 0.830hC | 58.3 | 27.8 |
| 2_ImC | 0.237 | 0.494 | 0.540hPt + 0.842hC | 58.1 | 26.4 |
| 3_ImC | 0.244 | 0.495 | 0.538hPt + 0.843hC | 58.3 | 27.2 |
| 4_ImC | 0.260 | 0.497 | 0.535hPt + 0.845hC | 56.9 | 23.6 |
1 Referring to the CO2 carbon atom.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



