Submitted:
30 October 2023
Posted:
31 October 2023
You are already at the latest version
Abstract
Aspergillus montevidensis is an important domesticated fungus that has been applied to produce many traditional fermented foods under high osmotic conditions. However, the detailed mechanisms of tolerance to osmotic stress remain largely unknown. Here, we construct a target-deleted strain (ΔLeuRS) of A. montevidensis and found that the ΔLeuRS mutants grew slowly and suppressed the development of the cleistothecium compared to the wide-type strains (WT) under salt-stressed conditions. Furthermore, differentially expressed genes (P<0.0001) governed by LeuRS were involved in salt tolerance, ABC transporter, amino acid metabolism, sugar metabolism, and reproduction process. The ΔLeuRS strains compared to WT strains under short- and long-term salinity stress especially altered accumulation levels of metabolites, such as amino acids and derivatives, carbohydrates, organic acids, and fatty acids. The study provides new insights into the underlying mechanisms of salinity tolerance and lays a foundation for favor improvement of foods fermented with A. montevidensis.
Keywords:
Leucyl-tRNA synthetase
; transcriptome
; metabolome
; Aspergillus montevidensis
; osmotic tolerance
1. Introduction
The filamentous Aspergillus is a complex group of ascomycetes comprising about 340 officially recognized species, many of which are widely used in food fermentation [1]. Among Aspergillus species, A. montevidensis is an important domesticated fungus that has been used to produce or isolated from fermented and ripened katsuobushi [2]. A. amstelodami (the anamorph of A. montevidensis) is commonly isolated from meju, a brick of fermented dried soybeans, and is frequently occurred in fermented cocoa beans [3,4]. Furthermore, these species dominate the microbial community involved in post-fermented Chinese dark teas, including Pu-erh tea, Fu brick tea, and Liupao tea [5,6,7]. During the production processes of these foods, high osmolarity conditions enhance the dominance of A. montevidensis, which can result in improved quality and taste of fermented foods. Therefore, it is of great significance for understanding the osmotolerant mechanism of A. montevidensis.
We previously found that A. montevidensis is highly dependent on the fine regulation of morphological, transcriptional, and metabolic responses to manage osmotic stress tolerance [8]. Under salinity stress, A. montevidensis widely developed yellow fruiting bodies (cleistothecia), which are known as 'golden flower' in China' brick tea, and hypha growth was obviously promoted [9]. Salinity-induced mycelia significantly expressed hundreds of genes that controlled cellular processes, such as oxidative stress response, amino acid transport and metabolism, glycolysis and TCA cycles, and fatty acid β-oxidation, for the intracellular accumulation of a variety of amino acids, soluble sugars and fatty acids. Among these significantly expressed genes, a unigene encoding leucyl-tRNA synthetase (LeuRS), which routinely catalyzes the specific attachment of leucine to tRNALeu in protein synthesis [10], robustly increased its expression by a factor of 11.52 [8], suggesting that LeuRS could influence osmotic stress tolerance and reproduction process of A. montevidensis.
Sexual and asexual development in Aspergillus species is a highly complex biological process which is affected by both environmental factors and complex intrinsic signals [11]. Extensive studies in A. nidulans showed that the sexual cycle was influenced by many environmental variables, such as light, pH values, temperature, and atmospheric gases [12]. The formation of cleistothecia of A. niger, A. japonicus, and A. ochraceus was greatly affected by the concentrations of different carbon and nitrogen sources [12]. To date, at least 78 genes are required for the modulation of sexual process of Aspergillus. However, there are 20 regulators controlling hundreds of genes, which are involved in asexual development of in A. nidulans [13]. These sex-related genes are linked to perception of environmental signals, signal transduction, transcriptional activators, and maturation of ascospores and conidiophores [11].
Recent studies have shown many previously unknown functions of LeuRS, for example, it serves as a leucine sensor for the mTORC1 signaling pathway in eukaryotes, which subsequently regulates protein synthesis, cell growth, ribosome biogenesis, nutrient uptake, and autophagy [14,15,16]. LeuRS also mediates tolerance to glucose starvation and norvaline-induced stress in yeast and mammals [17,18]. Here, we found that deletion of LeuRS in A. montevidensis influenced mycelial growth rate, antioxidant capacity, significantly suppressed the formation of cleistothecia, and prompted conidial development under salt stress. ΔLeuRS mutants changed expression patterns of multiple genes for substance transport, and amino acid and sugar metabolism, etc. Together, our findings highlighted that LeuRS play a key role in salinity tolerance of A. montevidensis.
2. Materials and methods
2.1. Fungal strains and culture conditions
The wide-type A. montevidensis (no: CGMCC 3.15762) used in this study was maintained routinely on tryptone dextrose agar slants (YPD: 1% yeast extract, 2% tryptone, 2% glucose, 1.5% agar, pH 6.5) supplemented with the NaCl final concentration (1.5 M). The morphological and growth rates of WT and ΔLeuRS mutant were examined on YPD media with 0 M, and 1.5 M NaCl at 28 °C for 14 days. The WT and mutants were cultivated in YPD liquid media with or without 1.5 M NaCl at 28 °C for 14 days (long-term salt stress). These strains were also grown in YPD liquid media without salt at 28 °C for 14 days and then were stressed by the addition of salt (1.5 M) for the short-term culture (1 h). Control samples were obtained from the 14-day cultures untreated with salt. Salt-treated and untreated cultures were washed twice with pre-chilled PBS solution. The mycelia were collected by centrifugation at 4 °C, 12,000 rpm for 10 min, and used in examination of transcriptome, metabolome, antioxidant enzyme (catalase, CAT; superoxide dismutase, SOD) activity [19].
2.2. Constructs for LeuRS knockdown and fungal transformation
We sequenced the whole genome of A. montevidensis and found a 3,501-bp single-copy gene, which encodes LeuRS [8,20]. The LeuRS gene of A. montevidensis (WT) was eliminated by homologous recombination. Briefly, the flanking fragments upstream and downstream (around 1500 bp) of the LeuRS gene were amplified from the wide-type strain, with the specific primer pairs LeuRS1/1F-1R (LeuRS1/1F (PstI): 5’-GCCTGCAGTCCCGATCTTTCACAGACTG-3’; LeuRS1/1R (XbaI): 5’-TCTCTAGACCGAAGAACTTGGGGTACTT-3’) and LeuRS2/2F-2R (LeuRS2/2F (SpeI): 5’-ACACTAGTGATGCTACTCGTATCGCTTT-3’; LeuRS2/2R (EcoRV): 5’-AGGATATCGGAACAGACATAGCGGTTTG-3’), respectively. The amplified DNA fragments were digested by PstI and XbaI, and SpeI and EcoRV, respectively, and then ligated into the plasmid pPK2 at the corresponding restriction sites. The construct pPK2-LeuRS1-Sur-LeuRS2 was transformed into the wide-type strain according to Agrobacterium tumefaciens-mediated transformation [21]. Transformants were selected on YPD agar containing 100 μg/ml of chlorimuron. The colonies were confirmed by PCR amplification using the primer pairs (LeuRS-YZ5f: 5’-CCGTTTTCGCTTTGGATGAG-3’; LeuRS-YZ3r:5’-TGACTCCTGACTCATACTGC-3’) and sequenced.
2.3. Transcriptome sequencing and data analysis
RNA extraction and library preparation of salt-treated and untreated cultures were performed as described by Ding et al. [8]. The cDNA libraries were sequenced on an Illumina Novaseq 6000 platform and 150-bp paired-end reads were generated. The raw data were processed by removing adaptor sequences, low quality reads and reads with unknown nucleotides > 5%. High-quality read assembly was performed using Trinity software (v2.4.0) [22]. Clean reads were assigned to the reference genome (accesssion number JAJFZZ000000000) [20]. The RPKM (reads per kb per million reads) of each unigene was calculated to analyze the expression level of genes between samples. Genes with a false discovery rate (FDR < 0.001) and log2 ratio ≥1 were identified as differentially expressed genes (DEGs) [23]. DEG enrichment analysis was performed in different databases using BLATx, including NCBI nonredundant protein (NR), Gene Ontology (GO), Swissprot, KOG and the Kyoto Encyclopedia of Genes and Genomes (KEGG) with E < 10−5. The biochemical pathways of DEGs were further implemented by KEGG mapping using the cluster Profiler (v3.4.4) [24].
2.4. UHPLC-Q-TOF/MS of metabolites and data analysis
The salinity-treated and non-treated fungal mycelia of the WT and the mutant strains (ΔLeuRS) were extracted with methanol-acetonitrile-water (2:2:1, v/v), which contained internal standard mixtures labeled with isotopes. The samples were vortexed, sonicated in an ice water bath for 4 min, incubated at -40 °C for 1 h, and then centrifuged at 4 °C for 15 min (12,000 rpm) to collect the supernatants. The extraction process was repeated three times. The supernatants were pooled, filtered through a 0.22-µm filter. Meanwhile, a quality control (QC) sample was prepared by mixing equal volumes of each extraction, and was injected to monitor the stability of the system. Metabolites were analyzed using an UHPLC system (Vanquish, Thermo Fisher Scientific) with a UPLC BEH Amide column (2.1 mm × 100 mm, 1.7 μm) coupled to a Q Exactive HFX mass spectrometer (Orbitrap MS, Thermo). The mobile phase consisted of water (pH 9.75) (containing 25 mmol/l ammonium acetate and 25 mmol/l ammonia hydroxide) and acetonitrile. The autosampler temperature was 4 °C and the injection volume was 3 μl. The elution gradient was carried out as follows: 0 min, 5% B; 3 min, 20% B; 9 min, 95% B; 13 min, 95% B; 13.1 min, 5% B; and 16 min, 5% B. A mass scan range of m/z 70-1000 was performed for full-scan analysis. The ESI source conditions were established as follows: sheath gas flow rate 30 arb, aux gas flow rate 25 arb, capillary temperature 350 °C, full MS resolution 60,000, collision energy 10/30/60, spray voltage 3.6 (positive) or -3.2kV (negative), respectively. The raw mass data were converted into mzXML format using ProteoWizard software. XCMS software was applied for peak extraction, peak alignment, and peak matching. Metabolites were then annotated using BiotreeDB (V2.1) software.
2.5. Statistical analysis
All experiments were conducted in independent triplicates, and data are reported as the mean value ± SD (standard deviation). Student's t-test was performed to examine the significant differences (P<0.05) by using SPSS 26.0 software (SPSS Inc., Chicago, IL, USA). Differentially-accumulated metabolites with absolute log2 FC ≥ 1 and P<0.05 were selected for further analysis. Principal component analysis (PCA) was performed by R package (version 4.0.0). Orthogonal projections to latent structures discriminant analysis (OPLS-DA) by SIMCA 14.1 [25] were conducted to screen characteristic components contributing to group discrimination. Differential metabolites were selected by the statistically significant variable importance threshold in projection values > 1 and P-values < 0.05. Significantly different data sets in transcriptomic and metabolomics analysis were used to generate metabolite-transcript pathways by KEGG mapping.
2.6. Accession number(s)
The raw sequence data have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under BioSample accessions: SRR24694851 to SRR24694851.
3. Results
3.1. LeuRS influences mycelial growth, cleistothecium formation, and stress tolerance of A. montevidensis
ΔLeuRS mutants were verified by PCR amplification and sequencing (Figure S1). The deletion of LeuRS resulted in a significant reduction in hyphal growth rates compared to WT strains under salinity or no-salt conditions (Figure S2). Mycelial growth showed a significant morphological difference between the WT strain and the LeuRS mutant on the YPD media supplemented with or without NaCl. The WT strain had yellow colonies, while the ΔLeuRS colonies were white under no-salt stress (Figure S3) or salinity condition for 14 days (Figure 1). The WT strain also produced a large number of yellow-colored spherical cleistothecia, while the formation of cleistothecium was strongly inhibited in the LeuRS mutant after salt stress for 14 days. Salt-stress causes oxidative stress in living cells [26]. Therefore, we examined antioxidative activities of LeuRS mutant and WT strain (Figure S4). CAT activity in the ΔLeuRS cultures induced with or without NaCl was significantly lower than that in the WT, and SOD activity in the mutant samples suffering from the long-term salinity stress was obviously lower compared to the corresponding WT group. These results indicated that LeuRS plays a crucial role in the regulation of mycelial growth, antioxidative capability, and sexual development of A. montevidensis.
3.2. Transcriptome profiles of the LeuRS mutant and the WT strain
We further investigated changes in gene expression profiles of the WT strain and the ΔLeuRS mutant treated without (ΔLeuRS _0 vs. WT_0) or with short-term (1 h) (ΔLeuRS _1h vs. WT_1h) or long-term (14 d) (ΔLeuRS_14d vs. WT_14d) saline stresses. With three biological replicates, transcriptomic sequencing generated 383,214,494 and 399,020,304 clean reads for the three comparison groups (18 libraries). We match unigene sequences with the NR, Swiss-prot, KEGG and KOG databases using blastx (E < 10−5). Of these, a total of 7,165 unigenes were functionally annotated. PCA indicated that all biological replicates were grouped together and PC1 and PC2 captured 60% of the variance in the data. This represents the difference in gene expression profiling between the WT strain and the ΔLeuRS mutant (Figure 2a). Furthermore, we identified significantly differentially expressed unigenes (DEGs) FDR <0.05 and the |log2 ratio | ≥ 1 as the threshold. Based on these criteria, 2114 DEGs were found in the comparison of ΔleuRS_0h vs. WT_0h, among which 716 were up-regulated and 1398 were down-regulated. Under saline stress, 2048 DEGs were identified in the comparison groups (ΔLeuRS_1h/WT_1h), with 822 enhanced and 1226 repressed. And 1578 DEGs were detected after comparison of ΔLeuRS_14d with WT_14d, including 700 up-regulated genes and 878 down-regulated genes (Figure 2b). Our results showed that LeuRS was of great significance in mediating gene expression in A. montevidensis in response to the change in salinity.
3.3. Analysis of DEGs
GO analysis showed that the upregulated DEGs of ΔLeuRS_0h vs. WT_0h were enriched in 10 GO terms (P <0.05), such as ATP binding, cellular nitrogen compound biosynthetic process, cellular biosynthetic process, and macromolecule biosynthetic process. However, the DEGs down-regulated in this group were assigned to 10 GO terms (P<0.05), such as ion transmembrane transporter activity, substrate-specific transmembrane transporter activity, arginine metabolic process, glutamine family amino acid metabolic process, cellular amino acid metabolic process (Figure S5). The KEGG annotation of these DEGs was significantly enriched (P<0.05) in ABC transporters (Figure 3).
Under salinity stress, the upregulated DEGs of ΔLeuRS_1h vs. WT_1h were significantly (P<0.05) associated with 10 GO terms, such as nucleic acid binding transcription factor activity, cellular nitrogen compound biosynthetic process, gene expression, and organic acid metabolic process. And the downregulated DEGs were significantly (P<0.05) enriched in 7 GO terms, such as transition metal ion binding, cellular amino acid metabolic process, arginine metabolic process, and glutamine family-amino acid metabolic process (Figure S5). Six significantly enriched KEGG pathways (P<0.05) of DEGs were glyoxylate and dicarboxylate metabolism, butanoate metabolism, degradation of aromatic compounds, alanine, aspartate and glutamate metabolism, linoleic acid metabolism, nitrogen metabolism, etc (Figure 3). On the contrary, the DEGs upregulated in the long-term treated groups (ΔLeuRS_14d vs. WT_14d) were related to 7 GO terms (P<0.05), such as endopeptidase activity, active transmembrane transporter activity, cofactor metabolic process, and glycolytic process. However, the downregulated DEGs were successfully assigned into 13 GO terms (P<0.05), which consisted of membrane-bounded organelle, mitochondrion, ion binding, substrate-specific transmembrane transporter activity, single-organism metabolic process, small molecule metabolic process, and oxidation-reduction process (Figure S5). These DEGs were significantly involved in 10 KEGG pathways (P<0.05), such as sugar metabolism, tryptophan metabolism, glyoxylate and dicarboxylate metabolism, arginine and proline metabolism, arginine biosynthesis and biosynthesis of unsaturated fatty acids (Figure 3).
3.4. DEGs significantly related to ABC transporters, nitrogen and carbon metabolism, and sexual reproduction
We further found that in the groups (ΔLeuRS_0h vs. WT_0h), the expression levels of 7 unigenes encoding ABC transporters were down-expressed by factors of 1.9 to 9.6, excluding two unigenes (putatively coding ABC transporter G family member 21, and brefeldin A efflux transporter), which were enhanced by factors of 5.2 to 9.2 (Figure 4a, Table S1). As expected, 16 genes (by factors of 1.0 to 13.2) in pairwise comparisons (ΔLeuRS_1h vs. WT_1h) were down-regulated in expression which encode nitrate reductase, nitrite reductase, aspartate aminotransferase, argininosuccinate synthase, etc., while 4 genes coding glutamine synthetase, aspartate carbamoyl transferase, and carbonic anhydrase were up-expressed in expression by factors of 1.4 to 7.3 (Figure 4b, Table S1). On the contrary, 18 genes of the comparisons (ΔLeuRS_14d vs. WT_14d) decreased their expression (by factors of 1.3 to 15.5), which were mainly associated with 3-(3-hydroxy-phenyl) propionate hydroxylase, ornithine carbamoyl transferase, argininosuccinate lyase, arginase, etc., while 7 genes encoding proteins such as dimethylglycine oxidase, ornithine decarboxylase, agmatine deiminase, and agmatinase were up-regulated by factors of 1.3 to 3.1 (Figure 4b, Table S1). Similarly, among DEGs involved in carbon metabolism (Figure 4c, Table S1), 25 genes were down-regulated (by factors of 1.1 to 10.6) and 12 genes were up-expressed (by factors of 1.3 to 7.8) in pairwise comparisons (ΔLeuRS_1h vs. WT_1h), while 10 (by factors of 1.8-9.6) and 20 genes (by factors of 1.5-9.8) were found to be down and up-regulated in the ΔLeuRS_14d vs. WT_14d groups, respectively. These significant DEGs were involved in the metabolism of sugars, glyoxylate and dicarboxylate, aromatic, and other carbon-containing substances, which were mainly composed of citrate synthase, alcohol dehydrogenase, MFS monosaccharide transporter, short chain dehydrogenase, glyceraldehyde 3-phosphate dehydrogenase, and hexokinase. We found that many known genes that encode sexual development activators such as VeA, wetA, velB, and sidC were depressed more than one-fold in the ΔLeuRS groups compared to WT (Table S1). However, ΔLeuRS mutants up-regulated asexual developmental genes (BrlA and Arp1/2) by factors of 1.9 to 7.8. Our findings demonstrated that LeuRS mediated many cellular processes (substance transport, nitrogen and carbon metabolism, and reproduction) of the fungus under salt stress.
3.5. Differentially accumulated metabolites regulated by LeuRS
Untargeted UHPLC-Q-TOF/MS analysis identified a total of 366 compounds, and PCA revealed significant differences in the metabolites responsible for separation between the ΔLeuRS and WT groups (Figure S6), indicating that the ΔLeuRS and WT samples had substantially different metabolite profiles. Under the non-stress condition, a total of 55 metabolites differed significantly (P<0.05, |log2 fold change |≥1) between the ΔLeuRS_0h vs. WT_0h groups. Metabolites were classified mainly into 5 categories and comprised 40 up-regulated and 15 down-regulated metabolites (Table S2). The most abundant types of metabolites were mainly associated with organic acids (40%), amino acid derivatives (18%), nucleotides and derivatives 18%), carbohydrates and derivatives (14.5%), and fatty acids (9%). Based on the classification of the KEGG pathway, differential metabolites were enriched in the top-15 pathways such as ABC transporters, tyrosine metabolism, D-amino acid metabolism, tryptophan metabolism, purine metabolism, aminobenzoate degradation, and alanine, aspartate and glutamate metabolism (Figure S7). The metabolites most significantly up- and down-regulated (P<0.05, |log2 fold change | ≥ 5) were D-alanyl-D-alanine, xanthohumol, (4E)-1,7-bis(4-hydroxyphenyl) hept-4-en-3-one, 4-nitrophenyl phosphate, salsolinol, adenine, allantoic acid, etc. (Figure 5a).
Under salt stress, a total of 115 metabolites between the ΔLeuRS_1h vs. WT_1h groups (P<0.05, |log2 fold change | ≥1) were identified: 41 and 74 metabolites were regulated up and down, respectively (Table S2). These significantly differentially accumulated metabolites (SDAMs) were assigned mainly into 5 types, including organic aids (30.4%), amino acid derivatives (27%), fatty acids (14.8%), nucleotides and derivatives (14.8%) and carbohydrates (13%). SDAMs were enriched mainly in 15 KEGG pathways, consisting of amino acid biosynthesis, ABC transporters, D-amino acid metabolism, protein digestion and absorption, aminoacyl-tRNA biosynthesis, 2-oxocarboxylic acid metabolism, phenylalanine metabolism, tryptophan metabolism, etc. (Figure S7). Among SDAMs, ten metabolites (N-acetylglutamine, trans-3-coumarate, 3-methylxanthine, ribothymidine, 3'-AMP, dibutyl phthalate, 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-3,4-dihydro-2H-1-benzopyran-4-one, methionine sulfoxide, D-alanyl-D-alanine and 4-hydroxyphenylpyruvate) increased by factors of 5.4 to 16.5 in the ΔLeuRS_1h samples, compared to those of the WT_1h groups (Table S2). However, 15 compounds (N-(2-furoyl) glycine, (4E)-1,7-bis (4-hydroxyphenyl) hept-4-en-3-one, quinolinic acid, NAD, gentisic acid, salsolinol, adrenosterone, 2,3-dihydroxybenzoic acid, 6β-hydroxytestosterone, tropic acid, allantoic acid, norvaline, 7-methylguanine, itaconic acid, and 4-nitrophenyl phosphate) decreased by factors of 5.2 to 12.1 in the ΔLeuRS_1h samples (Figure 5b).
The levels of 96 metabolites differed significantly between the ΔLeuRS_14d vs. WT_14d groups with the thresholds | log2 fold change | ≥1 and the value of P<0.05. The content of 44 SDEMs was upregulated, whereas that of 52 SDEMs was downregulated in ΔLeuRS_14d compared to WT_14d (Table S2). These SDEMs were assigned mainly to five types, including organic aids (38.5%), carbohydrate and derivatives (19.8%), fatty acids (14.6%), nucleotides and derivatives (13.5%), and amino acid derivatives (13.5%). The 15 most enriched KEGG pathways of SDAMs were involved in the biosynthesis of cofactors, purine metabolism, ABC transporters, phenylalanine metabolism, biosynthesis of amino acids, tyrosine metabolism, etc. (Figure S7). Among SDAMs, eight metabolites (norvaline, benzene-1,2,4-triol, citraconic acid, m-coumaric acid, indoleacetic acid, gluconic acid, kynurenic acid and 4-hydroxyphenylpyruvate) were strongly elevated by factors of 5.9 to 19.0 in the ΔLeuRS_14d samples compared to WT_14d samples, while nine (paeonol, dehydroascorbic acid, erythrono-1,4-lactone, succinic anhydride, ketoleucine, picolinic acid, D-ribose, paliperidone and hypoxanthine) decreased dramatically by factors of 5.3 to 13.7 in the ΔLeuRS_14d groups (Figure 5c). The differential metabolite profile regulated by LeuRS in A. montevidensis may contribute to improving the osmotic tolerance of the fungus.
4. Discussion
A. montevidensis is commonly used in the production of traditional fermented foods [4,7]. During fermentation processes, the transcriptional responses of A. montevidensis to osmotic conditions are necessary for the formation of flavor of foods. To reveal the functional roles of LeuRS (which was strikingly up-expressed in A. montevidensis under osmotic stress), we constructed a knockout mutant of LeuRS and found that the ΔLeuRS mutant of A. montevidensis retarded mycelial growth rate, decreased salt tolerance, and deactivated cleistothecium formation compared to the WT strain (Figure 1, Figure S2). Transcriptomic and metabolomic analysis revealed that LeuRS are widely related to ABC transporters, amino acid metabolism, carbohydrate metabolism, fatty acid metabolism, and sexual development of A. montevidensis responding to salinity stress.
Here, we detected that many DEGs between the ΔLeuRS_0h vs. WT_0h groups were down-regulated, which were significantly enriched in ATP-binding cassette (ABC) transporters (Table S1). For example, decreasing expression of genes encodes several subfamily B, D, and G members of ABC transporters. Extensive research showed that ABC transporters are evolutionarily conserved integral membrane proteins responsible for the allocation of a wide variety of substrates, including ions, sugars, amino acids, polypeptides, complex lipids, toxic metabolites, and even toxins [27,28]. In addition, previous reports indicated that the specific upregulation of some ABC transporters was involved in osmo-stress tolerance of cells [29,30]. Based on these findings, we conclude that LeuRS could play a vital role in the regulation of ABC transporter-mediated substance transport in the osmoadaptation of the halophilic fungus.
Specific regulation of amino acid metabolism is crucial for controlling cell growth and proliferation, osmotic adjustment of all living organisms [8,31,32]. As expected, 40 significant DEGs between the ΔLeuRS vs. WT groups under short- and long-term salt stress were associated with the metabolism pathways of many amino acids (Table S1). This showed that LeuRS is important for the regulation of amino acid metabolization of A. montevidensis in response to salinity stress. We further found that 14 DEGs were involved in the metabolism of alanine, aspartate, and glutamate, suggesting that LeuRS mediated the metabolism of amino acids (alanine, aspartate, and glutamate) for osmoadaptation. For example, ΔLeuRS mutants exposed to salt stress increased the expression of genes encoding glutamine synthetase and aspartate carbamoyl transferase, which were up-regulated in salinity-induced plants [33,34]. However, deletion mutations significantly inhibited the transcription of genes encoding enzymes such as aspartate aminotransferase, succinate-semialdehyde dehydrogenase, 1-pyrroline-5-carboxylate dehydrogenase and carbamoyl-phosphate synthase in the salt-induced and non-induced mycelia. Previous reports showed that up-expression of aspartate aminotransferase and 1-pyrroline-5-carboxylate dehydrogenase was important for the survival of plants in saline conditions [33,35]. The deletion of carbamoyl-phosphate synthase in Colletotrichum gloeosporioides led to a slow growth rate and extreme sensitivity to high osmotic stress [36]. Furthermore, we observed that ΔLeuRS mutants compared to WT strains under saline conditions increased the levels of some amino acids and their derivatives (eg, L-asparagine, L-glutamine, D-alanine, γ-glutamylalanine, N-acetylglutamine, and D-alanyl-D-alanine) (Table S2), which could play a crucial role in improving salt tolerance of fungi.
Previous studies indicated that activation of the arginine and proline metabolism pathway confers enhanced stress tolerance in different eukaryotes [8,37]. However, little is known about the regulatory mechanism of the pathway. Here, we found that 9 DEGs between the ΔLeuRS_14d vs. WT_14d groups were significantly associated with arginine and proline metabolism, especially in salt-induced samples (Figure 4, Table S1), indicating that LeuRS influences the proline and arginine metabolic pathway to enhance fungal adaptation to salinity. Under long-term salinity stress, ΔLeuRS mutants up-expressed genes encoding agmatinase, and ornithine decarboxylase, above controls. These genes participated in the biosynthesis of putrescine, which is a key metabolite that improves plant tolerance to salinity by scavenging free radicals [38]. And mutants increased the expression of D-amino acid oxidase, which could accelerate the process of proline transformation into 1-pyrroline-2-carboxylate in proline metabolism. A previous study showed that the enhancement of D-amino-acid oxidase activity was correlated with the heat tolerance of fungi [39]. However, ΔLeuRS mutants significantly decreased the transcription of two unigenes coding arginase (by a factor of more than 3 times) compared to WT strains in salt-treated and untreated controls. Up-regulation of arginase could alleviate damage to plants caused by salinity [40].
Furthermore, we found that ΔLeuRS mutants compared to WT groups under the 14-day salt induction significantly changed the expression patterns of 8 DEG involved in tryptophan metabolism (Figure 4, Table S1), suggesting that LeuRS could mediate tryptophan metabolism in fungi to counteract salt stress. This finding was partially supported by a study case in the plant, which activated the phenylalanine, tyrosine, and tryptophan pathway to improve osmoprotective ability [41]. We further observed that salt-induced and uninduced mycelia of the mutants were compared against the WT down-expressed kynureninase, amidase, L-amino-acid oxidase, and NADPH-cytochrome P450 reductase. However, wide-type A. montevidensis up-expressed L-amino-acid oxidase against high salinity stress [8], and the expression of NADPH-cytochrome P450 reductase in Bacillus subtilis was induced by acid stress [42]. Furthermore, deletion of LeuRS led to the differential accumulation of many metabolites (e.g., 4-hydroxyphenylpyruvate, kynurenic acid, L-phenylalanine, L-tyrosine, and quinolinic acid) from the tryptophan pathway in A. montevidensis under different salinity conditions (Figure 5, Table S2). 4-hydroxyphenylpyruvate was dramatically accumulated (by a factor of more than 16 times) in salt-induced ΔLeuRS strains, compared to controls, suggesting a potential protective role of this compound for mutants against salt stress. And application of L-tyrosine increased the tolerance to salinity of A. montevidensis [8].
Carbohydrate metabolism has been shown to play a key role in maintaining osmotic homeostasis and intracellular energy balance through carbon partitioning in microbes [43]. Here, we found that ΔLeuRS mutants upregulated many DEGs coding enzymes such as hexokinase, enolase, pyruvate decarboxylase, citrate synthase, and malate dehydrogenase in the glycolysis/glucoseogenesis and citrate cycle, especially for salt-induced cells (Table S1). Hexokinases control the first step of all major pathways of glucose utilization and therefore influence the extent and direction of glucose flux within the cell. Previous studies showed that the expression of hexokinases promoted tolerance to drought and salt stress in plants [44]. Similarly, increased gene transcriptional levels of enolase, citrate synthase, and malate dehydrogenase were also observed in halophytes exposed to hypersaline stress [45,46]. Furthermore, our study indicated that the ΔLeuRS mutants differentially expressed genes significantly involved in glyoxylate and dicarboxylate metabolism (which is related to carbohydrate metabolism). In agreement with our results, activation of this pathway was shown in sesames for tolerance to salinity [47]. Moreover, mutants under long-term salinity stress markedly increased the expression of many enzymes coding catalase (which catalyzes the dismutation of H2O2 into H2O and O2) and significantly decreased the expression of (S) -2-hydroxy acid oxidase (which transforms glycolate into H2O2 and glyoxylate), suggesting that LeuRS mediated reactive oxygen species scavenging for fungal osmoadaptation. And, the elimination of LeuRS led to the differential accumulation of many compatible sugars and organic acids in the salt-induced samples (Figure 5, Table S2), which was consistent with our previous study on A. montevidensis [8]. According to our findings, it can be inferred that LeuRS is of importance for the regulation of sugar metabolism in the defense of fungi against salt stress.
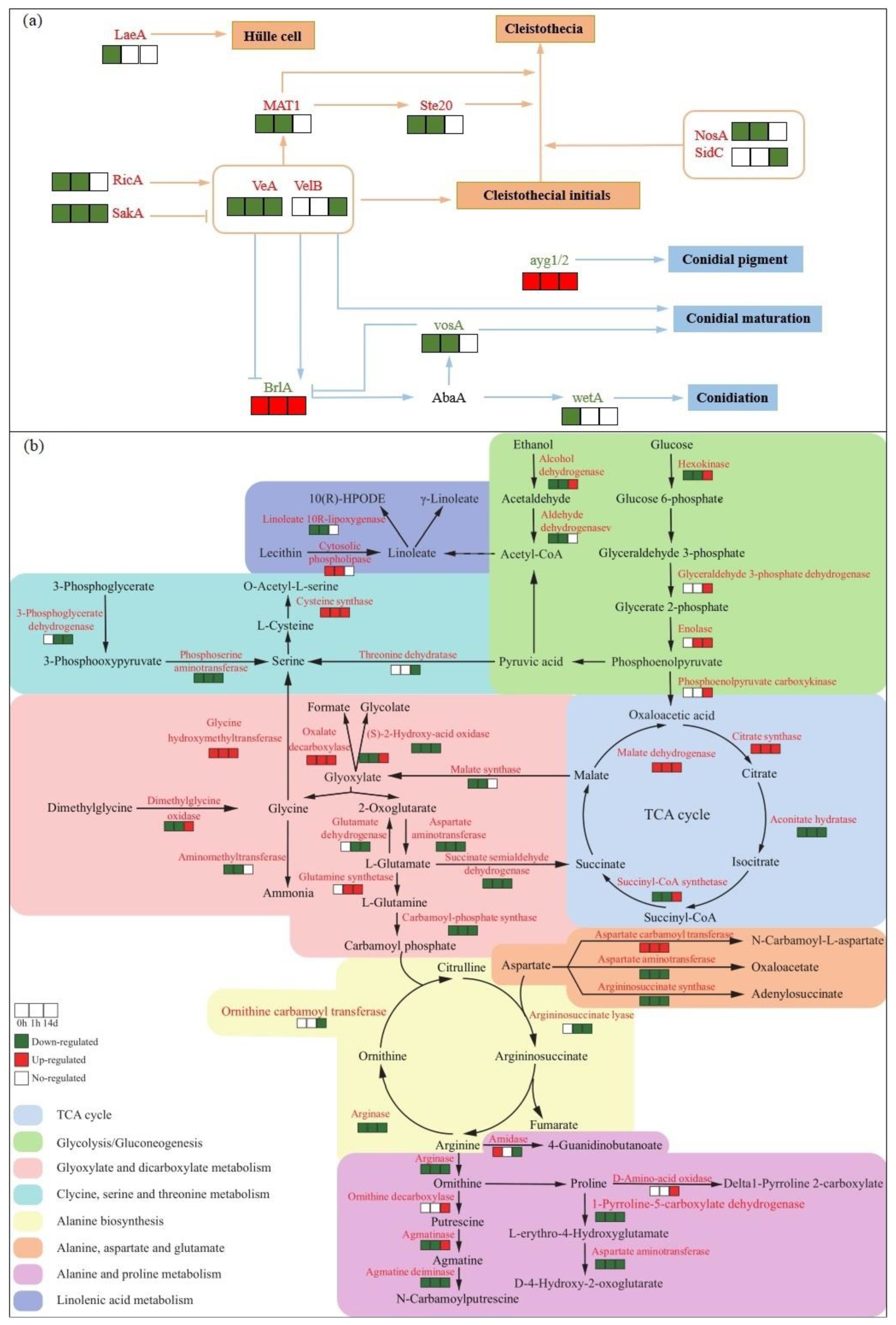
The development of sexual/asexual reproductive structures in Aspergillus species is regulated by complex intrinsic signals [12]. To date, multiple genes have been identified to play asexual and sexual reproduction roles in A. nidulans, A. fumigatus, A. flavus, and A. parasiticus [12,13]. Here, we found that LeuRS knockout dramatically varied the expression levels of many reproduction-related genes in aspergilli (Figure 6a, Table S1). Previous studies showed that the core ‘velvet’ proteins (VeA, VelB, and LaeA), together with the associated protein VosA, play a pivotal role in light/dark regulation of sexual development [48,49,50]. The removal of veA increased the sensitivities to high osmolarity, oxidation stress, and triggered the production of fungal conidiation [51]. The expression of MAT2, ste20, ricA, and sakA was involved in the mating processes and signal transduction in the aspergilli, and the elimination of individual genes (MAT2, ste20, ricA, nosA, and sidC) resulted in a severe loss of self-fertility [12,52]. Up-expression of BrlA, a central regulator, activates the conidial developmental pathway of A. nidulans [13]. Taken together, our finding showed that LeuRS could act as a prominent modulator for reproduction process in aspergilli.
5. Conclusion
We constructed the ΔLeuRS mutant and characterized the cellular function of LeuRS in A. montevidensis under osmolarity conditions. Our results demonstrated that deletion of LeuRS influenced the expression of multiple genes of the halophilic fungus. We observed that the mutants varied the expression levels of many genes governing sexual and asexual reproduction in fungi (Figure 6a). We found that DEGs of ΔLeuRS mutants vs. WT strains significantly enriched in KEGG pathways such as ABC transporters, alanine, aspartate and glutamate metabolism, arginine and proline metabolism, glycolysis/glucoseogenesis, and TCA cycle (Figure 6b). Metabolomic analyses showed that a range of metabolites were differentially accumulated in this pairwise comparison, such as amino acids, sugars, and organic acids. The findings will help reveal fungal osmoadaptation strategies and to elucidate the flavor formation mechanisms of foods fermented with A. montevidensis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Xiaowei Ding: Data curation, Writing–review & editing. Wanting Liu: Methodology, Writing–Original draft. Kaihui Liu: Conceptualization, Writing–Original draft, Supervision. Xiang Gao: Data curation, software, Yue Liu: Methodology. All authors read and approved the manuscript.
Funding
This work was co-financed by the National Natural Science Foundation of China (No. 31970120), the Key Research and Development Project of Shaanxi (No. 2023-YBSF-325).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
References
- Park, H.S.; Jun, S.C.; Han, K.H.; Hong, S.B.; Yu, J.H. Diversity, application, and synthetic biology of industrially important Aspergillus fungi. Adv. Appl. Microbiol. 2017, 100, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Nakabayashi, R.; Ogawa, C.; Kimura, Y.; Yokota, S.; Doi, M. Characterization of surface Aspergillus community involved in traditional fermentation and ripening of katsuobushi. Int. J. Food Microbiol. 2020, 327, 108654. [Google Scholar] [CrossRef] [PubMed]
- Copetti, M. V.; Iamanaka, B.T.; Frisvad, J.C.; Pereira, J.L.; Taniwaki, M.H. Mycobiota of cocoa: from farm to chocolate. Food Microbiol. 2011, 28, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.A.; Kim, E.; Yang, S.M.; Lee, S.; Yoon, S.R.; Jang, K.S.; Kim, H.Y. High-throughput sequencing of the microbial community associated with the physicochemical properties of meju (dried fermented soybean) and doenjang (traditional Korean fermented soybean paste). LWT 2021, 146, 111473. [Google Scholar] [CrossRef]
- Li, Z.; Mao, Y.; Teng, J.; Xia, N.; Huang, L.; Wei, B.; Chen, Q. Evaluation of mycoflora and citrinin occurrence in Chinese Liupao Tea. J. Agric. Food. Chem. 2020, 68, 12116–12123. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wei, B.Y.; Teng, J.W.; Huang, L.; Xia, N. Analyses of fungal community by Illumina MiSeq platforms and characterization of Eurotium species on Liupao tea, a distinctive post-fermented tea from China. Food Res. Int. 2017, 99, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Wang, Y.; Wen, J.; Liu, P.; Liu, Z.; Li, Z. Fungal community associated with fermentation and storage of Fuzhuan brick-tea. Int. J. Food Microbiol. 2011, 146, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Liu, K.; Lu, Y.; Gong, G. Morphological, transcriptional, and metabolic analyses of osmotic-adapted mechanisms of the halophilic Aspergillus montevidensis ZYD4 under hypersaline conditions. Appl. Microbiol. Biotechnol. 2019, 103, 3829–3846. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.H.; Ding, X.W.; Narsing Rao, M.P.; Zhang, B.; Zhang, Y.G.; Liu, F.H.; Liu, B.B.; Xiao, M.; Li, W.J. Morphological and transcriptomic analysis reveals the osmoadaptive response of endophytic fungus Aspergillus montevidensis ZYD4 to high salt stress. Front. Microbiol. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Tukalo, M.; Yaremchuk, A.; Fukunaga, R.; Yokoyama, S.; Cusack, S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat. Struct. Mol. Biol. 2005, 12, 923–930. [Google Scholar] [CrossRef]
- Krijgsheld, P.; Bleichrodt, R.; van Veluw, G.J.; Wang, F.; Müller, W.H.; Dijksterhuis, J.; Wösten, H.A.B. Development in aspergillus. Stud. Mycol. 2013, 74, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.S.; O’Gorman, C.M. Sexual development and cryptic sexuality in fungi: insights from Aspergillus species. FEMS Microbiol. Rev. 2012, 36, 165–192. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-López, M.; Chen, W.; Eagle, C.E.; Gutiérrez, G.; Jia, W.L.; Swilaiman, S.S.; Huang, Z.; Park, H.S.; Yu, J.H.; Cánovas, D.; Dyer, P.S. Evolution of asexual and sexual reproduction in the aspergilli. Stud. Mycol. 2018, 91, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mashkoor, M.; Grove, A. Yeast Crf1p: An activator in need is an activator indeed. Comput. Struct. Biotechnol. J. 2021, 20, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, J.H.; Yoon, I.; Lee, C.; Sichani, M.F.; Kang, J.S.; Kang, J.; Guo, M.; Lee, K.Y.; Han, G.; Kim, S.; Han, J.M. Coordination of the leucine-sensing Rag GTPase cycle by leucyl-tRNA synthetase in the mTORC1 signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5279–E5288. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.Q.; Fang, Z.P.; Ye, Q.; Chi, C.W.; Wang, E.D. Self-protective responses to norvaline-induced stress in a leucyl-tRNA synthetase editing-deficient yeast strain. Nucleic Acids Res. 2017, 45, 7367–7381. [Google Scholar] [CrossRef] [PubMed]
- Yoon, I.; Nam, M.; Kim, H.K.; Moon, H.S.; Kim, Sungmin, Jang, J.; Song, J.A.; Jeong, S.J.; Kim, S.B.; Cho, S., Kim, Y.H.; Lee, J.; Yang, W.S.; Yoo, H.C.; Kim, K., Kim, M.S.; Yang, A.; Cho, K.; Park, H.S.; Hwang, G.S.; Hwang, K.Y.; Han, J.M.; Kim, J.H.; Kim, S. Glucose-dependent control of leucine metabolism by leucyl-tRNA synthetase 1. Science 2020, 367, 205–210. [CrossRef]
- Yang, G.; Zhao, S.; Gong, J.; Huang, M.; Yu, W.; Zhang, K.; Hu, D. Dissipation and the effects of thidiazuron on antioxidant enzyme activity and malondialdehyde content in strawberry. J. Sci. Food Agric. 2019, 99, 4331–4337. [Google Scholar] [CrossRef]
- Liu, K.; Ding, X.; Wang, G.; Liu, W. Complete genome sequencing of halophilic endophytic Aspergillus montevidensis, strain ZYD4, isolated from alfalfa stems grown in saline-alkaline soils. Mol. Plant Microbe Interact. 2022, 35, 867–869. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.R.; Zhang, C.C.; Fan, H.F.; Guo, Z.Y.; Yang, H.Y.; Chen, M.; Han, J.J.; Xu, J.; Zhang, K.Q.; Liang, L.M. An efficient gene disruption system for the nematophagous fungus Purpureocillium lavendulum. Fungal Biol. 2019, 123, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; Chen, Z.; Mauceli, E.; Hacohen, N.; Gnirke, A.; Rhind, N.; Di Palma, F.; Birren, B.W.; Nusbaum, C.; Lindblad-Toh, K.; Friedman, N.; Regev, A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182-5. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Gostinčar, C.; Gunde-Cimerman, N. Overview of oxidative stress response genes in selected halophilic fungi. Genes (Basel) 2018, 9, 143. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Tampé, R. Structural and mechanistic principles of ABC transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef]
- Duarte-Delgado, D.; Dadshani, S.; Schoof, H.; Oyiga, B.C.; Schneider, M.; Mathew, B.; Léon, J.; Ballvora, A. Transcriptome profiling at osmotic and ionic phases of salt stress response in bread wheat uncovers trait-specific candidate genes. BMC Plant Biol. 2020, 20, 428. [Google Scholar] [CrossRef]
- Lee, D.W.; Hong, C.P.; Thak, E.J.; Park, S.G.; Lee, C.H.; Lim, J.Y.; Seo, J.A.; Kang, H.A. Integrated genomic and transcriptomic analysis reveals unique mechanisms for high osmotolerance and halotolerance in Hyphopichia yeast. Environ. Microbiol. 2021, 23, 3499–3522. [Google Scholar] [CrossRef]
- Bianchi, F.; van’t Klooster, J.S.; Ruiz, S.J.; Poolman, B. Regulation of amino acid transport in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2019, 83, e00024-19. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Turkyilmaz Unal, B.; García-Caparrós, P.; Khursheed, A.; Gul, A.; Hasanuzzaman, M. Osmoregulation and its actions during the drought stress in plants. Physiol. Plant 2021, 172, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, D.; Barvkar, V.T.; Nadaf, A.; Mundhe, S.; Kadoo, N.Y. Integrative omics analysis in Pandanus odorifer (Forssk.) Kuntze reveals the role of asparagine synthetase in salinity tolerance. Sci. Rep. 2019, 9, 932. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Yuan, Y.Z.; Ou, J.Q.; Lin, Q.H.; Zhang, C.F. Glutamine synthetase and glutamate dehydrogenase contribute differentially to proline accumulation in leaves of wheat (Triticum aestivum) seedlings exposed to different salinity. J. Plant Physiol. 2007, 164, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Yoshiba, Y.; Sanada, Y.; Yamaguchi-Shinozaki, K.; Wada, K.; Shinozaki, K. Characterization of the gene for delta1-pyrroline-5-carboxylate synthetase and correlation between the expression of the gene and salt tolerance in Oryza sativa L. Plant Mol. Biol. 1997, 33, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, A.; Tariq, M.; Ahmed, M.; Zhou, Z.; Ali, I.; Mahmood, R.T. Carbamoyl phosphate synthase subunit CgCPS1 is necessary for virulence and to regulate stress tolerance in Colletotrichum gloeosporioides. Plant Pathol. J. 2021, 37, 414–414. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Shi, L.; Yang, R.; Guo, H.; Zhang, S.; Geng, G. Transcriptome analysis of Auricularia fibrillifera fruit-body responses to drought stress and rehydration. BMC Genomics 2022, 23, 58. [Google Scholar] [CrossRef]
- González-Hernández, A.I.; Scalschi, L.; Vicedo, B.; Marcos-Barbero, E.L.; Morcuende, R.; Camañes, G. Putrescine: A key metabolite involved in plant development, tolerance and resistance responses to stress. Int. J. Mol. Sci. 2022, 23, 2971. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Wang, Q.; Lu, D.; Zhang, H.; Wang, J.; Fu, R. Transcriptional profiling provides new insights into the role of nitric oxide in enhancing Ganoderma oregonense resistance to heat stress. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- da Silva, C.J.; Batista Fontes, E.P.; Modolo, L.V. Salinity-induced accumulation of endogenous H2S and NO is associated with modulation of the antioxidant and redox defense systems in Nicotiana tabacum L. cv. Havana. Plant Sci. 2017, 256, 148–159. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, H.; Jin, J.; Ma, X.; Li, K. Physiological and transcriptome analysis on diploid and polyploid Populus ussuriensis Kom. under salt stress. Int. J. Mol. Sci. 2022, 23, 7529. [Google Scholar] [CrossRef] [PubMed]
- Thackray, P.D.; Moir, A. SigM, an extracytoplasmic function sigma factor of Bacillus subtilis, is activated in response to cell wall antibiotics, ethanol, heat, acid, and superoxide stress. J. Bacteriol. 185, 3491–3498. [CrossRef]
- Mattila, J.; Hietakangas, V. Regulation of carbohydrate energy metabolism in Drosophila melanogaster. Genetics 2017, 207, 1231–1253. [Google Scholar]
- Pérez-Díaz, J.; Batista-Silva, W.; Almada, R.; Medeiros, D.B.; Arrivault, S.; Correa, F.; Bastías, A.; Rojas, P.; Beltrán, M.F.; Pozo, M.F.; Araújo, W.L.; Sagredo, B. Prunus hexokinase 3 genes alter primary C-metabolism and promote drought and salt stress tolerance in Arabidopsis transgenic plants. Sci. Rep. 2021, 11, 7098. [Google Scholar] [CrossRef]
- Panda, A.; Rangani, J.; Parida, A.K. Comprehensive proteomic analysis revealing multifaceted regulatory network of the xero-halophyte Haloxylon salicornicum involved in salt tolerance. J. Biotechnol. 2020, 324, 143–161. [Google Scholar] [CrossRef]
- Piro, A.; Marín-Guirao, L.; Serra, I.A.; Spadafora, A.; Sandoval-Gil, J.M.; Bernardeau-Esteller, J.; Fernandez, J.M.R.; Mazzuca, S. The modulation of leaf metabolism plays a role in salt tolerance of Cymodocea nodosa exposed to hypersaline stress in mesocosms. Front. Plant Sci. 2015, 6, 464. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhou, R.; Wang, X.; Dossa, K.; Wang, L.; Zhang, Y.; Yu, J.; Gong, H.; Zhang, X.; You, J. Transcriptome and metabolome analyses of two contrasting sesame genotypes reveal the crucial biological pathways involved in rapid adaptive response to salt stress. BMC Plant Biol. 2019, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Ö.; Krappmann, S.; Ni, M.; Jin, W.B.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.; Kwon, N.J.; Keller, N.P.; Yu, J.H.; Braus, G.H. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Han, K.Y.; Kim, K.J.; Han, D.M.; Jahng, K.Y.; Chae, K.S. The veA gene activates sexual development in Aspergillus nidulans. Fungal Genetics and Biology 2002, 37, 72–80. [Google Scholar] [CrossRef]
- Sarikaya Bayram, O.; Bayram, O.; Valerius, O.; Park, H.S.; Irniger, S.; Gerke, J.; Ni, M.; Han, K.H.; Yu, J.H.; Braus, G.H. LaeA control of velvet family regulatory proteins for light-dependent development and fungal cell-type specificity. PLoS Genet. 2010, 6, 1–17. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, H.; Wang, Y.; Ge, Y.; Ren, X.; Ren, C.; Wang, Y.; Ren, X.; Liu, Y.; Liu, Z. The role of the veA gene in adjusting developmental balance and environmental stress response in Aspergillus cristatus. Fungal Biol. 2018, 122, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Kwon, N.J.; Park, H.S.; Jung, S.; Kim, S.C.; Yu, J.H. The putative guanine nucleotide exchange factor RicA mediates upstream signaling for growth and development in Aspergillus. Eukaryot. Cell 2012, 11, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Morphological characteristics of ΔLeuRS mutant and wide-type A. montevidensis (WT) strain grown on YPD with the 1.5 M NaCl for 14 days 28 °C. Colony morphology of mutant (a) and WT strain (d), in situ observation of microscopic morphological characteristics of mutant (b) and WT (e), the conidial head stained with crystal violet of mutant (c) and cleistothecia of WT (f).
Figure 1.
Morphological characteristics of ΔLeuRS mutant and wide-type A. montevidensis (WT) strain grown on YPD with the 1.5 M NaCl for 14 days 28 °C. Colony morphology of mutant (a) and WT strain (d), in situ observation of microscopic morphological characteristics of mutant (b) and WT (e), the conidial head stained with crystal violet of mutant (c) and cleistothecia of WT (f).
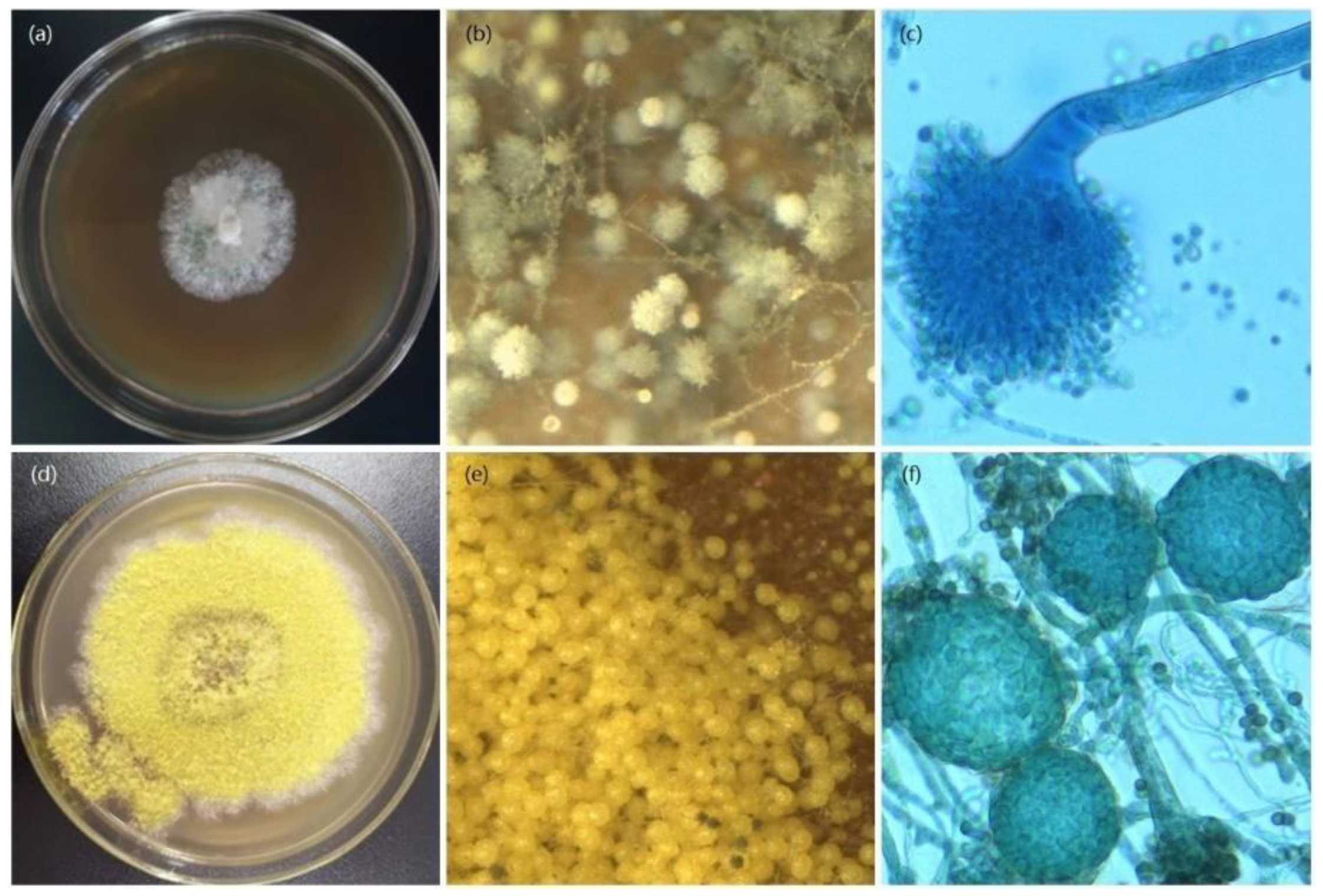
Figure 2.
Analysis of DEGs of ΔleuRS mutants compared with WT strains.
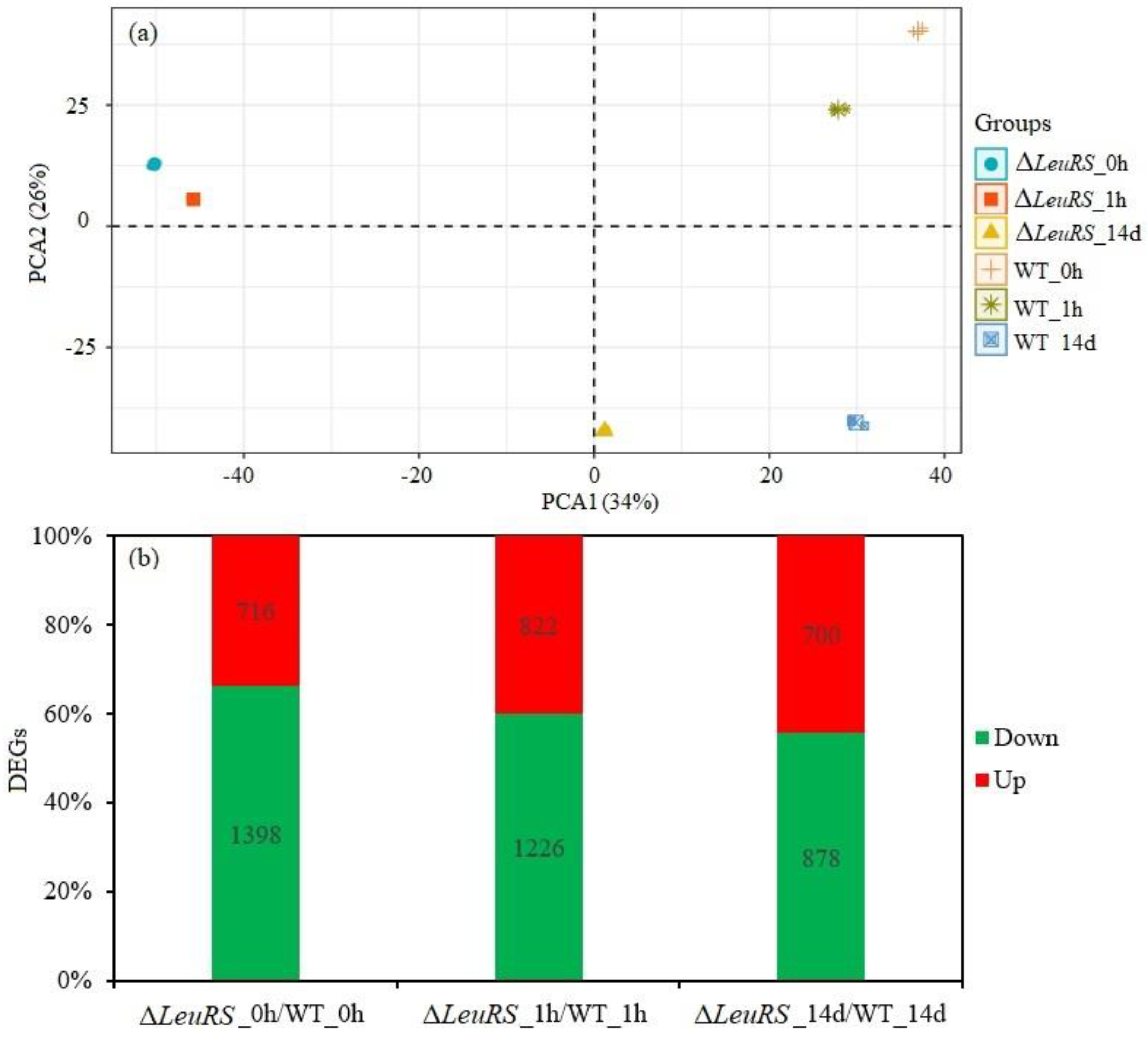
Figure 3.
KEGG enrichment analysis of DEGs involved in ABC transporters (a), metabolization of amino acids (b, c), glyoxylate and dicarboxylate, aromatic compounds, butanoate, sugars, organic acids, and fatty acids (d, e).
Figure 3.
KEGG enrichment analysis of DEGs involved in ABC transporters (a), metabolization of amino acids (b, c), glyoxylate and dicarboxylate, aromatic compounds, butanoate, sugars, organic acids, and fatty acids (d, e).
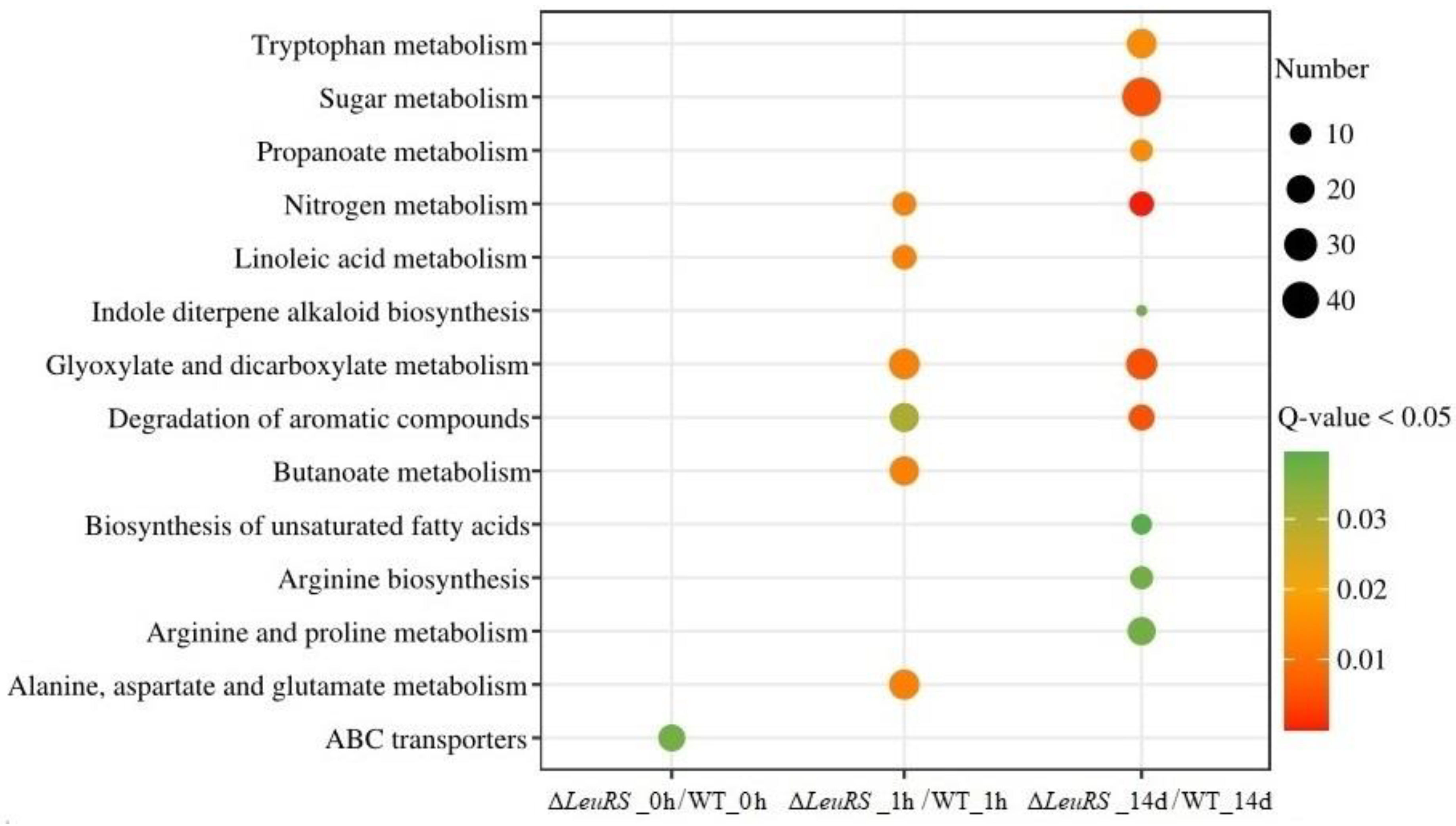
Figure 4.
Heat map analysis of some DEGs in compared groups of ΔleuRS mutants and WT strains treated with 1.5 M NaCl for 0h, 1h, and 14 days, respectively.
Figure 4.
Heat map analysis of some DEGs in compared groups of ΔleuRS mutants and WT strains treated with 1.5 M NaCl for 0h, 1h, and 14 days, respectively.
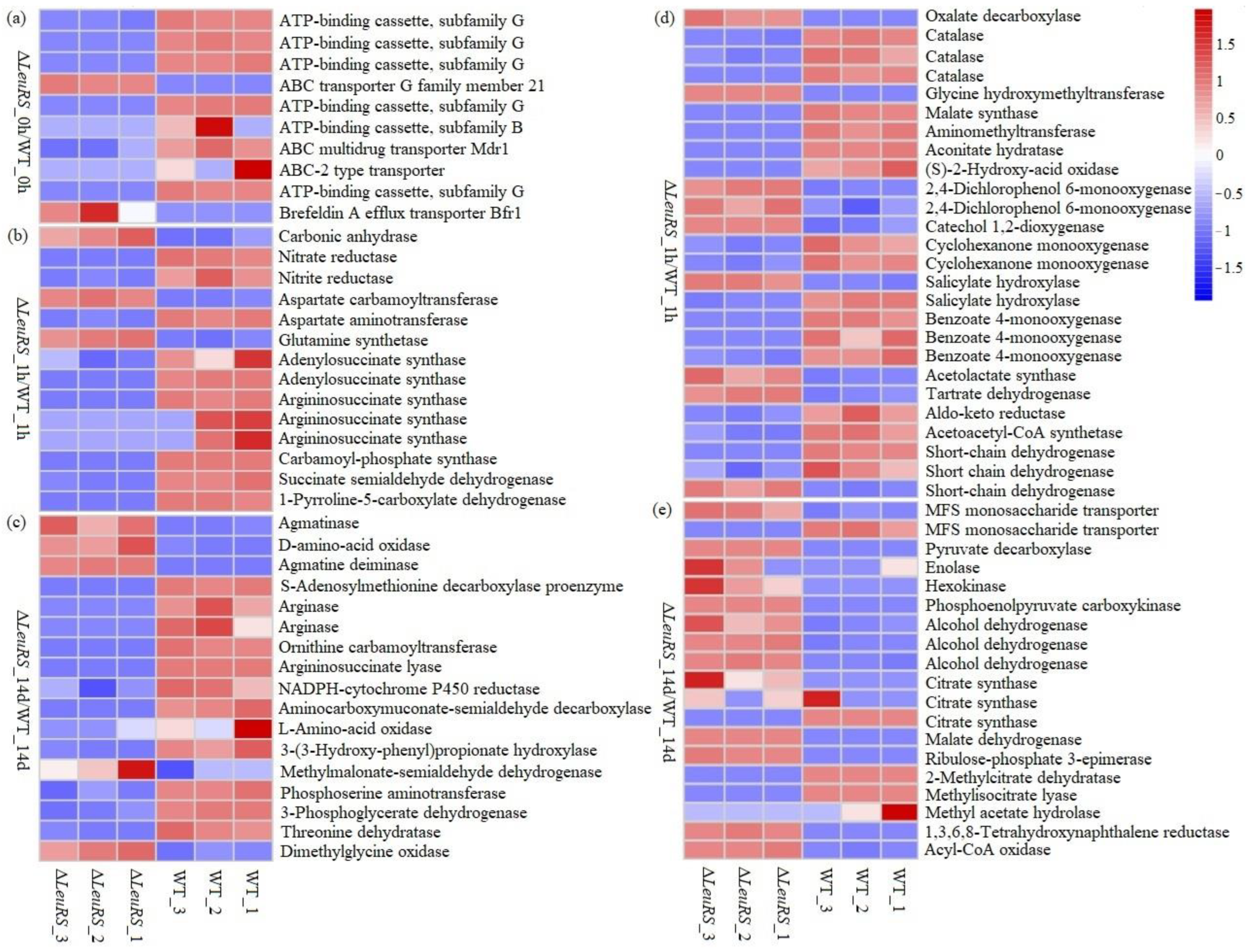
Figure 5.
Barplot analysis of discriminatory metabolites (P<0.05, log2 fold change >5.0) in compared groups of ΔleuRS mutants and WT strains treated with 1.5 M NaCl for 0h (a), 1h (b), and 14 days (c), respectively.
Figure 5.
Barplot analysis of discriminatory metabolites (P<0.05, log2 fold change >5.0) in compared groups of ΔleuRS mutants and WT strains treated with 1.5 M NaCl for 0h (a), 1h (b), and 14 days (c), respectively.
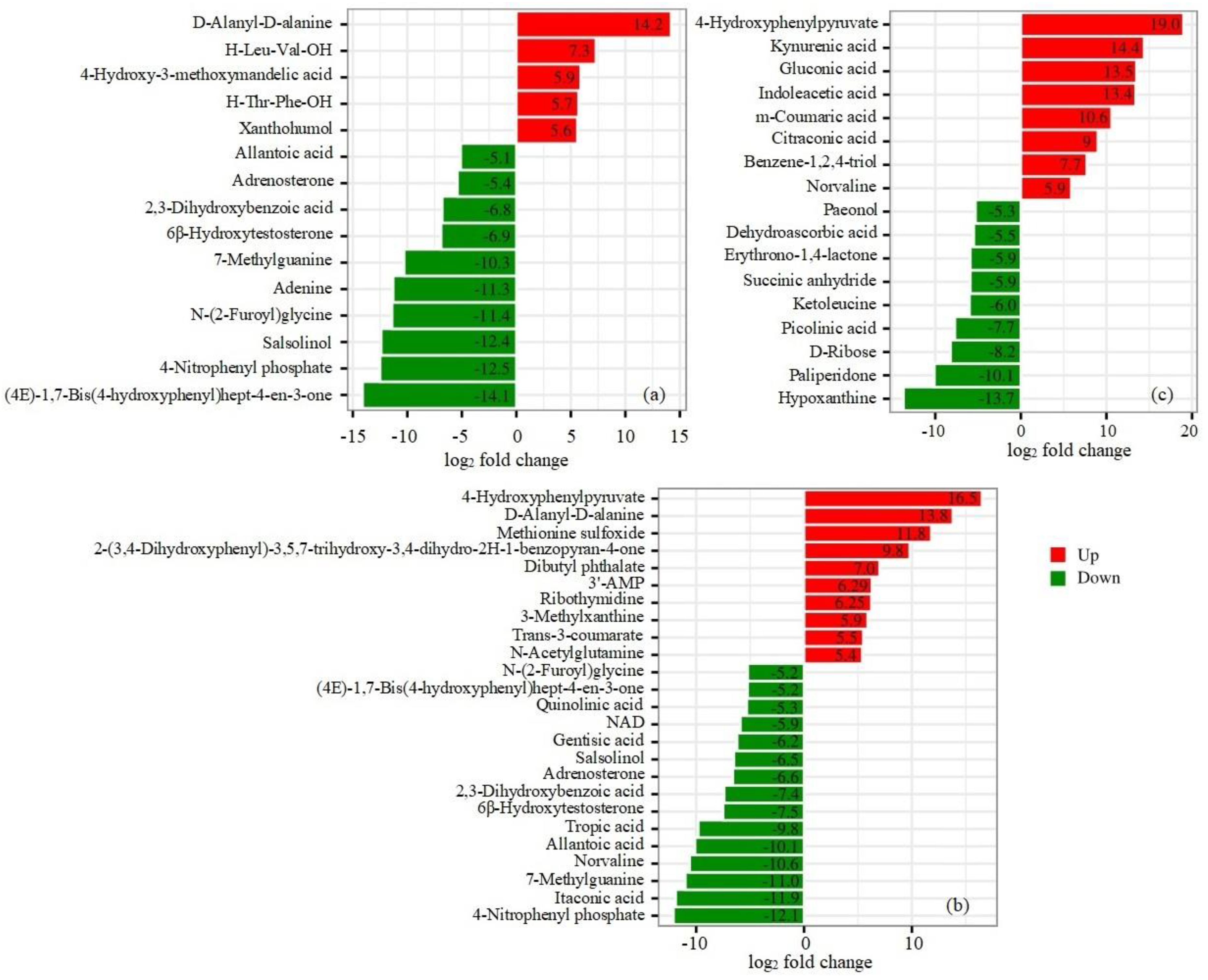
Figure 6.
Conceptual profile showing enriched pathways of DEGs and SDAMs between ΔleuRS mutants and WT strains. DEGs involved in sexual and asexual processes (a). DEGs and SDAMs were related to the metabolic pathways of amino acids, sugars, organic acids, and fatty acids (b). Genes marked with red or green color showed that these were respectively up- and down-regulated at different time points. .
Figure 6.
Conceptual profile showing enriched pathways of DEGs and SDAMs between ΔleuRS mutants and WT strains. DEGs involved in sexual and asexual processes (a). DEGs and SDAMs were related to the metabolic pathways of amino acids, sugars, organic acids, and fatty acids (b). Genes marked with red or green color showed that these were respectively up- and down-regulated at different time points. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



