
Submitted:
30 October 2023
Posted:
30 October 2023
You are already at the latest version
Abstract
Background: Although autophagy is a pro-survival process of tumor cells, in particular conditions and when differently regulated by specific signals it can stimulate cell death. We previously demonstrated that the selective stimulation of the M2 muscarinic receptor subtype (M2 mAChR) negatively controls cell proliferation and survival and causes oxidative stress and cytotoxic and genotoxic effects in both GBM cell lines and GBM stem cells (GSCs). In this work, we have evaluated whether autophagy was induced as a downstream mechanism of the observed cytotoxic processes induced by M2 mAChR activation by the orthosteric agonist APE or the dualsteric agonist N-8-Iper. Methods: To assess the activation of autophagy, we analyzed the expression of LC3B by Western blot analysis and in LC3B-EGFP transfected cell lines. Apoptosis was assessed by Caspases 3 and 9 protein expression. Results: Our data indicate that activation of M2 mAChR by N-8-Iper promotes autophagy in both U251 and GB7 cells lines as suggested by the LC3B-II expression level and analysis of the transfected cells by fluorescence microscopy. Autophagy induction by M2 mAChRs is regulated by the decreased activity of the PI3K/AKT/mTORC1 pathway and upregulated by the pAMPK expression. Downstream autophagy activation, the increase of apoptosis was also observed in both cell lines after treatment with the two M2 agonists. Conclusions: N-8-Iper treatment causes autophagy via pAMPK upregulation, followed by apoptosis in both investigated cell lines. In contrast, the absence of autophagy in APE-treated GSC cells seems to indicate that cell death could be triggered by mechanisms alternative to those observed for N-8-Iper.
Keywords:
Glioblastoma
; cancer stem cells
; M2 muscarinic receptor
; orthosteric and dualsteric muscarinic agonism
; autophagy
; apoptosis
; mTORC1
1. Introduction
Autophagy is an evolutionarily conserved catabolic process in all eukaryotes, from yeast to humans. In normal conditions, the autophagy is necessary to maintain cellular homeostasis through the degradation of dysfunctional organelles, protein aggregates or misfolded proteins, that are delivered to the lysosomes for recycling [1]. One of the classes of autophagic pathway consists of macro-autophagy [2], involving the sequestration of substrates within double-membrane cytosolic vesicles called “autophagosomes”. These organelles fuse with lysosomes, generating a single membrane organelle known as “autolysosome”, which represents the degradative compartment of the entire process [3,4]. Two main factors are involved in the regulation of the autophagic process, i.e., the TOR complex-1 (TORC1) and the AMP-activated protein Kinase (AMPK), which play opposite roles. Indeed, TORC1 negatively regulates autophagy, both in S. cerevisiae and in mammals [5]. Conversely, AMPK inhibits the TORC1 pathway through the phosphorylation of both its inhibitor protein TSC2 (tuberin) [6] and Raptor, one further TORC1 component [7]. In addition, AMPK activates the Unc-5-like kinase 1 (ULK1), the serine/threonine protein kinase that triggers autophagy [8].
Today, the role of autophagy in tumor remains a controversial issue. While numerous studies have shown that autophagy promote tumor suppression in the early stage of tumorigenesis and autophagy-associated death of cancer cells, other reports have highlighted the role of autophagy as a pro-tumor survival process under conditions of environmental stress [9,10].
Glioblastoma multiforme (GBM), the most lethal human brain tumor, has lower levels of protein markers of autophagy than low-grade astrocytic tumors [11,12]; in particular, ULK1 and ULK2 levels are significantly lower in GBM patients than in healthy controls [13]. An analysis conducted according to the Karnofsky Performance Scale index, which allows to classify the patients based on their functional impairment, reported that a high expression of Microtubule-associated protein 1A/1B-light chain 3 (LC3), a hallmark of autophagy, was correlated with an improved patient prognosis [14].
In addition, hyper-phosphorylation of AKT and mTOR has been detected in patient with grade-III and grade-IV gliomas. These factors, when active, are associated with downregulation of autophagic process and increased cell proliferation and stemness of glioma stem cells [15,16]. Considering this evidence on the role of autophagy as a tumor suppressor, drugs with synergistic action with the chemotherapy agents already in use, notably temozolomide, are being developed [10].
Since many years, our group has been studying the effects of M2 muscarinic acetylcholine receptor (M2 mAChR) activation in GBM. mAChRs are G protein coupled-receptors (GPCRs) involved in the regulation of several fundamental processes in both central and peripheral nervous systems [17]. Moreover, the expression of mAChRs in different tumors as well as their strategic role in the modulation of cancer cell proliferation, survival and migration have been demonstrated [18,19,20,21,22]. In this framework, we have demonstrated that the M2 mAChR activation by the orthosteric M2 agonist Arecaidine Propargyl Ester (APE) has negative effects on cell proliferation and survival, both in GBM cell lines and in GBM cancer stem cells (GSCs) [23,24,25,26]. Recently, we have also shown that treatment with an M2 agonist results in an increase of aberrant mitosis in U251 and U87 cell lines, caused by an altered mitotic spindle formation by reducing the ability of microtubules to bind chromosomes. GBM cell lines are unable to proliferate after APE treatment, due to the triggering of catastrophic mitotic events and apoptosis [27].
To improve the therapeutic potential of mAChR ligands in different pathological conditions such as Alzheimer's disease [28,29], schizophrenia [17,30] and cancer [18,21,23], novel hybrid molecules, named "dualsteric or bitopic agonists”, were designed, synthesized and tested. This kind of ligands, which have been reported also for various GPCR receptor families, recognize simultaneously the acetylcholine (orthosteric) binding site and an additional, allosteric site. Such a cooperative interaction may increase the degree of selectivity among the subtypes and also promote signaling bias, since binding to the allosteric site may favor a functionally relevant receptor structure that controls a specific signaling pathway [31,32,33].
Iper-8-naphthalimide (N-8-Iper) is a synthetic dualsteric agonist for the M2 mAChR previously characterized by our research group [33,34,35]. We demonstrated that N-8-Iper reduced GSCs proliferation in a comparable manner to APE in two GSC lines, GB7 cells and G166 cells. Quite interestingly, N-8-Iper is active at doses (3 μM for GB7 cells and 25 μM for G166 cells) that are significantly lower than that of APE (100 μM) [34,35].
Starting from our previous results, here we investigated the downstream molecular targets linked to the cytotoxic effects observed after M2 mAChR activation with APE and N-8-Iper [23,26,35,36]. Moreover, we analyzed the signal transduction pathways involved in these processes, that could be affected by the different recognition mode of the two investigated muscarinic agonists.
We detected an upregulation of the autophagy process both in the U251 cell line and in GB7 cells after N-8-Iper treatment at high and low doses, while APE triggered autophagy only in U251 cells. Moreover, we studied the downregulation of the phosphoinositide 3-kinase (PI3K)/AKT/TORC1 pathway and the upregulation of AMPKα expression, that have been identified as two of the main pathways involved in the modulation of the autophagy flux. Finally, the increased autophagy after M2 stimulation by N-8-Iper positively improved the apoptotic process, as shown by the increase in the cleaved form of Caspases 9 and 3.
2. Materials and Methods
2.1. Cell Cultures
The GSCs GB7 cells were obtained from human GBM biopsy [37,38,39]. The cells were cultured on laminin-coated dishes (1 μg/mL; Sigma-Aldrich, St. Louis, MO, USA) and maintained in serum free medium consisting of DMEM/F12 (Sigma-Aldrich, St. Louis, MO, USA) and Neurobasal medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) (1:1; v:v) supplemented with 1% streptomycin, 50 IU/mL penicillin (Sigma-Aldrich, St. Louis, MO, USA), 1% glutamine (Sigma-Aldrich, St. Louis, MO, USA), 1% N2 supplement (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 2% B27 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 20 ng/mL EGF (Recombinant Human Epidermal growth factor, Peprotec, London, UK), and 20 ng/mL FGF (Recombinant Human FGF-basic, ABM, Richmond, Canada). The GB7 cell culture was maintained at 37 °C in an atmosphere of 5% CO2/95% air. Human glioblastoma U251MG cell line was cultured in DMEM (Sigma-Aldrich, St. Louis, MO, USA) containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 50 μg/mL streptomycin, 50 IU/mL penicillin, 2 mM glutamine (Sigma-Aldrich, St. Louis, MO, USA), 1% non-essential amino acids (Sigma-Aldrich, St. Louis, MO, USA), and maintained at 37 °C in a 10% CO2 atmosphere.
2.2. Pharmacological Treatments
Arecaidine Propargyl Ester hydrochloride (APE, Sigma-Aldrich, Milan, Italy) is a synthetic compound obtained from modification of arecaidine, a natural alkaloid derived from areca nut. M2 agonist APE was used to selectively stimulate the M2 mAChR subtype. The ability of this agonist to bind the M2 mAChR subtype was previously demonstrated in GBM stable cell lines (U87 and U251 cell lines) and in GSCs (GB7 and GB8 cells) by pharmacological binding experiments and knockdown of the receptors by siRNA transfection pool [23,40]. Iper-8-naphthalimide (N-8-Iper) was synthesized according to a known literature procedure [41]. Its ability to selectively bind M2 mAChR subtype has been showed by pharmacological binding and M2 mAChR knockdown experiments [35]. To compare the activity of the two M2 receptor ligands, we first used both agonists at high concentration (100 μM), then adopted the lowest doses of N-8-Iper (25 μM and 3 μM) producing significant effects in U251 cell line and GB7 cells, as shown previously [34].
2.3. U251 cell line transfection and immunofluorescence analysis
Transfection was performed on the U251 cell line using the following expression vectors: EGFP-LC3B and mRFP-EGFP-LC3B. Cells were seeded on 6-well plates at the density of 2 x 105/well, and transfected at 80% confluence using LipofectamineTM 3000 Reagent (Invitrogen, Waltham, MA, USA), according to the manufacturer's protocol. 48 h after transfection, the cells were maintained in complete medium supplemented with geneticine (800μg/ml; Sigma-Aldrich, St. Louis, MO, USA) to select only antibiotic-resistant clones containing EGFP-LC3B fusion protein or mRFP-EGFP-LC3B fusion protein.
Stable U251-EGFP-LC3 or U251-mRFP-EGFP-LC3 cells were plated on coverslips arranged in 24-well plates at the density of 2 x 104 cells. Cells were treated with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper with or without 25 μM Chloroquine (CQ) for 72 h. Then, cells were washed 3 times with PBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. After 3 washes in PBS, cells were incubated with Hoechst 33342 (1:1000, Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature (RT), for nuclei counterstaining, and then washed 3 times with PBS. At the end, coverslips were fixed on microscope slides with a PBS-glycerol (3:1; v/v) solution. The images were acquired with a Apotome Zeiss fluorescence microscope through Zeiss Zen lite software (Zeiss, Oberkochen, Germany).
2.4. Cell Viability Assay
U251 cells were seeded on 96-well plates at the density of 1.5 × 104 cells/well. After 24 h, cells were treated with N-8-Iper at different times (ranging from 24 to 96 h). Cell proliferation was evaluated by colorimetric assay based on 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO, USA) metabolization. The MTT assay was performed according to the protocol optimized by Mosmann [42]. MTT was dissolved in PBS at 5 mg/ml. The MTT stock solution (10×) was added and diluted (1×) in each well and then incubated at 37°C for 3 h. Isopropanol (+ 0.04 M HCl + 1% Triton X-100) to dissolve the dark blue crystals. For each well, the optical density (OD) at 570 nm was measured by Multiskan FC (Thermofisher Scientific, Waltham, MA, USA).
2.5. Protein Extraction and Western Blot Analysis
Cells were harvested in lysis buffer (Tris-EDTA 10mM, 0.5% NP40, NaCl 150 mM), containing a protease inhibitor, boiled for 5 min at 90°C. The protein extracts were run on SDS-polyacrilamide gel (SDS- PAGE) and transferred to Polyvinylidene Difluoride (PVDF) sheets (Merck Millipore, Darmstadt, Germany). Membranes were blocked for 40 min in 5% of non-fat milk powder (Sigma-Aldrich, St. Louis, MO, USA) in PBS containing 0.1% Tween-20 (PBS-Tween), and then incubated overnight at 4°C with one of the following primary antibodies: anti-LC3B (1:1500, Sigma-Aldrich, St. Louis, MO, USA), anti-PI3 Kinase p85 (dilution 1:800, Cell Signaling, Danvers, MA, USA), anti-Phospho AKTThr308 (1:800, Cell Signaling, Danvers, MA, USA), anti-AKT pan (1:800, Cell Signaling, Danvers, MA, USA), anti-Phospho AMPK𝛼1/2Thr172 (dilution 1:800, Cell Signaling, Danvers, MA, USA), anti-AMPK𝛼1 (dilution 1:1000, Immunological Science, Milan, Italy), anti-Phospho p70 S6 KinaseThr389 (dilution 1:600, Immunological Science, Milan, Italy), anti-p70S6 Kinase (dilution 1:1000, Immunological Science, Milan, Italy), anti-Caspase 9 (dilution 1:2000, Immunological Science, Milan, Italy), anti-Caspase 3 (dilution 1:2000, Immunological Science, Milan, Italy), anti-β-Actin (1:2000, Immunological Science, Milan, Italy). β-Actin was used as reference protein for loading control.
2.6. Statistical Analysis
Data are presented as the mean ± SEM. Statistical analysis was performed by One-way ANOVA followed by Dunnett multiple comparison post-test. Data were considered statistically significant at * p < 0.05, ** p < 0.01 and *** p < 0.001. Data analyses were performed with GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA).
3. Results
3.1. Analysis of autophagy in U251 cell line and GB7 cells
We demonstrated that selective activation of M2 mAChR by the dualsteric agonist N-8-Iper for G166 and GB7 GSCs, causes a reduction in cell proliferation in a time- and dose-dependent manner [34,35]. For GB7 cells, the lowest concentration of N-8-Iper that produced a significant reduction in cell number was 3 μM [35]. Using the MTT assay, we evaluated the action of different concentrations of N-8-Iper after 24, 48, 72 and 96 h of treatment on the U251 cell line. As shown in Figure 1a, the first dose of N-8-Iper capable of significantly reducing U251 cell proliferation was 25 μM. The lowest doses analyzed 12.5 μM, 6 μM and 3 μM (data not shown) did not alter cell proliferation compared to untreated cells until 72 h. For this reason, a 25 μM dose was used as the first effective dose in U251 cell line for subsequent experiments.
To evaluate whether the M2 mAChR activation was able to induce autophagy, we analyzed the LC3 protein localization. LC3 is synthesized in its inactive form (pro-LC3) and subsequently converted to its active cytosolic form (LC3-I). LC3-I undergoes proteolytic cleavage at the C-terminal and is then converted to LC3-II by conjugation with phosphatidylethanolamine on the surface of nascent autophagosomes. The lipidated LC3 (LC3-II) is associated with autophagosomes and autolysosomes [43,44]. To observe and quantify the accumulation of LC3-II in autophagosomes, we stably transfected the U251 cells with the vector encoding the EGFP-LC3B fusion protein. Stably transfected cells were treated cells with 100 μM APE, 100 μM and 25 μM N-8-Iper for 72 h (Figure 2).
By fluorescence microscopy analysis (Figure 2a), green spots were observed in cells treated with M2 agonists, indicative of LC3 recruitment to the membrane of autophagosomes. In contrast, untreated cells showed predominantly uniformly scattered green fluorescence into the cells. A quantitative analysis was conducted by counting the green spots in each cell under the different treatment conditions. As shown in the graph in Figure 2b, a significant increase in the green spots present within the cells was observed after 72 h of 100 μM APE, and 100 μM or 25 μM N-8-Iper treatments compared with untreated cells, suggesting a significant increase of autophagy process after treatment with both M2 agonists.
To determine whether the accumulation of LC3B-II was the result of an increased de novo biosynthesis of autophagosomes or a blockage of the autophagy process, we analyzed LC3B protein turnover by fluorescence analysis in cell transfected with a pH-sensitive vector with double fluorescence (mRFP-EGFP-LC3B) and by Western blot analysis.
The mRFP-EGFP-LC3B vector allowed us to study the progression from autophagosome to autolysosome, exploiting the different pH of the two organelles and the different pH sensitivity of EGFP, which is sensitive to acidic pH, and RFP, which is resistant to pH changes. The U251 cell line was stably transfected with mRFP-EGFP-LC3B vector, then cells were treated with 100 μM APE and 100 μM N-8-Iper or 25 μM N-8-Iper for 72 h (Figure 3). In this experiment, the LC3B localization was analyzed in the presence or absence of 25 μM Chloroquine (CQ), an inhibitor of late-stage autophagy as it blocks the fusion of autophagosomes to lysosomes [45]. The U251-mRFP-EGFP-LC3B cells showed an increase in red fluorescence spots and the absence of green ones, sign of formation of autophagolysosomes evidenced by the persistence of red fluorescence and by the absence of green fluorescence, that can be detected only in an acidic pH environment (Figure 3a). The images in Figure 3a,b are high magnification of representative fields of the different experimental conditions. Fluorescence images at lower magnification can be appreciated in the supplementary Figure S1. Quantitative analysis performed by counting all the fluorescent cells, those with only red spots and those with green spots, showed that, after treatment with the M2 mAChR agonists, a significant increase in the percentage of cells with only red spots was observed. The absence of green spots indicates that the M2 agonists trigger the autophagy process and allow a proper fusion between autophagosomes and lysosomes (Figure 3c). After CQ treatment, an increase in green spots was observed in the cells, indicative of a block of the autophagy process at the autophagosome stage, where neutral pH allows the mRFP-EGFP-LC3B construct to express both green and red fluorescence (Figure 3b). However, quantitative analysis did not reveal any significant modulation in the percentage of cells with green spots between the different experimental conditions (Figure 3d).
Then we investigated the LC3B protein expression by Western blot analysis, not only in the U251 cell line but also extending the analysis to the previously characterized GSCs, GB7 cells [35]. By Western blot analysis we quantified the expression of LC3B-I and LC3B-II forms in both cell lines after treatment with M2 agonists. A 3 μM treatment with N-8-Iper was used in GB7 cells, since this was the lowest effective dose in reducing cell proliferation [34]. Cells were also treated in combination with 25 μM CQ to study the progression of autophagy flux in the presence or in absence of an autophagy inhibitor. In the U251 cell line, after 48 h, an increase in the LC3B-II form compared with LC3B-I was observed only with APE treatment (Figure 4a). Enhancement of the LC3B-II form, after treatment with N-8-Iper at high (100 μM) and low (25 μM) doses, became evident only after 72 h of treatment, when the increase of the LC3B-II/LC3B-I ratio in APE-treated cells was still meaningful (Figure 4c). After 72 h of treatment, the increased LC3B-II/LC3B-I ratio was also observed in U251 cells, in which M2 agonists were provided in combination with CQ compared with CQ alone (Figure 4d); on the other hand, no significant change of this ratio was revealed under the same experimental conditions after treatment for 48 h (Figure 4b). As shown in Figure 4e, in GB7 cells treatment with 100 μM APE for 48 h did not overexpress LC3B-II compared with untreated cells, whereas in N-8-Iper-treated cells there was an upregulation of LC3B-II compared with LC3B-I at both high (100 μM) and low (3 μM) doses. Overexpression of LC3B-II after APE treatment was not observed even by prolonging the treatment time (data not shown). Conversely, a significant increase in LC3B-II expression was measured after 48 h of treatment with the combination N-8-Iper (100 μM or 3 μM) and CQ, compared with cells treated with 25 μM CQ alone (Figure 4f).
3.2. Analysis of TORC1 and AMPK expression in U251 cell line and GB7 cells
With the aim to evaluate the molecular mechanism involved in the M2 receptor-triggered autophagy, we assessed the expression of proteins implicated in the PI3K/AKT pathway and of the two regulators of the catabolic process, AMPK and TORC1. Thus, by western blot analysis, we investigated the PI3K/AKT/mTOR signaling pathway upon APE or N-8-Iper treatments in both U251 cell line and GB7 cells. We examined the expression of PI3K p85 and AKT, phosphorylated in Threonine 308 (Thr 308), that is activated by Phosphoinositide-dependent kinase-1 (PDK1), which in turn is activated by PI3K. As shown in Figure 5a, in the U251 cell line PI3K p85 is downregulated after 72 h of treatment with both 100 μM APE and N-8-Iper at high (100 μM) and low (25 μM) doses. M2 agonists were able to downregulate the PI3K p85 protein even in the GB7 cells after 72 h of treatments (Figure 5b,c). The same trend was observed for the expression of the active form of AKT protein, phosphorylated in Thr 308; in fact APE and N-8-Iper caused significant downregulation of Phopho-AKT (Thr 308) expression, compared to AKT (pan) which does not discriminate between the active and inactive form, in both U251 cell line (Figure 5d) and GB7 cells (Figure 5e,f).
To evaluate whether downregulation of the PI3K/AKT pathway could affect TORC1 activity, we analyzed the protein expression of the downstream effector of TORC1, p70 S6K, phosphorylated at Thr389. In the U251 cell line, both 100 μM APE and N-8-Iper (100 μM and 25 μM) caused a decrease in the expression of Phospho-p70 S6K (Thr389) compared with the unphosphorylated form (Figure 5g). As shown in Figure 5h, a reduction in the Phospho-p70 S6K (Thr389) expression was observed in GB7 cells treated with high- or low-dose of N-8-Iper, whereas no significant change was observed after 72 h of APE treatment compared with untreated cells.
Another factor involved in the regulation of TORC1 is AMPK, which inhibits the activity of this complex. Western blot analysis showed that M2 mAChR activation with both agonists caused increased protein expression of Phospho-AMPKα (phosphorylated at Thr172), compared with inactive AMPKα in the U251 cell line (Figure 5i). In GB7 cells, we evidenced the same trend only after N-8-Iper treatments, whereas in the APE-treated cells the active AMPKα form was not significantly modulated respect to the control condition (Figure 5j).
3.3. Analysis of apoptosis induction following M2 mAChR activation
Since an excessive level of autophagy could promote cell death, including the apoptotic process [46], we evaluated the expression of proteins that are known to be activated during apoptosis, i.e., Caspase-9 and Caspase-3, also prolonging for additional 24 h the treatment with M2 agonists applied in the autophagy experimental protocol.
For the U251 cell line, we analyzed the expression of Caspase-9 and Caspase-3 proteins after 72 h and 96 h of treatment. As shown in Figure 6a, after 72 h of 100 μM APE treatment, Caspase-9 cleaved/Pro caspase-9 ratio increased compared with untreated cells (Ctrl). In contrast, no significant modulation was observed in cells treated with high-dose (100 μM) and low-dose (25 μM) of N-8-Iper. The same trend was monitored for Caspase-3 after 72 h of treatment with the M2 receptor agonists (Figure 6d). After 96 h of treatment, we detected an increased expression of the cleaved forms of Caspase-9 (Figure 6b) and Caspase-3 (Figure 6e) with 100 and 25 μM N-8-Iper doses. Similarly, after 72 h treatment with the M2 agonists (100 μM APE, 100 and 3 μM N-8-Iper), in GB7 cells, the active forms of Caspase-9 (Figure 6c) and Caspase-3 (Figure 6f) showed an enhanced expression compared with the control cells.
Representative western blot analysis of Caspase-3 cleaved /Pro caspase-3 expression after 72 h of treatment (d) and after 96 h of treatment (e) with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in the U251 cell line. (f) Western blot analysis of Caspase-3 cleaved /Pro caspase-3 expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Caspase-3 cleaved normalized with the bands of Pro Caspase-3 protein. Actin was used as internal reference protein. The data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p <0.001, ** p <0.01, * p <0.05).
To evaluate whether activation of the apoptotic process is a consequence of the M2 receptor-induced autophagy, western blot analysis for Caspase-9 and Caspase-3 was performed in both cell lines in the presence of CQ, either alone or in combination with the M2 agonists. Figure 7a,d showed that after 72 h of treatment in the U251 cells, the M2 agonist + CQ did not cause an increase of the cleaved form of both Caspases 3 and 9, at variance with what we had observed in cells treated with the M2 agonist alone (Figure 6a,d). The same trend was detected for Caspase-9 in the U251 cells, after 96 h of treatment (Figure 7b), while the cleaved form of Caspase-3 was completely absent under all experimental conditions (Figure 7e). In GB7 cells, the expression of Caspases after 100 μM APE + CQ condition was not evaluated, because the same concentration of APE alone did not cause an increase in autophagy (see Figure 4e). Also in these cells, the presence of CQ in combination with N-8-Iper both at high (100 μM) and low (3 μM) doses, did not induce the expression of the cleaved form of Caspase-9 that, in this case, is reduced respect to the control with CQ alone (Figure 7c). Like in U251 cells after 96h of treatment, in GB7 cells we did not detect the cleaved form of Caspase-3 but found a strong accumulation of Procaspase-3 in all experimental conditions (Figure 7f).
4. Discussion
Over the past decade, various studies have shown that activation of mAChRs is critical in the regulation of cell proliferation and cancer progression [47]. In this context, we have been studying the role of the M2 mAChR in cancer for many years, demonstrating that activation of this receptor subtype brings about anti-proliferative and cytotoxic effects on cells of different tumor types such as ovarian cancer [19], breast cancer [18], neuroblastoma [21,48], and GBM [34]. As far as GBM we showed that, in the U251 cell line, activation of M2 mAChR by the orthosteric agonist APE caused cell cycle arrest as well as progressive accumulation of the cells in the G2/M phase [25]. In GSCs cells, named GB7, APE and the dualsteric agonist N-8-Iper were capable to inhibit cell proliferation in a time- and dose-dependent manner [23,35]. Moreover, analysis of the U251 cell fraction with hypodiploid DNA content and higher granularity (SSC) obtained by flow cytometry, and the ELISA quantification of cytoplasmic nucleosomes, showed that treatment with 100 μM APE for 72 h caused an increase in apoptotic cells [25]. Once the same analysis was performed in GB7 cells after 48 h and 72 h treatment with 100 μM N-8-Iper, this M2 mAChR activator produced an enhanced percentage of apoptotic cells [35].
At present, the role of autophagy in cancer cells is receiving much attention since this process seems to have divergent effects depending on tumor type and/or grade. Indeed, it has been observed that autophagy plays a strategic role in the regulation of tumor progression, acting as a tumor suppressor in the early stages of tumor development and as a pro-survival mechanism in the later stages of tumorigenesis [1]. A recent study has showed that combined treatment with temozolomide and rapamycin, an mTOR pathway inhibitor, causes overexpression of Beclin-1 and LC3-II, thus considerably increasing autophagy-induced cell death of U251 cells [49]. Building on this evidence, by transfection of the constructs overexpressing LC3B-EGFP we demonstrated that the autophagic process is promoted after activation of M2 mAChRs in the U251 stable cell line by both M2 orthosteric (APE) and dualsteric (N-8-Iper) agonists (Figure 2). With the pH-sensitive expression construct, mRFP-EGFP-LC3B, we demonstrated that M2 agonists upregulate autophagy without causing its arrest. In fact, the increase in LC3 aggregates was observed only after addition of CQ, an autophagy flux blocker, and no-summative effect between CQ and the applied M2 agonist was put in evidence (Figure 3). The ability of M2 mAChR activation to promote autophagy was also confirmed by the analysis of LC3B protein expression by Western blotting (Figure 4). In this experiment, human GSCs were also assayed, to compare the effects of M2 agonists on a human established cell line (U251 cell line) with those on human GSCs (GB7 cells). Western blot analysis confirmed that N-8-Iper was able to induce autophagy both in U251 cell line and GB7 cells, while APE was able to increase LC3B-II compared with LC3B-I only in U251 cell line. The combined treatment with M2 agonists, that alone caused an increase in LC3B-II expression, and CQ significantly increased the LC3B-II/LC3B-I ratio when compared with the treatment with CQ alone (Figure 4d,f). These results, together with those from the cells transfected with LC3-EGFP and mRFP-GFP-LC3B vectors, confirm the ability of M2 agonists to promote the autophagic flux and allow the fusion between autophagosomes and lysosomes; as a matter of fact, only CQ blocked the autophagy process at the autophagosome stage. However, a difference between the two studied M2 agonists clearly emerged since APE was found to induce autophagy in U251 cells only, whereas N-8-Iper was efficacious in both U251 and GB7 cells.
To further explore the different modulation pattern of autophagy by the two M2 agonists in both cell lines, we analyzed the expression of TORC1 and AMPK, two main factors involved in the regulation of this process. Treatment with N-8-Iper for 72 h, at high and low doses, downregulated in both cell lines the protein expression for PI3K p85 (Figure 5a-c), the active form of AKT (phosphorylated at Thr308) (Figure 5d-f), and the active form of the downstream effector of TORC1, p70 S6K phosphorylated at Thr389 (Figure 5g-h). APE produced the same downstream effects but only in U251 cells, while in GB7 cells it negatively modulated the PI3K/AKT pathway but did not cause significant downregulation of the active form of p70 S6K, indicating a failure in TORC1 downregulation. Regarding the AMPKα protein level, an increase in the phosphorylated (active) form over the non-phosphorylated (inactive) one was observed after treatment with both APE and N-8-Iper, at high and low doses (Figure 5i). In GB7 cells, only N-8-Iper was able to significantly raise the Phospho-AMPKα protein levels, further confirming the inability of APE to promote autophagy in GSCs (Figure 5j). In Figure 8, we have summarized the signaling pathways of the M2 receptor-induced autophagy process emerging from the above discussed data. Worth highlighting is the upregulation of AMPKα and downregulation of TORC1 activity following treatment with the dualsteric muscarinic agonist N-8-Iper in both cell lines. Conversely, the orthosteric agonist APE engendered a parallel modulation profile of the autophagy flux only in U251 cells and not in GB7 cells.
As already underlined, several literature data suggest that the autophagic flux may lead to cell death, including apoptosis [46,50]. In previous investigations, we demonstrated that the two M2 agonists induce cytotoxic effects [26,35] and by Florescence Activated Cell Sorting (FACS) analysis we found that they caused cell cycle arrest and apoptotic cell death [25,35,36]. To further support these results and delve into the correlation between apoptotic cell death and M2 receptor-induced autophagy, we analyzed the expression of two of the proteins involved in the apoptotic process, i.e., Caspase-9 and Caspase-3 (Figure 6). A treatment of 72 h with APE in U251 cells induced the increase of the cleaved forms of both Caspase-9 (Figure 6a) and Caspase-3 (Figure 6d), confirming our previous data. It was necessary to extend the treatment to 96 h to observe the increase of these proteins in N-8-Iper-treated U251 cells (Figure 6b,e). In GB7 cells, the cleaved form of Caspases 9 (Figure 6c) and 3 (Figure 6f) were upregulated by N-8-Iper. The treatment duration is indicative of a direct sequence between autophagy and apoptotic events. Unlike N-8-Iper treatment, the upregulation of apoptotic proteins after APE treatment does not seem to be linked with autophagy in GB7 cells.
Treatment with the autophagy inhibitor CQ allowed to improve the correlation between autophagy and downstream apoptosis. Inhibition of the autophagic flux by CQ caused in fact a remarkable reduction or complete absence of caspases in their active form, differently from what we observed by treating cells with N-8-Iper or APE alone. These results suggest that prevention of the autophagy process, which is promoted by M2 agonists (N-8-Iper for the U251 cell line and GB7 cells, APE for U251 cells only), interferes with the activation of apoptosis.
According to the previous data both APE and N-8-Iper have antiproliferative effects, interfering with PI3K/AKT pathway, in both cell models [23,24,35]. Downregulation of the PI3K/AKT/TORC1 pathway, involved in regulating the proliferation, and activation of autophagy can lead to cell death by apoptosis in both GBM cell models after N-8-Iper treatments. Downregulation of the PI3K/AKT pathway after APE treatment in GB7 cells can explain the decreased cell proliferation previously described [36] but it is not able to inhibit TORC1 action, as also suggested by the lack of increase AMPK expression. This evidence explains the lack of upregulation of the autophagic process in GB7 cells after APE treatment. Western blot analysis of caspases and previous results from APE-treated GB7 cells, in which an increased percentage of annexin V-positive cells was assessed [36], suggest that 100 μM APE induces directly apoptosis without any correlation with autophagy [36]. This different APE behavior in GB7 cells compared with U251 may in part dependent on genetic bacjground of two cells line. GB7 cells present p53 wild type differently by U251 cells that are p53 mutated. Considering the anti-prolifertive and the strong cytotoxic effects produced by APE in cells where p53 is active, according to its function of genome guardian and regulator of the cell cycle, it may drive directly the cells to apotptosis.
Differently N8-Iper and APE seems to trace the activation of the same pathway inducing autophagy followed by the apoptotic process in U251 cells but in GB7 cells the same effcts was evident only with N8-Iper.
5. Conclusions
The results of this study confirm that selective M2 agonists may counteract cell survival in GSCs and stable GBM cell lines. Indeed, both N-8-Iper and APE were found to activate the autophagic process followed by apoptosis by downregulating the PI3K/AKT/TORC1 pathway and upregulating the AMPK protein expression. However,in GSCs, APE induces apoptosis but without promoting the autophagic process, and it probably triggers alternative mechanisms that could be mediated by the p53 protein. [26,27,40].
At present, we are unable to speculate on the different activity profiles characterizing the two types of muscarinic agonists, that could be related to some extent to a difference in the activation mode of the M2 mAChR. However, our overall data confirm the promising role of this receptor subtype as a strategic therapeutic target for GBM therapy. Albeit further analyses are necessary, the ability of dualsteric agonists like N-8-Iper to act at low concentrations and activate similar effects in stable and primary GBM cells highlights its relevance in view of a putative therapeutic perspective.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: A.M.T. and C.G: Methodology: C.G. and M.M. Dualsteric agonist synthesis: C.D., C.M. and M.D.A. Formal analysis: C.G. Stable cell transfection: A.I.; D.T. Investigation: C.G. and M.M. Data curation: C.G. GSCs cell line production: L.C.; Writing original draft preparation, C.G. and A.M.T. Writing—review and editing: A.M.T.; D.T., L.C., M.D.A.. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The research was supported by Ateneo Sapienza Funds to A.M.T. and by MIUR CIB project 2019.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a Molecular Target for Cancer Treatment. Eur. J. of Pharmac. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bulletin du Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. Autophagy: Supporting Cellular and Organismal Homeostasis by Self-Eating. Inter. J. of Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.-L. AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Mizushima, N. The Role of the Atg1/ULK1 Complex in Autophagy Regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.-Y.; et al. Autophagy. In Advances in Cancer Research; Elsevier, 2013; Vol. 118, pp. 61–95 ISBN 978-0-12-407173-5.
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Vial, D.; McKeown-Longo, P.J. Role of EGFR Expression Levels in the Regulation of Integrin Function by EGF: EGFR levels regulate integrin activation. Mol. Carcinog. 2016, 55, 1118–1123. [Google Scholar] [CrossRef]
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The Prognostic Role of Beclin 1 Protein Expression in High-Grade Gliomas. Autophagy 2009, 5, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Patric, I.R.P.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation Silencing of ULK2, an Autophagy Gene, Is Essential for Astrocyte Transformation and Tumor Growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N.; et al. Monitoring Autophagy in Glioblastoma with Antibody against Isoform B of Human Microtubule-Associated Protein 1 Light Chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Zhang, X.; Li, X.; Ren, Y.; Ma, X.; Li, X.; Wang, L. Association between AKT/mTOR Signalling Pathway and Malignancy Grade of Human Gliomas. J. Neurooncol. 2011, 103, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic Acetylcholine Receptors: Mutant Mice Provide New Insights for Drug Development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Español, A.J.; Salem, A.; Di Bari, M.; Cristofaro, I.; Sanchez, Y.; Tata, A.M.; Sales, M.E. The Metronomic Combination of Paclitaxel with Cholinergic Agonists Inhibits Triple Negative Breast Tumor Progression. Participation of M2 Receptor Subtype. PLoS ONE 2020, 15, e0226450. [Google Scholar] [CrossRef] [PubMed]
- Taggi, M.; Kovacevic, A.; Capponi, C.; Falcinelli, M.; Cacciamani, V.; Vicini, E.; Canipari, R.; Tata, A.M. The Activation of M2 Muscarinic Receptor Inhibits Cell Growth and Survival in Human Epithelial Ovarian Carcinoma. J. Cell. Biochem. 2022, 123, 1440–1453. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 Muscarinic Receptor Antagonists Inhibit Small Cell Lung Carcinoma Growth and Mitogen-Activated Protein Kinase Phosphorylation Induced by Acetylcholine Secretion. Cancer Res. 2007, 67, 3936–3944. [Google Scholar] [CrossRef]
- Lucianò, A.M.; Perciballi, E.; Fiore, M.; Del Bufalo, D.; Tata, A.M. The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 8433. [Google Scholar] [CrossRef]
- Calaf, G.M.; Crispin, L.A.; Muñoz, J.P.; Aguayo, F.; Bleak, T.C. Muscarinic Receptors Associated with Cancer. Cancers 2022, 14, 2322. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The Activation of M2 Muscarinic Receptor Inhibits Cell Growth and Survival in Human Glioblastoma Cancer Stem Cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Ponti, D.; Calogero, A.; Tata, A.M. M2 Muscarinic Receptors Inhibit Cell Proliferation in Human Glioblastoma Cell Lines. Life Sci. 2012, 91, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Bari, M.D.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 Receptor Activation Inhibits Cell Cycle Progression and Survival in Human Glioblastoma Cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Iorio, E.; Carpinelli, G.; Ricordy, R.; Fiore, M.; Degrassi, F.; et al. Cytotoxic and Genotoxic Effects Mediated by M2 Muscarinic Receptor Activation in Human Glioblastoma Cells. Neurochem. Int. 2015, 90, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Alessandrini, F.; Guerriero, C.; Fiore, M.; Asteriti, I.A.; Castigli, E.; Sciaccaluga, M.; Guarguaglini, G.; Degrassi, F.; et al. M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines. Cells 2021, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Fritz, J.J.; Wess, J.; Lah, J.J.; Levey, A.I. Deletion of M1 Muscarinic Acetylcholine Receptors Increases Amyloid Pathology in Vitro and in Vivo. J. Neurosci. 2010, 30, 4190–4196. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Kitazawa, M.; Caccamo, A.; Baglietto-Vargas, D.; Estrada-Hernandez, T.; Cribbs, D.H.; Fisher, A.; LaFerla, F.M. Loss of Muscarinic M1 Receptor Exacerbates Alzheimer’s Disease–Like Pathology and Cognitive Decline. The American Journal of Pathology 2011, 179, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Dencker, D.; Wörtwein, G.; Woldbye, D.P.D.; Cui, Y.; Davis, A.A.; Levey, A.I.; Schütz, G.; Sager, T.N.; Mørk, A.; et al. A Subpopulation of Neuronal M4 Muscarinic Acetylcholine Receptors Plays a Critical Role in Modulating Dopamine-Dependent Behaviors. J. Neurosci. 2010, 30, 2396–2405. [Google Scholar] [CrossRef]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-Coupled Receptor. J. Bio. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Mohr, K. Dualsteric GPCR Targeting and Functional Selectivity: The Paradigmatic M(2) Muscarinic Acetylcholine Receptor. Drug Discov. Today Technol. 2013, 10, e245–252. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, C.; Matera, C.; Del Bufalo, D.; De Amici, M.; Conti, L.; Dallanoce, C.; Tata, A.M. The Combined Treatment with Chemotherapeutic Agents and the Dualsteric Muscarinic Agonist Iper-8-Naphthalimide Affects Drug Resistance in Glioblastoma Stem Cells. Cells 2021, 10, 1877. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Spinello, Z.; Matera, C.; Fiore, M.; Conti, L.; De Amici, M.; Dallanoce, C.; Tata, A.M. Activation of M2 Muscarinic Acetylcholine Receptors by a Hybrid Agonist Enhances Cytotoxic Effects in GB7 Glioblastoma Cancer Stem Cells. Neurochem. Int. 2018, 118, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Alessandrini, F.; Spinello, Z.; Guerriero, C.; Fiore, M.; Caffarelli, E.; Laneve, P.; Dini, L.; Conti, L.; Tata, A.M. Cross Interaction between M2 Muscarinic Receptor and Notch1/EGFR Pathway in Human Glioblastoma Cancer Stem Cells: Effects on Cell Cycle Progression and Survival. Cells 2020, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Rispoli, R.; Conti, C.; Celli, P.; Caroli, E.; Carletti, S. Neural Stem Cells and Glioblastoma. Neuroradiol. J. 2014, 27, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Crisafulli, L.; Caldera, V.; Tortoreto, M.; Brilli, E.; Conforti, P.; Zunino, F.; Magrassi, L.; Schiffer, D.; Cattaneo, E. REST Controls Self-Renewal and Tumorigenic Competence of Human Glioblastoma Cells. PLoS ONE 2012, 7, e38486. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Bari, M.D.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 Receptor Activation Inhibits Cell Cycle Progression and Survival in Human Glioblastoma Cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef]
- Bock, A.; Merten, N.; Schrage, R.; Dallanoce, C.; Bätz, J.; Klöckner, J.; Schmitz, J.; Matera, C.; Simon, K.; Kebig, A.; et al. The Allosteric Vestibule of a Seven Transmembrane Helical Receptor Controls G-Protein Coupling. Nat. Commun. 2012, 3, 1044. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-Positive Structures Are Prominent in Autophagy-Deficient Cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Xue, Y.; Yan, X.; Li, J.; Wu, Y.; Guo, R.; Zhang, J.; Zhang, L.; Li, Y.; Liu, Y.; et al. Autophagy Inhibitor Chloroquine Induces Apoptosis of Cholangiocarcinoma Cells via Endoplasmic Reticulum Stress. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The Role of Interaction between Autophagy and Apoptosis in Tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.-P. Muscarinic Receptors and Ligands in Cancer. Am. J. Physiol. Cell Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef] [PubMed]
- Lucianò, A.M.; Mattei, F.; Damo, E.; Panzarini, E.; Dini, L.; Tata, A.M. Effects Mediated by M2 Muscarinic Orthosteric Agonist on Cell Growth in Human Neuroblastoma Cell Lines. Pure Appl. Chem. 2019, 91, 1641–1650. [Google Scholar] [CrossRef]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M. Effect and Molecular Mechanism of mTOR Inhibitor Rapamycin on Temozolomide-induced Autophagic Death of U251 Glioma Cells. Oncol. Lett. 2017. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
Figure 1.
Effect of N-8-Iper (12.5 μM, 25 μM, 50 μM and 100 μM) on U251 cell growth at different time points of treatment (ranging from 1 to 4 days in vitro, DIV). Data represent the mean (± SEM) of four different experiments performed in sextuplicate. ANOVA test was used followed by Dunnett’s post test (N-8-Iper treated cells vs. untreated cells, Ctrl. *** p < 0.001; ** p < 0.01; * p < 0.05).
Figure 1.
Effect of N-8-Iper (12.5 μM, 25 μM, 50 μM and 100 μM) on U251 cell growth at different time points of treatment (ranging from 1 to 4 days in vitro, DIV). Data represent the mean (± SEM) of four different experiments performed in sextuplicate. ANOVA test was used followed by Dunnett’s post test (N-8-Iper treated cells vs. untreated cells, Ctrl. *** p < 0.001; ** p < 0.01; * p < 0.05).
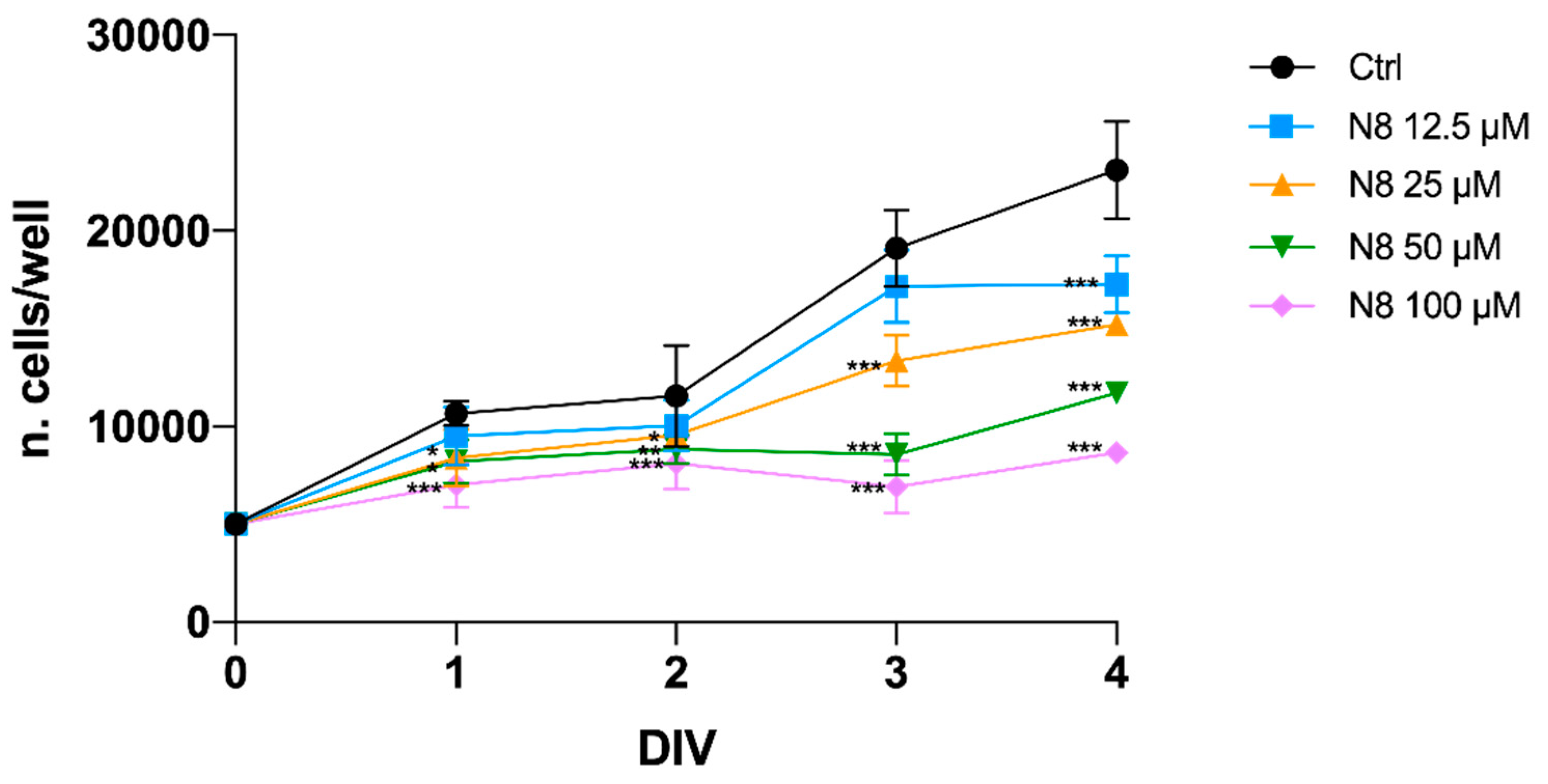
Figure 2.
(a) Representative images of fluorescence microscopy analysis of the U251 cell line transfected with the EGFP-LC3B expression vector, treated for 72 h with 100 μM APE, 100 μM N-8-Iper (N8) or 25 μM N-8-Iper (N8). Nuclei were stained with Hoechst 33342. White arrows indicate green spots. Scale bar = 10 μm. (b) The quantification has been performed counting the number of green spots within each cell in 13 photographic fields for each experimental condition. All the experiments were performed in triplicate. The values (mean ± SEM) are reported as number of green dots/cells. For each experimental condition, about 150 cells were analyzed. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p < 0.001).
Figure 2.
(a) Representative images of fluorescence microscopy analysis of the U251 cell line transfected with the EGFP-LC3B expression vector, treated for 72 h with 100 μM APE, 100 μM N-8-Iper (N8) or 25 μM N-8-Iper (N8). Nuclei were stained with Hoechst 33342. White arrows indicate green spots. Scale bar = 10 μm. (b) The quantification has been performed counting the number of green spots within each cell in 13 photographic fields for each experimental condition. All the experiments were performed in triplicate. The values (mean ± SEM) are reported as number of green dots/cells. For each experimental condition, about 150 cells were analyzed. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p < 0.001).
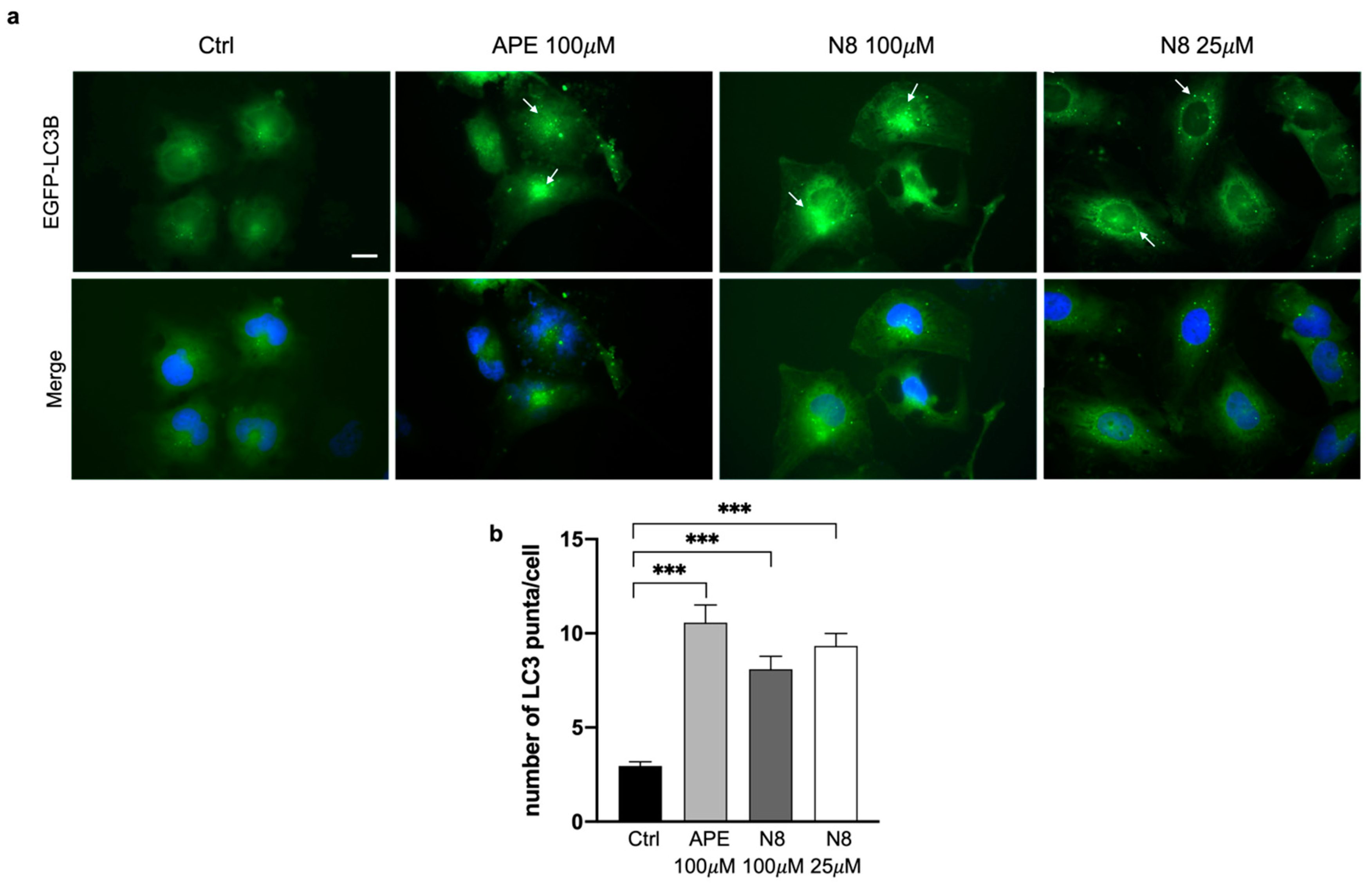
Figure 3.
Representative images of fluorescence microscopy analysis of the U251 cell line stably transfected with the mRFP-GFP-LC3B expression vector, treated for 72 h with 100 μM APE, 100 μM N-8-Iper (N8) or 25 μM N-8-Iper (N8) in (a) the absent or (b) presence of 25 μM CQ.. Scale bar=5 μm. (c, d) The quantification has been performed counting the number of cells without dots (black bars), with green dots or with red dots in 15 photographic fields for each experimental condition performed in triplicate. The values (mean ± SEM) are reported as percentage of red or green positive cells respect to total active cells. For each experimental condition, about 100 cells were analyzed. ANOVA test was used followed by Dunnett’s post test Ctrl (untreated cells) vs M2 agonists treated cells; *** p <0.001, * p <0.05. In d) any significant difference was observed between red and green dots for each experimental condition.
Figure 3.
Representative images of fluorescence microscopy analysis of the U251 cell line stably transfected with the mRFP-GFP-LC3B expression vector, treated for 72 h with 100 μM APE, 100 μM N-8-Iper (N8) or 25 μM N-8-Iper (N8) in (a) the absent or (b) presence of 25 μM CQ.. Scale bar=5 μm. (c, d) The quantification has been performed counting the number of cells without dots (black bars), with green dots or with red dots in 15 photographic fields for each experimental condition performed in triplicate. The values (mean ± SEM) are reported as percentage of red or green positive cells respect to total active cells. For each experimental condition, about 100 cells were analyzed. ANOVA test was used followed by Dunnett’s post test Ctrl (untreated cells) vs M2 agonists treated cells; *** p <0.001, * p <0.05. In d) any significant difference was observed between red and green dots for each experimental condition.
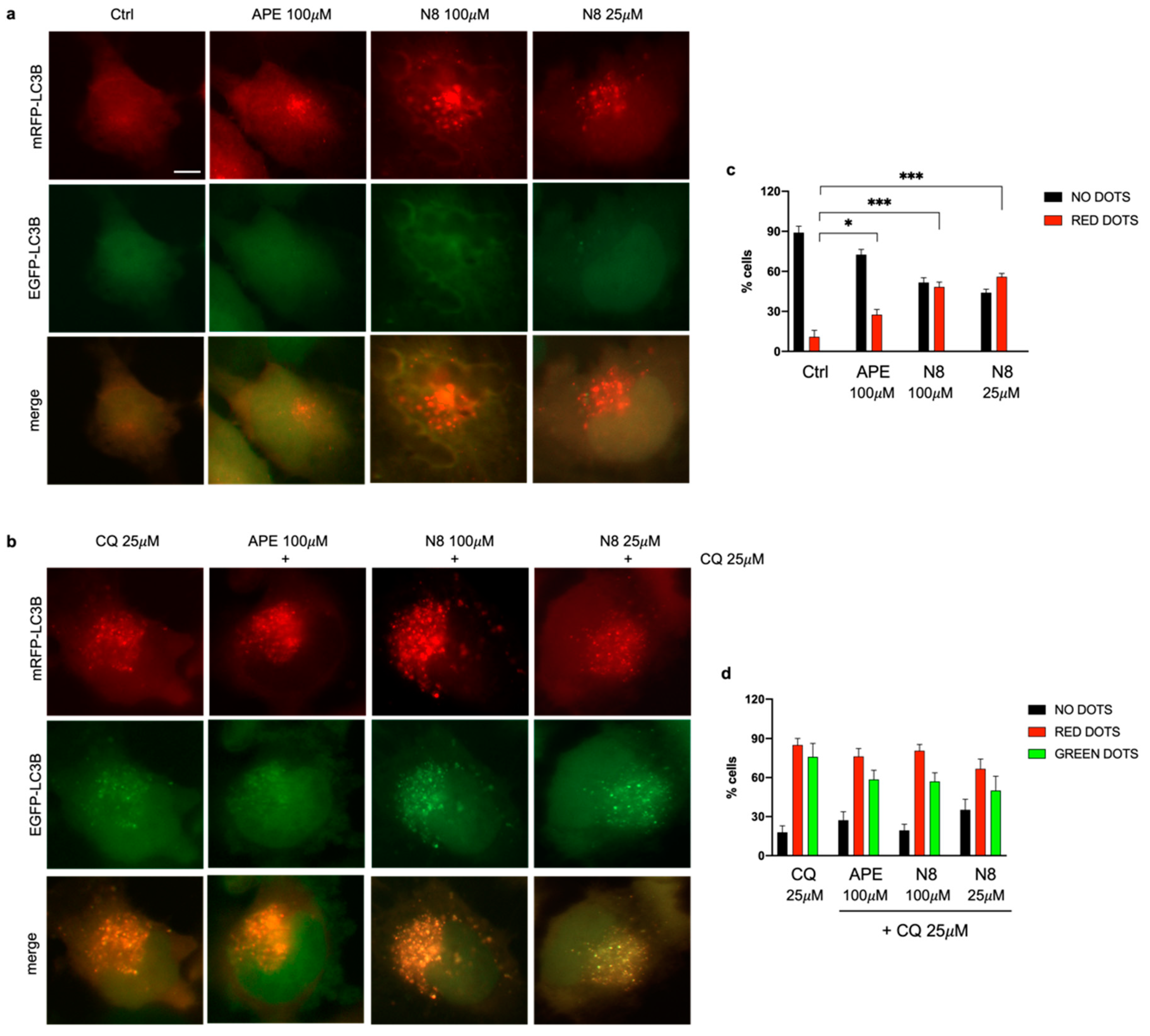
Figure 4.
Representative western blot relative to LC3B-II/I expression after 48 h (a) and 72h (c) of treatment with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper and after 48 h (b) and 72 h (d) of co-treatment with 25μM CQ and APE (100 μM) or N8 (100 μM and 25 μM) + CQ in the U251 cell line. (e) Representative western blot analysis of LC3-II/I expression after 48 h of treatment with 100 μM APE, 100 μM N-8-Iper, 3 μM N-8-Iper and (f) after 48 h of co-treatment with 25μM CQ and APE (100 μM) or N-8-Iper (100 μM and 3 μM) + CQ in the GB7 cells. Actin was used as internal reference protein. The graphs show the densitometric analysis of the bands of the western blot analysis for LC3B-II normalized with the bands of LC3B-I protein. The data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p <0.001, ** p <0.01, * p <0.05).
Figure 4.
Representative western blot relative to LC3B-II/I expression after 48 h (a) and 72h (c) of treatment with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper and after 48 h (b) and 72 h (d) of co-treatment with 25μM CQ and APE (100 μM) or N8 (100 μM and 25 μM) + CQ in the U251 cell line. (e) Representative western blot analysis of LC3-II/I expression after 48 h of treatment with 100 μM APE, 100 μM N-8-Iper, 3 μM N-8-Iper and (f) after 48 h of co-treatment with 25μM CQ and APE (100 μM) or N-8-Iper (100 μM and 3 μM) + CQ in the GB7 cells. Actin was used as internal reference protein. The graphs show the densitometric analysis of the bands of the western blot analysis for LC3B-II normalized with the bands of LC3B-I protein. The data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p <0.001, ** p <0.01, * p <0.05).
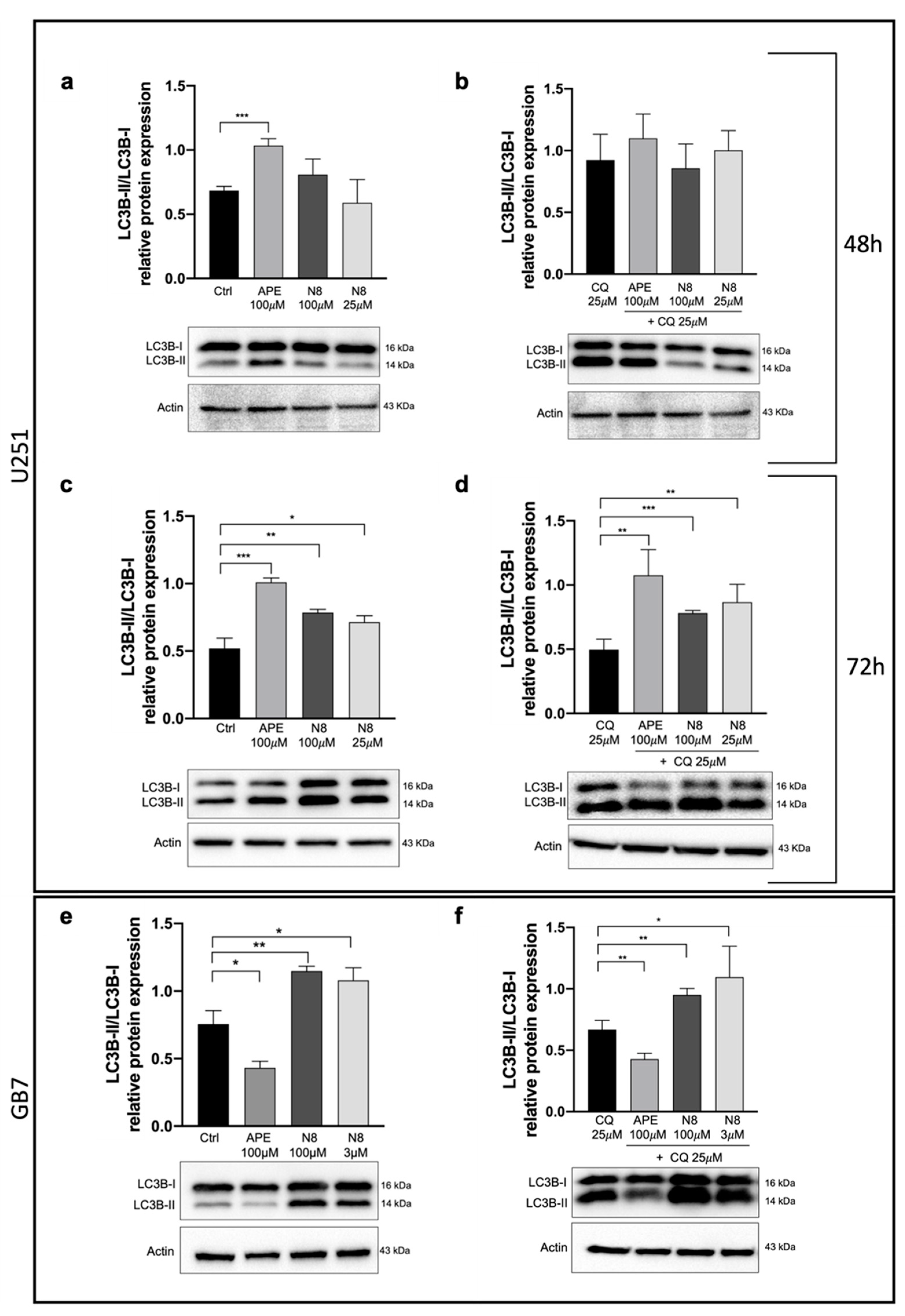
Figure 5.
(a) Representative western blot analysis for PI3K p85 expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper in U251 cell line and (b) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and (c) 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for PI3K p85 normalized with the bands of actin protein. (d) Representative western blot analysis of AKT phosphorylated in Thr 308 (Phospho-AKTThr308) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (e) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and (f) 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-AKTThr308 normalized with the bands of AKT pan. Actin was used as reference protein. (g) Representative western blot analysis of p70 S6K phosphorylated in Thr 389 (Phospho-p70 S6KThr389) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (h) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-p70 S6KThr389 normalized with the bands of p70 S6K. Actin was used as reference protein. (i) Representative western blot analysis of AMPK𝛼 phosphorylated in Thr 172 (Phospho-AMPK𝛼 Thr172) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (j) after 72 h of treatment in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-AMPK𝛼Thr172 normalized with the bands of AMPK𝛼. Actin was used as reference protein. The data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p <0.001, ** p <0.01, * p <0.05).
Figure 5.
(a) Representative western blot analysis for PI3K p85 expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper in U251 cell line and (b) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and (c) 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for PI3K p85 normalized with the bands of actin protein. (d) Representative western blot analysis of AKT phosphorylated in Thr 308 (Phospho-AKTThr308) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (e) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and (f) 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-AKTThr308 normalized with the bands of AKT pan. Actin was used as reference protein. (g) Representative western blot analysis of p70 S6K phosphorylated in Thr 389 (Phospho-p70 S6KThr389) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (h) after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper and 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-p70 S6KThr389 normalized with the bands of p70 S6K. Actin was used as reference protein. (i) Representative western blot analysis of AMPK𝛼 phosphorylated in Thr 172 (Phospho-AMPK𝛼 Thr172) expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 25 μM N-8-Iper in U251 cell line and (j) after 72 h of treatment in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Phospho-AMPK𝛼Thr172 normalized with the bands of AMPK𝛼. Actin was used as reference protein. The data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post test (untreated cells (Ctrl) vs M2 agonists treated cells; *** p <0.001, ** p <0.01, * p <0.05).
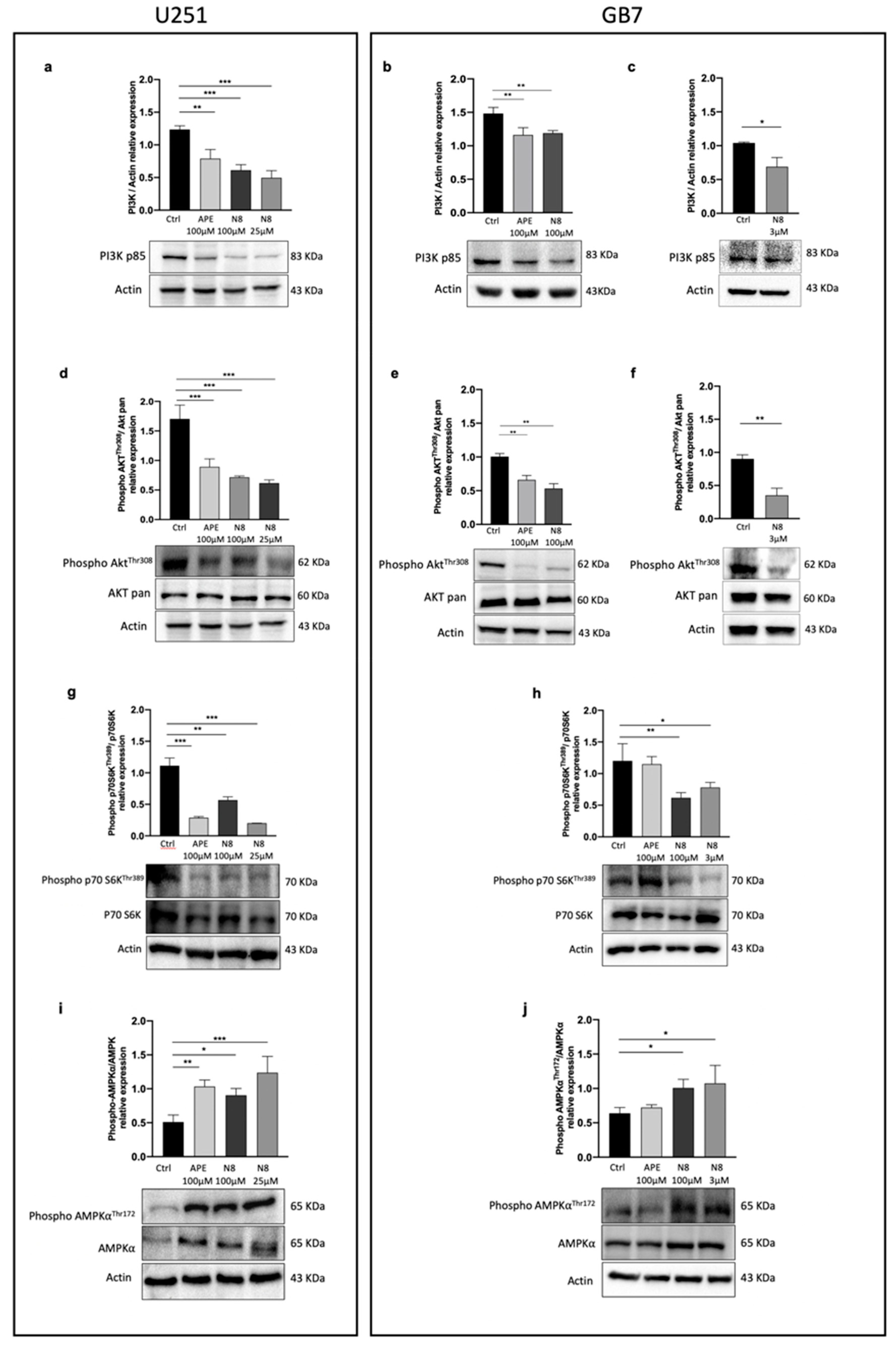
Figure 6.
Representative Western Blot analysis of Caspase-9 cleaved /Pro caspase-9 expression after 72 h of treatment (a) and after 96 h of treatment (b) with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper in the U251 cell line. (c) Representative western blot analysis of Caspase-9 cleaved /Pro caspase-9 expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Caspase-9 cleaved normalized with the bands of Pro Caspase-9 protein. Actin was used as internal reference protein.
Figure 6.
Representative Western Blot analysis of Caspase-9 cleaved /Pro caspase-9 expression after 72 h of treatment (a) and after 96 h of treatment (b) with 100 μM APE, 100 μM N-8-Iper (N8), 25 μM N-8-Iper in the U251 cell line. (c) Representative western blot analysis of Caspase-9 cleaved /Pro caspase-9 expression after 72 h of treatment with 100 μM APE, 100 μM N-8-Iper, 3 μM N-8-Iper in GB7 cells. The graphs show the densitometric analysis of the bands of western blot analysis for Caspase-9 cleaved normalized with the bands of Pro Caspase-9 protein. Actin was used as internal reference protein.
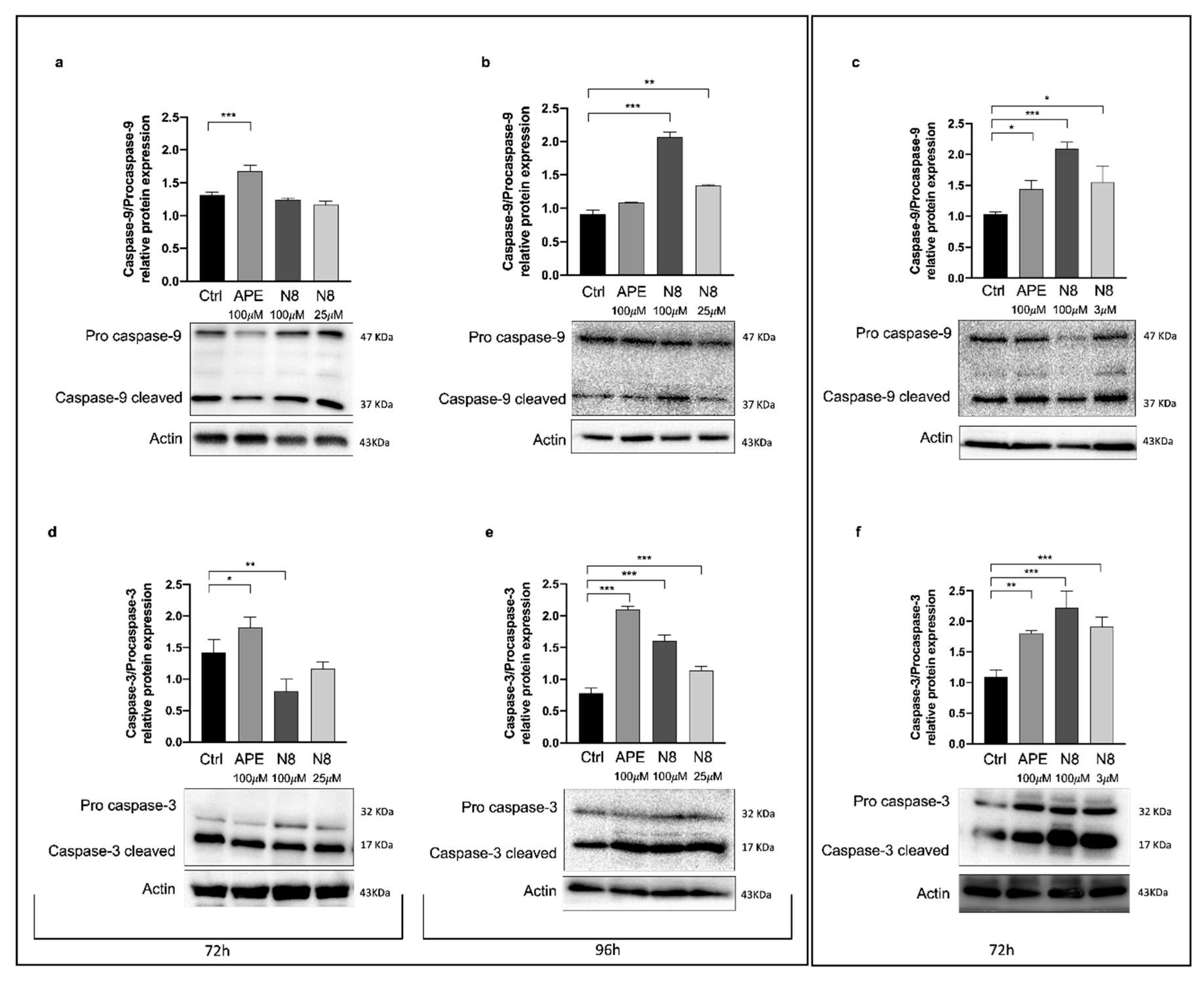
Figure 7.
Representative Western blot analysis of Caspase-9 cleaved/Pro caspase-9 expression after 72 h of treatment (a) and after 96 h of treatment (b) with 25 μM CQ, 100 μM APE + 25 μM CQ, 100 μM N-8-Iper (N8) + 25 μM CQ, 25 μM N8 + 25 μM CQ in the U251 cell line. (c) Representative Western blot analysis of Caspase-9 cleaved/Pro caspase-9 expression after 72 h of treatment with 25 μM CQ, 100 μM N8 + 25 μM CQ, 3 μM N8 + 25 μM CQ in GB7 cells. Graphs show the densitometric analysis of the bands of Western blot analysis for Caspase-9 cleaved normalized with the bands of Pro Caspase-9 protein. Actin was used as the internal reference protein. (d) Representative Western blot analysis of Caspase-3 cleaved/Pro caspase-3 expression after 72 h of treatment. The graph shows the densitometric analysis of the bands of western blot analysis for Caspase-3 cleaved normalized with the bands of Pro Caspase-3 protein. Representative western blot analysis of Caspase-3 cleaved/Pro caspase-3 expression (e) after 96 h of treatment with 25 μM CQ, 100 μM APE + 25 μM CQ, 100 μM N-8-Iper (N8) + 25 μM CQ, 25 μM N8 + 25 μM CQ in the U251 cell line and (f) after 72 h of treatment with 25 μM CQ, 100 μM N8 + 25 μM CQ, 3 μM N8 + 25 μM CQ in GB7 cells. Graphs show the densitometric analysis of the bands of Western blot analysis for Pro Caspase-3 normalized with the bands of actin protein. Actin was used as the internal reference protein. e, f) No statical differences have been observed between all experimental conditions (n.s. p > 0.05). Data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post-test (untreated cells (Ctrl) vs M2 agonists treated cells; ** p <0.01, * p <0.05).
Figure 7.
Representative Western blot analysis of Caspase-9 cleaved/Pro caspase-9 expression after 72 h of treatment (a) and after 96 h of treatment (b) with 25 μM CQ, 100 μM APE + 25 μM CQ, 100 μM N-8-Iper (N8) + 25 μM CQ, 25 μM N8 + 25 μM CQ in the U251 cell line. (c) Representative Western blot analysis of Caspase-9 cleaved/Pro caspase-9 expression after 72 h of treatment with 25 μM CQ, 100 μM N8 + 25 μM CQ, 3 μM N8 + 25 μM CQ in GB7 cells. Graphs show the densitometric analysis of the bands of Western blot analysis for Caspase-9 cleaved normalized with the bands of Pro Caspase-9 protein. Actin was used as the internal reference protein. (d) Representative Western blot analysis of Caspase-3 cleaved/Pro caspase-3 expression after 72 h of treatment. The graph shows the densitometric analysis of the bands of western blot analysis for Caspase-3 cleaved normalized with the bands of Pro Caspase-3 protein. Representative western blot analysis of Caspase-3 cleaved/Pro caspase-3 expression (e) after 96 h of treatment with 25 μM CQ, 100 μM APE + 25 μM CQ, 100 μM N-8-Iper (N8) + 25 μM CQ, 25 μM N8 + 25 μM CQ in the U251 cell line and (f) after 72 h of treatment with 25 μM CQ, 100 μM N8 + 25 μM CQ, 3 μM N8 + 25 μM CQ in GB7 cells. Graphs show the densitometric analysis of the bands of Western blot analysis for Pro Caspase-3 normalized with the bands of actin protein. Actin was used as the internal reference protein. e, f) No statical differences have been observed between all experimental conditions (n.s. p > 0.05). Data are the average (mean ± SEM) of three independent experiments. ANOVA test was used followed by Dunnett’s post-test (untreated cells (Ctrl) vs M2 agonists treated cells; ** p <0.01, * p <0.05).
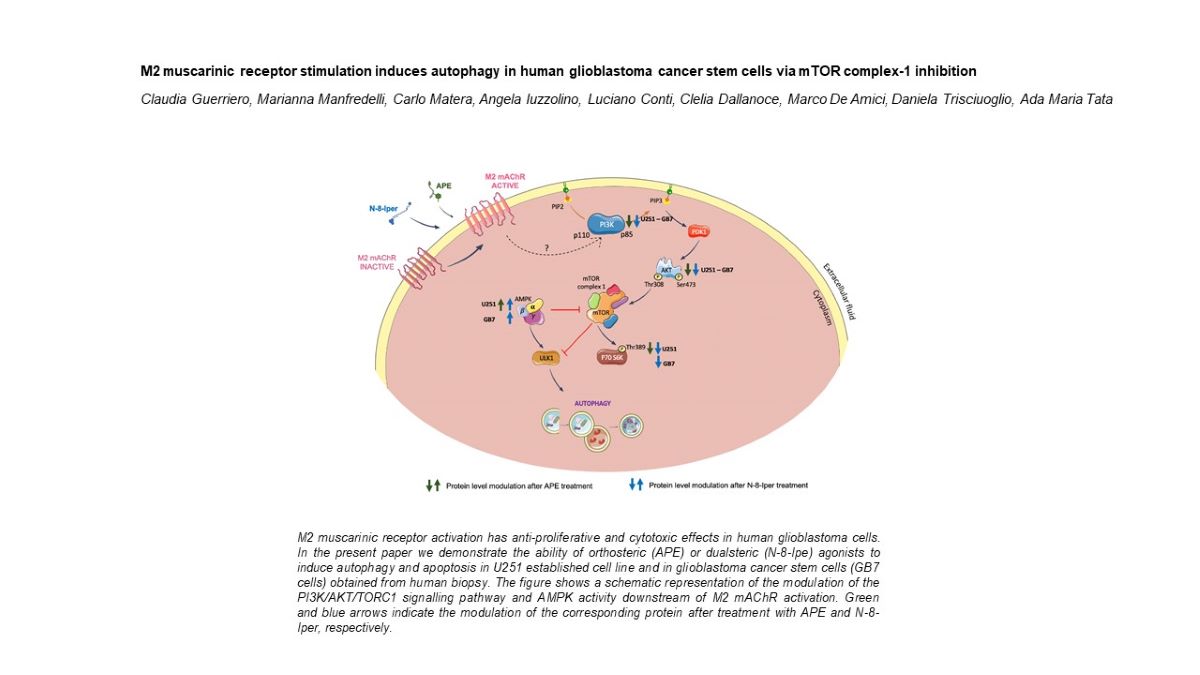
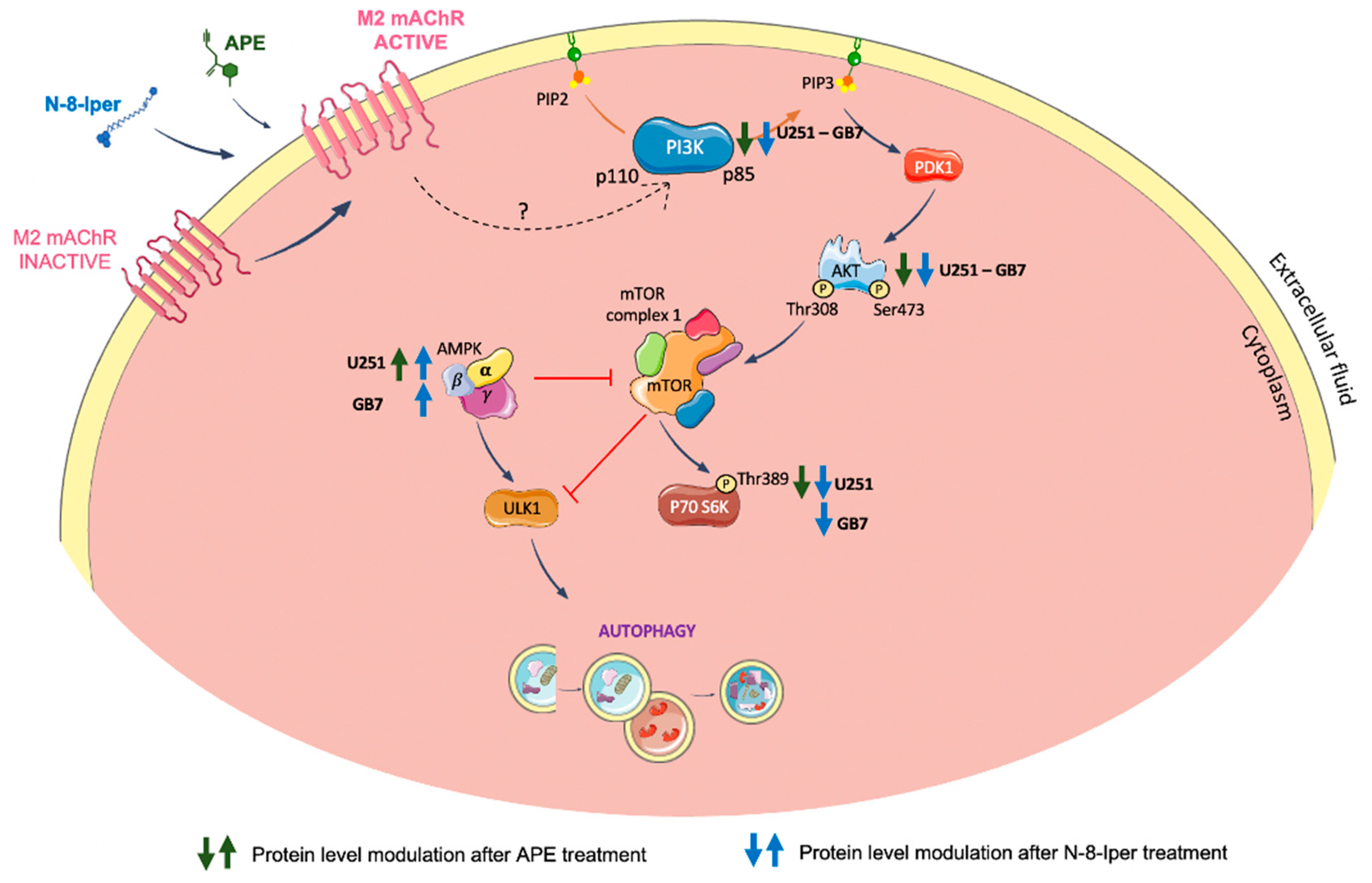
Figure 8.
Schematic representation of the modulation of PI3K/AKT/TORC1 signaling pathway and AMPK activity downstream of M2 mAChR activation by APE or N-8-Iper in U251 and GB7 cell lines. Green and blue arrows indicate the modulation of the corresponding protein after treatment with APE and N-8-Iper, respectively.
Figure 8.
Schematic representation of the modulation of PI3K/AKT/TORC1 signaling pathway and AMPK activity downstream of M2 mAChR activation by APE or N-8-Iper in U251 and GB7 cell lines. Green and blue arrows indicate the modulation of the corresponding protein after treatment with APE and N-8-Iper, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



