Submitted:
27 October 2023
Posted:
27 October 2023
You are already at the latest version
Abstract
African indigenous herbs and medicinal plants are extensively reported for their protective and therapeutic potential against Diabetes. However, most of the constituent phytochemicals that may account for the therapeutic activity are largely uninvestigated. Exploring novel phytochemical–target interactions in silico could help to provide useful insights into the mechanism underpinning the biological activities of these plants and their constituent phytochemicals. The aim of this study was to explore the interactions of terpene struc-tures previously reported from selected African antidiabetic with two emerging drug targets in diabetes. Structure-based virtual screening was used to screen 107 terpene structures against DPP-4 and PTP1B enzymes. The MD simulation, MM-GBSA free energy calculation and DFT were used to clarify the inter-actions of the in silico hits with the target enzymes. Molecular docking, post-docking Prime MM-GBSA analysis revealed the top terpenes with high binding affinity with the active site regions of DPP-4 and PTP1B. Ensemble docking revealed three triterpenes viz: cucurbitacin B (T1), 6-Oxoisoiguesterin (T4) and 20-Epi-isoiguesterinol (T2) as in silico hits which exhibit strong interaction potential with critical residues that define the catalytic triad (Ser630 and His740); oxyanion cavity (Ser631); hydrophobic S1 pocket (Tyr662) and the charged S2 pocket (Arg125) in the active site region of DPP-4. The interactions of these compounds with DPP-4 exhibited structural stability and conformational flexibility during 100 ns full atomistic MD simulation as indicated by the structure dynamics parameters including RMSD, RoG, SASA and hydrogen bond number. The post-MD MM/GBSA calculations further revealed the stability of the triterpene-DPP-4 complexes. Furthermore, Frontier molecular orbital calculations showed that, the triterpenes possess high interaction potential with the enzyme. The triterpenes showed desirable ADMET properties and drug-likeness. Therefore, cucurbitacin B, 6-Oxoisoiguesterin and 20-Epi-isoiguesterinol are recommended for experimental validation.
Keywords:
diabetes
; terpenes
; docking
; MD simulation
; MM-GBSA
; DFT
1. Introduction
Diabetes mellitus (DM) ranks among the most challenging public health burdens in the world as it affects about 463 million [1] and may increase to 552 million by the year 2030 and 700 million by 2045 as projected by the International Diabetes Federation (IDF) [1,2,3,4]. It is generally characterized by hyperglycemia and impaired glucose homeostasis resulting from defects in insulin secretion, insulin action, or both [5,6]. Type I diabetes mellitus (T1DM) is implicated in about 10% of the diabetic population while Type II diabetes mellitus (T2DM) contributes approximately 90% of diabetes-related cases [7]. While T1DM is often managed by insulin treatments and lifestyle modifications [8], an ideal treatment for T2DM is still elusive as the pathogenesis is highly complicated. Thus, many patients are still unable to meet glycemic goals [9,10]. Therefore, the persistent unaddressed needs of most patients remain an important factor driving the quest for better prevention and treatment options of diabetes mellitus.
Several promising therapeutic agents, which target various players in diabetes are promising oral agents for T2DM patients [11,12]. These include the starch blockers, insulin secretagogues, insulin mimickers and insulin sensitizers [12,13,14,15]. Acarbose, sulfonylureas, meglitinides, thiazolidinediones, biguanides aimed at blunting post-prandial glucose rise, suppressing hepatic glucose output, stimulating insulin release and increasing peripheral glucose utilization respectively are already approved for clinical use [1,16], but not without various adverse effects [1,17]. Beyond alpha-glucosidase, alpha-amylase and other starch blockers widely exploited for antidiabetic drug development [18], dipeptidyl peptidase-4 (DPP-4) enzyme has been recommended as target for antidiabetic agents as it also acts in carbohydrate metabolism by delaying gastric emptying, increasing insulin secretion, and reducing glucagon secretion [12,19]. The DPP-4 inhibitors stimulate the breakdown of endogenous incretin hormones and promote pancreatic glucose-dependent insulin secretion. The enzyme is a serine peptidase which act by rapidly degrading the incretin hormones, such as glucagon-like peptide 1 (GLP 1) that play an important role in blood glucose regulation, causing short life span of the enzyme. Therapeutic strategy that involves inhibiting DPP-4 would help to maintain the endogenous level of GLP 1 and increased β cell mass. Thus DPP-4 inhibitors are capable of enhancing glucose-dependent insulin secretion, slowing down gastric emptying, and reducing postprandial glucagon and of food intake [20,21,22,23]. Metformin is the first-choice medication which is most frequently recommended for treating T2D. This drug act by improving glucose metabolism through both AMP-activated kinase (AMPK) activation and enhanced GLP-1 release [24]. Thus, it improves insulin sensitivity of relevant tissues [25,26]. However, metformin like other antidiabetic drugs is often accompanied by moderate to severe side effects [27]. Another emerging target for enhancing insulin sensitivity in various cells is the Protein tyrosine phosphatase 1B (PTP1B). By improving the sensitivity of the insulin receptor, PTP1B inhibitors have the potential to cure insulin resistance-associated ailments [28,29]. The PTP1B acts by negatively regulating insulin receptor signaling pathway in cells [30]. Agents that inhibit this enzyme can activate phosphorylation of several insulin receptor kinase substrates which in turn stimulate the phosphoinositide-3-kinase as well as subsequent protein kinase B expression. This further promotes translocation of glucose transporter into the cell membrane from intracellular vesicles; and thereby promote uptake of glucose molecules by the cells for energy generation. Due to the major roles played by insulin disturbance in diabetes, insulin signaling is highly recommended for targeting diabetes and explored for developing new antidiabetic agents [10].
African flora possesses a very rich biodiversity of herbs, spices and medicinal which is widely exploited in traditional, alternative and complementary medicine for treating several diseases which include diabetes mellitus (DM) [13,31]. Most traditional healers in Africa who detain the ancestral heritage of these medicinal plants are illiterate with their ethnopharmacological knowledge transmitted verbally from generation to generation which is at risk of disappearing. Thus, the World Health Organization (WHO) has recommended scientific studies to document the folk knowledge and validate the acclaimed therapeutic potential of these plants from the perspective of developing improved medications (WHO, 2013). In this direction, the literature is replete with scientific reports on the anti-diabetic activity of African herbs, spices and medicinal as previously revealed by excellent review articles [31,32,33,34]. Most reported studies mainly focused on plant extracts and/or fractions, with only few having got scientific validation. Several ethnopharmacologically important antidiabetic herbs in Africa have shown potent activity against DPP-4 [31]. The extracts of Antidesma madagascariense Lam, whose leaf and stem bark decoctions are used in folk medicine in Madagascar for the management of DM, showed inhibitory activity against DPP-4 enzyme with an IC50 value of 79.2 mg/ml [35,36]. This inhibition indicated the ability of the extract to improve insulin sensitivity. In the same vein, several bioactive constituents of African indigenous herbs have been screened through several in vitro and in vivo studies for activity against PTP 1B. Ethyl acetate fraction and isolated pimarane diterpenes from Icacina oliviformis, a popular food spice native to the regions of West and Central Africa, have been reported to inhibit PTP1B [31,37,38]. Mohammed and Tajuddeen [31] revealed that the widely reported African antidiabetic plants contain several classes of compounds which include terpenes, alkaloids, flavonoids, and carotenoids. Owing to their wealth of chemical structures, such antidiabetic plants are considered as an inventory of bioactive compounds which might be useful as a basis for drug discovery and development in combating diabetes. Terpene structures, which belong to an important class of natural products have been extensively reported for antidiabetic activity and some of them are already under various stages of pre-clinical and clinical evaluation geared towards developing antidiabetic agents [10,14]. The antidiabetic potential of plant-derived terpenes could be attributed to varying mechanisms of action which include: increasing insulin sensitivity [14,39], inhibiting pancreatic amylase and glucosidase enzymes [40,41], reducing oxidative stress [42], and inhibiting the development of diabetic complications [43,44]. Several studies have revealed that, terpene-rich bioactive extracts of several African medicinal plants have shown antidiabetic activity by targeting key proteins associated with type II diabetes in vitro and in vivo as reviewed by Panigrahy, Bhatt [14]. Several terpene-rich plant extracts and terpene isolates have been widely reported to target DPP-4 [45], PTP1B [46,47] and other targets [14] in diabetes in vitro and in vivo.
Exploring novel phytochemical–target interactions in silico could help to provide useful insights into the mechanism underpinning the biological activities of medicinal plants and their constituent phytochemicals. It also plays an important role in compound screening and drug discovery. Drug-target interactions (DTI) characterize the binding of drug compounds to specific targets. Binding affinity of drugs refers to the strength of DTIs; and thereby greatly influences the drug's efficacy and therapeutic potential. Molecular modeling tools, which provide rapid, economically feasible and environmentally sound techniques for probing DTIs are valuable for screening un-investigated phytochemicals and predicting their interactions with molecular targets using integrated chemical informatics, bioinformatics and systems biology approaches. Employing these tools, the three-dimensional visualization of the electronic and steric molecular properties of the interaction between bioactive compounds and drug targets could be computed. The ultimate goal of such computational modeling in natural product research is to predict, in advance of any targeted laboratory testing, novel biologically active compounds. The modeling methodologies often employ the classical molecular mechanics (MM) and quantum mechanics (QM) computations [48]. Integrated molecular mechanics computations and quantum mechanics computations could provide an accurate and efficient theoretical description of a biomolecular system. Earlier studies have revealed the potential of in silico methods for predicting DPP-4 inhibitors [49,50,51] and PTP1B inhibitors [52,53] from natural sources. Terpenes, a class of compounds with compact molecular structure and less symmetry are known to bind multifarious drug targets to exhibit therapeutic potential in human diseases. Therefore, investigating terpene-target interactions hold great promise in antidiabetic drug discovery [14]. In spite of the reported antidiabetic activity of various African herbs and the therapeutic roles of terpene structure, many terpenes from African herbs remain uninvestigated. The aim of this study was to explore the interactions of terpene structures from African antidiabetic plants with two emerging drug targets in diabetes.
2. Results
2.1. Binding affinity and interactions of terpenes with DPP-4 enzyme
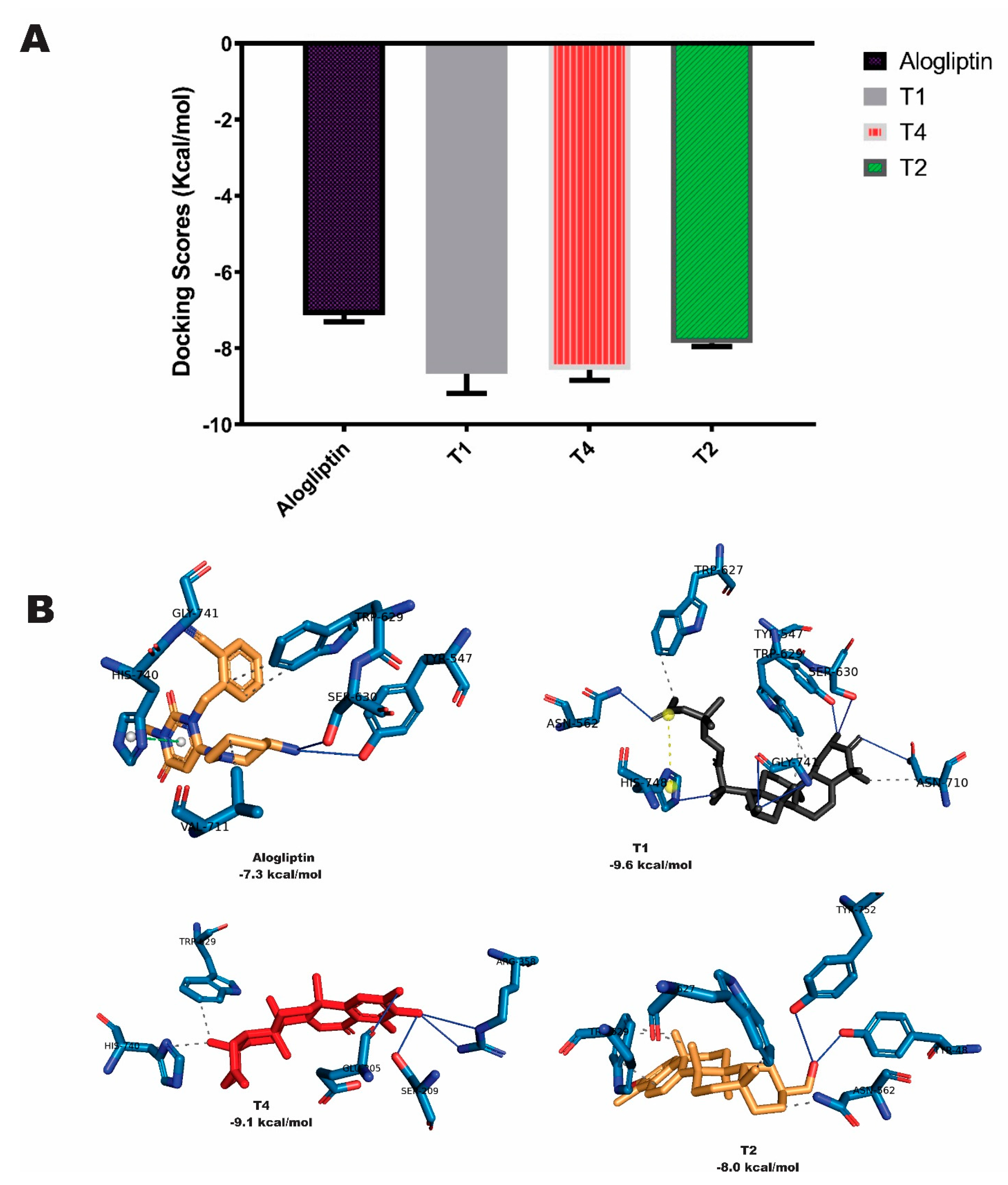
The initial screening based on the Larmackian Genetic Algorithm docking protocol revealed 37 terpene structures with docking scores (-7.7 to -10.0 Kcal/mol) lower or comparable to that of the co-crystalized alogliptin (-7.7 Kcal/mol) (Table S1). Ranking based on the docking scores and screening for favourable interactions with the target enzyme revealed the top 7 compounds (Table 1), most of which belong to the triterpenes sub-class of terpenes.
The topmost 7 terpene-DPP-4 complexes were subjected to post-docking MM-GBSA computations. While MM-GBSA (ΔGbind) of the reference alogliptin was -37.02Kcal/mol, the MM-GBSA (ΔGbind) of the top-docked terpene structure ranged from -47.80 kcal/mol (Cucurbitacin B) to -19.72 kcal/mol (1-Deacetylkhivorin) as shown in Table 1. It is worthy of note that, Cucurbitacin B with the lowest binding energy with DPP-4 also exhibited the lowest MM-GBSA free binding energy to the enzyme. While van der Waals, coulombic and non-polar solvation energies made the most notable free energy contributions to Cucurbitacin B binding to DPP-4, van der Waals and non-polar solvation made important contributions to binding of several other terpenes (Table S2). The terpene structures demonstrated high levels of binding affinity, suggesting exceptional potential for sustained interactions with the DPP-4 enzyme. Interaction analysis of the selected terpenes with the binding pocket of DPP-4 revealed that, cucurbitacin B, 6-Oxoisoiguesterin, and 20-Epi-isoiguesterinol had binding poses with DPP-4 similar to that of the co-crystalised alogliptin. These compounds also featured strong and favourable interactions with the critical amino acid residues in the active site of the enzyme in a similar manner to the native compound as shown in Table 2. Table 2 shows that, the hit triterpenes and alogliptin had strong interactions with various residues involved in the catalytic triad, oxyanion cavity, hydrophobic S1 pocket and charged S2 pocket in the binding pocket of DPP-4. The 3-D representations of the interactions is depicted in Figure 1.
2.2. Binding affinity and interactions of terpenes with PTP1B enzyme
Molecular docking, MM-GBSA post docking analysis and molecular interactions were also employed to screen the terpene structures for potential inhibitors of PTP1B enzyme. Although all the docked compounds had docking scores and MM-GBSA binding energy higher than those of the reference isothiazolidinone, the topmost compounds had favourable interactions with the binding pocket the protein. The Molecular docking scores and prime MM-GBSA scores of the top terpenes with PTP1B are shown in Table 3. Favourable interactions were also observed upon visualization of the top terpene-PTP1B complexes as shown in Table 4 and Figure 2.
2.3. Ensemble docking analysis of selected triterpenes against DPP-4
Based on the results from molecular docking and post docking MM-GBSA which showed higher affinity of top-docked terpenes with DPP-4, the top docked terpenes were further subjected to ensemble docking analysis in order to provide more flexibility in the docking experiment. The ensemble docking analysis revealed that, the average docking scores of the hit triterpenes were lower than that of the reference alogliptin.
Figure 3.
(A) Mean scores of docking of top terpenes with structure ensemble of DPP4 (n=4). (B) Interactions of top terpenes with a representative conformer of DPP4.
Figure 3.
(A) Mean scores of docking of top terpenes with structure ensemble of DPP4 (n=4). (B) Interactions of top terpenes with a representative conformer of DPP4.
2.4. Molecular dynamics of terpene-DPP-4 interactions
The dynamics simulation was employed to model the interactions of the top-docking terpenes with DPP-4 in a dynamic environment. The thermodynamic parameters that indicate stability of the DPP-4 enzyme back-bone C-α atoms in complex with T1, T4 and T2 were calculated and plotted as shown in Figure 4. The RMSD, a measure of the difference between two sets of coordinates, was computed and depicted in Figure 4a.
The RMSD plot (Figure 4a) revealed that, the DPP-4 backbone, T1-DPP-4, T4-DPP-4, T2-DPP-4 and S1-DPP-4 molecular systems showed a slight rising RMSD trend in initial 0.6 ns, 0.8 ns, 0.1 ns, 0.6 ns and 1.1 ns and then stabilized at mean values of 2.84 Å, 2.85 Å, 3.02 Å, 3.17 Å and 2.50 Å respectively. These values indicated that the terpene–DPP-4 complexes reached a stable conformational trend over 100 ns of the simulation with few conformational transitions as the RMSD values were maintained within 2 Å. Except for the major fluctuations observed with the trajectory of DPP-4-cucurbitacin B complex, all the complexes exhibited relatively stable atomic trajectories displaying minor and consistent fluctuations. Figure 4b shows that the average RoG values for DPP-4 backbone, T1-DPP-4, T4-DPP-4, T2-DPP-4 and S1-DPP-4 biomolecular systems are 26.00 Å, 26.36 Å, 26.25 Å, 26.02 Å and 26.57 Å respectively. The results also revealed that the RoG values were maintained within 2 Å. The result shown in Figure 4c revealed that, the SASA values of DPP-4 backbone, T1-DPP-4, T4-DPP-4, T2-DPP-4 and S1-DPP-4 biomolecular systems are 29052 Å2, 29797 Å, 29669 Å, 29115 Å2 and 30015 Å2 respectively. To provide more insight into the stability of the complexes, the number of H bonds found in the ligand-protein complexes was computed as depicted in Figure 4d. The results revealed a steady trend for the five biomolecular systems which span around 173, 180, 174, 172 and 184 bonds for the DPP-4 backbone, T1-DPP-4, T4-DPP-4, T2-DPP-4 and S1-DPP-4 biomolecular systems respectively. The plot demonstrates large variations in the amino acid residues between 220 and 260 as well as those around 680, with the RMSF reaching notable peaks. Other residues, in addition to these also feature some notable peaks.
2.5. Free energy simulation of DPP-4-terpene complexes
To further validate the docking scores of the ligands to the protein, the trajectories obtained from the MD simulations of the terpene-DPP-4 complexes were subjected to free energy simulation using MM-GBSA. Table 5 shows a number of solvation and molecular mechanics parameters and their energy contributions.
The results show a number of solvation and molecular mechanics parameters and their energy contributions. Among the terpenoids, T1 (ΔGtotal = -11.85 kJ/mol) exhibited the strongest binding affinity as indicated by the lowest free binding energy as compared to other terpenes but with higher free binding energy than the reference agloliptin. The results also revealed that, the main components of the free energy of the top complexes are van der waals energy and electrostatic energy. Amino acids occurring around the binding pocket showed significant contributions to the overall binding affinity to the terpen-DPP-4 complexes as indicated by the MM-PBSA free energy decomposition analysis (Figure 5).
2.6. Frontier molecular orbitals of top triterpenes
To further investigate the behavior of the selected terpene structures as DPP-4 inhibitors, density functional theory computation was performed. Table 6 shows that, the hit compounds had close band gaps which were quite low.
The HOMO and LUMO orbital of the hit compounds are given in Figure 6. The regions of the compounds with red and blue color denote the positive and negative signal/phase of the molecular orbital respectively.
The orbital stands for the likelihood of discovering an electron. The colours red and blue signify the nodal character of the orbital. The HOMO orbital appears to be centralized on the phenyl rings of the hit compounds while the LUMO orbital were predominantly of the side chains of the aromatic ring structures.
2.7. Predicted physicochemical and ADMET properties of hit triterpenes
The drug-likeness characteristics of the top triterpenes were assessed to ascertain the suitability of the compounds as drug candidates. The drug-likeness characteristics of the compunds revealed desirable drug-like properties (Table 7). Using the Lipinski rule of five assessments, it was shown that all the hit compounds are in accordance to Lipinski rule.
3. Discussion
In this study, we used integrated in-silico methods to explore the molecular interactions of terpene structures previously reported from African antidiabetic plants with two emerging drug targets in diabetes. Such methods, which are valuable for sample space minimization and resource maximization, can provide rapid, economically feasible and environmentally sound techniques for probing the biological activities of uninvestigated phytochemicals by identifying their molecular targets; and thereby provide insights into the mechanism underpinning their activity. Besides, they enable more targeted experimental and preclinical evaluations. Herein, structure-based virtual screening was used to screen 107 terpene structures against the 3D-structures of DPP-4 and PTP1B enzymes. Molecular dynamics (MD) simulation, Molecular mechanics with generalized Born and surface area solvation (MM/GBSA) free energy calculation and density functional theory (DFT) were used to clarify the interactions of top the terpenes with the target enzymes. The Structure-based virtual screening helped to identify top terpenes with higher binding affinity with the target enzymes than the reference drugs. Post-docking MMGBSA analysis of the top terpenes revealed good binding affinity scores. It is worthy of note that, Cucurbitacin B (T1) with the strongest binding affinity with DPP-4 as indicated by the lowest docking score also exhibited the lowest post-docking MM-GBSA free binding energy score to the enzyme. Molecular docking, MM-GBSA post docking analysis and molecular interactions also revealed top terpenes as potential inhibitors of PTP1B enzyme.
The interactions of the top terpenes were analyzed alongside the reference drugs. The results showed that, the interaction of the co-crystalized compound (alogliptin) with the binding site of DPP-4 involve several types of bonds including Conventional Hydrogen Bonds, Carbon Hydrogen Bonds, Pi-Cation Electrostatic interactions, Pi-Donor Hydrogen Bond, Pi-Sigma Hydrophobic interactions, Pi-Pi Stacked Hydrophobic interactions and Pi-Alkyl Hydrophobic interactions. Key interactions of the co-crystalized compound involve Carbon Hydrogen bond, Pi-Cation electrostatic interactions and Pi-Alkyl hydrophobic interaction with HIS740 as well as Pi-Donor hydrogen bond with SER630. The DPP-4 enzyme, which is catalytically active as dimers in both the soluble and cell-surface form, has 766 amino acids, containing a C-terminal hydrolase domain as well as the N-terminal propeller domain and a C-terminal hydrolase domain. In addition to the catalytic triad comprising Ser630, Asp708 and His740, DPP-4 contains Tyr47 and Ser631 associated with the oxyanion hole; Tyr631, Val656, Trp659, Tyr662, Tyr666, and Val711 functioning in the hydrophobic S1 pocket; and Arg125, Glu205, Glu206, Phe357, Ser209 and Arg358 in the and a charged S2 pocket [54,55]. Interaction analysis of the selected terpene structures with the binding pocket of DPP-4 revealed that, cucurbitacin B, 6-Oxoisoiguesterin (T4), and 20-Epi-isoiguesterinol (T2) had binding poses with DPP-4 similar to that of the co-crystalised alogliptin. Cucurbitacin B, a tetracyclic triterpene containing the cucurbitane nucleus skeleton, namely, 19-(10-9beta)-abeo-10alpha-lanost-5-ene with a several oxygen substitutions in different positions. It was observed that, the oxygen substitutions in the structure played key roles in the interaction of the compound with active site and hydrophobic S1 pocket in the binding site region of DPP-4. There was strong interaction with the catalytic triad residues as important carbonyl oxygen in the compound structure received a strong conventional hydrogen bond from the side chains of SER630 and another from HIS740 side chain. Also, the ether oxygen received a strong conventional hydrogen bond from the side chain of TYR631 in the hydrophobic S1 pocket. In this pocket, the side chain of TYR666 conducted a Pi-Sigma Hydrophobic interaction with a methyl group and Pi-Alky hydrophobic interaction with another methyl group in the compound. The side chain of TYR662 also conducted a Pi-Alkyl hydrophobic interaction with a methyl group in the compound. Cucurbitacin B, was derived from Cogniauxia podolaena [56], a flowering perennial plant that belongs to the family Cucurbitaceae. It grows primarily in the wet tropical biome. The genus Cogniauxia is native to Angola, Cabinda, Cameroon, Central African Republic, Congo, Equatorial Guinea, Gabon, Guinea, Zaïre. The species in this genus are known to possess antidiabetic activity [57]. Four bisnorterpenes derived from Ekebergia capensis [58] viz: 20-Epi-isoiguesterinol, isoiguesterin, 6-oxoisoiguesterin and isoiguesterinol were also observed to show stronger binding affinity than alogliptin as indicated by lower binding energies. The Ekebergia capensis is a flowering plant which belong the family Meliaceae is fondly called Cape ash and its range extends from the South African Eastern Cape to Sudan and Ethiopia. Its antidiabetic potential has been exploited in Eastern Cape KwaZulu-Natal, Limpopo and Mpumalanga [59]. The herbal formulations comprising infusion prepared from the leaves of this plant and taken orally has shown antidiabetic activity streptozotocin-induced diabetic rats [59]. The 6-Oxoisoiguesterin (T4), one of the compounds from this plant interacted with the active site of DPP-4 through a carbon Hydrogen Bond and Pi-Alkyl Hydrophobic interaction with HIS740. As shown in Table 2 and Figure 1. The 20-Epi-isoiguesterinol (T3), another bisnortriterpene, interacted with the catalytic trial through a Pi-Alkyl Hydrophobic interaction with HIS740 as depicted in Table 2 and Figure 1. As extensively reviewed by Hamid, Alqahtani [60], tetracyclic triterpenoids, a class of triterpenoids found in a variety of medicinal plants are notable for treating diabetes and its complications. In a previous study, botulin, a triterpene derived from samples of the root bark of Euclea undulate widely exploited traditionally to treat diabetes and other ailments in Southern Africa, has been reported to show a reduction in level of blood sugar as compared to reference drug glibenclamide [61,62]. This study suggests the molecular mechanism underpinning the activity of such drugs. Also, earlier studies on the structure-activity relationship of terpenes have revealed the few roles of the molecular structure of terpenes and terpenoids in manifesting their antidiabetic activity. For instance, Chen, Lu [63] revealed that, sesquiterpenes are highly effective as antidiabetic agents which may result from their structurally compact configuration, low ramification, and low symmetry. Another author revealed that, the triterpenes with hydroxyl and carboxyl functional groups exhibit high antidiabetic activity which is mediated through various mechanisms [64]. In the review by Panigrahy, Bhatt [14], terpene structures containing the most hydrogen bonds and hydrophobic interactions exhibit enhanced anti-diabetic activity. Other compounds with high affinity for DPP-4 are also reported in this study (Table 1). These include several acyclic triterpenes derived from Glossocalyx brevipes (Siparunaceae) [65]. The native range of Glossocalyx is Southern Nigeria to Western Central Tropical Africa. Glossocalyx brevipes has been documented in the treatment of diabetes and its interconnected diseases [66]. Two important limonoids (1-Deacetylkhivorin and 7-Deacetylkhivorin) derived from Khaya grandifoliola (Meliaceae) [67] also showed low binding energy. Khaya grandifoliola, which is also known as African mahogany belong to the family Meliaceae and found abundantly in the Republic of Benin, Nigeria, the Democratic Republic of the Congo, Ivory Coast, Ghana, Guinea, Sudan, Togo, and Uganda. This plant has been utilized in polyherbal formulations for the treatment of T2DM [68]. Structure-based virtual screening of selected terpenes with PTP1b also revealed strong binding interactions. Although all the docked compounds had docking scores and MM-GBSA binding energy higher than those of the reference isothiazolidinone, the topmost compounds had favourable interactions with the binding pocket the protein. The Beilshmiedic acid derivatives viz: Tsangibeilin B and Cryptobeilic acid C , which were isolated from Beilschmiedia cryptocaryoides (Lauraceae) [69] had the lowest binding score. Several bisnorterpenes (isoiguesterin, 6-Oxoisoiguesterin, isoiguesterinol and 20-Epi-isoiguesterinol) derived from Ekebergia capensis [58] also had low binding scores. In addition, galanolactone, a labdane diterpenoid derived from Aframomum arundinaceum (Zingiberaceae) [70] exhibited low binding energy. Van der Waals, non-polar solvation, and Coulombs energy terms made most significant contributions to the binding affinity of the ligands.
Based on our results from molecular docking and post docking MM-GBSA which showed higher affinity of top-docked terpenes with DPP-4, the top docked terpenes were further subjected to ensemble docking analysis. Ensemble docking which involves the use of multiple molecular docking simulations was employed to improve the accuracy and reliability of predicting the top terpenes binding mode and affinity with DPP-4 enzyme. To achieve this, a DPP-4 ensemble was constructed by subjecting the experimental structure to a 100 ns MD simulation which generated three clusters. A representative structure was selected for each cluster and docked against the hit triterpenes (T1, T4, T2) along with the reference compound (alogliptin) using AutoDock Vina software [71,72].The result of the ensemble docking are shown in Figure 3. The study showed that, docking of top triterpenes with the active site of DPP-4 conformational representatives was consistent with the docking result obtained from docking with the single DPP-4 experimental structure. It is also worthy of note that, interaction analysis of the docked conformation with representative clusters revealed that the amino acid interaction were preserved and reproducible (Figure 5).
The top terpene-DPP-4 complexes were subjected to MD simulation alongside the reference alogliptin and unliganded enzyme to capture a more comprehensive picture of biomolecular systems considering the inherent flexibility and dynamics of proteins. The MD simulation is often integrated with docking simulation and other molecular modeling techniques to further validate virtual screening through static docking and assess the stability of drug-receptor complexes. Computation of various thermodynamic parameters from the MD simulation trajectories revealed the structural stability and conformation flexibility of the ligand-DPP-4 complexes alongside the unliganded protein as depicted in Figure 4. The RMSD values which indicate the overall structural stability of the DPP-4 enzyme back-bone C-α atoms in complex with alogliptin, T1, T4 and T2 are shown in Figure 4a. Except for the major fluctuations observed with the trajectory of DPP-4-cucurbitacin B complex, all the complexes exhibited relatively stable atomic trajectories displaying minor and consistent fluctuations. During MD simulation, low levels of RMSD which are characterized by such consistent fluctuations indicate equilibration and stability of the system. However, higher fluctuations indicate low stability. However, highly deviated structures after equilibration such as the observed RMSD of DPP-4-cucurbitacin B complex may imply major conformational transitions by DPP-4 to attain stable conformation of the enzyme with cucurbitacin B. In the context of molecular modeling and structural biology, RMSD is often used to compare the structures of two molecules, such as a crystal structure and a molecular dynamics simulation trajectory. To calculate RMSD, one first aligns the two sets of coordinates, typically by superimposing them using a least-squares fitting algorithm. Then, the distance between each corresponding pair of atoms in the two sets of coordinates is calculated, and the average of the squared distances is taken. Finally, the square root of the average squared distance is computed to obtain the RMSD value. The RMSD is a widely used metric in molecular modeling and structural biology, as it provides a quantitative measure of the similarity between two structures. A low RMSD value indicates a high degree of similarity, while a high RMSD value indicates a greater difference between the two structures. The radius of gyration calculation helps to assess how spread out the mass of the complexes is from its rotation axis. It is often employed in MD simulation to estimate the compactness of the bound structures. The RoG values of the apoprotein and the protein-ligand complexes were maintained within 2 Å indicating stability of the molecular system. Also the SASA, a measure of the total surface area of a molecule that is available for interaction with other molecules, such as enzymes or receptors indicate stability of the complexes (Figure 4c). It is calculated by measuring the area of the molecule that is exposed to solvent, such as water. The SASA is important because it can help predict the binding affinity of a molecule to its target. Generally, molecules with a larger SASA are more likely to bind strongly to their targets because they have more surface area available for interaction. Since the RoG values indicate compactness of the complex structures and those of SASA reflect the extent of the solvent accessible surface tendency of the protein structure. Taken together, the terpene-DPP-4 complexes with reduced RoG and SASA values reflect a low tendency of unfolding deviation from their initial structure. Hydrogen bond number existing in the molecular systems was calculated in order to provide more insight into the stability of the protein-ligand complexes. The study revealed that, DPP-4-ligand complexes exhibit a high tendency to maintain compact and well-folded biomolecular structure characterized by stable intermolecular bonds. The high number of H-bonds observed in the ligand-protein complexes (Figure 4d) further indicate well bounded ligand and compact structures. In this study, the values of RMSF for DPP-4 enzyme residues towards the triterpenes were calculated and shown in Figure 4e. The plot demonstrates large variations in the amino acid residues between 220 and 260 as well as those around 680, with the RMSF reaching notable peaks. Other residues, in addition to these also feature some notable peaks. This suggests that they have a strong affinity for interacting with terpenes and that the structures may be able to bind strongly to the protein at these points. Computation of the RMSF from the trajectory files helps to evaluate the crucial contributions that a protein's amino acid residues make towards achieving stable conformations for protein-ligand complexes. The RMSF analysis often reveals how a portion of the protein deviates from its mean structure upon interactions with a ligand. The RMSF plot displays the degree of fluctuation seen for each protein residue, providing insight into the degree of adaptability they have in a changing environment. As a result, motifs or individual amino acid residues that exhibit higher RMSF and flexibility have a stronger propensity of interactions with the ligand.
Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) is often employed in molecular modeling investigations to assess the binding free energy of a compounds with a receptor. It is often considered as a post-processing technique for analyzing trajectories obtained from an MD simulation run. Given the limitations of molecular docking simulation, which is mostly reliant on molecular mechanics calculations, the trajectories obtained from the MD simulations of the terpene-DPP-4 complexes were subjected to free energy simulation using MM-GBSA, which combines conventional MM with implicit solvation models. The main components of the free energy of the top complexes are van der waals energy and electrostatic energy. Amino acids occurring around the binding pocket showed significant contributions to the overall binding affinity to the terpen-DPP-4 complexes as indicated by the MM-PBSA free energy decomposition analysis (Figure 5).
Density functional theory is a quantum mechanical approach that calculates the electronic density of a system, which can then be used to calculate other properties such as total energy, electronic charge distribution, and molecular geometry. The essential chemical characteristics of the terpene structures were analysed using frontier molecular orbital analysis of the completely optimised structures as shown in Table 6. According to FMO theory, one of the key factors that influence the biological activities of drug-like compounds is the energy level of their HOMO and LUMO orbitals [73]. The HOMO energy indicates the compound's ability to donate electrons, whereas the LUMO energy quantifies the compound's capacity to accept electrons. All the triterpene structures displayed higher HOMO energy than the reference alogliptin, which is consistent with the molecular docking, ensemble docking and post-docking MM-GBSA. In simple term, a compound with small energy gap is more polarizable and often associated with high reactivity [74]. These suggest that the hit compounds have high tendency of participating in molecular reactions. In a previous study, it was observed that clinically approved therapeutic showed good correlation with the experimental data having electron affinity value ranging between −1.5 and 2.0 eV [75]. The fact that all our hit compounds EA value lies within this range suggest a good druglikeness property. In addition, it was observed that the highest electrophilicity index was observed in cucurbitacin B, oxo-isoguesterin and Epi-isoguestrin. The electrophilicity index depicts information about electron system structure, stability, bonding, reactivity, and dynamics at ground and excited states [76]. Ganesan, Raja [76] further explained that, compounds with a high electrophilicity index are more likely to interact with biomolecules, suggesting a preferential reactivity of these compounds. Overall, all the hit compounds show promising reactivity indices, which emphasizes significant potential as lead candidates.
The drug-likeness characteristics of the top triterpenes were assessed to ascertain the suitability of the compounds as drug candidates. These compounds possess desirable properties (Table 7), which are important to the ultimate nutraceuticals and drug development. Using the Lipinski rule of five assessments, it was shown that all the hit compounds are in accordance to Lipinski rule. Based on Lipinski rule, orally active drugs have no more than one violation of the set criteria viz: less than 5 hydrogen bond donors, less than 10 hydrogen bond acceptors, molecular mass below 500 daltons and octanol-water partition coefficient less than 5 [77,78]. The fact that the compounds did not exceed one of the Lipinski violations suggest they can be readily absorbed and in therapeutic doses. The lead-likeness property was further corroborated by the Veber, Egan and Muegge rule. Further emphasizing the druglikeness property of the drugs is the result from pain (Pan-assay interference compounds) alert prediction. While the previous rules focused on the oral bioavailability of the drug, pain alert helps to identify the tendency of the compounds to participate in non-specific binding to multiple targets. The PAINS is one of the major results for false positive in drug development. Based on these prediction, it was observed that most of the top compounds do not contain scaffolds capable of non-specific binding as such the high binding affinity obtained is less likely due to false positive. Furthermore, most of hits were not predicted as p-glycoprotein substrate. The Pgp is responsible for binding foreign particles and facilitating their rapid elimination from the body. Since most of the compounds are not pgp substrate, the implication is the compounds are likely to have long circulating time thereby achieving maximum therapeucity before clearance from the body.
4. Materials and Methods
4.1. Preparation of protein structures
The 3-dimensional structure of human DPP-4 (PDB ID: 3G0B) which was co-crystallized with T22800 (alogliptin) and that of the human PTP1B (PDB ID: 2CM7), which was co-crystalized with isothiazolidinone, were retrieved from the Protein Data Bank (https://www.rcsb.org). From these protein structures, the co-crystalized compound and water molecules were deleted. Each hydrogen atom was integrated into protein the structures using Autodock Vina 4.2 program.
4.2. Ligand preparation
An in-house library of 107 terpenes previously reported from African medicinal plants was created after a thorough search of the literature. The structures of the terpenes from the in-house library and the protein native ligands were obtained in the structural data format (SDF) from the PubChem database at www.pubchem.ncbi.nlm.nih.gov. Discovery studio was used to convert SDF structures of ligands and the standard drugs into PDB chemical format. The ChemDraw version 19 was used to prepare unavailable structures, which were then converted to mol2 chemical format and then to PDB chemical format. As previously demonstrated, the non-polar hydrogen molecules were combined with the carbon atoms, and the polar Gasteiger-type hydrogen charges were assigned to the atoms in the chemical structure [79]. Internal degrees of freedom and torsions were both set to 0. The structures were then transformed using AutoDock Tools into the dockable PDBQT format.
4.3. Molecular docking calculations
The structures of the terpenes were imported through OpenBabel [80] into AutoDock Vina incorporated in PyRx 0.8 [71]. Energy minimization of the ligands was performed using Universal force field (UFF) using conjugate gradient descent as the optimization algorithm. The structures of the terpenes were then docked with the binding pockets of both DPP-4 and PTP1B. The active site regions of these enzymes were defined and selected using grid boxes with parameters depicted in table 1. The virtual screening was performed keeping all docking parameters and exhaustiveness as default. After the scoring, the docked poses were retrieved and the ligand-protein interactions visualized using Discovery Studio Visualizer version 16.

Table 8.
Grid box parameters for defining active site regions of DPP-4 and PTP1B.
| Dimensions | DPP-4 (Å) |
PTP1B (Å) |
|---|---|---|
| center_x | 34.88 | 14.79 |
| center_y | 29.17 | -3.40 |
| center_z | 16.00 | 1.69 |
| Size x | 21.92 | 16.50 |
| Size y | 17.77 | 19.72 |
| Size z | 22.83 | 27.64 |
4.4. Prime MM-GBSA post docking calculations
Post docking free energy simulation through MM-GBSA was applied to the top ligand-protein complexes in order validate the docking score. To achieve this, the MMGBSA panel in Maestro was employed to compute the binding affinity of the top terpenes with the target enzymes as demonstrated in a previous study [81]. The MMGSA helped to estimate the difference in free binding energy between the terpene structures and the enzymes in both the non-bound state and complexed form after the process of energy minimization. The OPLS3forcefield parameters were used for the MM-GBSA, while VSGB was employed to model the continuum solvent. The computations were done with all other options set as default. The binding free energy computations were undertaken based on the following equations
Where
E is the minimized energies for molecular systems
represents the non-polar contribution to the energy of solvation resulting from the surface area, while is the surface energy of the systems.
4.5. Ensemble docking analysis
Three different coordinates of DPP-4 resulting from clusterization of the MD simulation trajectories were docked against the selected top three terpenes (T1, T4, T2) along with the reference compound (alogliptin) using AutoDock Vina software [71,72]. The selected terpene structures were docked following the docking protocol. The docking complexes were analyzed using Protein-Ligand Interaction Profiler (PLIP) web server and visualized using PyMOL 2.4 software [82].
4.6. Molecular dynamics simulation
Initially, the human DPP-4 (PDBID: 3G0B) protein was subjected to a 100 ns full atomistic MD simulation in order to generate an ensemble of the protein for performing the ensemble docking analysis. Then, the apo enzyme, and the DPP-4-terpene complexes obtained from docking calculations were subjected to MD simulation. These were performed using GROMACS 2019.2 and GROMOS96 43a1 selected as forcefield on the WebGRO [83,84,85]. The preliminary topology files of the terpene structures were prepared using PRODRG webserver (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg) [86]. The apo DPP-4 and the DPP-4-terpene complexes were solvated within a cubic box of the transferable intermolecular potential using a four-point (TIP4P) water model. The periodic boundary conditions were applied and a physiological condition of 0.154 M concentration set by neutralized NaCl ions. The minimization of the apo DPP-4 system and the complex systems was carried out in 10000 steps employing the steepest descent algorithm and applying the NVT ensemble for 0.3 nanosecond. This was followed by 0.3 ns of equilibration under NPT condition. The system temperature was set up and maintained at 310 K with the velocity rescale. The system pressure was also set and maintained at 1 atm employing the Parrinello-Rahmanbarostat. The integrator used for the computation was the Leap-frog integrator with a time step of 2 femtosecond. During the 100 ns full atomistic MD simulation production run, 0.1 ns snapshot was saved with a total of 1000 frame for each biomolecular system. Various thermodynamic parameters viz: RMSD, RMSF, SASA, RoG, and hydrogen bond number were computed from the trajectory files obtained from the dynamic simulation runs using VMD TK console scripts [87].
4.7. Clustering analysis
The trajectory files obtained from the initial MD simulation of the DPP-4 protein was clustered to generate the ensemble of the protein which were used for the ensemble docking simulation. Also, the trajectory files obtained from the simulation runs of DPP-4-terpene complex systems were clustered. The clustering analysis was performed employing the elbow method using TTClust V 4.9.0. Following cluster generation, a representative structure of each cluster of the complexes was chosen for interaction analysis with the aid of the Protein-Ligand Interaction profiler (PLIP) [88].
4.8. Binding free energy calculation using MM-GBSA
The Gmx_MMPBSA algorithm was used to calculate the binding free energy for each complex using Molecular Mechanics Generalized Born Surface Area (MM-GBSA). The salt concentration and the solvation method were 0.154 and 5, respectively. The internal and external dielectric constants were set to 1.0 and 78.5, respectively with other options as default. The decomposition of free energy was calculated to find which amino acids within 10 Å contribute most to the binding [89,90]. The MM-GBSA approach utilized is depicted in equation (v):
The ‘< >’ indicates the means of the free energies of complex, protein, and terpene structure over the frames used for the calculations. Several frames (200) were used to calculate the free energy. Various energy terms were computed using Equations (vi) to (x) as follows:
Where:
∆H represents the enthalpy which is computed from gas-phase energy (Egas) and solvation-free energy (Esol). The T∆S term represents the entropy contributions to the total binding affinity which was not included because of the aim of comparing the relative binding free energies. Egas is comprised the electrostatic and van der Waals energy terms; Eele, EvdW, respectively. Esol was computed from the polar solvation energy (EGB) and nonpolar solvation energy (ESA) which was estimated from the solvent accessible surface area [91,92].
4.9. Density functional theory
On the basis of the electron density associated with the ligands, DFT asserts that ground state energy and other molecular attributes are determined only by this density. As a basis set for a DFT calculation using the B3LYP functional and 6-31G** as a single point energy calculation, the Jaguar panel of Maestro was employed in this work. A variety of molecular reactivity indicators, including electrophilicity, hardness, softness, electron affinity, ionization potential, and molecular electrostatic potential, were assessed. The following mathematical equations were used to perform the computations as discussed previously [93].
𝐸𝑙𝑒𝑐𝑡𝑟𝑜𝑛 𝑎𝑓𝑓𝑖𝑛i𝑡𝑦 (𝐸𝐴) ≈ − (LUMO)
𝐼𝑜𝑛𝑖𝑧𝑎𝑡𝑖𝑜𝑛 𝑝𝑜𝑡𝑒𝑛𝑡𝑖𝑎𝑙 (𝐼𝑃) ≈ − (HOMO)
Chemical potential (µ) = - (I + A)/2
Electrophilicity (ω) = µ2/2η
4.10. Physicochemical admetSAR analysis
The hit triterpene structures were passed through predictive physicochemical analysis. The drug-like properties were obtained from the SwissADME web werver platform (http://www.swissadme.ch/index.php) [94]. The ADMETox prediction was performed on the Protox-II webserver (https://tox-new.charite.de/protox_II/index.php) [95]. To perform these analyses, The SDF file format as well well as the canonical SMILES of the of the selected terpenes were retrieved from the PubChem Database and imported into the various webservers for druglikeness and ADMET computations using default parameters.
5. Conclusion
Drug-target investigations geared towards providing underlying mechanism of herbs, spices and medicinal plants are highly recommended for validating their acclaimed therapeutic potential. Catalogue of West African indigenous plants are extensively reported for protective and therapeutic potential against diabetes. However, most of the constituent phytochemicals that may account for the therapeutic potential are largely uninvestigated. We employed computational modeling in this study to assess the interactions of selected plant-derived terpenes with two emerging drug targets in diabetes. In this study, several triterpenes were reported to exhibit strong binding tendency with DPP-4 and PTP1B enzymes. Three hit triterpenes viz: Cucurbitacin B, 20-Epi-isoiguesterinol and 6-Oxoisoiguesterin are suggested as promising inhibitors of DPP-4. These compounds maintained strong binding affinity and stable interactions tendency with DPP4 as indicated by ensemble docking, post-docking MMGBSA, MD simulation, MD-based MMGBSA and density functional theory. The binding interactions of these triterpenes were sustained by interactions with several amino acid residues in the catalytic triad, oxyanion cavity, hydrophobic S1 pocket and charged S2 pocket in the active site region of DPP-4. In addition, the hit compounds exhibit good drug-likeness and moderate ADMET properties. Therefore, these triterpene structures are suggested for experimental validation towards their development as potent inhibitors of DPP-4 enzyme in diabetes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, O.M.O; methodology, O.M.O, G.A.G, F.O, IO, and I.M.I; software, O.M.O; validation, C.O.O.; formal analysis, O.M.O and M.M.F.; investigation, O.M.O. and G.A.B.; data curation, M.M.F.; writing—original draft preparation, O.M.O; writing—review and editing, O.M.O., O.S., G.E.B. and C.O.O.; visualization, O.M.O., G.A.B.; supervision, C.O.O.; project administration, O.M.O. and C.O.O.; funding acquisition, A.M.E. and G.E.B. All authors have read and agreed to the published version of the manuscript.”
Funding
the publication was supported through Researchers supporting Project number (RSP-2023-R406).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data associated with the this study are available from the corresponding author upon request.
Acknowledgments
The authors acknowledge with thanks the support of the Nutritional and Industrial Biochemistry Unit, department of Biochemistry, University of Ibadan, Nigeria. We also extend our gratitude to King Saud University (Riyadh, Saudi Arabia) for funding through Researchers supporting Project number (RSP-2023-R406).
Conflicts of Interest
The authors do not have conflicting interests.
Sample Availability
Not applicable.
References
- Kaur N, Kumar V, Nayak SK, et al. Alpha-amylase as molecular target for treatment of diabetes mellitus: A comprehensive review. Chemical Biology & Drug Design. 2021 2021/10/01;98,539-560. [CrossRef]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H. Q&A: Five questions on the 2015 IDF Diabetes Atlas. Diabetes Res. Clin. Pract. 2016, 115, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Reimann, M.; Bonifacio, E.; Solimena, M.; Schwarz, P.; Ludwig, B.; Hanefeld, M.; Bornstein, S. An update on preventive and regenerative therapies in diabetes mellitus. Pharmacol. Ther. 2009, 121, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Mourad, A.A.E.; Khodir, A.E.; Saber, S.; Mourad, M.A.E. Novel Potent and Selective DPP-4 Inhibitors: Design, Synthesis and Molecular Docking Study of Dihydropyrimidine Phthalimide Hybrids. Pharmaceuticals 2021, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes care. 2014 Jan;37 Suppl 1:S81-90. [CrossRef]
- Piłaciński, S.; Zozulińska-Ziółkiewicz, D.A. State of the art paper Influence of lifestyle on the course of type 1 diabetes mellitus. Arch. Med Sci. 2014, 1, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Chowta, M.; Chowta, N.; Shastry, R.; Kamath, P. The Proportion of Type 2 Diabetic Patients Achieving Treatment Goals and the Survey of Patients’ Attitude Towards Insulin In tiation ini Patient s with Inadequate Glycaemic Control with Oral Anti-diabetic Drugs. Curr. Diabetes Rev. 2021, 17, e110620182719. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, T.; Sampson, S.R. Activation of Insulin Signaling by Botanical Products. Int. J. Mol. Sci. 2021, 22, 4193. [Google Scholar] [CrossRef] [PubMed]
- Moller David, E. Metabolic Disease Drug Discovery— “Hitting the Target” Is Easier Said Than Done. Cell metabolism. 2012 2012/01/04/;15,19-24. [CrossRef]
- Kanwal, A.; Kanwar, N.; Bharati, S.; Srivastava, P.; Singh, S.P.; Amar, S. Exploring New Drug Targets for Type 2 Diabetes: Success, Challenges and Opportunities. Biomedicines 2022, 10, 331. [Google Scholar] [CrossRef]
- Kasole, R.; Martin, H.D.; Kimiywe, J. Traditional Medicine and Its Role in the Management of Diabetes Mellitus: "Patients' and Herbalists' Perspectives". Evidence-Based Complement. Altern. Med. 2019, 2019, 2835691. [Google Scholar] [CrossRef]
- Panigrahy, S.K.; Bhatt, R.; Kumar, A. Targeting type II diabetes with plant terpenes: the new and promising antidiabetic therapeutics. Biologia 2021, 76, 241–254. [Google Scholar] [CrossRef]
- Ogunyemi OM, Gyebi GA, Saheed A, et al. Inhibition mechanism of alpha-amylase, a diabetes target, by a steroidal pregnane and pregnane glycosides derived from Gongronema latifolium Benth [Original Research]. Frontiers in molecular biosciences. 2022 2022-August-10;9. [CrossRef]
- He, Z.; Zhou, Z.; Yang, Y.; Yang, T.; Pan, S.; Qiu, J.; Zhou, S. Overview of clinically approved oral antidiabetic agents for the treatment of type 2 diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2015, 42, 125–138. [Google Scholar] [CrossRef] [PubMed]
- A Tahrani, A.; Bailey, C.J.; Del Prato, S.; Barnett, A.H. Management of type 2 diabetes: new and future developments in treatment. Lancet 2011, 378, 182–197. [Google Scholar] [CrossRef]
- Ogunyemi, O.M.; Gyebi, A.G.; Adebayo, J.O.; Oguntola, J.A.; Olaiya, C.O. Marsectohexol and other pregnane phytochemicals derived from Gongronema latifolium as α-amylase and α-glucosidase inhibitors: in vitro and molecular docking studies. SN Appl. Sci. 2020, 2, 2119. [Google Scholar] [CrossRef]
- Nauck, M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes, Obes. Metab. 2016, 18, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Niedereichholz, U.; Ettler, R.; Holst, J.J.; Ørskov, C.; Ritzel, R.; Schmiegel, W.H.; Edgerton, D.S.; Kraft, G.; Smith, M.S.; et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am. J. Physiol. Metab. 1997, 273, E981–E988. [Google Scholar] [CrossRef]
- Koliaki, C.; Doupis, J. Incretin-based therapy: a powerful and promising weapon in the treatment of type 2 diabetes mellitus. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2011, 2, 101–121. [Google Scholar] [CrossRef]
- Salvo, F.; Moore, N.; Arnaud, M.; Robinson, P.; Raschi, E.; De Ponti, F.; Bégaud, B.; Pariente, A. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: systematic review and meta-analysis. BMJ 2016, 353, i2231. [Google Scholar] [CrossRef]
- Paternoster S, Falasca M. Dissecting the Physiology and Pathophysiology of Glucagon-Like Peptide-1 [Review]. Frontiers in Endocrinology. 2018 2018-October-11;9. [CrossRef]
- Glossmann, H.H.; Lutz, O.M. Pharmacology of metformin – An update. Eur. J. Pharmacol. 2019, 865, 172782. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Biondani G, Peyron J-F. Metformin, an Anti-diabetic Drug to Target Leukemia [Mini Review]. Frontiers in Endocrinology. 2018 2018-August-10;9. [CrossRef]
- McGovern, A.; Tippu, Z.; Hinton, W.; Munro, N.; Whyte, M.; de Lusignan, S. Comparison of medication adherence and persistence in type 2 diabetes: A systematic review and meta-analysis. Diabetes, Obes. Metab. 2017, 20, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Hussain H, Green IR, Abbas G, et al. Protein tyrosine phosphatase 1B (PTP1B) inhibitors as potential anti-diabetes agents: patent review (2015-2018). Expert Opinion on Therapeutic Patents. 2019 2019/09/02;29(9):689-702. [CrossRef]
- Teimouri, M.; Hosseini, H.; ArabSadeghabadi, Z.; Babaei-Khorzoughi, R.; Gorgani-Firuzjaee, S.; Meshkani, R. The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications. J. Physiol. Biochem. 2022, 78, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Gupta, S.J.; Chaudhary, A.; Garg, V.K. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents – A brief review. Bioorganic Chem. 2017, 70, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Tajuddeen, N. Antidiabetic compounds from medicinal plants traditionally used for the treatment of diabetes in Africa: A review update (2015–2020). South Afr. J. Bot. 2022, 146, 585–602. [Google Scholar] [CrossRef]
- Mohammed, A.; Ibrahim, M.A.; Islam, S. African Medicinal Plants with Antidiabetic Potentials: A Review. Planta Medica 2014, 80, 354–377. [Google Scholar] [CrossRef] [PubMed]
- Olaiya C, O Soetan K, M Esan A. The role of nutraceuticals, functional foods and value added food products in the prevention and treatment of chronic diseases. Afr J Food Sci. 2016;10,185-193. [CrossRef]
- Mohammed A, Ibrahim H, Islam MS. Chapter 8 - Plant-Derived Antidiabetic Compounds Obtained From African Medicinal Plants: A Short Review. In: Atta ur R, editor. Studies in Natural Products Chemistry. Vol. 54: Elsevier; 2017. p. 291-314.
- Mahomoodally, M.; Korumtollee, H.; Chady, Z. Ethnopharmacological uses of Antidesma madagascariense Lam. (Euphorbiaceae). J. Intercult. Ethnopharmacol. 2015, 4, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Beidokhti, M.; Lobbens, E.; Rasoavaivo, P.; Staerk, D.; Jäger, A. Investigation of medicinal plants from Madagascar against DPP-IV linked to type 2 diabetes. South Afr. J. Bot. 2018, 115, 113–119. [Google Scholar] [CrossRef]
- Catarino, L.; Havik, P.J.; Romeiras, M.M. Medicinal plants of Guinea-Bissau: Therapeutic applications, ethnic diversity and knowledge transfer. J. Ethnopharmacol. 2016, 183, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, Z.; Guo, B.; Sun, M.; Onakpa, M.M.; Yao, G.; Zhao, M.; Che, C.-T. Modified diterpenoids from the tuber of Icacina oliviformis as protein tyrosine phosphatase 1B inhibitors. Org. Chem. Front. 2020, 7, 355–367. [Google Scholar] [CrossRef]
- Chang, C.-I.; Chou, C.-H.; Liao, M.-H.; Chen, T.-M.; Cheng, C.-H.; Anggriani, R.; Tsai, C.-P.; Tseng, H.-I.; Cheng, H.-L. Bitter melon triterpenes work as insulin sensitizers and insulin substitutes in insulin-resistant cells. J. Funct. Foods 2015, 13, 214–224. [Google Scholar] [CrossRef]
- Majouli, K.; Hlila, M.B.; Hamdi, A.; Flamini, G.; Ben Jannet, H.; Kenani, A. Antioxidant activity and α-glucosidase inhibition by essential oils from Hertia cheirifolia (L.). Ind. Crop. Prod. 2016, 82, 23–28. [Google Scholar] [CrossRef]
- Panigrahy SK, Kumar A, Bhatt R. Hedychium coronarium Rhizomes: Promising Antidiabetic and Natural Inhibitor of α-Amylase and α-Glucosidase. Journal of dietary supplements. 2020;17,81-87. [CrossRef]
- Gong Y, Chen K, Yu SQ, et al. [Protective effect of terpenes from fructus corni on the cardiomyopathy in alloxan-induced diabetic mice]. Zhongguo ying yong sheng li xue za zhi = Zhongguo yingyong shenglixue zazhi = Chinese journal of applied physiology. 2012 Jul;28,378-80, 384.
- Ma TK, Xu L, Lu LX, et al. Ursolic Acid Treatment Alleviates Diabetic Kidney Injury By Regulating The ARAP1/AT1R Signaling Pathway. Diabetes, metabolic syndrome and obesity : targets and therapy. 2019;12:2597-2608. [CrossRef]
- Praveen Kumar M, Poornima, Mamidala E, et al. Effects of D-Limonene on aldose reductase and protein glycation in diabetic rats. Journal of King Saud University - Science. 2020 2020/04/01/;32,1953-1958. [CrossRef]
- Purnomo, Y.; Soeatmadji, D.W.; Sumitro, S.B.; Widodo, M.A. Anti-diabetic potential of Urena lobata leaf extract through inhibition of dipeptidyl peptidase IV activity. Asian Pac. J. Trop. Biomed. 2015, 5, 645–649. [Google Scholar] [CrossRef]
- Liang, L.-F.; Gao, L.-X.; Li, J.; Taglialatela-Scafati, O.; Guo, Y.-W. Cembrane diterpenoids from the soft coral Sarcophyton trocheliophorum Marenzeller as a new class of PTP1B inhibitors. Bioorganic Med. Chem. 2013, 21, 5076–5080. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-B.; Zhang, H.; Dong, S.-H.; Sheng, L.; Wu, Y.; Li, J.; Yue, J.-M. New triterpenoids with protein tyrosine phosphatase 1B inhibition from Cedrela odorata. J. Asian Nat. Prod. Res. 2014, 16, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ogunyemi, O.M.; Gyebi, G.A.; Ibrahim, I.M.; Esan, A.M.; Olaiya, C.O.; Soliman, M.M.; Batiha, G.E.-S. Identification of promising multi-targeting inhibitors of obesity from Vernonia amygdalina through computational analysis. Mol. Divers. 2022, 27, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Singla, R.; Jaitak, V. In Silico Study of Flavonoids as DPP-4 and α-glucosidase Inhibitors. Lett. Drug Des. Discov. 2018, 15, 634–642. [Google Scholar] [CrossRef]
- Chalichem, N.S.S.; Jupudi, S.; Yasam, V.R.; Basavan, D. Dipeptidyl peptidase-IV inhibitory action of Calebin A: An in silico and in vitro analysis. J. Ayurveda Integr. Med. 2021, 12, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Quek, A.; Kassim, N.K.; Lim, P.C.; Tan, D.C.; Latif, M.A.M.; Ismail, A.; Shaari, K.; Awang, K. α-Amylase and dipeptidyl peptidase-4 (DPP-4) inhibitory effects of Melicope latifolia bark extracts and identification of bioactive constituents using in vitro and in silico approaches. [CrossRef]
- Quy PT, Van Hue N, Bui TQ, et al. Inhibitory, biocompatible, and pharmacological potentiality of dammarenolic-acid derivatives towards α-glucosidase (3W37) and tyrosine phosphatase 1B (PTP1B). Vietnam Journal of Chemistry. 2022 2022/04/01;60,223-237. [CrossRef]
- Olawale, F.; Olofinsan, K.; Iwaloye, O.; Chukwuemeka, P.O.; Elekofehinti, O.O. Screening of compounds from Nigerian antidiabetic plants as protein tyrosine phosphatase 1B inhibitor. Comput. Toxicol. 2022, 21, 100200. [Google Scholar] [CrossRef]
- Zhang, Z.; Wallace, M.B.; Feng, J.; Stafford, J.A.; Skene, R.J.; Shi, L.; Lee, B.; Aertgeerts, K.; Jennings, A.; Xu, R.; et al. Design and Synthesis of Pyrimidinone and Pyrimidinedione Inhibitors of Dipeptidyl Peptidase IV. J. Med. Chem. 2011, 54, 510–524. [Google Scholar] [CrossRef]
- Antony, P.; Baby, B.; Aleissaee, H.M.; Vijayan, R. A Molecular Modeling Investigation of the Therapeutic Potential of Marine Compounds as DPP-4 Inhibitors. Mar. Drugs 2022, 20, 777. [Google Scholar] [CrossRef]
- Banzouzi JT, Soh PN, Mbatchi B, et al. Cogniauxia podolaena: bioassay-guided fractionation of defoliated stems, isolation of active compounds, antiplasmodial activity and cytotoxicity. Planta medica. 2008 Oct;74,1453-6. [CrossRef]
- Olarewaju, O.O.; Fajinmi, O.O.; Arthur, G.D.; Coopoosamy, R.M.; Naidoo, K.K. Food and medicinal relevance of Cucurbitaceae species in Eastern and Southern Africa. Bull. Natl. Res. Cent. 2021, 45, 208. [Google Scholar] [CrossRef]
- Murata, T.; Miyase, T.; Muregi, F.W.; Naoshima-Ishibashi, Y.; Umehara, K.; Warashina, T.; Kanou, S.; Mkoji, G.M.; Terada, M.; Ishih, A. Antiplasmodial Triterpenoids from Ekebergia capensis. J. Nat. Prod. 2008, 71, 167–174. [Google Scholar] [CrossRef]
- Nyakudya, T.T.; Tshabalala, T.; Dangarembizi, R.; Erlwanger, K.H.; Ndhlala, A.R. The Potential Therapeutic Value of Medicinal Plants in the Management of Metabolic Disorders. Molecules 2020, 25, 2669. [Google Scholar] [CrossRef]
- Hamid, K.; Alqahtani, A.; Kim, M.-S.; Cho, J.-L.; Cui, P.H.; Li, C.G.; Groundwater, P.W.; Li, G.Q. Tetracyclic triterpenoids in herbal medicines and their activities in diabetes and its complications. Curr. Top. Med. Chem. 2015, 15, 2406–2430. [Google Scholar] [CrossRef]
- Deutschländer, M.; Lall, N.; Van de Venter, M.; Hussein, A. Hypoglycemic evaluation of a new triterpene and other compounds isolated from Euclea undulata Thunb. var. myrtina (Ebenaceae) root bark. J. Ethnopharmacol. 2011, 133, 1091–1095. [Google Scholar] [CrossRef]
- Maroyi, A. Euclea undulata Thunb.: Review of its botany, ethnomedicinal uses, phytochemistry and biological activities. Asian Pacific journal of tropical medicine. 2017 2017/11/01/;10,1030-1036. [CrossRef]
- Chen, L.; Lu, X.; El-Seedi, H.; Teng, H. Recent advances in the development of sesquiterpenoids in the treatment of type 2 diabetes. Trends Food Sci. Technol. 2019, 88, 46–56. [Google Scholar] [CrossRef]
- Nazaruk, J.; Borzym-Kluczyk, M. The role of triterpenes in the management of diabetes mellitus and its complications. Phytochem. Rev. 2014, 14, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Mbah, J.A.; Tane, P.; Ngadjui, B.T.; Connolly, J.D.; Okunji, C.C.; Iwu, M.M.; Schuster, B.M. Antiplasmodial Agents from the Leaves ofGlossocalyx brevipes. Planta Medica 2004, 70, 437–440. [Google Scholar] [CrossRef] [PubMed]
- N, T.; E, F.; V, K.; R, J.; D, N.; Dj, S.; Telesphore, N.B. Ethnomedical and Ethnopharmacological Study of Plants Used by Indigenous People of Cameroon for The Treatments of Diabetes and its Signs, Symptoms and Complications. J. Mol. Biomarkers Diagn. 2016, 8, 310. [Google Scholar] [CrossRef]
- Bickii, J.; Njifutie, N.; Foyere, J.A.; Basco, L.K.; Ringwald, P. In vitro antimalarial activity of limonoids from Khaya grandifoliola C.D.C. (Meliaceae). J. Ethnopharmacol. 2000, 69, 27–33. [Google Scholar] [CrossRef]
- Kale, O.E.; Akinpelu, O.B.; Bakare, A.A.; Yusuf, F.O.; Gomba, R.; Araka, D.C.; Ogundare, T.O.; Okolie, A.C.; Adebawo, O.; Odutola, O. Five traditional Nigerian Polyherbal remedies protect against high fructose fed, Streptozotocin-induced type 2 diabetes in male Wistar rats. BMC Complement. Altern. Med. 2018, 18, 160. [Google Scholar] [CrossRef] [PubMed]
- Talontsi, F.M.; Lamshöft, M.; Bauer, J.O.; Razakarivony, A.A.; Andriamihaja, B.; Strohmann, C.; Spiteller, M. Antibacterial and Antiplasmodial Constituents of Beilschmiedia cryptocaryoides. J. Nat. Prod. 2013, 76, 97–102. [Google Scholar] [CrossRef]
- Wabo, H.K.; Tane, P.; Connolly, J.D. Diterpenoids and sesquiterpenoids from Aframomum arundinaceum. Biochem. Syst. Ecol. 2006, 34, 603–605. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Olawale, F.; Iwaloye, O.; Elekofehinti, O.O. Virtual screening of natural compounds as selective inhibitors of polo-like kinase-1 at C-terminal polo box and N-terminal catalytic domain. J. Biomol. Struct. Dyn. 2021, 40, 13606–13624. [Google Scholar] [CrossRef]
- Matuszek, A.M.; Reynisson, J. Defining Known Drug Space Using DFT. Mol. Informatics 2016, 35, 46–53. [Google Scholar] [CrossRef]
- Ganesan, M.; Raja, K.K.; Murugesan, S.; Kumar, B.K.; Rajagopal, G.; Thirunavukkarasu, S. Synthesis, biological evaluation, molecular docking, molecular dynamics and DFT studies of quinoline-fluoroproline amide hybrids. J. Mol. Struct. 2020, 1217, 128360. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Gyebi GA, Elfiky AA, Ogunyemi OM, et al. Structure-based virtual screening suggests inhibitors of 3-Chymotrypsin-Like Protease of SARS-CoV-2 from Vernonia amygdalina and Occinum gratissimum. Comput Biol Med. 2021 Sep;136:104671. [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Olawale, F.; Iwaloye, O.; Olofinsan, K.; Ogunyemi, O.M.; Gyebi, G.A.; Ibrahim, I.M. Homology modelling, vHTS, pharmacophore, molecular docking and molecular dynamics studies for the identification of natural compound-derived inhibitor of MRP3 in acute leukaemia treatment. Chem. Pap. 2022, 76, 3729–3757. [Google Scholar] [CrossRef]
- Salentin S, Schreiber S, Haupt V, et al. PLIP: Fully automated protein-ligand interaction profiler. Nucleic acids research. 2015 04/14;43. [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bekker H, Berendsen H, Dijkstra E, et al., editors. Gromacs-a parallel computer for molecular-dynamics simulations. 4th International Conference on Computational Physics (PC 92); 1993: World Scientific Publishing.
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallographica Section D: Biological Crystallography. 2004;60,1355-1363. [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Miller BR, 3rd, McGee TD, Jr., Swails JM, et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of chemical theory and computation. 2012 Sep 11;8,3314-21. [CrossRef]
- Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Xue, W.; Yang, F.; Wang, P.; Zheng, G.; Chen, Y.; Yao, X.; Zhu, F. What Contributes to Serotonin–Norepinephrine Reuptake Inhibitors’ Dual-Targeting Mechanism? The Key Role of Transmembrane Domain 6 in Human Serotonin and Norepinephrine Transporters Revealed by Molecular Dynamics Simulation. ACS Chem. Neurosci. 2018, 9, 1128–1140. [Google Scholar] [CrossRef]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert opinion on drug discovery. 2021 Nov;16,1233-1237. [CrossRef]
- Elekofehinti, O.O.; Iwaloye, O.; Olawale, F.; Chukwuemeka, P.O.; Folorunso, I.M. Newly designed compounds from scaffolds of known actives as inhibitors of survivin: computational analysis from the perspective of fragment-based drug design. Silico Pharmacol. 2021, 9, 47. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee P, Eckert AO, Schrey AK, et al. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic acids research. 2018 Jul 2;46(W1):W257-w263. [CrossRef]
Figure 1.
The 3D representation of the interactions of hit triterpenes and alogliptin with amino acid residues in the active site of DPP-4. Ligand structures are depicted in stick representations. Types of interaction are in dotted lines and are depicted in colour green (Hydrogen -bonds) and purple (hydrophobic interactions-pi-alkyl, alkyl, pi-stacking, pi-pi T-shaped, pi-sulfur and pi-stacking interactions). Three letter-code were used to represent amino acid residues.
Figure 1.
The 3D representation of the interactions of hit triterpenes and alogliptin with amino acid residues in the active site of DPP-4. Ligand structures are depicted in stick representations. Types of interaction are in dotted lines and are depicted in colour green (Hydrogen -bonds) and purple (hydrophobic interactions-pi-alkyl, alkyl, pi-stacking, pi-pi T-shaped, pi-sulfur and pi-stacking interactions). Three letter-code were used to represent amino acid residues.
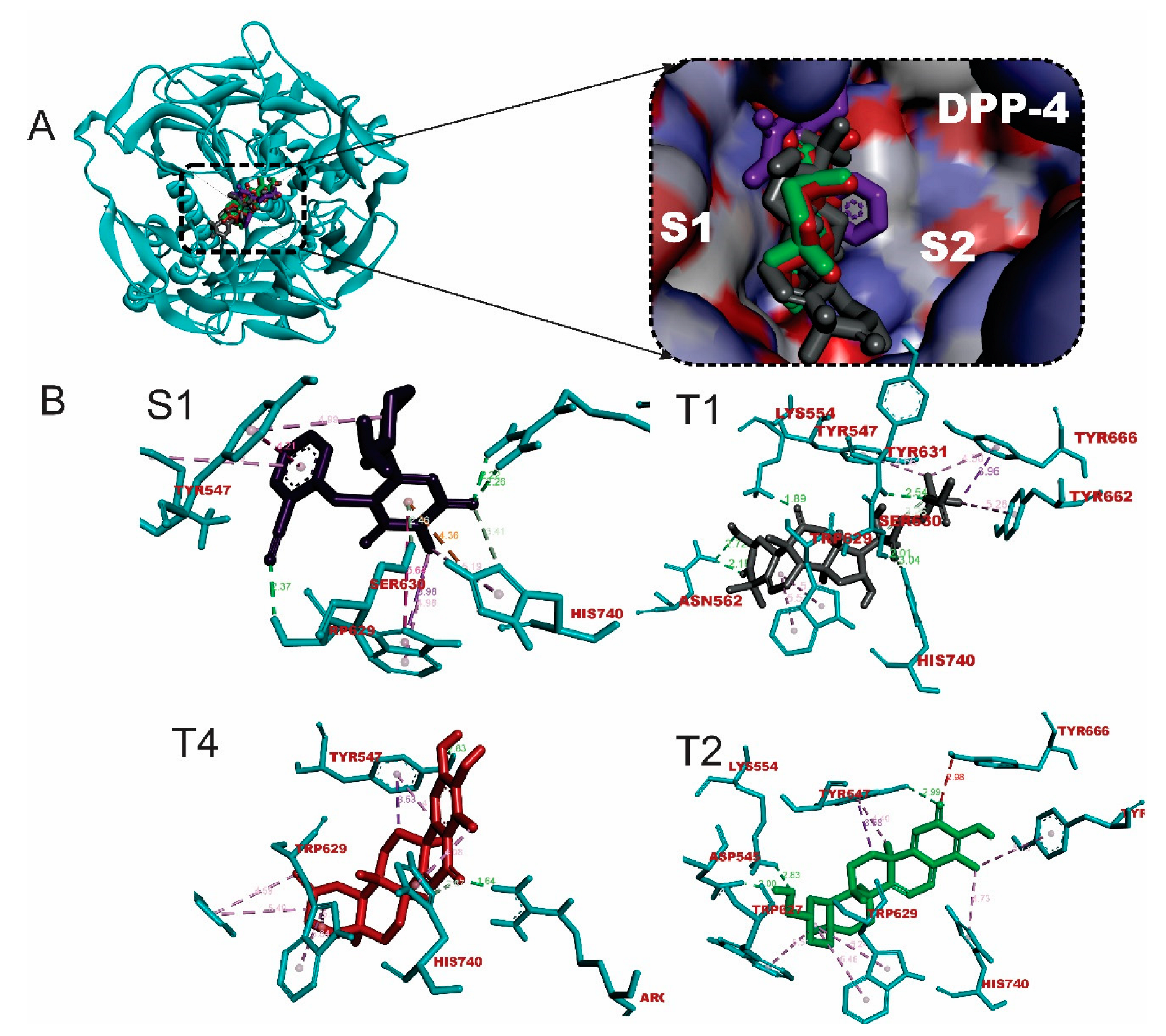
Figure 2.
The 3D representation of the interactions of hit triterpenes and isothiazolidinone with critical residues in the binding pocket of PTP1B. Ligand structures are depicted in stick representations. Types of interaction are in dotted lines and are depicted in colour green (Hydrogen -bonds) and purple (hydrophobic interactions-pi-alkyl, alkyl, pi-stacking, pi-pi T-shaped, pi-sulfur and pi-stacking interactions). Three letter-code were used to represent amino acid residues.
Figure 2.
The 3D representation of the interactions of hit triterpenes and isothiazolidinone with critical residues in the binding pocket of PTP1B. Ligand structures are depicted in stick representations. Types of interaction are in dotted lines and are depicted in colour green (Hydrogen -bonds) and purple (hydrophobic interactions-pi-alkyl, alkyl, pi-stacking, pi-pi T-shaped, pi-sulfur and pi-stacking interactions). Three letter-code were used to represent amino acid residues.
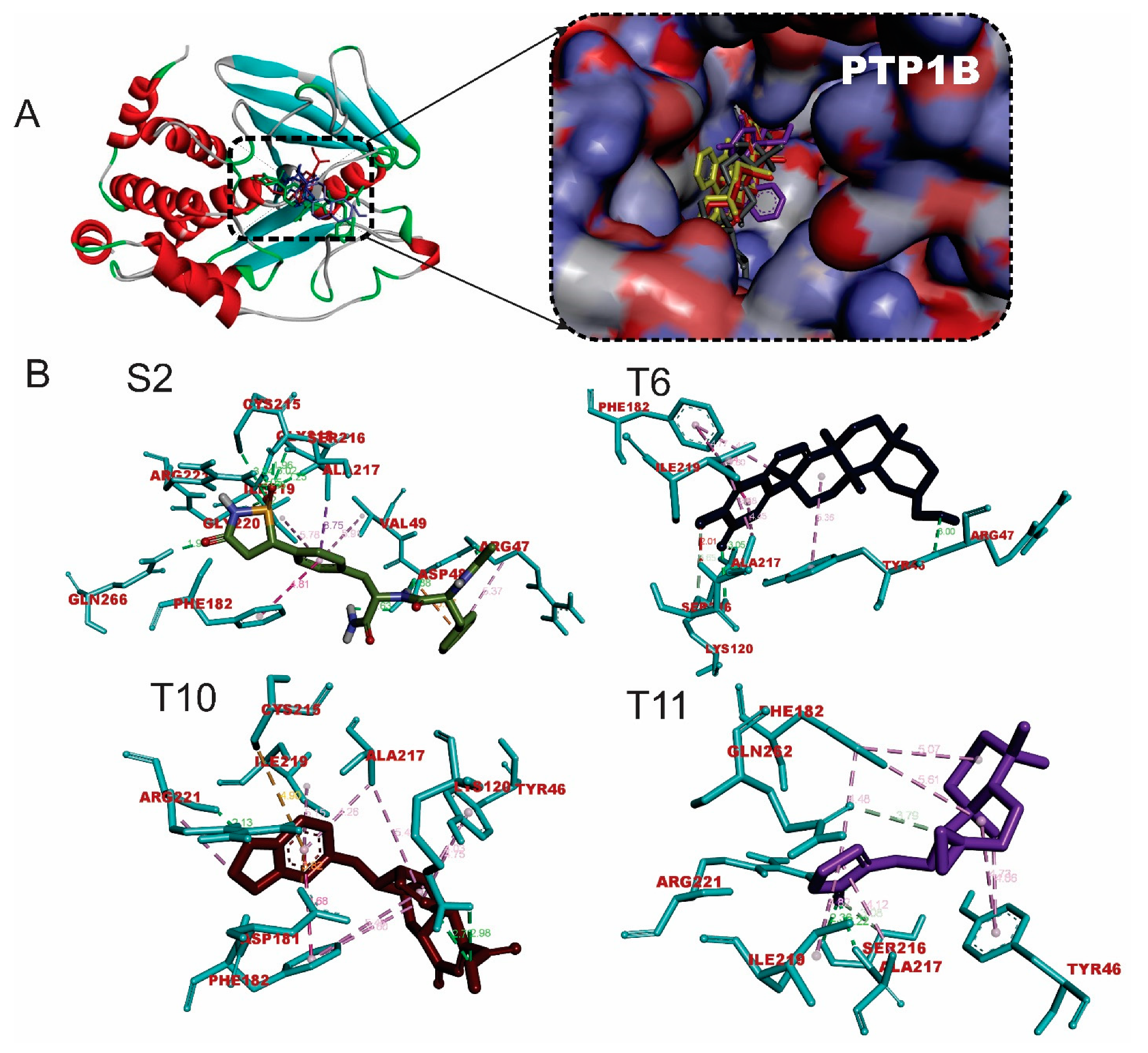
Figure 4.
Thermodynamic parameters computed from MD simulation trajectories.
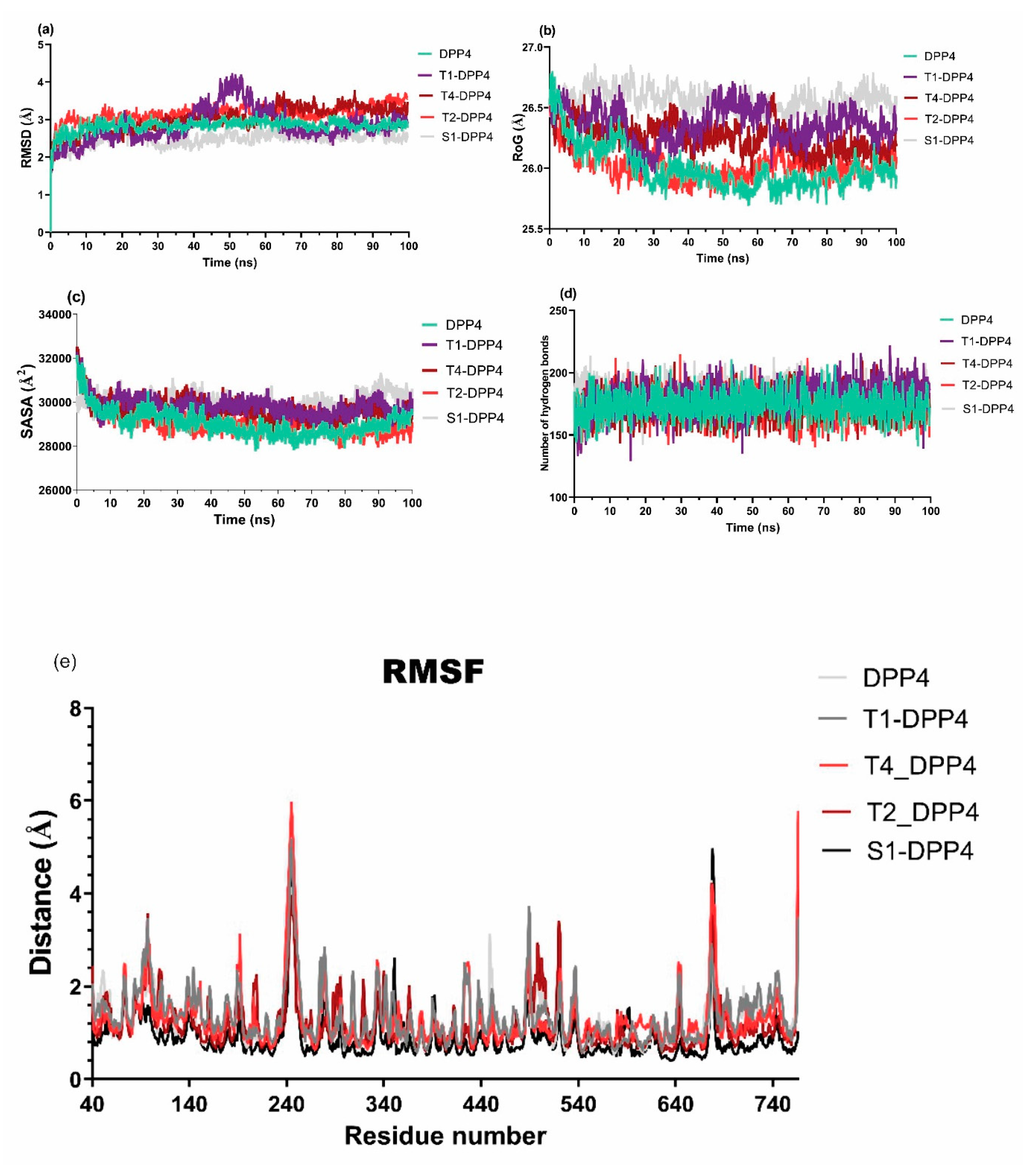
Figure 5.
Contribution of amino acid residues to the binding affinity of DPP-4 with the ligands.
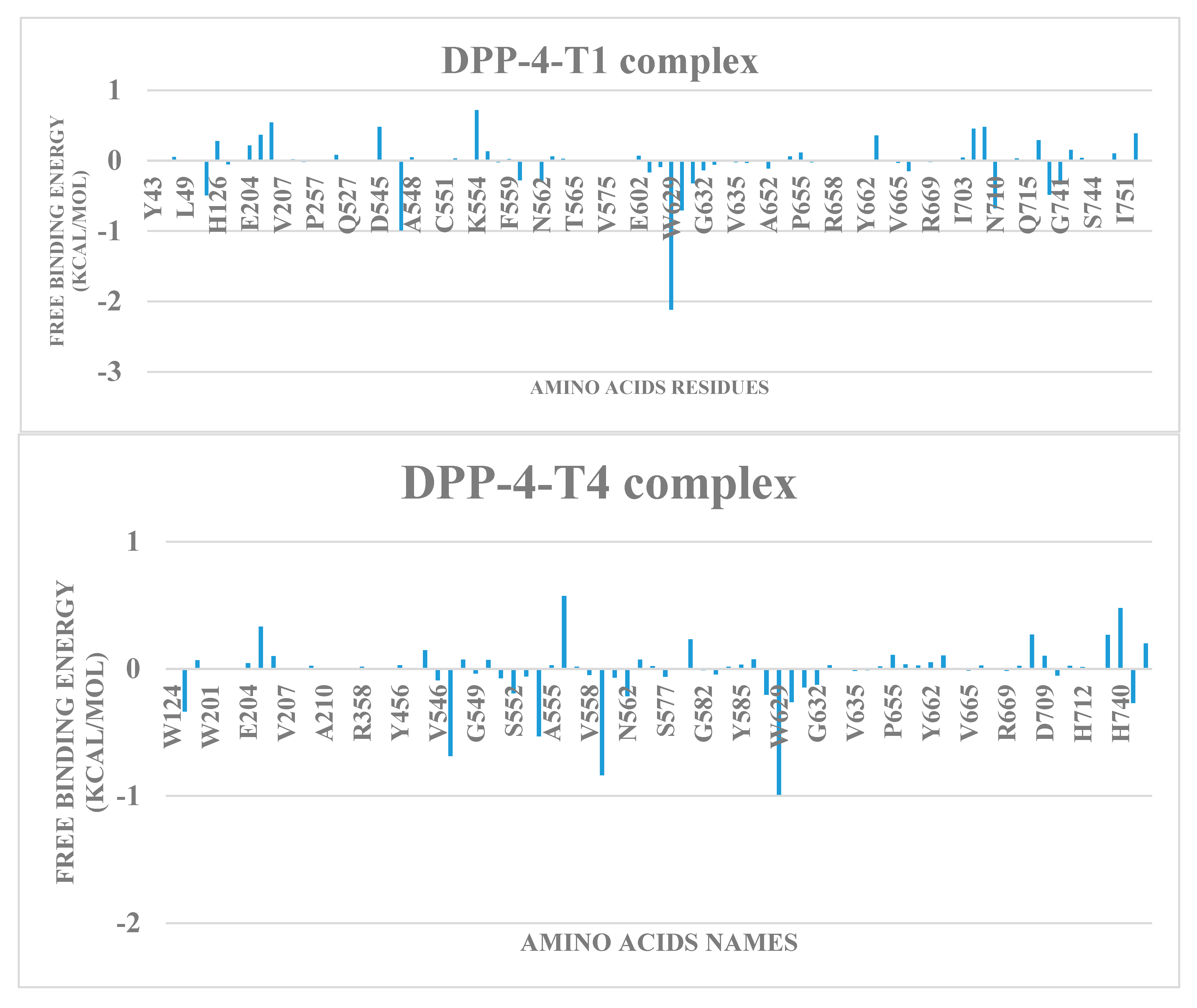
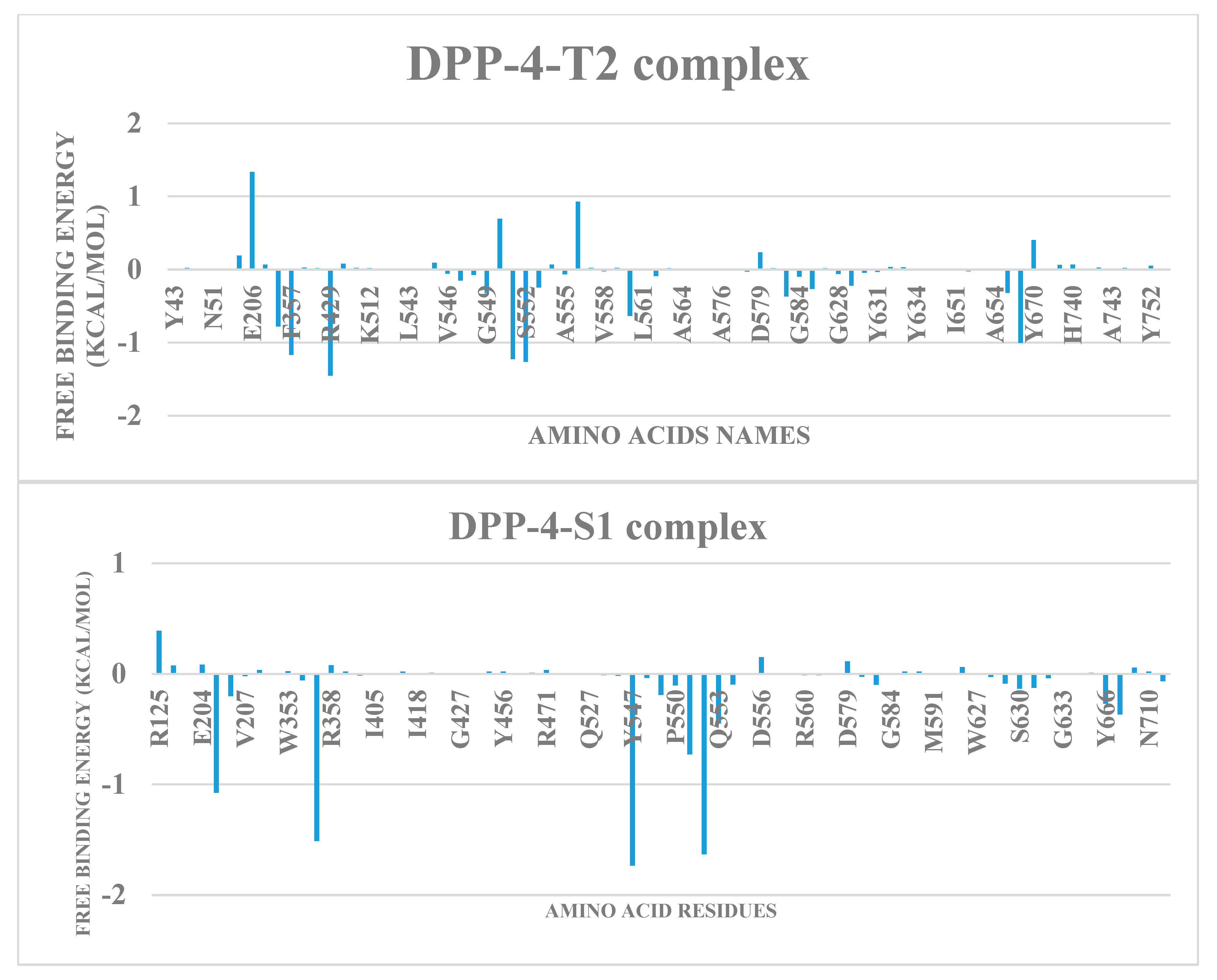
Figure 6.
Molecular orbital images and calculated bandgap of top triterpene structures.
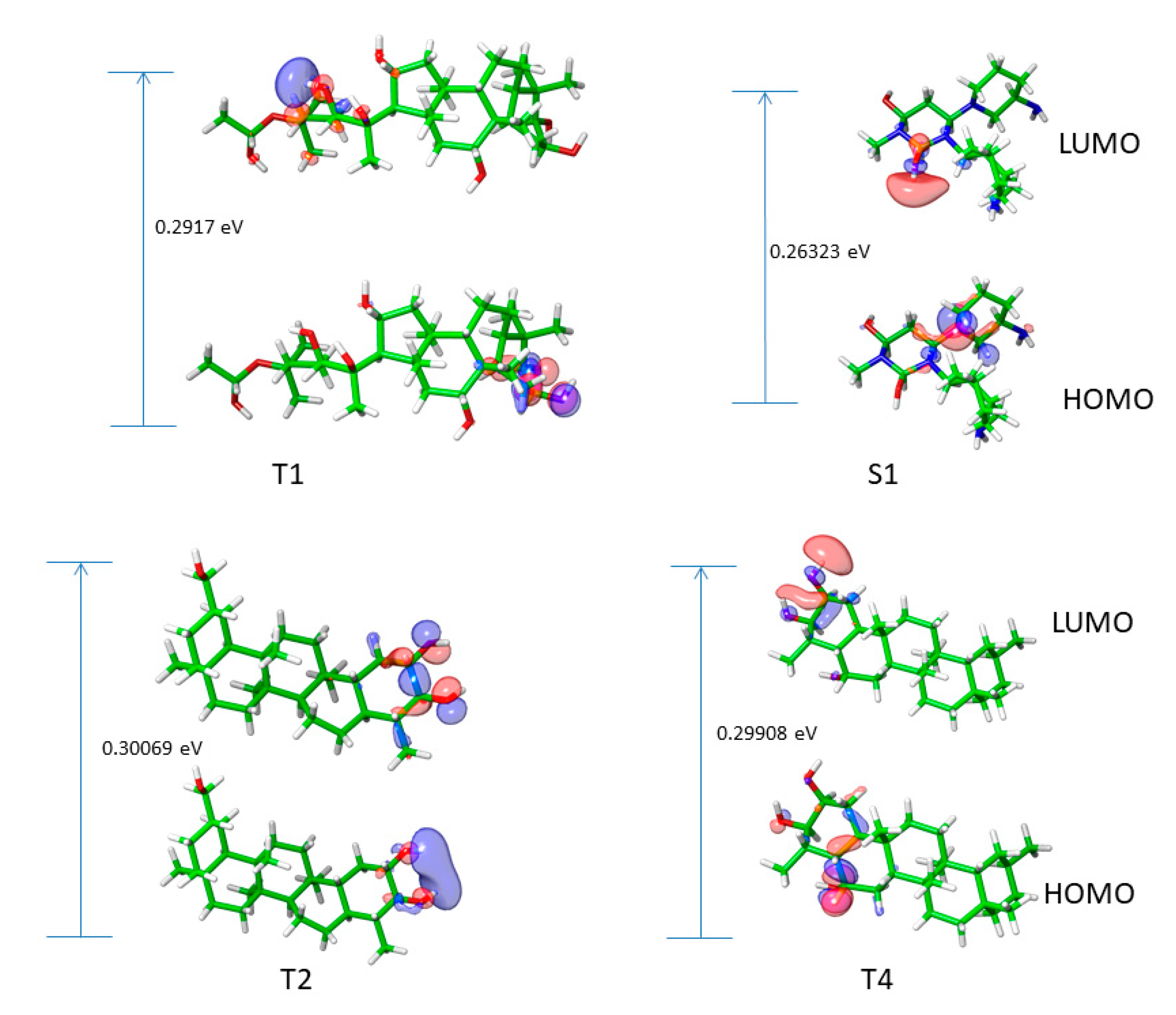
Table 1.
Docking scores of terpenes against DPP-4.
| S/N | Compounds | Class | Dockingscore (Kcal/mol) | PostDocking MM-GBSA (Kcal/mol) |
|---|---|---|---|---|
| S1 | Alogliptin | -7.7 | -37.02 | |
| T1 | Cucurbitacin B | tetracyclic triterpenes | -9.9 | -47.80 |
| T2 | 20-Epi-isoiguesterinol | Bisnortriterpenes | -9.9 | -29.78 |
| T3 | Isoiguesterin | Bisnortriterpenes | -9.7 | -28.75 |
| T4 | 6-Oxoisoiguesterin | Bisnortriterpenes | -9.3 | -32.90 |
| T6 | Isoiguesterinol | Bisnortriterpenes | -9.0 | -29.36 |
| T7 | 1-Deacetylkhivorin | Limonoids | -8.9 | -19.72 |
| T8 | 7-Deacetylkhivorin | Limonoids | -8.8 | -25.71 |
Table 2.
Amino acid interactions of DPP-4 with top-docked terpenes.
| Compounds |
Hydrogen bonds (Bond lenght Å) |
Hydrophobic Interaction | Other interactions | |||
|---|---|---|---|---|---|---|
| No | Residues | No | Residues | No | Residues | |
| Alogliptin | 5 | ARG125(2.22;2.26) TRP629(2.37) HIS740(3.41) SER630(2.46) | 7 | TRP629(3.99;5.64;4.98) TYR547(4.21;4.99) HIS740(5.19) LYS554 | 1 | HIS740 |
| T1 | 7 | LYS554 ASN562(2) SER630(2) TYR631 HIS740 | 6 | TYR666(2) TYR547 TRP629(2) TYR662 | 0 | None |
| T4 | 3 | ARG125 TYR547 HIS740 | 7 | TYR547(2) TRP627(2) TRP629(2) HIS740 | 0 | None |
| T2 | 3 | TYR547 LYS544 ASP545 | 7 | TYR547(2) TRP627 TRP629(2) TYR662 HIS740 | 0 | None |
NB: The residues that define the catalytic triad are presented in bold font.
Table 3.
Molecular docking and prime MM-GBSA binding energy of top terpenes with PTP1B.
| S/N | Compounds | Class | Dockingscore (Kcal/mol) | Prime MM-GBSA (Kcal/mol) |
|---|---|---|---|---|
| S2 | ISOTHIAZOLIDINONE | 9.8 | -85.23 | |
| T9 | Tsangibeilin B | Beilshmiedic acid derivatives | -8.4 | -42.32 |
| T10 | Cryptobeilic acid C | Beilshmiedic acid derivatives | -8.1 | -44.09 |
| T3 | Isoiguesterin | Bisnorterpenes | -7.5 | -42.07 |
| T4 | 6-Oxoisoiguesterin | Bisnorterpenes | -7.4 | -41.11 |
| T6 | Isoiguesterinol | Bisnorterpenes | -7.0 | -45.82 |
| T11 | Galanolactone | Clerodane and labdanediterpenoids | -6.5 | -43.56 |
| T2 | 20-Epi-isoiguesterinol | Bisnorterpenes | -6.2 | -29.06 |
Table 4.
Amino acid interactions of PTP1B with top hit terpenes.
| Compounds |
Hydrogen bonds (Bond length Å) |
Hydrophobic Interaction | Other interactions | |||
|---|---|---|---|---|---|---|
| No | Residues | No | Residues | No | Residues | |
| S2 |
11 |
ARG47(2.01) CYS215(3.04) SER216(1.96) ALA217(2.25) GLY218(3.02) ILE219(2.36) GLY220(2.02) ARG221(2.05) GLN266(1.99) ASP48(1.88; 2.63) | 3 | ALA217(3.75) PHE182(4.81) ARG47(5.37) | 1 | ASP48(3.87) |
| T6 |
4 |
ARG47 LYS120 SER216(2) | 7 | PHE182(3) ALA217(2) ILE219 TYR46 | 0 | None |
| T10 |
4 |
LYS120(2.76;2.98) ARG221(2.13; 2.06) | 8 | PHE182(4.68;5.45) ALA217(5.49; 4.26) ARG221(3.74) TYR46(4.75; 4.03) ILE219(5.15) | 2 | ASP181 (3.82) CYS215(4.90) |
| T11 |
5 |
SER216(2.36;3.08) ALA217(2.22) ARG221(2.28) GLN262(3.79) | 6 | ALA217(4.12) ILE219(4.82) TYR46(4.73; 4.06) PHE182 (5.07; 4.48) | 0 | None |
NB: The residues that define the catalytic triad are presented in bold font.
Table 5.
The MM-GBSA calculations for DPP-4 – triterpene complex MD trajectory.
| SYSTEM | ΔVDWAALS | ΔEEL | ΔEGB | ΔESURF | ΔGGAS | ΔGSOLV | ΔTOTAL |
|---|---|---|---|---|---|---|---|
| T1 | -34.67 ± 3.39 | -18.69 ± 7.22 | 46.29 ± 6.49 | -4.79 ± 0.43 | -53.35 ± 7.49 | 41.5 ± 6.35 | -11.85 ± 3.34 |
| T4 | -22.20 ± 3.38 | -7.25 ± 12.28 | 25.02 ± 10.43 | -2.82 ± 0.65 | -29.44 ± 13.02 | 22.2 ± 10.09 | -7.24 ± 4.62 |
| T2 | -30.73 ± 5.27 | 125.46 ± 28.01 | -93.17 ± 30.7 | -3.82 ± 0.66 | 94.73 ± 31.83 | -96.99 ± 30.24 | -2.26 ± 3.64 |
| S1 | -22.63 ± 4.15 | -304.75 ± 33.46 | 312.75 ± 28.86 | -3.28 ± 0.53 | -327.39 ± 33.19 | 309.48 ± 28.53 | -17.91 ± 6.42 |
Table 6.
Density functional theory analysis of top triterpenes.
| HOMO | LUMO | Band gap | Electronegativity | Ionization potential |
Electron affinity |
Hardness | softness | Chemical potential |
Electrophilicity | |
|---|---|---|---|---|---|---|---|---|---|---|
| S1 | -0.20306 | 0.06017 | 0.26323 | 0.071445 | 0.20306 | -0.06017 | 0.131615 | 3.798959 | -0.07145 | 0.002552 |
| T1 | -0.24354 | 0.04816 | 0.29170 | 0.097690 | 0.24354 | -0.04816 | 0.145850 | 3.428180 | -0.09769 | 0.004772 |
| T4 | -0.24676 | 0.05232 | 0.29908 | 0.09722 | 0.24676 | -0.05232 | 0.14954 | 3.343587 | -0.09722 | 0.004726 |
| T2 | -0.24692 | 0.05377 | 0.30069 | 0.096575 | 0.24692 | -0.05377 | 0.150345 | 3.325684 | -0.09658 | 0.004663 |
Table 7.
Physicochemical and ADMET characteristics of the hit triterpenes.
| Terpenoids | Cucurbitacin B | 20-Epi-isoiguesterinol | 6-Oxoisoiguesterin |
|---|---|---|---|
| MW | 558.7 | 422.6 | 420.58 |
| #Rotatable bonds | 6 | 1 | 0 |
| #H-bond acceptors | 8 | 3 | 3 |
| #H-bond donors | 3 | 2 | 2 |
| TPSA | 138.2 | 57.53 | 57.53 |
| ESOL Class | MS | MS | PS |
| Lipinski violations | 1 | 1 | 1 |
| Ghose violations | 3 | 1 | 1 |
| Veber violations | 0 | 0 | 0 |
| Egan violations | 1 | 1 | 1 |
| Muegge violations | 1 | 1 | 1 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 |
| PAINS alerts | 0 | 0 | 1 |
| Brenk alerts | 2 | 0 | 2 |
| Synthetic Accessibility | 6.79 | 6.29 | 5.21 |
| GI absorption | Low | High | High |
| BBB permeant | No | No | No |
| Pgp substrate | Yes | No | No |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | No | No | No |
| CYP2C9 inhibitor | No | Yes | Yes |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | Yes | Yes | Yes |
| Bioavailability Score | 0.55 | 0.55 | 0.55 |
S-Soluble; MS-Moderately Soluble; PS-Poorly Soluble.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



