
Submitted:
30 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Background: Currently, limited number of therapeutic options are available to treat Diabetes mellitus, and to find a potential candidate, laboratory work takes time and also needs animal studies. So, the aim of this study was to determine the efficacy of some natural phytocompounds with the aid of molecular docking, bioinformatics and in silico drug design approaches. Method: Two proteins (Human CYP3A4 linked to metformin and Human dipeptidyl peptidase-IV) were selected and molecular docking studies were conducted using Pymol, AutoDock Vina, PyRx, and Discovery Studio. Different important pharmacokinetic parameters like ADME and toxicity data were obtained from online databases SwissADME and pkCSM program. Results: It was found that human dipeptidyl peptidase-IV (PDB ID: 4A5S) has exhibited a maximal affinity of -9.7 Kcal/mol for bryonolic acid and hesperidin, but only -6.7 kcal/mol for metformin hydrochloride. Similarly, the highest affinity of hesperidin for human CYP3A4 bound to metformin (PDB ID: 5G5J) is -10.7 Kcal/mol compared to metformin hydrochlor (-6.3 Kcal/mol). Besides, all the compounds have been documented outstanding ADMET profile, and accepted by drug-likeness or Lipinski rule. Conclusions: The present study suggested that these compounds can be further investigated in vitro and in vivo to establish them as lead compounds against Diabetes mellitus..
Keywords:
Lagenaria siceraria
; In silico study
; Drug design
; Diabetes mellitus
; Molecular docking
; ADMET
1. Introduction
Diabetes mellitus type 2 is one of the major health concerns of the 21st century in the globe. According to data, the prevalence of diabetes has been doubled in both developed and developing countries in the last two decades [1]. By 2030, more than a half billion people are anticipated to have diabetes worldwide [2]. Diabetes can hinder productivity by causing illness and death at an early age, and its impacts are likely to get worse as the disease starts to affect younger people [3]. Edwin Gale referred to diabetes as “the diabetes apocalypse” due to its prominence on the public health agenda as a global epidemic and a danger to human health and the global economy [4]. The worldwide expenditure on diabetes was at least USD 376 billion in 2010, and is projected to reach USD 490 billion by 2030 [5]. The diabetes epidemic and its effects are caused by complex genetic and epigenetic systems which interact with a system that is made up of many behavioral and environmental factors [1].
Diabetes mellitus is caused by a deficit or lower efficacy of endogenous insulin or the inappropriate use of insulin by target cells, and therefore. is characterized by an abnormal metabolism of carbohydrates, hypertension, and vasculature-affecting complications [6]. It can directly influence blood lipid levels, resulting in diabetes dyslipidaemia [7]. The basic characteristics of diabetic dyslipidemia include elevated blood levels of triglyceride (TG), decreased high density lipoprotein (HDL), and elevated levels of low-density lipoprotein (LDL) [8,9].
Currently, insulin and several oral antidiabetic medicines such as sulfonylureas, thiazolidinediones, and glucosidase inhibitors are being used to treat diabetes. To improve glycemic control, these medications can be used alone or in combination [10,11]. Good glycemic control slows the onset or progression of diabetes complications, but does not eliminate them altogether. In addition to the limitations described above, each of the aforementioned antidiabetic medicines is linked with a variety of major side effects [12,13]. Consequently, antidiabetic discovery has moved its attention to medicinal plants in order to provide innovative, promising, and cost-effective medications with minimal side effects [14]. There are more than one thousand plant species that are utilized as traditional medicines for diabetes mellitus all over the world [15]. The Cucurbitaceae family has long been a source of therapeutic compounds in research. Several plant components, particularly fruits, from this family have been studied for their pharmacological activities [16].
Bottle gourd (Lagenaria siceraria (Molina) Standl.), commonly known as white-flowered gourd or calabash, is a diploid species (2n) of the genus Lagenaria of the Cucurbitaceae family [17,18]. The genus Lagenaria includes six species: L. siceraria (Molina) Standl., L. abyssinica (Hook f.) Jeffrey, L. guineensis (G. Don) Jeffrey, L. ruffa (Gilg.) Jeffrey, L. breviflora (Benth.) Roberty, and L. sphaerica (Sonder) Naudin. All six species are found in Africa, which is regarded to be the center of the genetic diversity of L. siceraria [19]. It can be differentiated from other types of pumpkin by its white blooms, distinctive fruit, seed, and leaf forms [20]. Lagenaria siceraria is the only economically valuable species grown globally for a variety of purposes, including food, medicinal, adornment, and the production of household goods, and musical instruments. This fruit is found in two varieties: sweet and bitter. The sweet type is typically eaten as a vegetable, but the bitter, wild species is favored for medicinal purposes [21].
Bottle gourd has been linked to a variety of health advantages, including cardioprotective, diuretic, anti-cancer, aphrodisiac, general tonic, antidote to some poisons and scorpion stings, alternate purgative, and cooling properties. Fresh bottle gourd fruit juice is used to treat a variety of ailments, including diabetes, hypertension, flatulence, liver problems, and as a diuretic [22,23,24]. Syrup made from the succulent fruits can be used to treat a variety of respiratory conditions, as well as pain, ulcers, fever, asthma, and other bronchial problems. The fruit’s pulp has several medicinal uses, including as a cough remedy, an antidote to poisons, and a cooling diuretic [23,25].
Numerous chemical substances, such as sterols, terpenoids, flavonoids, and saponins, have been identified from this species. The fruit is said to include the triterepenoide cucurbitacins B, D, G, and H, two sterols, fucosterol and campesterol, aerpene bryonolic acid, flavone-C glycosides (a ribosome inactivating protein), and lagenin [26]. These chemical components are known for giving anti-diabetic properties. Therefore, the primary purpose of this study was to develop a useful pharmaceutical agent derived from the natural source. In light of this, the aim of the study is to treat diabetic mellitus using chemical components derived from Lagenaria siceraria applying computational tools like molecular docking, ADMET characteristics, Lipinski Rules, etc. This new technique ushers in a new age for constructing effective drugs and designing novel biological substances in a short amount of time and at a reduced cost [27]. This study, therefore, can open a new door for the development of a cost-effective diabetes treatment with fewer side effects.
2. Results
2.1. Structure optimization
2.2. Lipinski rule, Pharmacokinetics and Drug likeness
The data related to Lipinski rule are predicted using the online data set SwissADME, and the derived values are displayed in Table 2. The acquired values revealed that the molecular weight ranges from 176.12 to 610.56 Dalton, the Hydrogen bond acceptor ranges from 01 to 15 and the Hydrogen bond donor ranges from 01 to 8. The bioavailability index of the ligands is found to range from 0.17 to 0.85. Hence, according to the gathered data, with the exception of ligand 7, all other ligands obey the Lipinski rule. As nearly all of the compounds have accepted the Lipinski value, they can be recommended for oral medications and additional investigations, such as molecular docking, ADMET, and other relevant analyses.
2.3. Molecular docking
Ten targeted compounds and standard (Metformin hydrochloride) have been incorporated into molecular docking studies against two receptor-binding proteins, Human CYP3A4 bound to metformin (PDB ID: 5G5J) and Human dipeptidyl peptidase-IV (PDB ID: 4A5S). Human dipeptidyl peptidase-IV exhibited a maximal affinity of -9.7 Kcal/mol for bryonolic acid and hesperidin, but only -6.7 Kcal/mol for Metformin hydrochloride. Similarly, the highest affinity of hesperidin for Human CYP3A4 bound to metformin (PDB ID: 5G5J) was found to be -10.7 Kcal/mol. In contrast, the conventional Metformin hydrochloride achieved a value of -6,3 Kcal/mol. The results have been shown in Table 3.
2.4. Ligand-protein interaction and molecular docking poses
The interaction between protein and ligand has been visualized by Biovia discovery studio 2021. After molecular docking studies, this section describes how a targeted protein interacts to phytomolecules and how many active sites can be detected. Typically, hydrogen bonds and hydrophobic bonds account for binding energy [29]. In Figure 1, bryonolic acid has been shown to interact with Human dipeptidyl peptidase-IV. In the 2D structure, it is clearly seen that hydrogen and hydrophobic bonds are present which are bonded with ligand and they are responsible for the pharmacological efficacy. The most potential active amino acid residues present in Human dipeptidyl peptidase-IV (PDB ID: 4A5S) with bryonolic acid are ASP A:545, TRP A:629, TYR A:547, VAL A: 711, TYR A:662, and HIS A:740.
2.5. ADMET studies
The ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties are calculated using the web server pkCSM and presented in Table 4. Except for ligand 2, the results of the water solubility test showed that the remaining ligands are highly soluble in aqueous media, with the maximum Caco-2 permeability being 1.21 for ligands 06, 08, and 10. The G.I. absorption refers to how quickly the medications break down after entering the digestive system. Most of the compounds in this study have an excellent bioavailability rate, ranging from 92.03% to 99.55%, but only a small number of them dissociate badly in the gastrointestinal tract, with ranges of 39.15% and 31.48%. In order to produce a prospective drug candidate, the overall absorption rate is, therefore, fulfilled. In addition, none of them can cross the BBB. The VDss level varies from -1.009 to +0.338, with a maximum total clearance rate of 0.631 mL/min/kg. No ligand produces positive result for CYP450 1A2 Inhibitor, CYP 450 2C9 Substrate, or CYP450 3A4 Inhibitor; however, for CYP450 3A4 Substrate, ligand 01, 03, 04, 06, 08, 09, and 10 exerted a positive result.
Maximum compounds are free from AMES toxicity, hepatotoxicity and skin sensitization, but bryonolic acid, bryononic acid, and oleanolic acid may possess hepatic toxicity, and cucurbitacin G may show AMES toxicity as shown in Table 5. The maximum tolerated dose is 1.598 mg/kg/day, the maximum oral rat acute toxicity level is 2.553mol/kg and oral rat chronic toxicity 3.186 mg/kg/day.
3. Discussion
Ten bioactive compounds of various types reported from Lagenaria siceraria have been selected for assessing anti-diabetic activity. Pharmacological drug-likeness is a revolutionary step of the bioavailability assessment of a specific compound as an oral treatment [30]. It is predicted that nine out of ten targeted pharmaceuticals are not changed transparently owing to their adverse effects, leading to significant drug expenses, time, and human resources being squandered. This issue arises from an inability to determine the precise pharmacological properties [31,32]. Yet, by applying Lipinski’s five-rule (Ro5), it is feasible to easily assess the characteristics of lead compounds, including their bioavailability and G.I. absorption [33]. Orally active medicines are required to adhere to this guideline, which states that there cannot be more than one violation of the predetermined criterion. Hence, it was determined whether or not each docking chemical satisfied Lipinski’s Ro5 requirements [34,35]. With the exception of L-ascorbic acid (2), all of the targeted compounds, including bryonolic acid (1), bryonolol (3), bryononic acid (4), cucurbitacin G (5), fucosterol (6), hesperidin (7), isofucosterol (8), oleanolic acid (9), and spinasterol (10) exhibited at least one violation towards Ro5. Hesperidin (7), on the other hand, exhibited a maximum of three violations, which implies that it cannot be employed for oral dosage form. According to this rule, it is also suggested that, if a drug has two or more Ro5 violations, such as hesperidin does, then it demonstrates poor solubility or permeability [36]. As a result, this rule serves as a foundation for making predictions regarding the high possibility of success or failure of the development of a medicine based on a single chemical exhibiting a certain pharmacological or biological function [37,38].
Molecular docking is becoming increasingly popular among researchers, drug developers, and scientists as a result of its low cost and what appears to be its ease of usage [39]. This approach makes it feasible to make accurate predictions regarding the interaction that takes place in the coordination sphere between a ligand and a receptor [40,41]. In order to successfully conduct out molecular docking, there are three primary requirements that must be met: bond intensity, molecular interactions, and bond characterization. Lead compounds exhibited relatively low binding energies, hydrogen bonding, and van der Waals interactions as well as a favorable ADME profile [42,43,44].
Any biologically active compounds are regarded to have the potential to have pharmacological effects if they have a minimum binding energy of -6.0 Kcal/mole [45,46]. Therefore, all ten ligands showed greater affinity than standard Metformin hydrochloride towards two receptor-binding proteins. But, the highest affinity (-10.7 Kcal/mol) was shown by Hesperidin for Human CYP3A4 bound to metformin. It follows that the targeted molecules exerted better affinity than the original Metformin hydrochloride.
Non-covalent interactions, including hydrogen bond and hydrophobic interactions, play an important role in maintaining energetically favorable ligands at the active site of a specific macromolecular structure, hence boosting improved binding affinity and drug efficacy [47]. In an effort to realize that the protein-ligand complex system remained in a stable state throughout the molecular docking simulation, it was determined that the system maintained a steady state [48]. This experiment has been conducted to analyze and compare the stability of these conceptual connection models. In the binding pocket of protein ligands, it is evident how they interacted during the synthesis of complex molecules. Thus, amino acid residues that function throughout the interaction may be the most important determinant of the binding affinity of protein-ligand complexes [49,50]. Lastly, a 2D representation of active amino acid residues revealed the presence of distinct kinds of amino acids with varying bond angles. ASP A:545, TRP A:629, TYR A:547, VAL A: 711, TYR A:662, and HIS A:740 are among the amino acid residues formed during the formation of the protein-bryonolic acid complex.
In modern years, the in silico ADMET studies of potential bioactive compounds have been exploited in drug development as a main screening technique for the early discovery phase [51,52]. Early ADMET research screening may lower the risk of failure during the clinical study [53]. The pkCSM is one of the greatest predictors of these necessary ADMET data [54]. Except for ligand 2, the majority of the chemicals in this study were very soluble in water. Maximum substances were discovered to have an absorption rate greater than 90%, but there were also compounds with poor solubility; these data showed that these compounds have varying levels of absorption ability in the gastrointestinal system. Caco-2 permeability values varied between -0.25 and 1.21. The ligands cannot cross the blood-brain barrier (BBB), whereas their volume of distribution (VDss) ranged from -1.009 to 0.996 log L/kg. Only ligands 01, 03, 04, 06, 08, 09, and 10 may block the CYP450 3A4 substrate during metabolism. The total clearance rate was determined as -0.081mL/min/kg to 0.63mL/min/kg. The majority of them were found to be devoid of AMES mutagenicity, skin sensitization, and hepatotoxicity, as predicted by toxicological modelling. The toxicological data implied that these compounds have good physiochemical and pharmacokinetics properties.
The molecular docking of selected compounds with Human CYP3A4 and Human dipeptidyl peptidase-IV demonstrated that all of the examined ligand molecules exhibited excellent binding energy, and bryonolic acid displayed hydrogen and hydrophobic bonding with several amino acids in the active pockets. Thus, bryonolic acid had the highest affinity for human dipeptidyl peptidase-IV and possessed substantial antihyperglycemic effects.
4. Materials and Methods
4.1. Optimization and Ligand preparation
4.2. Protein preparation and Molecular Docking study
Two proteins (Human CYP3A4 linked to metformin with PDB ID: 5G5 and Human dipeptidyl peptidase-IV with PDB ID: 4A5S) were selected from the RSCB Protein Data Bank and were retrieved as PDB files. It was then analyzed and cleaned by Pymol (version 2) of all the excess molecules such as water and ligands in order to obtain a fresh protein. The nonbonding interaction of drug-protein was then done using the AutoDock Vina docking software tool. Docking studies were carried out utilizing the configurable ligand and receptor grid boxes in PyRx and AutoDock Vina Wizard, respectively. Finally, the visual depiction of the 2D interaction and active amino acid residues specific for the ligand in protein structure was created using Discovery Studio as shown in Table 6.
4.3. Lipinski rule, Pharmacokinetics and Drug likeness
Christopher A. Lipinski was the first person to present a description of the Lipinski rule in 1997 [34]. This rule describes the pharmacokinetic properties of a molecule by emphasizing the significance of molecular characteristics. This rule applies specially to oral drugs. Then, these drug similarity attributes have been evaluated using the Swiss ADME online open access database. Lipinski rule includes - Molar mass of the compound, Number of hydrogen bond acceptors, Number of hydrogen bond donors, Number of rotatable bonds, Molar refractivity, Topological Polar Surface Area (TPSA), Solubility, Lipinski’s rule of five, and Bioavailability score [57].

4.4. ADMET Properties
When developing a novel drug, it is essential to perform a comprehensive analysis of the pharmacokinetic properties of the compound [60]. The pkCSM program package (http://biosig.unimelb.edu.au/pkcsm/) is an online application that aids in the evaluation of physicochemical descriptors, ADME parameter prediction, pharmacokinetic characteristics, and toxicity aspects of any chemical compound [54]. In this study, the Water solubility Log S, Caco-2 Permeability, volume of distribution (VDss) of human, CYP450 1A2 Inhibitor, CYP450 3A4 Substrate, CYP450 3A4 Inhibitor, CYP450 2C9 Substrate, and Total Clearance (mL/min/kg) have been enumerated. Another important parameter, toxicity was assessed. This parameter included AMES (Salmonella/microsome mutagenicity assay) toxicity, maximum tolerable dose for humans (mg/kg/day), oral rat acute toxicity (LD50), oral rat chronic toxicity (LOAEL), hepatotoxicity, and skin sensitization.
5. Conclusions
In this study, the mechanism underlying the antidiabetic effects of some natural compounds obtained from Lagenaria siceraria is confirmed by computational approaches such as molecular docking, ADMET and drug likeness. The findings revealed that some of the reported molecules have antidiabetics activity, which suggested that these compounds could be potential to manage diabetes. The overall study has been conducted based on the concept of in silico method. Overall, analysis has documented that these natural ligands are capable of establishing various H-bonds as well as hydrophobic and van der walls interactions and are strongly bonded with the target receptors. Finally, these compounds should, therefore, undergo extensive in vitro and in vivo tests in wet lab to establish them as potential drug candidates.
Author Contributions
Conceptualization, M.S.A.; methodology, A.S.; software, A.S., F.N. and M.L.A.; validation, M.Z.S., A.A.C. and S.K.; formal analysis, A.S.; investigation, M.S.A., M.Z.S. and A.S.; resources, M.S.A., Z.A.C., A.A.C., and S.K.; data curation, M.S.A.; writing—original draft preparation, A.S., F.N.; writing—review and editing, M.S.A.; visualization, M.L.A.; supervision, M.S.A., M.Z.S., Z.A.C., A.A.C., and S.K.; project administration, M.S.A.; funding acquisition, M.S.A. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was supported by a grant from the Ministry of Science and Technology, Government of the People’s Republic of Bangladesh. (Project ID: SRG-224557, Dated: 14 November, 2022)
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to large size.
Acknowledgments
We express our gratitude to the authority of the Department of Pharmaceutical Chemistry for using the computers and other facilities of the Molecular Pharmacology and Herbal Drug Research Laboratory established under the HEQEP Project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zimmet, P.Z.; Magliano, D.J.; Herman, W.H.; Shaw, J.E. Diabetes: A 21st Century Challenge. Lancet Diabetes Endocrinol 2014, 2, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global Estimates of the Prevalence of Diabetes for 2011 and 2030. Diabetes Res Clin Pract 2011, 94, 311–321. [Google Scholar] [CrossRef]
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The Worldwide Epidemiology of Type 2 Diabetes Mellitus—Present and Future Perspectives. Nat Rev Endocrinol 2012, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Valodara, A.; Johar SR, K. TYPE 2 DIABETES MELLITUS AND ITS VASCULAR COMPLICATIONS. Towards Excellence 2022, 14. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, X.; Brown, J.; Vistisen, D.; Sicree, R.; Shaw, J.; Nichols, G. Global Healthcare Expenditure on Diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010, 87, 293–301. [Google Scholar] [CrossRef]
- Gad-Elkareem, M.A.M.; Abdelgadir, E.H.; Badawy, O.M.; Kadri, A. Potential Antidiabetic Effect of Ethanolic and Aqueous-Ethanolic Extracts of Ricinus Communis Leaves on Streptozotocin-Induced Diabetes in Rats. PeerJ 2019, 7, e6441. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.-Q.; Huang, W.; Wang, Y. SDF-1α and CXCR4 as Therapeutic Targets in Cardiovascular Disease. Am J Cardiovasc Dis 2012, 2, 20. [Google Scholar]
- Ramen, C.B.; Manas, C. An Overview on Management of Diabetic Dyslipidemia. Journal of Diabetes and Endocrinology 2013, 4, 27–36. [Google Scholar]
- Wu, L.; Parhofer, K.G. Diabetic Dyslipidemia. Metabolism 2014, 63, 1469–1479. [Google Scholar] [CrossRef]
- Nabi, S.A.; Kasetti, R.B.; Sirasanagandla, S.; Tilak, T.K.; Kumar, M.V.J.; Rao, C.A. Antidiabetic and Antihyperlipidemic Activity of Piper Longum Root Aqueous Extract in STZ Induced Diabetic Rats. BMC Complement Altern Med 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Turner, R.C.; Cull, C.A.; Frighi, V.; Holman, R.R.; Group, U.K.P.D.S. (UKPDS); Group, U.K.P.D.S. (UKPDS). Glycemic Control with Diet, Sulfonylurea, Metformin, or Insulin in Patients with Type 2 Diabetes Mellitus: Progressive Requirement for Multiple Therapies (UKPDS 49). JAMA 1999, 281, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Erejuwa, O.O. Oxidative Stress in Diabetes Mellitus: Is There a Role for Hypoglycemic Drugs and/or Antioxidants. Oxidative stress and diseases 2012, 217, 246. [Google Scholar]
- Toma, A. Recent Advances on Novel Dual-Acting Peroxisome Proliferator-Activated Receptor Alpha and Gamma Agonists. Int J Pharm Sci Res 2013, 4, 1644. [Google Scholar]
- Marles, R.J.; Farnsworth, N.R. Antidiabetic Plants and Their Active Constituents. Phytomedicine 1995, 2, 137–189. [Google Scholar] [CrossRef]
- Rao, M.U.; Sreenivasulu, M.; Chengaiah, B.; Reddy, K.J.; Chetty, C.M. Herbal Medicines for Diabetes Mellitus: A Review. Int J PharmTech Res 2010, 2, 1883–1892. [Google Scholar]
- Minocha, S. An Overview on Lagenaria Siceraria (Bottle Gourd). Journal of Biomedical and Pharmaceutical Research 2015, 4, 4–10. [Google Scholar]
- Beevy, S.S.; Kuriachan, P. Chromosome Numbers of South Indian Cucurbitaceae and a Note on the Cytological Evolution in the Family. J Cytol Genet 1996, 31, 65–71. [Google Scholar]
- Morimoto, Y.; Maundu, P.; Fujimaki, H.; Morishima, H. Diversity of Landraces of the White-Flowered Gourd (Lagenaria Siceraria) and Its Wild Relatives in Kenya: Fruit and Seed Morphology. Genet Resour Crop Evol 2005, 52, 737–747. [Google Scholar] [CrossRef]
- Mashilo, J.; Shimelis, H.; Odindo, A. Phenotypic and Genotypic Characterization of Bottle Gourd [Lagenaria Siceraria (Molina) Standl.] and Implications for Breeding: A Review. Sci Hortic 2017, 222, 136–144. [Google Scholar] [CrossRef]
- Cutler, H.C.; Whitaker, T.W. Cucurbits from the Tehuacan Caves; University of Texas Press, 1967. [Google Scholar]
- Upaganlawar, A.; Balaraman, R. Bottle Gourd (Lagenaria Siceraria) A Vegetable Food for Human Health-a Comprehensive Review. Pharmacol online 2009, 1, 209–226. [Google Scholar]
- Agrawal, S.S.; Mohale, D.S.; Ghule, B. v; Saoji, A.N.; Yeole, P.G. Studies on the Antihyperlipidemic Activity of Flavonoidal Fraction of Lagenaria Siceraria. International Journal of Chemical Sciences 2008, 6, 751–760. [Google Scholar]
- Duke, J.A. Handbook of Biologically Active Phytochemicals and Their Activities; CRC Press, Inc., 1992; ISBN 0849336708. [Google Scholar]
- Nadkarni, K.M. Indian Materia Medica, Bombay Popular Prakashan. Mumbai VI 1954, 629. [Google Scholar]
- van Wyk, B.-E.; Gericke, N. People’s Plants: A Guide to Useful Plants of Southern Africa; Briza publications, 2000; ISBN 1875093192. [Google Scholar]
- Abdin, M.Z.; Arya, L.; Saha, D.; Sureja, A.K.; Pandey, C.; Verma, M. Population Structure and Genetic Diversity in Bottle Gourd [Lagenaria Siceraria (Mol.) Standl.] Germplasm from India Assessed by ISSR Markers. Plant systematics and evolution 2014, 300, 767–773. [Google Scholar]
- Akash, S.; Chakma, U.; Chandro, A.; Matin, M.M.; Howlader, D. The Computational Screening of Inhibitor for Black Fungus and White Fungus by D-Glucofuranose Derivatives Using in Silico and SAR Study. Org. Commun 2021, 14, 336–353. [Google Scholar]
- Nicholls, A.; McGaughey, G.B.; Sheridan, R.P.; Good, A.C.; Warren, G.; Mathieu, M.; Muchmore, S.W.; Brown, S.P.; Grant, J.A.; Haigh, J.A. Molecular Shape and Medicinal Chemistry: A Perspective. J Med Chem 2010, 53, 3862–3886. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T.; Baker, D. A Simple Physical Model for Binding Energy Hot Spots in Protein–Protein Complexes. Proceedings of the National Academy of Sciences 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Ahire, E.D.; Sonawane, V.N.; Surana, K.R.; Talele, G.S. Drug Discovery, Drug-Likeness Screening, and Bioavailability: Development of Drug-Likeness Rule for Natural Products. In Applied pharmaceutical practice and nutraceuticals; Apple Academic Press, 2021; pp. 191–208. ISBN 1003054897. [Google Scholar]
- Kumer, A.; Chakma, U.; Rana, M.M.; Chandro, A.; Akash, S.; Elseehy, M.M.; Albogami, S.; El-Shehawi, A.M. Investigation of the New Inhibitors by Sulfadiazine and Modified Derivatives of α-d-Glucopyranoside for White Spot Syndrome Virus Disease of Shrimp by in Silico: Quantum Calculations, Molecular Docking, ADMET and Molecular Dynamics Study. Molecules 2022, 27, 3694. [Google Scholar] [CrossRef]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to Identify Potential Natural Antiviral for COVID-19 Treatment. J Biomol Struct Dyn 2021, 39, 4225–4233. [Google Scholar] [CrossRef]
- Rashid, M. Design, Synthesis and ADMET Prediction of Bis-Benzimidazole as Anticancer Agent. Bioorg Chem 2020, 96, 103576. [Google Scholar] [CrossRef]
- Walters, W.P. Going Further than Lipinski’s Rule in Drug Design. Expert Opin Drug Discov 2012, 7, 99–107. [Google Scholar] [CrossRef]
- Hill, R.; Kenakin, T.; Blackburn, T. Pharmacology for Chemists: Drug Discovery in Context; Royal Society of Chemistry, 2017; ISBN 1782621423. [Google Scholar]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T. Bin Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (M pro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica (Cairo) 2020, 2020. [Google Scholar]
- Lionta, E.; Spyrou, G.; K Vassilatis, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr Top Med Chem 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angewandte Chemie International Edition 2013, 52, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; An, J.; Zhao, J.; Zhao, S.; Lv, H.; Wang, S. Drug-Target Interaction Prediction Based on Multisource Information Weighted Fusion. Contrast Media Mol Imaging 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.A.; Ayyad, R.R.A.; Zayed, M.F.; Abulkhair, H.S.; Elkady, H.; El-Adl, K. Design, Synthesis, in Silico ADMET Profile and GABA-A Docking of Novel Phthalazines as Potent Anticonvulsants. Arch Pharm (Weinheim) 2019, 352, 1800387. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase in Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Schulz-Gasch, T.; Stahl, M. Binding Site Characteristics in Structure-Based Virtual Screening: Evaluation of Current Docking Tools. J Mol Model 2003, 9, 47–57. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein− Ligand Complexes. J Med Chem 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Kumer, A.; Chakma, U.; Matin, M.M.; Akash, S.; Chando, A.; Howlader, D. The Computational Screening of Inhibitor for Black Fungus and White Fungus by D-Glucofuranose Derivatives Using in Silico and SAR Study. Organic Communications 2021, 14. [Google Scholar] [CrossRef]
- Rahman, M.; Islam, M.; Akash, S.; Mim, S.; Rahaman, M.; Bin Emran, T.; AKKOL, E.; Sharma, R.; Alhumaydhi, F.; Sweilam, S. In Silico Investigation and Potential Therapeutic Approaches of Natural Products for COVID-19: Computer-Aided Drug Design Perspective. Front Cell Infect Microbiol 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Pagano, B.; Cosconati, S.; Gabelica, V.; Petraccone, L.; De Tito, S.; Marinelli, L.; La Pietra, V.; Saverio di Leva, F.; Lauri, I.; Trotta, R. State-of-the-Art Methodologies for the Discovery and Characterization of DNA G-Quadruplex Binders. Curr Pharm Des 2012, 18, 1880–1899. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I. A Simple Principle for Understanding the Combined Cellular Protein Folding and Aggregation. Curr Protein Pept Sci 2020, 21, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J Chem Inf Comput Sci 2001, 41, 856–864. [Google Scholar] [CrossRef]
- Gohlke, H.; Klebe, G. Approaches to the Description and Prediction of the Binding Affinity of Small-molecule Ligands to Macromolecular Receptors. Angewandte Chemie International Edition 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Liu, G.; Tang, Y. In Silico ADMET Prediction: Recent Advances, Current Challenges and Future Trends. Curr Top Med Chem 2013, 13, 1273–1289. [Google Scholar] [CrossRef]
- Daoud, N.E.-H.; Borah, P.; Deb, P.K.; Venugopala, K.N.; Hourani, W.; Alzweiri, M.; Bardaweel, S.K.; Tiwari, V. ADMET Profiling in Drug Discovery and Development: Perspectives of in Silico, in Vitro and Integrated Approaches. Curr Drug Metab 2021, 22, 503–522. [Google Scholar] [CrossRef]
- Borah, P.; Hazarika, S.; Deka, S.; Venugopala, K.N.; Nair, A.B.; Attimarad, M.; Sreeharsha, N.; Mailavaram, R.P. Application of Advanced Technologies in Natural Product Research: A Review with Special Emphasis on ADMET Profiling. Curr Drug Metab 2020, 21, 751–767. [Google Scholar] [CrossRef]
- Pires, D.E. V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Milne, G.W.A. Software Review of ChemBioDraw 12.0 2010.
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J Cheminform 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov Today Technol 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Sevrioukova, I.F.; Denisov, I.G.; Zhang, X.; Chiu, T.-L.; Thomas, D.G.; Hanse, E.A.; Cuellar, R.A.D.; Grinkova, Y.V.; Langenfeld, V.W. Heme Binding Biguanides Target Cytochrome P450-Dependent Cancer Cell Mitochondria. Cell Chem Biol 2017, 24, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.M.; Clark, D.E.; Dunsdon, S.J.; Fenton, G.; Fillmore, A.; Harris, N.V.; Higgs, C.; Hurley, C.A.; Krintel, S.L.; MacKenzie, R.E. Novel Heterocyclic DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. Bioorg Med Chem Lett 2012, 22, 1464–1468. [Google Scholar] [CrossRef]
- Shi, S. Biologics: An Update and Challenge of Their Pharmacokinetics. Curr Drug Metab 2014, 15, 271–290. [Google Scholar] [CrossRef]
Figure 1.
Human dipeptidyl peptidase-IV (PDB ID: 4A5S) with Bryonolic acid.
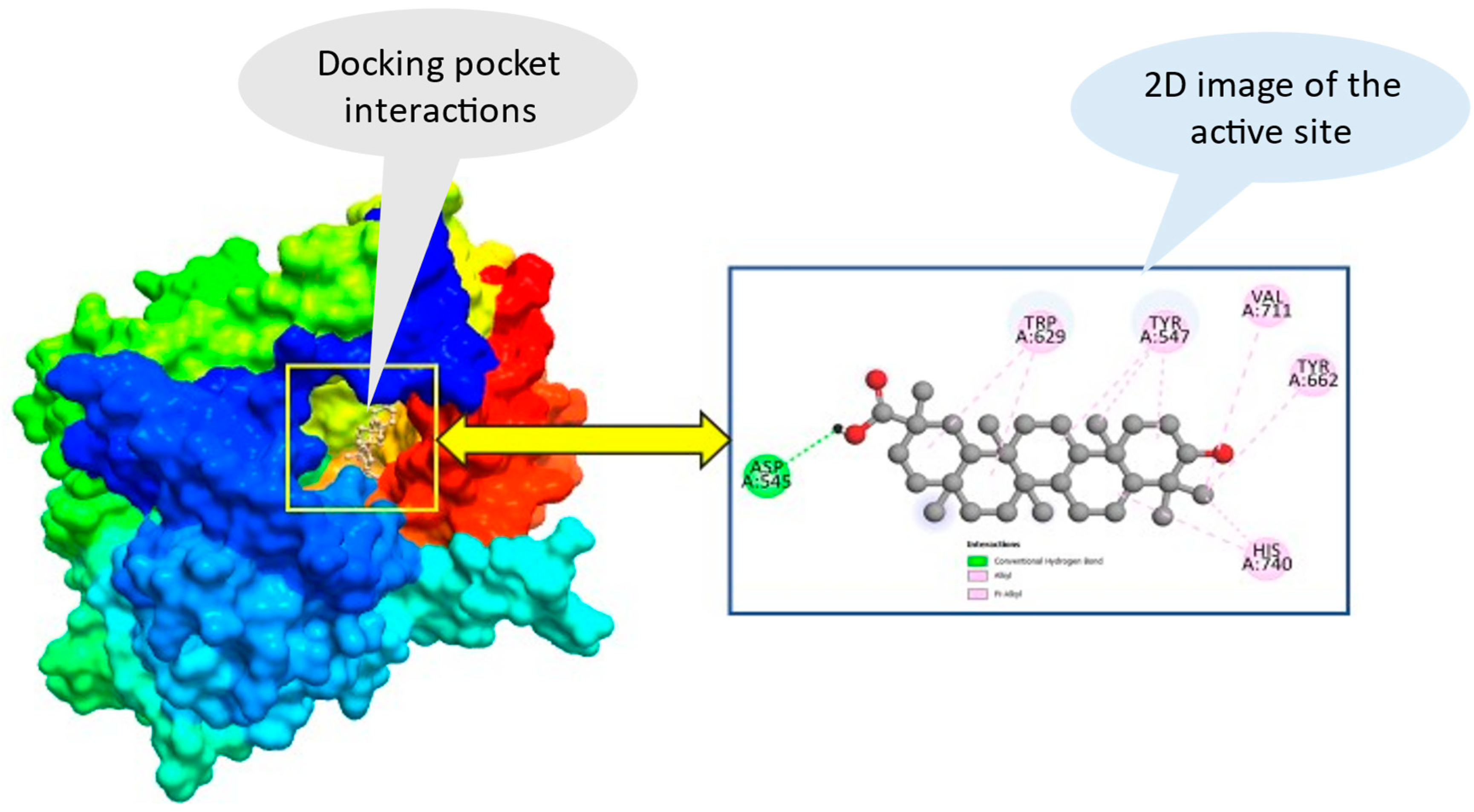
Table 1.
Optimized chemical structures of phytoconstituents reported from Lagenaria siceraria.
Bryonolic acid
|
L-ascorbic acid
|
Bryonolol
|
Bryononic acid |
Cucurbitacin G
|
Fucosterol
|
Hesperidin
|
Isofucosterol
|
Oleanolic acid
|
Spinasterol
| ||
Table 2.
Data of Lipinski rule, Pharmacokinetics and Drug likeness.
| Ligand No. | Parameters of Lipinski rule, Pharmacokinetics and Drug likeness | |||||||
| PubChem ID | Molecular weight (Dalton) | Hydrogen bond acceptor | Hydrogen bond donor | Topological polar surface area (Ų) | Lipinski rule | Bioavailability Score | ||
| Result | violation | |||||||
| 01 | 159970 | 456.7 | 3 | 2 | 57.53 | Yes | 1 | 0.85 |
| 02 | 54670067 | 176.12 | 6 | 4 | 107.22 | Yes | 0 | 0.55 |
| 03 | 15756408 | 442.72 | 2 | 2 | 40.46 | Yes | 1 | 0.55 |
| 04 | 472768 | 456.7 | 3 | 2 | 57.53 | Yes | 1 | 0.85 |
| 05 | 441818 | 534.68 | 8 | 5 | 152.36 | Yes | 1 | 0.55 |
| 06 | 5281328 | 412.69 | 1 | 1 | 20.23 | Yes | 1 | 0.55 |
| 07 | 10621 | 610.56 | 15 | 8 | 234.29 | No | 3 | 0.17 |
| 08 | 5281326 | 412.69 | 1 | 1 | 20.23 | Yes | 1 | 0.55 |
| 09 | 10494 | 456.7 | 3 | 2 | 57.53 | Yes | 1 | 0.85 |
| 10 | 5281331 | 412.69 | 1 | 1 | 20.23 | Yes | 1 | 0.55 |
Table 3.
Summary of molecular docking.
| No | Name | PubChem CID | Human CYP3A4 bound to metformin (PDB ID: 5G5J) | Human dipeptidyl peptidase-IV (PDB ID: 4A5S) |
| Binding affinity (Kcal/mol) | Binding affinity (Kcal/mol) | |||
| 01 | Bryonolic acid | 159970 | -9.3 | -9.7 |
| 02 | L-ascorbic acid | 54670067 | -9.9 | -9.1 |
| 03 | Bryonolol | 15756408 | -8.3 | -9.5 |
| 04 | Bryononic acid | 472768 | -9.7 | -9.2 |
| 05 | Cucurbitacin G | 441818 | -9.6 | -9.0 |
| 06 | Fucosterol | 5281328 | -9.9 | -9.1 |
| 07 | Hesperidin | 10621 | -10.7 | -9.7 |
| 08 | Isofucosterol | 5281326 | -9.2 | -9.3 |
| 09 | Oleanolic acid | 10494 | -9.1 | -9.1 |
| 10 | Spinasterol | 5281331 | -9.5 | -9.2 |
| Metformin hydrochloride | 14219 | -6.3 | -6.7 | |
Table 4.
ADME studies of selected compounds reported from L. siceraria.
| Sl. No | Absorption | Distribution | Metabolism | Excretion | ||||||||
| Water solubility LogS | Caco-2 permeability | Human Intestinal Absorption (%) | VDss (human) | BBB Permeability | CYP450 1A2 Inhibitor | CYP 450 2C9 Substrate | CYP450 3A4 Substrate | CYP450 3A4 Inhibitor | Total Clearance (mL/min/kg) |
Renal OCT2 substrate | ||
| 01 | -3.45 | 1.14 | 98.11 | -0.853 | No | No | No | Yes | No | -0.036 | No | |
| 02 | -1.55 | -0.25 | 39.15 | 0.218 | No | No | No | No | No | 0.631 | No | |
| 03 | -6.33 | 1.1 | 92.03 | 0.338 | No | No | No | Yes | No | 0.067 | No | |
| 04 | -3.46 | 1.15 | 98.42 | -0.799 | No | No | No | Yes | No | -0.036 | No | |
| 05 | -4.42 | 0.43 | 69.47 | -0.398 | No | No | No | No | No | 0.322 | No | |
| 06 | -6.75 | 1.21 | 94.64 | 0.179 | No | No | No | Yes | No | 0.619 | No | |
| 07 | -3.01 | 0.50 | 31.48 | 0.996 | No | No | No | No | No | 0.211 | No | |
| 08 | -6.71 | 1.21 | 94.64 | 0.179 | No | No | No | Yes | No | 0.619 | No | |
| 09 | -3.26 | 1.16 | 99.55 | -1.009 | No | No | No | Yes | No | -0.081 | No | |
| 10 | -6.68 | 1.21 | 94.97 | 0.178 | No | No | No | Yes | No | 0.611 | No | |
Table 5.
Toxicity prediction of selected compounds reported from L. siceraria.
| Sl. No. | Compound name | AMES toxicity | Max. tolerated dose (human) mg/kg/day |
Oral Rat Acute Toxicity (LD50) (mol/kg) | Oral Rat Chronic Toxicity (mg/kg/day) |
Hepatotoxicity | Skin Sensitization |
| 01 | Bryonolic acid | No | 0.098 | 2.294 | 2.065 | Yes | No |
| 02 | L-ascorbic acid | No | 1.598 | 1.063 | 3.186 | No | No |
| 03 | Bryonolol | No | -0.863 | 2.213 | 2.031 | No | No |
| 04 | Bryononic acid | No | 0.098 | 2.294 | 2.065 | Yes | No |
| 05 | Cucurbitacin G | Yes | -0.461 | 2.502 | 2.001 | No | No |
| 06 | Fucosterol | No | -0.653 | 2.553 | 0.89 | No | No |
| 07 | Hesperidin | No | 0.525 | 2.506 | 3.167 | No | No |
| 08 | Isofucosterol | No | -0.653 | 2.553 | 0.89 | No | No |
| 09 | Oleanolic acid | No | 0.094 | 2.196 | 2.109 | Yes | No |
| 10 | Spinasterol | No | -0.664 | 2.54 | 0.872 | No | No |
| Metformin hydrochloride | Yes | 0.874 | 2.465 | 2.122 | No | Yes | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



