
Submitted:
30 September 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
SORL1 gene encodes LR11/SorLA, a protein that binds -amyloid precursor protein (bAPP) and drives its intracellular trafficking. SORL1 mutations occurring frequently in a subset of familial cases of Alzheimer’s disease (AD) have been documented but their pathogenic potential is not yet clear and questions remain concerning their putative influence on the physiopathological processing of bAPP. We have assessed the influence of three SORL1 mutations that were described as likely disease-causing and that were associated with either benign (SorLA924) or severe (SorLA141 and SorLA511) AD phenotypes. We examined the influence of wild-type and mutants SorLA in transiently transfected HEK293 cells expressing either wild-type or Swedish mutated bAPP on bAPP expression, secreted Ab and sAPPa levels, intracellular Ab40 and Ab42 peptides, APP-CTFs (C99 and C83) expressions, a-, b- and g-secretases expressions and activities as well as Ab and CTFs-degrading enzymes. These paradigms were studied in control conditions or after pharmacological proteasomal modulation. We also established stably transfected CHO cells expressing wild-type SorLA and established the colocalization of wild-type SorLA and bAPP. Overall, although we mostly confirmed previous data concerning the influence of wild-type SorLA on bAPP processing, we were unable to evidence significant alterations triggered by our set of SorLA mutants, whatever the cells or pharmacological conditions examined. Our study however does not rule out the possibility that other AD-linked SORL1 mutations could indeed affect bAPP processing and that pathogenic mutations examined in the present study could interfere with other cellular pathways/triggers in AD.
Keywords:
SORL1
; mutations
; secretases
; bAPP
; Ab peptides
; C-terminal fragments
; neprilysin
; proteasome
; degradation
; cellular localization
; transient and stable expressions.
1. Introduction
Alzheimer’s disease (AD) is a complex pathology characterized by either early or late onsets. Most of early onset (EOAD) cases are of familial origin and follow an autosomal dominant transmission [1,2,3] while late onset (LOAD) cases usually referred to as sporadic AD cases have been shown to be associated to various risk factors [4,5]. The recent occurrence of genome wide associations studies allowed elucidating some of these risk factors [6,7]. Indeed, cohorts built with growing numbers of cases coupled to powerful bioinformatics means, allowed analysis of this bulk of data and led to a still increasing number of gene candidates that could underlie individual susceptibility to AD pathology.
Contradicting the apparent dichotomy between familial and sporadic AD cases, SORL1 gene has been implicated in both EOAD and LOAD. Thus, Rogaeva and Colleagues [8] first evidenced a genetic link between SORL1 and AD. This was further corroborated by independent meta analyses confirming the link between SORL1 variants and sporadic AD cases [9,10,11]. Interestingly, exome sequencing also delineated SORL1 mutations in both EOAD and LOAD cases [12,13].
SORL1 encodes a sortilin-related receptor with A-type repeats named SorLA (also referred to as LR11) [8]. This protein has been characterized as a neuronal sorting receptor for β-amyloid precursor protein (βAPP), the precursor of Aβ peptides that accumulate in AD-affected brains [14]. The ability of SorLA to physically interact with βAPP drives its lysosomal sorting and was shown to be affected by familial AD mutations [13,15]. This was reported to yield functional consequences on the levels of Aβ load in cells [13,16] and animals [15]. However, it appears that the phenotypic alterations on Aβ peptides levels as well as the nature of Aβ peptides modified varied according to the degree of rarity of the SORL1 variants [16] as well as cellular and experimental settings. Furthermore, very few studies examined the direct influence of SorLA mutations on other steps of the βAPP physiopathological maturation.
Here we exhaustively examined the influence of three frequent SORL1 mutations linked to either benign of severe autosomal dominant cases of AD on the amyloidogenic and non amyloidogenic βAPP products, secretases expressions and activity and Aβ degradation processes.
2. Materials and Methods
2.1. Constructs and site-directed mutagenesis
Wild-type human SORL1 cDNA, cloned in the pcDNA3.1 (+) vector, was provided by one of us (P. St. G-H). Site-directed mutagenesis kits from Agilent (Santa Clara, QuikChange II) and mutagenesis primers (Eurogentec, Belgium) were used to obtain the SorLA141, SorLA511 and SorLA924 mutants (Table 1). All the nucleotidic modifications were verified by full sequencing of the constructs.
2.2. Cell cultures and transfections
Human Embryonic Kidney 293 cells (HEK293) expressing wild-type βAPP (wt-APP) or swedish-mutated βAPP (swe-APP) were cultured (5% CO2, 37°C) in DMEM (Invitrogen, Carlsbad) containing Fetal Bovine Serum (FBS 10%, Sigma, Saint Louis) and antibiotics (50 U/ml penicillin / 50 μg/ml streptomycin, Invitrogen). Wild-type or mutant SORL1 cDNA were transiently transfected (24h) using jetPRIME reagent (Polyplus, Strasbourg) (2μg or 10μg of cDNA in 35-mm or 100-mm dishes, respectively) in control or inhibitors conditions (lactacystin, 5μM). In alkalizing conditions, cells were treated for 16h at 37°C with NH4Cl (10mM). Chinese Hamster Ovary cells (CHO) expressing wt-APP were obtained by stable transfection of pcDNA4 vector [17]. Cells were maintained in DMEM containing 10% FBS, sodium hypoxanthine-thymidine supplement, and 300 μM proline. Cells were stably transfected with 2 μg of wild-type or mutant SORL1 cDNA according to Lipofectamine protocols (Thermo Fisher Scientific). Clones were selected with 250 μg/ml Zeocin (Invitrogen).
2.3. Cells immunostaining
CHO cells grown on coverslips were fixed in paraformaldehyde 4% solution for 10 min, permeabilized withTriton-X 100 (0.1%) for 10 min, saturated in BSA (5%) / Tween20 (0.1%), and probed for 1h with appropriate primary antibodies: β-Amyloid (clone 6E10, Sigma, Saint Louis) mouse monoclonal (1:1000) or SorLA rabbit polyclonal (1:1000, Sigma, Saint Louis). After washes, coverslips were incubated for 1h with Alexa Fluor-488 and Alexa Fluor-594 conjugated antibodies (Molecular Probes, 1:1000) and DAPI (1:20000, Roche) staining. Finally, the sections were washed with PBS, then mounted onto glass slides and cover-slipped. The stained slices were kept at 4°C before analysis with confocal microscopy (Zeiss LSM 780 with 63X Objective) [18].
2.4. sAPPα secretion and total secreted Aβ Immunoprecipitation
Cells were grown in 6-well plates and allowed to secrete Aβ for 16h in OptiMEM (1ml, Invitrogen, Carlsbad) + 1% FBS (Sigma, Saint Louis) containing phosphoramidon (10μM, Sigma, Saint Louis) in order to prevent Aβ degradation by neprilysin as described [19]. Media were collected, completed with one tenth of 10X RIPA buffer (Tris-HCl pH 8.0 (100mM) containing NaCl (1.5M) and EDTA (50mM)). sAPPα secretion was measured in 20μl of medium collected and deposited on an 8% Tris-Glyine gel [20]. For Aβ immunoprecipitation, the remaining secretate was incubated overnight with a 100-fold dilution of 6E10 (Eurogentec, Belgium) and protein A agarose beads (Invitrogen, Carlsbad). Beads were washed twice with 1X RIPA and subjected to Tris/tricine 16.5% polyacrylamide gel. Proteins were transferred onto nitrocellulose membranes and incubated overnight with the 6E10 monoclonal antibody at a 1/1000 dilution. Immunological complexes were detected with a goat anti-mouse peroxidase-conjugated antibody (1/2000 dilution). Chemiluminescence was recorded using a Luminescence Image Analyser LAS-4000 (FujiFilm, Tokyo) and quantifications were performed using the MultiGAUGE analyser software.
2.5. Sandwich ELISA of secreted and intracellular Aβ
Cells were grown in 6-well plates and allowed to secrete Aβ as described above. Intracellular Aβ peptides were recovered as described [21]. Aβ40 and Aβ42 were measured using human Aβ40 and Aβ42 ELISA kits (Invitrogen, Carlsbad) as described [22] . The minimal detectable amount of human Aβ42 is <10pg/ml and human Aβ40 is <6pg/ml.
2.6. Western blotting
SorLA, neomycin, βAPP, sAPPα, CTFs and β-tubulin were separated on Tris-glycine (8%) or Tris-tricine (16.5%) gels. Proteins were transferred onto Hybond-C membranes (GE Healthcare, Boston) and then probed with the following antibody: anti-SorLA (antibodies-online, Aachen), anti-neomycin (Merck Millipore, Darmstadt), 22C11 (anti-N-terminal sequence of βAPP), 2H3 (anti-human Aβ targeting sequence 1-12 that reacts with the N-terminus of sAPPα but not sAPPβ); BR188 (anti-C-terminal sequence of βAPP, provided by Dr. M. Goedert); anti-PS1-NT raised against residues 1–65 of human PS1 (kind gift from Dr. Fraser; anti-PS2-Loop raised against residues 269–394 from the intracellular loop region of human PS2 (provided by Dr. Thinakaran; anti-APH1aL raised against the C-terminal region of human APH1aL (kind gift from one of us, PSGH); anti-PEN2 raised against the last 24 amino acids of human PEN2 (CR8, Covance); anti nicastrin was a goat polyclonal antibody raised against the N terminus of human nicastrin (sc-14369, Santa Cruz Biotechnology, Inc.); anti-β-tubulin (Sigma, Saint Louis). Immunological complexes were revealed with either anti-mouse or anti-rabbit peroxidase antibodies (Beckman Coulter, Fullerton), followed by electrochemiluminescence.
2.7. In vitro γ-secretase Assay
γ-secretase assay by means of reconstituted membranes was carried out as described previously [23]. An equal amount of membranes preparations was incubated overnight with a recombinant C100-FLAG corresponding to the β-secretase-derived βAPP fragment harboring a methionine residue in position 1 [23]. Aβ and AICD-FLAG were detected by Western blotting on a 16.5% Tris/tricine gel and revealed with either anti-Aβ 2H3 or anti-FLAG antibodies, respectively.
2.8. BACE1 fluorimetric Assay
BACE1 activity was followed with (7-methoxycoumarin-4-yl)acetyl-SEVNLDAEFRK(2,4-dinitrophenyl)-RRNH2; 10μM, R&D Systems, Minneapolis) in absence or in the presence of β-secretase inhibitor I (50μM, PromoCell, Heidelberg) as described previously [24]. BACE1 activity corresponds to the β-secretase inhibitor-sensitive fluorescence recorded at 320 and 420 nm as excitation and emission wavelengths, respectively.
2.9. a-secretase activity on intact cells
Plated confluent HEK293 cells were pretreated for 30 min at 37°C with 1ml of PBS supplemented with or without the zinc metalloprotease inhibitor o-phenanthroline (100 μM) then the quenched fluorimetric α-secretase substrate JMV2770 (10 μM, provided by Dr. Hernandez), was directly added to the cultured cells for various times as described [25]. At each incubation time period, 100μl of medium were collected and fluorescence was recorded in a 96-well plates at 320 and 420 nm as excitation and emission wavelengths, respectively. After removal of the last sample, cells were resuspended in lysis buffer (10 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 0.5% deoxycholate, and 5 mM EDTA), protein concentrations were determined by the Bradford method [26], and all fluorimetric values were normalized according to protein contents.
2.10. Neprilysin activity measurements.
Neprilysin (NEP) activity was followed as described previously [27]. Briefly, cell homogenate samples (50μg of proteins) were incubated in a final volume of 100μl containing NEP substrate (Suc-Ala-Ala-Phe-7AMC, 20μM, Sigma, Saint Louis) in the absence or presence of the NEP inhibitor phosphoramidon (10μM, Sigma, Saint Louis). NEP activity was considered as the phosphoramidon-sensitive fluorescence recorded at 390 and 460 nm as excitation and emission wavelengths.
2.11. In vitro cathepsin B activity assay
HEK293 cells were lysed mechanically in homogenization buffer (250mM sucrose, 1mM EDTA, 5mM Hepes pH 7.4) using firstly a Dounce homogenizer then with a seringe. The cell suspension was centrifuged for 5 min at 850 x g then the supernatant was further centrifuged for 90 min at 20000 x g. The pellet (membrane-enriched fraction) was resuspended in Tris-HCl (10mM, pH 7.5) and all samples were adjusted to 6μg/μl before analysis. Cathepsin B activity was monitored as described [21] by incubating samples (60μg of protein extracts) in a final volume (100μl) of acetate buffer (25mM, pH 5.5, L-cysteine HCl, 8mM) containing cathepsin B substrate (carboxybenzoyl-Arg-Arg-7-Amido-4-methylcoumarin, (100μM, Sigma, Saint Louis) in the absence or presence of leupeptin (10μM, Sigma, Saint Louis). Specific cathepsin B activity was considered as the leupeptin-sensitive fluorescence recorded at 320 nm (excitation) and 420 nm (emission) using a fluorescence plate reader (FLUOstar Omega, BMG Labtech, Ortenberg). Fluorescence was recorded every 5 min during 150 min and cathepsin B activity was calculated as the slope in initial velocity conditions, i.e in the linear part of the curve corresponding to the initial 30 min.
2.12. Statistical Analysis
Statistical analyses were performed with PRISM Software (Graph-Pad Software, San Diego) by using the unpaired Student’s t-test for pairwise comparisons.
3. Results
Expression and fate of SorLA mutant proteins in wt-APP- and swe-APP-expressing cells.
Since SorLA protein is involved in βAPP lysosomal sorting [28] and because wt-APP and swe-APP traffic differently [29], we have first examined the influence of SORL1 mutations on βAPP physiopathological maturation in both wt-APP- and swe-APP-expressing HEK293 cells. First, we assessed wt and mutant SorLA expressions after SORL1 cDNA transient transfection. We show that wt, SorLA511 and SorLA924 were similarly expressed in wt- or swe-APP cells while SorLA141 was not detected (Figure 1A, upper panel). The lack of SorLA141 detection was not due to inefficient transfection process as indicated by efficient and similar neomycin expression (Figure 1A, lower panel). Thus, we envisioned the possibility that the absence of SorLA141 could be linked to its instability due to rapid degradation as we previously established for some parkin mutants [30]. Lactacystin, a proteasome inhibitor [31] that potentiates recovery of AD-related proteins [32,33] indeed enhances wt, SorLA511 and SorLA924 expressions in both wt- and swe-APP cells but failed to uncover SorLA141 (Figure 1B).
Interestingly, as previously described [16], wt-APP expression appeared increased by wt-SorLA expression but this augmentation was not potentiated by SorLA mutations (Figure 2A).
In order to confirm these observations, we established stable transfected CHO cells expressing either wild-type or mutated SorLA. In agreement with the above described data, we detected clones expressing wt-SorLA, SorLA511 and SorLA924 proteins (selected clones for further analyses are indicated in red circles, see Supplementary Figure S1A–C, but none of them express significant levels of SorLA141 protein (Supplementary Figure S1A–C). Of note, as was observed in transiently transfected cells, SorLA, SorLA511 and SorLA924 expressions were potentiated upon proteasomal inhibition while this pharmacological treatment did not allow unravelling SorLA141 protein (Supplementary Figure S1D).
These stably transfected cells also proved useful to examine the putative co-localization of SorLA proteins with wt-APP (see expressions of wt-APP in Supplementary Figure S1A–C). Supplementary Figure S1A–C (lower panels) indicates that wt-SorLA, SorLA511 and SorLA924 were readily detectable in these cells and colocalized with wt-APP (Supplementary Figure S1A-B, see merge panels) while immunohistochemical analyses confirmed the lack of in situ expression of SorLA141 (Supplementary Figure S1C). Closer analysis of wt-APP and wt-SorLA colocalization indicates that both are readily detectable in intracellular vesicular structures, identifiable by ponctiform labeling in the cell cytoplasm (Supplementary Figure S2, right panels) and that this colocalization appears to be partly disrupted when SorLA is mutated at 511 or 924 (Supplementary Figure S2, left panel). This above set of data allows concluding that: 1) wt, SorLA511 and SorLA924 expressions are not modulated by the nature of the βAPP expressed (wt- or Swe-APP) nor by the cell type and transfection procedures (transient transfection in HEH293 or stable transfection in CHO); 2) wt, SorLA511 and SorLA924 expressions are potentiated by proteasome inhibitors; 3) wt-APP and SorlA co-localize at a subcellular level and 511 and 924 SorLA mutations reduce the ability of SorLA to colocalize with wt-APP. 4) SorLA141 is not detectable in both transient and stable transfectants, even after proteasomal pharmacological blockade.
3.1. Influence of wild-type SorLA and its mutants on endogenous βAPP expression and on its non amyloidogenic proteolysis.
Since SorLA modulates APP trafficking, we first examined whether cellular expressions of wt-APP and/or swe-APP could be affected by SorLA mutations in transiently transfected HEK293 cells. We first observed a slight but statistically significant increase in endogenous wt-APP expression triggered by wt-SorLA expression (Figure 2A). These data appear similar in stably transfected cells, which display an increased βAPP expression linked to wt-SorLA expression (Supplementary Figure S3A). However, neither endogenous APP (Figure 2A), overexpressed wt-APP (Figure 3A) nor swe-APP (Figure 3B) expressions were affected by SorLA mutations in both transient (Figs.2 and 3) and stable (Supplementary Figure S3A) transfectants.
βAPP undergoes both constitutive and regulated non amyloidogenic processing by α-secretase, that leads to sAPPα secretion [20] and concomitant formation of its C-terminal counterpart C83 [34]. We show that endogenous secreted sAPPα was reduced in cells overexpressing wt-SorLA (Figure 2B). As expected, wt-APP expression increases sAPPα in stably transfected cells (compare mock and DNA3 lanes in Supplementary Figure S3B). Of note, the recovery of endogenous sAPPα (Figure 2B) or sAPPα recovered in both stably (compare DNA3 and mutants lanes in Supplementary Figure S3B) and transiently (Figure 4A) transfected cells were similarly reduced by wt and mutated SorLA. SorLA mutations also did not affect sAPPα recovery in swe-APP-expressing cells (Figure 4B). This set of data indicating a lack of influence of SorLA mutations on the non-amyloidogenic pathwway of βAPP was confirmed by direct fluorimetric measurement of α-secretase activity by recording the phenanthroline–sensitive JMV2770-hydrolyzing activity [25] on intact wt-and swe-APP cells. Thus, no difference in α-secretase activity in wt- and swe-APP plated cells was observed (Figure 4C,D).
3.2. Influence of wild-type SorLA and its mutants on Aβ peptides and γ-secretase expression and activity.
We aimed at examining the influence of SorLA on Aβ production/recovery in our transient and stably transfected cells.
We first measured total Aβ peptides recovery in secretates of wt- APP-expressing cells by immunoprecipitation (Supplementary Figure S4A) in the presence of the inhibitor phosphoramidon in order to prevent neprilysin-dependent degradation of Aβ peptides [27,35,36,37]. As previously reported, wt-SorLA reduces total Aβ, the recovery of which was similarly triggered by SorLA511 and SorLA141 (Supplementary Figure S4A). It is well known that Aβ is mainly a mix of Aβ40 and Aβ42 [34], that the latter accounts for about 10% of total Aβ and that slight modifications of Aβ42/40 ratio could drive toxic phenotypes in cells [38]. Since, tiny modulation of Aβ42 that could have been underscored in total Aβ immunoprecipitation procedure, we have delineated the respective levels of Aβ40 and Aβ42 by sensitive ELISA. As previously described, Aβ42 levels correspond to about 10-15% of Aβ40 (compare values in wt condition in Supplementary Figure S4A,B). However, we did not observe SorLA-related modifications of Aβ40 and Aβ42 recoveries except for the 924 SorLA mutant that triggers a faint increase of secreted Aβ42 (Supplementary Figure S4B) but not Aβ40 (Supplementary Figure S4A). Overall, the above data were confirmed in wt-APP and Swe-APP-expressing transient transfectants. Thus neither total Aβ (Figure 5A) nor Aβ40 and Aβ42 (Figure 5B) measured in secretates by ELISA were affected by SorLA mutations in both wt-APP or swe-APP expressing cells (Figure 5A,B).
Aβ peptides can also aggregate and settle intracellularly [39]. Noticeably, Aβ42 is particularly prone to aggregation [40] and its intracellular proportion relative to total Aβ is augmented. We thus examined the levels of both intracellular Aβ40 and Aβ42 peptides in transiently transfected cells. As expected, intracellular Aβ42 levels relative to Aβ40 are drastically increased (compare black and empty bars in wt condition in Figure 5C). However, here again, wt- and mutant SorLA-expressions did not affect intracellular levels of Aβ40 and Aβ42, whatever the APP species examined (Figure 5C).
The last proteolytic step yielding Aβ peptides is accounted for by γ-secretase, an heterotetrameric complex composed of ApH1, nicastrin, Pen-2 and presenilin 1 or 2 that harbor the catalytic core [41,42,43,44,45]. Thus, we attempted to confirm the lack of effect of SorLA proteins on secreted and intracellular Aβ by examining the expression of all components of the γ-secretase complex and by measuring its catalytic activity. Figure 6A clearly shows that none of the components expression was affected by wt- of SorLA mutants, in both wt- and swe-APP-expressing cells. Finally, we have previously reported a procedure aimed at reconstituting functional γ-secretase in cell membranes and by measuring its activity by means of recombinant C100 [23]. This fragment that corresponds to β-secretase-derived cleavage of βAPP (to which a N-terminal methionine has been added, see Experimental procedure) allows monitoring Aβ peptides and their corresponding C-terminal counterpart AICD [46] productions upon γ-secretase cleavage only [23]. Figure 6B indicates that expressions of wt and mutated SorLA does not affect γ-secretase activity measured in swe-APP-expressing cells.
Overall, the above-described data obtained by multiple and complementary approaches consistently demonstrate that SorLA mutations do not affect secreted Aβ and intracellular Aβ40/42 and do not modulate γ-secretase expression and activity.
3.3. Influence of wild-type SorLA and its mutants on βAPP C-terminal fragments and β-secretase activity.
The amyloidogenic pathway occurring on βAPP always consists in a rate-limiting catalytic step by the β-secretase BACE1 [47] that yields a C-terminal (CTF) fragment referred to as C99. C99 can undergo an α-secretase-mediated hydrolysis generating another CTF called C83 [48,49,50]. Subsequently, both C99 and C83 produce the transcription factor AICD upon γ-secretase-mediated cleavage [46]. We examined the putative influence of wt- and mutated SorLA on CTFs expressions in wt-expressing stably transfected cells. As expected, in control condition, wt-APP increases the levels of both C83 and C99 (compare mock and DNA3 lanes in Supplementary Figure S3) but C83 expression was more abundant than C99 in
(Supplementary Figure S3C), in accordance with our previous observation that significant part of C83 production derived from C99 [48]. In these cells, wt- and mutated SorLA similarly reduce the expressions of C83 and C99 (Supplementary Figure S3C).
As expected, expressions of both CTFs were enhanced by the Swedish mutation (compare wt (-) lanes in Figure 7A,B). in agreement with previous reports on the influence of this APP mutation on β-secretase activity [51,52]. In both wt- and swe-APP-expressing cells, wt- and mutated SorLA similarly reduce the expressions of C83 and C99 (Figure 7A,B).
A previous report also indicated that CTFs expression could be enhanced by alcalinization [53] likely through protection against proteolysis by acidic hydrolases [21,54]. This was clearly confirmed in Figure 7A,B where NH4Cl drastically enhanced both C99 and C83 expressions. This was apparently not due to a blockade of g-secretase cleavage since NH4Cl did not affect g-secretase components expressions in both cell lines, whatever the nature of SorLA exmained (Figure 6A,B).
One should note the puzzling reduction of CTFs observed in SorLA141- expressing cells that could likely be due to aspecific but yet unexplained mechanisms.
Both Aβ (Figure 5) and C99 (Figure 7) productions that require BACE1-mediated βAPP cleavage are insensitive to SorLA mutations. This consistent set of data was further strengthened by direct fluorimetric analysis of BACE1 activity [24]. Thus, we did not detect any modulation of BACE1 by wt- or mutated SorLA in both wt-APP- and swe-APP-expressing cells (Figure 7C).
3.4. Influence of wild-type SorLA and its mutants on neprilysin and cathepsin B.
In sporadic AD, Aβ and CTF accumulation are not due to increased production but rather to age-related defects in their catabolic processes. Several enzymes are implicated in Aβ degradation but neprilysin appears consistently proposed as the main degrading enzyme [36,55,56]. Concerning C99 and C83, we and others have documented their destruction in lysosomal compartment by acidic hydrolases, including cathepsin B [21,54]. In this context, in order to complete our global view of both maturation and degradation processes, we assessed the putative influence of wt- and mutated SorLA on neprilysin and cathepsin B activities. Figure 8 illustrates the lack of influence of SorLA mutations on neprilysin (Figure 8A) and cathepsin B (Figure 8B) activities in both wt-APP- and swe-APP-expressing cells.
4. Discussion
Alzheimer’s disease is a neurodegenerative pathology with complex etiology. A subset of cases, with early onset and rapid progression are due to rare autosomal dominant mutations on βAPP and presenilins 1 and 2 while most of cases occur lately and are of sporadic origin. However, sporadic cases can be also influenced by a genetic component since some appear linked to risk factors associated to mutations on a still growing number of genes.
SORL1 gene is the only gene to date that has been proposed to contribute to both EOAD and LOAD [8]. This gene encodes a protein SorLA, that drives βAPP to the cell surface [16] and that is involved in the targeting of Aβ peptides to the lysosomal compartment [15]. Overall, this suggested that SORL1-associated mutations could well contribute to AD pathology by modulating βAPP routing and proteolytic processing. However close examination of data precluded to draw firm conclusions concerning the mechanisms that are defective in mutated SorLA-expressing cells or animal models. Particularly, there exists a discrepancy between cellular and in vivo modulation of βAPP expression triggered by SorLA [15]. Further, functional consequences on βAPP processing and more precisely on Aβ40 and Aβ42 recoveries appeared highly variable according to the nature of the SORL1 mutation and Aβ species examined [16]. This led us to examine in depth the putative influence of SORL1 mutations on the physiopathological maturation of βAPP as well as on the fate of various βAPP catabolites.
Because SorLA is involved in βAPP trafficking [57] and because βAPP routing is affected by the Swedish mutations, we first carried our study on cells expressing either wt- or swe-APP. First, in order to bring insights on expressions and fate of SorLA mutants, we assessed the influence of the proteasome inhibitor lactacystin [58]. This pharmacological treatment was previously shown to affect βAPP or secretases-related proteins [33,59]. We observed a potentiation of SorLA511 and SorLA924 upon proteasome inhibition in both wt- and swe-APP-expressing cells. It should be noted that SorLA141 was never detectable, even after pharmacological blockade of the proteasome although transfection efficiency was checked as normal. This should be explained by an extreme lability as we previously documented for a parkin mutant [30]. This data has been confirmed in stably transfected CHO cells that do not express detectable levels of SorLA141, even after proteasomal inhibition.
SorLA511 and SorLA924 expressions did not affect βAPP expression in both transiently or stably transfected cell models when compared to wt-SorLA. Our data envisioned the whole amount of βAPP because we considered that overall, this would reflect both intracellular and membrane-associated counterparts and thus, consists in a final readout of expression, traffic and clearance of βAPP. Our data agree with a previous study showing that SORL1 mutants did not affect total βAPP expression in HEK293 cells [16]. However, it was of importance to confirm the cellular localization of wt-APP and SorLA. Our immunohistochemical analysis established first, that wt-APP and wt-SorLA indeed co-localized in stably transfected CHO cells and, second, that the SorLA mutations moderately alter this co-localization.
Although wt-SorLA reduced the levels of secreted sAPPα in both transfectant models, we did not observe SorLA variants-associated modulation of sAPPα. Previous data concerning secreted α- and β-secretases-derived APP fragments led to contrasted observations. Thus, Vardarajan and coll. reported on an enhancement of sAPPβ by common and rare SorLA variants with no change linked to wt-SorLA [16]. Caglayan et al. showed that in vivo expression of wt-SorLA did not nor alter sAPPα levels [15]. Conversely, Cuccaro et al. showed a drastic reduction of sAPPα triggered by wt-SorLA and an increase observed after expression of two SorLA mutants linked to EOAD [13]. This could not be accounted for by distinct cell models as both studies were carried out on HEK293 cells expressing swe-APP. This emphasizes the fact that the influence on α-secretase-mediated processing of βAPP could be differently affected by the nature of the SorLA mutations. In our study, to strengthen our observations, we directly measured α-secretase activity on plated cells. We confirm that α-secretase activity and C83 (the sAPPα C-terminal APP counterpart also referred to as α-CTF) levels were not modulated by SorLA511 and SorLA924 expressions. Previous studies on the influence of SorLA mutants on Aβ levels also led to contrasted conclusions. For instance, Cuccaro and colleagues reported on very faint increases of secreted Aβ42 triggered by two SorLA mutants to levels that remained lower or close to that recovered in mock-transfected cells [13]. Some of these mutants (SorLA T588I) even did not modify secreted levels of Aβ40. Whatever the mutant studied, sAPPβ levels remain similar to those recovered in mock-transfected swe-APP-expressing cells [13]. In another study, the level of secreted Aβ40 was affected by rare but not by common SorLA variants while secreted Aβ42 was modulated by both variants [16]. These discrepant data could be explained by the nature of the mutation examined but also by the fact that Aβ secreted represents only a subset of total Aβ and that it is important to also assess the levels of intracellular Aβ. Overall, our study indicates that wt-SorLA as well as SorLA511 and SorLA924 similarly reduced both intracellular and secreted Aβ40 and Aβ42 in both transiently and stably transfected cells. This conclusion was corroborated by three independent lines of results: 1) β-secretase activity was not modulated by SorLA variants; 2) γ-secretase activity and expressions of its four proteic components were not changed upon SorLA variant expressions; 3) the expression of C99, that is the precursor of Aβ, was not affected by SorLA mutations. It should be noted here that some of these data are supported by previous observations obtained with other variants. Thus, Vardarajan and colleagues did not observe any modulation of PS1 (the catalytic core of γ-secretase) levels upon expressions of variants linked to both EOAD and LOAD [16].
Finally, we analyzed the putative influence of SorLA mutants on the events taking place downstream to βAPP catabolites production. Aβ is mainly processed by neprilysin while CTFs (C99 and C83) undergo proteolysis by acidic lysosomal proteases, including cathepsin B. As would be predictable from our above-described data, SorLA mutants did not modify these neprilysin and cathepsin B activities. Thus, neither production not clearing mechanisms are affected by the SorLA mutations examined in our work.
Our study does not rule out the possibility that additional mutations could well influence βAPP physiopathological processing. However, we can conclude that our studied mutations reported to be linked to benign or severe AD cases do not trigger their putative pathogenic phenotype through modulation of βAPP physiopathological maturation. Again, this does not rule out the possibility that these mutations trigger pathogenic phenotypes by influencing other cellular pathways. Particularly, further study remains to be performed to assess the possible influence of these variants on Tau-related pathology or on hippocampal atrophy as has been described for a subset of SORL1 mutants [60].
5. Conclusions
Our study does not rule out the possibility that additional mutations could well influence βAPP physiopathological processing. However, we can conclude that our studied mutations reported to be linked to benign or severe AD cases do not trigger their putative pathogenic phenotype through modulation of βAPP physiopathological maturation. Again, this does not rule out the possibility that these mutations trigger pathogenic phenotypes by influencing other cellular pathways. Particularly, further study remains to be performed to assess the possible influence of these variants on Tau-related pathology or on hippocampal atrophy as has been described for a subset of SORL1 mutants [60].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Expression and fate of wt-SorLA and its mutants in stably transfected CHO cells; Figure S2: Immunohistochemical analysis of the co-localization of wt-APP and wt-SorlA of SorLA mutants in CHO stably transfected cells; Figure S3: βAPP, sAPPα and C-terminal fragments C83 and C99 expressions in stably transfected CHO cells expressing wt-APP and wt-SorLA of SorLA mutants; Figure S4: Secreted Aβ peptides produced by stably transfected CHO cells expressing wt-APP and wt-SorlA of SorLA mutants.
Author Contributions
CB performed and analyzed the experiments, ED designed and characterized SORLA mutants, PStGH provided wild-type SORL1 cDNA and corrected the manuscript, FC designed the study, analyzed data and wrote the manuscript.
Funding
This work has been developed and supported through the LABEX (excellence laboratory, program investment for the future) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer’s disease.
Acknowledgments
We wish to acknowledge Dr. J.F.Hernandez (UMR5247 CNRS, Montpellier, France) for providing us with JMV2770. We would like to warmly thank the anonymous donor who contributed significantly with several gifts to our study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Progress in molecular biology and translational science 2012, 107, 79–100. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.H.; Petit, A. Molecular biology and genetics of Alzheimer’s disease. Comptes rendus biologies 2005, 328, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s & dementia the journal of the Alzheimer’s Association 2016, 12, 733–748. [Google Scholar] [CrossRef]
- Pihlstrom, L.; Wiethoff, S.; Houlden, H. Genetics of neurodegenerative diseases: an overview. Handbook of clinical neurology 2017, 145, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lu, Y. Genes associated with Alzheimer’s disease: an overview and current status. Clinical interventions in aging 2016, 11, 665–681. [Google Scholar] [CrossRef]
- Lambert, J.C.; Amouyel, P. Genetics of Alzheimer’s disease: new evidences for an old hypothesis? Current opinion in genetics & development 2011, 21, 295–301. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 2007, 39, 168–177. [Google Scholar] [CrossRef]
- Jin, C.; Liu, X.; Zhang, F.; Wu, Y.; Yuan, J.; Zhu, J.; Zhang, F.; Wang, G.; Cheng, Z. An updated meta-analysis of the association between SORL1 variants and the risk for sporadic Alzheimer’s disease. J Alzheimers Dis 2013, 37, 429–437. [Google Scholar] [CrossRef]
- Reitz, C.; Cheng, R.; Rogaeva, E.; Lee, J.H.; Tokuhiro, S.; Zou, F.; Bettens, K.; Sleegers, K.; Tan, E.K.; Kimura, R.; et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol 2011, 68, 99–106. [Google Scholar] [CrossRef]
- Wang, Z.; Lei, H.; Zheng, M.; Li, Y.; Cui, Y.; Hao, F. Meta-analysis of the Association between Alzheimer Disease and Variants in GAB2, PICALM, and SORL1. Molecular neurobiology 2016, 53, 6501–6510. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Hannequin, D.; Coutant, S.; Rovelet-Lecrux, A.; Wallon, D.; Rousseau, S.; Legallic, S.; Paquet, C.; Bombois, S.; Pariente, J.; et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 2012, mp201215. [Google Scholar] [CrossRef] [PubMed]
- Cuccaro, M.L.; Carney, R.M.; Zhang, Y.; Bohm, C.; Kunkle, B.W.; Vardarajan, B.N.; Whitehead, P.L.; Cukier, H.N.; Mayeux, R.; St George-Hyslop, P.; et al. SORL1 mutations in early- and late-onset Alzheimer disease. Neurology. Genetics 2016, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Normal and abnormal biology of the beta-amyloid precursor protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef]
- Caglayan, S.; Takagi-Niidome, S.; Liao, F.; Carlo, A.S.; Schmidt, V.; Burgert, T.; Kitago, Y.; Fuchtbauer, E.M.; Fuchtbauer, A.; Holtzman, D.M.; et al. Lysosomal Sorting of Amyloid-beta by the SORLA Receptor Is Impaired by a Familial Alzheimer’s Disease Mutation. Sci Transl Med 2014, 6, 223ra220. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Zhang, Y.; Lee, J.H.; Cheng, R.; Bohm, C.; Ghani, M.; Reitz, C.; Reyes-Dumeyer, D.; Shen, Y.; Rogaeva, E.; et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015, 77, 215–227. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Sunyach, C.; Ferreira, S.T.; Marzolo, M.P.; Bauer, C.; Thevenet, A.; Checler, F. alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem 2012, 287, 5021–5032. [Google Scholar] [CrossRef]
- Lauritzen, I.; Becot, A.; Bourgeois, A.; Pardossi-Piquard, R.; Biferi, M.G.; Barkats, M.; Checler, F. Targeting gamma-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Translational neurodegeneration 2019, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Chami, L.; Buggia-Prevot, V.; Duplan, E.; Delprete, D.; Chami, M.; Peyron, J.F.; Checler, F. Nuclear factor-kappaB regulates betaAPP and beta- and gamma-secretases differently at physiological and supraphysiological Abeta concentrations. J Biol Chem 2012, 287, 24573–24584. [Google Scholar] [CrossRef] [PubMed]
- Ancolio, K.; Marambaud, P.; Dauch, P.; Checler, F. a-secretase-derived product of b-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer’s disease. J. Neurochem. 1997, 69, 2494–2499. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci 2012, 32, 16243–16255. [Google Scholar] [CrossRef] [PubMed]
- Afram, E.; Lauritzen, I.; Bourgeois, A.; El Manaa, W.; Duplan, E.; Chami, M.; Valverde, A.; Charlotte, B.; Pardossi-Piquard, R.; Checler, F. The eta-secretase-derived APP fragment etaCTF is localized in Golgi, endosomes and extracellular vesicles and contributes to Abeta production. Cell Mol Life Sci 2023, 80, 97. [Google Scholar] [CrossRef] [PubMed]
- Sevalle, J.; Amoyel, A.; Robert, P.; Fournie-Zaluski, M.C.; Roques, B.; Checler, F. Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem 2009, 109, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Andrau, D.; Dumanchin-Njock, C.; Ayral, E.; Vizzavona, J.; Farzan, M.; Boisbrun, M.; Fulcrand, P.; Hernandez, J.F.; Martinez, J.; Lefranc-Jullien, S.; et al. BACE1- and BACE2-expressing human cells: characterization of beta-amyloid precursor protein-derived catabolites, design of a novel fluorimetric assay, and identification of new in vitro inhibitors. J Biol Chem 2003, 278, 25859–25866. [Google Scholar] [CrossRef] [PubMed]
- Cisse, M.A.; Gandreuil, C.; Hernandez, J.F.; Martinez, J.; Checler, F.; Vincent, B. Design and characterization of a novel cellular prion-derived quenched fluorimetric substrate of alpha-secretase. Biochem Biophys Res Commun 2006, 347, 254–260. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–259. [Google Scholar] [CrossRef]
- Pardossi-Piquard, R.; Petit, A.; Kawarai, T.; Sunyach, C.; Alves da Costa, C.; Vincent, B.; Ring, S.; D’Adamio, L.; Shen, J.; Muller, U.; et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 2005, 46, 541–554. [Google Scholar] [CrossRef]
- Andersen, O.M.; Reiche, J.; Schmidt, V.; Gotthardt, M.; Spoelgen, R.; Behlke, J.; von Arnim, C.A.; Breiderhoff, T.; Jansen, P.; Wu, X.; et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 2005, 102, 13461–13466. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D. The Swedish mutation causes early-onset Alzheimer’s disease by b-secretase cleavage within the secretory pathway. Nature Medicine 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nature cell biology 2009, 11, 1370–1375. [Google Scholar] [CrossRef]
- Fenteany, G.; Schreiber, S.L. Lactacystin, proteasome function, and cell fate. J Biol Chem 1998, 273, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Ancolio, K.; Lopez-Perez, E.; Checler, F. Proteasome inhibitors prevent the degradation of familial Alzheimer’s disease-linked presenilin 1 and trigger increased Ab42 secretion by human cells. Mol. Medicine 1998, 4, 147–157. [Google Scholar] [CrossRef]
- Dunys, J.; Kawarai, T.; Giaime, E.; Wilk, S.; Herrant, M.; Auberger, P.; St George-Hyslop, P.; Alves da Costa, C.; Checler, F. Study on the putative contribution of caspases and the proteasome to the degradation of Aph-1a and Pen-2. Neurodegener Dis 2007, 4, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Checler, F. Processing of the b-amyloid precursor protein and its regulation in Alzheimer’s disease. J. Neurochem. 1995, 65, 1431–1444. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Turner, A.J. b-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases. J. Neurochem. 2002, 81, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Baig, S.; Palmer, J.; Palmer, L.E.; Kehoe, P.G.; Love, S. Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 2008, 18, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Shirotani, K.; Tsubuki, S.; Iwata, N.; Takaki, Y.; Harigaya, W.; Maruyama, K.; Kiryu-Seo, S.; Kiyama, H.; Iwata, H.; Tomita, T.; et al. Neprilysin degrades both amyloid b peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 2001, 276, 21895–21901. [Google Scholar] [CrossRef]
- Kuperstein, I.; Broersen, K.; Benilova, I.; Rozenski, J.; Jonckheere, W.; Debulpaep, M.; Vandersteen, A.; Segers-Nolten, I.; Van Der Werf, K.; Subramaniam, V.; et al. Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J 2010, 29, 3408–3420. [Google Scholar] [CrossRef]
- Chui, D.H.; Dobo, E.; Makifuchi, T.; Akiyama, H.; Kawakatsu, S.; Petit, A.; Checler, F.; Araki, W.; Takahashi, K.; Tabira, T. Apoptotic neurons in Alzheimer’s disease frequently show intracellular Abeta42 labeling. J Alzheimers Dis 2001, 3, 231–239. [Google Scholar] [CrossRef]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s A4/b amyloid peptide analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [CrossRef]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the g-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Haass, C. Take five-BACE and the g-secretase quartet conduct Alzheimer’s amyloid b-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and nicastrin with presenilin generate an active g-secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Pardossi-Piquard, R.; Checler, F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem 2012, 120 (Suppl. 1), 109–124. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease Beta-secretase enzyme, BACE1. Mol Neurodegener 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Flammang, B.; Pardossi-Piquard, R.; Sevalle, J.; Debayle, D.; Dabert-Gay, A.S.; Thevenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-beta protein precursor intracellular domain, AICD, derives from beta-secretase-generated C-terminal fragment. J Alzheimers Dis 2012, 30, 145–153. [Google Scholar] [CrossRef]
- Goodger, Z.V.; Rajendran, L.; Trutzel, A.; Kohli, B.M.; Nitsch, R.M.; Konietzko, U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci 2009, 122, 3703–3714. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Kellett, K.A.; Beckett, C.; Makova, N.Z.; Revett, T.J.; Nalivaeva, N.N.; Hooper, N.M.; Turner, A.J. The Transcriptionally Active Amyloid Precursor Protein (APP) Intracellular Domain Is Preferentially Produced from the 695 Isoform of APP in a {beta}-Secretase-dependent Pathway. J Biol Chem 2010, 285, 41443–41454. [Google Scholar] [CrossRef]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the b-amyloid precursor protein in familial Alzheimer’s disease increases b-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Squazzo, S.L.; Koo, E.H. Enhanced release of amyloid b-protein from codon 670/671 “Swedish” mutant b-amyloid precursor protein occurs in both secretory and endocytic pathways. J. Biol. Chem. 1996, 271, 9100–9107. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gele, P.; Drobeck, H.; Begard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J Biol Chem 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol 2016. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Fisk, L.R.; Belyaev, N.D.; Turner, A.J. Amyloid-degrading enzymes as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 2008, 5, 212–224. [Google Scholar] [CrossRef]
- Hersh, L.B.; Rodgers, D.W. Neprilysin and amyloid beta peptide degradation. Curr Alzheimer Res 2008, 5, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, L.; Madsen, P.; Jacobsen, C.; Nielsen, M.S.; Gliemann, J.; Petersen, C.M. Activation and functional characterization of the mosaic receptor SorLA/LR11. J Biol Chem 2001, 276, 22788–22796. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Standaert, R.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Alves da Costa, C.; Ancolio, K.; Lopez-Perez, E.; Marambaud, P. Role of the Proteasome in Alzheimer’s disease. Biochimica Biophysica Acta 2000, 1502, 133–138. [Google Scholar] [CrossRef]
- Louwersheimer, E.; Ramirez, A.; Cruchaga, C.; Becker, T.; Kornhuber, J.; Peters, O.; Heilmann, S.; Wiltfang, J.; Jessen, F.; Visser, P.J. , et al. The influence of genetic variants in SORL1 gene on the manifestation of Alzheimer’s disease. Neurobiol Aging 2015, 36, 1605 e1613-1620. [Google Scholar] [CrossRef]
Figure 1.
Wild-type and mutated SorLA expressions and fate in wt-APP- and swe-APP-expressing HEK293 cells. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP- expressing HEK293 cells in absence (-) or in the presence (+) of lactacystin (B) as described in experimental procedure. Twenty-four hours after transfection, cells were harvested then SorLA and tubulin expressions were monitored by western blot as described in the procedures. Gels correspond to one representative blot of 2 to 5 independent analyses. All full gels are shown in a supplementary material.
Figure 1.
Wild-type and mutated SorLA expressions and fate in wt-APP- and swe-APP-expressing HEK293 cells. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP- expressing HEK293 cells in absence (-) or in the presence (+) of lactacystin (B) as described in experimental procedure. Twenty-four hours after transfection, cells were harvested then SorLA and tubulin expressions were monitored by western blot as described in the procedures. Gels correspond to one representative blot of 2 to 5 independent analyses. All full gels are shown in a supplementary material.
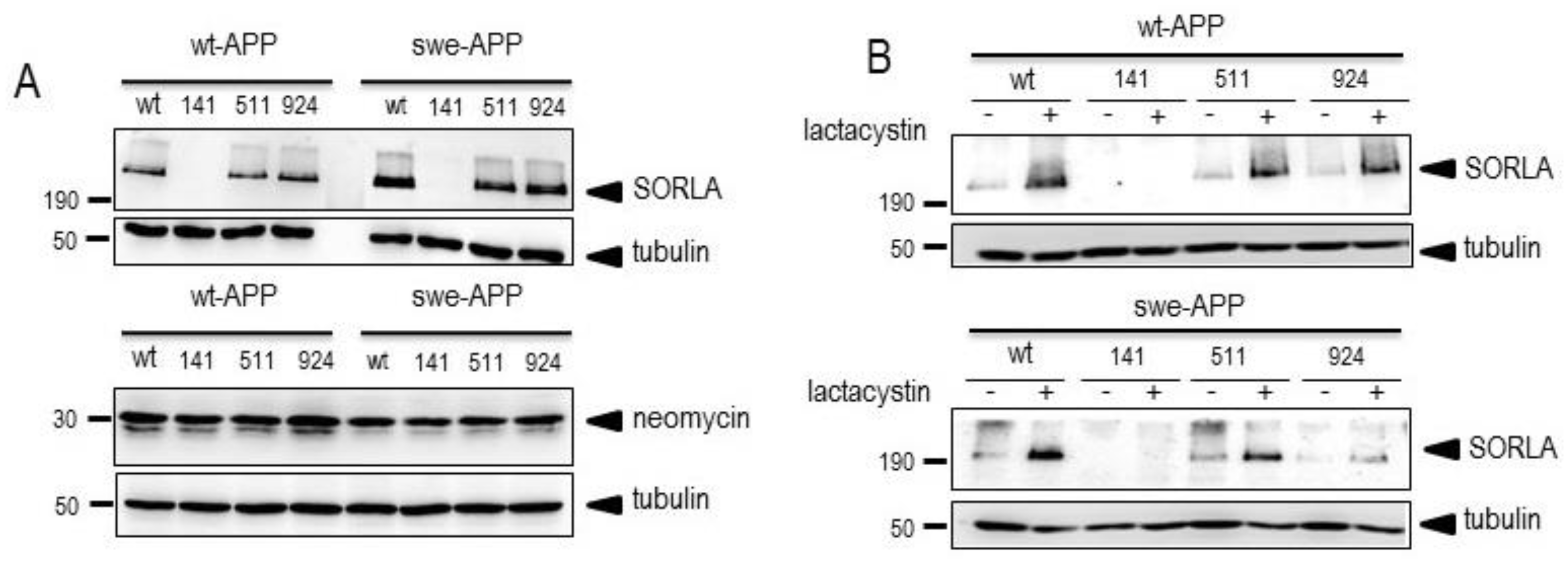
Figure 2.
Influence of wild-type and mutated SorLA on endogenous wt-APP expression and secreted sAPPα. cDNA encoding empty cDNA, wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP-expressing HEK293 cells (A). Twenty-four hours after transfection, secretates were recovered then cells were harvested and sAPPa and βAPP expressions were monitored by western blot as described in experimental procedures. Bars are densitometric analyses expressed as percent of control (mock (DNA3)-transfected cells taken as 100 and are the means ± SEM of 3 (A) or 4 (B) independent determinations. ns, non statistically significant. All full gels are shown in supplementary materials.
Figure 2.
Influence of wild-type and mutated SorLA on endogenous wt-APP expression and secreted sAPPα. cDNA encoding empty cDNA, wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP-expressing HEK293 cells (A). Twenty-four hours after transfection, secretates were recovered then cells were harvested and sAPPa and βAPP expressions were monitored by western blot as described in experimental procedures. Bars are densitometric analyses expressed as percent of control (mock (DNA3)-transfected cells taken as 100 and are the means ± SEM of 3 (A) or 4 (B) independent determinations. ns, non statistically significant. All full gels are shown in supplementary materials.
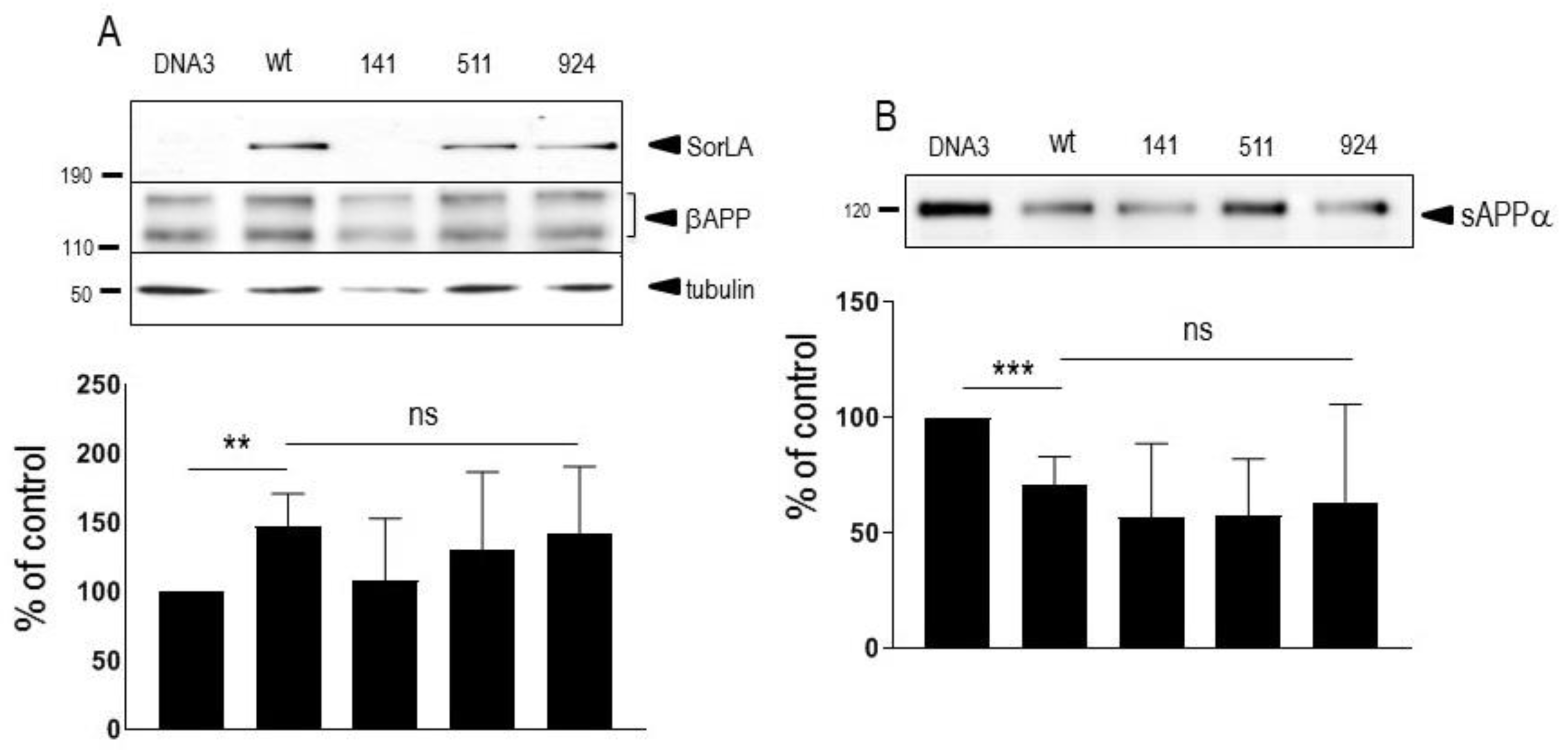
Figure 3.
Influence of wild-type and mutated SorLA on wt-APP and swe-APP expressions. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A) or swe-APP- (B) expressing HEK293 cells. Twenty-four hours after transfection, cells were harvested then βAPP expression was monitored by western blot as described in experimental procedures. Bars are densitometric analyses expressed as percent of control (SorLAwt cDNA transfection in corresponding cells) and are the means ± SEM of 3 (A) or 4 (B) independent determinations. ns, non statistically significant. All full gels are shown in supplementary materials.
Figure 3.
Influence of wild-type and mutated SorLA on wt-APP and swe-APP expressions. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A) or swe-APP- (B) expressing HEK293 cells. Twenty-four hours after transfection, cells were harvested then βAPP expression was monitored by western blot as described in experimental procedures. Bars are densitometric analyses expressed as percent of control (SorLAwt cDNA transfection in corresponding cells) and are the means ± SEM of 3 (A) or 4 (B) independent determinations. ns, non statistically significant. All full gels are shown in supplementary materials.
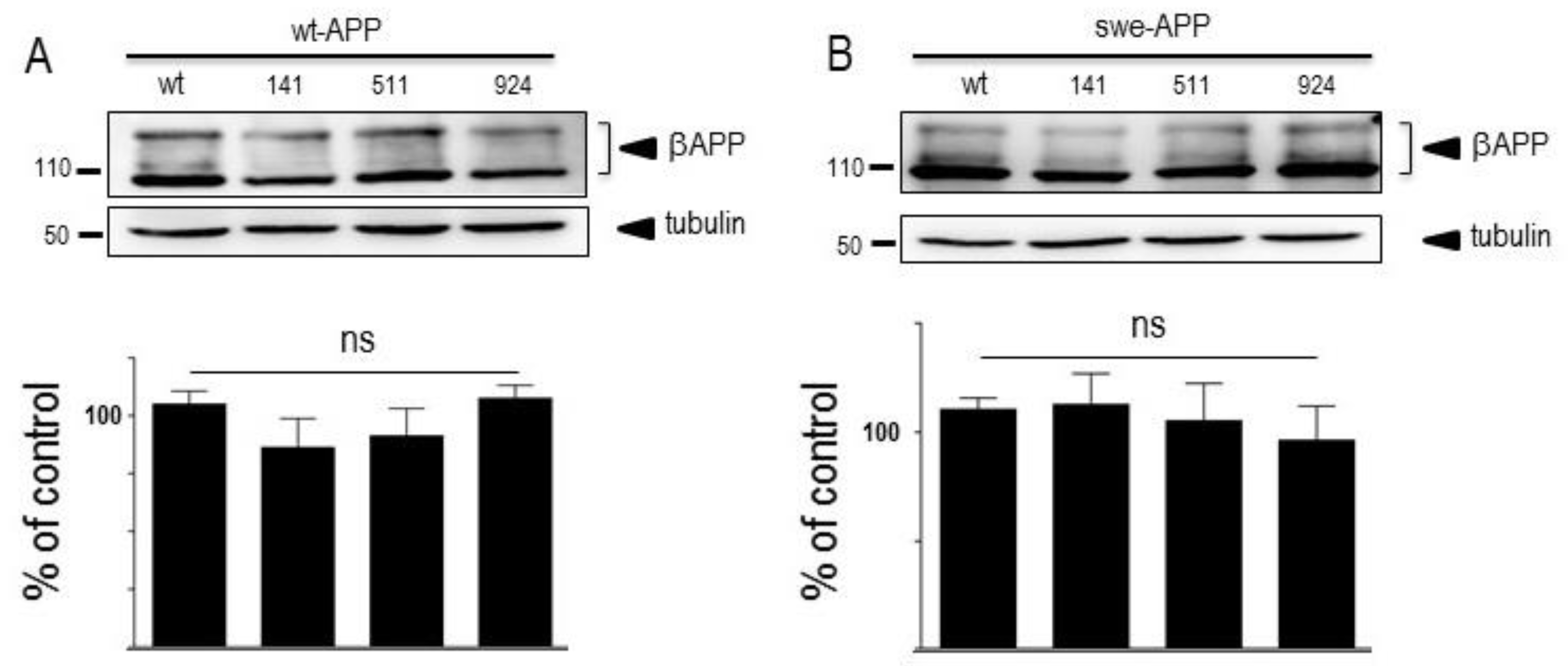
Figure 4.
Influence of wild-type and mutated SorLA on secreted sAPPα and α-secretase activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A,C) or swe-APP- (B,D) expressing HEK293 cells. Twenty-four hours after transfection, cells were harvested then sAPPα expression was measured in secretates (A,B) by western blotting as described in the experimental procedures. Bars correspond to densitometric analysis of sAPPα expressed as percent of control (sAPPα in SorLAwt -expressing cells) and are the means ± SEM of 5 (A) or 4 (B) independent experiments. In C and D, α-secretase activity was fluorimetrically recorded on plated cells as described in the experimental procedure. Bars correspond to the phenanthroline-sensitive JMV2770-hydrolysing activity and are the means ± SEM of 9 (C) and 5 (D) independent experiments. ns, non statistically significant.
Figure 4.
Influence of wild-type and mutated SorLA on secreted sAPPα and α-secretase activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A,C) or swe-APP- (B,D) expressing HEK293 cells. Twenty-four hours after transfection, cells were harvested then sAPPα expression was measured in secretates (A,B) by western blotting as described in the experimental procedures. Bars correspond to densitometric analysis of sAPPα expressed as percent of control (sAPPα in SorLAwt -expressing cells) and are the means ± SEM of 5 (A) or 4 (B) independent experiments. In C and D, α-secretase activity was fluorimetrically recorded on plated cells as described in the experimental procedure. Bars correspond to the phenanthroline-sensitive JMV2770-hydrolysing activity and are the means ± SEM of 9 (C) and 5 (D) independent experiments. ns, non statistically significant.
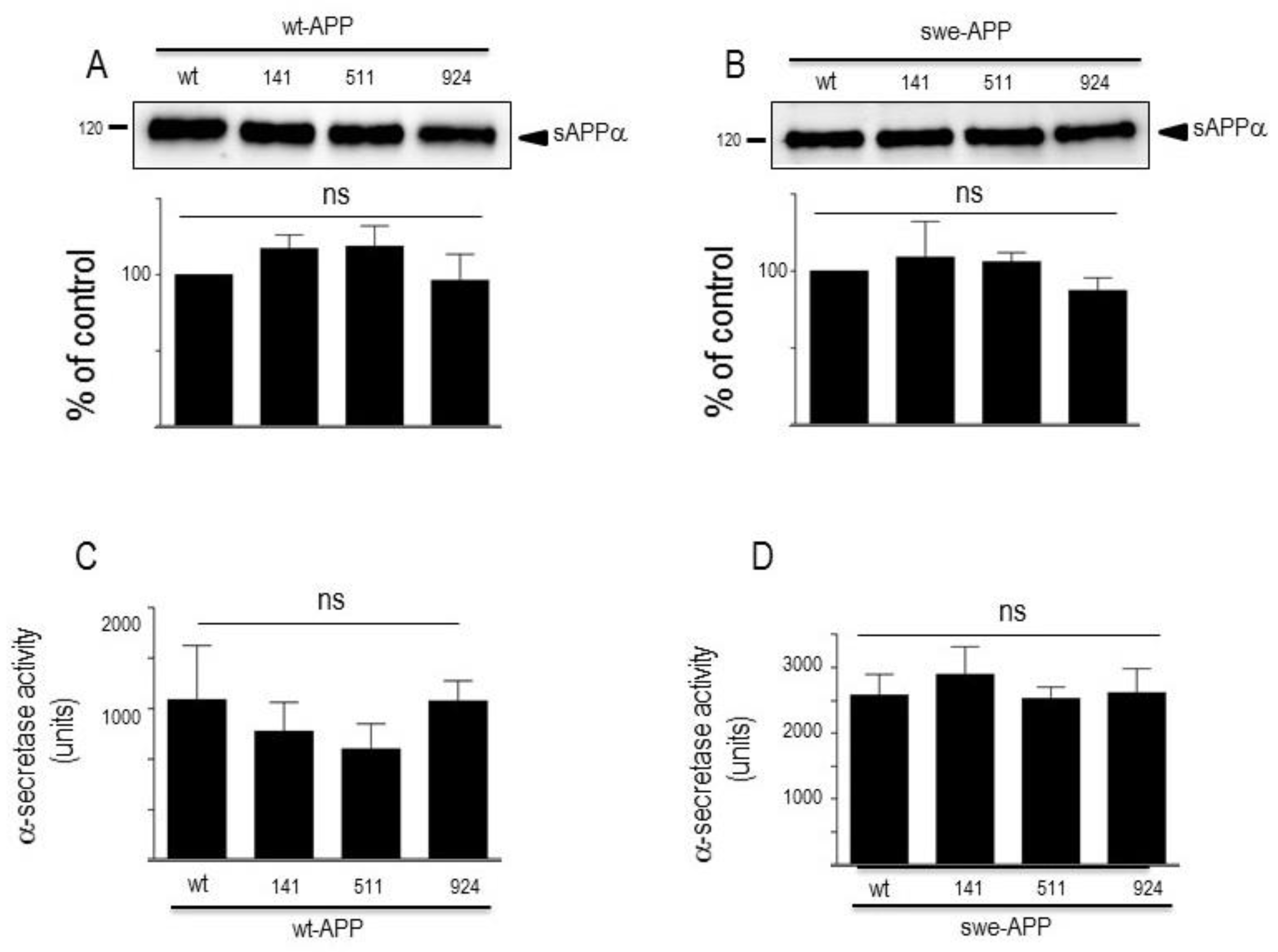
Figure 5.
Influence of wild-type and mutated SorLA on secreted and intracellular Aβ peptides. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP-expressing HEK293 cells (A-C). Twenty-four hours after transfection, total Aβ (A) recovered in medium was analyzed by immunoprecipitation (A). Bars are densitometric analyses expressed as percent of control (densitometries in SorLAwt -expressing cells) and are the means ± SEM of 3 experiments. In B and C, cells were analyzed for secreted (B) or intracellular (C) Aβ40 and Aβ42 (C) by ELISA as described in the experimental procedures. ELISA quantifications are the means ± SEM of 5 independent experiments. ns, non statistically significant.
Figure 5.
Influence of wild-type and mutated SorLA on secreted and intracellular Aβ peptides. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP-expressing HEK293 cells (A-C). Twenty-four hours after transfection, total Aβ (A) recovered in medium was analyzed by immunoprecipitation (A). Bars are densitometric analyses expressed as percent of control (densitometries in SorLAwt -expressing cells) and are the means ± SEM of 3 experiments. In B and C, cells were analyzed for secreted (B) or intracellular (C) Aβ40 and Aβ42 (C) by ELISA as described in the experimental procedures. ELISA quantifications are the means ± SEM of 5 independent experiments. ns, non statistically significant.
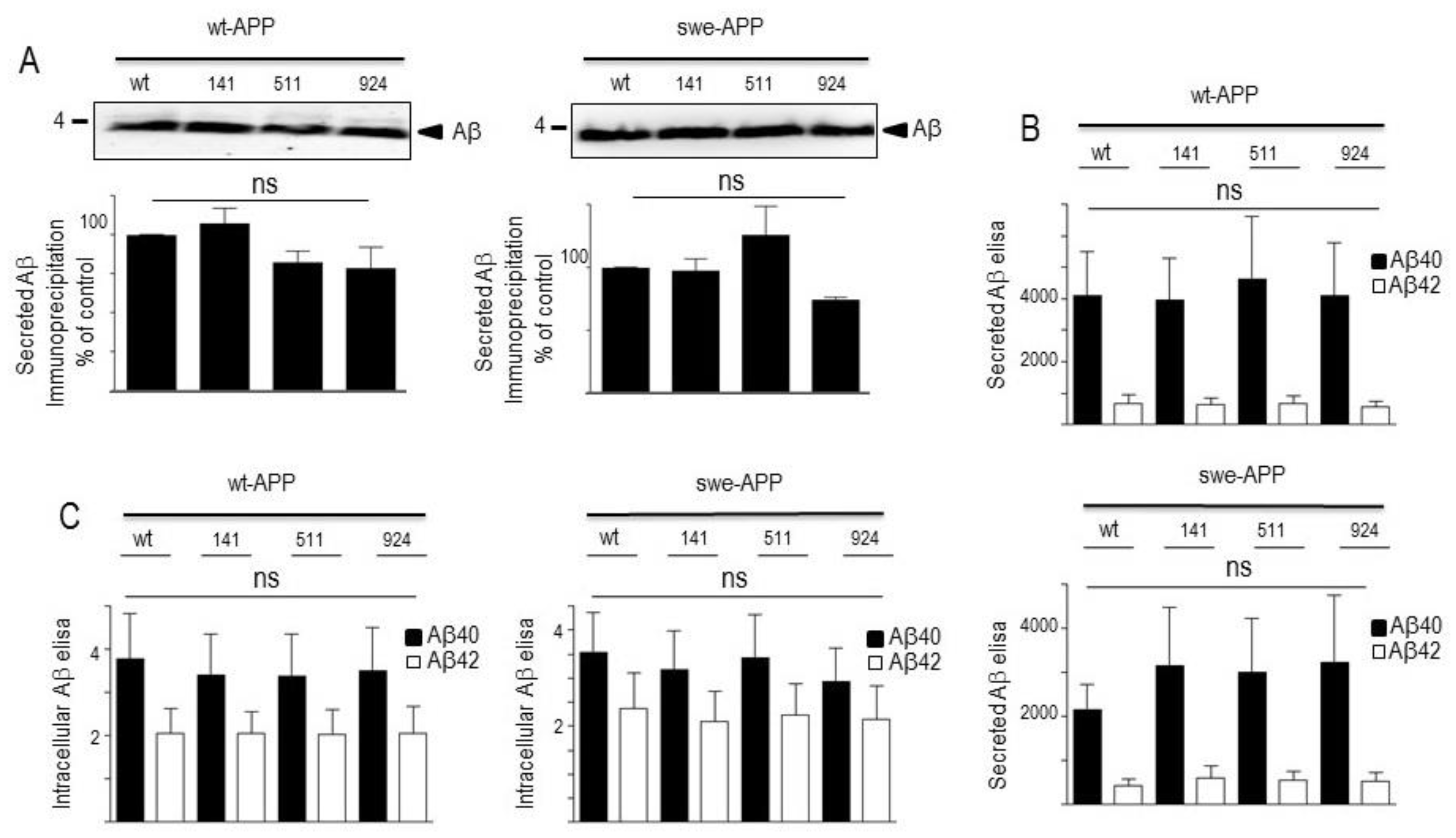
Figure 6.
Influence of wild-type and mutated SorLA on γ-secretase expression and activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A, left panel) or swe-APP- (A, right panel) expressing HEK293 cells, in absence (-) or in the presence (+) of NH4Cl. Twenty-four hours after transfection, the expressions of presenilin 1 (PS1), presenilin 2 (PS2), Aph1, Pen2, nicastrin and tubulin were analyzed by western blotting as described in the experimental procedure. In B, in vitro γ-secretase activity was measured in reconstituted membranes prepared from cells expressing SorLAwt or SorLA mutants as described in the experimental procedures. Bars are densitometric analyses of Aβ and AICD expressions and are the means ± SEM of 6 independent experiments.
Figure 6.
Influence of wild-type and mutated SorLA on γ-secretase expression and activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A, left panel) or swe-APP- (A, right panel) expressing HEK293 cells, in absence (-) or in the presence (+) of NH4Cl. Twenty-four hours after transfection, the expressions of presenilin 1 (PS1), presenilin 2 (PS2), Aph1, Pen2, nicastrin and tubulin were analyzed by western blotting as described in the experimental procedure. In B, in vitro γ-secretase activity was measured in reconstituted membranes prepared from cells expressing SorLAwt or SorLA mutants as described in the experimental procedures. Bars are densitometric analyses of Aβ and AICD expressions and are the means ± SEM of 6 independent experiments.
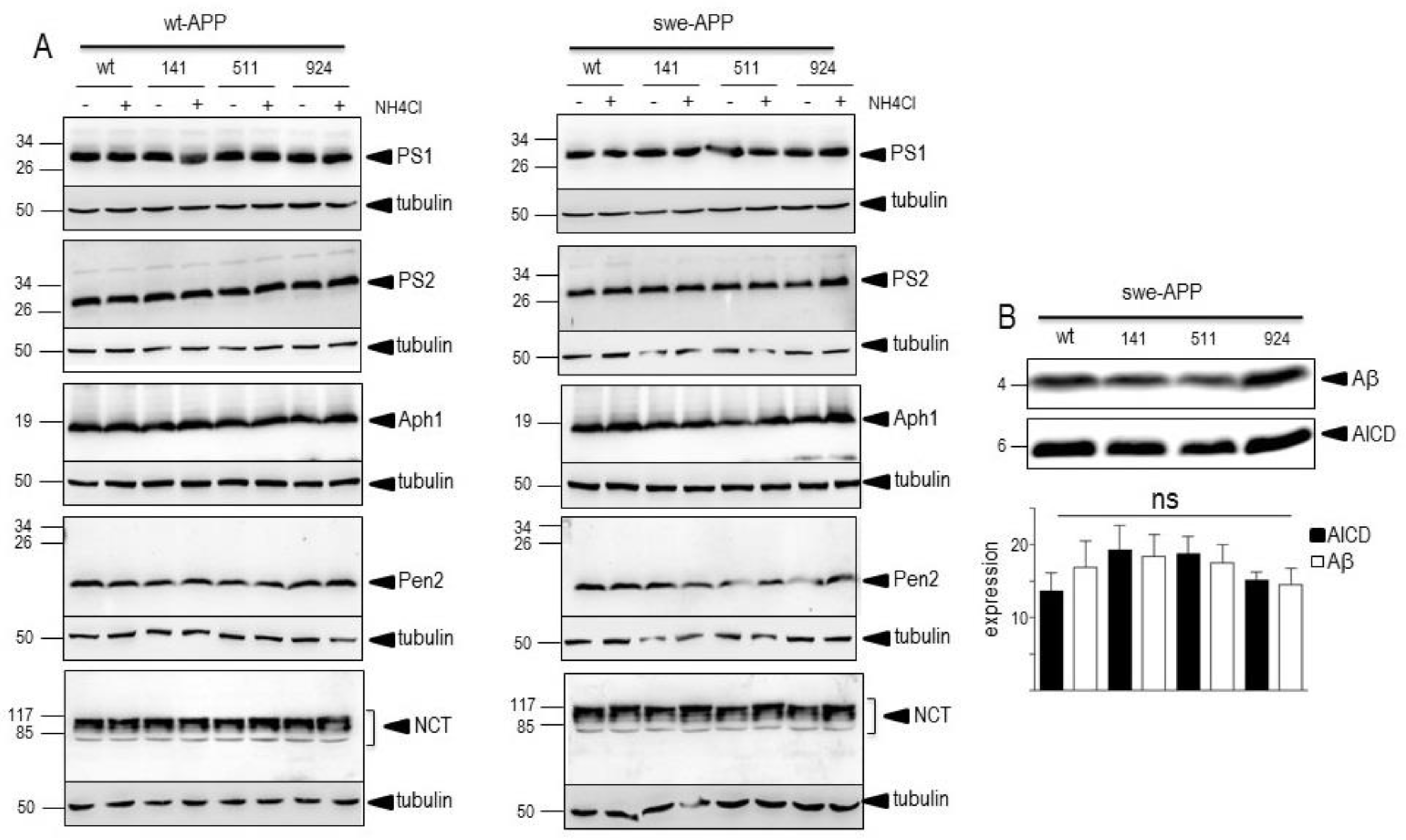
Figure 7.
Influence of wild-type and mutated SorLA on CTFs expression and BACE1 activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A) or swe-APP- (B) expressing HEK293 cells, in absence (-) or in the presence (+) of NH4Cl. Twenty-four hours after transfection, the expressions of CTFs (C83 and C99) and tubulin were analyzed by western blot as described in the experimental procedure. Bars are densitometric analyses of C99 expression and are the means ± SEM of 7 (A) or 6 (B) independent experiments. In C, BACE1 activity was fluorimetrically measured in cell homogenates as described in the experimental procedures. Bars represent the β-secretase inhibitor I-sensitive (7-methoxycoumarin-4-yl) acetyl-SEVNL-DAEFR K (2,4-dinitrophenyl)-RRNH2-hydrolyzing activity and are the means ± SEM of 5 independent experiments. ns, non statistically significant.
Figure 7.
Influence of wild-type and mutated SorLA on CTFs expression and BACE1 activity. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- (A) or swe-APP- (B) expressing HEK293 cells, in absence (-) or in the presence (+) of NH4Cl. Twenty-four hours after transfection, the expressions of CTFs (C83 and C99) and tubulin were analyzed by western blot as described in the experimental procedure. Bars are densitometric analyses of C99 expression and are the means ± SEM of 7 (A) or 6 (B) independent experiments. In C, BACE1 activity was fluorimetrically measured in cell homogenates as described in the experimental procedures. Bars represent the β-secretase inhibitor I-sensitive (7-methoxycoumarin-4-yl) acetyl-SEVNL-DAEFR K (2,4-dinitrophenyl)-RRNH2-hydrolyzing activity and are the means ± SEM of 5 independent experiments. ns, non statistically significant.
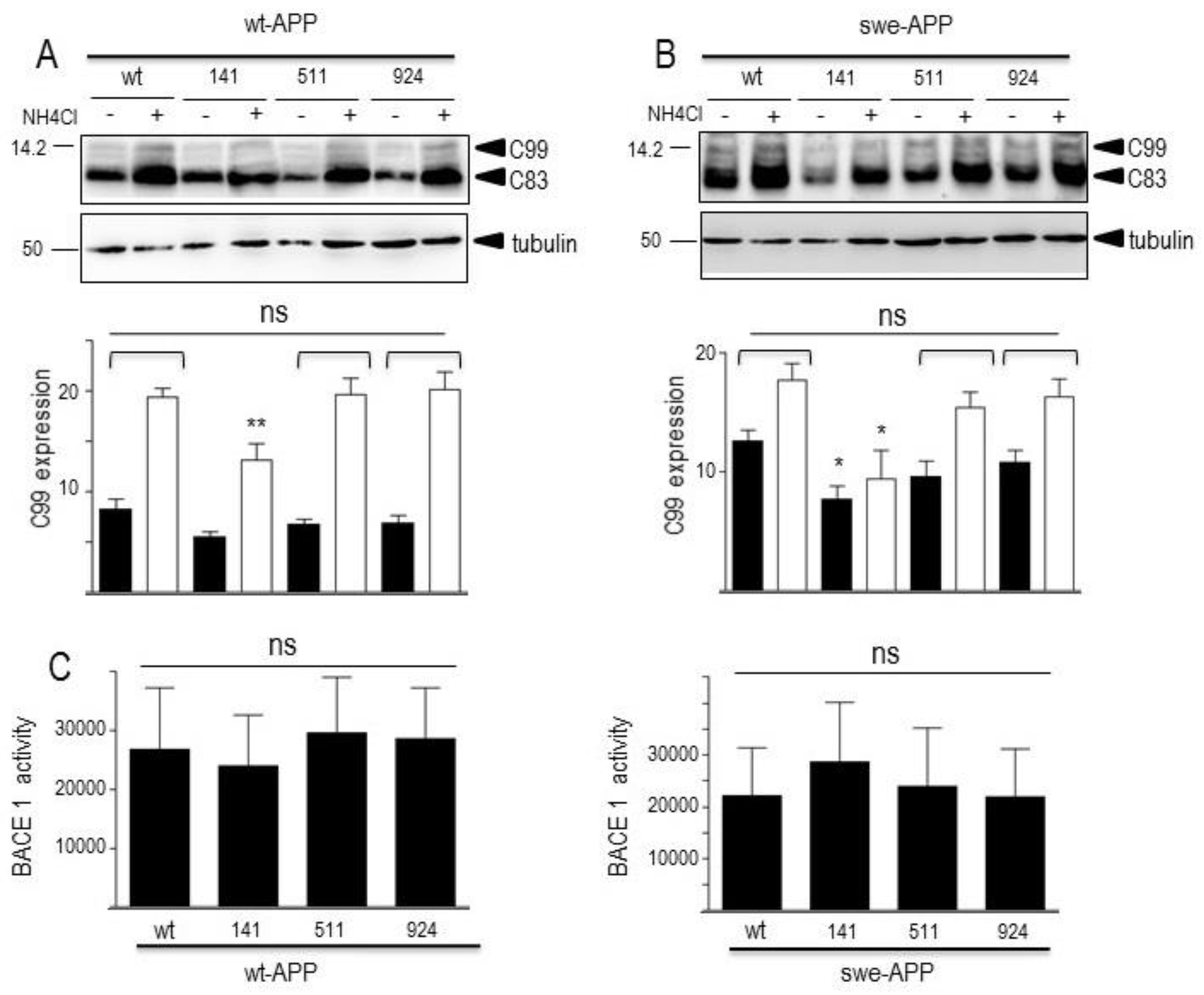
Figure 8.
Influence of wild-type and mutated SorLA on neprilysin and cathepsin B activities. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP-expressing HEK293 cells. Twenty-four hours after transfection, neprilysin (A) and cathepsin B (B) activities were fluorimetrically measured with either Suc-Ala-Ala-Phe-7AMC (in the absence or presence of the NEP inhibitor phosphoramidon) or with carboxybenzoyl-Arg-Arg-7-Amido-4-methylcoumarin (without or with leupeptin) for neprilysin and cathepsin B, respectively. Bars correspond to the inhibitor-sensitive hydrolyzing activities and are the means ± SEM of 5 independent experiments. ns, non statistically significant.
Figure 8.
Influence of wild-type and mutated SorLA on neprilysin and cathepsin B activities. cDNA encoding wild-type (SorLAwt), SorLA141, SorLA511 and SorLA924 cDNA were transiently transfected in wt-APP- or swe-APP-expressing HEK293 cells. Twenty-four hours after transfection, neprilysin (A) and cathepsin B (B) activities were fluorimetrically measured with either Suc-Ala-Ala-Phe-7AMC (in the absence or presence of the NEP inhibitor phosphoramidon) or with carboxybenzoyl-Arg-Arg-7-Amido-4-methylcoumarin (without or with leupeptin) for neprilysin and cathepsin B, respectively. Bars correspond to the inhibitor-sensitive hydrolyzing activities and are the means ± SEM of 5 independent experiments. ns, non statistically significant.
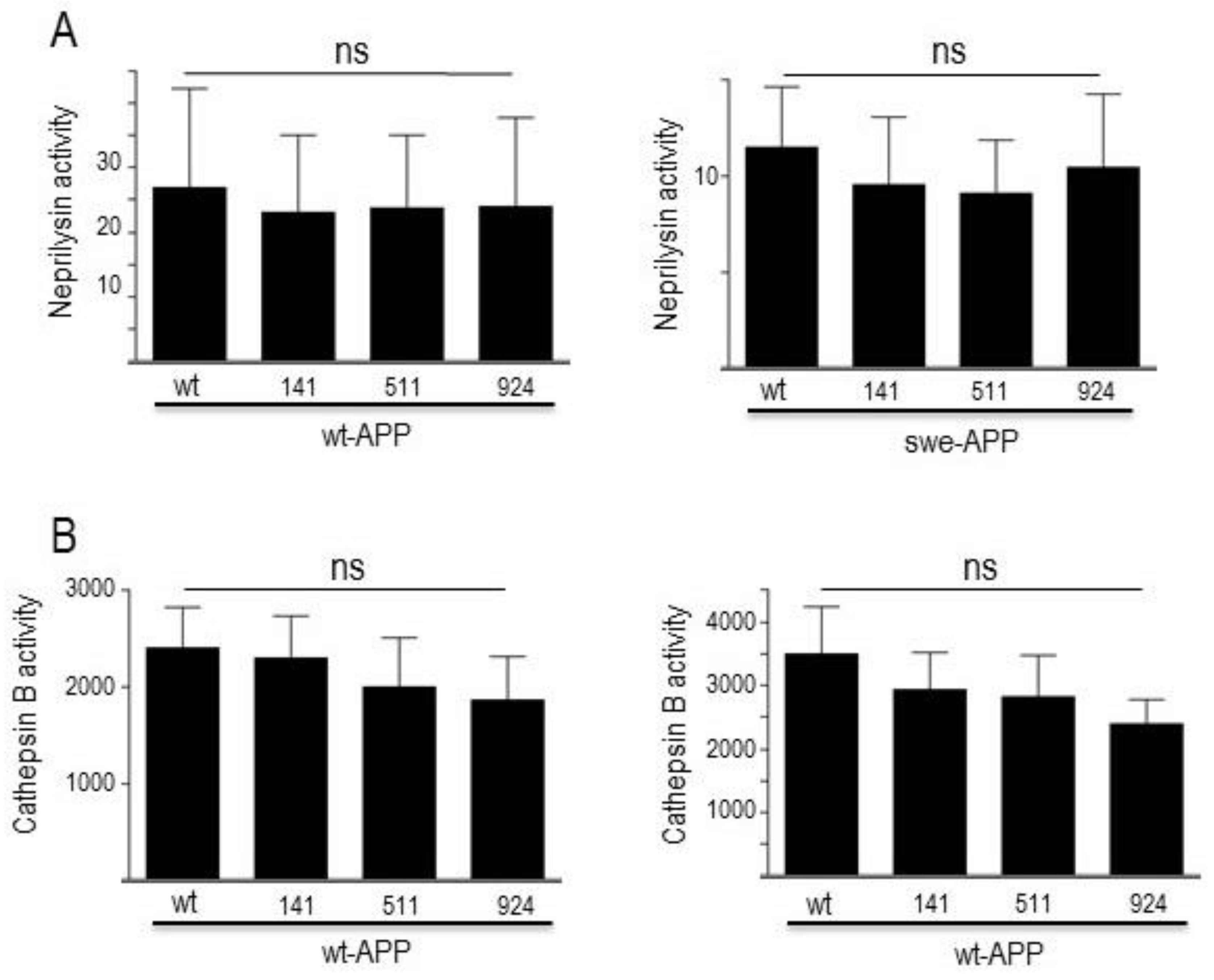

Table 1.
Point mutations in bold and corresponding mutant.
| hSORL1 point mutation (nucleotide) |
Forward Primer | HSorLA point mutation (amino acid) |
|---|---|---|
| A422G | 5′-GTG-TCT-TAC-GAC-TGT-GGA-AAA-TCA-TTC-3′ | Y141C |
| G1531C | 5′-GGC-TCA-GTG-CGA-AAG-AAC-TTG-GCT-AGC-AA-3′ | G511R |
| A2771G | 5′-GAT-GTG-AAG-TGG-CCC-AGT-GGC-ATC-TCT-GTG-3′ | N924S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



