Submitted:
05 April 2023
Posted:
06 April 2023
Read the latest preprint version here
Abstract
The brain is a complex organ that controls body functions and homeostasis through the action of neuronal and glial cells. Neuronal activity is highly energy-dependent and requires large numbers of functional mitochondria to provide substantial amount of energy via mitochondrial oxidative phosphorylation (OXPHOS), the most efficient metabolic process to generate adenosine triphosphate (ATP). Under stress conditions, neurons are particularly vulnerable to mitochondrial dysfunction, leading to decreased ATP synthesis, excessive generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), and intracellular Ca2+ dyshomeostasis, although not necessarily in this order. Alzheimer's disease (AD) and Parkinson’s disease (PD) are the two most common neurodegenerative diseases in elderly and are characterized by the presence of abnormal protein aggregates and the progressive and irreversible loss of neurons in specific brain regions. The exact mechanisms underlying the etiopathogenesis of AD or PD remain unknown, but there is extensive evidence indicating that compromised mitochondrial energy metabolism along with a depleted antioxidant system play a vital role in the pathophysiology of these neurological disorders. Toxic accumulation of proteins such as amyloid β peptides (Aβ) or amyloid precursor protein (APP) in AD and α-synuclein (α-syn) or leucine-rich repeat kinase 2 (LRRK2) in PD cause mitochondrial deficits through direct inhibition of electron transport chain (ETC) assembly and function, thereby resulting in further generation of ROS/RNS and disturbance of Ca2+ influx. Due to the improvement in life expectancy, the incidence of age-related neurodegenerative diseases has significantly increased. There is no effective protective treatment or therapy available but rather only very limited palliative treatment. There is an urgent need for the development of preventive strategies and disease-modifying therapies (both neuroprotective and neurorestorative interventions) to treat AD/PD. Here, we review the capability of some heterocyclic compounds to modulate Ca2+ homeostasis and signaling with a potential role in regulating mitochondrial function and associated free radical production during the development and onset of AD or PD. Moreover, we have included the chemical synthesis of a series of heterocycles and their derivatives.
Keywords:
Alzheimer’s disease
; Parkinson’s disease
; mitochondria
; oxidative stress
; calcium
; heterocyclic compounds
Introduction
The brain is particularly prone to oxidative insult due to a complex interconnected myriad of reasons, such as a high metabolic activity, neurotransmitter autoxidation, elevated content of redox active transition metals, modest antioxidant defense, glutamate excitotoxicity, and altered calcium (Ca2+) influx and signaling processes.[1] An impaired antioxidant system or aberrant and sustained free radical formation can result in redox balance variations and concomitant alteration in redox-sensitive signaling pathways, leading to significant changes in the state or activity of a neuron. At physiological levels, brain reactive oxygen species (ROS) and reactive nitrogen species (RNS) are second messengers involved in intracellular signaling but an elevated concentration of free radicals causes harmful effects to biological macromolecules that contribute to the aging process and the pathogenesis of neurodegenerative diseases.[2,3,4,5] Lipid peroxidation (LPO) consists in the abstraction of allylic hydrogen atoms from side chains of polyunsaturated fatty acids (PUFAs) by ROS and RNS. A major deleterious outcome of LPO is the generation of a variety of reactive aldehyde species, such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE). The reaction between O2•− and NO• leads to the formation of peroxynitrite, which targets tyrosine residues in proteins via free radical addition to generate 3-nitrotyrosine (3-NT). Protein carbonylation is an oxidative stress-driven non-enzymatic and irreversible post-translational modification (PTM). The synthesis of protein carbonyls normally responds to the oxidative deamination of alkaline amino acids such as arginine, lysine, and histidine. Advanced glycation end products (AGEs) are formed in a non-enzymatic reaction among lipids, proteins or nucleic acids and reducing sugars. The interaction of AGEs with their receptors RAGEs elicits oxidative stress. ROS/RNS may also interact with nucleobases of the DNA (e.g., guanine) to form 8-hydroxy-2-deoxyguanosine (8-OHdG) while oxidative RNA damage induces the production of 8-hydroxyguanosine (8-OHG).
Compelling evidence has demonstrated that mitochondrial electron transport chain (ETC) is the major endogenous source of ROS/RNS generation, although the endoplasmic reticulum (ER) and peroxisomes can be also an important site of free radical formation, showing a redox interplay between these organelles.[6] Specifically, respiratory chain complexes I (NADH: ubiquinone oxidoreductase) and III (ubiquinol: cytochrome c oxidoreductase) produce high rates of O2•−. The ETC also comprises membrane-embedded proteins in the inner mitochondrial membrane that shuttle electrons from NADH and FADH2 to molecular oxygen. Simultaneously, protons are pumped from the mitochondrial matrix to the intermembrane space, thereby resulting in the reduction of oxygen to water. The energy released from these redox reactions is stored as a mitochondrial potential used to drive the phosphorylation of adenosine diphosphate (ADP) to form adenosine triphosphate (ATP). Moreover, PTMs involve enzyme-mediated covalent addition of specific functional groups (such as phosphorylation, ubiquitination, glycosylation, nitration, and methylation) to proteins after their synthesis. Redox-related PTMs can modulate the activity of proteins implicated in a variety of cellular signaling pathways, such as protein folding and degradation, transcription factor expression and activity, and energy metabolism by regulating the tricarboxylic acid cycle (TCA) and glycolytic enzymes, fatty acid metabolism, and protein cysteine thiol nitrosation, sulfenylation or glutathionylation of the mitochondrial ETC complexes.[7,8,9,10] Oxidative DNA damage of the gene promoter encoding subunits of the F1 and F0 domains of ATP synthase has also been observed during aging and neurodegeneration.[11] Mitochondria along with the ER play an important role in controlling intracellular Ca2+ homeostasis, which regulates several vital neuronal processes, including synaptic plasticity, metabolic regulation, proliferation, gene expression, and apoptosis through modulation of a number of enzymes such as phospholipases, proteases, and nucleases. During aging and in neurodegenerative diseases, Ca2+ can be converted from a physiological signal into a pathological effector. Cytosolic and organelle Ca2+ overload promotes Ca2+ mitochondrial accumulation, which triggers the opening of the mitochondrial permeability transition pore (mPTP) and the disruption of the mitochondrial membrane potential (ΔΨm). Sustained mPTP opening leads to Ca2+ release, mitochondrial depolarization, OXPHOS disruption, compromised structural and functional integrity of the inner mitochondrial membrane, and release of cytochrome c and other apoptogenic proteins from the outer mitochondrial membrane.[12]
AD and PD are neurodegenerative diseases that share common pathological features, including protein misfolding and aggregation, synaptic impairment, mitochondrial deficits, axonopathy, and aberrant free radical production. In addition, altered Ca2+ homeostasis and signaling may contribute to accelerating the pathogenesis of AD/PD. Ca2+ overload is mainly mediated by Aβ and tau in AD and α-synuclein (α-syn) and leucine-rich repeat kinase 2 (LRRK2) in PD.[13,14,15,16] Modifications in neuronal Ca2+ influx via voltage-gated Ca2+ channels (VOCCs) and glutamate receptors promote excitotoxic Ca2+ accumulation and concomitant defective neurotransmission, impaired synaptic plasticity and damaged mitochondrial function, including increased ROS/RNS production, activation of mitochondrial permeability transition, stimulation of mitophagy and decreased ATP synthesis.[16] Therefore, targeting aberrant Ca2+ homeostasis may represent a plausible option for the prevention and therapy of neurodegenerative diseases. We have reviewed a number of heterocyclic compounds that modulate Ca2+ signaling and homeostasis and can serve as a therapeutic target in both AD and PD.
Mitochondrial deficits and oxidative stress as close partners in Alzheimer’s disease brain damage
AD is the most common neurodegenerative disorder characterized by brain atrophy and impaired cognitive performance. Neuropathological studies have observed an extracellular accumulation of amyloid beta (Aβ) and an intraneuronal deposition of insoluble neurofibrillary tangles (NFTs) containing hyperphosphorylated tau protein.[17] A large body of research has demonstrated that impaired mitochondrial function (and associated energy failure) is a causative factor of AD and occurs before development of Aβ plaques and NFTs, indicating this is an early event in the pathogenesis of the disease. Mitochondrial pathological changes drive disease progression, leading to an increased oxidative burden, synaptic degeneration, dysregulated Ca2+ homeostasis, and neuronal loss. This is consistent with the “mitochondrial cascade hypothesis” that postulates that bioenergetic deficits mediates AD.[18,19,20] Brain metabolic variations would be primarily responsible for mitochondrial dysfunction in AD. Glucose deprivation leads to reduced activity in the default mode network, an area that includes the posterior cingulate cortex, the precuneus, the medial prefrontal cortex, the inferior parietal cortex, and the medial temporal lobe and that preferentially associated with atrophy and amyloid and tau deposition in AD.[21,22,23] Positron emission tomography (PET) with [18F]-fluro-2-deoxyglucose analyses have shown a progressive decline in cerebral glucose metabolism in AD patients.[24,25,26] Glucose metabolism is connected to thiamine-dependent pathways, including the Krebs cycle and the pentose phosphate pathway, which are compromised in AD.[27,28,29] In addition, oxidation of glucose by substrate-level and oxidative phosphorylation produces ATP molecules and results in a synergistic effect with mitochondria in metabolic pathways.
Another specific event linked to mitochondrial failure in AD is the development of phenotypic changes in these organelles. Defective mitochondrial function is characterized by the abnormal formation of a subset of swollen mitochondria with distorted cristae. Indeed, ultrastructural examination confirmed the presence of mitochondrial morphometric abnormalities in postmortem brain specimens from individuals with AD.[30,31] Postmortem examination of AD patients revealed a significant decrease in the numbers of intact mitochondria.[30] Furthermore, mitochondrial dysfunction correlates with certain enzyme deficiencies, such as the α-ketoglutarate dehydrogenase complex (α-KGDH), pyruvate dehydrogenase complex (PDHC), and transketolase.[29,32,33,34,35] There is a direct relationship between reduced brain regional glucose metabolism and downregulated thiamine-dependent enzyme activities (such as α-KGDH).[27,34] The most common feature found in AD is a deficiency in complex IV (cytochrome c oxidase, COX), which has been reported in the cortical and hippocampal brain regions[36,37,38,39,40] and platelets[36,41,42,43] of patients. Downregulated complex IV activity has been also detected in AD cybrid cells.[44] Cognitively normal subjects with a parental history of late-onset AD exhibited COX decreased activity in platelet mitochondria, suggesting a role for mitochondrial DNA (mtDNA) in maternal transmission, since no differences in COX activity were seen between paternal history of late-onset AD and controls.[45] All these empirical observations undeniably implicate mitochondrial damage in the pathophysiology of AD.
Together with mitophagy, mitochondrial dynamics – a specialized type of mitochondrial autophagy – serves as a quality control mechanism for the maintenance of mitochondrial integrity and functionality and is crucial for neuronal homeostasis and survival. Mitochondrial dynamics are tightly regulated by the fusion-fission machinery that promotes the generation or degradation of a mitochondrial syncytium. The molecular process of fusion is driven by the GTPases optic atrophy type 1 (Opa1) and mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) whereas dynamin-related protein (Drp1) interacts with the mitochondrial fission 1 protein (Fis1), the mitochondrial fission factor (Mff), and the mitochondrial dynamics proteins of 49 and 51 kDa (MiD49/51) to mediate mitochondrial fission.[46,47,48] Axonal transport is a cellular mechanism that controls the active trafficking of proteins, lipids, organelles, and neurotransmitters, and is critical for the maintenance of the neuronal network function and viability. Kinesin motor proteins are responsible for the anterograde transport, which carries new synthesized material from the cell body to distal axons. Retrograde transport is required for efficient distribution of cargoes from the axon terminals toward the soma and is mediated by dynein. It has been described a crosstalk between mitochondrial fusion and fission events and axonal transport integrity.[5,49,50] Mitochondrial deficits and free radical production, changes in redox homeostasis, and apoptosis correlate with abnormalities in mitochondrial dynamics and axonal transport.[5,51] An imbalance between mitochondrial fusion and fission rates has been documented in AD. Mitochondrial axonal transport is tightly interconnected with mitochondrial dynamics and play a prominent role in preserving mitochondrial morphology and quality control. mRNA and protein levels of Opa1, Mfn1 and Mf2 were reduced while gene expression and protein content of Drp1 and Fis1 were upregulated in postmortem brain samples from individuals with different Braak AD stages.[52] In primary neurons from AβPP mice, mRNA levels of genes involved in mitochondrial fusion were downregulated while the expression of mitochondrial fission-related genes were augmented, suggesting an excessive mitochondrial fragmentation.[53] In the same study, AβPP neurons exhibited decreased mitochondrial anterograde axonal transport although retrograde mitochondrial motility remained unchanged in axonal projections. Mitochondrial axonal trafficking deficits, abnormal mitochondrial distribution, and increased content of 4-HNE and Ca2+-induced H2O2 were detected in transgenic mice overexpressing human APP/Aβ (Tg mAPP mice).[54]
Aβ aggregation may also determine mitochondrial function. Enhanced Aβ burden has been detected in mitochondria from autopsy specimens of late onset AD as in transgenic mice overexpressing mutant amyloid precursor protein (APP).[55,56] Aβ1-40 and Aβ1-42 mitochondrial internalization is mediated by the receptor components TOM20, TOM40 and TOM70 of the translocase of the outer membrane (TOM) complex and this translocation is independent of the ΔΨm.[57] The APP N-terminal fragment contains a mitochondrial targeting sequence that creates stable intermediate complexes with TOM and TIM complexes.[56] Moreover, Aβ can interact with Aβ-binding alcohol dehydrogenase (ABAD) in cerebral cortex mitochondria of sporadic individuals with AD and in cultured neurons from transgenic mice overexpressing ABAD and exposed to Aβ, resulting in a leakage of ROS.[58] Aβ (mainly oligomeric Aβ and Aβ42) can interact with cyclophilin D, a regulatory component of the mitochondrial mPTP, leading to synaptic pathology, mitochondrial stress and neuronal death in both the temporal cortex of AD patients and transgenic APP mice.[59] Aβ/cyclophilin D-mediated toxicity might involve a Ca2+ signaling mechanism.
The Aβ25-35 peptide induced a rapid, specific and dose-dependent downregulation of COX activity in non-synaptic mitochondria isolated from rat brain.[60] Impaired energy metabolism, including inhibition of COX and several dehydrogenase activities together with deficiencies in mitochondrial respiration were reported in Aβ-treated non-synaptic rat brain mitochondria.[61] Synaptic mitochondria exhibited a larger age-dependent accumulation of Aβ and mitochondrial abnormalities compared with non-synaptic mitochondria, indicating that synaptic mitochondria are more prone to Aβ pathology.[54] Several familial and amyloid-based animal models of AD, including triple transgenic AD, APP, Thy1, and COXd/AD mice display systemic mitochondrial dysfunction, including downregulated COX activity, impaired mitochondrial respiration, augmented glycolysis and marked oxidative insult.[62,63,64,65] Apolipoprotein E (APOE), the major genetic risk factor for late-onset AD, may directly interact with mitochondria, affecting its function, dynamics and motility. COX immunoreactivity was significantly depleted in post-mortem cortical samples and posterior cingulate cortex of young-adult APOE ε4 carriers.[66,67] In addition, proteins involved in the regulation of ketone and glucose metabolism were also affected.[67] Aβ-induced persistent mitochondrial fission causes deleterious effects. Exposure to Aβ or overexpression of APP resulted in excessive mitochondrial fragmentation and abnormal mitochondrial distribution of in neuronal cultures.[68,69,70] Similar results were obtained when crossing Drp1+/− mice with APP transgenic mice; partial reduction of Drp1 protected against APP/Aβ-induced mitochondrial and synaptic impairment.[71] In addition, Aβ can induce S-nitrosylation of Drp1, which increases its translocation into mitochondria.[72] Ca2+ signaling and oxidative stress are important contributors to Aβ-induced mitochondrial fragmentation. Aβ promotes mitochondrial Ca2+ influx and Ca2+/calmodulin-dependent protein kinase II (CAMKII)-mediated protein kinase B (Akt) activation, thereby causing Drp1 phosphorylation and increasing its mitochondrial translocation.[73]
Tandem mass tag multiplexed quantitative mass spectrometry revealed that tau protein can interact with a subset of mitochondrial proteins.[74] Intracellular accumulation of tau can disrupt mitochondrial function by downregulating complex I activity, diminishing ATP synthesis and interrupting mitochondrial dynamics.[75] P301S tau mice exhibited reduced complex I (NADH: ubiquinone oxidoreductase), α-KGDHC and transketolase enzyme activity accompanied of lower mtDNA copy number and increased mitochondrial fission in the cerebral cortex.[35] The same study demonstrated that advanced glycation end products were attenuated in tau mice, which showed an important oxidative and nitrosative damage. Tau can also cooperate with Aβ to induce a synergistic detrimental effect on mitochondria. Indeed, tau and Aβ interaction can exacerbate respiratory capacity abnormalities, inhibit both complex I and 4 activities, and disturb energy metabolism on isolated mitochondria from the cerebral cortex of triple transgenic AD mice.[76] The interplay between the NH2-truncated tau peptide and Aβ mediated mitochondrial dysfunction through the impairment of the adenine nucleotide translocator type 1 (ANT-1), located in the inner mitochondrial membrane and responsible to catalyze the adenosine diphosphate-adenosine triphosphate (ADP/ATP) exchange.[77]
Pathological p-tau exhibits lower affinity for the microtubule network, resulting in increased fission events. It has been shown that transgenic P301L tau mice display an unbalanced concentration of mitochondrial dynamics-associated proteins in the hippocampi, with a diminished immunoreactivity of fusion proteins and elevated fluorescence signal of fission proteins.[78] Moreover, ablation of tau ameliorates mitochondrial function by preserving mitochondrial dynamics and structure and reducing oxidative insult. In particular, genetic deletion of tau caused a shift of mitochondrial dynamics towards fusion and upregulated both 4-HNE and Nrf2 mRNA levels in the mouse hippocampus.[79] In addition, genetic tau ablation blocks Aβ-mediated activation of glycogen synthase kinase 3β (GSK3β) – a kinase responsible for tau phosphorylation – which leads to an improvement of the anterograde axonal transport of mitochondria in primary hippocampal neuron cultures from transgenic hAPP mice.[80] Tau mice expressing the R406W mutation showed axonal transport deficits that causes an accumulation of insoluble tau in the neuronal perikarya and subsequent development of NFTs.[81] Diverse pathogenic isoforms of tau inhibited kinesin-based fast axonal transport by activating the PP1-GSK3 signaling pathway in isolated squid axoplasm.[82]
Sustained mitochondrial dysfunction is a primary cause of an excessive generation of ROS/RNS in AD brains and leads to Aβ aggregation and toxicity.[83] Aggregation of Aβ or tau within mitochondria not only compromises the function of crucial mitochondrial proteins but also instigates oxidative stress. ROS/RNS promote Aβ and tau pathology via activation of p38 mitogen-activated protein kinase (MAPK).[84] The connection between Aβ1-40 and Aβ1-42 and mitochondrion interfered with its function and led to mitochondrial oxidative damage in N2a cells overexpressing human wild-type APP and Tg2576 AD mice, which showed a downregulation in COX enzymatic activity and elevated content of H2O2 before Aβ plaque formation.[68] Aβ attenuated mitochondrial respiration and ∆Ψ generation induced by various substrates of complexes I and IV. The Aβ1–42 peptide enhanced the levels of ROS by inhibiting complex I activity and membrane LPO associated with complex IV deficiencies. Furthermore, a sharp increase in the GSSG/GSH ratio was observed in postmortem AD specimens, indicating a defective antioxidant defense system.[85]
Free radical-mediated chain of reactions that results in an oxidative deterioration of PUFAs is a key feature of AD. The LPO product malondialdehyde (MDA) is robustly increased in the cerebral cortex and hippocampus of AD patients.[86,87] Levels of 4-HNE have been reported in different AD brain regions, including the temporal and entorhinal cortex and the hippocampus compared with age-matched controls.[88,89] Increased immunoreactivity of 4-HNE parallel to reduced levels of antioxidant proteins and enzymes such as catalase, glutathione peroxidase, superoxide dismutase and peroxiredoxin were described in the superior temporal gyrus from APOE ε4 cases.[90] Moreover, high levels of the LPO markers 4-hydroxy-2-nonenal (4-HNE), F2-isoprostanes, and 8,12-iso-iPF2α-VI were found in the cerebral spinal fluid (CSF) specimens of AD patients.[91,92,93] Redox proteomic analyses revealed a significant lipoxidation and nitration of several key mitochondrial enzymes, including the ATP synthase in the hippocampus of AD subjects.[94,95] ROS-induced functional variations in the F1Fo-ATP synthase may represent a potential mechanism of OXPHOS deficiency in AD.[96] A large body of literature has shown that protein nitration is a feature of AD. The number of 3-NT-positive neurons was robustly enhanced in postmortem brain samples from AD cases.[97,98,99,100] Protein carbonyls are also upregulated in subjects with AD though the expression pattern varies between different brain regions.[101] Carbonylation of proteins and AGEs plasma levels were specifically elevated in male AD patients.[102] The content of DNA strand breaks was higher in cerebral cortex and hippocampus specimens from individuals with AD compared to controls.[103,104] HPLC analysis revealed a prominent increase in 8-OHdG levels in DNA isolated from the brains of idiopathic AD cases while elevated 8-OHG immunoreactivity was described in temporal cortex neurons of preclinical early-onset individuals with AD.[105,106]
Impaired mitochondrial function and associated oxidative damage in Parkinson’s disease
PD is a chronic neurodegenerative disease characterized by the loss of DA neurons in the substantia nigra (SN) and their axonal projections to the striatum. A histopathological hallmark of the disease is the presence of neuronal cytoplasmic inclusions termed Lewy bodies (LB), which are predominantly composed of α-syn and, to a lesser extent, ubiquitin.[107] α-Syn can undergo PTMs, including nitration, ubiquitination, glycosylation and phosphorylation (serine 129 (pS129) is the dominant pathological modification of α-syn).[5] SNCA gene point mutations or multiplications cause familial dominant PD. Based on PET and single photon emission computed tomography studies, a significant glucose hypometabolism was detected in the cerebral cortex of individuals with early-stage PD.[108] Using [18F]-fluro-2-deoxyglucose PET imaging, a robust attenuation in glucose metabolism was found in the hippocampus and in the temporo-parietal and occipital regions of PD dementia (PDD) patients.[109] An independent study confirmed that PD subjects have decreased glucose metabolism in the occipital and inferior parietal lobes in comparison to the control group.[110] There is a correlation between cerebral glucose intake and synaptic density in individuals with LB disease, but progressive glucose hypometabolism cannot be fully explained by synaptic degeneration.[111] Neurons metabolize glucose predominantly through the pentose phosphate pathway (instead of using glycolysis) to preserve their redox status. Assessment of the levels of nicotinamide adenine dinucleotide phosphate (NADPH), an enzyme produced by the glucose-6-phosphate dehydrogenase, was performed in brain biopsy samples of mild PD cases (with low LB deposition) and moderate-to-severe PD cases (with an important LB pathology). The findings suggested that perturbed glucose metabolism is an early event in idiopathic PD.[112] Interestingly, in vitro and in vivo studies revealed that α-syn may play a central role in glucose uptake through the activation of the LPAR2/Gab1/PI3K/Akt signaling pathway.[113]
Mitochondrial defects in PD also involve morphologically abnormal mitochondria, evidenced by organelle swelling and reduced cristae size and number. In SN neurons of patients with PD, around 80% of total mitochondria exhibited an irregular shape and swollen morphology with deranged cristae patterns.[114] Using electron microscopy, subsets of mitochondria appeared swollen and rounded or enlarged in cybrid cell cultures prepared from platelet-derived mtDNA of sporadic PD cases.[115] Thiamine is an important cofactor for various critical enzymes involved in brain oxidative metabolism, including α-KGDH, PDHC, and transketolase. In contrast to AD, the levels of thiamine remain unchanged in the plasma of PD cases.[116] Nevertheless, free thiamine content was significantly reduced in lumbar CSF specimens of PD subjects.[117] The cerebellar enzymatic activity (but no protein concentration) of α-KGDH declined by 50% in PD patients and was independent of an overall decrease in mitochondrial numbers.[118] However, Mizuno and colleagues found an increased α-KGDH immunoreactivity in the SN of patients with idiopathic PD that correlates with disease severity.[119] Pyruvate dehydrogenase E1 subunit alpha 1 (PDHA1) regulates PDHC through reversible phosphorylation. Individuals with sporadic PD displayed lower PDHA1 fluorescence in both the putamen and SN relative to the control group.[120] The activity of α-KGDH, PDHC, and succinate dehydrogenase (SDH) and the respiratory function were inhibited in PC12 cells overexpressing monoamine oxidase B (MAO B).[121]
Systemic deficiencies in complex I assembly and decreased activity might result in impaired oxidative capacity, ensuing ROS/RNS overproduction, and progressive mitochondrial deficiencies, a major culprit in the degenerative process of DA neurons. Disturbances in mitochondrial complex I were initially seen in SN tissue of postmortem human samples.[122,123] Noteworthy, inhibition of complex I was detected in the SN pars compacta (but not in SN reticulata).[124] Impaired catalytic activity of complex I has been found in the frontal cortex and in peripheral tissues such as, skeletal muscle, platelets and lymphocytes of late-onset PD subjects.[122,125,126,127,128] Nevertheless, a significant downregulation in the enzymatic activity of complex I/III was also reported in untreated patients with early-onset PD.[129] Progressive and permanent loss of complex I in mouse DA neurons resulted in impaired behavioral outcome and early axonal damage, which diminishes retrograde transport of striatal trophic factors and induces bioenergetic failure.[130]
Mounting evidence supports the notion that defects in mitochondrial dynamics are likely a common mechanism leading to mitochondrial dysfunction and neurodegenerative process in PD.[131] Perturbations in mitochondrial dynamics and disrupted motor-cargo interactions have been observed in individuals with PD. Indeed, SN tissue from patients with sporadic PD displayed a significant immunoreactivity attenuation of both the short and long OPA1 isoforms, though no changes were noticed in MFN1 protein concentration.[114] p.A495V and p.G488R heterozygous OPA1 missense mutations were identified in individuals with parkinsonism and subclinical optic neuropathy.[132] OPA1 is further linked to non-syndromic, idiopathic PD associated with aberrant changes in cristae structure and disrupted mitochondrial networks.[133] Altered trafficking along axons may represent a slow but steady feature disrupting mitochondrial homeostasis, since mitochondria undergo bidirectional transport along microtubule and actin filaments. Impaired axonal transport has been reported in clinical PD. In particular, SN DA neurons displayed low levels of kinesin heavy chain (KHC) and kinase light chain (KLC1) in subjects with early-onset PD while DYNLT3 immunoreactivity was markedly diminished in patients with late-onset PD.[134] Moreover, parkinsonized rats exhibited decreased content in mitochondrial fusion proteins (OPA1 and MFN2), increased levels of mitochondrial fission proteins (DRP1) and attenuated anterograde (KHC and KLC1) retrograde (DYNLT3) axonal transport.[2,135] Mitochondrial quality control also includes mitophagy, a mitochondrion-selective autophagic process to degrade dysfunctional or defective mitochondria.[136,137]
α-Syn not only localizes in the cytosol but also at or in mitochondria of DA neurons in different systems, including cell cultures, rodent midbrain, and human subjects with PD.[5,138] Tom40 is an essential protein-conducting pore that directly interacts with α-syn for its import into mitochondria.[138] Protein levels of Tom40 (but not TOM20) were significantly attenuated in PD brains and transgenic mice overexpressing wild-type human α-syn. In addition, depletion of TOM40 promoted α-syn accumulation, oxidative insult, and DNA damage but stereotaxic delivery of TOM40 reversed α-syn-induced pathological events.[139] The data suggest that α-syn interaction with the mitochondrial protein import machinery might be an upstream effect in α-syn-mediated toxicity. A specific association between wild-type α-syn and mitochondria-associated ER membranes was found in cells and transgenic mice expressing α-syn, a finding supported by the fact that α-syn predominantly binds to lipid rafts and acidic phospholipid-rich membrane domains.[140] Di Maio et al. revealed a high-affinity binding between specific post-translationally modified α-syn and TOM20 in midbrain DA neurons of PD cases and rotenone-injected rats.[141] Such association was not detected with TOM22, TOM40 or the component of the translocase of the inner membrane TIM23.
Mitochondria-targeted α-syn may cause structural damage to the organelle. Differentiated DA cells transduced with α-syn exhibited compromised mitochondrial structural integrity, including massive swelling, abnormal morphology, and distorted cristae.[142] Inhibition of complex I activity and altered levels of complex I-related proteins also occur as a consequence of α-syn mitochondrial import, which can further increase endogenous content of α-syn, thereby initiating a feed-forward cycle. Thus, accumulation of α-syn led to 3-fold decrement in complex I activity in the SN of postmortem PD brains.[138] Selective reduction in complex I immunoreactivity was observed in midbrain homogenates of AAV-A53T α-syn-transduced rats.[2] The fluorescence intensity of Ndufs3, a subunit of complex I, arose downstream α-syn-TOM20 association in cultured cells.[141] A significant decrease of the complex I subunit NDUFB8 was reported in α-syn transgenic mice.[139]
α-Syn plays an important role in controlling mitochondrial integrity by regulating dynamics, transport, and clearance. A53T and A30P mutations in α-syn elicited mitochondrial fragmentation via a DRP1-independent pathway and increased OPA1 cleavage in crude mitochondrial fractions from M17 cells.[140] Mitochondrial fragmentation mediated by α-syn can occur in either a Drp1-independent or -dependent mechanism in HeLa cells.[143] The authors also showed that overexpression of MFN1 or MFN2 fusion proteins did not prevent fragmentation, supporting the idea that α-syn plays a selective role in fission events. α-Syn-induced mitochondrial fragmentation precedes OXPHOS disturbances, including mitochondrial respiration and ΔΨm.[143] Preformed α-syn fibrils perturbed mitochondrial fission-fusion cycle by attenuating OPA1 levels and increasing DRP1 immunoreactivity in cultured rat ventral midbrain DA neurons.[5] Combined exposure to α-syn fibrils and rotenone resulted in an additive toxicity. Human neural stem cells overexpressing A53T mutant α-syn increased the amount of short round-shaped (fragmented) mitochondria despite the concentration of both MFN and Drp1 remained unchanged, supporting a physiological role for α-syn in regulating mitochondrial morphology probably linked to an association between the N-terminal region and the mitochondrial membrane.[144] Furthermore, the C-terminal domain of α-syn triggered mitochondrial fragmentation and oxidation.[145] α-Syn-induced fragmented mitochondrial phenotype was reversed following co-expression of DJ-1, PINK1 or parkin.[146]
Primary ventral midbrain neuronal cultures incubated with synthetic α-syn fibrils displayed axonal transport deficiencies, with variations in kinesin and dynein markers.[5] Live-imaging analyses revealed no differences in anterograde and retrograde mitochondrial content between wilt-type or mutated α-syn and the control group.[144] Nevertheless, overexpression of α-syn (especially A53T α-syn) in embryonic stem cells led to alterations in mitochondrial flux, suggesting an imbalance in mitochondrial distribution along axons. AAV delivery of human α-syn in the medulla oblongata gradually spread to more rostral brain regions – following a stereotypical pattern that may reflect neuron-to-neuron transmission – and accumulated in dystrophic axons, where was distributed and propagated in both ipsilateral and contralateral sides via axonal transport.[147] Inhibition of LRRK2 kinase activity promoted α-syn movement toward the presynaptic terminal in primary hippocampal neuron cultures transfected with α-syn.[148] Reduced levels of pS935 LRRK2 (an indirect assessment of LRRK2 kinase activity) mediated α-syn accumulation in presynaptic terminals of the mouse SN and striatum.[148] The C-terminal truncation of α-syn is responsible for retrograde motility of mitochondria.[145] In addition, membrane potential is required for α-syn transport, a process dependent on the mitochondrial ATP pool.[138] Cells transfected with α-syn promoted Ca2+ trafficking from the ER to the mitochondria via mitochondria-associated ER membranes.[149] However, α-syn depletion inhibits agonist-stimulated Ca2+ entry into mitochondrial matrix.
Higher OH• concentration was detected in isolated mitochondrial fractions from primary DA neuron cultures transfected with α-syn relative to the control group.[138] Protein thiol oxidation was a feature of cell cultures incubated in the presence of oligomeric, pS129, or DA-modified α-syn.[141] Moreover, knockdown of endogenous α-syn expression resulted in a robust reduction of protein thiol oxidation in SN DA neurons. Elevated MitoSox-red fluorescence was observed in cell lines expressing α-syn, which was mitigated by SIRT3.[150] Exposure to exogenous α-syn fibrils augmented ROS/RNS production by increasing the MitoSox (O2•–) signal and the levels of 3-NT in primary midbrain neuronal cultures.[5] AAV-driven overexpression of human mutated A53T α-syn into the rat SN elicited systemic oxidative insult, including LPO and protein nitration in the ipsilateral hemisphere.[2] Either wild-type or A53T α-syn accumulation in isolated mitochondria from human neuroblastoma cells induced peroxidation of lipids and NO• formation, the later sensitive to intramitochondrial ionized Ca2+.[151] Cells expressing α-syn displayed an aberrant content of mtDNA deletions and oxidative DNA lesions.[139] In summary, specific derangements in complex I are responsible for α-syn conformational changes, impaired mitochondrial function and biogenesis, exacerbated oxidative stress and Ca2+ dysregulation, leading to DA neuron degeneration.
Ca2+ dysregulation and downstream effects in Alzheimer’s disease
Ca2+ signaling in neurons is often started by activation of plasma membrane channels including voltage-gated Ca2+ channels (VOCCs), receptor-operated Ca2+ channels (ROCCs) as, for instance, NMDA receptors or other Ca2+-permeable such as transient receptor channels (TRP) or store-operated channels (SOCs) driven by Stim and Orai protein family members (Figure 1) whose opening induces Ca2+ influx into cells due to the large electrochemical gradient for Ca2+. Ca2+ signaling also starts following activation of Ca2+ release channels in the ER such as IP3 receptors after G protein-coupled receptor mediated phospholipase C activation induced mainly by glutamate metabotropic receptors or acetylcholine muscarinic receptors. Ca2+ release also takes place through ryanodine receptor channels activated by different messengers, including Ca2+ itself, which is responsible of the Ca2+-induced, Ca2+ release (CICR) mechanism, a process known to be primed by chemicals like caffeine. Ca2+ influx and Ca2+ release mechanisms enhance the cytosolic free intracellular Ca2+ concentration ([Ca2+]cyt) leading to neuron cell activation as stated above. This process is limited by endogenous Ca2+ buffers, particularly Ca2+ binding proteins such as calbindin, and Ca2+ extrusion systems, including plasma membrane Ca2+ ATPases (PMCAs), sarcoplasmic and ER Ca2+ ATPases (SERCAs), and Na+/Ca2+ exchangers (NCXs) that remove Ca2+ from cytosol back to the extracellular space and/or to intracellular organelles, principally, the ER. Mitochondria also work as Ca2+ removing organelles. In this case, Ca2+ is not pumped in or exchanged but enters the mitochondrial matrix through the mitochondrial Ca2+ uniporter (MCU) complex, containing an activated Ca2+ channel that promotes mitochondrial Ca2+ uptake in favor of a massive electromotive force, the ΔΨm (Figure 1). In fact, subtle modifications in ΔΨm may considerably influence mitochondrial Ca2+ uptake ability.[152] Ca2+ uptake by mitochondria activated the Krebs cycle and energy production but excess leading to mitochondrial Ca2+ overload may enhance oxidant stress and mPTP, leading to apoptosis. In contrast to other organelles, mitochondria are not Ca2+ stores and accordingly, mitochondrial Ca2+ excess returns to the cytosol in a substantially slow fashion, in exchange for Na+ through mitochondrial Na+-Li+/Ca2+ exchangers (NCLX).
The MCU is a low affinity Ca2+ channel. Therefore, only very high Ca2+ concentrations limited in time and space (Ca2+ hot spots or microdomains) efficiently activate MCU and mitochondrial Ca2+ uptake. As a consequence, only those mitochondria closely located to sites of generation of these Ca2+ microdomains such as IP3 receptor channels at the ER and some Ca2+ channels at the plasma are able to efficiently take up Ca2+.[153] We used mitochondria-targeted aequorin to monitor, for the first time, the coupling of Ca2+ release and mitochondrial Ca2+ uptake in the soma and neurites of neurons in primary cultures.[154,155] This configuration limits mitochondrial Ca2+ overload involved in mPTP. However, if Ca2+ influx or Ca2+ release mechanisms are enhanced and/or sustained, or Ca2+ buffers or extrusion systems are saturated, then mitochondrial Ca2+ overload may occur leading to neuron cell death. In the last few years, a large body of evidence indicates that changes in intracellular Ca2+ homeostasis may contribute to neuron damage in AD and PD.[156]
Early evidence suggested that neurons obtained from mouse models of AD displayed changes in intracellular Ca2+ homeostasis, including alterations in resting levels of intracellular Ca2+ and Ca2+ stores.[157] These changes could be associated to the effects of mutations in the APP and presenilins PS1 and PS2, which process APP either directly or indirectly via excessive production of Aβ species. Mounting evidence indicates that presenilins may work as ER leak channels, so presenilin mutations involved in familial AD cases would lead to variations in Ca2+ store content and Ca2+ release.[158] In addition, elevated generation of Aβ species tend to oligomerize and eventually aggregate, thereby resulting in the formation of toxic Aβ species. The mechanism by which Aβ species promote neuron damage is not totally understood. First indications suggested that Aβ proteins could integrate in lipid bilayers and form Ca2+-permeable pores termed amyloid channels.[159] We showed that Aβ1-42 oligomers, the assembly state that probably correlates better with cognitive deficits in AD, increase Ca2+ influx in rat cerebellar and hippocampal neurons, followed by mitochondrial Ca2+ overload as monitored by bioluminescence imaging in individual neurons expressing mitochondria-targeted aequorin.[160,161] Mitochondrial Ca2+ overload induced by Aβ1-42 oligomers promote mPTP opening followed by release of cytochrome c and apoptosis. Importantly, prevention of mitochondrial Ca2+ uptake without affecting the rise in cytosolic Ca2+ protected neurons. For instance, small mitochondrial depolarization – that reduces the driving force for mitochondrial Ca2+ uptake – diminishes the mitochondrial Ca2+ overload and the ensuing release of cytochrome c and apoptosis.[160]
Interestingly, a number of non-steroidal anti-inflammatory drugs (NSAIDs), such as the aspirin metabolite salicylate, and over the counter NSAIDs such as sulindac sulfide, indomethacin, ibuprofen and its enantiomer R-flurbiprofen (that partially depolarize mitochondria at low concentrations) resulted in the inhibition of both the mitochondrial Ca2+ overload and the resulting apoptosis. Thus, Aβ neurotoxicity depends largely on mitochondrial Ca2+ overload and any compound targeting MCU, or ΔΨm may protect against Aβ oligomer-induced pathology. Therefore, mitochondrial Ca2+ emerged as a potential new target to prevent AD[162] that could be targeted by different compounds, like the antibiotic minocycline, able to prevent neurotoxicity[163] or drugs like methadone that promote neurotoxicity.[164] Interestingly, clinical data suggested that some of these compounds, particularly several NSAIDs, showed a neuroprotective effect in several animal models of AD. R-Flurbiprofen, an enantiomeric form of flurbiprofen lacking anti-inflammatory activity was selected for a large AD clinical trial. Unfortunately, this trial failed probably because selected patients undergo large neuron cell death at the time of recruitment. The question remains whether NSAIDs and mitochondria are still good candidates for developing new drugs against AD.[165] Overall, data suggest a critical role for intracellular Ca2+ dyshomeostasis in AD that is amenable for the development of novel pharmacological agents to be tested in further clinical trials.[166]
In addition to the contribution of excitotoxicity, mutations in APP and/or presenilins and the overproduction of Aβ oligomers, a critical component that is often overlooked is the critical influence of aging. We have developed an in vitro model of neuronal aging based on long-term culture of rat hippocampal neurons that, under specific conditions, acquire an aging phenotype. We reported that Ca2+ responses induced by activation of VOCCs after plasma membrane depolarization and neurotoxins, including the glutamate receptor agonist NMDA and Aβ oligomeric forms, were significantly elevated in aged neurons compared to young neurons.[167,168] These functional changes were associated to changes in the expression of NMDA receptor subunits. Interestingly, NMDA and Aβ oligomers increased mitochondrial Ca2+ overload and apoptosis only in aged neurons but not in young cells, possibly because the cytosolic Ca2+ responses evoked by the neurotoxins were high enough to induce mitochondrial Ca2+ overload only in aged neurons, thus implying a critical role of mitochondrial Ca2+ in cell dead induced by these agonists.
Neuroinflammation also plays a pivotal role in the etiopathogenesis of AD. A critical component of inflammation is the activation of Toll-like receptors (TLRs), transmembrane pattern-recognition receptors of the innate immune system that recognize diverse pathogen-derived and tissue damage-related ligands and induce the corresponding immune response in specific cells. Recent data suggest that TLR signaling contributes to the pathogenesis of AD and other age-related, neurodegenerative diseases.[169] Consistently, we recently demonstrated that the TLR4 agonist lipopolisacharide (LPS) induces Ca2+ responses and apoptosis in hippocampal aged neurons in vitro while no effects were observed on young neurons.[170] These effects were prevented by administration of TLR4 antagonists. Consistently, TLR4 expression is significantly increased in aged neurons relative to young cells. Treatment of aged neurons with Aβ oligomers enhanced TLR4 expression as well as LPS-mediated Ca2+ responses and neuron cell loss. These data indicate that both aging and Aβ oligomers may contribute to increase the susceptibility to neuroinflammation in rat hippocampal neurons.[171]
The data also suggest that aging neurons undergo significant changes in expression and/or activity of molecular players involved in Ca2+ transport that renders them more vulnerable to damage. We reported that long-term cultures of rat hippocampal neurons underwent such Ca2+ remodeling.[172] Specifically, aged neurons show enhanced resting [Ca2+]cyt and Ca2+ store content and release from ER, together with increased Ca2+ transfer from the ER into mitochondria. Moreover, aged neurons exhibited a significant decrease in the so-called store-operated Ca2+ entry (SOCE), a pathway that has been related to dendritic spine stability and memory storage. Therefore, these changes in Ca2+ homeostasis found in aging neurons may favor energy production at the risk of increased susceptibility to mitochondrial Ca2+ overload and cell death as well as reduced spine stability. These functional changes correlated with altered expression of the ER IP3 receptor, mitochondrial MCU, NMDA and TLR4 receptors, and the plasma membrane molecular players involved in SOCE.[173] Interestingly, treatment of aged neurons with Aβ oligomers further exacerbated most of the changes involved in Ca2+ remodeling associated to aging and the susceptibility to cell death, including resting Ca2+, Ca2+ store content, and Ca2+ responses to NMDA and TLR4.[173] Administration of Aβ oligomers also decreases further SOCE in aged neurons. We proposed that neuronal aging is associated to Ca2+ remodeling that favors energy production at the expense of increased susceptibility to damage and decreased ability for memory formation. In addition, this process is exacerbated by the generation of Aβ oligomers, leading to pathological aging that contributes to the development of AD.[174]
The mechanism by which Aβ peptide species, particularly oligomers, promote neuron damage is not totally understood. The proposal that Aβ species form Ca2+ permeable pores or channels responsible for Ca2+ entry is not widely accepted.[166] Alternatively, it has been proposed that Aβ oligomers bind an activate plasma membrane Ca2+ channels, particularly NMDA receptor channels.[175] We have recently shown that Ca2+ responses to Aβ oligomers were highly dependent on synaptic networking.[176] In particular, we demonstrated that generation of spontaneous, synchronous [Ca2+]cyt oscillations in neurons are abolished by many different blockers of synaptic transmission (such as NMDA receptor antagonists) and blockers of action potential propagation (tetrodotoxin). In the absence of networking activity, Ca2+ responses to the Aβ oligomers are smaller and are inhibited only by NMDA receptor antagonists and blockers of the formation of amyloid channels (such as NA7). In addition, combination of these two blockers essentially abolished Ca2+ responses induced by Aβ oligomers.[176] These findings suggest the involvement of both NMDA receptors and the amyloid channels in the primary response to Aβ oligomers that are further enhanced by networking activity. In support of this notion, we also showed that non-neuronal cells expressing NMDA receptors exhibited Ca2+ responses to oligomers, in contrast to cells lacking NMDA. Expression of NMDA receptor subunits NR1/NR2A and NR1/NR2B in these cells restored Ca2+ responses to NMDA but not to Aβ oligomers. These data suggest that Aβ oligomers may promote Ca2+ entry via both amyloid channels and NMDA receptors. Thus, NMDA receptors appear necessary but not sufficient for Ca2+ responses to oligomers. Furthermore, Aβ oligomers may activate distant neurons intertwisted by synaptic connections, thus favoring the spreading of excitation by the recruiting of further NMDA receptors and specific VOCCs, leading to massive Ca2+ entry, excitotoxicity, and neuron degeneration in AD.[176]
Accordingly, intracellular Ca2+ homeostasis and its dysregulation play a pivotal role in the susceptibility to neuron damage during aging, neuroinflammation, excessive production of amyloid peptides, excitotoxicity, and mutated presenilins, all of them processes involved in AD. We are currently working on the development of novel drugs targeting these pathways, for example, the synthesis of marine heterocyclic compounds.[177]
Altered Ca2+ homeostasis and concomitant neurotoxicity in Parkinson’s disease
Damage of SN DA is considered a main mechanism responsible for PD. These neurons are under extreme bioenergetic demand because of three particular features: (i) the maintenance of a giant axonal tree that requires anterograde/retrograde transport of cytosolic metabolites and organelles, (ii) the maintenance and propagation of action potentials (APs) and the restoring of ionic gradients, and (iii) the large number of synaptic vesicles cycling. Related to the elevated energy requirement, SN DA neurons display large [Ca2+]cyt oscillations that can boost mitochondrial OXPHOS activity, which may render them vulnerable to damage when neurons are challenged by different stressors like genetic mutations, mitochondrial toxins or defective aging (Figure 1).
In 2007, Surmeier and cols. proposed for the first time that reliance of DA neurons on L-type VOCCs sensitive to dihydropyridines or Cav1.3 channels, for rhythmic pacemaking rendered them vulnerable to different stressors known to promote PD. These authors demonstrated that reliance of pacemaking activity on these VOCCs increased in a time-dependent manner and differed from young SN DA neurons, that apparently, use alternative mechanisms for pacemaking activity common to other neuron types unaffected in PD.[178] Interestingly, it appears that these juvenile mechanisms remain latent in aged neurons, such that inhibition of Cav1.3 turn on the pacemaking mechanism to the juvenile form, thus protecting DA neurons from damage. Specifically, treatment with isradipine, a blocker of Cav1.3 channels, reversed the pacemaking phenotype and protected against different PD toxins. Data prompted the consideration of dihydropyridines, used for decades as hypertensive treatment, could be a novel approach for PD.[179] The present view is that spike-activated Ca2+ influx mediated by Cav1.3 channels may trigger Ca2+ release from the ER that is transferred to mitochondria to stimulate mitochondrial OXPHOS by two Ca2+-dependent mechanisms: a mechanism mediated by the MCU-dependent Ca2+ uptake and enhanced Krebs cycle and mitochondrial respiration, and another mechanism dependent of the malate-aspartate shuttle. The disruption of any of the two mechanisms results in the impairment of the ability of DA neurons to sustain a spike activity. Thus, although this feedforward mechanism attends DA neurons bioenergetic demands linked to sustained spiking, it also underlies the increased oxidative stress and damage with aging or disease.[180]
Whether targeting mitochondria oxidant stress is enough to prevent PD progression has been extensively examined. Unfortunately, these attempts have failed so far to show efficacy. Recent epidemiological studies provided evidence of a significant correlation between low PD risk and the use of Cav1 channel inhibitors such as dihydropyridines. A recent Phase III clinical trial using isradipine did not show beneficial effects on PD patients. The reasons for the trial failure may be associated to the extensive damage of DA neurons in patients already at the time of diagnosis and/or recruitment, and/or the low level of isradipine concentration achieved in vivo in the central nervous system due to large drug clearance.[181] Despite this failure, there is still a substantial interest in developing novel drugs selectively targeting Ca2+ signaling in DA neurons.
For instance, cyclic α-aryl β-dicarbonyl derivatives are important scaffolds in medicinal chemistry that have been used for developing new candidate agents for treating PD. Specifically, a palladium-catalyzed coupling reactions of haloarenes were conducted recently with diverse five- to seven-membered cyclic β-dicarbonyl derivatives that include barbiturate, pyrazolidine-3,5-dione, and 1,4-diazepane-5,7-dione. The coupling reactions of different para- or meta-substituted aryl halides were efficient when Pd(tBu3P)2, Xphos, and Cs2CO3 are used in 1,4-dioxane reflux conditions. Consequently, several 5-aryl barbiturates were synthesized to be used as new scaffolds of Cav1.3 channel inhibitors with an IC50 at 1.42 μM.[182]
Modulation of calcium signaling and homeostasis by heterocyclic compounds in Alzheimer’s disease and Parkinson’s disease
Extensive evidence has demonstrated that dysregulation of Ca2+ homeostasis and signaling are mechanisms that play an essential role in the etiopathogenesis of AD and PD. Aβ peptides are involved in the excessive accumulation of intracellular Ca2+ through the formation of Ca2+-permeable pores in the plasma membrane or by increasing Ca2+ influx via activation of L-type VOCCs and NMDA or AMPA receptors.[14,15] On the other hand, genetic mutations in presenilin or activation of inositol 1,4,5-trisphosphate (IP3) and ryanodine receptors promote Ca2+ leak in the ER, which produces an elevation of both cytosolic and mitochondrial Ca2+ concentration.[183,184,185] Mutations in the gene encoding for LRRK2, which are associated with both familial and sporadic PD, elicited transcriptional upregulation of the mitochondrial Ca2+ uniporter and the mitochondrial Ca2+ uptake 1 protein in post-mortem human brain, fibroblasts, and primary mouse cortical neurons.[16] Pathological mutant LRRK2 enhanced mitochondrial Ca2+ uptake and dendritic injury but increased mitochondrial Ca2+ release restored Ca2+ homeostasis and was neuroprotective. These findings demonstrated that LRRK2-driven neurodegeneration raises susceptibility to mitochondrial Ca2+ overload and implicates mitochondrial Ca2+ dyshomeostasis in the pathogenesis of PD. Moreover, knockdown or inhibition of IP3 kinase B led to α-syn aggregation and phosphorylation in primary cortical neurons from mice incubated with α-syn fibrils, which increases (i) Ca2+ release from the ER, (ii) mitochondrial Ca2+ content, and (iii) mitochondrial respiration.[13] We have found some heterocyclic compounds with the capability of modulating Ca2+ uptake that can be considered as promising pharmacological agents for the treatment of AD and PD (Table 1).
1. ANAVEX2-73
ANAVEX2-73 (Blarcamesine) is a non-selective muscarinic acetylcholine receptor (mAChR) and alfa-1 receptor (S1R) ligand that exhibits an important affinity for its pharmacological targets at micromolar concentration.[186] σ1 Receptors are ubiquitously expressed in the central nervous system (CNS) and are located at mitochondria-associated ER membranes, where they interact with IP3 receptors to regulate Ca2+ exchange between the ER and mitochondria, thereby resulting in reduced mitochondrial stress, regulation of ion channels, activation of the nuclear erythroid 2-related factor 2 (NRF2) / antioxidant response element (ARE) pathway, and limited apoptosis.[187,188] IP3 receptor-mediated Ca2+ release correlates with variations in the availability of mitochondrial Ca2+ and ATP synthesis.[189] A formal concept analysis (FCA) combined with the Knowledge Extraction and Management (KEM) environment was able to identify ANAVEX2-73 (tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride) as a potential genomic biomarker of disease and therapeutic response in a phase IIa clinical trial.[190] An ongoing randomized, double-blind phase III trial of this compound is evaluating the bioavailability, safety, tolerability, and effectiveness of the treatment in patients with AD (Nct03790709), PD with dementia (NCT03774459) and Rett syndrome (NCT03758924).
Intraperitoneal administration of ANAVEX2-73 (0.3-1 mg/kg) reversed scopolamine- and dizocilpine-induced learning impairments one week after intracerebroventricular injection of the neurotoxic Aβ25-35 peptide in mice.[191] Using the same mouse model, treatment with ANAVEX2-73 inhibited the phosphatidyl-inositol 3-kinase (PI3K)/Akt pathway, thereby resulting in the activation of GSK-3β, improvement of behavioral deficits, and limitation of Aβ seeding and tau-induced pathology.[192] Moreover, the drug preserved mitochondrial integrity and function in isolated mitochondria from the hippocampal brain region of Aβ25-35 injected mice by increasing the activity of complex IV and oxygen consumption at all states.[193] Exposure to ANAVEX2-73 also decreased the peroxidation of lipids, Bax/Bcl-2 ratio (that determines the cell susceptibility to apoptosis) and cytochrome c release. In an independent study, ANAVEX2-73 restored the respiratory control ratio and preserved complex IV levels from Aβ toxicity in a Ca2+-dependent fashion that regulates several tricarboxylic acid cycle (TCA) enzymes including α-ketoglutarate dehydrogenase complex and isocitrate dehydrogenase, responsible for NADH production, a substrate for complex I.[194] ANAVEX2-73 displayed a synergic effect with donepezil (but not with memantine) and restored spontaneous alternation and passive avoidance response in a non-transgenic mouse model of AD.[195] Incubation with ANAVEX2-73 elicited the accumulation of LC3-II-positive puncta and succeeding autophagic flux in cell cultures. C. elegans expressing GFP-LGG-1 (a marker of autophagy) treated with ANAVEX2-73 exhibit increased autophagic activity. In addition, administration of ANAVEX2-73 to Aβ42-expressing nematodes upregulated proteostasis capacity, ultimately resulting in a dissociation and clearance of Aβ42 aggregates.[196]
In summary, ANAVEX2-73 modulates Ca2+ release after its interaction with IP3 receptors and prevents mitochondrial failure in multiple ways, including through the activation of the NRF2/ARE pathway that directly controls the expression of several antioxidant and anti-inflammatory genes. In addition, the molecule promotes autophagosome biogenesis and autophagic flux to degrade aggregated proteins and damaged organelles (such as mitochondria).
ANAVEX2-73 (1-(2,2-diphenyltetrahydro-3-furanyl)-N,N-dimethylmethanamine hydrochloride) 1 unique total synthesis was reported by Foscolos and co-workers.[197] The key step during the synthesis is the reduction-opening of lactone 7 to provide 1,4-diol 8, which was subsequently converted by acid-catalyzed cyclodehydration to ANAVEX2-73 1 (Scheme 1).
2. Caffeine
Caffeine (Mateine), a purine alkaloid present in several plants (Coffea, Camellia, Cola, Cirus, Ilex, Paullinia, and Theobroma), is the most consumed psychoactive substance in the world.[198] Coffee contains a variety of compounds such as caffeine, chlorogenic acid, cafestol, diterpenes and kahweol.[199] Moreover, coffee is a rich source of bioactive components that contribute to its biological activity, including potassium (K+), magnesium (Mg2+), niacin and potent antioxidants (chlorogenic acid and tocopherol).[200] Research studies have established a strong relationship between frequent caffeine consumption and reduced risk of developing AD and PD, with no detectable adverse effects in the CNS in an exposed population. A large body of literature has shown that mutations in presenilins are linked with Ca2+ signaling dysregulation and elevated Ca2+ release from the ER in neuronal cultures expressing mutant PS,[201,202,203] transgenic mice engineered to overexpress mutant PS1,[204,205] and fibroblasts from subjects with AD.[206,207,208] PS induces excessive Ca2+ accumulation and release in part via its biochemical interaction with the IP3 receptor Ca2+ release channel, thereby resulting in an anomalous regulation of Ca2+ signaling pathways and stimulation of APP processing and Aβ synthesis,[209,210,211] even prior the formation of plaques and NFTs.[212] Ca2+ storage content is higher in mutant presenilin-1 (PS1) knock-in neurons primary cortical neurons from a triple transgenic mouse model of AD (3xTg-AD) compared to non-transgenic cells.[213] Caffeine exposure altered Ca2+ signaling and promoted its release, which was independent of extrusion mechanisms or variations in the steady-state concentration of specific Ca2+-binding proteins. However, this effect was attributed to the activation of the ryanodine receptors (RyRs) that become sensitized to so-called process of Ca2+-induced release by caffeine.
The CAIDE longitudinal epidemiological study suggested an association between mid-life moderate coffee (but no tea) consumption and late-life reduced risk of dementia and AD.[214] Specifically, caffeine intake (3-5 cups per day) reversed cognitive impairment (assessed by Mini-Mental State Examination) in a gender-specific manner (women age 65 and older). A recent mendelian randomization study found that genetically predicted higher caffeine content in the plasma correlates with diminished risk of AD.[215] Following confounding adjustment, it has been described that long-term coffee consumption (≥ 2 cups/day) was associated with a significant cognitive decline decrease in pathological Aβ deposition. Nevertheless, no changes were observed in cortical thickness, cerebral white matter hyperintensities or cerebral glucose metabolism, which are features also related to the neurodegenerative process.[216] The Honolulu Heart Program provided the first evidence showing a potential beneficial effect of caffeine intake in PD patients. Coffee drinkers (28 oz or more per day) had 5-fold lower incidence of developing PD compared to non-caffeine drinkers following an adjustment for both age and pack-years of cigarette smoking.[217] This study was further supported by Ascherio and colleagues.[218] In addition, consumption of decaffeinated coffee was not associated with decreased risk of PD, suggesting that caffeine, rather than other coffee components, accounts for the inverse association observed. The findings showed a significant negative correlation between caffeine intake and risk of developing PD in men but a U-shaped relationship among women. The risk of PD was similar in women using hormones and women who never used hormones. Interestingly, caffeine diminished the risk of PD in menopausal women that did not take hormone replacement but there is a higher risk (4-fold) among hormone users with high caffeine.[219] The risk of PD was even lower when coffee intake is combined with cigarette smoking and nonsteroidal anti-inflammatory drug use, resulting in a cumulative effect.[220]
A different clinical trial confirmed that caffeine consumption mitigates the risk of PD, but its neuroprotective properties may vary depending on its interaction with other factors, such as obesity and low serum cholesterol levels, which can modify the risk of having PD.[221] A meta-analysis involving a large number of participants found a non-linear relationship between coffee consumption and the incidence of PD that achieves the maximum protective effect at 3 cups per day.[222] However, the authors described a linear dose relationship of reduced risk of PD with both tea and caffeine consumption, especially in men compared to women and in European and Asian population relative to USA residents. Based on an open-label, dose-escalation study, caffeine had potential motor and non-motor benefits in subjects with PD, with the maximum tolerated dose of 100-200 mg/day bis in die (BID) without affecting sleep quality.[223] In a randomized controlled trial, administration of 200 mg caffeine BID for six weeks did not improve daytime somnolence in PD patients but possessed the potential to reverse motor symptoms.[224] Transgenic mice overexpressing the human APP gene carrying the Swedish mutation (APPsw) treated with 0.3 mg/mL caffeine in the drinking water for 5 months exhibited an improvement in multiple behavioral measurements.[225] In addition, enzyme-linked immunosorbent assay showed a significant downregulation of both Aβ40 and Aβ42 levels and decreased PS1 and β-secretase (BACE) protein concentration in the hippocampus of these AD mice. Oral supplementation with caffeine for 4 months restored motor performance, anxiety and memory deficits, prevented neuronal death in the CA1 pyramidal layer of the hippocampus, and promoted neurogenesis in the absence of detectable pathological effects on the Aβ pathology in transgenic Tg4-42 and 5xFAD mouse models of AD.[226] Caffeine did not alter the optical density or mRNA expression levels of A1 or A2A receptors in the mouse cerebral cortex or hippocampus.
Aβ burden was reduced in both brain and plasma of APPsw and APP/PS1 mice (which contain human transgenes for both APP bearing the Swedish mutation and PSEN1 containing a L166P mutation) following either acute or chronic caffeine administration.[227] However, amyloid plasma levels were not correlated with (i) Aβ brain content, (ii) cognitive impairment, and (iii) pro-inflammatory cytokine levels in aged mice. Animals receiving caffeine also display an enhanced memory performance. It has been also demonstrated that long-term administration of caffeine is protective in the THY-Tau22 transgenic mouse model of progressive AD-like tau pathology by limitation of spatial memory abnormalities, tau phosphorylation, oxidative stress, and inflammation.[228] Since persistent (but no acute) administration of caffeine increases cerebrospinal fluid (CSF) secretion in rats in comparison to the control-treated group, it has been proposed that chronic caffeine intake can promote CSF production combined with an elevated cerebral blood flow velocity and Na+/K+-ATPase levels.[229] Protein kinase A (PKA) is a heterotetrametric enzyme comprised of two regulatory and two catalytic subunits that plays an essential role in cell proliferation with an anti-apoptotic activity. The cyclic adenosine monophosphate (cAMP)-response element binding protein (CREB) is a transcription factor that modulates a subset of genes implicated in cognition and neuron survival. c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) are mitogen-activated protein kinases that can trigger apoptotic signaling by the upregulation of pro-apoptotic genes through the activation of specific transcription factors or directly, by catalyzing protein phosphorylation. Administration of caffeine upregulated PKA activity, induced CREB phosphorylation, and reduced the levels of phosphorylated JNK and ERK in the striatum (but no frontal cortex) of APPsw mice, suggesting that caffeine pro-apoptotic signaling.[230] Caffeine intake resulted in memory capacity improvement and increased hippocampal brain neurotrophic derived factor (BNDF) and tyrosine receptor kinase B (TrkB) immunoreactivity in PS1/APP double transgenic mice and rats treated with aluminum chloride.[231,232]
Caffeine also has a neuroprotective role in PD. Exposure to caffeine preserved the degeneration of the nigrostriatal DAergic pathway in neurotoxin-based animal models of PD, such as rotenone, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and 6-hydroxydopamine (6-OHDA). Histopathological and immunohistochemical characterization of brain tissue from rotenone-exposed rats show a significant degeneration of SN DA neurons and their projections into the striatum. However, administration of caffeine provided a dose-dependent therapeutic effect against rotenone-mediated neurotoxicity.[233] Injection of caffeine through a peritoneal route improved the behavioral phenotype, normalized brain enzymatic activities of both acetylcholinesterase (AChE) and Na+/K+-ATPase (which play an important role in memory and learning) and mitigated neuroinflammation and oxidative damage in rotenone-treated rats.[234] Caffeine modulated striatal protein content and catalytic activity of cytochrome P450 (a membrane-bound hemoprotein that plays a central role in the detoxification of xenobiotics), glutathione-S-transferases (a family of phase II detoxification enzymes involved in the protection of macromolecules from attack by ROS, reactive electrophiles, environmental carcinogens, and chemotherapeutic agents) and vesicular monoamine transporter-2 (VMAT-2, an integral presynaptic protein that controls the packaging and release of DA and other neurotransmitters from synaptic vesicles) in mice receiving MPTP for 4 weeks.[235] Using ex vivo 1H-[13C]-NMR spectroscopy, Bagga et al. found that pretreatment with caffeine increased neuronal glutamatergic and GABAergic metabolic activity, and neurotransmission in MPTP-injected mice.[236] Oral supplementation with the coffee component eicosanoyl-5-hydroxytryptamide attenuated MPTP-induced nigrostriatal DAergic cell loss in mice, displayed both antioxidant and anti-inflammatory properties and normalized phosphoprotein phosphatase 2A (PP2A) methylation and activity.[237]
Intraperitoneal injection of 10-20 mg/kg day of caffeine improved motor dysfunction and increased catecholamine levels in a rat model of 6-OHDA-induced striatal lesion.[238] Striatal injection of 6-OHDA elicited apomorphine-induced rotation and impaired locomotor activity in parallel with a loss of DA immunoreactivity and an inflammatory response, but administration of caffeine ameliorated the behavioral and pathological PD-like phenotype. Moreover, it has been also reported that administration of caffeine in the drinking water can exert neuroprotective effects by decreasing the number of inclusions positive for pS129 α-syn, content of TUNEL-stained apoptotic cells, LC3-mediated macroautophagy, and lysosome-associated membrane protein type 2A (LAMP2A) chaperone-related autophagy in mice that received an intracerebral injection of synthetic α-syn fibrils with the A53T missense mutation.[239] Regarding its potential mechanism of action, caffeine is an antagonist of the adenosine-2A receptor (A2AR), which are predominantly localized in the GABAergic striatopallidal neurons projecting from the caudate nucleus and the putamen to the external segment of the globus pallidus (indirect pathway).[240] A2ARs colocalize with DA2 receptors to form heteromeric complexes that mediate the allosteric antagonism between adenosinergic and DAergic transmission.[241] Noteworthy, a double-blind, randomized, crossover study showed that treatment with caffeine improved the pharmacokinetic profile of levodopa by reducing its plasma concentration-time profile and by prolonging its effect.[242]
In summary, while caffeine has complex biological and pharmacological profiles, experimental evidence indicates that caffeine readily crosses the blood-brain barrier and exerts its action by antagonizing A2ARs and confers neuroprotection by stimulating mitochondrial function and by attenuating excitatory neurotransmitter release, oxidative insult, and neuroinflammation. Clinical trials have shown that caffeine significantly enhances AD-related memory loss and improves motor symptoms in PD patients. In addition, caffeine has a well-established long-term safety profile. Together with its low cost and high availability, caffeine is a promising therapeutic agent for the treatment of neurodegenerative diseases.
The most recent total caffeine (1,3,7-trimethyl-3,7-dihydro-1H-purine-2,6-dione) 2 synthesis was reported by Narayan in 2003 from uracil 9.[243] N-Methylation in the presence of a strong base such as sodium hydride in dimethylsulfoxide (DMSO) produced 1,3-dimethyluracil 10, which was nitrated and subsequently reduced to 5-amino-1,3-dimethyluracil 12, using iron and hydrochloric acid. Compound 13 was obtained following two conventional steps and resulted in the formation of theophylline 14 after a reduction process and an intramolecular heterocyclization reaction with iron and acetic acid. The final methylation at position 7 led to the generation of caffeine 2. Therefore, synthesis of caffeine needed seven reaction steps with an ~10% overall yield (Scheme 2).
In recent years, new and improved methodologies for the N-methylation of theophylline 14 have been published. A novel technique uses the Q-tube apparatus in water at over boiling temperature as a green solvent.[244] In a different study, Schnürch’s group optimized the employment of quaternary ammonium salts as monoselective N-methylation reagents (Scheme 3).[245]
3. Diltiazem
Diltiazem (Cardizem) is a non-dihydropyridine Ca2+ channel blocker with antihypertensive, antiarrhythmic and vasodilation properties. The drug selectively targets the VOCCs, which are the primary mediators of Ca2+ influx into neurons in response to membrane depolarization. P/Q- and N-type VOCCs regulate neurotransmitter release upon arrival of the action potential to the axon terminal in the presynaptic neuron. Glutamate release promotes postsynaptic Ca2+ trafficking by activation of NMDA receptors (NMDAR) or through an indirect pathway involving L-type VOCCs.[246] Reduced Ca2+ transient at presynaptic or postsynaptic sites can mitigate glutamate-induced excitotoxicity. Therefore, administration of Ca2+ channel blockers has become an interesting approach for the treatment of neurodegenerative diseases. PET and postmortem analyses of different brain regions (such as cerebral cortex and amygdala) from AD subjects revealed a marked inhibition of AChE enzymatic activity, responsible for the hydrolytic metabolism of the neurotransmitter acetylcholine into acetate and choline.[247,248,249] In contrast, administration of Aβ25-35 peptide upregulated the activity of AChE through modulation of the L-type VOCCs (increasing intracellular Ca2+ concentration) in embryonal carcinoma P19 cells.[250] When cultures were incubated in the presence of diltiazem, the authors found a 75% loss of AChE enzyme activity.
Epidemiological data obtained from individuals with PD indicate that long-term treatment with Ca2+ channel blockers targeting Ca2+ channels of DA neurons may represent a potential therapeutic strategy, reducing the risk for developing the disease.[251,252,253] A larger study involving 65,001 patients, reinforced the connection between Ca2+ channel blockers and diminished incidence of PD.[254] In addition, centrally acting Ca2+ channel blockers prevent nigrostriatal DAergic degeneration in parkinsonian mice and non-human primates and improve survival rate in vitro.[178,255,256] There is evidence supporting a beneficial effect of diltiazem against DA toxicity in human neuroblastoma cells.[257] Cav1.2 and Cav1.3 are L-type VOCCs regulate DAergic neuron spontaneous firing activity in the SN region of the brain. DA neurons fire either in a low frequency single-spike pattern or transiently, in a high-frequency so-called burst mode. Data from animal models support clinical observations showing that diltiazem has positive effects on AD-induced pathology. Ca2+ channel blockers, such as diltiazem, protect neurons from Aβ-induced influx of Ca2+ ions and downstream toxicity as well as decrease the amyloid content by facilitating the clearance rate.[258,259] Excessive neuronal Ca2+ influx contributes to neuronal dysfunction and degeneration that underlies cognitive disturbances in AD. Neuronal cultures incubated with Aβ peptides led to an enhanced Ca2+ entry.[260] Furthermore, endogenous accumulation of oligomeric Aβ led to an upregulated expression of L-type VOCCs (Cav1.2).[259] Diltiazem improved survival rate and decreased Ca2+ intracellular concentration by blocking L-type Ca2+ channels in vitro. Diltiazem protected MC65 neuroblastoma cells from the toxicity mediated by the APP C-terminal fragment by improving cell survival.[261]
In addition, the Aβ25-35 fragment decreased both secreted APP and Aβ while promoted cell-associated APP in chick sympathetic neurons. Enhanced APP secretion with no variations in the amount of cell-associated APP occurred in response to diltiazem.[262] The accumulation of exogenous Aβ significantly attenuated cell viability and proliferation (determined by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide, MTT) in primary hippocampal neuronal cultures, which was reversed by exposure to diltiazem.[13] This study also provided evidence supporting that Aβ toxicity shows an early Ca2+-independent phase and a late Ca2+-dependent phase. Spontaneous alternation behavior, a reliable measure of short-term of spatial working memory, was significantly increased in mice treated with diltiazem.[263] Chronic exposure to aluminum or any other of its forms in the drinking water has been associated with higher risk of developing AD.[264,265,266] Aluminum accumulation has been detected in the brain of AD cases, where causes neurofibrillary degeneration in neurons.[267] Bouras et al. found a specific increased content of aluminum within NFTs in both the inferior temporal cortex and hippocampus of individuals with AD.[268] Aluminum induces Aβ protein conformational modifications and aggregation (including amyloid fibrillation) either in vitro or in vivo.[269,270,271,272] Aluminum chloride has been reported to cause dementia (AD accounts for the ~70% of cases) but administration of diltiazem reversed learning and memory deficits, AChE upregulation, and oxidative damage in mice.[273] Ginkgo biloba augmented the bioavailability of diltiazem by modulating cytochrome P450 3A activity in both the rat liver and small intestine.[274]
It has been reported that Cav1.2 and Cav1.3 L-type VOCCs are susceptible of degeneration during the progression of PD.[275,276,277] During spontaneous action potentials, L-type VOCCs contribute to Ca2+ oscillations in the soma and dendrites of constantly active midbrain DA neurons. The extended opening of L-type VOCCs throughout autonomous pacemaking induces prolonged cytoplasmic Ca2+ trafficking and its overload in SN DA neurons, leading to mitochondrial oxidative stress.[275,277] L-type VOCCs-mediated alteration of the steady-state of DA levels is responsible for causing downstream oxidative stress and α-syn-induced toxicity.[278] In contrast, some studies suggest that administration of Ca2+ channel blockers (including diltiazem) could be associated with the development of parkinsonian symptoms, particularly, in subjects that already had subclinical idiopathic PD pathology.[279,280] Despite there is a lack of research investigating the potential antioxidant effect of diltiazem in AD or PD, it has been found that its intraperitoneal injection decreased the levels of nitrite (an indirect assessment of nitric oxide) and MDA (a lipid peroxidation marker) and upregulated the activity of several antioxidant enzymes such as reduced glutathione, catalase, and superoxide dismutase in a streptozotocin-induced rat model of diabetes.[281] High-performance liquid chromatography analysis showed a significant decline of MDA content in perfused rabbit hearts treated with diltiazem.[282] Oral supplementation with diltiazem for two weeks reduced thiobarbituric acid-reactive species (TBARS, a by-product of LPO) and nitrite content and upregulated superoxide dismutase (SOD) and reduced glutathione enzymatic activities in an aluminum chloride mouse model of AD.[273] Because of their autonomous pacemaking activity coupled to action potentials, SN DA neurons have particularly high energetic (ATP) demands and may be subjected to elevated levels of basal oxidative stress.[283]
Likewise, the anti-inflammatory effect of diltiazem in patients with AD or PD has not yet been examined. Diltiazem produced a dose-dependent inhibitory effect on T-cell proliferation.[284] Individuals undergoing cardiopulmonary bypass surgery displayed at increase in IL-6 content, which was significantly attenuated following diltiazem administration.[285] Enhanced levels of the anti-inflammatory cytokine IL10 were observed in subjects with unstable angina receiving diltiazem.[286] Moreover, intraperitoneally injected diltiazem reduced IL-10 and TNF-α (but no nitrite/nitrate and IL-6) plasma concentration in BALB/c mice injected with bacterial lipopolysaccharide (LPS).[287] Synthetic Aβ25-35 fragments added to human fetal brain cell cultures activates microglia and increases the levels of intracellular Ca2+ in a time-dependent fashion.[288] Treatment with the L-type VSCCs antagonist diltiazem resulted in a transmembrane diminished Ca2+ influx that abolishes microglial stimulation. Cav1.2 VOCCs (particularly α1 subunit) are highly expressed in reactive astrocytes of mice overexpressing Aβ protein precursor (AβPP), which is specifically linked to an Aβ plaque deposition.[289] Cav1.2 channels are also expressed in microglia. Variations in the balance between M1 (pro-inflammatory) and M2 (anti-inflammatory) microglial phenotypes are related to PD. It has been demonstrated that diltiazem-induced blockade of L-type VOCCs promotes pro-inflammatory M1 transition and decreases anti-inflammatory M2 macrophage polarization in mouse microglia-derived MG6 cells, resulting in an upregulated iNOS mRNA expression and downregulated arginase levels.[290] In addition, knockdown of microglial Cav1.2 led to behavioral impairment, DA neuron degeneration in the SN of MPTP-injected mice compared to control group, and reduce polarization toward the M1 phenotype.
In summary, the L-type VOCC diltiazem is a Ca2+ antagonist that can be used to treat dementia and dementia-related diseases due to its capability of improving cognitive deficits, such as learning and memory capacity. The drug also improves neuron survival and exhibits both antioxidant and anti-inflammatory properties. Diltiazem can cause nausea and vomiting, pulmonary oedema, and renal failure. Diltiazem, together with verapamil, are the most toxic Ca2+ channel blockers in overdose and their toxicity is associated with significant cardiovascular collapse, metabolic problems (e.g., hyperglycemia) and vascular smooth muscle tone deficits.
The first racemic synthesis of diltiazem (2-(2-dimethylaminoethyl)-5-(4-methoxyphenyl)-3-oxo-6-thia-2-azabicyclo[5.4.0]undeca-7,9,11-trien-4-yl]ethanoate) 3 was described in 1990.[291] The first asymmetric total synthesis of (+)-diltiazem was reported by Naito and co-workers, using a diastereoface differentiating nucleophilic addition as the key step to create the two contiguous stereogenic centers.[292] Schwartz used the separation of diastereomeric glycidic esters (15 and 16) by direct crystallization as the key step, based on their marked difference in solubility.[293] Enantiomerically pure glycidic ester 15, the required isomer for the synthesis of (+)-diltiazem 3, was the major product obtained (54% yield) (Scheme 4).
In 1994, Jacobsen et al. used a manganese-catalyzed asymmetric epoxidation of cinnamate esters for the preparation of an enantiomerically pure key intermediate 18 in the synthesis of 3 (Scheme 5).[294]
To date, more synthetic routes to prepare (+)-diltiazem 3 from a chiral epoxide intermediate have been reported. The enantiomerically pure epoxide has been obtained by the introduction of a chiral auxiliary in the reaction[295,296], utilization of Yang’s catalyst[297], addition of metal complexes[298] or by direct acquisition.[299]
A useful synthetic methodology employed to induce chirality in the product is the utilization of lipase[300,301] or baker’s yeast catalysed reactions.[302] The most recent approach employed for total synthesis of (+)-diltiazem 3 was reported by Chen and colleagues in 2022.[303] Racemic keto ester 19, obtained in two steps from commercially available reagents, is converted into enantiomerically pure hydroxy ester 20 by ketoreductase-catalyzed dynamic reductive kinetic resolution. Two conventional steps (ring closure and ring opening) are further taken to produce an intermediate 21, which is readily subjected to intramolecular acid-catalyzed amidation. Two additional steps from intermediate benzothiazepinone 22 are required to obtain (+)-diltiazem 3. This chemoenzymatic synthesis involves eight steps to obtain (+)-diltiazem 3 in ~45% overall yield (Scheme 6).
4. Latrepirdine
Latrepirdine (Dimebon) is a carboline that blocks H1 histamine receptor (H1R) activity. A broad spectrum of effects on neurologically relevant targets was proposed based on various cell- and animal-based studies. The compound is a potent reversible and competitive inhibitor of both AChE and butyrylcholinesterase, which leads to elevated acetylcholine content and concomitant boosting of cognitive performance.[304,305] Besides its anti-histaminic properties, latrepirdine can modulate a wide range of other neurotransmitter receptors (such as DA, serotonin, glutamate, α-adrenergic, and imidazole).[306,307] Studies carried out in rat cerebral neurons confirmed the inhibitory effect of latrepirdine on NMDA receptors and involve the polyamine site of the NMDA NR2B subunit (the target for histamine) as a potential binging site.[304,308] Latrepirdine has also been shown to interact with Ca2+ channels; for example, its administration effectively blocked the activity of L-type VOCCs (IC50 = 57 μM) in cerebellar granule neurons.[309] A pilot clinical trial carried out in patients with mild-to-moderate AD given latrepirdine for 8 weeks showed an important reduction of neuropsychiatric symptoms (depression) and a significant cognitive function enhancement together with a lack of hematological and biochemical disturbances.[304] Drug safety and efficacy were also examined in moderate-to-severe AD subjects (NCT00912288). Data from a phase II randomized, double-blind, placebo-controlled clinical study showed a significant improvement in cognition, function, and behavioral outcome following 60 mg/day latrepirdine for 26 and 52 weeks (6 months extension phase) in individuals with mild-to-moderate AD with no adverse effects recorded.[310] CONNECTION and CONTACT phase III trials (NCT00838110) failed to show significant improvement in any primary or secondary outcome measures of cognition in patients treated with latrepirdine.[311] A phase II randomized, placebo-controlled trial of latrepirdine showed a protective effect against cognitive impairment in subjects with mild-to-moderate Huntington’s disease.[312] Results from the double-blind, placebo-controlled Phase III HORIZON trial (NCT00920946) revealed that latrepirdine failed to achieve statistical significance for co-primary endpoints in individuals with mild-to-moderate Huntington's disease. A meta-analysis study revealed that latrepirdine did not ameliorate overall cognition, but the molecule enhanced the neuropsychiatric inventory scale, used to assess psychopathology in dementia patients.[306]
Cerebellar granule neurons incubated with the Aβ25-35 peptide displayed morphological alterations, cell loss, and dysregulated intracellular Ca2+ homeostasis.[309] Nevertheless, exposure to latrepirdine preserved neurons against Aβ-mediated toxicity through inhibition of the L-type VOCCs current. Intraperitoneal administration of latrepirdine reversed learning and memory loss following chronic partial deprivation of cerebral cholinergic functions, which causes dementia in rats.[305] Latrepirdine had the capability of modulating cognitive ability in 5xFAD mice, though it was unable to mitigate Aβ-associated pathology.[313] P301S tau mice consistently performed better on both the inverted grid and accelerating rotarod tests after latrepirdine treatment, suggesting that the molecule is associated with enhanced motor performance.[314] Treatment with latrepirdine 15 min prior scopolamine-induced memory impairment significantly ameliorated delayed matching-to-sample task accuracy (to assess memory) in young adult and aged Rhesus macaques.[315] Treatment with latrepirdine in the drinking water for 4 months resulted in improved cognitive function in TgCRND8 mice although did not modify neither Aβ40 or Aβ42 content, suggesting APP processing as a direct target of the drug.[316] FDG-PET studies revealed an increased cerebral glucose utilization in aged mice in response to latrepirdine exposure.[317] Augmented succinate dehydrogenase activity, ΔΨm, and ATP synthesis were normalized following latrepirdine administration in differentiated SH-SY5Y cells and primary neuron cultures from mouse cerebral cortex with no differences in the mtDNA copy number, thereby suggesting a restoration of mitochondrial function.[318] Preservation of mitochondrial shape, mass, and respiratory chain complex activities were seen following latrepirdine treatment in cells incubated with Aβ.[319] The compound can also preserve mitochondrial function by targeting the mPTP opening induced by the Aβ25-35 peptide and MPP+ (1-methyl-4-phenylpyridinium).[320] Mitochondrial accumulation of Ca2+ induces depolarization of the mitochondrial membrane and, if large and sustained enough, a subsequent irreversible opening of the mPTP. Moreover, no changes were observed in pivotal metabolic enzymes, suggesting that latrepirdine’s neuroprotective effect is independent of promoting mitochondrial energy metabolic pathways.[309]
It has been shown that short-term exposure to latrepirdine causes: (i) Aβ accumulation and increased secretion of APP metabolites in mouse N2a neuroblastoma cells overexpressing APPsw; (ii) higher levels of Aβ42 in isolated cortical synaptoneurosome preparations from TgCRND8 mice; and (iii) enhanced Aβ40 levels in the hippocampal interstitial fluid of Tg2576 AD mice.[321] Either Ca2+ or Aβ25-35 toxic fragment promoted mitochondrial LPO but administration of latrepirdine inhibited oxidative damage to lipids.[320,322] In addition, the molecule has been shown to prevent LPO induced by tert-butyl hydroperoxide in rat brain mitochondrial homogenates.[323] In addition, administration of the drug diminished the content of hyperphosphorylated tau-positive dystrophic neurons in the mouse spinal cord while no changes in the levels of inflammatory markers were detected. Latrepirdine can also have a pro-autophagic activity, with a selective autophagic elimination of protein aggregates. Latrepirdine induces MTOR- and ATG5-dependent autophagy, which results in decreased levels of intracellular APP metabolites, including Aβ in the neocortex and hippocampus of TgCRND8 AD mice.[324] Latrepirdine can also stimulate autophagy-mediated degradation of α-syn in differentiated SH-SY5Y neurons and in the mouse brain.[325] Immunohistochemical analyses found a significant decrease in the number of TDP-43-like inclusions in cells transfected with TDP-43.[326] α-Syn pathogenic and toxic effect was markedly reduced in S. cerevisiae, SH-SY5Y cells, and the mouse brain through a selective autophagic protein degradation.[325] Administration of latrepirdine attenuated methamphetamine- (but not MPTP) induced cytotoxicity in mice in a body temperature-independent manner, suggesting a neurotoxin-specific protective effect.[13] However, the authors only measured the concentration of DA in the mouse striatum, with a lack of assessment of other additional key factors associated with neurodegenerative processes. Increased lifespan and motor performance parallel to reduced γ-syn aggregation, number of proteinaceous inclusions, and the inflammatory response were observed in transgenic mice overexpressing γ-syn treated with latrepirdine in the drinking water.[314] In contrast, exposure to the drug failed to protect DAergic neurons in C. elegans and mouse models of PD.[327] Fourteen-old-month α-syn transgenic mice receiving latrepirdine did not exhibit neurochemical, behavioral, or histopathological variations relative to the control group. Noteworthy, these mice recapitulate some pathological features typically manifested at the early onset of the disease.[328]
In summary, psychiatric symptoms (such as depression) and learning and cognitive measures were restored in AD patients undergoing latrepirdine treatment, in part due to its ability to increase acetylcholine concentration by inhibition of either AChE or histamine receptors. Latrepirdine may elicit neuroprotective activity by promoting mitochondrial function and clearance of a range of intracellular inclusions through the stimulation of autophagy. The molecule may also regulate several targets involved in AD pathology, such as neurotransmitter receptor activity, stabilization of Ca2+ signaling, and LPO. Toxicological studies have shown that latrepirdine is safe and a well-tolerated drug. A dosage exceeding the therapeutic range by 100 times for a period of 2 months did not cause any physiological changes or pathology in guinea pigs, rats, or dogs.[304,329]
Latrepirdine (2,3,4,5-tetrahydro-2,8-dimethyl-5-[2-(6-methyl-3-pyridinyl)ethyl]-1H-pyrido[4,3-b] indole) 4 was originally synthesized using the Fischer-indole reaction.[330] A more recent synthesis reported by Zheng et al. employed p-toluidine 23 and 2-methyl-5-vinylpyridine 24 as commercial starting materials and achieved the desired product 4 in 16% yield over four reactions steps.[331] Moreover, cyclization of compound 26 and 1-methylpiperidin-4-one 27 was carried out with 80% HAc in a one-pot two steps reaction instead of using benzene and HCl/EtOH (Scheme 7).
The synthesis of latrepirdine 4 has also been performed using the chemistry of ruthenium (III) catalysts in a concise and efficient manner.[332] The reaction comprised six steps, which required three purification processes only with an ~47% overall yield. The key step involved the stereoselective formation of γ-carboline 32 from ortho-substituted aryl azide 31 catalysed by RuCl3·nH2O (Scheme 8).
5. Nifedipine
Nifedipine (Procardia) is a first-generation dihydropyridine Ca2+ channel blocker used to treat hypertension and to control angina pectoris. Nifedipine is a selective antagonist of the L-type VOCCs that plays an essential role in neuronal processes triggered by membrane depolarization, thereby contributing to Ca2+-mediated events activated by signaling pathways and diverse stimuli, including neurotransmitter release, rhythmic firing, gene expression, etc.[333] There is growing evidence that nifedipine may be an effective therapeutic agent for the treatment of neurodegenerative diseases, including AD and PD. APOE ε4 peptide-induced increase in the concentration of intracellular Ca2+ and transcriptional activity of cAMP-response element-binding protein (CREB) in rat hippocampal neuronal cultures was reduced after administration of nifedipine, suggesting that L-type VOCCs are involved in neuron responses to APOE ε4.[334] In culture of primary mouse hippocampal neurons, Aβ enhanced the content of cytosolic Ca2+ and concomitant phosphorylation of serine-880 in the AMPA-selective glutamate receptor 2 (GluR2), which leads to attenuated synaptic activity.[335] In contrast, exposure to nifedipine diminished Ca2+ levels and GluR2 phosphorylation and increased cell surface GluR2. It has been reported that Aβ-mediated toxicity promotes the activation of the Ca2+-calmodulin kinase II (CaMKII)/AMP-activated protein kinase (AMPK) pathway.[336,337] Exposure to nifedipine mitigated Aβ toxicity by preserving cell viability in SH-SY5Y cells stably transfected with an empty vector or expressing the cellular prion protein, indicating that Ca2+ influx via L-type VOCCs is involved in Aβ-induced neurotoxicity.[338] Administration of nifedipine significantly diminished the content of secreted Aβ1-42 and key components of the gamma secretase complex (e.g., PS-1) in H4 neuroglioma cells overexpressing APP, without triggering cell death.[339] Primary CNS neurons cultured from PS1-deficient mice showed higher susceptibility to oxidative stress but exposure to nifedipine increased the survival rate, suggesting that L-type VOCCs-mediated Ca2+ influx via was responsible for the neuronal loss.[340]
It has been suggested that deficits in APP−/− mice are associated with alterations in Ca2+ homeostasis. Treatment with nifedipine (but not the NMDAR blocker 2-amino-5-phosphonovaleric acid) restored post-hypoxic related damage, leading to an improvement of neuron functional deficiencies such as population spike amplitude and depolarized resting ΔΨm in hippocampal slices from mice lacking APP.[341] To determine the specific pathways involved in altered Ca2+ homeostasis, primary cortical neuron cultures from 3xTg-AD or APPsw mice were incubated with intracellular Aβ.[342] Quantitative measurements displayed a marked boost in resting [Ca2+]cyt concentration (further enhanced by extracellular Ca2+ efflux) that was blocked by nifedipine, suggesting that elevated Ca2+ influx occurs through L-type VOCCs. An age-dependent increase of L-type VOCC amplitude was described in CA1 pyramidal neurons (but not CA3 or dentate granule neurons) in 3xTgAD mice relative to wild-type mice, consistent with the notion that CA1 neurons are prone to p-tau/NFT pathology due to an excessive Ca2+ trafficking. Nevertheless, Ca2+ current was limited by nifedipine, which attributes to the L-type VOCCs the enhanced intracellular Ca2+ pool.[343] The stromal interaction molecule 1 (STIM1) is a type I transmembrane protein that plays a pivotal role in Ca2+ influx. Stim1 positively modulates Orai1 channels for store-operated Ca2+ entry and negatively modulates L-type VOCCs. STIM1 protein levels were decreased in the medium frontal gyrus of patients diagnosed with AD.[344] STIM1-KO differentiated cells displayed an elevated Ca2+ entry in response to membrane depolarization, which was nifedipine-sensitive. Primary hippocampal neuronal cultures from 5xFAD mice exposed to nifedipine inhibit abnormal VOCC and store operated Ca2+ (SOC) entry, a process regulated mainly by STIM1. In contrast, STIM2 was responsible to modulate depolarization-mediated Ca2+ entry through VOCCs into cells with full Ca2+ stores.[345] Intracellular Ca2+ responses to membrane depolarization were potentiated by the V337M mutant tau in diverse cell cultures, which were inhibited following administration of nifedipine. These findings suggest that upregulation of L-type VOCCs-mediated Ca2+ influx results from destabilization of microtubules triggered by tau mutations.[346] Late insoluble (but not soluble or early insoluble) tau aggregates activated sensitive Ca2+ channels, thereby increasing Ca2+ signaling, cell loss, and oxidative damage (O2•− levels) in primary cultures of rat cortical neurons and astrocytes.[347] The rate of ROS production was significantly diminished in the presence of the NADPH oxidase inhibitor AEBSF (2-aminoethyl) benzenesulfonyl fluoride hydrochloride), indicating that ROS formation was NADPH oxidase-dependent. In addition, administration of nifedipine limited tau-mediated Ca2+ influx, generation of ROS, and overall toxicity. The data demonstrated that: (i) tau effect depends on its aggregation state, and (ii) late insoluble aggregates incorporate into the membranes, resulting in an ionic current alteration and VOCC stimulation.
The subthalamic nucleus (STN) has been proposed to play a central role in the disrupted function of the basal ganglia circuitry associated with PD. Distinct activity patterns, such as increased burst firings, have been observed in STN neurons and represent a pathognomonic electrophysiological feature linked to PD. High-frequency stimulation of the STN or pharmacological blockade of the subthalamopallidal network in monkeys and rats improved motor symptoms.[348,349,350] Patch-clamp studies performed in rat brain slices showed that around 50% of STN neurons display the intrinsic property of switching from single-spike activity to burst-firing mode.[351] Treatment with nifedipine caused an irreversible reduction in burst frequency and abolished burst firing. Furthermore, the role of VOCCs was investigated in STN neurons. Based on nifedipine effects on the frequency and current curve, it was established that both short- and long-duration rebound bursting neurons contain nifedipine-sensitive Cav1.2-1.3 channels, which only contribute to rebound activity in STN neurons with long-lasting rebounds.[352] High-frequency stimulation-induced oscillations in the STN resulted at least in part from Ca2+ entry through the high-threshold potential nifedipine-sensitive L-type VOCCs.[353] Pasternak et al. reported that nifedipine was not linked to significantly lower risk of developing PD, but the small sample size undermined the findings.[354] Nifedipine exposure did not have a significant effect on DA neuron survival but exhibited a stimulatory impact on neurite length. The actions of cholinesterases such as AChE and butyrylcholinesterase on Ca2+ conductance may be responsible for the trophic effect on neurite outgrowth in embryonic ventral midbrain cultures.[355] Nifedipine limited both glutamate- and NMDA-related CREB phosphorylation. Stimulation of D1 receptors or cAMP pathway in primary striatal neuron cultures produced cytosolic accumulation of Ca2+ that interacted with nifedipine, resulting in Ca2+-mediated CREB phosphorylation and c-fos gene expression.[356]
Injection of nifedipine into the dorsal striatum did not affect apomorphine-induced rotational behavior, indicating that it had no effect on DAergic transmission.[357] In a different study, microinjection of 6-OHDA resulted in a significant upregulation of mRNA levels of the Cav1.2 Ca2+ channel α1 subunit in the ipsilateral SN.[358] Subcutaneous injection of 3.5 mg/kg nifedipine significantly decreased apomorphine-induced rotation and partially restored striatal DA concentration in 6-OHDA-lesioned rats. These findings imply that L-type VOCCs are directly connected with DA neurodegeneration. Nifedipine prevented nobiletin- (a natural polymethoxy flavonoid extracted from the fruit peel of citrus) induced DA release in the CA1 region of the hippocampus of MPTP-injected mice.[359] The high-threshold Ca2+ spike (HTS) and the slow oscillatory potential (SOP) are diverse Ca2+ conductances that play an important role in the generation of action potentials in SN DA neurons. While nifedipine showed a slight inhibitory effect on HTS, the molecule was able to block SOP. Moreover, nifedipine steadily abolished the spontaneous firing pattern.[360] Quinpirole is a D2 receptor agonist that inhibits Ca2+ current in both 6-OHDA-lesioned and reserpine-injected rats.[361] However, quinpirole inhibitory effect was reversed by nifedipine. These data suggest that DA-depletion leads to a rearrangement of the high voltage-activated (HVA) Ca2+ current profile, an outcome also observed in monogenic forms of PD (DJ1 mice). Rotenone-treated SH-SY5Y cells incubated with nifedipine showed a concentration-dependent decrease in Ca2+ trafficking, suggesting that the neurotoxin activates the L-type VOCCs opening.[362] The results also demonstrated that Ca2+ is involved in rotenone-mediated apoptosis and ROS production. Pretreatment with nifedipine increased cell survival, synaptic vesicle exocytosis, and neurite outgrowth as well as mitigated DA release and generation of ROS (using the dichlorodihydrofluorescein diacetate (DCFH-DA) probe) in PC12 treated with rotenone.[363] These results indicated that intracellular Ca2+ plays an important role in rotenone-induced DA toxicity. Quantitative assessment in organotypic sagittal vibrosections from p10 rat brain, showed that L-type VOCC inhibitor nifedipine do not exert a neurotoxic activity on DAergic or cholinergic neurons.[364] Even though nifedipine did not protect cholinergic neurons, the drug counteracted axotomy-mediated DA neuron degeneration in the SN but not in the ventral tegmental area, possibly by regulating proinflammatory cytokine release.
In summary, nifedipine is an antagonist of L-type VOCCs antagonist involved in Aβ and tau pathology, neurotoxin-induced DA degeneration, Ca2+ homeostasis and signaling, synaptic function, oxidative insult, and apoptotic cell death. The primary manifestation of nifedipine-related toxicity is hypotension secondary to loss of systemic vascular resistance. Subacute and subchronic toxicity studies indicated that oral administration of nifedipine has a safety profile at doses of up to 50 mg/kg in rats over a period of thirteen weeks. In dogs, no damage was detected up to 100 mg/kg dosage for a period of four weeks. Rats tolerated daily intravenous administration of 2.5 mg/kg nifedipine over a period of three weeks while dogs tolerated up to 0.1 mg/kg nifedipine for six days. An overdose of nifedipine can induce severe hypotension, systemic vasodilation, and reflex tachycardia.
The first total synthesis of nifedipine 5 was reported in 1989 by Singh, who used an acid-catalyzed reaction of an enamine with two perhydro-heterocycles.[365] Other synthetic procedures were reported in the following years using different methodologies.[366,367,368,369,370] Solid phase synthesis was also applied [371,372] as well as one-pot solvent-free synthesis.[373,374,375] The most common synthetic procedure to obtain pyridines is using the Hantzsch reaction.[376,377,378,379,380,381,382,383] The preparation of 1,4-dihydropyridines requires two equivalents of β-keto ester, an aldehyde and nitrogen donor. Sudalai and co-workers utilized dimethylmalonate 35 as the β-keto ester, 2-nitro-benzaldehyde 36 for the aldehyde and ammonium acetate as the nitrogen donor (Scheme 9).[384]
This reaction has a great number of modifications, such as the utilization of different homogeneous[385,386,387,388] or heterogeneous[389,390,391] catalysts, and the variation of the solvent.[392] In 2021, nifedipine (3,5-dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate) 5 was synthesised through a photoinduced iron-catalysed ipso-nitration via single-electron transfer.[393] Aryl iodine 37 changed the iodine to nitro substituent to obtain 5 with good yield induced by photocatalysis (Scheme 10).
The first flow multicomponent synthesis of nifedipine 5 was reported elsewhere.[394] Methanol solutions of compounds 36, 38 and 39 were loaded to a heated 10 mL stainless steel coil reactor (150 °C) at 0.167 mL min−1 to obtain nifedipine 5 in 71% yield (Scheme 11).
More recently, Xiang et al. described a green methodology for the synthesis of nifedipine 1.[395] A metal-free multicomponent cycloaddition of ketones with an ammonium cation under a CO2 atmosphere is used to obtain the desired dihydropyridine derivatives. Thus, using methyl acetoacetate 38 and o-nitrobenzaldehyde 36, in the presence of ammonium chloride (NH4Cl) and CO2 in aqueous solution, 5 was obtained in 32% yield (Scheme 12).
6. Nimodipine
Nimodipine (Nimotop) is a Ca2+ channel blocker utilized to prevent brain damage as a consequence of reduced blood flow. The molecule antagonizes the Cav1.2-1.3 L-type VOCCs, showing a greater affinity for the Cav1.2 channel.[396] As a highly lipophilic compound, nimodipine can easily cross the BBB and reaches elevated concentrations in the CSF. The drug has specific binding affinity for dihydropyridine receptors, in contrast to nifedipine that shows predominantly peripheral effects. Nimodipine has been proposed to be of potential therapeutic utility in clinical trials of subjects with AD or PD. The beneficial effects attributable to nimodipine are related to clinical symptoms, cognitive function, and overall physical activity. The drug was shown to be more effective in primary degenerative dementia rather than in multi-infarct dementia.[397] A multicenter, double-blind, placebo controlled, randomized clinical trial of nimodipine (30 mg/kg tris in die) for 3 months demonstrated its efficacy to prevent behavioral, cognitive or affective impairments in patients with primary degenerative dementia of the Alzheimer's type.[398,399] An identical dose of nimodipine resulted in a marked improvement of the global functional state in individuals with mental deterioration of degenerative or vascular origin.[400] A review article of several double-blind, placebo-controlled clinical trials using nimodipine demonstrated its beneficial properties in elderly subjects suffering from a cognitive impairment syndrome.[401] The first randomized, double-blind, controlled trial focusing on subcortical vascular dementia was carried out in 2005 and concluded that nimodipine might exert a protective effect against cardiovascular comorbidities.[402]
Clinical observations have been confirmed by experimental research. Nimodipine possess the potential to reverse learning and memory defects in both young and aged hypertensive rats.[403,404] Bilateral injection of Aβ1-42 in the entorhinal cortex of rats led to delayed acquisition in a spatial reference memory task and decreased excitatory transmission. Long-term treatment with nimodipine prevented aged-related attenuation of learning and memory through modulation of synaptosome Ca2+-binding proteins.[405] Administration of nimodipine improved reversal spatial learning impairment and synaptic current defects associated with Aβ pathology.[406,407] Age-related increase of the L-type Ca2+ channel protein α1D in the CA1 region of the hippocampus correlated with higher working memory defect, but nimodipine-treated rats for several months exhibited lower α1D immunoreactivity and improved spatial working memory.[408] Nimodipine ameliorated scopolamine-induced cognitive decline via its regulatory effect on the brain-derived neurotrophic factor (BDNF) and acetylcholine, which were elevated in both the cortex and hippocampus.[409] Nimodipine also ameliorated memory failure in aged rhesus monkeys.[410] Sustained depolarization-mediated increase of cytosolic levels of Ca2+ (but not released from the ER) produce substantial amounts of intraneuronal Aβ1-42 and cell death in primary rat cortical neuron cultures. Exposure to nimodipine reduced Ca2+ influx and associated Aβ1-42 accumulation.[411] Nimodipine restored the neurophysiological responses of ghrelin on the ΔΨm in young but not older neurons from Tg2576 female mice incubated with the oligomeric Aβ1-42 fragment, indicating that Aβ-mediated intracellular Ca2+ dysregulation is only reversible during the early stages of Aβ pathology.[412] MTT assay and morphometric cell counting showed that treatment with nimodipine protected both rat cortical and hippocampal neurons from Aβ25-35-induced cell death.[413] In primary rat hippocampal neuron cultures, Aβ25-35 fragment triggered reversible enhancement in intracellular Ca2+ content and bursts of action potentials, which were exacerbated after extracellular Mg2+ removal. Treatment with the L-type blocker nimodipine inhibited Aβ25-35-mediated toxic effects in both cortical and hippocampal neuronal cultures.[414,415] A different study using the same cultures confirmed that addition of nimodipine limited Aβ25-35-related Ca2+ uptake and apoptotic effect.[416,417] Patch-clamp recording studies demonstrated that nimodipine inhibits Aβ25-35 peptide excessive Ca2+ current density. Nevertheless, exposure to nimodipine did not mitigate inhibit oxidative damage associated to Aβ25-35. These results suggest that Aβ significantly enhances Ca2+ trafficking via nimodipine-sensitive L-type VSCCs, which leads to free radical-induced neuronal loss.
Okadaic acid promoted phosphorylation of tau by increasing Ca2+ influx through L-type VOCCs, since nimodipine attenuates phospho-tau levels in SH-SY5Y neuroblastoma cells.[418] H4/APPsw cells or primary neuronal cultures derived from the cerebellum of Tg2576 AD mice incubated with nimodipine displayed an augmented Aβ42 secretion. In addition, a comparable effect was described in Tg2576 mice injected with nimodipine, which showed a marked increase in Aβ42 plasma concentration. Thus, the drug may modulate the release of Aβ42 through an elusive mechanism rather than its capacity to inhibit Ca2+-influx pathways.[419] Driving tau expression in M4/6 neurons led to increased sensitivity of Ca2+ transients to nimodipine. This effect was ablated in M4/6 neurons co-expressing tau and Ca-α1D-RNAi.[420] Moreover, the authors demonstrated that knockdown of the Drosophila L-type Ca2+ channel Ca-α1D reverses tau-induced olfactory memory abnormalities by restoring Ca2+ handling. Elevated Ca2+ concentration aggravated Aβ-mediated behavioral impairment and defective chemotaxis, resulting in a shortened lifespan in C. elegans overexpressing Aβ1-42.[421] Exposure to nimodipine extended lifespan and rescued motor lesions, synaptic deficiencies, and DAergic degeneration in worms. Furthermore, nimodipine limited Aβ aggregation via through upregulation of glutathione S-transferase activity, resulting in an attenuation of oxidative damage. LPS-treated rats exhibited important memory deficits, enhanced synaptosomal Ca2+ uptake, and microglial (but not astrocytic) stimulation with subsequent cytokine storm but administration of nimodipine reversed LPS-induced toxicity.[422] A dose-dependent depletion of Aβ-stimulated IL-1β content and release was detected following nimodipine exposure in both the N13 microglia cell line and cultures of primary mouse microglia together with reduced cell loss.[423] In addition, the molecule mitigated Aβ-induced intracellular accumulation of IL-1β in mice receiving a stereotaxic hippocampal injection of oligomeric Aβ1-42 peptide.
A more recent study reported that nimodipine combined with piracetam results in a significant improvement of cognitive abilities and quality of life scores in patients with vascular dementia after an ischemic stroke.[424] Sadleir et al. showed that oral administration of nimodipine to 5XFAD mice did not have a beneficial effect on amyloid pathology, since the molecule did not prevent neuritic dystrophy or reduce cortical or hippocampal Aβ content.[425] However, the treatment did not exacerbate the AD phenotype, suggesting a safety and tolerability profile. It has been described that iron uptake, which competes with Ca2+ for entry into neurons through the L-type VOCCs, can be inhibited with nimodipine in a dose-dependent fashion in potassium chloride stimulated neuronal cells.[426] The data suggest that, under cellular iron overload conditions, iron uptake occurs through L-type VOCCs. Subcutaneous delivery of nimodipine reversed behavioral abnormalities, preserved the number of DAergic neurons in the locus coeruleus, and attenuated microglial activation in LPS-infused rats.[427] Exposure to nimodipine blocked DA neuron pacemaker activity, which can be restored by virtual Cav1.3 channels.[428] In contrast, virtual NMDAR were not capable of restoring regular pacing in nimodipine-silenced DA neurons. In vitro and in vivo studies demonstrates that incubation with nimodipine abolishes autonomous pacemaking in DA neurons and the underlying ΔΨm oscillations.[178]
Primary culture of cerebellar granule neurons incubated with MPP+ led to prominent cell death but nimodipine limited the effect of the neurotoxin.[429] MPP+ elevated cytosolic Ca2+ content and induced cell death parallel to mitochondrial depolarization and fragmentation in vitro.[430] However, exposure to nimodipine diminished Ca2+ levels, improved cell survival rate, and restored mitochondrial morphology. Nimodipine also ameliorated MPTP-induced behavioral phenotype and limited striatal DA depletion, SN neuronal loss, and oxidative insult without regulating MAO-B activity. The data indicated that nimodipine has the ability to improve mitochondrial function and integrity and involves L-type VOCCs in MPTP-mediated nigrostriatal DA neurodegeneration. In a different study, the same authors reported defective function in proteins involved in the modulation of intracellular Ca2+ homeostasis, including calbindin and calpain. This abnormal function was rescued by nimodipine treatment.[431] A different report showed that nimodipine had no effect on behavioral impairment and striatal DA depletion but preserved DA neurons from death in marmosets injected with MPTP.[256] Similar findings were observed in MPTP-treated mice, in which treatment with nimodipine protected DA neurons in the SN but no changes were found in the striatum.[255]
Nimodipine simultaneously upregulated DA release whilst suppress AChE release in both rat cerebral cortex and striatum.[432] Nimodipine prevented dendritic spine loss and behavioral abnormalities but not associated rotational asymmetry in DA-grafted rats.[433] Moreover, exposure to nimodipine did not impact DA graft survival but promoted graft reinnervation of striatum. Continuous-release pellets of nimodipine prevented locomotor disturbances, in unilateral 6-OHDA mesencephalic lesions.[434] Intrastriatal injection of 6-OHDA in rats caused a significant loss of retrogradely fluorogold and DAergic labelled neurons in the SN at 1-month post-injection. Nimodipine treatment failed to improve behavioral phenotype or nigrostriatal DA degeneration.[435] Exposure to nimodipine increased survival of SN (but not VTA) DA neurons in axotomy-induced rat model.[364] Although the precise mechanism remains elusive, the ability of the drug to counteract the inflammatory processes may be crucial for mitigating axotomy-induced neurodegeneration. Knockdown of Homer1, a postsynaptic density scaffold protein that regulates synaptic plasticity and Ca2+ signaling, preserved DA neurons from MPP+ toxicity.[436] This protective effect was linked to attenuated Ca2+-mediated ROS generation, which in turn, was dependent on the modulatory activities on ER Ca2+ trafficking and release through plasma membrane Ca2+ channels. Nimodipine significantly decreased the amount of NO• and LPS-activated microglia, which releases pro-inflammatory mediators such as interleukin-1β (IL-1β), prostaglandin E2 (PGE2), and tumor necrosis factor-α (TNF-α).[437] In addition, in the absence of microglia, pretreatment with nimodipine did not exert a neuroprotective effect against MPP+-mediated DA toxicity. Nimodipine also downregulated DA uptake in neuron-glia cultures from mice lacking functional NADPH oxidase (an enzyme involved in the production of O2•−) incubated with LPS. Taken together, these findings indicate that nimodipine protects DAergic neurons by mitigating the inflammatory response and by inhibiting NADPH oxidase-ROS signaling pathway.
In summary, the L-type Ca2+ channel blocker antagonist nimodipine protects from Aβ and presenilin pathology and from LRRK2- and α-syn-induced toxicity. The molecule ameliorates behavioral outcome, increases synaptic transmission and neuron survival, improves mitochondrial function, and attenuates oxidative stress and inflammation. Nimodipine is, in general, well tolerated although sensations of warmth and skin reddening can occur. High concentrations of the drug can result in reduced blood pressure, headache, nausea, muscle weakness, and gastrointestinal complaints.[397,398,399] Isolated CNS symptoms, such as insomnia, tachycardia, and increased motor activity have been reported. Nimodipine exhibits a low incidence of severe side-effects.
A solid-phase synthesis was reported by Gordeev in 1996 for the preparation of nimodipine (3-(2-Methoxyethyl) 5-propan-2-yl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate) 6 and other bioactive dihydropyridines.[438] Condensation of the immobilized N-tethered enamino component 42 with the corresponding 2-benzylidene α-keto ester 43 provided the conjugated enamine 44. This intermediate was treated with trifluoroacetic acid to obtain the free enamino ester which spontaneously cyclizes in solution to afford the desired product 6 (Scheme 13).
More recently, due to a considerable demand of nimodipine in the Russian market, Pharm. Sintez Co. has developed a new technology to produce the drug in pilot batches.[439] The approach includes the production of 1-methylethyl-3-amino-crotonate 48 and 2-methoxyethyl-2-(3-nitrobenzyl-idene)acetoacetate 49. Cyclocondensation of both compounds, 48 and 49, employing iPrOH as the solvent and in the presence of hydrochloric acid, afforded the final product 6 in high yield and purity (Scheme 14).
Abbreviations
Ac: acetate; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; DMAP: 4-dimethylaminopyridine; DMF: dimethylformamide; DMSO = dimethyl sulfoxide; Dppf = 1,1'-bis(diphenylphosphino)ferrocene; GDH: glutamate dehydrogenase; NADP+: nicotinamide-adenine dinucleotide phosphate; Tf: trifluoromethanesulfonate; TFA: trifluoroacetic acid; THF: tetrahydrofuran; Tos = Ts: toluenesulfonyl.
Conclusions and Future Directions
Multiple candidate drugs for treating AD or PD have failed over the last years. Further research is still needed to determine the therapeutic time window and the stage of the disease that should be used in clinical trials. Neuropathological and biochemical assessments support the notion that mitochondrial dysfunction causes an increase of free radicals and Ca2+-induced toxicity, which are important factors in the neurodegenerative process described in AD and PD. Based on the combination of preclinical and clinical data, there is a general consensus that development of therapeutic interventions targeting mitochondrial Ca2+ signaling may slow or stop the progression of neurodegenerative diseases, including AD and PD. A set of strategies to normalize mitochondrial Ca2+ homeostasis and signaling has been described. One potential approach involves Ca2+ uptake blockade through the L-type VOCCs, which inhibits Ca2+ transient at either presynaptic or postsynaptic sites and attenuates glutamate-mediated excitotoxicity. Selective antagonists of Cav1.2-1.3 L-type VOCCs also exhibit beneficial properties since prolonged opening of L-type channels results in augmented Ca2+ release and influx and its accumulation in DA neurons. Chemical synthesis, also revised here, may lead to the development of novel derivatives with increased efficiency and/or availability. The capability of heterocyclic agents of modulating mitochondrial Ca2+ content and signaling and the ability to preserve mitochondrial biogenesis and function provides a plausible biological basis for their neuroprotective effect. In summary, decreasing the susceptibility of neurons to injury should not only reduce the incidence of AD or PD but also slow the progression, broadening the therapeutic window for patients with early-stage disease.
Funding support
This work was supported by the Excellence Program #CCVC8485 from Junta de Castilla y León, Spain, and by grant #RTI2018-099298-B-100 from Ministry of Science and Innovation, Spain, to CV and LN, and grant #VA294P18 from Junta de Castilla y León, Spain, to LN. VT is supported by the Maria Zambrano’s Excellence Program (#CL-EI-2021 IBGM) from the Ministry of Science and Innovation and the University of Valladolid, Spain, as well as by the Internationalization program of Junta de Castilla y León, Spain.
Author contributions
VT, LN, and CV participated in the conceptualization of the article, wrote the initial manuscript, prepared the figures and tables, and contributed to revisions. AB, PGA, and LFP discussed the chemical synthesis, prepared the chemical equations and structural formulas, and revised the manuscript. All authors read and approved the final version of the manuscript.
References
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 2018, 15, 490-503. [CrossRef]
- Beal, M.F.; Chiluwal, J.; Calingasan, N.Y.; Milne, G.L.; Shchepinov, M.S.; Tapias, V. Isotope-reinforced polyunsaturated fatty acids improve Parkinson's disease-like phenotype in rats overexpressing alpha-synuclein. Acta Neuropathol Commun 2020, 8, 220. [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer's disease. Neurobiol Dis 2020, 138, 104795. [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol Neurobiol 2016, 53, 4094-4125. [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol Life Sci 2017, 74, 2851-2874. [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis 2018, 9, 331. [CrossRef]
- van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat Commun 2018, 9, 1581. [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968-983 e924. [CrossRef]
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal 2013, 6, rs1. [CrossRef]
- Song, I.K.; Lee, J.J.; Cho, J.H.; Jeong, J.; Shin, D.H.; Lee, K.J. Degradation of Redox-Sensitive Proteins including Peroxiredoxins and DJ-1 is Promoted by Oxidation-induced Conformational Changes and Ubiquitination. Sci Rep 2016, 6, 34432. [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883-891. [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta 2014, 1842, 1267-1272. [CrossRef]
- Abe, K.; Kimura, H. Amyloid beta toxicity consists of a Ca(2+)-independent early phase and a Ca(2+)-dependent late phase. J Neurochem 1996, 67, 2074-2078.
- Corona, C.; Pensalfini, A.; Frazzini, V.; Sensi, S.L. New therapeutic targets in Alzheimer's disease: brain deregulation of calcium and zinc. Cell Death Dis 2011, 2, e176. [CrossRef]
- Espana, J.; Valero, J.; Minano-Molina, A.J.; Masgrau, R.; Martin, E.; Guardia-Laguarta, C.; Lleo, A.; Gimenez-Llort, L.; Rodriguez-Alvarez, J.; Saura, C.A. beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci 2010, 30, 9402-9410. [CrossRef]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J Neurosci 2017, 37, 11151-11165. [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer's disease. N Engl J Med 2010, 362, 329-344. [CrossRef]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer's disease mitochondrial cascade hypothesis: an update. Exp Neurol 2009, 218, 308-315. [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med Hypotheses 2004, 63, 8-20. [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis 2010, 20 Suppl 2, S265-279. [CrossRef]
- Buckner, R.L.; Snyder, A.Z.; Shannon, B.J.; LaRossa, G.; Sachs, R.; Fotenos, A.F.; Sheline, Y.I.; Klunk, W.E.; Mathis, C.A.; Morris, J.C.; et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005, 25, 7709-7717. [CrossRef]
- Greicius, M.D.; Srivastava, G.; Reiss, A.L.; Menon, V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A 2004, 101, 4637-4642. [CrossRef]
- Putcha, D.; Eckbo, R.; Katsumi, Y.; Dickerson, B.C.; Touroutoglou, A.; Collins, J.A. Tau and the fractionated default mode network in atypical Alzheimer's disease. Brain Commun 2022, 4, fcac055. [CrossRef]
- Kim, E.J.; Cho, S.S.; Jeong, Y.; Park, K.C.; Kang, S.J.; Kang, E.; Kim, S.E.; Lee, K.H.; Na, D.L. Glucose metabolism in early onset versus late onset Alzheimer's disease: an SPM analysis of 120 patients. Brain 2005, 128, 1790-1801. [CrossRef]
- Smith, G.S.; de Leon, M.J.; George, A.E.; Kluger, A.; Volkow, N.D.; McRae, T.; Golomb, J.; Ferris, S.H.; Reisberg, B.; Ciaravino, J.; et al. Topography of cross-sectional and longitudinal glucose metabolic deficits in Alzheimer's disease. Pathophysiologic implications. Arch Neurol 1992, 49, 1142-1150. [CrossRef]
- De Santi, S.; de Leon, M.J.; Rusinek, H.; Convit, A.; Tarshish, C.Y.; Roche, A.; Tsui, W.H.; Kandil, E.; Boppana, M.; Daisley, K.; et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging 2001, 22, 529-539. [CrossRef]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P.; Baker, A.; Carlson, K.C.; Harding, B.; Perrino, P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol 1988, 45, 836-840. [CrossRef]
- Ke, Z.J.; Gibson, G.E. Selective response of various brain cell types during neurodegeneration induced by mild impairment of oxidative metabolism. Neurochem Int 2004, 45, 361-369. [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 2005, 57, 695-703. [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 2001, 21, 3017-3023. [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Cardoso, S.M.; Swerdlow, R.H. Mitochondrial abnormalities in Alzheimer's disease: possible targets for therapeutic intervention. Adv Pharmacol 2012, 64, 83-126. [CrossRef]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 2010, 1802, 122-134. [CrossRef]
- Gibson, G.E.; Zhang, H.; Sheu, K.F.; Bogdanovich, N.; Lindsay, J.G.; Lannfelt, L.; Vestling, M.; Cowburn, R.F. Alpha-ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann Neurol 1998, 44, 676-681. [CrossRef]
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain alpha-ketoglutarate dehydrogenase complex activity in Alzheimer's disease. J Neurochem 1993, 61, 2007-2014. [CrossRef]
- Tapias, V.; Jainuddin, S.; Ahuja, M.; Stack, C.; Elipenahli, C.; Vignisse, J.; Gerges, M.; Starkova, N.; Xu, H.; Starkov, A.A.; et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum Mol Genet 2018, 27, 2874-2892. [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging 2002, 23, 371-376. [CrossRef]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 2001, 57, 260-264. [CrossRef]
- Cottrell, D.A.; Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer's disease. Neuropathol Appl Neurobiol 2002, 28, 390-396. [CrossRef]
- Maurer, I.; Zierz, S.; Moller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging 2000, 21, 455-462. [CrossRef]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem 1994, 63, 2179-2184. [CrossRef]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology 1990, 40, 1302-1303. [CrossRef]
- Parker, W.D., Jr.; Mahr, N.J.; Filley, C.M.; Parks, J.K.; Hughes, D.; Young, D.A.; Cullum, C.M. Reduced platelet cytochrome c oxidase activity in Alzheimer's disease. Neurology 1994, 44, 1086-1090. [CrossRef]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, D.G.; et al. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion 2006, 6, 323-330. [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology 1997, 49, 918-925. [CrossRef]
- Mosconi, L.; de Leon, M.; Murray, J.; E, L.; Lu, J.; Javier, E.; McHugh, P.; Swerdlow, R.H. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer's disease. J Alzheimers Dis 2011, 27, 483-490. [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 2014, 15, 634-646. [CrossRef]
- Tapias, V. Editorial: Mitochondrial Dysfunction and Neurodegeneration. Front Neurosci 2019, 13, 1372. [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062-1065. [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651-666. [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson's Disease and Huntington's Disease. Front Neurosci 2018, 12, 342. [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener 2020, 15, 30. [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 2011, 20, 2495-2509. [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet 2011, 20, 4515-4529. [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 2010, 107, 18670-18675. [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 2006, 26, 9057-9068. [CrossRef]
- Pavlov, P.F.; Hansson Petersen, C.; Glaser, E.; Ankarcrona, M. Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med 2009, 13, 4137-4145. [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 2008, 105, 13145-13150. [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004, 304, 448-452. [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 2008, 14, 1097-1105. [CrossRef]
- Canevari, L.; Clark, J.B.; Bates, T.E. beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett 1999, 457, 131-134. [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 2002, 80, 91-100. [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2009, 106, 14670-14675. [CrossRef]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2007, 104, 14163-14168. [CrossRef]
- Hauptmann, S.; Scherping, I.; Drose, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 2009, 30, 1574-1586. [CrossRef]
- Ronnback, A.; Pavlov, P.F.; Mansory, M.; Gonze, P.; Marliere, N.; Winblad, B.; Graff, C.; Behbahani, H. Mitochondrial dysfunction in a transgenic mouse model expressing human amyloid precursor protein (APP) with the Arctic mutation. J Neurochem 2016, 136, 497-502. [CrossRef]
- Perkins, M.; Wolf, A.B.; Chavira, B.; Shonebarger, D.; Meckel, J.P.; Leung, L.; Ballina, L.; Ly, S.; Saini, A.; Jones, T.B.; et al. Altered Energy Metabolism Pathways in the Posterior Cingulate in Young Adult Apolipoprotein E varepsilon4 Carriers. J Alzheimers Dis 2016, 53, 95-106. [CrossRef]
- Valla, J.; Yaari, R.; Wolf, A.B.; Kusne, Y.; Beach, T.G.; Roher, A.E.; Corneveaux, J.J.; Huentelman, M.J.; Caselli, R.J.; Reiman, E.M. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer's susceptibility gene. J Alzheimers Dis 2010, 22, 307-313. [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 2006, 15, 1437-1449. [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 2009, 29, 9090-9103. [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 2008, 105, 19318-19323. [CrossRef]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum Mol Genet 2016, 25, 5148-5166. [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102-105. [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta 2016, 1863, 2820-2834. [CrossRef]
- Liu, C.; Song, X.; Nisbet, R.; Gotz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J Biol Chem 2016, 291, 8173-8188. [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 2016, 6, 24756. [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci U S A 2009, 106, 20057-20062. [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Abeta in Alzheimer's disease mitochondria contributes to the synaptic deterioration. Neurobiol Aging 2012, 33, 833 e831-825. [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 2018, 27, 30-40. [CrossRef]
- Jara, C.; Aranguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol 2018, 18, 279-294. [CrossRef]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.Q.; Mucke, L. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol 2015, 209, 419-433. [CrossRef]
- Zhang, B.; Higuchi, M.; Yoshiyama, Y.; Ishihara, T.; Forman, M.S.; Martinez, D.; Joyce, S.; Trojanowski, J.Q.; Lee, V.M. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci 2004, 24, 4657-4667. [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci 2011, 31, 9858-9868. [CrossRef]
- Selfridge, J.E.; E, L.; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer's disease. Neurobiol Dis 2013, 51, 3-12. [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol 2014, 2, 873-877. [CrossRef]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298-311. [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology 1995, 45, 1594-1601. [CrossRef]
- McIntosh, L.J.; Trush, M.A.; Troncoso, J.C. Increased susceptibility of Alzheimer's disease temporal cortex to oxygen free radical-mediated processes. Free Radic Biol Med 1997, 23, 183-190. [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging 1998, 19, 33-36. [CrossRef]
- Montine, K.S.; Reich, E.; Neely, M.D.; Sidell, K.R.; Olson, S.J.; Markesbery, W.R.; Montine, T.J. Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. J Neuropathol Exp Neurol 1998, 57, 415-425. [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer's disease. Sci Rep 2018, 8, 11553. [CrossRef]
- Pratico, D.; Clark, C.M.; Lee, V.M.; Trojanowski, J.Q.; Rokach, J.; FitzGerald, G.A. Increased 8,12-iso-iPF2alpha-VI in Alzheimer's disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol 2000, 48, 809-812.
- Quinn, J.F.; Montine, K.S.; Moore, M.; Morrow, J.D.; Kaye, J.A.; Montine, T.J. Suppression of longitudinal increase in CSF F2-isoprostanes in Alzheimer's disease. J Alzheimers Dis 2004, 6, 93-97. [CrossRef]
- Selley, M.L.; Close, D.R.; Stern, S.E. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer's disease. Neurobiol Aging 2002, 23, 383-388. [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis 2006, 22, 76-87. [CrossRef]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis 2008, 30, 107-120. [CrossRef]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer's disease. Nat Commun 2016, 7, 11483. [CrossRef]
- Good, P.F.; Werner, P.; Hsu, A.; Olanow, C.W.; Perl, D.P. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol 1996, 149, 21-28.
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci 1997, 17, 2653-2657. [CrossRef]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem 2003, 85, 1394-1401. [CrossRef]
- Reed, T.T.; Pierce, W.M., Jr.; Turner, D.M.; Markesbery, W.R.; Allan Butterfield, D. Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J Cell Mol Med 2009, 13, 2019-2029. [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem 1995, 65, 2146-2156. [CrossRef]
- Sharma, A.; Weber, D.; Raupbach, J.; Dakal, T.C.; Fliessbach, K.; Ramirez, A.; Grune, T.; Wullner, U. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson's and Alzheimer's disease. Redox Biol 2020, 34, 101546. [CrossRef]
- Mullaart, E.; Boerrigter, M.E.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging 1990, 11, 169-173. [CrossRef]
- Anderson, A.J.; Su, J.H.; Cotman, C.W. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci 1996, 16, 1710-1719. [CrossRef]
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W., Jr.; Tabaton, M.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol 2012, 71, 233-241. [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol 1994, 36, 747-751. [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839-840. [CrossRef]
- Borghammer, P. Perfusion and metabolism imaging studies in Parkinson's disease. Dan Med J 2012, 59, B4466.
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Ray Chaudhuri, K.; Walker, Z.; Turkheimer, F.E.; Brooks, D.J. Microglia, amyloid, and glucose metabolism in Parkinson's disease with and without dementia. Neuropsychopharmacology 2013, 38, 938-949. [CrossRef]
- Firbank, M.J.; Yarnall, A.J.; Lawson, R.A.; Duncan, G.W.; Khoo, T.K.; Petrides, G.S.; O'Brien, J.T.; Barker, R.A.; Maxwell, R.J.; Brooks, D.J.; et al. Cerebral glucose metabolism and cognition in newly diagnosed Parkinson's disease: ICICLE-PD study. J Neurol Neurosurg Psychiatry 2017, 88, 310-316. [CrossRef]
- Andersen, K.B.; Hansen, A.K.; Schacht, A.C.; Horsager, J.; Gottrup, H.; Klit, H.; Danielsen, E.H.; Poston, K.L.; Pavese, N.; Brooks, D.J.; et al. Synaptic Density and Glucose Consumption in Patients with Lewy Body Diseases: An [(11) C]UCB-J and [(18) F]FDG PET Study. Mov Disord 2023. [CrossRef]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging 2014, 35, 1111-1115. [CrossRef]
- Rodriguez-Araujo, G.; Nakagami, H.; Hayashi, H.; Mori, M.; Shiuchi, T.; Minokoshi, Y.; Nakaoka, Y.; Takami, Y.; Komuro, I.; Morishita, R.; et al. Alpha-synuclein elicits glucose uptake and utilization in adipocytes through the Gab1/PI3K/Akt transduction pathway. Cell Mol Life Sci 2013, 70, 1123-1133. [CrossRef]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson's disease human samples and cellular models. Neurochem Int 2018, 118, 61-72. [CrossRef]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W.D., Jr. Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines. Exp Neurol 2000, 162, 37-50. [CrossRef]
- Gold, M.; Hauser, R.A.; Chen, M.F. Plasma thiamine deficiency associated with Alzheimer's disease but not Parkinson's disease. Metab Brain Dis 1998, 13, 43-53. [CrossRef]
- Jimenez-Jimenez, F.J.; Molina, J.A.; Hernanz, A.; Fernandez-Vivancos, E.; de Bustos, F.; Barcenilla, B.; Gomez-Escalonilla, C.; Zurdo, M.; Berbel, A.; Villanueva, C. Cerebrospinal fluid levels of thiamine in patients with Parkinson's disease. Neurosci Lett 1999, 271, 33-36. [CrossRef]
- Gibson, G.E.; Kingsbury, A.E.; Xu, H.; Lindsay, J.G.; Daniel, S.; Foster, O.J.; Lees, A.J.; Blass, J.P. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson's disease. Neurochem Int 2003, 43, 129-135. [CrossRef]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson's disease. Ann Neurol 1994, 35, 204-210. [CrossRef]
- Miki, Y.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of mitochondrial protein PDHA1 in Lewy body disease and PARK14. Biochem Biophys Res Commun 2017, 489, 439-444. [CrossRef]
- Mallajosyula, J.K.; Chinta, S.J.; Rajagopalan, S.; Nicholls, D.G.; Andersen, J.K. Metabolic control analysis in a cellular model of elevated MAO-B: relevance to Parkinson's disease. Neurotox Res 2009, 16, 186-193. [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990, 54, 823-827. [CrossRef]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol 1994, 36, 876-881. [CrossRef]
- Janetzky, B.; Hauck, S.; Youdim, M.B.; Riederer, P.; Jellinger, K.; Pantucek, F.; Zochling, R.; Boissl, K.W.; Reichmann, H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Neurosci Lett 1994, 169, 126-128. [CrossRef]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H. Platelet mitochondrial function in Parkinson's disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol 1992, 32, 782-788. [CrossRef]
- Yoshino, H.; Nakagawa-Hattori, Y.; Kondo, T.; Mizuno, Y. Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson's disease. J Neural Transm Park Dis Dement Sect 1992, 4, 27-34. [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson's disease frontal cortex. Brain Res 2008, 1189, 215-218. [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.A.; Cartlidge, N.E.; Parker, W.D., Jr.; Turnbull, D.M. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson's disease. J Neurol Sci 1991, 104, 203-208. [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson's disease. Ann Neurol 1995, 37, 714-722. [CrossRef]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650-656. [CrossRef]
- Vives-Bauza, C.; Tocilescu, M.; Devries, R.L.; Alessi, D.M.; Jackson-Lewis, V.; Przedborski, S. Control of mitochondrial integrity in Parkinson's disease. Prog Brain Res 2010, 183, 99-113. [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 2015, 78, 21-38. [CrossRef]
- Lynch, D.S.; Loh, S.H.Y.; Harley, J.; Noyce, A.J.; Martins, L.M.; Wood, N.W.; Houlden, H.; Plun-Favreau, H. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to OPA1 mutations. Neurol Genet 2017, 3, e188. [CrossRef]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain 2012, 135, 2058-2073. [CrossRef]
- Tapias, V.; McCoy, J.L.; Greenamyre, J.T. Phenothiazine normalizes the NADH/NAD(+) ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson's disease. Redox Biol 2019, 24, 101164. [CrossRef]
- Goiran, T.; Eldeeb, M.A.; Zorca, C.E.; Fon, E.A. Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson's Disease. Cells 2022, 11. [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener 2020, 15, 20. [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 2008, 283, 9089-9100. [CrossRef]
- Bender, A.; Desplats, P.; Spencer, B.; Rockenstein, E.; Adame, A.; Elstner, M.; Laub, C.; Mueller, S.; Koob, A.O.; Mante, M.; et al. TOM40 mediates mitochondrial dysfunction induced by alpha-synuclein accumulation in Parkinson's disease. PLoS One 2013, 8, e62277. [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 2014, 34, 249-259. [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med 2016, 8, 342ra378. [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Hollerhage, M.; Hoglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by alpha-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis 2019, 10, 865. [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 2011, 286, 20710-20726. [CrossRef]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marin-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. alphaSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson's disease. Sci Rep 2017, 7, 5042. [CrossRef]
- Krzystek, T.J.; Banerjee, R.; Thurston, L.; Huang, J.; Swinter, K.; Rahman, S.N.; Falzone, T.L.; Gunawardena, S. Differential mitochondrial roles for alpha-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis 2021, 12, 796. [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010, 29, 3571-3589. [CrossRef]
- Ulusoy, A.; Rusconi, R.; Perez-Revuelta, B.I.; Musgrove, R.E.; Helwig, M.; Winzen-Reichert, B.; Di Monte, D.A. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med 2013, 5, 1119-1127. [CrossRef]
- Brzozowski, C.F.; Hijaz, B.A.; Singh, V.; Gcwensa, N.Z.; Kelly, K.; Boyden, E.S.; West, A.B.; Sarkar, D.; Volpicelli-Daley, L.A. Inhibition of LRRK2 kinase activity promotes anterograde axonal transport and presynaptic targeting of alpha-synuclein. Acta Neuropathol Commun 2021, 9, 180. [CrossRef]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 2012, 287, 17914-17929. [CrossRef]
- Park, J.H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol Neurodegener 2020, 15, 5. [CrossRef]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci 2008, 65, 1272-1284. [CrossRef]
- Valero, R.A.; Senovilla, L.; Nunez, L.; Villalobos, C. The role of mitochondrial potential in control of calcium signals involved in cell proliferation. Cell Calcium 2008, 44, 259-269. [CrossRef]
- Villalobos, C.; Nunez, L.; Montero, M.; Garcia, A.G.; Alonso, M.T.; Chamero, P.; Alvarez, J.; Garcia-Sancho, J. Redistribution of Ca2+ among cytosol and organella during stimulation of bovine chromaffin cells. FASEB J 2002, 16, 343-353. [CrossRef]
- Nunez, L.; Senovilla, L.; Sanz-Blasco, S.; Chamero, P.; Alonso, M.T.; Villalobos, C.; Garcia-Sancho, J. Bioluminescence imaging of mitochondrial Ca2+ dynamics in soma and neurites of individual adult mouse sympathetic neurons. J Physiol 2007, 580, 385-395. [CrossRef]
- Núñez, L.; Villalobos, C.; García-Sancho, J. Coupling or not coupling of mitochondria to Ca2+ sources in neurones. Soma and neurites differ. Physiol News. 2008, 70, 23-24.
- Parys, J.B.; Pereira, C.F.; Villalobos, C. The Eighth ECS Workshop on "Calcium Signaling in Aging and Neurodegenerative Diseases". Int J Mol Sci 2019, 20. [CrossRef]
- Camandola, S.; Mattson, M.P. Aberrant subcellular neuronal calcium regulation in aging and Alzheimer's disease. Biochim Biophys Acta 2011, 1813, 965-973. [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer's disease - A therapeutic opportunity? Biochem Biophys Res Commun 2017, 483, 998-1004. [CrossRef]
- Arispe, N.; Diaz, J.C.; Simakova, O. Abeta ion channels. Prospects for treating Alzheimer's disease with Abeta channel blockers. Biochim Biophys Acta 2007, 1768, 1952-1965. [CrossRef]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 2008, 3, e2718. [CrossRef]
- Caballero, E.; Calvo-Rodriguez, M.; Gonzalo-Ruiz, A.; Villalobos, C.; Nunez, L. A new procedure for amyloid beta oligomers preparation enables the unambiguous testing of their effects on cytosolic and mitochondrial Ca(2+) entry and cell death in primary neurons. Neurosci Lett 2016, 612, 66-73. [CrossRef]
- Calvo-Rodriguez, M.; Nunez, L.; Villalobos, C. Non-steroidal anti-inflammatory drugs (NSAIDs) and neuroprotection in the elderly: a view from the mitochondria. Neural Regen Res 2015, 10, 1371-1372. [CrossRef]
- Garcia-Martinez, E.M.; Sanz-Blasco, S.; Karachitos, A.; Bandez, M.J.; Fernandez-Gomez, F.J.; Perez-Alvarez, S.; de Mera, R.M.; Jordan, M.J.; Aguirre, N.; Galindo, M.F.; et al. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol 2010, 79, 239-250. [CrossRef]
- Perez-Alvarez, S.; Solesio, M.E.; Cuenca-Lopez, M.D.; Melero-Fernandez de Mera, R.M.; Villalobos, C.; Kmita, H.; Galindo, M.F.; Jordan, J. Pharmacological Characterization of the Mechanisms Involved in Delayed Calcium Deregulation in SH-SY5Y Cells Challenged with Methadone. Int J Cell Biol 2012, 2012, 642482. [CrossRef]
- Sanz-Blasco, S.; Calvo-Rodriguez, M.; Caballero, E.; Garcia-Durillo, M.; Nunez, L.; Villalobos, C. Is it All Said for NSAIDs in Alzheimer's Disease? Role of Mitochondrial Calcium Uptake. Curr Alzheimer Res 2018, 15, 504-510. [CrossRef]
- Atwood, C.; Bacskai, B.; Kuchibhotla, K.; Bezprozvanny, I.; Chakroborty, S.; Fagan, T.; Foskett, K.; Green, K.; Goussakov, I.; Moyer, J.; et al. Alzheimer research forum live discussion: calcium in AD pathogenesis. J Alzheimers Dis 2009, 16, 909-917. [CrossRef]
- Calvo, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Nunez, L. Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: role of mitochondrial calcium. J Neurochem 2015, 132, 403-417. [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. Aging Enables Ca2+ Overload and Apoptosis Induced by Amyloid-beta Oligomers in Rat Hippocampal Neurons: Neuroprotection by Non-Steroidal Anti-Inflammatory Drugs and R-Flurbiprofen in Aging Neurons. J Alzheimers Dis 2016, 54, 207-221. [CrossRef]
- Pascual, M.; Calvo-Rodriguez, M.; Nunez, L.; Villalobos, C.; Urena, J.; Guerri, C. Toll-like receptors in neuroinflammation, neurodegeneration, and alcohol-induced brain damage. IUBMB Life 2021, 73, 900-915. [CrossRef]
- Calvo-Rodriguez, M.; de la Fuente, C.; Garcia-Durillo, M.; Garcia-Rodriguez, C.; Villalobos, C.; Nunez, L. Aging and amyloid beta oligomers enhance TLR4 expression, LPS-induced Ca(2+) responses, and neuron cell death in cultured rat hippocampal neurons. J Neuroinflammation 2017, 14, 24. [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Rodriguez, C.; Villalobos, C.; Nunez, L. Role of Toll Like Receptor 4 in Alzheimer's Disease. Front Immunol 2020, 11, 1588. [CrossRef]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca(2+) cross talk and loss of store-operated Ca(2+) entry (SOCE) in rat hippocampal neurons. Biochim Biophys Acta 2016, 1863, 2637-2649. [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca(2+) Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca(2+) Remodeling. Front Cell Neurosci 2019, 13, 22. [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Lopez-Vazquez, S.; Nunez, J.; Villalobos, C.; Nunez, L. Remodeling of Intracellular Ca(2+) Homeostasis in Rat Hippocampal Neurons Aged In Vitro. Int J Mol Sci 2020, 21. [CrossRef]
- Texido, L.; Martin-Satue, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184-190. [CrossRef]
- Caballero, E.; Hernando-Perez, E.; Tapias, V.; Calvo-Rodriguez, M.; Villalobos, C.; Nunez, L. Amyloid Beta Oligomers-Induced Ca(2+) Entry Pathways: Role of Neuronal Networks, NMDA Receptors and Amyloid Channel Formation. Biomedicines 2022, 10. [CrossRef]
- Gonzalez-Andres, P.; Fernandez-Pena, L.; Diez-Poza, C.; Villalobos, C.; Nunez, L.; Barbero, A. Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches. Mar Drugs 2021, 19. [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 2007, 447, 1081-1086. [CrossRef]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson's disease. Mov Disord 2010, 25 Suppl 1, S63-70. [CrossRef]
- Zampese, E.; Wokosin, D.L.; Gonzalez-Rodriguez, P.; Guzman, J.N.; Tkatch, T.; Kondapalli, J.; Surmeier, W.C.; D'Alessandro, K.B.; De Stefani, D.; Rizzuto, R.; et al. Ca(2+) channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci Adv 2022, 8, eabp8701. [CrossRef]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson's Disease. Cells 2020, 9. [CrossRef]
- Yun, J.; Jeong, D.; Xie, Z.; Lee, S.; Kim, J.; Surmeier, D.J.; Silverman, R.B.; Kang, S. Palladium-Catalyzed alpha-Arylation of Cyclic beta-Dicarbonyl Compounds for the Synthesis of Ca(V)1.3 Inhibitors. ACS Omega 2022, 7, 14252-14263. [CrossRef]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005, 102, 105-110. [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 2006, 126, 981-993. [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits. Acta Neuropathol 2017, 134, 749-767. [CrossRef]
- Vamvakides, A. [Anticonvulsant and forced swim anti-immobility effects of tetrahydro-N, N-dimethyl-2,2-diphenyl-3-furanemethanamine (AE37): common action mechanism?]. Ann Pharm Fr 2002, 60, 88-92.
- Hayashi, T.; Maurice, T.; Su, T.P. Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J Pharmacol Exp Ther 2000, 293, 788-798.
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596-610. [CrossRef]
- Willems, P.H.; Valsecchi, F.; Distelmaier, F.; Verkaart, S.; Visch, H.J.; Smeitink, J.A.; Koopman, W.J. Mitochondrial Ca2+ homeostasis in human NADH:ubiquinone oxidoreductase deficiency. Cell Calcium 2008, 44, 123-133. [CrossRef]
- Hampel, H.; Williams, C.; Etcheto, A.; Goodsaid, F.; Parmentier, F.; Sallantin, J.; Kaufmann, W.E.; Missling, C.U.; Afshar, M. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer's disease therapy: Analysis of the blarcamesine (ANAVEX2-73) Phase 2a clinical study. Alzheimers Dement (N Y) 2020, 6, e12013. [CrossRef]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (sigma1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J Psychopharmacol 2011, 25, 1101-1117. [CrossRef]
- Lahmy, V.; Meunier, J.; Malmstrom, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyperphosphorylation and Abeta(1)(-)(4)(2) generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and sigma(1) receptor agonist, in a nontransgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 2013, 38, 1706-1723. [CrossRef]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer's disease model. Front Cell Neurosci 2015, 8, 463. [CrossRef]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox Res 2019, 35, 1-18. [CrossRef]
- Maurice, T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav Brain Res 2016, 296, 270-278. [CrossRef]
- Christ, M.G.; Huesmann, H.; Nagel, H.; Kern, A.; Behl, C. Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo. Cells 2019, 8. [CrossRef]
- Foscolos, G.B.; Kolocouris, N.; Fytas, G.; Marakos, P.; Pouli, N.; Vamvakides, A. Synthesis and pharmacological study of some new beta-(dialkylaminomethyl)- gamma-butyrolactones and their tetrahydrofuran analogues. Farmaco 1996, 51, 19-26. [CrossRef]
- Ashihara, H.; Suzuki, T. Distribution and biosynthesis of caffeine in plants. Front Biosci 2004, 9, 1864-1876. [CrossRef]
- Ludwig, I.A.; Clifford, M.N.; Lean, M.E.; Ashihara, H.; Crozier, A. Coffee: biochemistry and potential impact on health. Food Funct 2014, 5, 1695-1717. [CrossRef]
- Hall, S.; Desbrow, B.; Anoopkumar-Dukie, S.; Davey, A.K.; Arora, D.; McDermott, C.; Schubert, M.M.; Perkins, A.V.; Kiefel, M.J.; Grant, G.D. A review of the bioactivity of coffee, caffeine and key coffee constituents on inflammatory responses linked to depression. Food Res Int 2015, 76, 626-636. [CrossRef]
- Cedazo-Minguez, A.; Popescu, B.O.; Ankarcrona, M.; Nishimura, T.; Cowburn, R.F. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J Biol Chem 2002, 277, 36646-36655. [CrossRef]
- Smith, I.F.; Boyle, J.P.; Vaughan, P.F.; Pearson, H.A.; Cowburn, R.F.; Peers, C.S. Ca(2+) stores and capacitative Ca(2+) entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer's disease presenilin-1 mutation. Brain Res 2002, 949, 105-111. [CrossRef]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem 2000, 275, 18195-18200. [CrossRef]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 2000, 149, 793-798. [CrossRef]
- Barrow, P.A.; Empson, R.M.; Gladwell, S.J.; Anderson, C.M.; Killick, R.; Yu, X.; Jefferys, J.G.; Duff, K. Functional phenotype in transgenic mice expressing mutant human presenilin-1. Neurobiol Dis 2000, 7, 119-126. [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer's disease families. Neurobiol Dis 1998, 5, 37-45. [CrossRef]
- Hirashima, N.; Etcheberrigaray, R.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Govoni, S.; Alkon, D.L. Calcium responses in human fibroblasts: a diagnostic molecular profile for Alzheimer's disease. Neurobiol Aging 1996, 17, 549-555. [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A 1994, 91, 534-538. [CrossRef]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871-883. [CrossRef]
- Leissring, M.A.; LaFerla, F.M.; Callamaras, N.; Parker, I. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis 2001, 8, 469-478. [CrossRef]
- Leissring, M.A.; Parker, I.; LaFerla, F.M. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem 1999, 274, 32535-32538. [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 2004, 24, 508-513. [CrossRef]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem 2005, 94, 1711-1718. [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer's disease. J Alzheimers Dis 2010, 20 Suppl 1, S167-174. [CrossRef]
- Larsson, S.C.; Woolf, B.; Gill, D. Plasma Caffeine Levels and Risk of Alzheimer's Disease and Parkinson's Disease: Mendelian Randomization Study. Nutrients 2022, 14. [CrossRef]
- Kim, J.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Jung, G.; Lee, H.N.; Sohn, B.K.; Lee, J.Y.; Kim, Y.K.; et al. Coffee intake and decreased amyloid pathology in human brain. Transl Psychiatry 2019, 9, 270. [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674-2679. [CrossRef]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol 2001, 50, 56-63. [CrossRef]
- Ascherio, A.; Chen, H.; Schwarzschild, M.A.; Zhang, S.M.; Colditz, G.A.; Speizer, F.E. Caffeine, postmenopausal estrogen, and risk of Parkinson's disease. Neurology 2003, 60, 790-795. [CrossRef]
- Powers, K.M.; Kay, D.M.; Factor, S.A.; Zabetian, C.P.; Higgins, D.S.; Samii, A.; Nutt, J.G.; Griffith, A.; Leis, B.; Roberts, J.W.; et al. Combined effects of smoking, coffee, and NSAIDs on Parkinson's disease risk. Mov Disord 2008, 23, 88-95. [CrossRef]
- Saaksjarvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Mannisto, S. Prospective study of coffee consumption and risk of Parkinson's disease. Eur J Clin Nutr 2008, 62, 908-915. [CrossRef]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson's disease. Geriatr Gerontol Int 2014, 14, 430-439. [CrossRef]
- Altman, R.D.; Lang, A.E.; Postuma, R.B. Caffeine in Parkinson's disease: a pilot open-label, dose-escalation study. Mov Disord 2011, 26, 2427-2431. [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology 2012, 79, 651-658. [CrossRef]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer's mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941-952. [CrossRef]
- Stazi, M.; Lehmann, S.; Sakib, M.S.; Pena-Centeno, T.; Buschgens, L.; Fischer, A.; Weggen, S.; Wirths, O. Long-term caffeine treatment of Alzheimer mouse models ameliorates behavioural deficits and neuron loss and promotes cellular and molecular markers of neurogenesis. Cell Mol Life Sci 2021, 79, 55. [CrossRef]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer's disease transgenic mice. J Alzheimers Dis 2009, 17, 681-697. [CrossRef]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Muller, C.E.; Buee, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer's disease-like tau pathology. Neurobiol Aging 2014, 35, 2079-2090. [CrossRef]
- Han, M.E.; Kim, H.J.; Lee, Y.S.; Kim, D.H.; Choi, J.T.; Pan, C.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; et al. Regulation of cerebrospinal fluid production by caffeine consumption. BMC Neurosci 2009, 10, 110. [CrossRef]
- Zeitlin, R.; Patel, S.; Burgess, S.; Arendash, G.W.; Echeverria, V. Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer's transgenic mice. Brain Res 2011, 1417, 127-136. [CrossRef]
- Ghoneim, F.M.; Khalaf, H.A.; Elsamanoudy, A.Z.; Abo El-Khair, S.M.; Helaly, A.M.; Mahmoud el, H.M.; Elshafey, S.H. Protective effect of chronic caffeine intake on gene expression of brain derived neurotrophic factor signaling and the immunoreactivity of glial fibrillary acidic protein and Ki-67 in Alzheimer's disease. Int J Clin Exp Pathol 2015, 8, 7710-7728.
- Han, K.; Jia, N.; Li, J.; Yang, L.; Min, L.Q. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer's disease. Mol Med Rep 2013, 8, 737-740. [CrossRef]
- Soliman, A.M.; Fathalla, A.M.; Moustafa, A.A. Dose-dependent neuroprotective effect of caffeine on a rotenone-induced rat model of parkinsonism: A histological study. Neurosci Lett 2016, 623, 63-70. [CrossRef]
- Khadrawy, Y.A.; Salem, A.M.; El-Shamy, K.A.; Ahmed, E.K.; Fadl, N.N.; Hosny, E.N. Neuroprotective and Therapeutic Effect of Caffeine on the Rat Model of Parkinson's Disease Induced by Rotenone. J Diet Suppl 2017, 14, 553-572. [CrossRef]
- Singh, S.; Singh, K.; Patel, S.; Patel, D.K.; Singh, C.; Nath, C.; Singh, M.P. Nicotine and caffeine-mediated modulation in the expression of toxicant responsive genes and vesicular monoamine transporter-2 in 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease phenotype in mouse. Brain Res 2008, 1207, 193-206. [CrossRef]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective effects of caffeine in MPTP model of Parkinson's disease: A (13)C NMR study. Neurochem Int 2016, 92, 25-34. [CrossRef]
- Lee, K.W.; Im, J.Y.; Woo, J.M.; Grosso, H.; Kim, Y.S.; Cristovao, A.C.; Sonsalla, P.K.; Schuster, D.S.; Jalbut, M.M.; Fernandez, J.R.; et al. Neuroprotective and anti-inflammatory properties of a coffee component in the MPTP model of Parkinson's disease. Neurotherapeutics 2013, 10, 143-153. [CrossRef]
- Aguiar, L.M.; Nobre, H.V., Jr.; Macedo, D.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Cunha, G.M.; Sousa, F.C.; Viana, G.S. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol Biochem Behav 2006, 84, 415-419. [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front Neurosci 2018, 12, 301. [CrossRef]
- Schwarzschild, M.A.; Chen, J.F.; Ascherio, A. Caffeinated clues and the promise of adenosine A(2A) antagonists in PD. Neurology 2002, 58, 1154-1160. [CrossRef]
- Fuxe, K.; Ferre, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol Neurosci 2005, 26, 209-220. [CrossRef]
- Deleu, D.; Jacob, P.; Chand, P.; Sarre, S.; Colwell, A. Effects of caffeine on levodopa pharmacokinetics and pharmacodynamics in Parkinson disease. Neurology 2006, 67, 897-899. [CrossRef]
- Zajac, M.A.; Zakrzewski, A.G.; Kowal, M.G.; Narayan, S. A Novel Method of Caffeine Synthesis from Uracil. Synth. Commun. 2003, 33, 3291-3297. [CrossRef]
- Scimmi, C.; Cardinali, M.; Abenante, L.; Amatista, M.; Nacca, F.G.; Lenardao, E.J.; Sancineto, L.; Santi, C. Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water. Chemistry 2021, 3, 1126-1137. [CrossRef]
- Templ, J.; Gjata, E.; Getzner, F.; Schnürch, M. Monoselective N-Methylation of Amides, Indoles, and Related Structures Using Quaternary Ammonium Salts as Solid Methylating Agents. Org. Lett. 2022, 24, 7315-7319. [CrossRef]
- Nejatbakhsh, N.; Feng, Z.P. Calcium binding protein-mediated regulation of voltage-gated calcium channels linked to human diseases. Acta Pharmacol Sin 2011, 32, 741-748. [CrossRef]
- Kuhl, D.E.; Koeppe, R.A.; Minoshima, S.; Snyder, S.E.; Ficaro, E.P.; Foster, N.L.; Frey, K.A.; Kilbourn, M.R. In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer's disease. Neurology 1999, 52, 691-699. [CrossRef]
- Davis, K.L.; Mohs, R.C.; Marin, D.; Purohit, D.P.; Perl, D.P.; Lantz, M.; Austin, G.; Haroutunian, V. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA 1999, 281, 1401-1406. [CrossRef]
- Shinotoh, H.; Fukushi, K.; Nagatsuka, S.; Tanaka, N.; Aotsuka, A.; Ota, T.; Namba, H.; Tanada, S.; Irie, T. The amygdala and Alzheimer's disease: positron emission tomographic study of the cholinergic system. Ann N Y Acad Sci 2003, 985, 411-419.
- Sberna, G.; Saez-Valero, J.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein of Alzheimer's disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J Neurochem 1997, 69, 1177-1184. [CrossRef]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 2010, 67, 600-606. [CrossRef]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of antihypertensives and the risk of Parkinson disease. Neurology 2008, 70, 1438-1444. [CrossRef]
- Mullapudi, A.; Gudala, K.; Boya, C.S.; Bansal, D. Risk of Parkinson's Disease in the Users of Antihypertensive Agents: An Evidence from the Meta-Analysis of Observational Studies. J Neurodegener Dis 2016, 2016, 5780809. [CrossRef]
- Lee, Y.C.; Lin, C.H.; Wu, R.M.; Lin, J.W.; Chang, C.H.; Lai, M.S. Antihypertensive agents and risk of Parkinson's disease: a nationwide cohort study. PLoS One 2014, 9, e98961. [CrossRef]
- Kupsch, A.; Gerlach, M.; Pupeter, S.C.; Sautter, J.; Dirr, A.; Arnold, G.; Opitz, W.; Przuntek, H.; Riederer, P.; Oertel, W.H. Pretreatment with nimodipine prevents MPTP-induced neurotoxicity at the nigral, but not at the striatal level in mice. Neuroreport 1995, 6, 621-625. [CrossRef]
- Kupsch, A.; Sautter, J.; Schwarz, J.; Riederer, P.; Gerlach, M.; Oertel, W.H. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res 1996, 741, 185-196. [CrossRef]
- Mena, M.A.; Garcia de Yebenes, M.J.; Tabernero, C.; Casarejos, M.J.; Pardo, B.; Garcia de Yebenes, J. Effects of calcium antagonists on the dopamine system. Clin Neuropharmacol 1995, 18, 410-426. [CrossRef]
- Paris, D.; Bachmeier, C.; Patel, N.; Quadros, A.; Volmar, C.H.; Laporte, V.; Ganey, J.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Crawford, F.; et al. Selective antihypertensive dihydropyridines lower Abeta accumulation by targeting both the production and the clearance of Abeta across the blood-brain barrier. Mol Med 2011, 17, 149-162. [CrossRef]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: the case for isradipine. Biochim Biophys Acta 2011, 1812, 1584-1590. [CrossRef]
- Yu, J.T.; Chang, R.C.; Tan, L. Calcium dysregulation in Alzheimer's disease: from mechanisms to therapeutic opportunities. Prog Neurobiol 2009, 89, 240-255. [CrossRef]
- Anekonda, T.S.; Quinn, J.F.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer's disease. Neurobiol Dis 2011, 41, 62-70. [CrossRef]
- Mok, S.S.; Clippingdale, A.B.; Beyreuther, K.; Masters, C.L.; Barrow, C.J.; Small, D.H. A beta peptides and calcium influence secretion of the amyloid protein precursor from chick sympathetic neurons in culture. J Neurosci Res 2000, 61, 449-457. [CrossRef]
- Quartermain, D.; Garcia deSoria, V. The effects of calcium channel antagonists on short- and long-term retention in mice using spontaneous alternation behavior. Neurobiol Learn Mem 2001, 76, 117-124. [CrossRef]
- Levallois, P. Alzheimer's disease and aluminum. Neurology 1997, 48, 1141-1142. [CrossRef]
- Rondeau, V.; Commenges, D.; Jacqmin-Gadda, H.; Dartigues, J.F. Relation between aluminum concentrations in drinking water and Alzheimer's disease: an 8-year follow-up study. Am J Epidemiol 2000, 152, 59-66. [CrossRef]
- Gauthier, E.; Fortier, I.; Courchesne, F.; Pepin, P.; Mortimer, J.; Gauvreau, D. Aluminum forms in drinking water and risk of Alzheimer's disease. Environ Res 2000, 84, 234-246. [CrossRef]
- Crapper, D.R.; Krishnan, S.S.; Dalton, A.J. Brain aluminum distribution in Alzheimer's disease and experimental neurofibrillary degeneration. Science 1973, 180, 511-513. [CrossRef]
- Bouras, C.; Giannakopoulos, P.; Good, P.F.; Hsu, A.; Hof, P.R.; Perl, D.P. A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: comparison with Alzheimer's disease. Eur Neurol 1997, 38, 53-58. [CrossRef]
- Kawahara, M.; Kato, M.; Kuroda, Y. Effects of aluminum on the neurotoxicity of primary cultured neurons and on the aggregation of beta-amyloid protein. Brain Res Bull 2001, 55, 211-217. [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem 1993, 61, 1171-1174. [CrossRef]
- Pratico, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 2002, 16, 1138-1140. [CrossRef]
- Ricchelli, F.; Drago, D.; Filippi, B.; Tognon, G.; Zatta, P. Aluminum-triggered structural modifications and aggregation of beta-amyloids. Cell Mol Life Sci 2005, 62, 1724-1733. [CrossRef]
- Rani, A.; Neha; Sodhi, R.K.; Kaur, A. Protective effect of a calcium channel blocker "diltiazem" on aluminum chloride-induced dementia in mice. Naunyn Schmiedebergs Arch Pharmacol 2015, 388, 1151-1161. [CrossRef]
- Ohnishi, N.; Kusuhara, M.; Yoshioka, M.; Kuroda, K.; Soga, A.; Nishikawa, F.; Koishi, T.; Nakagawa, M.; Hori, S.; Matsumoto, T.; et al. Studies on interactions between functional foods or dietary supplements and medicines. I. Effects of Ginkgo biloba leaf extract on the pharmacokinetics of diltiazem in rats. Biol Pharm Bull 2003, 26, 1315-1320. [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 2009, 29, 11011-11019. [CrossRef]
- Kang, S.; Cooper, G.; Dunne, S.F.; Dusel, B.; Luan, C.H.; Surmeier, D.J.; Silverman, R.B. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat Commun 2012, 3, 1146. [CrossRef]
- Liss, B.; Striessnig, J. The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson's Disease. Annu Rev Pharmacol Toxicol 2019, 59, 263-289. [CrossRef]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218-229. [CrossRef]
- Graham, D.F.; Stewart-Wynne, E.G. Diltiazem-induced acute parkinsonism. Aust N Z J Med 1994, 24, 70. [CrossRef]
- Jeong, S.; Cho, H.; Kim, Y.J.; Ma, H.I.; Jang, S. Drug-induced Parkinsonism: A strong predictor of idiopathic Parkinson's disease. PLoS One 2021, 16, e0247354. [CrossRef]
- Anjaneyulu, M.; Chopra, K. Diltiazem attenuates oxidative stress in diabetic rats. Ren Fail 2005, 27, 335-344.
- Koller, P.T.; Bergmann, S.R. Reduction of lipid peroxidation in reperfused isolated rabbit hearts by diltiazem. Circ Res 1989, 65, 838-846. [CrossRef]
- Surmeier, D.J. Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol 2007, 6, 933-938. [CrossRef]
- Marx, M.; Weber, M.; Merkel, F.; Meyer zum Buschenfelde, K.H.; Kohler, H. Additive effects of calcium antagonists on cyclosporin A-induced inhibition of T-cell proliferation. Nephrol Dial Transplant 1990, 5, 1038-1044. [CrossRef]
- Fansa, I.; Gol, M.; Nisanoglu, V.; Yavas, S.; Iscan, Z.; Tasdemir, O. Does diltiazem inhibit the inflammatory response in cardiopulmonary bypass? Med Sci Monit 2003, 9, PI30-36.
- Dubey, L.; Hesong, Z. Anti-inflammatory action of diltiazem in patients with unstable angina. Postgrad Med J 2006, 82, 594-597. [CrossRef]
- Szabo, C.; Hasko, G.; Nemeth, Z.H.; Vizi, E.S. Calcium entry blockers increase interleukin-10 production in endotoxemia. Shock 1997, 7, 304-307. [CrossRef]
- Silei, V.; Fabrizi, C.; Venturini, G.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Lauro, G.M. Activation of microglial cells by PrP and beta-amyloid fragments raises intracellular calcium through L-type voltage sensitive calcium channels. Brain Res 1999, 818, 168-170. [CrossRef]
- Daschil, N.; Obermair, G.J.; Flucher, B.E.; Stefanova, N.; Hutter-Paier, B.; Windisch, M.; Humpel, C.; Marksteiner, J. CaV1.2 calcium channel expression in reactive astrocytes is associated with the formation of amyloid-beta plaques in an Alzheimer's disease mouse model. J Alzheimers Dis 2013, 37, 439-451. [CrossRef]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1.2 Ca(2+) channel exacerbates the symptoms in a Parkinson's disease model. Sci Rep 2019, 9, 9138. [CrossRef]
- Gizur, T.; Harsànyi, K. Some Applications of Isopropenyl Acetate To O-, N-and C-Acetylation. Synthetic Communications 1990, 20, 2365-2371. [CrossRef]
- Miyata, O.; Shinada, T.; Ninomiya, I.; Naito, T. Asymmetric induction at two contiguous stereogenic centers by diastereoface differentiating nucleophilic addition reaction. Tetrahedron Letters 1991, 32, 3519-3522. [CrossRef]
- Schwartz, A.; Madan, P.B.; Mohacsi, E.; O'Brien, J.P.; Todaro, L.J.; Coffen, D.L. Enantioselective synthesis of calcium channel blockers of the diltiazem group. The Journal of Organic Chemistry 1992, 57, 851-856. [CrossRef]
- Jacobsen, E.N.; Deng, L.; Furukawa, Y.; Martínez, L.E. Enantioselective catalytic epoxidation of cinnamate esters. Tetrahedron 1994, 50, 4323-4334. [CrossRef]
- Nangia, A.; Rao, P.; Madhavi, N. ChemInform Abstract: Carbohydrates as Chiral Auxiliaries in Asymmetric Darzens Reactions: Enantioselective Synthesis of the Benzothiazepine Ring System of Diltiazem. ChemInform 2010, 27. [CrossRef]
- Imashiro, R.; Kuroda, T. Asymmetric synthesis of methyl (2R,3S)-3-(4-methoxyphenyl) glycidate, a key intermediate of diltiazem, via Mukaiyama aldol reaction. Tetrahedron Letters 2001, 42, 1313-1315. [CrossRef]
- Seki, M. A Practical Synthesis of a Key Chiral Drug Intermediate via Asymmetric Organocatalysis. Synlett 2008, 2008, 164-176. [CrossRef]
- Mordant, C.; Caño de Andrade, C.; Touati, R.; Ratovelomanana-Vidal, V.; Hassine, B.B.; Genêt, J.-P. Stereoselective Synthesis of Diltiazem via Dynamic Kinetic Resolution. Synthesis 2003, 2003, 2405-2409. [CrossRef]
- Gizur, T.; Harsányi, K.; Fogassy, E. Studies of the resolution of racemates in the Synthesis of Diltiazem. Journal für Praktische Chemie/Chemiker-Zeitung 1994, 336, 628-631. [CrossRef]
- Desai, S.B.; Argade, N.P.; Ganesh, K.N. Remarkable Chemo-, Regio-, and Enantioselectivity in Lipase-Catalyzed Hydrolysis: Efficient Resolution of (±)-threo-Ethyl 3-(4-Methoxyphenyl)-2,3-diacetoxypropionate Leading to Chiral Intermediates of (+)-Diltiazem. The Journal of Organic Chemistry 1996, 61, 6730-6732. [CrossRef]
- Yamada, S.-i.; Tsujioka, I.; Shibatani, T.; Yoshioka, R. Efficient Alternative Synthetic Route to Diltiazem via (2R, 3S)-3-(4-Methoxyphenyl)glycidamide. CHEMICAL & PHARMACEUTICAL BULLETIN 1999, 47, 146-150. [CrossRef]
- Tokdar, P.; Ranadive, P.; George, S.; Upare, A.; Vishwasrao, S.; Roy, M.; Sivaramakrishnan, H. Bakers' Yeast Mediated Stereo Selective Reduction of Cis (+) Diketolactam to Cis (+) Hydroxylactam, a Key Intermediate for Diltiazem Synthesis in a Laboratory Scale Bioreactor. International Journal of Chemical Engineering and Applications 2011, 2, 386. [CrossRef]
- Yue, X.; Li, Y.; Liu, M.; Sang, D.; Huang, Z.; Chen, F. Biocatalytic dynamic reductive kinetic resolution of aryl α-chloro β-keto esters: divergent, stereocontrolled synthesis of diltiazem, clentiazem, and siratiazem. Chemical Communications 2022, 58, 9010-9013. [CrossRef]
- Bachurin, S.; Bukatina, E.; Lermontova, N.; Tkachenko, S.; Afanasiev, A.; Grigoriev, V.; Grigorieva, I.; Ivanov, Y.; Sablin, S.; Zefirov, N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann N Y Acad Sci 2001, 939, 425-435. [CrossRef]
- Lermontova, N.N.; Lukoyanov, N.V.; Serkova, T.P.; Lukoyanova, E.A.; Bachurin, S.O. Dimebon improves learning in animals with experimental Alzheimer's disease. Bull Exp Biol Med 2000, 129, 544-546. [CrossRef]
- Cano-Cuenca, N.; Solis-Garcia del Pozo, J.E.; Jordan, J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: a meta-analysis. J Alzheimers Dis 2014, 38, 155-164. [CrossRef]
- Wu, J.; Li, Q.; Bezprozvanny, I. Evaluation of Dimebon in cellular model of Huntington's disease. Mol Neurodegener 2008, 3, 15. [CrossRef]
- Grigorev, V.V.; Dranyi, O.A.; Bachurin, S.O. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull Exp Biol Med 2003, 136, 474-477. [CrossRef]
- Lermontova, N.N.; Redkozubov, A.E.; Shevtsova, E.F.; Serkova, T.P.; Kireeva, E.G.; Bachurin, S.O. Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) channels. Bull Exp Biol Med 2001, 132, 1079-1083. [CrossRef]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; dimebon, i. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207-215. [CrossRef]
- Bharadwaj, P.R.; Bates, K.A.; Porter, T.; Teimouri, E.; Perry, G.; Steele, J.W.; Gandy, S.; Groth, D.; Martins, R.N.; Verdile, G. Latrepirdine: molecular mechanisms underlying potential therapeutic roles in Alzheimer's and other neurodegenerative diseases. Transl Psychiatry 2013, 3, e332. [CrossRef]
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch Neurol 2010, 67, 154-160. [CrossRef]
- Peters, O.M.; Shelkovnikova, T.; Tarasova, T.; Springe, S.; Kukharsky, M.S.; Smith, G.A.; Brooks, S.; Kozin, S.A.; Kotelevtsev, Y.; Bachurin, S.O.; et al. Chronic administration of Dimebon does not ameliorate amyloid-beta pathology in 5xFAD transgenic mice. J Alzheimers Dis 2013, 36, 589-596. [CrossRef]
- Bachurin, S.O.; Shelkovnikova, T.A.; Ustyugov, A.A.; Peters, O.; Khritankova, I.; Afanasieva, M.A.; Tarasova, T.V.; Alentov, II; Buchman, V.L.; Ninkina, N.N. Dimebon slows progression of proteinopathy in gamma-synuclein transgenic mice. Neurotox Res 2012, 22, 33-42. [CrossRef]
- Webster, S.J.; Wilson, C.A.; Lee, C.H.; Mohler, E.G.; Terry, A.V., Jr.; Buccafusco, J.J. The acute effects of dimebolin, a potential Alzheimer's disease treatment, on working memory in rhesus monkeys. Br J Pharmacol 2011, 164, 970-978. [CrossRef]
- Wang, J.; Ferruzzi, M.G.; Varghese, M.; Qian, X.; Cheng, A.; Xie, M.; Zhao, W.; Ho, L.; Pasinetti, G.M. Preclinical study of dimebon on beta-amyloid-mediated neuropathology in Alzheimer's disease. Mol Neurodegener 2011, 6, 7. [CrossRef]
- Day, M.; Chandran, P.; Luo, F.; Rustay, N.R.; Markosyan, S.; LeBlond, D.; Fox, G.B. Latrepirdine increases cerebral glucose utilization in aged mice as measured by [18F]-fluorodeoxyglucose positron emission tomography. Neuroscience 2011, 189, 299-304. [CrossRef]
- Zhang, S.; Hedskog, L.; Petersen, C.A.; Winblad, B.; Ankarcrona, M. Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J Alzheimers Dis 2010, 21, 389-402. [CrossRef]
- Eckert, S.H.; Eckmann, J.; Renner, K.; Eckert, G.P.; Leuner, K.; Muller, W.E. Dimebon ameliorates amyloid-beta induced impairments of mitochondrial form and function. J Alzheimers Dis 2012, 31, 21-32. [CrossRef]
- Bachurin, S.O.; Shevtsova, E.P.; Kireeva, E.G.; Oxenkrug, G.F.; Sablin, S.O. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann N Y Acad Sci 2003, 993, 334-344; discussion 345-339. [CrossRef]
- Steele, J.W.; Kim, S.H.; Cirrito, J.R.; Verges, D.K.; Restivo, J.L.; Westaway, D.; Fraser, P.; Hyslop, P.S.; Sano, M.; Bezprozvanny, I.; et al. Acute dosing of latrepirdine (Dimebon), a possible Alzheimer therapeutic, elevates extracellular amyloid-beta levels in vitro and in vivo. Mol Neurodegener 2009, 4, 51. [CrossRef]
- Shevtzova, E.F.; Kireeva, E.G.; Bachurin, S.O. Effect of beta-amyloid peptide fragment 25-35 on nonselective permeability of mitochondria. Bull Exp Biol Med 2001, 132, 1173-1176. [CrossRef]
- Ustyugov, A.; Shevtsova, E.; Bachurin, S. Novel Sites of Neuroprotective Action of Dimebon (Latrepirdine). Mol Neurobiol 2015, 52, 970-978. [CrossRef]
- Steele, J.W.; Gandy, S. Latrepirdine (Dimebon(R)), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy 2013, 9, 617-618. [CrossRef]
- Steele, J.W.; Ju, S.; Lachenmayer, M.L.; Liken, J.; Stock, A.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine stimulates autophagy and reduces accumulation of alpha-synuclein in cells and in mouse brain. Mol Psychiatry 2013, 18, 882-888. [CrossRef]
- Yamashita, M.; Nonaka, T.; Arai, T.; Kametani, F.; Buchman, V.L.; Ninkina, N.; Bachurin, S.O.; Akiyama, H.; Goedert, M.; Hasegawa, M. Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett 2009, 583, 2419-2424. [CrossRef]
- De Jesus-Cortes, H.; Xu, P.; Drawbridge, J.; Estill, S.J.; Huntington, P.; Tran, S.; Britt, J.; Tesla, R.; Morlock, L.; Naidoo, J.; et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci U S A 2012, 109, 17010-17015. [CrossRef]
- Shelkovnikova, T.A.; Ustyugov, A.A.; Millership, S.; Peters, O.; Anichtchik, O.; Spillantini, M.G.; Buchman, V.L.; Bachurin, S.O.; Ninkina, N.N. Dimebon does not ameliorate pathological changes caused by expression of truncated (1-120) human alpha-synuclein in dopaminergic neurons of transgenic mice. Neurodegener Dis 2011, 8, 430-437. [CrossRef]
- Golubeva, M.I.; Shashkina, L.F.; Proinova, V.A.; Fedorova, E.A.; Nechushkina, L.V. [Preclinical study of the safety of the antihistaminic preparation dimebon]. Farmakol Toksikol 1985, 48, 114-119.
- Shadurski, K.; Danusevich, I.; Kost, A.; Vinogradova, E. Patent No. 1138164, 1985. In Proceedings of the Chem. Abstr, 1985; p. 198010.
- Gao, M.; Wang, M.; Hutchins, G.D.; Zheng, Q.-H. [11C]Dimebon, radiosynthesis and lipophilicity of a new potential PET agent for imaging of Alzheimer’s disease and Huntington’s disease. Bioorganic Med. Chem. Lett. 2010, 20, 2529-2532. [CrossRef]
- Dong, H.; Latka, R.T.; Driver, T.G. Ruthenium-Catalyzed γ-Carbolinium Ion Formation from Aryl Azides; Synthesis of Dimebolin. Org. Lett. 2011, 13, 2726-2729. [CrossRef]
- Helton, T.D.; Xu, W.; Lipscombe, D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci 2005, 25, 10247-10251. [CrossRef]
- Ohkubo, N.; Mitsuda, N.; Tamatani, M.; Yamaguchi, A.; Lee, Y.D.; Ogihara, T.; Vitek, M.P.; Tohyama, M. Apolipoprotein E4 stimulates cAMP response element-binding protein transcriptional activity through the extracellular signal-regulated kinase pathway. J Biol Chem 2001, 276, 3046-3053. [CrossRef]
- Liu, S.J.; Gasperini, R.; Foa, L.; Small, D.H. Amyloid-beta decreases cell-surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J Alzheimers Dis 2010, 21, 655-666. [CrossRef]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J 2011, 434, 503-512. [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 2012, 75, 824-837. [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer's amyloid-beta(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis 2014, 5, e1569. [CrossRef]
- Lovell, M.A.; Abner, E.; Kryscio, R.; Xu, L.; Fister, S.X.; Lynn, B.C. Calcium Channel Blockers, Progression to Dementia, and Effects on Amyloid Beta Peptide Production. Oxid Med Cell Longev 2015, 2015, 787805. [CrossRef]
- Nakajima, M.; Miura, M.; Aosaki, T.; Shirasawa, T. Deficiency of presenilin-1 increases calcium-dependent vulnerability of neurons to oxidative stress in vitro. J Neurochem 2001, 78, 807-814. [CrossRef]
- Hefter, D.; Kaiser, M.; Weyer, S.W.; Papageorgiou, I.E.; Both, M.; Kann, O.; Muller, U.C.; Draguhn, A. Amyloid Precursor Protein Protects Neuronal Network Function after Hypoxia via Control of Voltage-Gated Calcium Channels. J Neurosci 2016, 36, 8356-8371. [CrossRef]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; Laferla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting [Ca2+] in adult Alzheimer's disease mice. J Neurochem 2008, 105, 262-271. [CrossRef]
- Wang, Y.; Mattson, M.P. L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol Aging 2014, 35, 88-95. [CrossRef]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer's disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca(2+) entry. J Mol Med (Berl) 2018, 96, 1061-1079. [CrossRef]
- Skobeleva, K.; Shalygin, A.; Mikhaylova, E.; Guzhova, I.; Ryazantseva, M.; Kaznacheyeva, E. The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer's Disease. Int J Mol Sci 2022, 23. [CrossRef]
- Furukawa, K.; Wang, Y.; Yao, P.J.; Fu, W.; Mattson, M.P.; Itoyama, Y.; Onodera, H.; D'Souza, I.; Poorkaj, P.H.; Bird, T.D.; et al. Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J Neurochem 2003, 87, 427-436. [CrossRef]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J 2021, 288, 127-141. [CrossRef]
- Graham, W.C.; Robertson, R.G.; Sambrook, M.A.; Crossman, A.R. Injection of excitatory amino acid antagonists into the medial pallidal segment of a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treated primate reverses motor symptoms of parkinsonism. Life Sci 1990, 47, PL91-97. [CrossRef]
- Brotchie, J.M.; Mitchell, I.J.; Sambrook, M.A.; Crossman, A.R. Alleviation of parkinsonism by antagonism of excitatory amino acid transmission in the medial segment of the globus pallidus in rat and primate. Mov Disord 1991, 6, 133-138. [CrossRef]
- Benazzouz, A.; Gross, C.; Feger, J.; Boraud, T.; Bioulac, B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci 1993, 5, 382-389. [CrossRef]
- Beurrier, C.; Congar, P.; Bioulac, B.; Hammond, C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci 1999, 19, 599-609. [CrossRef]
- Hallworth, N.E.; Wilson, C.J.; Bevan, M.D. Apamin-sensitive small conductance calcium-activated potassium channels, through their selective coupling to voltage-gated calcium channels, are critical determinants of the precision, pace, and pattern of action potential generation in rat subthalamic nucleus neurons in vitro. J Neurosci 2003, 23, 7525-7542. [CrossRef]
- Garcia, L.; Audin, J.; D'Alessandro, G.; Bioulac, B.; Hammond, C. Dual effect of high-frequency stimulation on subthalamic neuron activity. J Neurosci 2003, 23, 8743-8751. [CrossRef]
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol 2012, 175, 627-635. [CrossRef]
- Whyte, K.A.; Greenfield, S.A. Effects of acetylcholinesterase and butyrylcholinesterase on cell survival, neurite outgrowth, and voltage-dependent calcium currents of embryonic ventral mesencephalic neurons. Exp Neurol 2003, 184, 496-509. [CrossRef]
- Eaton, M.E.; Macias, W.; Youngs, R.M.; Rajadhyaksha, A.; Dudman, J.T.; Konradi, C. L-type Ca2+ channel blockers promote Ca2+ accumulation when dopamine receptors are activated in striatal neurons. Brain Res Mol Brain Res 2004, 131, 65-72. [CrossRef]
- Belforte, J.E.; Magarinos-Azcone, C.; Armando, I.; Buno, W.; Pazo, J.H. Pharmacological involvement of the calcium channel blocker flunarizine in dopamine transmission at the striatum. Parkinsonism Relat Disord 2001, 8, 33-40. [CrossRef]
- Wang, R.; Ma, Z.; Wang, J.; Xie, J. L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotox Res 2012, 21, 266-270. [CrossRef]
- Yabuki, Y.; Ohizumi, Y.; Yokosuka, A.; Mimaki, Y.; Fukunaga, K. Nobiletin treatment improves motor and cognitive deficits seen in MPTP-induced Parkinson model mice. Neuroscience 2014, 259, 126-141. [CrossRef]
- Nedergaard, S.; Flatman, J.A.; Engberg, I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 1993, 466, 727-747.
- Martella, G.; Madeo, G.; Schirinzi, T.; Tassone, A.; Sciamanna, G.; Spadoni, F.; Stefani, A.; Shen, J.; Pisani, A.; Bonsi, P. Altered profile and D2-dopamine receptor modulation of high voltage-activated calcium current in striatal medium spiny neurons from animal models of Parkinson's disease. Neuroscience 2011, 177, 240-251. [CrossRef]
- Wang, X.J.; Xu, J.X. Possible involvement of Ca2+ signaling in rotenone-induced apoptosis in human neuroblastoma SH-SY5Y cells. Neurosci Lett 2005, 376, 127-132. [CrossRef]
- Sai, Y.; Chen, J.; Ye, F.; Zhao, Y.; Zou, Z.; Cao, J.; Dong, Z. Dopamine Release Suppression Dependent on an Increase of Intracellular Ca(2+) Contributed to Rotenone-induced Neurotoxicity in PC12 Cells. J Toxicol Pathol 2013, 26, 149-157. [CrossRef]
- Daschil, N.; Humpel, C. Nifedipine and nimodipine protect dopaminergic substantia nigra neurons against axotomy-induced cell death in rat vibrosections via modulating inflammatory responses. Brain Res 2014, 1581, 1-11. [CrossRef]
- Singh, H.; Singh, K. Carbon transfer reactions with heterocycles - V. A facile synthesis of nifedipine and analogues. Tetrahedron 1989, 45, 3967-3974. [CrossRef]
- Hayashi, M.; Yamauchi, K.; Kinoshita, M. N-Alkylation of Nitrogen Heterocyclic Compounds with Dialkyl Phosphites. Bulletin of the Chemical Society of Japan 1977, 50, 1510-1512. [CrossRef]
- Palacios, F.; Herrán, E.; Rubiales, G. Reaction of N-Vinylic Phosphazenes Derived from β-Amino Acids with Aldehydes. Azadiene-Mediated Synthesis of Dihydropyridines, Pyridines, and Polycyclic Nitrogen Derivatives. The Journal of Organic Chemistry 1999, 64, 6239-6246. [CrossRef]
- Khadilkar, B.; Chitnavis, A. ChemInform Abstract: Rate Enhancement in the Synthesis of Some 4-Aryl-1,4-dihydropyridines Using Methyl 3-Aminocrotonate under Microwave Irradiation. ChemInform 2010, 26. [CrossRef]
- Anniyappan, M.; Muralidharan, D.; Perumal, P.T. SYNTHESIS OF HANTZSCH 1,4-DIHYDROPYRIDINES UNDER MICROWAVE IRRADIATION. Synthetic Communications 2002, 32, 659-663. [CrossRef]
- Khadilkar, B.M.; Gaikar, V.G.; Chitnavis, A.A. Aqueous hydrotrope solution as a safer medium for microwave enhanced hantzsch dihydropyridine ester synthesis. Tetrahedron Letters 1995, 36, 8083-8086. [CrossRef]
- Gordeev, M.F.; Patel, D.V.; Gordon, E.M. Approaches to Combinatorial Synthesis of Heterocycles: A Solid-Phase Synthesis of 1,4-Dihydropyridines. The Journal of Organic Chemistry 1996, 61, 924-928. [CrossRef]
- Balogh, M.; Gerstmans, A.; Hermecz, I. SOLID ACID CATALYZED REACTION OF AMINALS WITH METHYL 3-AMINOCROTONATE. Heterocyclic Communications 1999, 5, 89-92. [CrossRef]
- Sivamurugan, V.; Kumar, R.S.; Palanichamy, M.; Murugesan, V. Synthesis of hantzsch 1,4-dihydropyridines under solvent-free condition using zn[(L)proline]2 as lewis acid catalyst. Journal of Heterocyclic Chemistry 2005, 42, 969-974. [CrossRef]
- Maheswara, M.; Siddaiah, V.; Rao, Y.K.; Tzeng, Y.-M.; Sridhar, C. A simple and efficient one-pot synthesis of 1,4-dihydropyridines using heterogeneous catalyst under solvent-free conditions. Journal of Molecular Catalysis A: Chemical 2006, 260, 179-180. [CrossRef]
- Gupta, R.; Gupta, R.; Paul, S.; Loupy, A. Covalently Anchored Sulfonic Acid on Silica Gel as an Efficient and Reusable Heterogeneous Catalyst for the One-Pot Synthesis of Hantzsch 1,4-Dihydropyridines under Solvent-Free Conditions. Synthesis-stuttgart 2007, 2007, 2835-2838. [CrossRef]
- Arumugam, P.; Perumal, P. ChemInform Abstract: Hantzsch Synthesis of Polyhydroquinolines — A Simple, Efficient and Neat Protocol. ChemInform 2008, 39. [CrossRef]
- Kumar, A.; Maurya, R.A. Efficient Synthesis of Hantzsch Esters and Polyhydroquinoline Derivatives in Aqueous Micelles. Synlett 2008, 2008, 883-885. [CrossRef]
- Subudhi, B.B.; Sahoo, S.P. Synthesis and Evaluation of Antioxidant, Anti-inflammatory and Antiulcer Activity of Conjugates of Amino Acids with Nifedipine. Chemical and Pharmaceutical Bulletin 2011, 59, 1153-1156. [CrossRef]
- Bridgwood, K.L.; Veitch, G.E.; Ley, S.V. Magnesium Nitride as a Convenient Source of Ammonia: Preparation of Dihydropyridines. Organic Letters 2008, 10, 3627-3629. [CrossRef]
- Blajovan, R.; Modra, D. THE STUDY SYNTHESIS OF NIFEDIPINE - CALCIUM ANTAGONIST. Annals of West University of Timisoara. Series of Chemistry 2013, 22, 47-56.
- Prasanthi, G.; Prasad, K.V.S.R.G.; Bharathi, K. Synthesis, anticonvulsant activity and molecular properties prediction of dialkyl 1-(di(ethoxycarbonyl)methyl)-2,6-dimethyl-4-substituted-1,4-dihydropyridine-3,5-dicarboxylates. European Journal of Medicinal Chemistry 2014, 73, 97-104. [CrossRef]
- El-Moselhy, T.F.; Sidhom, P.A.; Esmat, E.A.; El-Mahdy, N.A. Synthesis, Docking Simulation, Biological Evaluations and 3D-QSAR Study of 1,4-Dihydropyridines as Calcium Channel Blockers. Chemical and Pharmaceutical Bulletin 2017, 65, 893-903. [CrossRef]
- Sohal, H.; Goyal, A.; Sharma, R.; Khare, R. One-pot, multicomponent synthesis of symmetrical Hantzsch 1, 4-dihydropyridine derivatives using glycerol as clean and green solvent. European Journal of Chemistry 2014, 5, 171-175. [CrossRef]
- Paraskar, A.; Sudalai, A. Cu(OTf)2 Catalyzed High Yield Synthesis of Hantzsch 1,4-Dihydropyridines. Cheminform 2007, 38. [CrossRef]
- Fedorova, O.V.; Koryakova, O.V.; Valova, M.S.; Ovchinnikova, I.G.; Titova, Y.A.; Rusinov, G.L.; Charushin, V.N. Catalytic effect of nanosized metal oxides on the Hantzsch reaction. Kinetics and Catalysis 2010, 51, 566-572. [CrossRef]
- Siddaiah, V.; Basha, G.M.; Rao, G.P.; Prasad, U.V.; Rao, R.S. PEG-Mediated Catalyst-Free Synthesis of Hantzsch 1,4-Dihydropyridines and Polyhydroquinoline Derivatives. Synthetic Communications 2012, 42, 627-634. [CrossRef]
- Maurya, M.R.; Saini, N.; Avecilla, F. Study of temperature dependent three component dynamic covalent assembly via Hantzsch reaction catalyzed by dioxido- and oxidoperoxidomolybdenum(vi) complexes under solvent free conditions. RSC Advances 2016, 6, 12993-13009. [CrossRef]
- Rekunge, D.S.; Khatri, C.K.; Chaturbhuj, G.U. Sulfated polyborate: An efficient and reusable catalyst for one pot synthesis of Hantzsch 1,4-dihydropyridines derivatives using ammonium carbonate under solvent free conditions. Tetrahedron Letters 2017, 58, 1240-1244. [CrossRef]
- Mohammadi, B.; Mohammad, S.; Jamkarani, H.; A. Kamali, T.; Nasrollahzadeh, M.; Mohajeri, A. Sulfonic acid-functionalized silica: A remarkably efficient heterogeneous reusable catalyst for the one-pot synthesis of 1, 4-dihydropyridines. Turkish Journal of Chemistry 2010, 34. [CrossRef]
- Bitaraf, M.; Amoozadeh, A.; Otokesh, S. A Simple and Efficient One-pot Synthesis of 1,4-dihydropyridines Using Nano-WO3- supported Sulfonic Acid as an Heterogeneous Catalyst under Solvent-free Conditions. Journal of the Chinese Chemical Society 2016, 63, 336-344. [CrossRef]
- Palermo, V.; Sathicq, Á.G.; Constantieux, T.; Rodríguez, J.; Vázquez, P.G.; Romanelli, G.P. First Report About the Use of Micellar Keggin Heteropolyacids as Catalysts in the Green Multicomponent Synthesis of Nifedipine Derivatives. Catalysis Letters 2016, 146, 1634-1647. [CrossRef]
- Wu, X.Y. Facile and Green Synthesis of 1,4-Dihydropyridine Derivatives in n-Butyl Pyridinium Tetrafluoroborate. Synthetic Communications 2012, 42, 454-459. [CrossRef]
- Wu, C.; Bian, Q.; Ding, T.; Tang, M.; Zhang, W.; Xu, Y.; Liu, B.; Xu, H.; Li, H.-B.; Fu, H. Photoinduced Iron-Catalyzed ipso-Nitration of Aryl Halides via Single-Electron Transfer. ACS Catalysis 2021, 11, 9561-9568. [CrossRef]
- Guidi, M.; Moon, S.; Anghileri, L.; Cambié, D.; Seeberger, P.H.; Gilmore, K. Combining radial and continuous flow synthesis to optimize and scale-up the production of medicines. Reaction Chemistry & Engineering 2021, 6, 220-224. [CrossRef]
- Xiang, S.; Fan, W.; Zhang, W.; Li, Y.; Guo, S.; Huang, D. Aqueous CO2 fixation: construction of pyridine skeletons in cooperation with ammonium cations. Green Chemistry 2021, 23, 7950-7955. [CrossRef]
- Xu, W.; Lipscombe, D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 2001, 21, 5944-5951. [CrossRef]
- Lopez-Arrieta, J.M.; Birks, J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev 2002, CD000147. [CrossRef]
- Ban, T.A.; Morey, L.; Aguglia, E.; Azzarelli, O.; Balsano, F.; Marigliano, V.; Caglieris, N.; Sterlicchio, M.; Capurso, A.; Tomasi, N.A.; et al. Nimodipine in the treatment of old age dementias. Prog Neuropsychopharmacol Biol Psychiatry 1990, 14, 525-551. [CrossRef]
- Tollefson, G.D. Short-term effects of the calcium channel blocker nimodipine (Bay-e-9736) in the management of primary degenerative dementia. Biol Psychiatry 1990, 27, 1133-1142. [CrossRef]
- Parnetti, L.; Senin, U.; Carosi, M.; Baasch, H. Mental deterioration in old age: results of two multicenter, clinical trials with nimodipine. The Nimodipine Study Group. Clin Ther 1993, 15, 394-406.
- Schmage, N.; Bergener, M. Global rating, symptoms, behavior, and cognitive performance as indicators of efficacy in clinical studies with nimodipine in elderly patients with cognitive impairment syndromes. Int Psychogeriatr 1992, 4 Suppl 1, 89-99.
- Pantoni, L.; del Ser, T.; Soglian, A.G.; Amigoni, S.; Spadari, G.; Binelli, D.; Inzitari, D. Efficacy and safety of nimodipine in subcortical vascular dementia: a randomized placebo-controlled trial. Stroke 2005, 36, 619-624. [CrossRef]
- Meneses, A.; Terron, J.A.; Ibarra, M.; Hong, E. Effects of nimodipine on learning in normotensive and spontaneously hypertensive rats. Behav Brain Res 1997, 85, 121-125. [CrossRef]
- Paroczai, M.; Kiss, B.; Karpati, E. Effect of RGH-2716 on learning and memory deficits of young and aged rats in water-labyrinth. Brain Res Bull 1998, 45, 475-488. [CrossRef]
- Batuecas, A.; Pereira, R.; Centeno, C.; Pulido, J.A.; Hernandez, M.; Bollati, A.; Bogonez, E.; Satrustegui, J. Effects of chronic nimodipine on working memory of old rats in relation to defects in synaptosomal calcium homeostasis. Eur J Pharmacol 1998, 350, 141-150. [CrossRef]
- Gholami Pourbadie, H.; Naderi, N.; Janahmadi, M.; Mehranfard, N.; Motamedi, F. Calcium channel blockade attenuates abnormal synaptic transmission in the dentate gyrus elicited by entorhinal amyloidopathy. Synapse 2016, 70, 408-417. [CrossRef]
- Gholamipour-Badie, H.; Naderi, N.; Khodagholi, F.; Shaerzadeh, F.; Motamedi, F. L-type calcium channel blockade alleviates molecular and reversal spatial learning and memory alterations induced by entorhinal amyloid pathology in rats. Behav Brain Res 2013, 237, 190-199. [CrossRef]
- Veng, L.M.; Mesches, M.H.; Browning, M.D. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res 2003, 110, 193-202. [CrossRef]
- Topcu, A.; Saral, S.; Ozturk, A.; Saral, O.; Kaya, A.K. The effect of the calcium channel blocker nimodipine on hippocampal BDNF/Ach levels in rats with experimental cognitive impairment. Neurol Res 2023, 1-10. [CrossRef]
- Sandin, M.; Jasmin, S.; Levere, T.E. Aging and cognition: facilitation of recent memory in aged nonhuman primates by nimodipine. Neurobiol Aging 1990, 11, 573-575. [CrossRef]
- Pierrot, N.; Ghisdal, P.; Caumont, A.S.; Octave, J.N. Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem 2004, 88, 1140-1150. [CrossRef]
- Ishii, M.; Hiller, A.J.; Pham, L.; McGuire, M.J.; Iadecola, C.; Wang, G. Amyloid-Beta Modulates Low-Threshold Activated Voltage-Gated L-Type Calcium Channels of Arcuate Neuropeptide Y Neurons Leading to Calcium Dysregulation and Hypothalamic Dysfunction. J Neurosci 2019, 39, 8816-8825. [CrossRef]
- Ueda, K.; Shinohara, S.; Yagami, T.; Asakura, K.; Kawasaki, K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 1997, 68, 265-271. [CrossRef]
- Brorson, J.R.; Bindokas, V.P.; Iwama, T.; Marcuccilli, C.J.; Chisholm, J.C.; Miller, R.J. The Ca2+ influx induced by beta-amyloid peptide 25-35 in cultured hippocampal neurons results from network excitation. J Neurobiol 1995, 26, 325-338. [CrossRef]
- Weiss, J.H.; Pike, C.J.; Cotman, C.W. Ca2+ channel blockers attenuate beta-amyloid peptide toxicity to cortical neurons in culture. J Neurochem 1994, 62, 372-375. [CrossRef]
- Yagami, T.; Nakazato, H.; Ueda, K.; Asakura, K.; Kuroda, T.; Hata, S.; Sakaeda, T.; Sakaguchi, G.; Itoh, N.; Hashimoto, Y.; et al. Prostaglandin E2 rescues cortical neurons from amyloid beta protein-induced apoptosis. Brain Res 2003, 959, 328-335. [CrossRef]
- Yagami, T.; Ueda, K.; Asakura, K.; Kuroda, T.; Hata, S.; Sakaeda, T.; Kambayashi, Y.; Fujimoto, M. Effects of endothelin B receptor agonists on amyloid beta protein (25-35)-induced neuronal cell death. Brain Res 2002, 948, 72-81. [CrossRef]
- Ekinci, F.J.; Ortiz, D.; Shea, T.B. Okadaic acid mediates tau phosphorylation via sustained activation of the L-voltage-sensitive calcium channel. Brain Res Mol Brain Res 2003, 117, 145-151. [CrossRef]
- Facchinetti, F.; Fasolato, C.; Del Giudice, E.; Burgo, A.; Furegato, S.; Fusco, M.; Basso, E.; Seraglia, R.; D'Arrigo, A.; Leon, A. Nimodipine selectively stimulates beta-amyloid 1-42 secretion by a mechanism independent of calcium influx blockage. Neurobiol Aging 2006, 27, 218-227. [CrossRef]
- Higham, J.P.; Hidalgo, S.; Buhl, E.; Hodge, J.J.L. Restoration of Olfactory Memory in Drosophila Overexpressing Human Alzheimer's Disease Associated Tau by Manipulation of L-Type Ca(2+) Channels. Front Cell Neurosci 2019, 13, 409. [CrossRef]
- Zheng, Z.; Wu, K.; Ruan, Q.; Li, D.; Liu, W.; Wang, M.; Li, Y.; Xia, J.; Yang, D.; Guo, J. Suppression of Selective Voltage-Gated Calcium Channels Alleviates Neuronal Degeneration and Dysfunction through Glutathione S-Transferase-Mediated Oxidative Stress Resistance in a Caenorhabditis elegans Model of Alzheimer's Disease. Oxid Med Cell Longev 2022, 8287633. [CrossRef]
- Hopp, S.C.; D'Angelo, H.M.; Royer, S.E.; Kaercher, R.M.; Crockett, A.M.; Adzovic, L.; Wenk, G.L. Calcium dysregulation via L-type voltage-dependent calcium channels and ryanodine receptors underlies memory deficits and synaptic dysfunction during chronic neuroinflammation. J Neuroinflammation 2015, 12, 56. [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Colaianna, M.; Zotti, M.; Ferrari, D.; Trabace, L.; Zuliani, G.; Di Virgilio, F. Nimodipine inhibits IL-1beta release stimulated by amyloid beta from microglia. Br J Pharmacol 2012, 167, 1702-1711. [CrossRef]
- Zongfang, Z.; Wenjing, L.; Zhaomin, C.; Lei, Z. Therapeutic effect of piracetam with nimodipine on vascular dementia after cerebral infarction. Pak J Pharm Sci 2020, 33, 2405-2411.
- Sadleir, K.R.; Popovic, J.; Khatri, A.; Vassar, R. Oral nimodipine treatment has no effect on amyloid pathology or neuritic dystrophy in the 5XFAD mouse model of amyloidosis. PLoS One 2022, 17, e0263332. [CrossRef]
- Gaasch, J.A.; Geldenhuys, W.J.; Lockman, P.R.; Allen, D.D.; Van der Schyf, C.J. Voltage-gated calcium channels provide an alternate route for iron uptake in neuronal cell cultures. Neurochem Res 2007, 32, 1686-1693. [CrossRef]
- Hopp, S.C.; Royer, S.E.; D'Angelo, H.M.; Kaercher, R.M.; Fisher, D.A.; Wenk, G.L. Differential neuroprotective and anti-inflammatory effects of L-type voltage dependent calcium channel and ryanodine receptor antagonists in the substantia nigra and locus coeruleus. J Neuroimmune Pharmacol 2015, 10, 35-44. [CrossRef]
- Putzier, I.; Kullmann, P.H.; Horn, J.P.; Levitan, E.S. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci 2009, 29, 15414-15419. [CrossRef]
- Klimaviciusa, L.; Klusa, V.; Duburs, G.; Kaasik, A.; Kalda, A.; Zharkovsky, A. Distinct effects of atypical 1,4-dihydropyridines on 1-methyl-4-phenylpyridinium-induced toxicity. Cell Biochem Funct 2007, 25, 15-21. [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem Int 2016, 99, 221-232. [CrossRef]
- Singh, A.; Verma, P.; Raju, A.; Mohanakumar, K.P. Nimodipine attenuates the parkinsonian neurotoxin, MPTP-induced changes in the calcium binding proteins, calpain and calbindin. J Chem Neuroanat 2019, 95, 89-94. [CrossRef]
- Nordstrom, O.; Braesch-Andersen, S.; Bartfai, T. Dopamine release is enhanced while acetylcholine release is inhibited by nimodipine (Bay e 9736). Acta Physiol Scand 1986, 126, 115-119. [CrossRef]
- Soderstrom, K.E.; O'Malley, J.A.; Levine, N.D.; Sortwell, C.E.; Collier, T.J.; Steece-Collier, K. Impact of dendritic spine preservation in medium spiny neurons on dopamine graft efficacy and the expression of dyskinesias in parkinsonian rats. Eur J Neurosci 2010, 31, 478-490. [CrossRef]
- Schuster, S.; Doudnikoff, E.; Rylander, D.; Berthet, A.; Aubert, I.; Ittrich, C.; Bloch, B.; Cenci, M.A.; Surmeier, D.J.; Hengerer, B.; et al. Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry 2009, 65, 518-526. [CrossRef]
- Sautter, J.; Kupsch, A.; Earl, C.D.; Oertel, W.H. Degeneration of pre-labelled nigral neurons induced by intrastriatal 6-hydroxydopamine in the rat: behavioural and biochemical changes and pretreatment with the calcium-entry blocker nimodipine. Exp Brain Res 1997, 117, 111-119. [CrossRef]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.H.; Zheng, X.R.; Fei, Z.; Jiang, X.F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson's disease. Cell Signal 2013, 25, 2863-2870. [CrossRef]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580-589. [CrossRef]
- Gordeev, M.F.; Patel, D.V.; Gordon, E.M. Approaches to Combinatorial Synthesis of Heterocycles: A Solid-Phase Synthesis of 1,4-Dihydropyridines. J. Org. Chem. 1996, 61, 924-928. [CrossRef]
- Balaev, A.N.; Osipov, V.N.; Fedorov, V.E. Development of nimodipine production technology. Pharm. Chem. J. 2012, 46, 285-287. [CrossRef]
Figure 1.
Heterocycles target Ca2+ dyshomeostasis leading to mitochondrial Ca2+ overlad, oxidative damage and neuronal degeneration in AD and PD.
Figure 1.
Heterocycles target Ca2+ dyshomeostasis leading to mitochondrial Ca2+ overlad, oxidative damage and neuronal degeneration in AD and PD.
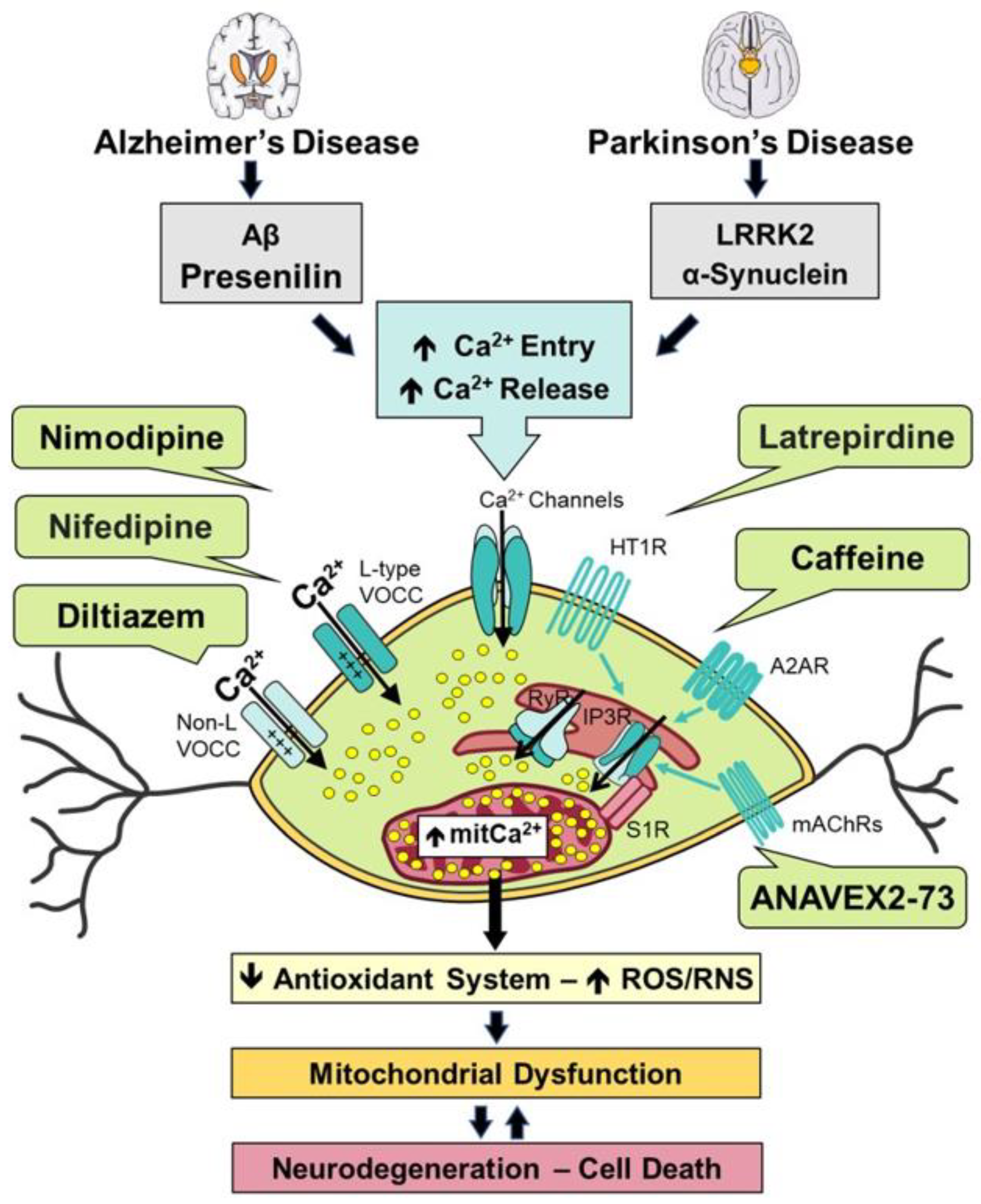
Scheme 1.
Unique total synthesis of ANAVEX2-73 1 reported by Foscolos and co-workers.
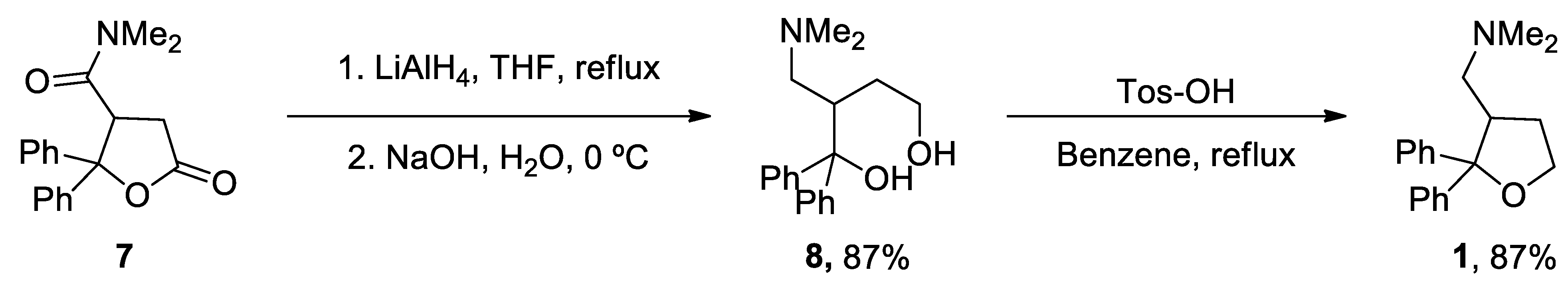
Scheme 2.
Synthesis of caffeine 2 reported by Narayan in 2003 from uracil 9.
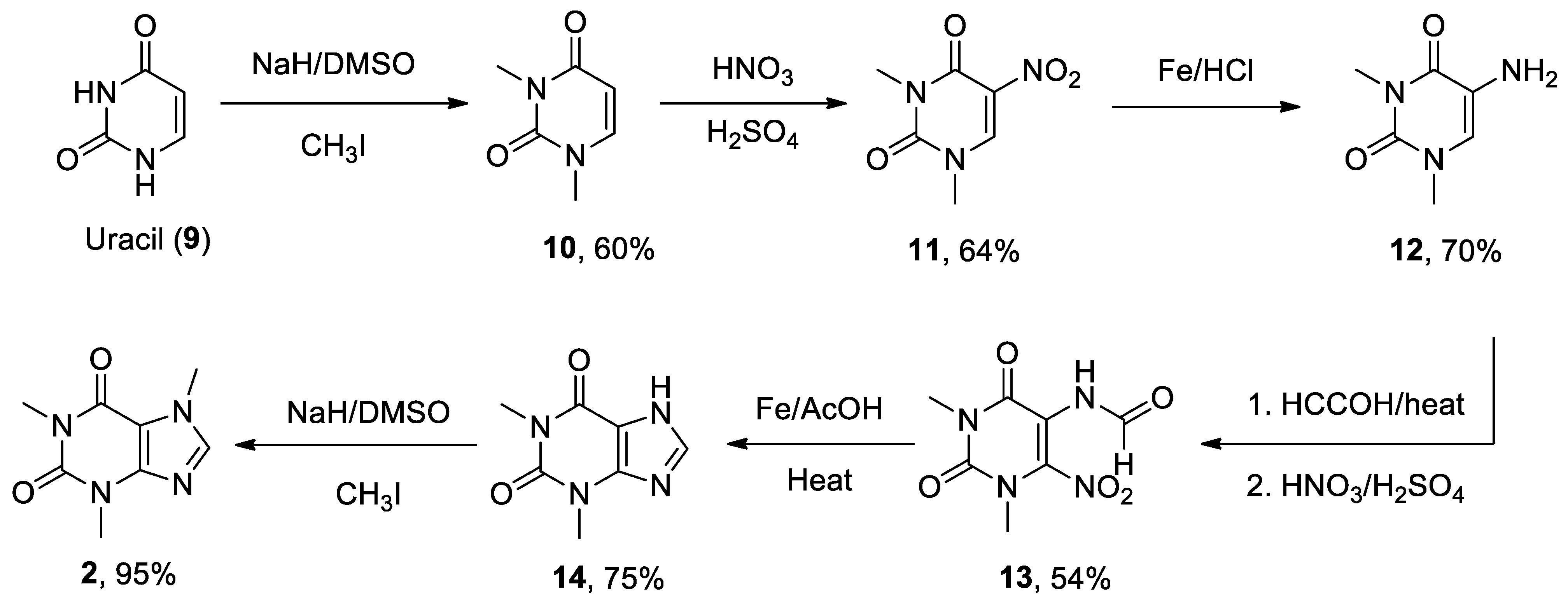
Scheme 3.
New methodologies applied for the N-methylation of theophylline 14.
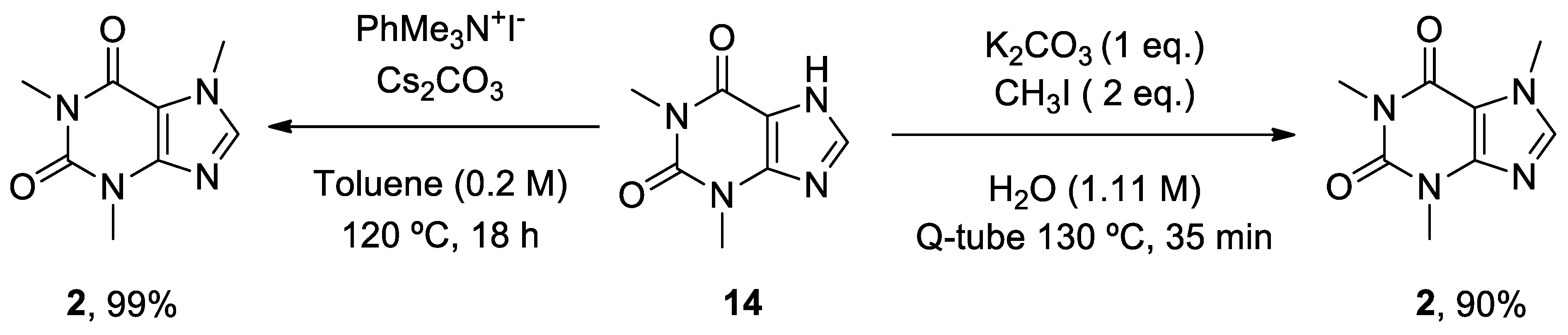
Scheme 4.
Schwartz’s strategy for the synthesis of (+)-diltiazem 3.
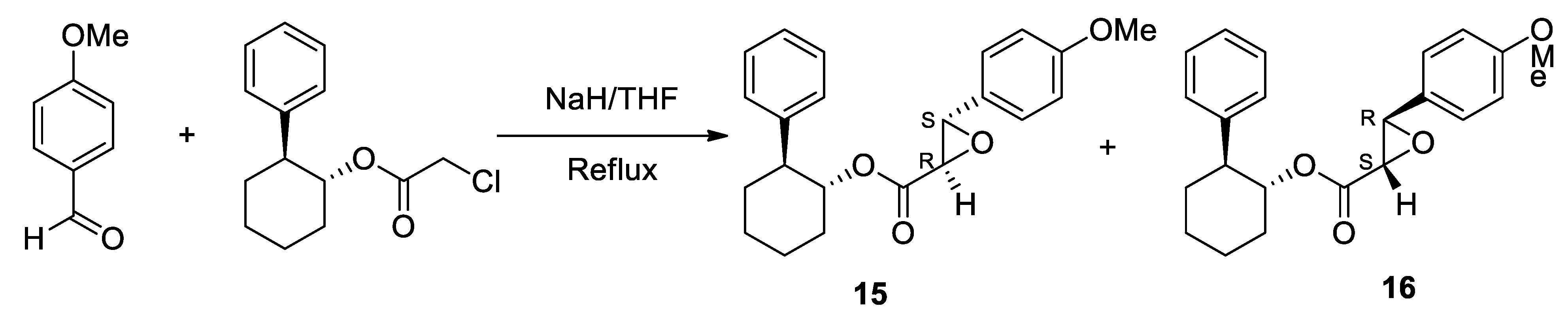
Scheme 5.
Preparation of the enantiomerically pure key intermediate 18 for the synthesis of (+)-diltiazem 3.
Scheme 5.
Preparation of the enantiomerically pure key intermediate 18 for the synthesis of (+)-diltiazem 3.
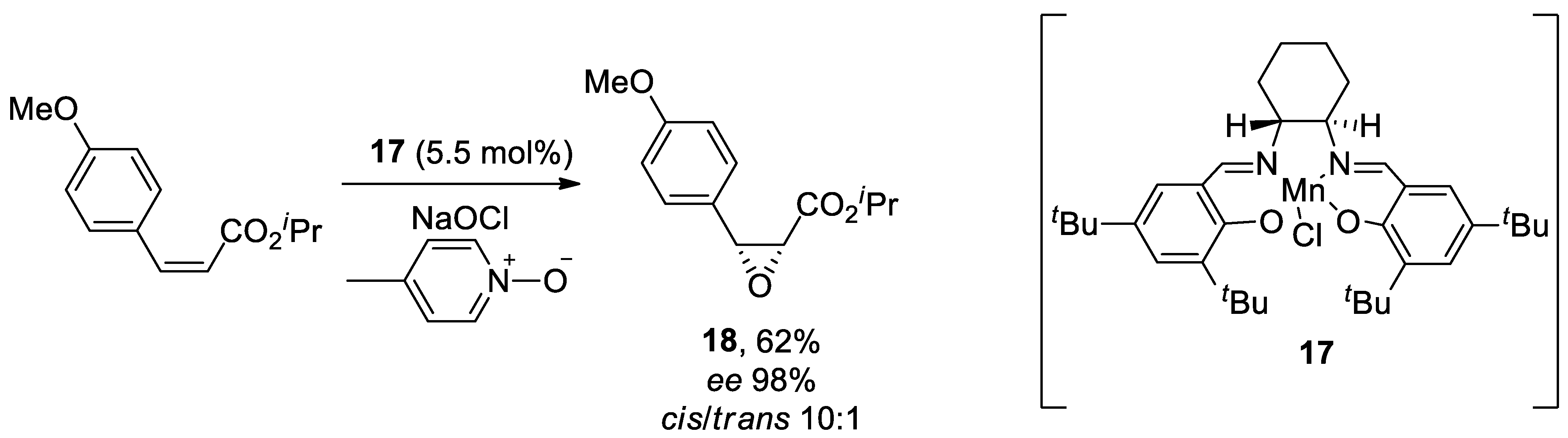
Scheme 6.
Chemoenzymatic synthesis of (+)-diltiazem 3 proposed by Chen and colleagues in 2022.
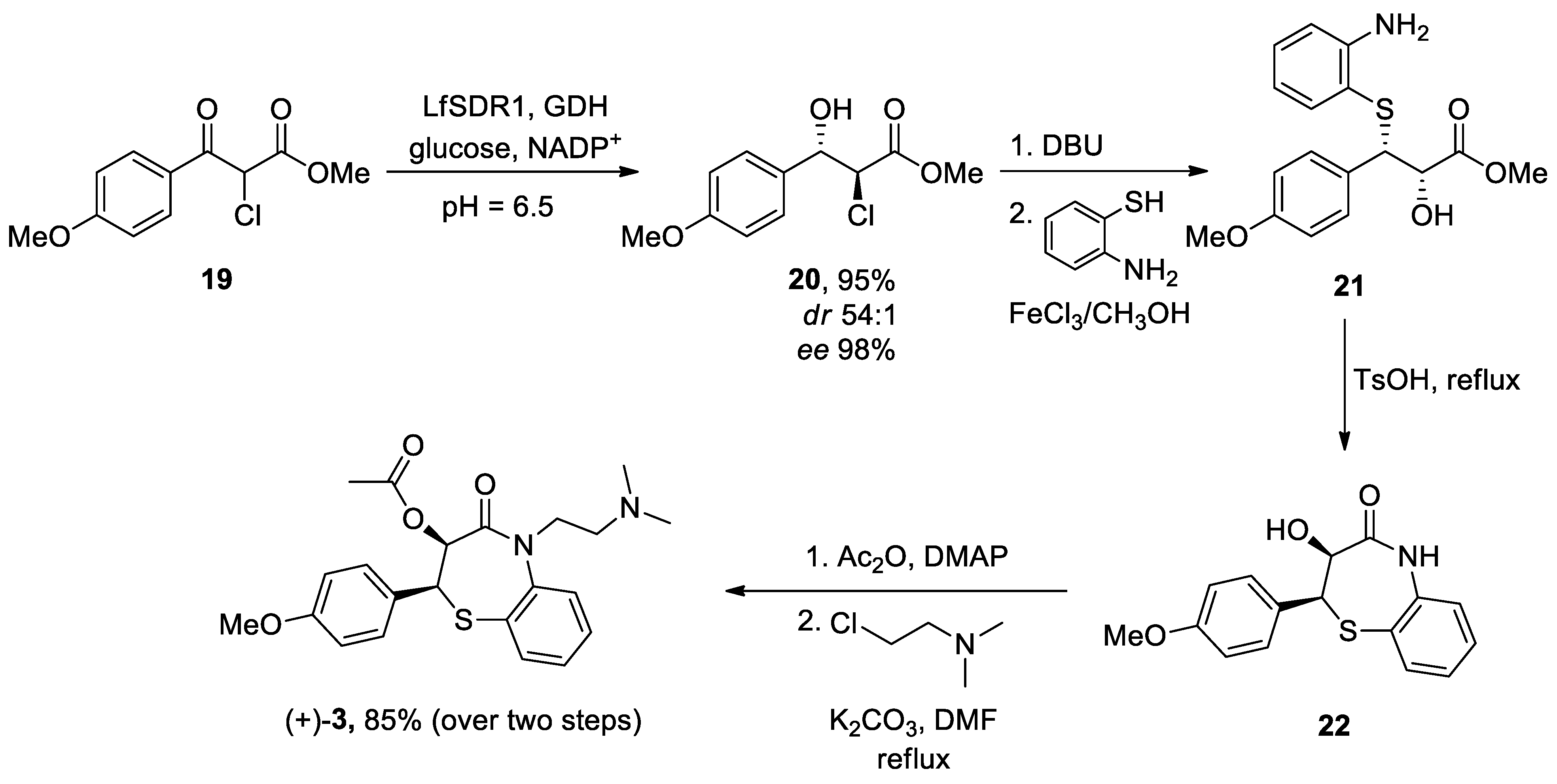
Scheme 7.
Synthesis of latrepirdine 4 reported by Zheng et al. 2022.
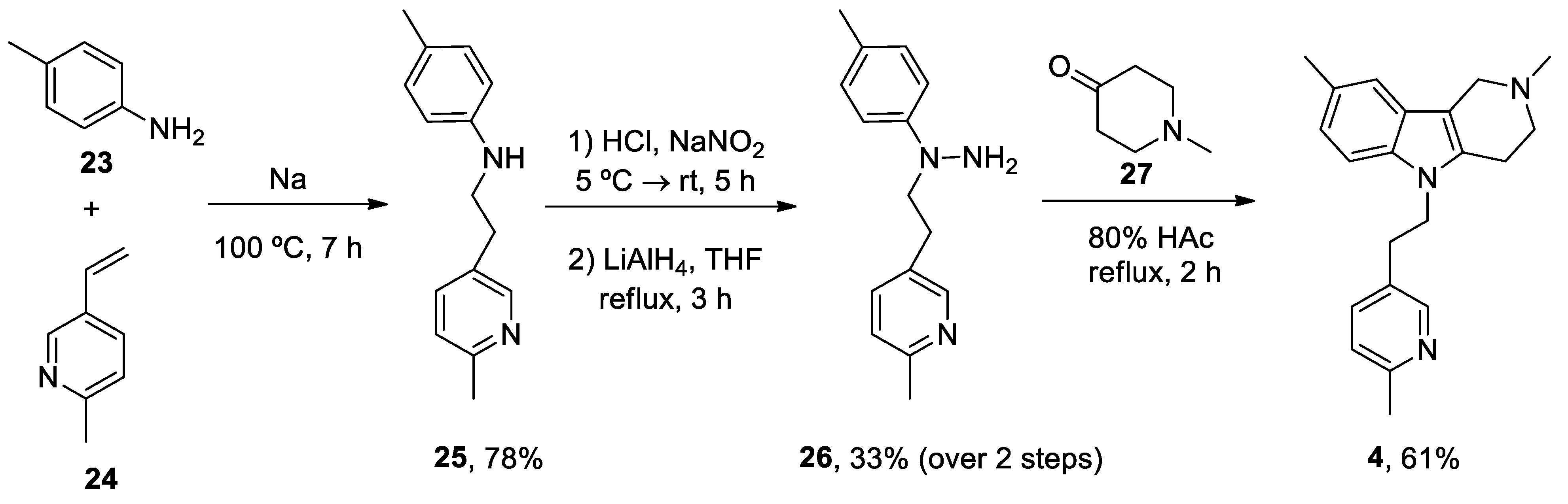
Scheme 8.
Ruthenium (III) catalysis used in the synthesis of latrepirdine 4.
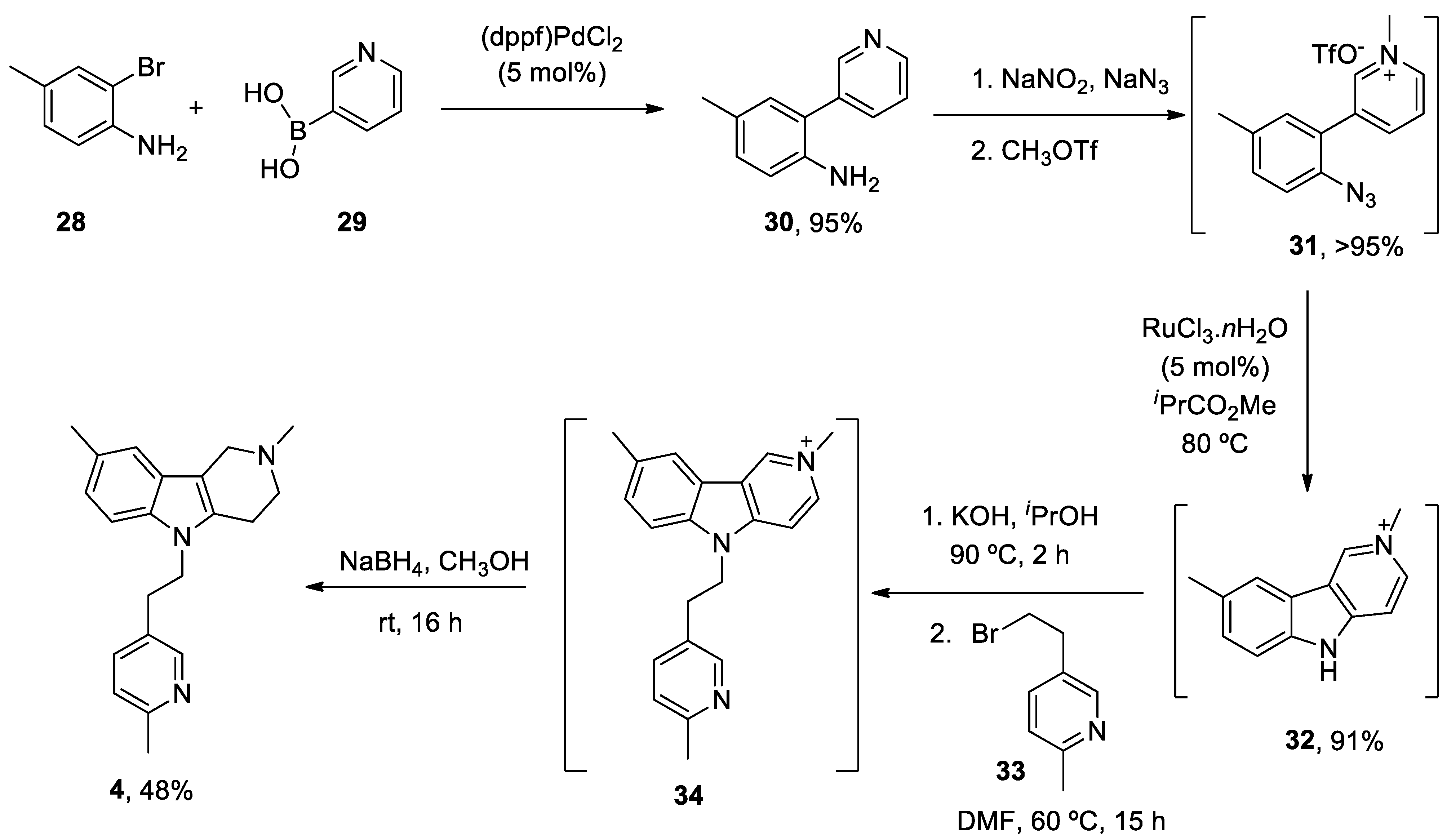
Scheme 9.
Hantzsch reaction used in the preparation of nifedipine 5.
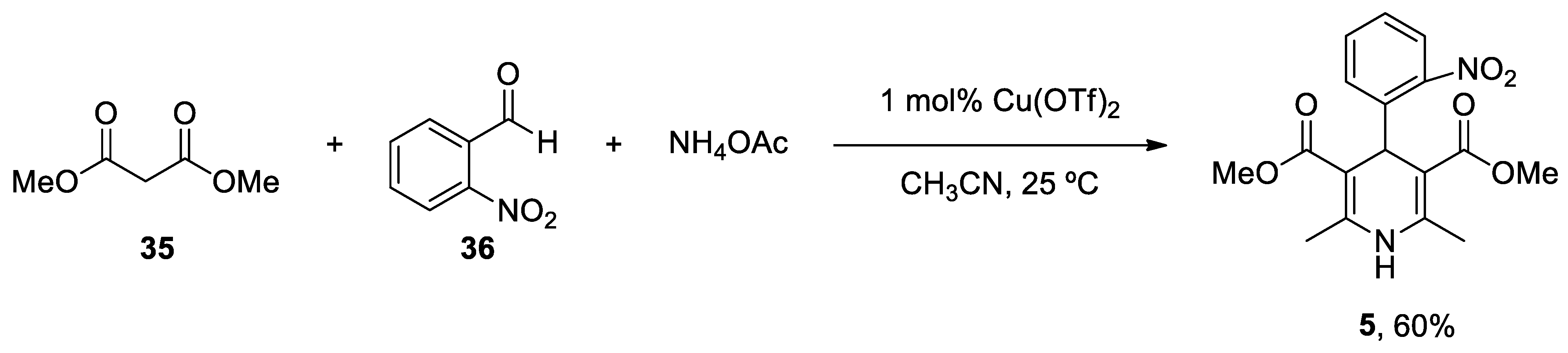
Scheme 10.
Synthesis of nifedipine 5 through a photoinduced iron-catalysed ipso-nitration via single-electron transfer.
Scheme 10.
Synthesis of nifedipine 5 through a photoinduced iron-catalysed ipso-nitration via single-electron transfer.
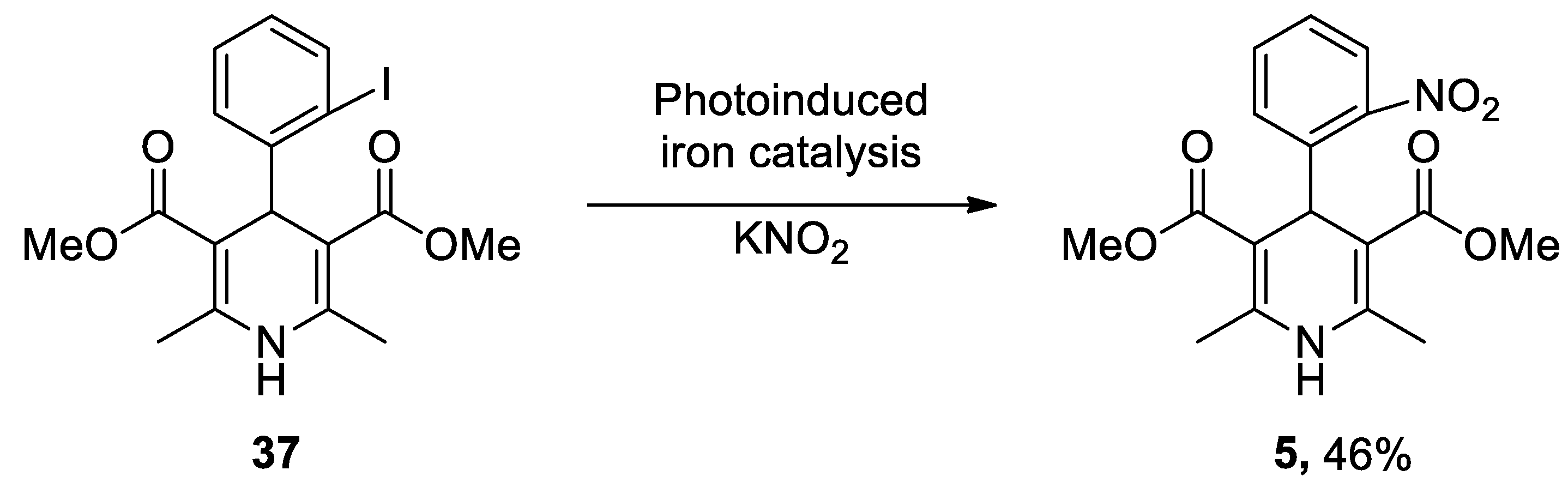
Scheme 11.
First flow multicomponent synthesis of nifedipine 5.
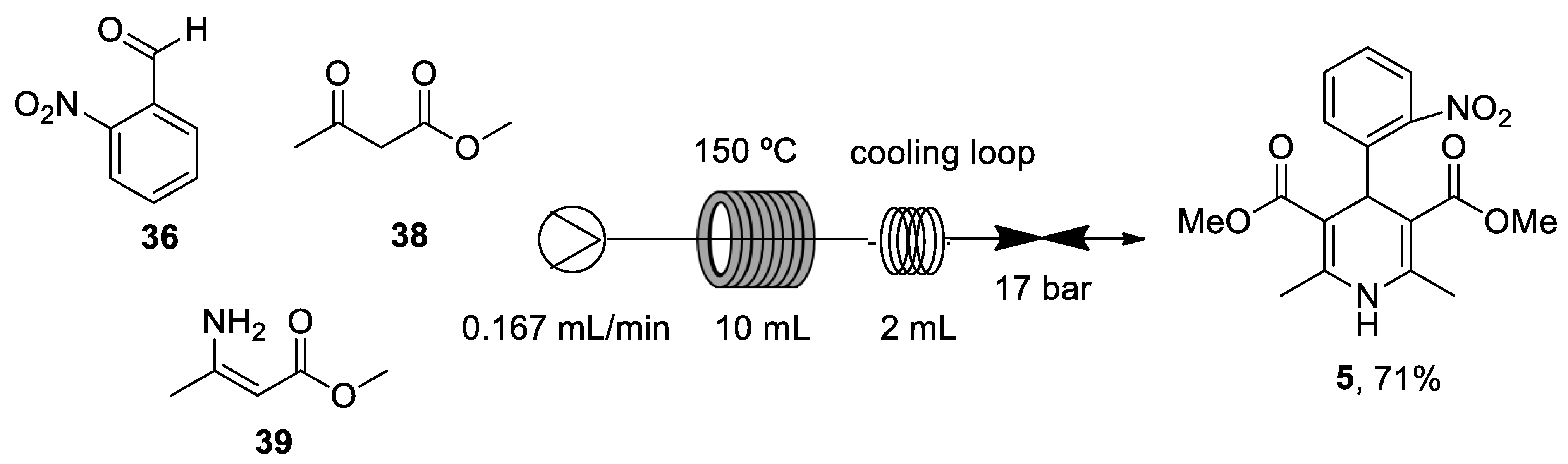
Scheme 12.
Green methodology for the synthesis of nifedipine 5 reported by Xiang et al. 2021.
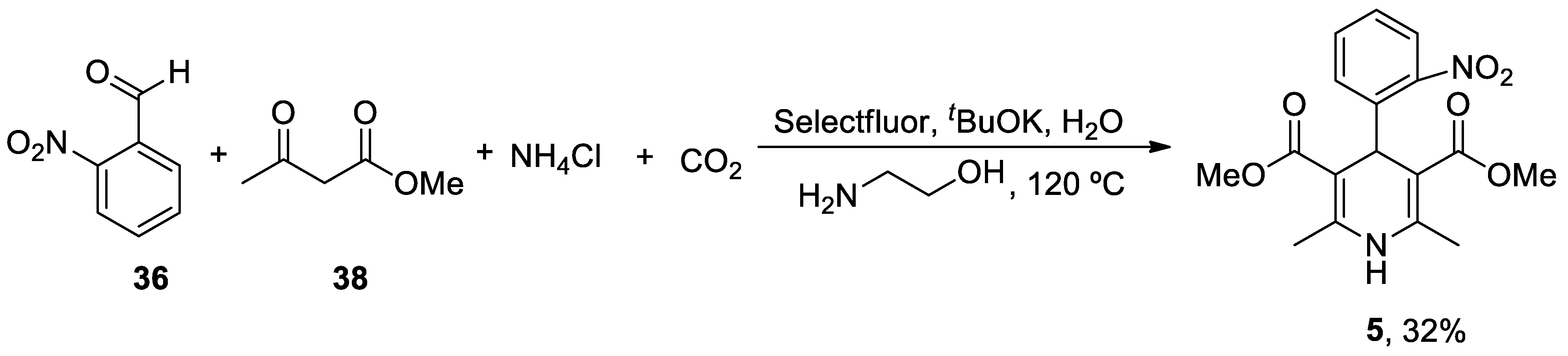
Scheme 13.
Solid-phase synthesis of nimodipine 6 reported by Gordeev.
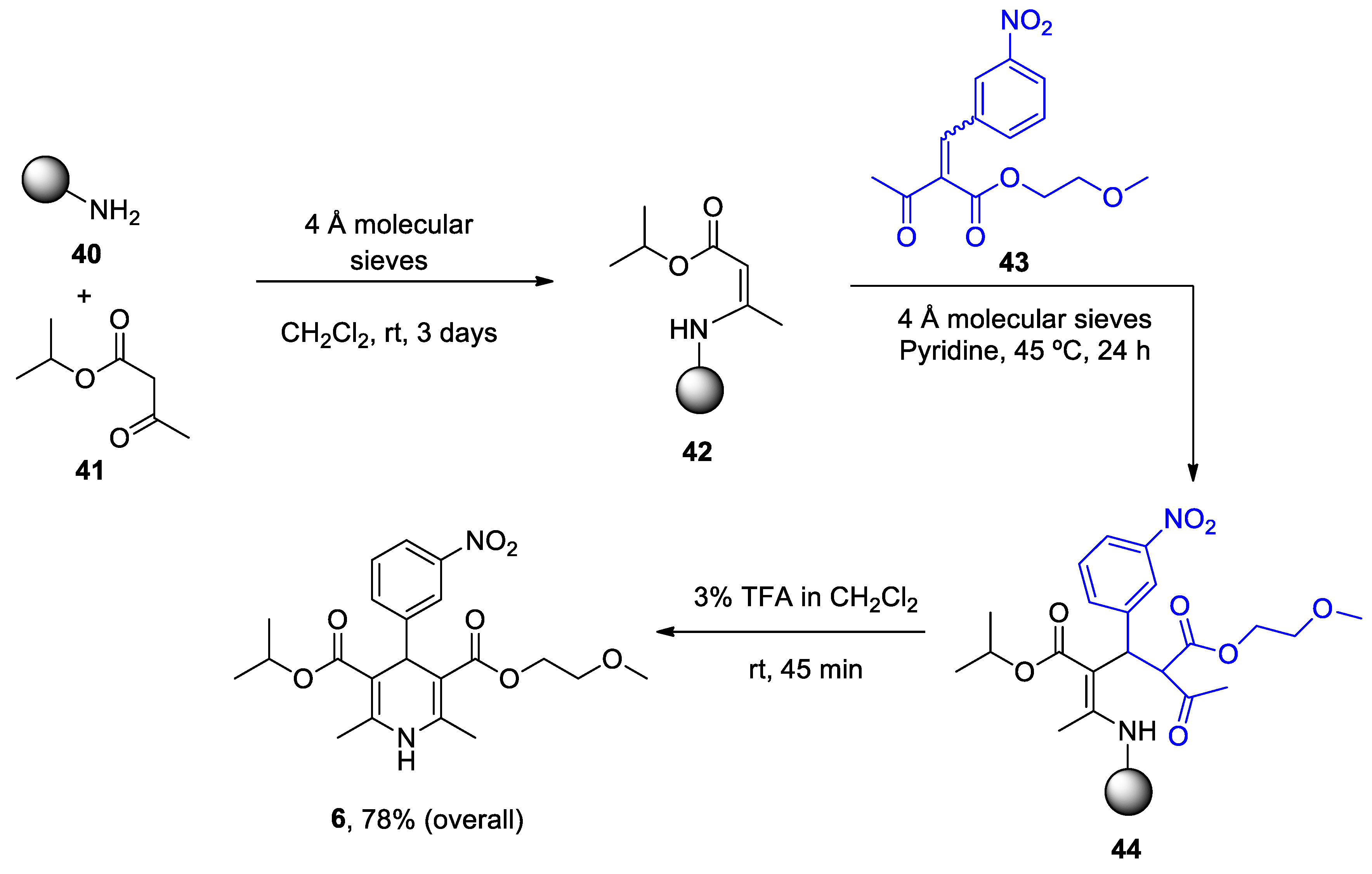
Scheme 14.
Production of nimodipine 5 in pilot batches by Pharm. Sintez. Co.
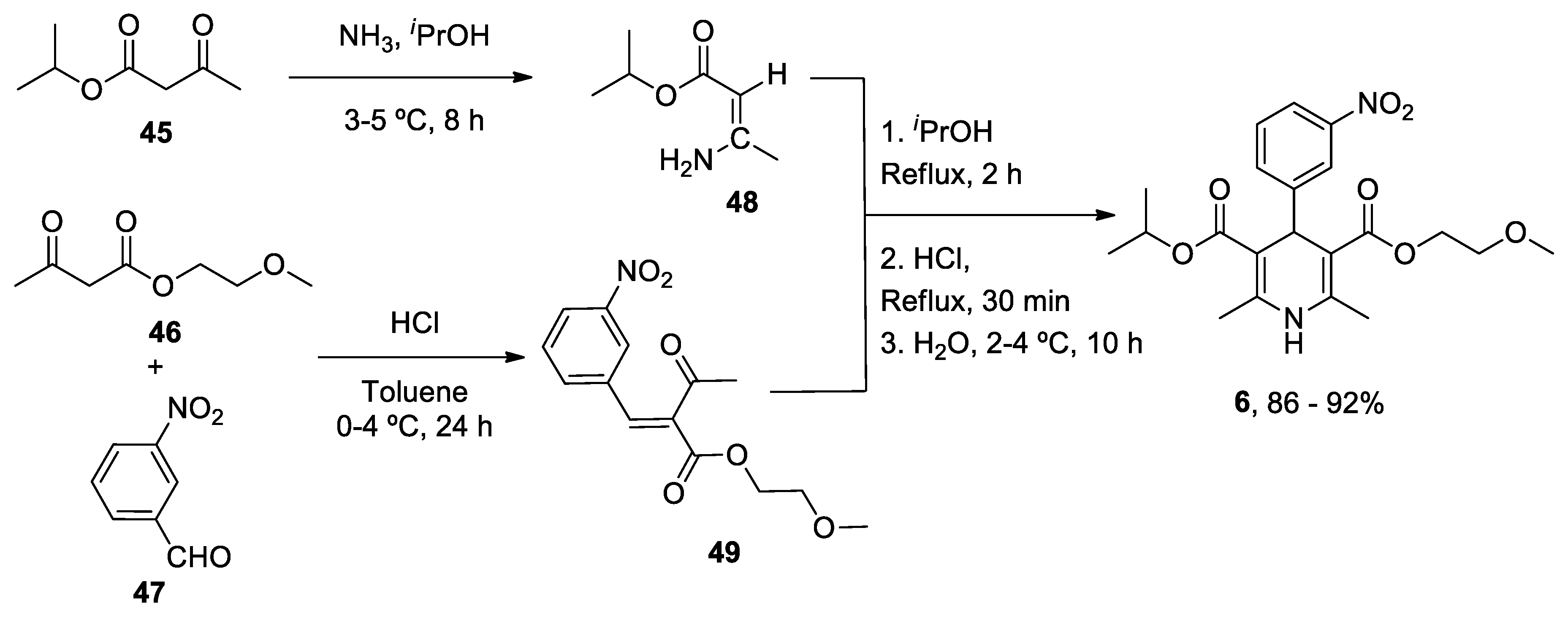

Table 1.
Summary of heterocycle compounds effects on AD and PD. Status: https://clinicaltrials.gov/.
Table 1.
Summary of heterocycle compounds effects on AD and PD. Status: https://clinicaltrials.gov/.
| Heterocyclic Compound | Chemical Name | Target | Proposed Mechanism | Effect on Neurodegenerative Diseases. Status of Clinical Trials |
|---|---|---|---|---|
|
1. ANAVEX2-73 (Blarcamesine) |
Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride |
mAChRs S1R |
|
Memory improvement and neuroprotective effects in AD and PD.
|
|
2. Caffeine (Mateine) |
1,3,7-Trimethyl-3,7-dihydro-1H-purine-2,6-dione |
A2AR RyR (+) |
|
Reduced risk of developing AD and PD Epidemiological studies: PD, possible motor benefits |
|
3. Diltiazen (Cardizem) |
(2S,3S)-5-(2-(Dimethylamino)ethyl)-2-(4-methoxyphenyl)-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]thiazepin-3-yl acetate | Non-dihydro-pyridine VOCC |
|
Reduced risk of developing AD and PD Epidemiological & Randomization Studys |
|
4. Latrepirdine (Dimebon) |
3,6-Dimethyl-9-(2-methyl-pyridyl-5)-ethyl-1,2,3,4-tetrahydro-γ-carboline dihydrochloride |
H1R Other Ca2+ Channels |
|
Cognitive and psychiatric benefit in ADImprovement in cognitive decline in HD Status:
|
|
5. Nifedipine (Procardia) |
3,5-Dimethyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate | L-Type VOCC |
|
Reducing risk developing AD and PD Epidemiological & Randomization Studies |
|
6. Nimodipine (Nimotop) |
3-isopropyl 5-(2-methoxyethyl) 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate | L-Type VOCC |
|
Reducing risk developing AD and PD Epidemiological & Randomization Studies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



