Submitted:
14 May 2026
Posted:
15 May 2026
You are already at the latest version
Abstract
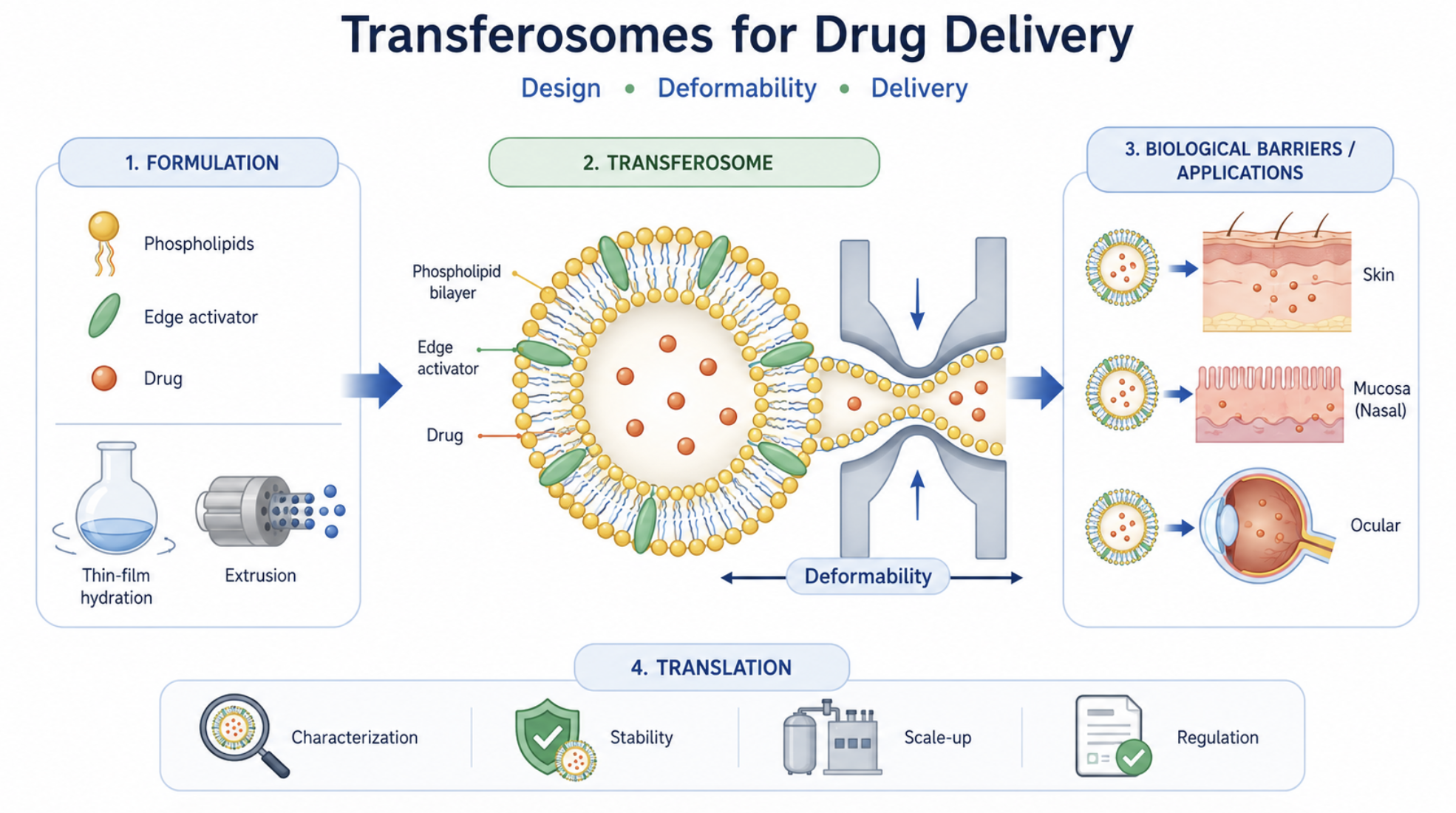
Transferosomes have emerged as one of the most extensively investigated ultradeformable vesicular systems for drug delivery, particularly for non-invasive administration across biological barriers. Their distinctive architecture, typically composed of phospholipids and edge activators, confers enhanced membrane flexibility compared with conventional liposomes, enabling improved adaptation to restrictive biological environments. Despite the growing body of literature, the field remains characterized by inconsistent terminology, heterogeneity in formulation strategies, and significant variability in characterization methods, which hinder meaningful comparison across studies and limit translational progress. This review provides a critical and integrated analysis of transferosomes, focusing on the relationship between formulation design, vesicle properties, deformability, and biological performance. The structural basis of transferosomes is examined with emphasis on the role of phospholipids, edge activators, and auxiliary components in modulating membrane organization, encapsulation behavior, colloidal stability, and drug release. Key quality attributes, including vesicle size, size distribution, surface charge, morphology, encapsulation efficiency, and physical stability, are discussed together with the main analytical approaches used for their evaluation. Deformability is addressed as the central functional feature of transferosomes, highlighting current experimental methods, sources of variability, and limitations affecting reproducibility and inter-study comparability. The interaction of transferosomes with biological barriers is critically examined, including the ongoing debate regarding intact vesicle penetration versus drug release prior to permeation. Major therapeutic applications are summarized, and transferosomes are positioned in comparison with conventional liposomes, ethosomes, and transethosomes within a context-dependent framework. Finally, key translational challenges are analyzed, including limited standardization, scalability constraints, storage instability, and regulatory uncertainty. In this context, this review establishes a structured framework linking formulation design, deformability, and biological performance, and identifies the critical parameters that must be controlled to enable reproducible, scalable, and clinically relevant transferosome-based drug delivery systems.
Keywords:
transferosomes
; ultradeformable vesicles
; drug delivery systems
; transdermal delivery
; biological barriers
; edge activators
; deformability
; lipid vesicles
; encapsulation efficiency
; nanocarriers
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



