Submitted:
29 April 2026
Posted:
06 May 2026
You are already at the latest version
Abstract
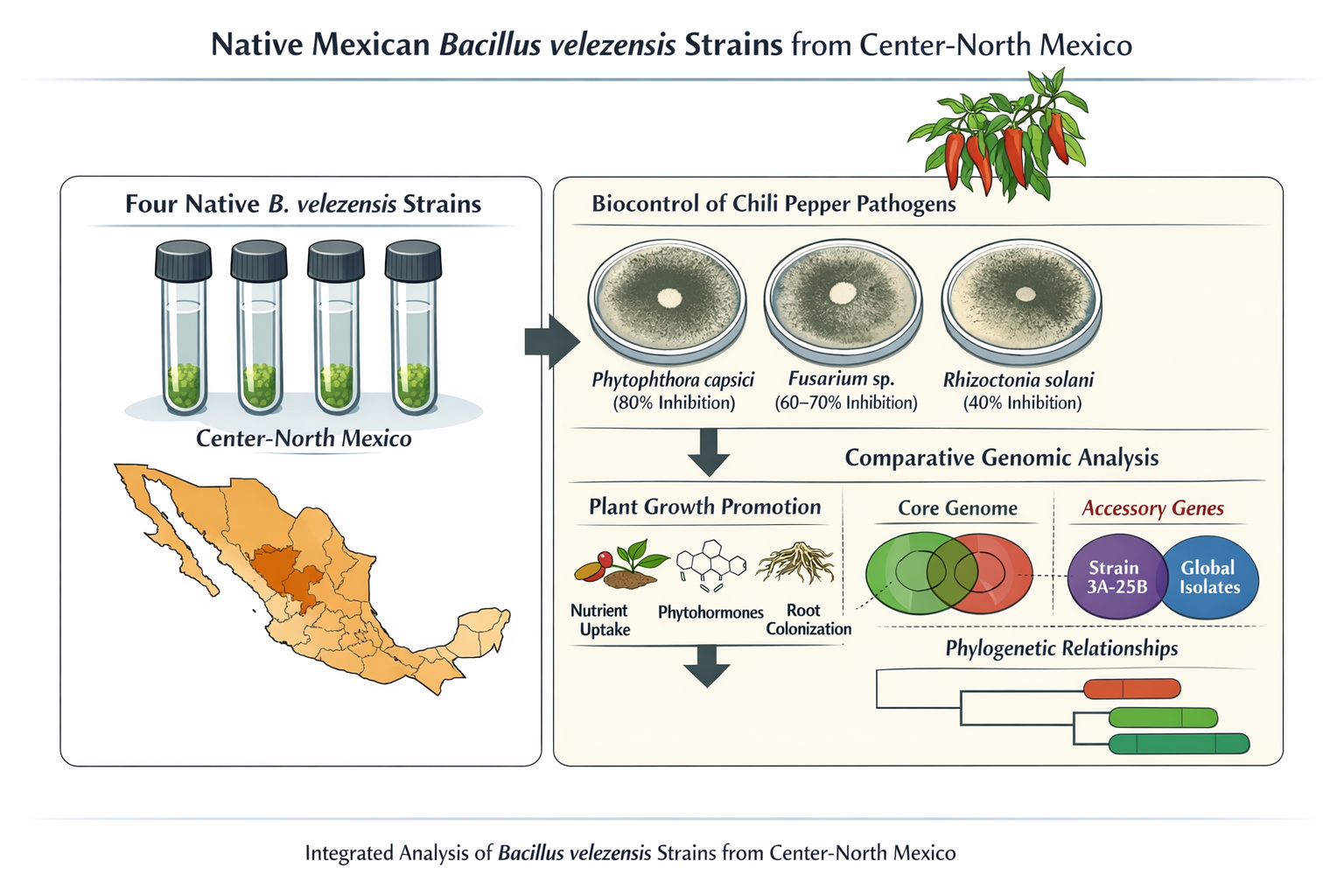
Bacillus velezensis is a rhizosphere-associated bacterium widely recognized for its roles in biological control and plant growth promotion; however, its functional diversity and evolutionary structure across scales remain incompletely understood. This study evaluated strains 2A-2B, 3A-6A, 2A-10A and 3A-25B from the Center-North of Mexico through integrated phenotypic assays and comparative genomics. Antagonistic activity was assessed via dual confrontation assays against major chili pepper phytopathogens, and plant–bacteria compatibility was examined in vitro. Genome-based analyses included pan-genome reconstruction, phylogenetic inference, and functional annotation, incorporating the screening of plant-associated genetic traits using the PLaBAse platform. All strains consistently inhibited phytopathogens (40–80%), with no significant differences among them, and displayed non-pathogenic interactions with the host plant. Genomic analyses revealed highly conserved core features alongside variation in accessory and strain-specific genes, with strain 3A-25B showing the highest divergence. Pan-genome analyses at regional and global scales indicated an open structure shaped by geography. Phylogenetically, three strains clustered together, whereas strain 3A-25B grouped with distant lineages. All genomes encoded extensive plant growth–promoting traits, while a substantial fraction of genes remained unannotated. These findings highlight functional consistency despite genomic divergence and support the ecological versatility and biotechnological potential of native B. velezensis strains.
Keywords:
rhizobacteria
; comparative genomics
; PGPR traits
; antifungal activity
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



