Submitted:
05 May 2026
Posted:
05 May 2026
You are already at the latest version
Abstract
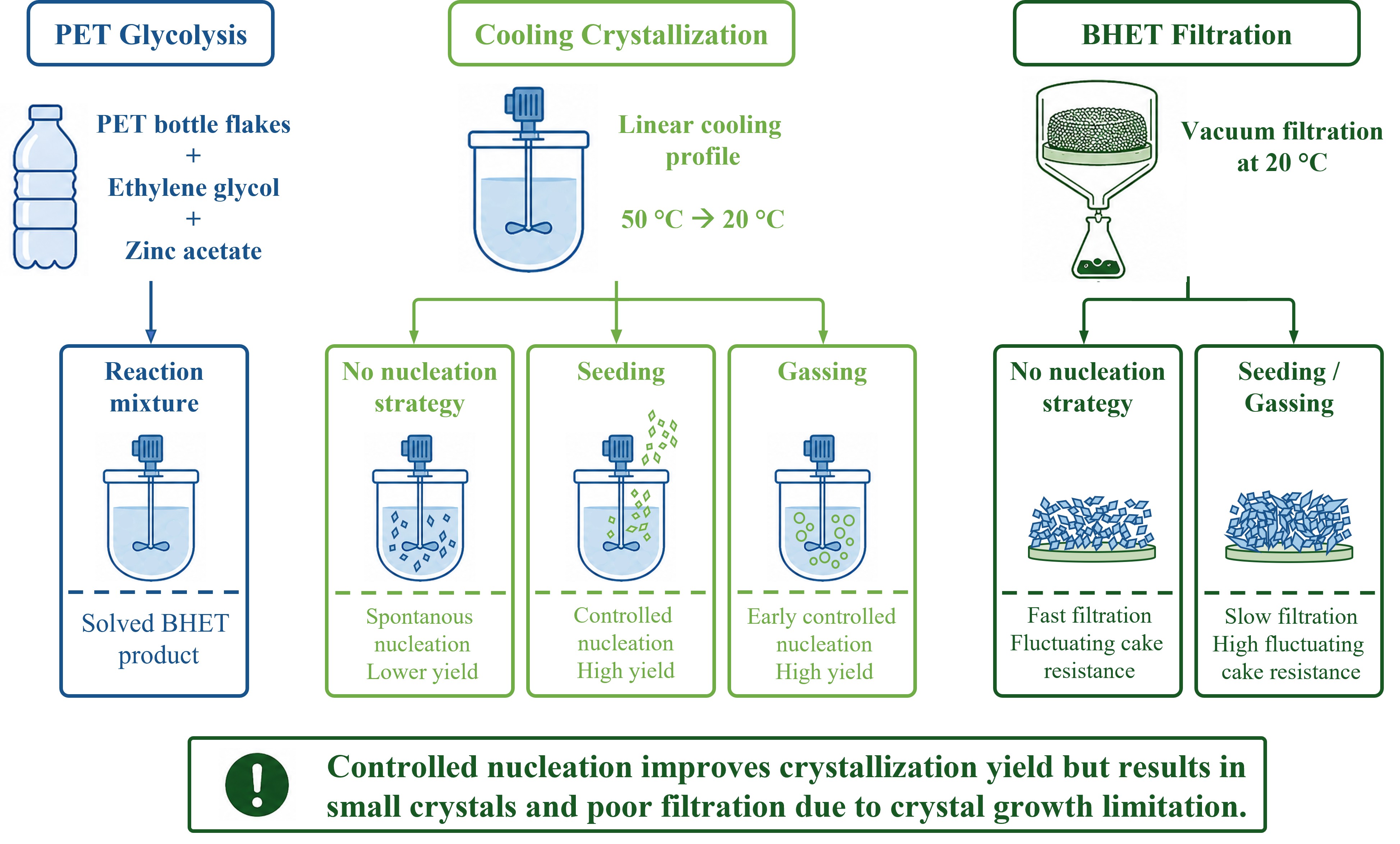
The recycling of polyethylene terephthalate (PET) is gaining increasing importance, as it enables the conversion of plastic waste into valuable raw materials and contributes to a circular economy. Recent research has primarily focused on optimizing the depolymerization step of PET glycolysis, while downstream processes often overlooking the at least equally critical downstream steps in recovering the monomer bis(2-hydroxyethyl) terephthalate (BHET). The implementation of a water‑free PET glycolysis process eliminates challenges related to internal solvent and homogeneous catalyst recycling that commonly occur in conventional processes. This study therefore focuses on BHET crystallization and filtration as key downstream unit operations. Two nucleation strategies, gassing and seeding, were investigated and compared with experiments without a nucleation strategy. The aim was to achieve reproducible process control during crystallization and to obtain crystals with good filterability, which is essential for efficient washing and high product purity. Experiments without a nucleation strategy showed poor reproducibility. In contrast, gassing and seeding improved crystallization control, particularly regarding nucleation temperature and relative crystallization yield. However, these strategies also resulted in significantly prolonged filtration times due to differences in filter cake properties. The anisotropic crystals exhibited a broad particle size distribution with a high fraction of fine particles, leading to small and heterogeneous pores in the filter cake. Limited crystal growth was identified as the main cause of the unfavorable filtration behavior.
Keywords:
glycolysis
; green chemistry
; chemical recycling
; sustainability
; material circularity
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



