Submitted:
17 April 2026
Posted:
21 April 2026
You are already at the latest version
Abstract
The gastrointestinal and pulmonary systems are severely affected in both homozygous and heterozygous cystic fibrosis (CF) patients. Cystic fibrosis (CF) is a genetic disorder affecting epithelial ion transport and multiple organ systems. While bacterial and fungal comorbidities are well studied, parasitic infections remain underexplored. This narrative review addresses an unexplored area, inspired by the common gastrointestinal symptoms observed in both CF and certain intestinal parasites. It aims to explore potential links between CF and intestinal parasitic diseases, based on clinical case reports, literature reviews, and research articles. In this study, we used various search engines, including PubMed, ScienceDirect, Elsevier, Wiley, ResearchGate, and Google Scholar. This was constructed by employing keywords such as Cystic fibrosis, CFTR, cystic fibrosis associated with pathogens, intestinal inflammation, bowel disease, intestinal protozoa, and helminthic infections. Once similar symptoms and molecular/immune mechanisms were detected, our keywords extended to include specific parasites associated with CF and/or CFTR, as well as chronic lung inflammation, pancreatitis, or diabetes. This review also examines immunomodulation in CF, with or without parasitic infection, and the influence of CF on predisposition to parasitic infection, diagnosis, or treatment. In a nutshell, we found only a few case reports on CF-protozoan infection comorbidities in non-Middle Eastern countries, and even those are dated two decades ago. This indicates the underestimation of the possibility of CF-comorbidity, which might be life-threatening. Hence, CF diagnosis in patients with intestinal parasitic infections, family pedigree reviews before treatment, and in vivo/ex vivo research studies are recommended.
Keywords:
cystic fibrosis co-morbidity
; protozoa
; helminths
; CFTR
; immune dysfunction
; gastrointestinal parasitic infection
1. Introduction
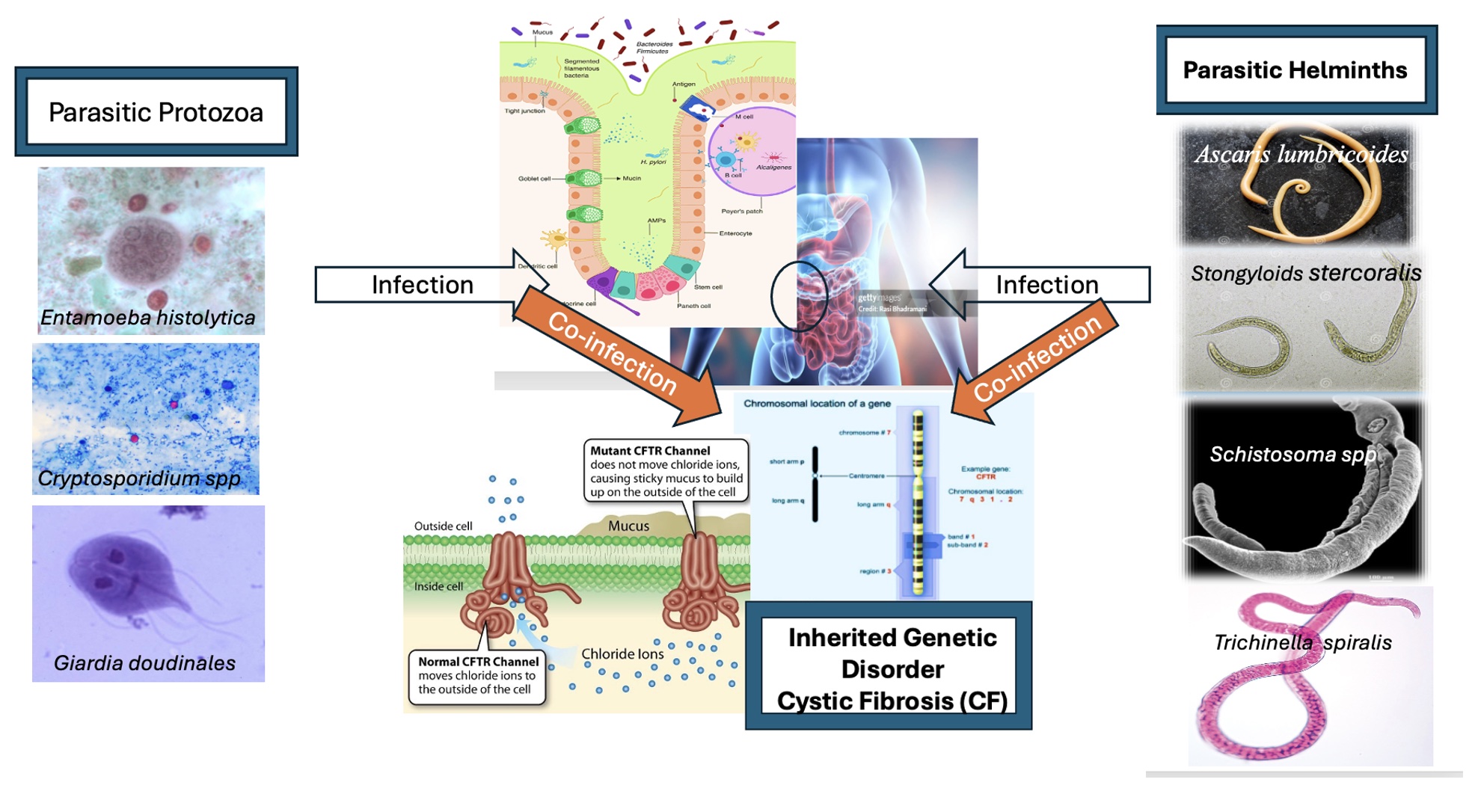
Cystic fibrosis (CF) is among the most common life-shortening genetic disorders in Europe, affecting approximately 30% of live births [1]. Recently, global mapping data have identified CF in many countries across Asia and Africa [2,3]. The disease results from mutations in the CFTR gene, located on chromosome 7. This gene encodes a chloride channel regulated by cyclic AMP (cAMP)-dependent phosphorylation, which is highly expressed on the apical surface of lung and intestinal epithelial cells, pancreatic ductal cells, cholangiocytes, and the reproductive tract [4,5,6]. Dysfunction of CFTR in the lungs leads to dehydrated mucus, impaired mucociliary clearance, chronic bacterial colonisation such as Pseudomonas aeruginosa, and progressive tissue damage [6]. In the gastrointestinal (GI) tract, CF symptoms include meconium ileus, pancreatic insufficiency, malabsorption, distal intestinal obstruction syndrome (DIOS), and changes in intestinal microbiota [7,8,9,10,11]. The bidirectional communication within the gut–lung axis in cystic fibrosis (CF) presents a complex pathophysiological paradigm, as the microbial ecosystems of both anatomical sites are fundamentally compromised by the same underlying aetiology, which is the loss of CFTR protein function. Although the resulting dysbiosis in the gastrointestinal and respiratory tracts develops somewhat independently—driven by distinct, organ-specific microenvironments characterized by highly viscous mucus and hyperinflammation—the two microbiomes remain highly interconnected [12]. Most notably, the composition of the pulmonary microbiome is heavily influenced by the intestinal microbiome through the systemic exchange of microbial metabolites [13]. In the context of CF, the gut–lung axis is further impaired by a marked reduction in microbial populations capable of synthesizing short-chain fatty acids (SCFAs) [14,15]. Because SCFAs possess critical immunomodulatory properties, this intestinal dysbiosis is directly correlated with defects in pulmonary immune homeostasis [16,17]. The profound interdependence of these mucosal microbiotas has been corroborated in vivo using a murine model of CF; specifically, Bazett and his colleagues [16] demonstrated that antibiotic-induced disruption of the intestinal microbiota precipitates pulmonary hyper-reactivity. Consequently, the depletion of gut microbial diversity and its associated functional capacity—often a result of recurrent antibiotic therapies initiated in early childhood—may be a significant exacerbating factor in the progression of pulmonary disease among patients with CF [17].
Despite extensive research on bacterial and fungal infections in CF [18], the relationship between CF and parasitic infections, particularly protozoa and helminths, remains poorly comprehended and underreported. Parasitic diseases, including intestinal protozoa and soil-transmitted helminths, affect over 18% of people worldwide, mainly in tropical and subtropical regions [19]. Due to the limited number of comprehensive studies on CF and intestinal parasite co-infections, this review adopts a narrative, non-systematic approach. Intestinal parasitic infections discussed in this review are those which meet one or more of the following criteria: (i) reported or plausible clinical relevance to CF patients; (ii) the availability of peer-reviewed publications that enable meaningful synthesis of clinical or pathophysiological insights; and (iii) the representation of pathogens that are clinically significant or likely to influence the course of disease in CF. It is worth noting that the global burden of parasitic infections, especially in low- and middle-income countries, may lead to underdiagnosis of CF, whilst CF-parasitic comorbidity is rarely addressed in high-income countries with advanced hygiene standards. With increased global mobility and longer life expectancy among CF patients due to modern treatments like CFTR modulators, clinicians now encounter CF patients in regions where parasitic infections are prevalent and receive referrals from these areas [20,21,22]. This review focuses on clinically significant infections worldwide to provide a balanced, comprehensive, and evidence-based overview of current knowledge, while highlighting gaps and areas for further research.
2. Pathophysiological Basis of Cystic Fibrosis and Mucosal Immunity
Over 2,500 CFTR variants have been classified into six functional groups. Mutations in classes I–III and VI typically result in little to no functional protein at the cell surface and are associated with severe disease, whereas classes IV and V retain partial channel activity and generally produce milder clinical phenotypes [16]. Under physiological conditions, CFTR regulates chloride and bicarbonate levels through Na+-derived Cl-/HCO3- and H+ ions transport across epithelial membranes, thereby maintaining fluid balance and optimal luminal pH (Figure 1). It also modulates pH changes induced by glucose metabolism in pancreatic islet β-cells [16,17]. CFTR dysfunction results in the most prevalent pulmonary disease observed in adulthood and impacts intestinal function across the individual’s entire lifespan [23]. It also modulates pH changes induced by glucose metabolism in pancreatic islet β-cells [23,24]. Mutated CFTR mediates CF, the most common lung disease in adulthood, which affects the intestine throughout a patient’s lifetime [25]. In the lungs, CFTR mutations mechanistically cause intracellular chloride accumulation and increased sodium absorption, water withdrawal from the airway surface, dehydration of the liquid layer, and mucus thickening [26,27,28]. This impairs mucociliary clearance, facilitating chronic pathogen colonisation and recurrent pulmonary exacerbations [29]. CF is associated with a dysregulated immune profile characterized by excessive neutrophilic inflammation, elevated pro-inflammatory cytokines [30,31], and a shift toward Th2- and Th17-mediated responses, particularly during exacerbations and direct bacterial (P. aeruginosa) infection [32]. Some studies have also revealed altered macrophage responses, impaired dendritic cell function, and reduced T-regulatory (Treg) cell activity in cystic fibrosis models [31,32]. These immune deviations may influence the host responses to intestinal parasitic infections in CF patients. In the gastrointestinal tract, CFTR gene expression on the apical surface of epithelial cells plays a vital role in regulating water and salt flow into the lumen, thereby maintaining the balance between absorption and secretion of intestinal fluids [33]. CFTR mutations have been suggested to lead to reduced luminal hydration, impaired bicarbonate secretion, altered Cl- secretion, intestinal pH, and mucus concentration [25,34,35,36,37]; similar to their aforementioned impacts on the pulmonary airways. As a consequence, gut microbiome disturbance, bacterial overgrowth, and alterations in host defence against enteric pathogens were suggested [38,39]
3. Pathobiological Basis of Major Human Intestinal Protozoa and Mucosal Immunity
Protozoan parasites exemplify a significant threat to human health, frequently colonising the gastrointestinal tract and resulting in severe clinical manifestations. Among the most clinically relevant species are Giardia lamblia, Entamoeba histolytica, and Cryptosporidium parvum, each associated with distinct pathogenic mechanisms [40]. While both C. parvum and G. lamblia inhabit the small intestine—specifically the duodenum, jejunum, and ileum—and are recognised as the leading etiologies of chronic diarrhoea (cryptosporidiosis and giardiasis, respectively) [41] , E. histolytica exhibits a distinct pathogenic profile [39]. This organism primarily colonises the large intestine and can penetrate the colonic mucosa [42]. Such invasion facilitates systemic dissemination to extraintestinal sites, most notably the liver, potentially leading to fatal amebiasis [42,43]. The three protozoa share common symptoms such as persistent diarrhea, abdominal pain and severe malabsorption [41,44]. Their foundational pathophysiology is driven by disease mechanisms that actively increase intestinal permeability and trigger chloride secretion [45].
At the cellular level, pathogen attachment and colonization induce host epithelial apoptosis, the physical rupture of critical epithelial tight junctions [46], and the shortening of brush-border microvilli [44,47], which directly causes deficiencies in disaccharidases and other essential digestive enzymes [48,49,50]. Molecularly, these parasites utilize virulence factors such as cysteine proteases, which degrade host immunoglobulins and chemokines, deplete protective intestinal mucus and drive microbiome dysbiosis to support their survival [51,52]. The host defense system is triggered to combat these parasitic protozoa, through activation of innate and adaptive immune pathways, beginning with the localized activation of CD8+ lymphocytes [53], and the stimulation of tuft cells by parasite-excreted metabolites [54]. This cascades into an adaptive immune response mediated by IL-6-producing dendritic and mast cells [55], alongside CD4+ T-cells that release pro-inflammatory cytokines such as IL-17 and TNF-alpha [56]. Ultimately, the pathogen’s proliferation is suppressed through clearance mechanisms, including the mucosal release of cytotoxic secretory IgA [57], and nitric oxide (NO) generated by both epithelial and immune cells [58,59,60], while localized mast cell degranulation enhances intestinal peristalsis to physically expel the pathogens [39].
4. Is There a Correlation Between CF and Intestinal parasites?
The symptoms of G. lamblia infection may overlap with GI manifestations of CF [6,7,8,9,10], making diagnosis challenging. G. duodenalis pathophysiology was studied in a human organoid system and proved to down-regulate the CFTR-mediated chloride secretion early [60], which is synergistic with the absence or impairment of CFTR-mediated Cl-/HCO-3 exchanger in CF patients [37]. Therefore, absence or dysfunction of CFTR may theoretically increase the susceptibility and long-term infection of G. lamblia. Several studies [61,62,63]found that the prevalence of giardiasis was higher in patients with CF or chronic pancreatitis than in control groups. A 20-year-old male CF patient, who was on a continuous treatment course of pancreatic enzyme and vitamin supplements, lost weight and underwent severe malabsorption due to an infection with Giardia. These symptoms recovered after a 5-day treatment course of an anti-giardiasis drug [61]. It is indicated that giardiasis reduced the efficacy of supplements in CF patients and worsened malabsorption [61]. It seems that CF treatment with pancreatic enzymes and vitamin supplements [64]is less effective than human duodenal and upper jejunal fluid, which has been shown to kill G. lamblia in vitro [65]. Altogether, altered intestinal pH in CF, along with mucus viscosity and reduced luminal flow, may facilitate Giardia adherence and persistence. In vitro studies suggest that Giardia may exploit host mucins for attachment [60], a mechanism that may be enhanced in CF due to mucin hypersecretion [37]. Giardia was also shown to activate the AKT pathways, destabilising glucose and insulin levels and worsening the “gut-metabolism” cycle [66]. Furthermore, Giardia was experimentally demonstrated to produce specialised thiol proteinases that degrade intestinal tight junctions, causing fluid leakage [66]. Therefore, a co-infection with CF may result in an accelerated “leaky gut” phenotype, further promoting systemic inflammation.
Although Cryptosporidium spp. is one of the most significant pathogens causing waterborne outbreaks [40,45] and is life-threatening to immunocompromised individuals [66,67,68], its prevalence and impact in cystic fibrosis (CF) patients remain relatively underreported. Cryptosporidium spp. are coccidian protozoa that reside intracellularly within a parasitophorous vacuole, located between the cell membrane and the cytoplasm, causing gastroenteric diseases characterised by watery diarrhoea [69,70,71]. Furthermore, Cryptosporidium spp. can cause respiratory infection through transmission by coughing and respiratory secretions via inhalation of aerosolised droplets [72]. In our investigation, we found only one study [73] on Google Scholar indicating co-infection with Cryptosporidium in a CF patient with a bilateral lung transplant. He experienced severe sepsis, renal failure, diarrhoea, and electrolyte disturbance caused by Cryptosporidium [73]. The authors suggested that lung transplant recipients, including those with CF, are at an elevated risk for severe, life-threatening cryptosporidiosis due to the intensive immunosuppressive regimens required to prevent allograft rejection [73]. Interestingly, Cryptosporidium was also shown to express a structure similar to CFTR, a 200-kDa integral protein localised at the host-parasite interface [74]. In this case, we assume that even in the absence or dysfunction of CFTR, the parasite still has a feeder organelle and can disseminate. Despite the severity of amoebic colitis [75], liver cirrhosis [75], and microbiome degradation [76,77,78], we have not found any case report or experimental model describing a comorbidity with CF.
5. Research Gaps
CF is underdiagnosed in low- and middle-income countries (LMICs), where parasitic infections are endemic, and misdiagnosis of CF as “chronic malnutrition” or “recurrent pneumonia” may delay genetic testing (Figure 2). Simultaneously, parasitic infections in undiagnosed CF patients could be fatal. The key research gaps include a lack of documentation on the prevalence of protozoa and helminthic parasitic infections in CF cohorts globally, the impact of parasites on CFTR modulator efficacy and pharmacokinetics, and mechanistic studies of parasite-CFTR interactions using organoid or murine models. Furthermore, there is no evidence regarding the long-term outcomes of co-infection to indicate whether parasitism acts synergistically or antagonistically in CF patients.
6. Conclusion
Parasitic infections constitute an underappreciated aspect of cystic fibrosis (CF) care, particularly in regions where such pathogens are endemic. Incorporating parasitological considerations into diagnostic and management frameworks may enhance clinical outcomes. Accordingly, future research should emphasize mechanistic investigations, strengthened epidemiological surveillance, and the development of region-specific clinical guidelines. Although bacterial and fungal infections predominate in the CF literature, protozoan and helminthic diseases remain underrecognized comorbidities with important global clinical implications. The altered epithelial physiology, immune dysregulation, and gastrointestinal dysfunction characteristic of CF may increase susceptibility to parasitic infections or modulate disease severity. Conversely, chronic parasitic infections may influence CF-associated inflammation and responsiveness to therapy. Case reports underscore diagnostic complexities and potential therapeutic risks, particularly in immunocompromised individuals with helminth infections. As life expectancy in CF continues to improve and global health disparities persist, the integration of parasitology into CF care through targeted screening, clinician education, and focused research is increasingly warranted. Advancing understanding of these interactions may inform novel therapeutic approaches and ultimately improve outcomes for individuals with CF worldwide.
Funding
This research received no specific grant from any funding agency.
Conflicts of Interest
The authors declare no conflict of interest.
References
- CFF. Annual report. 2023. Available online: https://www.cff.org/about-us/2023-annual-report.
- Abubakar Bobbo, K.; Ahmad, U.; Chau, D.M.; Nordin, N.; Abdullah, S. A comprehensive review of cystic fibrosis in Africa and Asia. Saudi J Biol Sci. 2023, 30(7), 103685. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- El Makhzen, N.; Daimi, H.; Bouguenouch, L.; Abriel, H. The burden of cystic fibrosis in North Africa. Front Genet. 2024, 14, 1295008. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science Erratum in: Science 1989 Sep 29;245(4925):1437. PMID: 2475911. 1989, 245(4922), 1066–73. [Google Scholar] [CrossRef]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N Engl J Med 2005, 352(19), 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015, 16(1), 45–56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013, 3(9), a009753. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yibcharoenporn, C.; Kongkaew, T.; Worakajit, N.; Khumjiang, R.; Saetang, P.; Satitsri, S.; Rukachaisirikul, V.; Muanprasat, C. Inhibition of CFTR-mediated intestinal chloride secretion by nornidulin: Cellular mechanisms and anti-secretory efficacy in human intestinal epithelial cells and human colonoids. PLoS One. 2024, 19(12), e0314723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Esposito, S.; De Palma, F.D.E.; Cernera, G.; Zarrilli, F.; Scialò, F.; Maiuri, M.C.; Amato, F.; Castaldo, G.; Villella, V.R. Mouse intestine as a useful model for CFTR electrophysiology function analysis. Methods Cell Biol. 2025, 197, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Curley, C.E.; Mobbs, C.L.; Mroz, M.S.; McLean, M.H.; Keely, S.J. The secondary bile acid, lithocholic acid, inhibits cystic fibrosis transmembrane conductance regulator expression and activity in colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2026, 330(2), G110–G122. [Google Scholar] [CrossRef]
- Ballout, J.; Waeber, N.B.; Diener, M. Ionic dependency, sensitivity to transport inhibitors and signal transduction of propionate-induced anion secretion in rat caecum. Eur J Pharmacol. 2026, 30;1013, 178511. [Google Scholar] [CrossRef] [PubMed]
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes (Basel) 2020, 11(5), 536. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marsland, B.J.; Gollwitzer, E.S. Host–microorganism interactions in lung diseases. Nat. Rev Immunol. 2014, 14, 827–835. [Google Scholar] [CrossRef]
- Anand, S.; Mande, S.S. Diet, microbiota and gut-lung connection. Front. Microbiol. 2018, 9, 2147. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Wang, N.; Tan, H.-Y.; Zhang, Z.; Feng, Y. The cross-talk between gut microbiota and lungs in common lung diseases. Front. Microbiol. 2020, 11, 301. [Google Scholar] [CrossRef]
- Bazett, M.; Bergeron, M.-E.; Haston, C.K. Streptomycin treatment alters the intestinal microbiome, pulmonary T cell profile and airway hyperresponsiveness in a cystic fibrosis mouse model. Sci. Rep. 2016, 6, 19189. [Google Scholar] [CrossRef]
- Nielsen, S.; Needham, B.; Leach, S.T.; Day, A.S.; Ja e, A.; Thomas, T.; Ooi, C.Y. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. Sci. Rep. 2016, 6, 24857. [Google Scholar] [CrossRef]
- Stanton, B.A.; Coutermarsh, B.; Barnaby, R.; Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PLOS ONE 2015, 10(5), e0127742. [Google Scholar] [CrossRef]
- World Health Organisation (WHO). Global report on neglected tropical diseases, 2025; ISBN 978-92-4-011404-3 (electronic version) ISBN 978-92-4-011405-0 (print version).
- Álvarez Fernández, A.; Traversi, L.; Polverino, E. The ageing of Cystic Fibrosis patients with new modulators: current gaps and challenges. Expert Review of Respiratory Medicine 2023, 17(12), 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Kley, A.C.; White, A.C., Jr. Parasitic Infections in Pulmonary and ICU Patients: Presentation, Diagnosis, and Treatment. Chest 2025, 167(3), 686–693. [Google Scholar] [CrossRef] [PubMed]
- Felipe Montiel, A.; Parisi, G.F.; Papale, M.; Pecora, G.; Presti, S.; Tosto, M.; Mulé, E.; Ornato, V.; Aloisio, D.; Leonardi, S. Evolving Cystic Fibrosis Care: Lung Immunology and Emerging Health Challenges in the Era of CFTR Modulators. Biomolecules 2025, 15(10), 1460. [Google Scholar] [CrossRef]
- Slae, M.; Wilschanski, M. Cystic fibrosis and the gut. Frontline Gastroenterol 2020, 12(7), 622–628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, H.; Antinozzi, P.A.; Hagenfeldt, K.A.; Maechler, P.; Wollheim, C.B. Molecular targets of a human HNF1α mutation responsible for pancreatic β-cell dysfunction. The EMBO Journal 2000, 19(16), 4257–64. [Google Scholar] [CrossRef]
- Scull CE, Luo M, Jennings S, Taylor CM, & Wang G. Cftr deletion in mouse epithelial and immune cells differentially influences the intestinal microbiota. Communications Biology 2022, 5(1), 1130. [CrossRef]
- Delpire, E. Advances in the development of novel compounds targeting cation-chloride cotransporter physiology. American Journal of Physiology-Cell Physiology 2021, 320(3), C324–C340. [Google Scholar] [CrossRef]
- Alberini G, Alexis Paz S, Corradi B, Abrams CF, Benfenati F, & Maragliano L. Molecular dynamics simulations of ion permeation in human voltage-gated sodium channels. Journal of Chemical Theory and Computation 2023, 19(10), 2953–2972. [CrossRef]
- Castagna A, Mango G, Martinelli N, Marzano L, Moruzzi S, Friso S, & Pizzolo F. Sodium chloride cotransporter in hypertension. Biomedicines 2024, 12(11), 2580. [CrossRef]
- Aksel-Uylar, A.A.; Topcuoglu, C.; Saglam, M.; Vardar-Yagli, N. Effects of inspiratory muscle training on physical fitness in cystic fibrosis: a systematic review and meta-analysis. BMC Sports Sci Med Rehabil. 2025, 17(1), 384. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aziz, W.; Sultana, H.; Kumar, V.; Tyagi, A. The Relationship Between NETosis and Biofilm Formation in Chronic Infections. Biomolecules 2025, 15(12), 1692. [Google Scholar] [CrossRef]
- Bateman, G.; Guo-Parke, H.; Harvey, C.; Rodgers, A.; Krasnodembskaya, A.; Linden, D.; et al. Bronchial epithelial cell-derived extracellular vesicles drive inflammasome activation and NTHi infection in COPD. Front Immunol. 2026, 2;16, 1713012. [Google Scholar] [CrossRef]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2013, 187(6), 621–9. [Google Scholar] [CrossRef] [PubMed]
- Scull, C.E.; Hu, Y.; Jennings, S.; Wang, G. Normalization of Cystic Fibrosis Immune System Reverses Intestinal Neutrophilic Inflammation and Significantly Improves the Survival of Cystic Fibrosis Mice. Cell Mol Gastroenterol Hepatol. 2025, 19(2), 101424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goswami, D.; Gabriel, S.E.; Cotton, C.U. Bicarbonate and CFTR: Implications for cystic fibrosis. Journal of Cystic Fibrosis 2017, 16 (Suppl 2), S45–S51. [Google Scholar] [CrossRef]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol. 2018, 19(6), 758–767. [Google Scholar] [CrossRef]
- Lazzarotto, E.S.; Vasco, J.F.M.; Führ, F.; Riedi, C.A.; Filho, N.A.R. Systematic review on fecal calprotectin in cystic fibrosis. J Pediatr (Rio J). 2023, 99(1), 4–10. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Reid, E.K.; Pohl, J.F.; Yuzyuk, T.K.; Padula, L.M.; Vavrina, K.; Altman, K. Cystic fibrosis and fat malabsorption: Pathophysiology of the cystic fibrosis gastrointestinal tract and the impact of highly effective CFTR modulator therapy. Nutr Clin Pract. 2024, 39 Suppl 1, S57–S77. [Google Scholar] [CrossRef] [PubMed]
- Haack, A.; Aragão, G.G.; Novaes, M.R. Pathophysiology of cystic fibrosis and drugs used in associated digestive tract diseases. World J Gastroenterol. 2013, 19(46), 8552–61. [Google Scholar] [CrossRef]
- Hemphill, A.; Müller, N.; Müller, J. Comparative Pathobiology of the Intestinal Protozoan Parasites Giardia lamblia, Entamoeba histolytica, and Cryptosporidium parvum. Pathogens. 2019, 8(3), 116. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chabe, M.; Lokmer, A.; Segurel, L. Gut protozoa: Friends or foes of the human gut microbiota? Trends Parasitol. 2017, 33, 925–934. [Google Scholar] [CrossRef]
- Huang, D.B.; White, A.C. An updated review on Cryptosporidium and Giardia. Gastroenterol. Clin. North Am. 2006, 35, 291–314. [Google Scholar] [CrossRef]
- Nakada-Tsukui, Kumiko; Nozaki, Tomoyoshi. Immune Response of Amebiasis and Immune Evasion by Entamoeba histolytica. Frontiers in Immunology 2016, 7. [Google Scholar] [CrossRef]
- Gonzalez-Ruiz, A.; Wright, S.G. Disparate amoebae. Lancet 1998, 351, 1672–1673. [Google Scholar] [CrossRef]
- Van Voorhis, W. Protozoan infections. Sci. Am. Med. 2014. [Google Scholar] [CrossRef]
- Checkley, W.; White, A.C., Jr.; Jaganath, D.; Arrowood, M.J.; Chalmers, R.M.; Chen, X.M.; et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet. Infect. Dis. 2015, 15, 85–94. [Google Scholar] [CrossRef]
- Koh, W.H.; Geurden, T.; Paget, T.; O’Handley, R.; Steuart, R.F.; Thompson, R.C.; Buret, A.G. Giardia duodenalis assemblage-specific induction of apoptosis and tight junction disruption in human intestinal epithelial cells: Effects of mixed infections. J. Parasitol. 2013, 99, 353–358. [Google Scholar] [CrossRef]
- Cotton, J.A.; Beatty, J.K.; Buret, A.G. Host parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 2011, 41, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.G.; Logan, M.R.; Klammer, G.M.; Teoh, D.A.; Buret, A.G. Jejunal brush border microvillous alterations in Giardia muris-infected mice: Role of T lymphocytes and interleukin-6. Infect. Immun. 2000, 68, 3412–3418. [Google Scholar] [CrossRef]
- Dunn, N.; Juergens, A.L. Giardiasis: Diagnosis, Pathogenesis and Treatment. StatPearls / International Journal of Health & Medical Research 2024, 3(6). [Google Scholar]
- Ribeiro, M.R.S.; Carvalho, C.F.; Gomes, M.A.; Cassali, G.D.; Oliveira, D.R. Fructooligosaccharides (FOS) intake modulates oxidative stress and restores gut morphology in Giardia lamblia-infected gerbils. An Acad Bras Cienc. 2025, 97(4), e20250346. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.Y.; Singer, S.M. The Intersection of Immune Responses, Microbiota, and Pathogenesis in Giardiasis. Trends Parasitol. 2017, 33, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Allain, T.; Fekete, E.; Buret, A.G. Giardia cysteine proteases: The teeth behind the smile. Trends Parasitol. 2019, 35, 636–648. [Google Scholar] [CrossRef]
- Cotton, J.A.; Beatty, J.K.; Buret, A.G. Host-parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 2011, 41, 925–933. [Google Scholar] [CrossRef]
- Schneider, C.; O’Leary, C.E.; von Moltke, J.; Liang, H.E.; Ang, Q.Y.; Turnbaugh, P.J.; et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 2018, 174, 271–284. [Google Scholar] [CrossRef]
- Kamda, J.D.; Nash, T.E.; Singer, S.M. Giardia duodenalis: Dendritic cell defects in IL-6-deficient mice contribute to susceptibility to intestinal infection. Exp. Parasitol. 2012, 130, 288–291. [Google Scholar] [CrossRef]
- Saghaug, C.S.; Sornes, S.; Peirasmaki, D.; Svard, S.; Langeland, N.; Hanevik, K. Human memory CD4+ T cell immune responses against Giardia lamblia. Clin. Vaccine Immunol. 2015, 23, 11–18. [Google Scholar] [CrossRef]
- Stäger, S.; Gottstein, B.; Sager, H.; Jungi, T.W.; Müller, N. Influence of antibodies in mother’s milk on antigenic variation of Giardia lamblia in the murine mother-o spring model of infection. Infect. Immun. 1998, 66, 1287–1292. [Google Scholar] [CrossRef]
- Lopez-Romero, G.; Quintero, J.; Astiazaran-Garcia, H.; Velazquez, C. Host defenses against Giardia lamblia. Parasite Immunol. 2015, 37, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Bénéré, E.; Van Assche, T.; Cos, P.; Maes, L. Intrinsic susceptibility of Giardia duodenalis assemblage subtypes A(I), A(II), B and E(III) for nitric oxide under axenic culture conditions. Parasitol. Res. 2012, 110, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Holthaus, D.; Kraft, M.R.; Krug, S.M.; Wolf, S.; Müller, A.; Delgado Betancourt, E.; et al. Dissection of Barrier Dysfunction in Organoid-Derived Human Intestinal Epithelia Induced by Giardia duodenalis. Gastroenterology. 2022, 162(3), 844–858. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, V.P.; Nash, T.E.; Hubbard, V.S. Severe giardiasis in a patient with cystic fibrosis. J Pediatr Gastroenterol Nutr. 1985, 4(2), 320–2. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Craft, J.C.; Mather, F.J.; Davis, S.H.; Wright, J.A., Jr. Prevalence of giardiasis in patients with cystic fibrosis. The Journal of Paediatrics 1988, 112(4), 555–559. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dickson, J.A.; Variend, S.; Taylor, C.J. Intestinal disease in cystic fibrosis. Arch Dis Child. 1988, 63(12), 1496–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lopez, J.; Wright, A.J.; Hammer, R.A.; Atilla, E. Chronic Pancreatitis is Associated with a High Prevalence of Giardiasis. Canadian Journal of Gastroenterology 1992, 6, 73–76. [Google Scholar] [CrossRef]
- Hernell, O.; Ward, H.; Bläckberg, L.; Pereira, M.E. Killing of Giardia lamblia by human milk lipases: an effect mediated by lipolysis of milk lipids. J Infect Dis. 1986, 153(4), 715–20. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, S.; Packi, K.; Rudek, A.; Wenclewska, S.; Kurowski, M.; Kurczabińska, D.; Śliwińska, A. The Influence of the Protozoan Giardia lamblia on the Modulation of the Immune System and Alterations in Host Glucose and Lipid Metabolism. Int J Mol Sci. 2024, 25(16), 8627. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahmadpour, E.; Safarpour, H.; Xiao, L.; Zarean, M.; Hatam-Nahavandi, K.; Barac, A.; et al. Cryptosporidiosis in HIV-positive patients and related risk factors: A systematic review and meta-analysis. Parasite 2020, 27, 27. [Google Scholar] [CrossRef]
- Piazzesi, A.; Pane, S.; Russo, A.; Del Chierico, F.; Francalanci, P.; Cotugno, N. Case Report: The impact of severe cryptosporidiosis on the gut microbiota of a pediatric patient with CD40L immunodeficiency. Front Cell Infect Microbiol. 2023, 13, 1281440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dupuy, F.; Valot, S.; Dalle, F.; Sterin, A.; L’Ollivier, C. Disseminated Cryptosporidium infection in an infant with CD40L deficiency. IDCases 2021, 24, e01115. [Google Scholar] [CrossRef]
- Fayer, R.; Ungar, B.L. Cryptosporidium spp. and cryptosporidiosis. Microbiol Rev. 1986, 50(4), 458–83. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, X.M.; Keithly, J.S.; Paya, C.V.; LaRusso, N.F. Cryptosporidiosis. N Engl J Med. 2002, 346(22), 1723–1731. [Google Scholar] [CrossRef]
- Bouzid, M.; Hunter, P.R.; Chalmers, R.M.; Tyler, K.M. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev. 2013, 26(1), 115–134. [Google Scholar] [CrossRef] [PubMed]
- Sponseller, J.K.; Griffiths, J.K.; Tzipori, S. The evolution of respiratory Cryptosporidiosis: evidence for transmission by inhalation. Clin Microbiol Rev. 2014, 27(3), 575–86. [Google Scholar] [CrossRef]
- Chaudhary, H.; Cochrane, A.B.; Tanna, S.; Aryal, S.; Kattih, Z.; Wilkinson, J.D.; et al. A Case Series of Lung Transplant Recipients Who Developed Life-Threatening Diarrhoea from Cryptosporidium Contaminated Water Supply. Archives of Clinical and Medical Case Reports 2026, 10, 06–10. [Google Scholar] [CrossRef]
- Perkins, M.E.; Riojas, Y.A.; Wu, T.W.; Le Blancq, S.M. CpABC, a Cryptosporidium parvum ATP-binding cassette protein at the host-parasite boundary in intracellular stages. Proc Natl Acad Sci U S A 1999, 96(10), 5734–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Turkeltaub, J.A.; McCarty, TR3rd; Hotez, P.J. The intestinal protozoa: Emerging impact on global health and development. Curr. Opin. Gastroenterol. 2015, 31, 38–44. [Google Scholar] [CrossRef]
- Iyer, L.R.; Verma, A.K.; Paul, J.; Bhattacharya, A. Phagocytosis of Gut Bacteria by Entamoeba histolytica. Front. Cell. Infect. Microbiol. 2019, 9, 34. [Google Scholar] [CrossRef]
- Guillen, N. The interaction between Entamoeba histolytica and enterobacteria sheds light on an ancient antibacterial response. Cell. Microbiol. 2019, 21, e13039. [Google Scholar] [CrossRef] [PubMed]
- Leon-Coria, A.; Kumar, M.; Chadee, K. The delicate balance between Entamoeba histolytica, mucus and microbiota. Gut Microbes 2020, 11, 118–125. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The Na+ driven Cl-/ HCO3 exchanger (NCBE) as a master regulator of intracellular pH (pHi) in fibro-inflammatory and parasitic pathologies. The schematic depicts the secondary active transport of 2Na+ and 2HCO3- into the cytosol in exchange for the efflux of 1Cl- and 1H+, a process that restores cellular homeostasis by shifting pHi from acidic 6.8 to alkaline 7.2 levels. In the context of chronic disease, this NCBE-mediated alkalinization is a critical determinant of the myofibroblast transition; persistent pHi dysregulation can promote a pro-fibrotic environment through altered protein folding and enhanced extracellular matrix deposition. Furthermore, in the intestinal epithelium, NCBE activity maintains the pH-dependent integrity of the mucus barrier, where its impairment during intestinal parasitic infections can facilitate pathogen colonization and trigger an inflammatory-fibrotic cascade. Consequently, the NCBE represents a pivotal link between ion rheostasis, host-parasite dynamics, and the progression of tissue fibrosis.
Figure 1.
The Na+ driven Cl-/ HCO3 exchanger (NCBE) as a master regulator of intracellular pH (pHi) in fibro-inflammatory and parasitic pathologies. The schematic depicts the secondary active transport of 2Na+ and 2HCO3- into the cytosol in exchange for the efflux of 1Cl- and 1H+, a process that restores cellular homeostasis by shifting pHi from acidic 6.8 to alkaline 7.2 levels. In the context of chronic disease, this NCBE-mediated alkalinization is a critical determinant of the myofibroblast transition; persistent pHi dysregulation can promote a pro-fibrotic environment through altered protein folding and enhanced extracellular matrix deposition. Furthermore, in the intestinal epithelium, NCBE activity maintains the pH-dependent integrity of the mucus barrier, where its impairment during intestinal parasitic infections can facilitate pathogen colonization and trigger an inflammatory-fibrotic cascade. Consequently, the NCBE represents a pivotal link between ion rheostasis, host-parasite dynamics, and the progression of tissue fibrosis.

Figure 2.
Global Health Implications and Research Gaps in Cystic Fibrosis (CF) and Parasitic Co-infections. (A) Diagnostic Disparities: In LMICs, CF is often underdiagnosed due to symptoms overlapping with chronic malnutrition and recurrent pneumonia. In HICs, parasitic co-infections are generally sporadic and often associated with travel or post-transplant immunosuppression. (B) Clinical Impact in Endemic Regions: Protozoan infections may cause “diagnostic shadowing” by concealing malabsorption. Helminths pose mechanical risks (worsening DIOS) and pharmacological vulnerabilities (e.g., steroid-induced hyperinfection). (C) Key Research Gaps: Data are scarce regarding the global prevalence of co-infections, their effect on CFTR modulator efficacy/pharmacokinetics, and mechanistic interactions in organoid or murine models. (D) Future Directions: A risk-stratified approach is recommended, prioritising screening of high-risk subgroups (e.g., immunosuppressed patients). Large, multicentre studies across South Asia, Sub-Saharan Africa, and Latin America are urgently required to determine whether parasitism accelerates or slows long-term CF progression. Image generated by https://www.biorender.com/.
Figure 2.
Global Health Implications and Research Gaps in Cystic Fibrosis (CF) and Parasitic Co-infections. (A) Diagnostic Disparities: In LMICs, CF is often underdiagnosed due to symptoms overlapping with chronic malnutrition and recurrent pneumonia. In HICs, parasitic co-infections are generally sporadic and often associated with travel or post-transplant immunosuppression. (B) Clinical Impact in Endemic Regions: Protozoan infections may cause “diagnostic shadowing” by concealing malabsorption. Helminths pose mechanical risks (worsening DIOS) and pharmacological vulnerabilities (e.g., steroid-induced hyperinfection). (C) Key Research Gaps: Data are scarce regarding the global prevalence of co-infections, their effect on CFTR modulator efficacy/pharmacokinetics, and mechanistic interactions in organoid or murine models. (D) Future Directions: A risk-stratified approach is recommended, prioritising screening of high-risk subgroups (e.g., immunosuppressed patients). Large, multicentre studies across South Asia, Sub-Saharan Africa, and Latin America are urgently required to determine whether parasitism accelerates or slows long-term CF progression. Image generated by https://www.biorender.com/.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



