Submitted:
11 March 2026
Posted:
12 March 2026
Read the latest preprint version here
Abstract
While falling costs have increased access to genomic sequencing, the impact of clinical sequencing is often hindered by the challenge of interpreting complex genetic data. The high prevalence of variants of unknown significance (VUSs) can lead to false reassurance or psychological distress, as patients and non-expert clinicians may misinterpret inconclusive results. We propose that artificial intelligence (AI) may serve as a critical clinical decision-support tool to improve the efficiency of genetic testing, especially in variant analysis. We advocate integrating AI throughout the genetic diagnostic workflow and outline current approaches to AI-assisted variant analysis to enable efficient personalized treatment. We also discuss anticipated challenges in this pursuit and offer recommendations to ensure precision, accuracy, reproducibility, and transparency.
Keywords:
variant analysis
; clinical decision support
; genomics
; explainable ai
; precision medicine
; diagnostic odyssey
Introduction
The average time to receive a genetic diagnosis across high-income countries ranges from 4 to 19 years [1,2]. Current practices force patients with genetic diseases into a ‘diagnostic odyssey,’ subjecting them to rounds of unnecessary clinic visits, procedures, and medications. This process closes or narrows their window of intervention, ultimately enabling disease progression and long-term disease damage. One critical component in addressing the diagnostic odyssey is high-throughput genetic testing, which has become widely accessible and cost-effective [3]. Even out-of-pocket sequencing costs are a fraction of the overall odyssey, which produces $86,000 to $516,000 in avoidable costs per patient [4].
Early disease identification and therapeutic intervention should be the norm. However, physicians report limitations in their genetics training [5,6,7], and many express reduced interest in genetic screening due to the rarity of genetic conditions [8]. On the contrary, 25-30 million Americans have a genetic disease (~ 1 in 11) [9]; therefore, health care providers should widely adopt genetic testing to fulfill this demand.
Variant analysis—the rate-limiting step in genetic testing [10]—classifies variants by their pathogenicity to guide clinical decision-making. Inaccurate interpretation at this stage fundamentally alters patient management, preventing the use of targeted therapies, initiating surveillance, or performing preventive procedures [11]. These errors also extend to the family, obscuring the need for cascade screening or preimplantation genetic diagnosis [12]. Consequently, misinterpreted variants contribute to avoidable morbidity and mortality through missed preventative interventions, while simultaneously inflicting psychological harm via false reassurance or unnecessary anxiety [13]. The standard of care uses ACMG/AMP and/or ESHG guidelines [14,15] for variant interpretation, but the process as a whole remains labor-intensive and relies heavily on experts. Results can be inconsistent and often yield variants that lack sufficient evidence to be classified as benign or pathogenic [11,16,17], complicating patient care. However, automation that leverages all available clinical, molecular, and population data in a standardized, reproducible manner could help reduce these issues. Artificial intelligence (AI), tools with ”human-like reasoning” built from a variety of machine learning (ML) models and/or large language models (LLMs) (reviewed in [18,19,20,21,22]), can optimize labor- and knowledge-intensive steps throughout the genetic testing process.
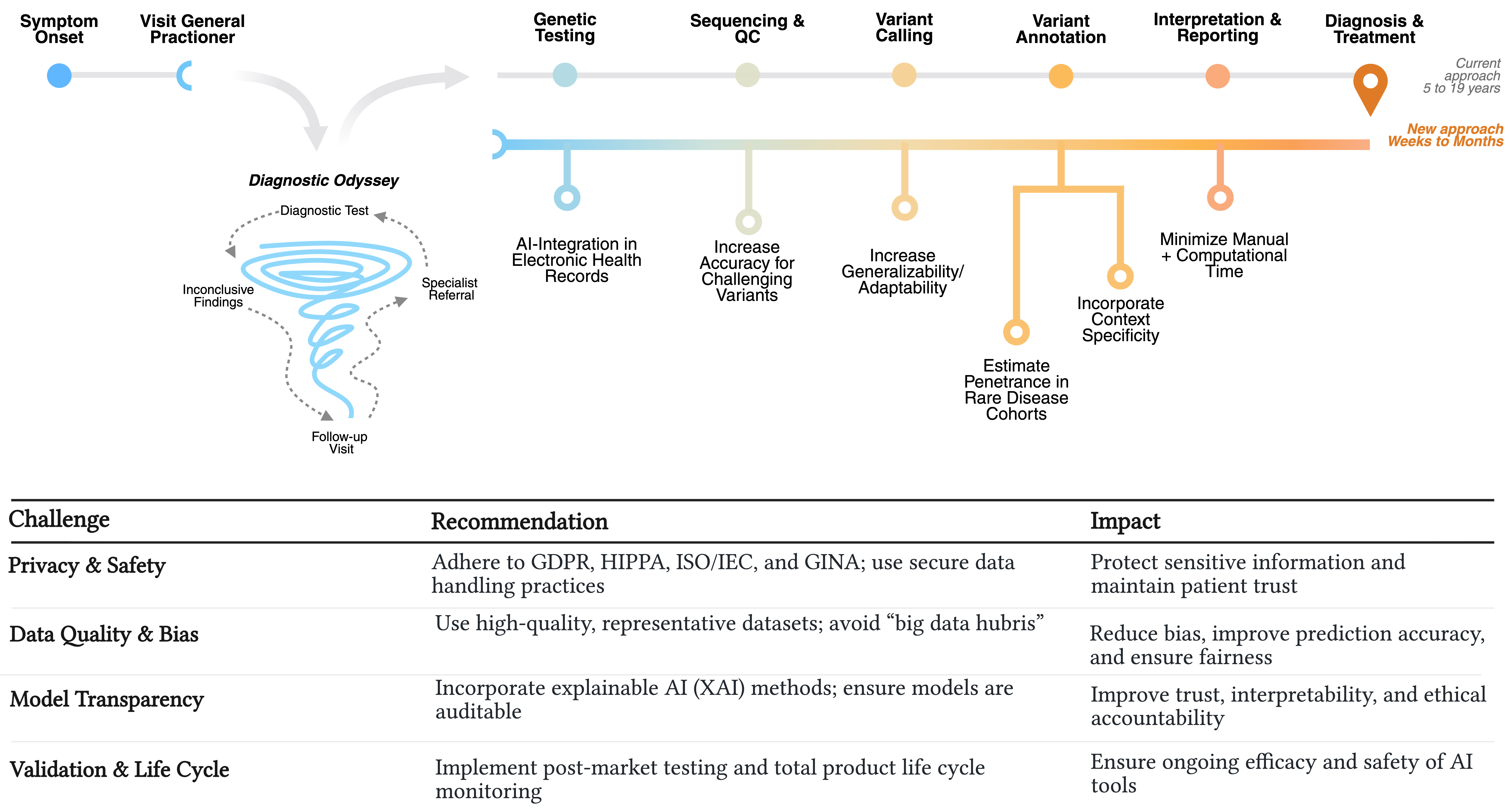
The schematic contrasts the slow, cyclic traditional approach with targeted AI opportunities that accelerate variant analysis and shorten time to diagnosis. The table summarizes potential challenges using our proposed AI-assisted approach, along with recommended solutions and their expected clinical impact. Created in BioRender. Taluri and Wilk. (2026) https://BioRender.com/6xxko8q
Approach to Variant Analysis
AI is emerging at a time when clinical genetics faces its greatest gap between knowledge and practice.
To prevent the diagnostic odyssey, physicians must first recognize patients who would benefit from genetic testing. Genetic diseases typically present with a constellation of signs (e.g., dysmorphism, early-onset, and/or multi-system involvement); therefore, AI can assist in determining when genetic testing may be appropriate (e.g., FACE2GENE [23]). For instance, AI-integrations in EHRs could detect potential patients, even those with subtle clinical presentations [24,25]. AI can also support physicians' continuing education through adaptive educational modules that account for each individual’s time constraints, goals, and baseline knowledge [26].
After sequencing, variant analysis processes the data in four key steps: variant calling, annotation, prioritization, and interpretation. AI/ML tools have already streamlined variant calling by reducing manual filtering and improving scalability. Examples of this include Google’s DeepVariant [27], DNAscope [28,29], DeepTrio [30], Clair3 [31], Medaka [32], and HELLO [33]. These tools offer speed and generalizability across sequencing platforms [34,35,36]. Following variant identification, variant annotation contextualizes a patient’s variants using sequence data, conservation, population frequency, and functional impact. This step requires synthesizing information across diverse databases. LLMs, a subtype of AI models that process and generate human language [18], excel at automating this process. Mining resources like ClinVar and gnomAD (i.e., large databases of patient variants) have been assessed in the context of their genetic sequences to predict a variant’s consequences on the primary structure (e.g., SpliceAI, AlphaMissense, and Evo2) [37,38]. Other ML models have enhanced variant annotation through feature-based learning (e.g., REVEL, CADD, PrimateAI-3D) [39,40,41]
Full-stack variant analysis pipelines, including AI-MARRVEL [42], Qiagen’s Franklin [43], Illumina’s Emedgene [44], and Nostos Genomics [45], have already automated variant interpretation and prioritization. Despite these advances, variants of uncertain significance (VUSs) remain the most common variant classification, accounting for ~35-37% of variants associated with rare diseases and cancer [46,47,48]. This ambiguity presents a critical clinical challenge; non-experts may misinterpret a VUS as 'normal' (false reassurance) or as a definitive diagnosis (unnecessary anxiety), leading to inappropriate care [13]. Reclassification is inherently difficult, as assigning a variant to benign, likely benign, likely pathogenic, or pathogenic annotations requires ≥90% certainty of its clinical relevance [14]. This threshold is challenging to meet, especially when context-specific data are limited and/or when considering non-coding (e.g., regulatory sequences [49] and splice sites [50]), low-penetrance, or hypomorphic variants [14,51]. Emerging tools aim to address this, such as DYNA, a disease-specific LLM that compares context-specific networks to score pathogenicity of coding and non-coding variants [52]. In a study of >17k cardiomyopathy VUSs from ClinVar, DYNA reclassified ~9% as pathogenic, likely pathogenic, benign, or likely benign [52]. Another promising approach to improving classification is to estimate penetrance. In rare diseases, small cohorts make it difficult, or even impossible, to calculate penetrance using traditional methods. However, Forrest et al. [53], developed disease-specific ML models to calculate disease probability and penetrance using EHR and genetic data.
AI-assisted variant analysis can clarify genetic test results (e.g., AI-enabled ACMG scoring within EHR and clinical trial eligibility screening [54]), enabling clinicians to weigh genomic evidence alongside clinical findings. With data-driven rationales to support clinical diagnostics, clinicians are better-equipped to make more efficient and accurate decisions. Clinicians can thereby reduce trial-and-error prescribing by linking variants to targeted therapies and trials. Ultimately, AI-assistance will increase genetic screening rates, preventing delays in care.
Challenges and Recommendations
Integrating AI into clinical genetics shows great promise, but we expect challenges ahead (Figure 1).
Trust in scientists is declining in the US [55], and global opinion toward AI remains cautious [56]. To restore public confidence, developers should collaborate with patients and clinicians when designing AI tools, leveraging their domain-specific expertise to improve model performance and ensure relevance [57,58].
Genetic data has historically raised significant legal, ethical, and privacy concerns due to its uniquely identifiable nature. Using this data with AI could raise additional concerns; therefore, training data and software must comply with national/international laws and standards [59,60,61,62,63]. Models for variant analysis should also adhere to established clinical standards from reputable organizations, such as ACMG, AMP, CAP [14], and ESHG [15,64].
A major shortcoming of many AI tools stems from the data they are trained on. Overreliance on large, uncurated datasets can introduce bias, inaccuracies, and outdated information, leading to large errors in predictions [65,66,67,68] and AI “hallucinations” [69]. Instead, datasets should be reliable and representative of the affected patient population [57,70,71,72,73,74,75]. This is especially critical in biomedical applications, where underrepresentation can perpetuate disparities [71,72,73,76,77,78,79]. However, implementing retrieval-augmented generation (RAG) systems (curated knowledgebases) has already aided biomedical applications and reduced AI hallucinations [80,81].
ML/AI models offer powerful capabilities for streamlining variant analysis by integrating multimodal data (e.g., genetic sequences, EHRs, biomedical knowledge graphs, and large-scale text mining) but often at the cost of interpretability, with many functioning as a “black box” [60,82]. To ensure fairness and accuracy, especially in clinical contexts, models must be auditable and explainable. An auditable model acts as a “glass box,” where processes can be systematically examined and traced (e.g., by logging decision logic [83] and data sources used as evidence [44], [45], [84], [85] [83], [86]. Explainable AI (XAI) techniques further enable users to dissect models and their predictions to assess the influence of individual features. Numerous XAI approaches are currently available–even for complex LLMs–despite their scale of parameters and training [87,88,89]. Some AI-assisted variant analyses and workflows already incorporate explainable AI (XAI) methods, such as scoring and ranking the importance of features that drive their predictions [44,53,90,91].
Confirming the correctness and translatability of AI-prioritized variants requires multi-tiered validation and continual monitoring. Models must be benchmarked and tested against high-quality, expert-curated datasets (e.g., ClinVar or specific disease cohorts) to ensure high sensitivity (>90%) in real-world scenarios [90], and predictions should be verified through orthogonal biological tests. Potential orthogonal evidence-based methods include segregation analysis [92], confirming variant tracks with phenotypes in a family, and in vivo or in vitro functional assays [11,92], providing experimental evidence supporting variant damage to a gene product. AI tools should follow a full product lifecycle approach, including international predetermined change control plans (PCCPs) for ML-enabled medical devices [93], with real-world performance tracked for safety and efficacy. Because models evolve, outputs may change and even contradict earlier reports; this should be expected and documented so clinicians and patients can modify care as needed [93].
Conclusions
Incorporating ML and AI into variant analysis can transform and expedite the genetic testing process with actionable clinical intelligence, enabling earlier diagnostics and potentially life-saving interventions. When designed with transparency and community engagement, these tools accelerate variant interpretation without compromising clinical judgement or patient trust. By prioritizing ethical design, high-quality data, and explainable models, AI-assisted genomics advances the principle of beneficence by improving accuracy and efficiency, while ensuring nonmaleficence through bias control, ultimately streamlining the path from genetic discovery to bedside treatment and, ultimately, improving long-term patient outcomes.
Funding
This work was funded by the UAB Pilot Center for Precision Animal Modeling (C-PAM) (U54-OD030167), the UAB Childhood Cystic Kidney Disease Center (UAB-CCKDC) - Informatic and Data Analytics Resource (U54-DK126087), and M.M. was in part supported by the U.S. Department of Veterans Affairs (1-I01-BX006266-01).
References
- Faye, F; Crocione, C; Anido de Peña, R; et al. Time to diagnosis and determinants of diagnostic delays of people living with a rare disease: results of a Rare Barometer retrospective patient survey. Eur J Hum Genet 2024, 32(9), 1116–26. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C; Parkinson, A; Namsrai, T; et al. Time to diagnosis for a rare disease: managing medical uncertainty. A qualitative study. Orphanet J Rare Dis 2024, 19(1), 297. [Google Scholar] [CrossRef] [PubMed]
- Kris A. Wetterstrand MS. DNA Sequencing Costs: Data [Internet]. Genome.gov. 2019 [cited 2025 Nov 5];Available from: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data.
- Delayed Diagnosis Study [Internet]. EveryLife Foundation for Rare Diseases. 2023 [cited 2025 Nov 18];Available from: https://everylifefoundation.org/delayed-diagnosis-study/#about-study.
- Rasouly, HM; Balderes, O; Marasa, M; et al. The effect of genetic education on the referral of patients to genetic evaluation: Findings from a national survey of nephrologists. Genet Med 2023, 25(5), 100814. [Google Scholar] [CrossRef] [PubMed]
- Peabody, J; DeMaria, L; Tamandong-LaChica, D; Florentino, J; Acelajado, MC; Burgon, T. Low rates of genetic testing in children with developmental delays, intellectual disability, and autism spectrum disorders. Glob Pediatr Health 2015, 2, 2333794X15623717. [Google Scholar] [CrossRef]
- Kneifati-Hayek, JZ; Zachariah, T; Ahn, W; et al. Bridging the gap in genomic implementation: Identifying user needs for precision nephrology. Kidney Int Rep 2024, 9(8), 2420–31. [Google Scholar] [CrossRef]
- Pasquier, L; Minguet, G; Moisdon-Chataigner, S; et al. How do non-geneticist physicians deal with genetic tests? A qualitative analysis. Eur J Hum Genet 2022, 30(3), 320–31. [Google Scholar] [CrossRef]
- Rare Diseases [Internet]. National Institutes of Health (NIH). [cited 2025 Nov 18];Available from: https://www.nih.gov/about-nih/nih-turning-discovery-into-health/promise-precision-medicine/rarediseases.
- Tagliafico, E; Bernardis, I; Grasso, M; et al. Workload measurement for molecular genetics laboratory: A survey study. PLoS One 2018, 13(11), e0206855. [Google Scholar] [CrossRef]
- Agaoglu, NB; Unal, B; Akgun Dogan, O; et al. Consistency of variant interpretations among bioinformaticians and clinical geneticists in hereditary cancer panels. Eur J Hum Genet 2022, 30(3), 378–83. [Google Scholar] [CrossRef]
- McNeill, A. A new system for variant classification? Eur J Hum Genet 2022, 30(2), 137–8. [Google Scholar] [CrossRef]
- Campeau, PM. An all-encompassing variant classification system proposed. Eur J Hum Genet 2022, 30(2), 139. [Google Scholar] [CrossRef]
- Richards, S; Aziz, N; Bale, S; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17(5), 405–24. [Google Scholar] [CrossRef]
- Houge, G; Laner, A; Cirak, S; de Leeuw, N; Scheffer, H; den Dunnen, JT. Stepwise ABC system for classification of any type of genetic variant. Eur J Hum Genet 2022, 30(2), 150–9. [Google Scholar] [CrossRef]
- Zukin, E; Culver, JO; Liu, Y; et al. Clinical implications of conflicting variant interpretations in the cancer genetics clinic. Genet Med 2023, 25(7), 100837. [Google Scholar] [CrossRef] [PubMed]
- Lin, L; Pan, H; Qi, Y; Ma, Y; Qiu, L. Reasons and Resolutions for Inconsistent Variant Interpretation. Human Mutation 2023, 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Machine Learning vs Deep Learning vs LLMs vs GenAI: Explained and How are they Different from Each Other? [Internet]. Cloud4C. 2024 [cited 2025 Nov 14];Available from: https://www.cloud4c.com/blogs/genai-vs-machine-learning-vs-deep-learning-vs-llms.
- Russell S, Norvig P. Artificial intelligence: A modern approach, global edition [Internet]. 4th ed. London, England: Pearson Education; 2021 [cited 2025 Oct 22]. Available from: https://elibrary.pearson.de/book/99.150005/9781292401171.
- Janiesch, C; Zschech, P; Heinrich, K. Machine learning and deep learning. Electron Mark 2021, 31(3), 685–95. [Google Scholar] [CrossRef]
- Koteluk, O; Wartecki, A; Mazurek, S; Kołodziejczak, I; Mackiewicz, A. How do machines learn? Artificial intelligence as a New Era in medicine. J Pers Med 2021, 11(1), 32. [Google Scholar] [CrossRef]
- Nichols, JA; Herbert Chan, HW; Baker, MAB. Machine learning: applications of artificial intelligence to imaging and diagnosis. Biophys Rev 2019, 11(1), 111–8. [Google Scholar] [CrossRef]
- Gurovich, Y; Hanani, Y; Bar, O; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med 2019, 25(1), 60–4. [Google Scholar] [CrossRef]
- Ye, J; Woods, D; Jordan, N; Starren, J. The role of artificial intelligence for the application of integrating electronic health records and patient-generated data in clinical decision support. AMIA Summits Transl Sci Proc 2024, 2024, 459–67. [Google Scholar]
- Yang, X; Chen, A; PourNejatian, N; et al. A large language model for electronic health records. NPJ Digit Med 2022, 5(1), 194. [Google Scholar] [CrossRef]
- Hajek, C; Hutchinson, AM; Galbraith, LN; et al. Improved provider preparedness through an 8-part genetics and genomic education program. Genet Med 2022, 24(1), 214–24. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R; Chang, P-C; Alexander, D; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 2018, 36(10), 983–7. [Google Scholar] [CrossRef]
- Freed D, Pan R, Chen H, Li Z, Hu J, Aldana R. DNAscope: High accuracy small variant calling using machine learning [Internet]. bioRxiv. 2022 [cited 2025 Oct 14];2022.05.20.492556. Available from: https://www.biorxiv.org/content/10.1101/2022.05.20.492556v1.abstract.
- Hu J, Freed D, Feng H, Chen H, Li Z, Chen H. Accelerated, Accurate, Hybrid Short and Long Reads Alignment and Variant Calling [Internet]. Bioinformatics. 2025;Available from: https://www.biorxiv.org/content/10.1101/2025.04.15.648987v3.full.pdf.
- Hu, X; Feng, C; Zhou, Y; Harrison, A; Chen, M. DeepTrio: a ternary prediction system for protein-protein interaction using mask multiple parallel convolutional neural networks. Bioinformatics 2022, 38(3), 694–702. [Google Scholar] [CrossRef]
- Su, J; Zheng, Z; Ahmed, SS; Lam, T-W; Luo, R. Clair3-trio: high-performance Nanopore long-read variant calling in family trios with trio-to-trio deep neural networks. Brief Bioinform 2022, 23(5), bbac301. [Google Scholar] [CrossRef]
- medaka: Sequence correction provided by ONT Research [Internet]. Github; [cited 2025 Oct 14]. Available from: https://github.com/nanoporetech/medaka.
- Ramachandran, A; Lumetta, SS; Klee, EW; Chen, D. HELLO: improved neural network architectures and methodologies for small variant calling. BMC Bioinformatics 2021, 22(1), 404. [Google Scholar] [CrossRef]
- Abdelwahab, O; Belzile, F; Torkamaneh, D. Performance analysis of conventional and AI-based variant callers using short and long reads. BMC Bioinformatics 2023, 24(1), 472. [Google Scholar] [CrossRef] [PubMed]
- Brand, F; Guski, J; Krawitz, P. Extending DeepTrio for sensitive detection of complex de novo mutation patterns. NAR Genom Bioinform 2024, 6(1), lqae013. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, O; Torkamaneh, D. Artificial intelligence in variant calling: a review. Front Bioinform 2025, 5, 1574359. [Google Scholar] [CrossRef]
- Tordai, H; Torres, O; Csepi, M; Padányi, R; Lukács, GL; Hegedűs, T. Analysis of AlphaMissense data in different protein groups and structural context. Sci Data 2024, 11(1), 495. [Google Scholar] [CrossRef]
- Brixi G, Durrant MG, Ku J, et al. Genome modeling and design across all domains of life with Evo 2 [Internet]. bioRxiv. 2025 [cited 2025 Oct 10];2025.02.18.638918. Available from: https://www.biorxiv.org/content/10.1101/2025.02.18.638918v1.abstract.
- Ioannidis, NM; Rothstein, JH; Pejaver, V; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016, 99(4), 877–85. [Google Scholar] [CrossRef]
- Kircher, M; Witten, DM; Jain, P; O’Roak, BJ; Cooper, GM; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014, 46(3), 310–5. [Google Scholar] [CrossRef]
- Gao, H; Hamp, T; Ede, J; et al. The landscape of tolerated genetic variation in humans and primates. Science 2023, 380(6648), eabn8153. [Google Scholar] [CrossRef]
- Mao D, Liu C, Wang L, et al. AI-MARRVEL - A knowledge-driven AI system for diagnosing Mendelian disorders. NEJM AI [Internet] 2024 [cited 2025 Oct 10];1(5). [CrossRef]
- Franklin - Bioinformatics Software [Internet]. Bioinformatics Software | QIAGEN Digital Insights. 2025 [cited 2025 Oct 14];Available from: https://digitalinsights.qiagen.com/franklin/.
- Meng, L; Attali, R; Talmy, T; et al. Evaluation of an automated genome interpretation model for rare disease routinely used in a clinical genetic laboratory. Genet Med 2023, 25(6), 100830. [Google Scholar] [CrossRef] [PubMed]
- Nostos Genomics – AI-driven genetic analysis platform [Internet]. [cited 2025 Oct 14];Available from: https://www.nostos-genomics.com/.
- Balmaña, J; Digiovanni, L; Gaddam, P; et al. Conflicting interpretation of genetic variants and cancer risk by commercial laboratories as assessed by the Prospective Registry of Multiplex Testing. J Clin Oncol 2016, 34(34), 4071–8. [Google Scholar] [CrossRef] [PubMed]
- Zawar, A; Manoj, G; Nair, PP; Deshpande, P; Suravajhala, R; Suravajhala, P. Variants of uncertain significance: At the crux of diagnostic odyssey. Gene 2025, 962(149587), 149587. [Google Scholar] [CrossRef] [PubMed]
- Fowler, DM; Rehm, HL. Will variants of uncertain significance still exist in 2030? Am J Hum Genet 2024, 111(1), 5–10. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Ž; Agarwal, V; Visentin, D; et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods 2021, 18(10), 1196–203. [Google Scholar] [CrossRef]
- Jaganathan, K; Kyriazopoulou Panagiotopoulou, S; McRae, JF; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176(3), 535–48.e24. [Google Scholar] [CrossRef]
- Fiorini, MR; Dilliott, AA; Farhan, SMK. Evaluating the utility of REVEL and CADD for interpreting variants in amyotrophic lateral sclerosis genes. Hum Mutat 2023, 2023(1), 8620557. [Google Scholar] [CrossRef]
- Zhan H, Zhang Z. DYNA: Disease-specific language model for variant pathogenicity [Internet]. arXiv [qbio. GN]. 2024 [cited 2025 Oct 9];Available from: http://arxiv.org/abs/2406.00164.
- Forrest, IS; Vy, HMT; Rocheleau, G; et al. Machine learning-based penetrance of genetic variants. Science 2025, 389(6763), eadm7066. [Google Scholar] [CrossRef]
- Jin, Q; Wang, Z; Floudas, CS; et al. Matching patients to clinical trials with large language models. Nat Commun 2024, 15(1), 9074. [Google Scholar] [CrossRef]
- Kennedy B, Tyson A. Americans’ Trust in Scientists, Positive Views of Science Continue to Decline [Internet]. Pew Research Center. 2023 [cited 2025 Nov 6];Available from: https://www.pewresearch.org/science/2023/11/14/confidence-in-scientists-medical-scientists-and-othergroups- and-institutions-in-society/.
- Poushter J, Fagan M, Corichi M. How People Around the World View AI [Internet]. Pew Research Center. 2025 [cited 2025 Nov 6];Available from: https://www.pewresearch.org/global/2025/10/15/how-peoplearound- the-world-view-ai/.
- Tomašev, N; Cornebise, J; Hutter, F; et al. AI for social good: unlocking the opportunity for positive impact. Nat Commun 2020, 11(1), 2468. [Google Scholar] [CrossRef] [PubMed]
- Erikson, SL. Cell phones ≠ self and other problems with big data detection and containment during epidemics. Med Anthropol Q 2018, 32(3), 315–39. [Google Scholar] [CrossRef] [PubMed]
- Genetic Information Nondiscrimination Act of 2008 (GINA) [Internet]. 2008 [cited 2025 Nov 13]. Available from: https://www.eeoc.gov/statutes/genetic-information-nondiscrimination-act-2008.
- Ruiz J. Machine learning and the right to explanation in GDPR [Internet]. Open Rights Group. [cited 2025 Oct 7];Available from: https://www.openrightsgroup.org/blog/machine-learning-and-the-right-toexplanation- in-gdpr/.
- European Union. Regulation (EU) 2016/679 General Data Protection Regulation (GDPR) [Internet]. Official Journal of the European Union; 2016 [cited 2025 Oct 7]. Available from: https://gdpr-info.eu/.
- Office for Civil Rights (OCR). Summary of the HIPAA Privacy Rule [Internet]. [cited 2022 Mar 16];Available from: https://www.hhs.gov/hipaa/for-professionals/privacy/laws-regulations/index.html.
- International Organization for Standardization, International Electrotechnical Commission. ISO/IEC 27001:2022 Information security, cybersecurity and privacy protection — Information security management systems — Requirements [Internet]. Geneva: ISO; 2022 [cited 2025 Nov 13]. Available from: https://www.iso.org/standard/27001.
- Houge, G; Bratland, E; Aukrust, I; et al. Comparison of the ABC and ACMG systems for variant classification. Eur J Hum Genet 2024, 32(7), 858–63. [Google Scholar] [CrossRef]
- Lazer, D; Kennedy, R; King, G; Vespignani, A. Big data. The parable of Google Flu: traps in big data analysis. Science 2014, 343(6176), 1203–5. [Google Scholar] [CrossRef]
- Ross C, Swetlitz I. IBM’s Watson supercomputer recommended “unsafe and incorrect” cancer treatments, internal documents show [Internet]. STAT. 2018 [cited 2025 Oct 7];Available from: https://www.statnews.com/2018/07/25/ibm-watson-recommended-unsafe-incorrect-treatments/.
- Xing S, Hong J, Wang Y, et al. LLMs can get “Brain Rot”! [Internet]. arXiv [cs.CL]. 2025 [cited 2025 Nov 3];Available from: http://arxiv.org/abs/2510.13928.
- Fieldhouse R. Too much social media gives AI chatbots “brain rot.” Nature [Internet] 2025 [cited 2025 Nov 3];Available from: http://dx.doi.org/10.1038/d41586-025-03542-2. [CrossRef]
- Beutel, G; Geerits, E; Kielstein, JT. Artificial hallucination: GPT on LSD? Crit Care 2023, 27(1), 148. [Google Scholar] [CrossRef]
- PIH’s Five S’s: Essential Elements for Strong Health Systems [Internet]. Partners In Health. [cited 2025 Nov 3];Available from: https://www.pih.org/article/pihs-five-ss-essential-elements-strong-health-systems.
- Nakayama, LF; Kras, A; Ribeiro, LZ; et al. Global disparity bias in ophthalmology artificial intelligence applications. BMJ Health Care Inform 2022, 29(1), e100470. [Google Scholar] [CrossRef] [PubMed]
- Daneshjou, R; Vodrahalli, K; Novoa, RA; et al. Disparities in dermatology AI performance on a diverse, curated clinical image set. Sci Adv 2022, 8(32), eabq6147. [Google Scholar] [CrossRef]
- Delgado, J; de Manuel, A; Parra, I; et al. Bias in algorithms of AI systems developed for COVID-19: A scoping review. J Bioeth Inq 2022, 19(3), 407–19. [Google Scholar] [CrossRef]
- Center for Devices, Radiological Health. Good Machine Learning Practice for Medical Device Development: Guiding Principles [Internet]. U.S. Food and Drug Administration. 2025 [cited 2025 Oct 7];Available from: https://www.fda.gov/medical-devices/software-medical-device-samd/good-machinelearning- practice-medical-device-development-guiding-principles.
- McCoy, LG; Bihorac, A; Celi, LA; et al. Building health systems capable of leveraging AI: applying Paul Farmer’s 5S framework for equitable global health. BMC Glob Public Health 2025, 3(1), 39. [Google Scholar] [CrossRef]
- Dastin J. Insight - Amazon scraps secret AI recruiting tool that showed bias against women [Internet]. Reuters. 2018 [cited 2025 Oct 7];Available from: https://www.reuters.com/article/world/insight-amazonscraps- secret-ai-recruiting-tool-that-showed-bias-against-women-idUSKCN1MK0AG/.
- Larson J, Angwin J, Kirchner L, Mattu S. How We Analyzed the COMPAS Recidivism Algorithm [Internet]. #creator. 2016 [cited 2025 Oct 7];Available from: https://www.propublica.org/article/how-we-analyzed-thecompas- recidivism-algorithm.
- Diaz M, Johnson I, Lazar A, Piper AM, Gergle D. Addressing age-related bias in sentiment analysis [Internet]. In: Proceedings of the 2018 CHI Conference on Human Factors in Computing Systems. New York, NY, USA: ACM; 2018 [cited 2025 Oct 7]. Available from: http://dx.doi.org/10.1145/3173574.3173986. [CrossRef]
- Obermeyer, Z; Powers, B; Vogeli, C; Mullainathan, S. Dissecting racial bias in an algorithm used to manage the health of populations. Science 2019, 366(6464), 447–53. [Google Scholar] [CrossRef] [PubMed]
- Leiser, F; Guse, R; Sunyaev, A. Large language model architectures in health care: Scoping review of research perspectives. J Med Internet Res 2025, 27, e70315. [Google Scholar] [CrossRef]
- Lee, J; Cha, H; Hwangbo, Y; Cheon, W. Enhancing large language model reliability: Minimizing hallucinations with dual retrieval-augmented generation based on the latest diabetes guidelines. J Pers Med 2024, 14(12), 1131. [Google Scholar] [CrossRef]
- Gosiewska, A; Kozak, A; Biecek, P. Simpler is better: Lifting interpretability-performance trade-off via automated feature engineering. Decis Support Syst 2021, 150(113556), 113556. [Google Scholar] [CrossRef]
- Sina Gräupner, O; Pawlaszczyk, D; Hummert, C. Basics of Auditable AI Systems. 2023 Congress in Computer Science, Computer Engineering, & Applied Computing (CSCE), 2023; IEEE; pp. 2355–62. [Google Scholar]
- Mercurio, SA; Chunn, LM; Khursigara, G; et al. ENPP1 deficiency: A clinical update on the relevance of individual variants using a locus-specific patient database. Hum Mutat 2022, 43(12), 1673–705. [Google Scholar] [CrossRef]
- Allot, A; Wei, C-H; Phan, L; et al. Tracking genetic variants in the biomedical literature using LitVar 2.0. Nat Genet 2023, 55(6), 901–3. [Google Scholar] [CrossRef] [PubMed]
- May W, Berghoff C, Böddinghaus J, et al. May 2022 Towards Auditable AI Systems From Principles to Practice [Internet]. 2022 [cited 2025 Oct 22];Available from: https://www.semanticscholar.org/paper/Whitepaper-%7C-May-2022-Towards-Auditable-AI-Systems- May-Berghoff/2af03e944a0c0e905ccf1b24e795f53cc780ea36#related-papers.
- Zhao, H; Chen, H; Yang, F; et al. Explainability for large language models: A survey. ACM Trans Intell Syst Technol 2024, 15(2), 1–38. [Google Scholar] [CrossRef]
- Chen, X; Wang, L; You, M; et al. Evaluating and enhancing large language models’ performance in domain-specific medicine: Development and usability study with DocOA. J Med Internet Res 2024, 26(1), e58158. [Google Scholar] [CrossRef]
- Peng, M; Guo, X; Chen, X; et al. LC-LLM: Explainable lane-change intention and trajectory predictions with Large Language Models. Communications in Transportation Research 2025, 5(100170), 100170. [Google Scholar] [CrossRef]
- AI & Variant Interpretation: From Data Tsunami to Diagnostic Clarity [Internet]. [cited 2025 Oct 7];Available from: https://www.nostos-genomics.com/news/ai-and-variant-interpretation-from-datatsunami- to-diagnostic-clarity.
- Lundberg S, Lee S-I. A unified approach to interpreting model predictions [Internet]. arXiv [cs.AI]. 2017;Available from: http://arxiv.org/abs/1705.07874.
- Kim, YE; Ki, CS; Jang, MA. Challenges and considerations in sequence variant interpretation for Mendelian disorders. Ann Lab Med 2019, 39(5), 421–9. [Google Scholar] [CrossRef] [PubMed]
- Center for Devices, Radiological Health. Predetermined Change Control Plans for Machine Learning- Enabled Medical Devices: Guiding Principles [Internet]. U.S. Food and Drug Administration. 2025 [cited 2025 Oct 7];Available from: https://www.fda.gov/medical-devices/software-medical-devicesamd/ predetermined-change-control-plans-machine-learning-enabled-medical-devices-guidingprinciples.
Figure 1.
Comparing the years-long diagnostic odyssey to an AI-enabled streamlined pathway.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



