Submitted:
05 March 2026
Posted:
09 March 2026
You are already at the latest version
Abstract
Glucose transporter type 4 (GLUT4), encoded by the SLC2A4 gene, is the final effector of insulin-stimulated glucose uptake in insulin-sensitive tissues: skeletal muscle, adipose tissue, and cardiac muscle. Its dynamic localization, retained intracellularly under basal conditions and massively translocated to the plasma membrane upon stimulation, makes it a master regulator of glycemic homeostasis. While the canonical insulin pathway (PI3K/Akt/TBC1D4) is the most potent and specific mechanism, its dysfunction is associated with insulin resistance and type 2 diabetes. Crucially, there are robust signaling pathways that are completely independent of insulin and regulate GLUT4 synthesis and translocation. Among these, those activated by muscle contraction are prominent, employing calcium signals (via CaMKII), mechanical/metabolic stress (via p38 MAPK γ/δ), and AMP-activated protein kinase (AMPK) activation. This review critically and comprehensively integrates current knowledge, from the molecular architecture of GLUT4 and its facilitated transport mechanism to the complex signaling networks converging on its regulation. The hierarchy, redundancy, and interdependence of these pathways are emphasized. It discusses how understanding insulin-independent mechanisms offers promising therapeutic opportunities for metabolic diseases, particularly for mimicking the benefits of exercise. Finally, future research directions are proposed to translate this molecular knowledge into novel clinical interventions.
Keywords:
GLUT4
; SLC2A4
; vesicular translocation
; insulin signaling
; PI3K/Akt
; AMPK
; exercise
; muscle contraction
; CaMKII
; p38 MAPK
; insulin resistance
; Type 2 diabetes
1. Introduction
Glucose is the primary energy source for mammalian cells. The American Diabetes Association indicates that in healthy individuals, fasting blood glucose should be less than 100 mg/dL (range: 70-99 mg/dL) (Spartano et al., 2025). Glucose is internalized into the cell via the glucose transporter type 4 (UniProt P14672, human GLUT4), which is specific to adipose tissue, skeletal muscle, and cardiac muscle. GLUT4 was initially cloned and sequenced in 1989 from rat adipocytes, skeletal muscle, and heart (Charron et al., 1989; James et al., 1989). Under basal conditions, GLUT4 is internalized into intracellular clusters, and its translocation cycle is regulated by factors that promote or inhibit its movement between the membrane and intracellular compartments. Hormones and factors that activate the translocation, endocytosis, and exocytosis cycles of this protein are insulin (Leto & Saltiel, 2012), leptin (Minokoshi et al., 2002), adiponectin (Yamauchi et al., 2002), as well as muscle contraction (Richter & Hargreaves, 2013), and hypoxia (Chen et al., 2001); these cycles are inhibited by cortisol (Dimitriadis et al., 1997), glucagon (Yang et al., 2021), catecholamines (Barth et al., 2007), and fatty acids (Shulman, 2000).
The peculiarity of GLUT4 lies in its apparently simple yet complexly regulated pattern of exocytosis and endocytosis. Unlike GLUT1, which is constitutively localized at the plasma membrane to ensure basal glucose uptake, more than 99% of GLUT4 under fasting or resting conditions is internalized into different regions of the cytoplasm, as described long years ago (Slot et al., 1991). Both insulin and muscle contraction independently trigger the massive translocation of these proteins to the plasma membrane, increasing their surface density and glucose uptake capacity by 20-40 times (Huang & Czech, 2007). This dynamic reserve system enables a rapid, amplifiable metabolic response to physiological glucose requirements. Alteration of this process leads to various pathologies, such as insulin resistance and type 2 diabetes mellitus (T2DM). Under these conditions, insulin signaling is compromised, leading to impaired GLUT4 translocation, chronic hyperglycemia, and its complications (Kahn, 1996). However, other robust signaling pathways, independent of insulin stimulation, are now known to help translocate GLUT4 to the cytoplasmic membrane for glucose uptake, including muscle contraction. Physical exercise is considered the second most important stimulus for maintaining this recycling dynamic, even in severe states of insulin resistance (Kennedy et al., 1999), with profound implications for the management of T2DM and other metabolic diseases.
This review aims to provide an updated summary of the state of the art on the biochemical and physiological mechanisms underlying GLUT4-mediated glucose uptake. We will address the structural principles governing its function as a transporter, as well as the signaling cascades regulating its synthesis and vesicular traffic. We will explain the hierarchy and interaction between the canonical (insulin) pathway and the multiple alternative pathways, with special attention to mechanisms activated by exercise. Finally, we will discuss the pathophysiological implications of these mechanisms and some future perspectives, including targeted therapies.
2. Brief Description of Mechanisms Involved in Insulin Exocytosis by the Pancreatic Beta Cell
Glucose-stimulated insulin secretion (GSIS) is a fundamental process for maintaining glycemic homeostasis and constitutes the essential mechanism by which pancreatic beta cells respond to postprandial hyperglycemia by releasing insulin into the circulation. The process begins with glucose entering the beta cell via the facilitative glucose transporters (SLC2A) family, in proportion to its extracellular concentration [17]. To understand the mechanism of insulin-mediated glycemic control, we will briefly review the function of pancreatic beta cells and their transporters (Figure 1).
In the human pancreatic beta cell, glucose uptake and excretion are mediated by distinct SLC2A family members [17]. GLUT1 (SLC2A1) and GLUT3 (SLC2A3) are the main transporters responsible for glucose entry into human beta cells [18]. Their high affinity for glucose (Km ~ 1–5 mM) enables efficient uptake even at low concentrations (<5 mM). GLUT1 is the most abundant in human islets, is considered the key and possibly quantitatively dominant transporter in human beta cells, and thus plays a fundamental role in glucose uptake at basal and moderate levels, maintaining the cell's energy metabolism healthy during fasting states. GLUT2 (Km ~15-20 mM) can also contribute to uptake, especially at higher glucose concentrations (≥10 mM) [19], making it well suited to respond to wide ranges of postprandial plasma glucose concentrations, but its quantitative contribution is less than that of GLUT1/GLUT3 [20]. Functional studies confirm that GLUT2 is indispensable for a complete secretory response to high glucose concentrations, ensuring that glucose influx into the cell is proportional to its extracellular concentration [17]. The uptake process is activated when glycemia rises (e.g., after a meal). Extracellular glucose enters cells passively through facilitative transporters GLUT1 and GLUT3 along their concentration gradients, whereas GLUT2 mediates massive cellular entry in response to a hyperglycemic stimulus [20]. Its constitutive expression is vital for basal cell function. Its dysregulation is linked to beta-cell dysfunction in type 2 diabetes.
Once in the cytosol of β cells, glucose is phosphorylated by glucokinase, which prevents its exit and commits it to the glycolytic pathway; subsequently, its products continue the Krebs cycle in mitochondria. This oxidative metabolism significantly increases the intracellular ATP/ADP ratio. The increase in ATP acts as a key metabolic signal that closes ATP-sensitive potassium channels (KATP) in the plasma membrane [21]. The closure of these channels reduces potassium conductance, depolarizing the cell membrane. When the membrane potential exceeds a critical threshold (~ -40 mV), voltage-dependent calcium channels (mainly L-type) are activated, allowing a massive influx of Ca²⁺ ions from the extracellular space into the cytosol [22]. This sudden increase in cytosolic Ca²⁺ concentration ([Ca²⁺]i) is the final trigger that initiates insulin granule exocytosis. Mature granules containing insulin are mobilized to the plasma membrane, where SNARE proteins (such as SNAP-25, syntaxin-1, and VAMP2) mediate vesicle fusion with the membrane, releasing insulin into the pericellular space and, finally, into the blood capillaries [22]. This stimulus-secretion coupling mechanism is not merely a linear event but is finely modulated by amplifying signals from metabolites (such as glutamate and malonyl-CoA) and incretin hormones (such as GLP-1), which enhance the secretory response by activating second-messenger pathways (cAMP, PKA, EPAC) [23]. Furthermore, GSIS exhibits a characteristic biphasic pattern: a rapid and transient first phase, corresponding to the release of immediately available granules (pool of granules ready for release), and a sustained second phase, dependent on the mobilization and recruitment of reserve granules from a deeper intracellular pool [23]. The integrity of this complex process is crucial; its dysfunction, whether due to defects in glucose sensitivity (GLUT2), mitochondrial ATP generation, KATP channel regulation, or Ca²⁺-dependent exocytosis machinery, is directly associated with the pathogenesis of type 2 diabetes mellitus, a mechanism discussed elsewhere.
3. Structure of GLUT4
Human GLUT4, encoded by the SLC2A4 gene, is a member of the SLC2A facilitator superfamily (Figure 2). It is a passive facilitator that transports glucose down its concentration gradient, without directly coupling to ATP hydrolysis [24]. Determining the complete atomic structure of GLUT4 has been a challenge since its discovery in 1988 (Charron et al., 1989; James et al., 1989). Currently, due to advances in cryo-electron microscopy (cryo-EM), artificial intelligence modeling (AlphaFold), and its high sequence homology (>65%) with other members of the SLC2A superfamily (especially GLUT1 (PDB: 4PYP) and GLUT3 (PDB: 4ZWC), whose structures were previously solved) [24,25], this protein has been modeled with an accuracy of 0.95 Å and a 95% confidence level [26]. Its primary structure of 509 amino acids adopts a barrel conformation formed by 12 transmembrane domains (TMDs), organized into two sheets of 6 alpha helices each (N-terminal sheet and C-terminal sheet), connected by a long intracellular loop that creates a central hydrophilic pore for glucose passage [27]. The N- and C-termini project into the cytosol. The C-terminus, in particular, is a disordered region of approximately 50 residues that acts as a platform for interaction with vesicular trafficking regulatory proteins [28]. GLUT4 oscillates between an outward-facing (exofacial) conformation, which captures glucose, and an inward-facing (endofacial) conformation, which releases it into the cytosol, without direct ATP expenditure [29].
Schematic representation of the 12-transmembrane domain arrangement of the GLUT4 transporter in the lipid bilayer. Transmembrane helices 1, 5, 7, and 11 (highlighted in red) form the central substrate translocation pathway that constitutes the glucose channel. The main cytosolic segments involved in its post-translational regulation and vesicular trafficking are indicated, including the N-terminal motif associated with internalization, the intracellular helical domain, and the C-terminal region that interacts with retention and recycling proteins.
3.1. Kinetics of Glucose Through the GLUT4 Pore: Rocking-Bundle Mechanism
The Km of GLUT4 (5.4 mM) is very close to the fasting blood glucose level (~5 mM), and the Vmax is 3.7 µm/mg/min-1 [30]. Insulin, by increasing the concentration of GLUT4 at the membrane surface, increases the Vmax for glucose entry or exit by up to 10 times, but not its Km [31]. The glucose influx and efflux system is kinetically symmetrical, with equal Km and Vmax [31]. Glucose transport follows the rocking-bundle mechanism (Figure 3). Essentially, the two 6-helix domains function as a rocking gate: they oscillate coordinately between three main conformational states: 1. Outward-open: The binding cavity is exposed to the extracellular space, allowing glucose entry. 2. Occluded: After substrate binding, the pore closes on both sides of the membrane, trapping the glucose. It ensures that the transported molecule is not released prematurely, thereby preventing a futile leak of ions or substrates. 3. Inward-open: The pore opens towards the cytosol, releasing glucose [27]. This cycle is reversible and depends solely on the glucose concentration gradient across the membrane [24].
3.2. Key Residues in the Binding Pore
Unlike other SLC2As, the glucose-binding site of human GLUT4 is formed by a constellation of amino acid residues from multiple transmembrane helices that create a hydrophilic and hydrophobic cavity through which glucose passes. Crystallography and cryo-electron microscopy studies place it at the center of GLUT4, bound to the small-molecule inhibitor cytochalasin B (CCB) (Yuan et al., 2022b), approximately 10-12 Å from the membrane surface, and it is formed by conserved residues from transmembrane domains (TMs) 1, 4, 5, 7, 10, and 11. The most recent and direct structural evidence, obtained by cryo-EM, indicates that the glucose-binding site in GLUT4 is formed by a network of amino acids that includes Asn176, Gln298, Gln299, Trp404, Phe38, Ile42, Ile180, Ile184, Ile303, Phe307, Phe395, Pro401, and Trp428 (Yuan et al., 2022b). Other studies, using mutagenesis and structural homology, identified Trp⁴¹¹ (TM10), Gln¹⁶¹ (TM4), Asn³¹⁷ (TM7), and Trp¹⁶⁸ (TM5) [24]. Glucose (in its D-glucopyranose form) interacts with GLUT4 primarily via its hydroxyl groups (C1, C2, and C3 inward; C4 and C6 outward to the pore), forming a precise hydrogen-bonding network (Ser310, Gln64, and Tyr309) with the C1, C2, and C6 hydroxyls of sugar within the occluded central site [32]. This specific polar environment confers selectivity for D-glucose over other hexoses.
Schematic representation of the three conformational phases of the transport cycle. In phase I (outward-open conformation), the exofacial cavity facilitates the initial capture of D-glucopyranose via polar interactions that promote its entry and partial occlusion of the substrate. In phase II (occluded state), the transporter adopts a closed conformation on both sides of the membrane, stabilizing glucose at the central binding site through a transient network of hydrophilic interactions and aromatic contacts that orient the substrate for translocation. In phase III (inward-open conformation), the endofacial opening reduces substrate affinity and favors its release into the cytosol.
The pore of GLUT4 provides a partially polar environment that stabilizes glucose during transport. The oxygen atoms of glucose's hydroxyl groups form specific hydrogen bonds with Gln64, Gln177, Asn176, Gln298, Gln299, and Ser310 residues [32]. As glucose progresses, the hydrocarbon ring undergoes hydrophobic stacking interactions, notably with Trp404 and Met420 [32,33]. This coordinated network of transient polar and hydrophobic interactions strategically reduces the energy barrier for glucose translocation, facilitating its passive diffusion across the lipid bilayer [32]. For substrate release into the cytosol in the inward-open conformation, characterized by rearrangement of TM5 and TM11, the side chain of Lys²⁰⁰ on the endofacial side of TM5 projects into the binding pocket to electrostatically and sterically perturb the hydrogen-bonding network, lowering glucose affinity and promoting its release into the cytosol [27].
3.4. GSV Storage Sites and Their Binding to the Cytoskeleton
GLUT4 proteins are stored and transported within specialized organelles called GLUT4 Storage Vesicles (GSVs), also known as Insulin-Responsive Vesicles (IRVs, Figure 4) [34,35]. These vesicles form a distinct, preformed pool within fat and muscle cells, designed for rapid mobilization to the cell surface in response to insulin [35]. They appear as small, spherical, or tubulo-vesicular structures with a diameter of approximately 50–80 nm [30,35]. In unstimulated cells, GSVs are retained and tethered intracellularly, often near the Golgi apparatus and endoplasmic reticulum [35]. The function of GSVs depends on their specific molecular composition, which includes a core set of cargo proteins and trafficking and fusion machinery.
The journey of GSVs to the cell surface is a tightly regulated, multi-step process. 1) Release from retention: In basal conditions, GSVs are anchored via TUG proteins. Insulin signaling triggers the cleavage of TUG, releasing the vesicles for transit [35]. 2) Transport to the membrane: The released vesicles are transported along the cytoskeleton. The kinesin motor protein KIF5B is involved in moving them to the cell periphery [35]. 3) Docking and fusion: At the plasma membrane, tethering is mediated by the t-SNARE proteins syntaxin4 and SNAP23. Final fusion requires the GSV's v-SNARE, VAMP2, to bind these t-SNAREs, thereby merging the vesicular membrane with the plasma membrane to expose GLUT4 [36,37].
Under hormonal stimulation, GLUT4 vesicles are rapidly transported to the plasma membrane and fuse with it, enabling rapid glucose uptake from the blood [38]. This intracellular transport and membrane fusion are finely regulated by a complex phosphorylation cascade [39], many of whose components remain unknown. Its sequestration in specific storage vesicles (GSVs) under basal conditions depends on sequence motifs in the intracellular loops at the C-terminal end. The FQQI motif (Phe⁵⁸⁹-Gln⁵⁹⁰-Gln⁵⁹¹-Ile⁵⁹²) at the C-terminus is critical for internalization and intracellular retention, and it interacts with components of the endocytosis complex, such as clathrin [40]. Furthermore, GLUT4 dynamically associates with actin filaments and microtubules via adapter proteins such as TUG (Aspscr1) and the motor protein dynein, which anchor GSVs to the cytoskeleton, preventing their spontaneous fusion with the plasma membrane [28].
4. Factors that Activate GLUT4 Translocation, Endocytosis, and Exocytosis
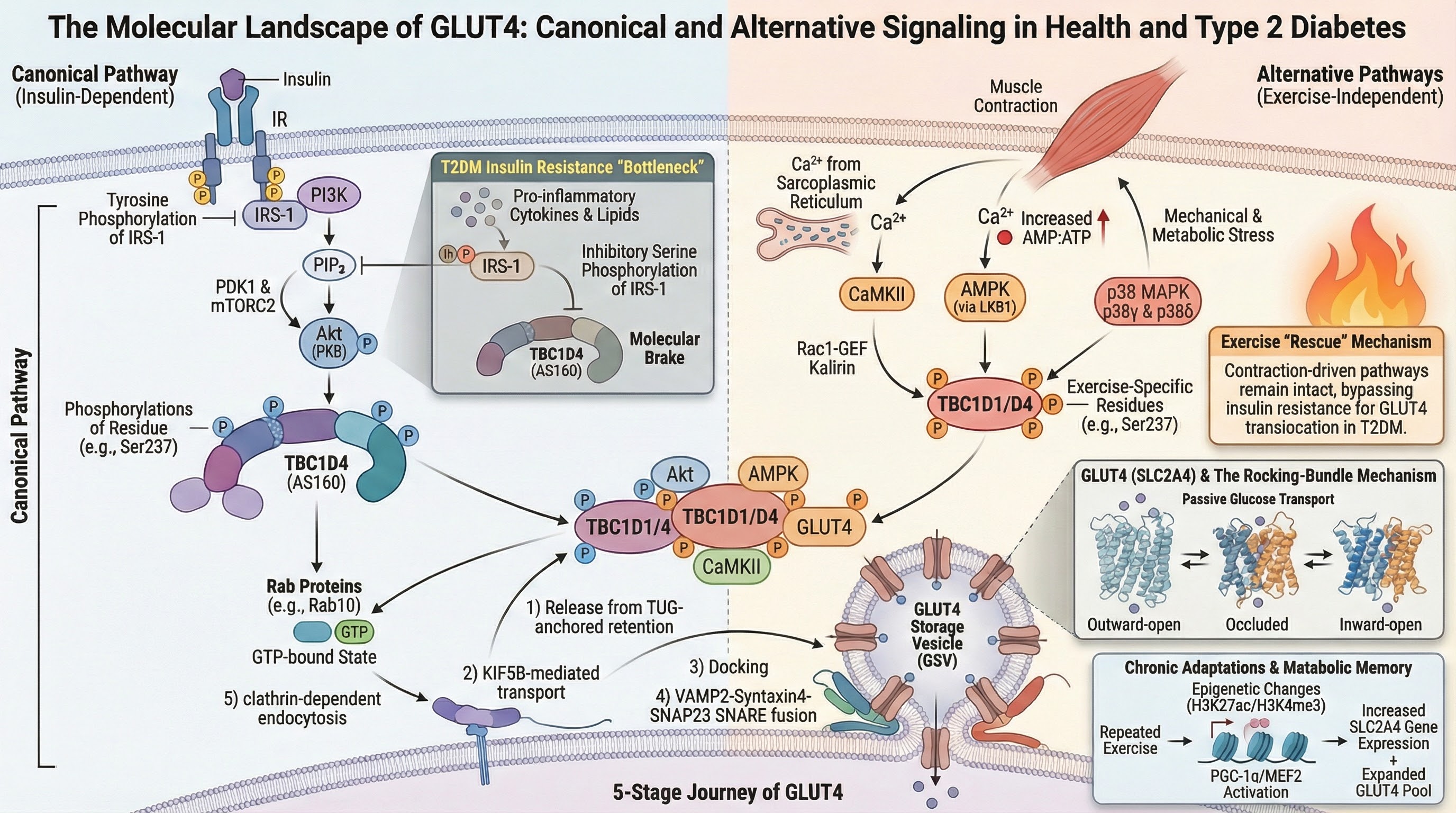
GSV translocation to the cell membrane and its invagination are the rate-limiting steps in glucose uptake, primarily occurring in skeletal muscle and adipose tissue (Figure 4 and Figure 5). The translocation occurs through several finely coupled mechanisms, both insulin-dependent and insulin-independent. These mechanisms activate a set of proteins involved in a signaling and phosphorylation cascade. These proteins intersect with two key substrates: AMP-activated protein kinase (AMPK) and AS160 (TBC1D4), which will be described below.
4.1. Intracellular Localization and Retention Motifs
A distinctive characteristic of GLUT4 is its dynamic recycling, which is regulated by specific sequence motifs (Figure 4). The N-terminal F⁵QQI motif is a key trafficking signal [41]. It serves a dual role by interacting with the μ subunits of different adaptor protein complexes: it binds μ2 of the AP-2 complex to mediate clathrin-dependent endocytosis [40,42], and it binds μ1 of the AP-1 complex for post-endosomal sorting [41]. This motif functions as a suboptimal internalization signal, together with a switch in endocytic pathways, to help fine-tune GLUT4 surface levels in response to insulin [43]. A separate di-leucine motif (LL489⁴⁹⁰) in the C-terminal domain is involved in intracellular retention and sorting from compartments such as the TGN [40]. This interaction recruits the clathrin-mediated endocytosis machinery, leading to constitutive GLUT4 internalization. This is the main mechanism of surface inhibition, or removal, with a membrane half-life of ~5-10 minutes in the absence of insulin. The contiguous C-terminal region (residues 495-509) harbors essential signaling sites that mediate its membrane retention in response to insulin and subsequent intracellular sequestration (recycling). It interacts with resident proteins in GSVs such as Insulin-Regulated Aminopeptidase (IRAP) and the anchoring protein TUG (AS160). The interaction with TUG is key for retaining GSVs in a specialized compartment, possibly anchoring them to the cytoskeleton [28]. This domain also interacts with components of the exocytosis machinery, such as the SNARE proteins Syntaxin 4 and SNAP23, facilitating specific fusion in response to the signal [44]. The dynamic and competitive interaction between the FQQI motif and the C-terminal domain determines the steady-state equilibrium of GLUT4 between the plasma membrane and the intracellular compartment.
4.2. Canonical Pathway of the Insulin-Stimulated Activation Cascade for Glucose Uptake (PI3K/Akt/TBC1D4)
Insulin secreted by pancreatic β cells is the primary stimulus for GLUT-4 translocation to the membrane and cellular glucose uptake, with skeletal muscle being the predominant tissue that uses glucose as fuel (~80%), while adipose and cardiac muscle have a lower contribution [39,45]. Insulin binding to its receptor activates a phosphorylation cascade culminating in the activation of the protein kinase Akt, also known as PKB. Akt phosphorylates effector proteins, such as AS160 (TBC1D4), inactivating their GAP (GTPase-activating protein) activity towards small Rab proteins. Activated (GTP-bound) Rab10 and Rab14 then recruit GSVs to the plasma membrane [4]. The fusion site with the membrane involves specific interactions between SNARE proteins on the vesicle (such as VAMP2) and on the plasma membrane (such as Syntaxin4 and SNAP23), facilitating transporter exposure on the cell surface [46]
This is the best-characterized endocrine pathway, essential for glucose homeostasis in the postprandial state. Its activation leads, within minutes, to massive GLUT4 translocation (Figure 5). It begins with insulin binding to the α subunit of its tyrosine kinase receptor (IR) on the cell membrane's external face, inducing a conformational change in the β subunit that exposes activation and autophosphorylation sites on tyrosine residues [47]. IRS proteins (1-4) are recruited to the phosphorylated domains of the IR via their PTB (phosphotyrosine-binding) domains and are phosphorylated on multiple tyrosine residues. Phosphorylation of Tyr612 and Tyr632 residues on IRS-1 recruits and activates class Ia PI3K (regulated by the p85 subunit, catalyzed by p110) [48]. Activated PI3K phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP₂) at the 3-position of the inositol ring, generating phosphatidylinositol (3,4,5)-trisphosphate (PIP₃). PIP₃ acts as a lipid second messenger that recruits proteins with pleckstrin-homology (PH) domains to the plasma membrane [49]. Both PDK1 (3-phosphoinositide-dependent protein kinase-1) and protein kinase B (Akt/PKB) possess PH domains that bind PIP₃ with high affinity, recruiting them to the plasma membrane. PDK1 phosphorylates Akt at the key residue Thr308 in the activation loop. This phosphorylation is necessary but not sufficient for maximum activity (Figure 5). A second phosphorylation, typically on Ser473 (in the C-terminal hydrophobic domain), is mediated by the mTORC2 complex (mammalian target of rapamycin complex 2) and is essential for full activation and substrate specificity of Akt [50].
4.3. TBC1D4 (AS160) as a Basal Translocation Suppressor
The most relevant Akt substrate for GLUT4 translocation is TBC1D4, also known as AS160 (Akt substrate of 160 kDa). In skeletal muscle, its close relative, TBC1D1, plays a parallel and often predominant role [51]. TBC1D4 is a GTPase-activating protein (GAP). Its GAP domain specifically targets a subset of small Rab GTPases, including Rab8A, Rab10, Rab13, and Rab14. Under basal conditions, the GAP activity of TBC1D4 catalyzes the hydrolysis of GTP bound to these Rabs to GDP, keeping them inactive (GDP-bound). Since GTP-bound Rabs are master regulators that recruit effectors for vesicle movement, anchoring, and fusion, this GAP activity constitutes a molecular brake that keeps GSVs immobilized and sequestered [52]. Akt phosphorylates TBC1D4 on at least 6 residues (Ser318, Ser341, Ser570, Ser588, Thr642, Ser751, in humans). Phosphorylation at Ser588 and Thr642 appears to be particularly critical [53]. The phosphorylation of Ser588 and Thr642 induces a conformational change that inhibits TBC1D4's GAP activity. As a result, GSV-associated Rabs remain active (GTP-bound) for longer.
4.4. Release of GSVs and Their Membrane Fusion
The release of the brake imposed by TBC1D4 allows GTP-bound Rabs to orchestrate the final steps of GLUT4 exocytosis: the insulin signal, possibly through Akt, induces ubiquitination and proteasomal degradation of the TUG protein. TUG acts as a physical chain that retains GSVs anchored to intracellular structures (possibly the Golgi or the cytoskeleton). Its removal releases the vesicles for transport [28].
GTP-bound Rabs (particularly Rab8A and Rab10) recruit motor complexes that act on the cytoskeleton. Rab10 recruits myosin Va, a motor that moves along actin filaments, transporting GSVs towards the cell cortex underlying the plasma membrane [54]. A role for microtubules and the CLASP2 protein in this transport is also suggested [55]. Subsequently, GTP-bound Rabs recruit anchoring complexes, such as the large octameric exocyst complex. This 8-subunit complex acts as a scaffold that physically brings the vesicle closer to the plasma membrane and maintains it in proximity, preparing it for fusion [56]. However, evidence suggests that not all GSVs are identical. There may be subpopulations with distinct protein composition or intracellular location that respond with greater or lesser preference to one stimulus or another. Furthermore, insulin and contraction may activate specific Rabs with distinct affinities. While insulin appears to critically depend on Rab10 in adipocytes [57], muscle contraction may make more prominent use of Rab8A and Rab13 [57]. Simultaneous activation of multiple Rabs could recruit a broader set of motor and fusion effectors
The fusion of GSVs to the membrane, mediated by Soluble NSF Attachment Protein Receptors (SNARE), is the final and irreversible step. SNAREs located on GSVs (v-SNAREs) and the target membrane (t-SNAREs) form a stable 4-alpha-helix bundle. The formation of the trans-SNARE complex (VAMP2-Syntaxin4-SNAP23) between the two membranes exerts a mechanical force that overcomes the repulsion between lipid bilayers, fusing them. The regulatory SM protein Munc18c is essential: it acts as a scaffold and chaperone, facilitating the correct folding of Syntaxin 4 and its interaction with VAMP2 [58]. After fusion, GLUT4 is incorporated into the plasma membrane; its transport pore is exposed to the extracellular space, allowing it to begin transporting glucose. This process is highly efficient and recruits most of the intracellular GLUT4 pool within 10-30 minutes.
5. AMPK-Independent GLUT4 Translocation Mechanisms
Regarding GLUT4 synthesis, insulin plays a less potent, indirect role than exercise. While the Akt/mTORC1 pathway can stimulate global protein translation and, in some contexts, modulate transcription factors, its direct effect on transcription of the GLUT4 gene (SLC2A4) is minimal. Paradoxically, chronic hyperinsulinemia associated with insulin resistance reduces GLUT4 expression, especially in adipose tissue, through mechanisms involving the transcription factor FOXO1 [59]. Therefore, it is crucial to highlight the existence of pathways completely independent of AMPK for GLUT4 translocation and glucose uptake. Mice with a double muscle knockout of the catalytic α1 and α2 subunits of AMPK show completely normal insulin-stimulated glucose uptake, both in vivo and ex vivo [60,61]. This demonstrates that AMPK is not a necessary intermediary in insulin signaling. (Figure 6).
5.1. Insulin-Independent Mechanisms: The Muscle Contraction Pathway
Muscle contraction is the most relevant physiological stimulus for increasing glucose uptake in skeletal muscle, independently of insulin. This pathway is complex and redundant and converges on the same endpoint as insulin, AMPK, and TBC1D1:
- -
- AMPK Pathway: The increase in AMP/ATP during exercise activates AMPK. AMPK phosphorylates and activates AS160 (TBC1D4), which inhibits its GAP (GTPase-activating) activity towards Rab proteins, thus allowing active Rabs (such as Rab8A, Rab13, Rab14) to promote GSV translocation [62].
- -
- Ca²⁺/CaMK Pathway: The calcium flux during contraction activates calcium/calmodulin-regulated kinase (CaMK). CaMK can also phosphorylate AS160 and another related protein, TBC1D1, which disinhibits GLUT4 translocation.
- -
- p38 MAPK Pathway: Mechanical and metabolic stress activate this pathway, which contributes to GLUT4 translocation and transcriptional regulation
The inhibition of the Rab-GTPase-activating proteins (GAPs) TBC1D1 and TBC1D4 is a critical mechanism for increasing glucose uptake in skeletal muscle, and this inhibition is achieved by distinct kinases with unique kinetics and specificity [62]. During muscle contraction, T-tubule membrane depolarization activates dihydropyridine receptors (DHPR), which in turn activate ryanodine receptors (RyR) on the sarcoplasmic reticulum, releasing a large amount of Ca²⁺ ions into the cytosol. The increase in Ca²⁺ binds to calmodulin (CaM), and the Ca²⁺/CaM complex activates CaMKII kinase. CaMKII is a serine/threonine kinase with a unique multimeric structure that allows it to retain activation memory through autophosphorylation at Thr287 after repeated Ca²⁺ pulses, making it ideally suited to respond to changes in contraction frequency [63]
While CaMKII is activated by contraction, recent evidence indicates it does not directly phosphorylate TBC1D1 at its key regulatory site. Instead, the energy-sensing AMP-activated protein kinase (AMPK) is the primary kinase responsible for phosphorylating TBC1D1 on specific residues in response to contraction, including Ser231 and Ser660 in mice (equivalents to Ser237 and Ser700 in humans) [64,65]. This phosphorylation inhibits TBC1D1's GAP activity, promoting GLUT4 translocation to the cell membrane. Experimental evidence from mice with a muscle-specific AMPK kinase-dead mutation shows that contraction-stimulated phosphorylation of TBC1D1 at these sites is greatly reduced [64,65]. Furthermore, mice expressing a non-phosphorylatable TBC1D1 mutant (Ser231Ala) display an impaired glycemic response to the AMPK activator AICAR, although interestingly, their response to actual exercise is preserved, suggesting compensatory mechanisms exist in vivo [66].
CaMKII contributes to contraction-stimulated glucose uptake through a different, indirect mechanism. Research now demonstrates that CaMKII activates a signaling cascade involving the Rac1-GEF Kalirin and the small GTPase Rac1, which subsequently leads to Akt phosphorylation [63]. Using specific CaMKII inhibitors (such as KN-93) or expressing dominant-negative CaMKII in muscle cells significantly reduces contraction-induced Akt phosphorylation and glucose uptake, confirming the physiological relevance of this pathway [63]. Thus, contraction engages two parallel signaling axes: an AMPK-TBC1D1 pathway that directly regulates GLUT4 vesicle trafficking and a CaMKII-Kalirin-Rac1-Akt pathway that modulates the actin cytoskeleton and vesicle translocation [63].
Regarding mechanical stress, muscle contraction, particularly eccentric or high-intensity contraction, generates mechanical forces, relative hypoxia, and reactive oxygen species (ROS). These distinct but concurrent signals converge to activate the mitogen-activated protein kinase (MAPK) family [67]. Unlike the ubiquitously expressed p38α (MAPK14) and p38β (MAPK11) isoforms, which are often associated with inflammatory and stress responses, the p38γ (MAPK12) and p38δ (MAPK13) isoforms are abundantly expressed in skeletal muscle and are preferentially activated by contractile stimuli [68,69]. This specific activation links contractile stress to the regulation of metabolic proteins, including the Rab-GTPase activating proteins TBC1D1 and TBC1D4 (also known as AS160). Evidence suggests at least two mechanisms by which this signaling axis promotes glucose uptake:
- -
- Direct phosphorylation of TBC1D1/D4: p38 MAPK phosphorylates TBC1D1 and TBC1D4 at sites distinct from those targeted by Akt and CaMKII. Human studies have identified exercise-specific residues, such as Ser237 and Ser660 in TBC1D1, that contribute to a unique exercise-induced phosphorylation code not replicated by insulin [70,71].
- -
- Potential activation of downstream kinases: p38 MAPK is a canonical activator of MAPK-activated protein kinase 2 (MK2). It is hypothesized that MK2, once activated, may further phosphorylate TBC1D1 on additional residues (e.g., Ser695), thereby amplifying the signal from the contracting muscle [72].
Phosphoproteomic studies of human muscle biopsies confirm that exercise induces a unique phosphorylation signature on TBC1D1, distinct from that induced by insulin [71]. The physiological relevance of this pathway is underscored by rodent studies showing that muscle-specific deletion of p38γ results in a substantial defect in contraction-stimulated glucose uptake (e.g., a reported ~45% reduction), without impairing insulin signaling [70]. This establishes p38γ as a bona fide and essential mediator of exercise-induced glucose metabolism, independent of the canonical insulin pathway.
Nitric oxide (NO) produced by neuronal nitric oxide synthase (nNOS) during contraction may promote glucose uptake by S-nitrosylation (covalent modification of cysteine residues) of proteins involved in glucose trafficking. An identified target is Vasodilator-Stimulated Phosphoprotein (VASP), whose S-nitrosylation at Cys137 facilitates actin remodeling, necessary for vesicular transport [73]. On the other hand, ROS, traditionally considered harmful, act as signaling molecules in this context. ROS can directly activate p38 MAPK and, to a lesser extent, AMPK. Their generation during exercise contributes to pro-translocation signaling [74]. These mechanisms ensure that, even in states of insulin resistance (where the PI3K/Akt pathway is attenuated), muscle can recruit GLUT4 to the membrane to meet its energy demand.
Representation of the human AMPK complex with detail of the α2 subunit activation loop. The magnification shows Thr172 in its phosphorylated state (pThr172), with the phosphate group coordinated by hydrogen bonds that stabilize the activation segment. This post-translational modification is essential for the kinase domain to adopt the catalytically competent conformation.
5.2. Genetic Evidence of Insulin- and AMPK-Independent Glucose Uptake
The most eloquent experimental evidence for this mechanism comes from genetically modified mouse models. Double knockout mice for AMPKα1 and AMPKα2 (dKO) show that glucose uptake induced by muscle contraction ex vivo (or by exercise in vivo) is reduced by only 40-50% [60]. This demonstrates that at least half of this response is mediated by mechanisms totally independent of AMPK. In insulin resistance models (such as ob/ob mice or high-fat diets), the capacity of exercise to stimulate glucose uptake is maintained or even potentiated, while the response to insulin is clearly attenuated [7]. These findings underscore that contraction pathways (CaMKII, p38 MAPK) constitute an essential and robust reserve system, evolutionarily conserved to ensure energy supply to working muscle, independent of hormonal status. On the other hand, AMP-activated protein kinase (AMPK) is an evolutionarily conserved master sensor that acts as the guardian of metabolic balance [75]. Its activation coordinates a broad catabolic response to restore ATP levels while suppressing costly anabolic processes. Although AMPK can be activated during muscle contraction, it represents a parallel, separable, and highly regulated signaling pathway that functions primarily as an integrator of cellular energy status rather than as a simple intermediary of insulin or contraction [60].
5.3. Canonical Activation Mechanism by Adenine Nucleotides
AMPK exists as a heterotrimer composed of a catalytic α subunit, a scaffolding β subunit, and a regulatory γ subunit, with multiple isoforms of each (α1, α2; β1, β2; γ1, γ2, γ3), conferring tissue and functional diversity. The α subunit contains the N-terminal kinase domain, an autoinhibitory domain (AID) that keeps the enzyme inactive under basal conditions, and a binding domain for the β/γ subunits at its C-terminal end. The β subunit acts as a central scaffold, containing a carbohydrate-binding module (CBM) that directs AMPK to glycogen granules, localizing it near its substrate and energy sources [76]. The γ subunit is the energy-sensing module. It contains four CBS (cystathionine β-synthase) motifs that form three binding sites (sites 1, 3, and 4) for adenine nucleotides. The competitive binding of these ligands constitutes the core of canonical regulation [77]. However, the central activation event of AMPK is the phosphorylation of the Thr172 residue in the activation loop of the α subunit. The degree of phosphorylation at Thr172 directly determines catalytic activity and is governed by a dynamic balance between upstream kinases and phosphatases.
The canonical mechanism is triggered by a cellular energy deficit, characterized by increased AMP:ATP and ADP:ATP ratios (e.g., during exercise, ischemia, or hypoxia). This increase has three cooperative and synergistic effects: (1) Direct allosteric activation, in which AMP binding to sites on the γ subunit induces a conformational change that partially counteracts autoinhibition by the AID, directly increasing kinase activity (~5 times), even without increased Thr172 phosphorylation [78]. (2) Facilitation of Thr172 phosphorylation: The binding of AMP (and, to a lesser extent, ADP) makes the AMPK complex a much better substrate for upstream regulatory kinases, such as LKB1, favoring stabilization of the phosphorylated conformation. (3) AMP/ADP binding makes the phosphorylated complex a worse substrate for protein phosphatases (inhibition of dephosphorylation), mainly PP2A and PP2C, which would otherwise dephosphorylate and inactivate AMPK by removing the phosphate from Thr172 [75]. ATP, at high concentrations, acts as an antagonist, competing for the same binding sites on the γ subunit; it favors an inactive conformation and promotes dephosphorylation. This exquisite system makes AMPK a sensor that responds to minimal changes (even 1-2%) in cellular adenylate charge
5.4. Upstream Regulatory Kinases
Upstream phosphorylation of AMPK α at Thr172 is absolutely essential for full activation (>100x) [77]. Its phosphorylation is carried out by at least three main kinases, whose relevance depends on cell type and stimulus: (1) Liver Kinase B1 (LKB1/STK11): It is the most important constitutive kinase for the response to energy stress. LKB1 forms a stable, obligatory complex with the accessory proteins STRAD and MO25, which are essential for its activity and localization [61]. LKB1 is constitutively active and monitors cellular energy status, phosphorylating AMPK when AMP/ADP binding favors an active conformation. Metformin, which mildly inhibits mitochondrial respiratory chain complex I, subtly increases the AMP:ATP ratio and activates AMPK in an LKB1-dependent manner [79] . Fasting, caloric restriction, and exercise contribute significantly to AMPK activation during contraction, especially in response to ATP consumption. (2) During muscle contraction, the increase in intracellular calcium ([Ca²⁺]i), independently of changes in adenine nucleotides, activates Calcium/Calmodulin-dependent Protein Kinase Kinase β (CaMKKβ), which in turn activates AMPK. This pathway is particularly relevant in neurons, where depolarization and Ca²⁺ entry activate AMPK to meet the energy demand of synaptic signaling. During muscle contraction, it works in synergy with LKB1. While LKB1 responds to ATP deficit, CaMKKβ responds to the initial Ca²⁺ signal released during contraction. Studies in mice with muscle-specific LKB1 deletion show that approximately 60% of contraction-induced AMPK activation is preserved, mainly due to CaMKKβ [80].
5.5. Role of AMPK in GLUT4 Regulation: Contextual Contribution
As already mentioned, AMPK is not the main pathway for insulin- or contraction-mediated activation but rather a modulatory contributor that augments other signals. Acutely, AMPK activates direct phosphorylation of TBC1D1 at residue Ser237 (Ser231 in mouse) [81]. This phosphorylation inhibits TBC1D1 GAP activity, facilitating Rab activation and GLUT4 translocation. Its effect is additive to phosphorylation mediated by CaMKII and p38 MAPK during contraction. In muscle, the contribution of AMPK to contraction-induced glucose uptake is approximately 30-40%, as demonstrated by the partial reduction observed in AMPKα1/α2 dKO mice [60]. AMPK can chronically induce GLUT4 gene expression in the long term through indirect mechanisms, such as activation of PGC-1α, since AMPK phosphorylates and activates PGC-1α, the master coactivator of mitochondrial biogenesis and metabolic gene expression, including SLC2A4 [82], and epigenetic modulation: AMPK phosphorylates and inhibits histone deacetylases like HDAC5, alleviating chromatin repression on the GLUT4 promoter and other metabolic genes [83].
AMPK is also not simply an exercise mimetic. Its chronic and systemic activation can have adverse effects, such as (1) lack of tissue specificity, as classic activators (AICAR, metformin) act in multiple organs. (2) Undesired cardiac activation, since potent AMPK activation in the heart (e.g., with MK-8722) can induce cardiac hypertrophy and glycogen accumulation, effects not observed with exercise [84]. (3) Inhibition by counterregulatory hormones, such as glucagon and adrenaline, activates PKA, which phosphorylates AMPKα at Ser173 (adjacent to Thr172), inhibiting AMPKα activation by LKB1. This prevents AMPK from activating glucose uptake when the body needs to mobilize energy [85]. Therefore, targeting AMPK lies in developing complex-specific allosteric activators (e.g., acting only on complexes containing the muscle α1β1γ1 subunit) or safer indirect modulators. AMPK activation by exercise may be slightly attenuated in obesity and T2DM, possibly due to inflammation or endoplasmic reticulum stress. However, its activation through drugs like metformin remains effective and constitutes one basis of therapy [86].
6. GLUT4 Synthesis and Regulation of Exercise-Induced Translocation Pathways
Physical exercise and stress are well-recognized stimuli for increasing glucose uptake in skeletal muscle via insulin-independent GLUT4 translocation (Figure 7). Both the intensity and duration of exercise modulate this process. Numerous studies have demonstrated that acute and chronic exercise, including both moderate- and high-intensity protocols, increases GLUT4 content at the cell membrane, thereby improving insulin sensitivity and glycemic control in various populations, including those with metabolic disorders [87,88,89]. This effect is mediated by several signaling pathways, most notably AMPK and Akt, as well as by changes in transcription factors such as myocyte enhancer transcription factor 2A (MEF2A) [87,90]. The extent and duration of GLUT4 translocation can vary with exercise variables, with indications that higher-intensity or longer-duration exercise may result in greater or more sustained elevations [88,89]. Well, some studies note that these effects can be transient or influenced by underlying health conditions [91]. Overall, the literature supports a robust link between exercise (both intensity and duration) and enhanced GLUT4 trafficking to the cell membrane. High-intensity protocols tend to elicit greater acute responses than moderate-intensity ones; however, regular moderate activity also produces significant cumulative benefits over time [88,89]. The underlying molecular mechanisms involve rapid activation of AMPK during energetic stress (intense/long-duration activity), as well as chronic upregulation of transcriptional regulators such as MEF2A that enhance overall cellular capacity for glucose transport [87].
Membrane depolarization and excitation-contraction coupling generate calcium signals, energy stress, and oxidative stress, thereby activating CaMKII, AMPK, and p38γ/δ. These kinases converge on the phosphorylation of TBC1D1/4, promoting the activation of Rab GTPases and the translocation of GLUT4 to the plasma membrane in an insulin-independent manner.
The total amount of GLUT4 protein in the cell is the primary determinant of the maximum capacity for glucose uptake. In addition to triiodothyronine (T3), muscle contraction and endurance exercise are the most potent stimuli for increasing SLC2A4 gene expression in skeletal muscle [92,93]. This effect is mediated by a well-defined signaling cascade that links acute and chronic contraction events to lasting changes in gene expression. The increase in total GLUT4 content is a fundamental adaptation to chronic aerobic training, expanding the system's reserve capacity. This regulation is predominantly transcriptional and operates on a timescale of hours to days.
It has been shown that each exercise session activates the signaling pathways described above: increased Ca²⁺ (via calcineurin and CaMK), activation of p38 MAPK γ/δ, and AMPK. These kinases converge on the regulation of the transcriptional coactivator PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha), considered the master regulator of mitochondrial and metabolic adaptations to exercise (Handschin & Spiegelman, 2006). p38 MAPK directly phosphorylates PGC-1α, increasing its stability and transcriptional activity. AMPK phosphorylates PGC-1α and also induces its expression at the transcriptional level. Ca²⁺ signals activate enzymes such as calcineurin and CaMKIV, which dephosphorylate and activate transcription factors (such as CREB) that, in turn, induce the expression of the PPARGC1A gene (which encodes PGC-1α). PGC-1α alone has limited DNA-binding capacity. Its main function is to recruit specific promoters by interacting with anchor transcription factors. For SLC2A4, the most important factor is MEF2 (Myocyte enhancer factor 2), mainly isoforms A and D. MEF2 binds to specific response elements in the SLC2A4 promoter. PGC-1α binds to MEF2 and acts as a bridge, recruiting a vast assembly of coactivator machinery, including chromatin remodeling complexes such as SWI/SNF, which displace nucleosomes, making DNA accessible. Histone-modifying enzymes, such as histone acetyltransferases (HATs) p300 and CBP, which add activation marks (acetylation at H3K27) to create a permissive chromatin environment [81]. And the basal transcription complex (RNA Pol II and associated factors). All of these drastically enhance transcription [81] and epigenetic changes (metabolic memory): repeated exercise induces lasting modifications in the chromatin of the SLC2A4 promoter, including increases in the activation marks H3K27ac and H3K4me3. These changes prime the gene for a faster and more sustained transcriptional response in subsequent exercise sessions [94]. As a result, this enhanced PGC-1α/MEF2 complex efficiently recruits RNA polymerase II, leading to a dramatic increase (up to 2-3-fold) in GLUT4 mRNA transcription. This additional mRNA is translated into a new GLUT4 protein, expanding the total pool of available GSVs for future stimuli (both insulin and contraction). This effect peaks between 24 and 48 hours post-exercise and can remain elevated for several days with regular training.
6.1. Exercise-Induced Metabolic Memory
Recent research has revealed that exercise not only activates transcription but also induces lasting epigenetic modifications at the SLC2A4 gene locus, creating a molecular memory that facilitates faster and more potent responses in subsequent sessions [95]. In the case of GLUT4, a single exercise session triggers GLUT4 translocation from intracellular depots to the plasma membrane and T-tubules, a process orchestrated by insulin-independent signaling pathways involving molecules like AMPK, Ca²⁺, nitric oxide, and distal activation of SNARE proteins and the cytoskeleton [7]. Critically, this acute translocation pathway remains intact even in states of insulin resistance, such as type 2 diabetes, where insulin-dependent signaling is compromised, thereby allowing exercise to remain a potent stimulus for glucose uptake [16]. An original study demonstrated that a single exercise session in humans induces a rapid increase in H3K4me3 methylation (a mark associated with active promoters ready for transcription) and H3K27ac acetylation (an active enhancer mark) at the SLC2A4 promoter [94].
The chronic dimension of metabolic memory arises from the ability of repeated physical training to sustainably increase the total expression of the GLUT4 protein in muscle fibers, thereby amplifying the pool available for future contraction and insulin stimulation [7,95]. This long-term adaptive effect is governed by profound epigenetic and transcriptional mechanisms. Exercise-sensitive kinases, like AMPK and CaMKII, regulate GLUT4 gene (SLC2A4) expression through the HDAC4/5-MEF2 axis; these kinases phosphorylate and promote nuclear export of class II histone deacetylases (HDAC4/5), relieving transcriptional repression and leading to histone hyperacetylation at the GLUT4 promoter, facilitating a permissive chromatin state and increased gene transcription [7,96]. Therefore, physical training consolidates the molecular memory by inducing stable epigenetic modifications and increasing basal GLUT4 content, thereby expanding and sustaining the capacity to recruit transporters to the membrane. Together, these acute (efficient translocation via intact pathways) and chronic (epigenetically regulated transcriptional upregulation) adaptations of GLUT4 induced by exercise synergize to establish an enhanced metabolic reserve. This functional memory not only explains the sustained improvements in insulin sensitivity and glucose homeostasis observed after training but also underscores the superiority of exercise as an intervention to counteract specific defects in insulin signaling in metabolic diseases, offering a resilient and potentially effective mechanism for glycemic control [7,16,97]. These epigenetic changes can persist for hours or even days after the acute stimulus ceases, priming the gene for more efficient activation. With chronic training, these marks can become more stable, contributing to the sustained phenotypic adaptation observed in many athletes.
7. Molecular Pathophysiology of T2DM
T2DM is not a state of generalized failure of glucose uptake, but rather a specific imbalance in certain regulatory pathways. Insulin resistance in skeletal muscle, a central defect in type 2 diabetes and obesity, is characterized by an early and selective alteration of the canonical PI3K/Akt signaling pathway necessary for the translocation of the glucose transporter GLUT4, thereby compromising insulin-stimulated glucose uptake [4,98]. This impairment frequently begins at the insulin receptor (IR) and its substrates (IRS), where phosphorylation at tyrosine residues (which activates the signal) is decreased, while inhibitory phosphorylation at serine residues of IRS-1 (e.g., Ser307) is increased, mediated by lipid-activated kinases (PKCθ) and inflammation (JNK, IKKβ) [98,99]. Another inhibitory phosphorylation of IRS-1 is promoted by intracellular lipids (e.g., diacylglycerol, ceramides) and inflammatory cytokines (e.g., TNF-α), which activate kinases such as PKCθ, JNK, and IKKβ that phosphorylate IRS-1 on serine residues (e.g., Ser307, Ser312) [100]. Serine phosphorylation prevents insulin receptor-mediated tyrosine phosphorylation and promotes IRS-1 degradation [99,101]
This inhibitory modification impairs the interaction between IRS-1 and the IR, reduces recruitment and activation of phosphatidylinositol 3-kinase (PI3K), and consequently decreases the generation of the key second messenger, PIP3, at the plasma membrane [4]. The decrease in PIP3 reduces Akt kinase (PKB) activation, particularly the phosphorylation of its Thr308 residue, which is crucial for its full activity [98]. Inhibited Akt kinase reduces phosphorylation of its critical substrates involved in GLUT4 trafficking, such as the GAP protein TBC1D4 (AS160). Under normal conditions, phosphorylation of TBC1D4 by Akt inhibits its GAP activity, allowing Rab GTPases (like Rab10) to remain in their active state (GTP-bound) and promoting the transport, anchoring, and fusion of GLUT4 storage vesicles (GSVs) with the plasma membrane [4,102]. Therefore, a compromised Akt signal leads to excessive GAP activity of the 160-kDa Akt substrate (TBC1D4), keeping Rabs inactive and creating a bottleneck that prevents massive GLUT4 translocation to the cell surface.
Another alteration is the pro-inflammatory state associated with chronic obesity, a central driver of insulin resistance. Furthermore, evidence suggests that intrinsic and early defects in vesicular trafficking machinery, such as inefficient SNARE fusion or altered exocyst recruitment, may coexist and exacerbate glucose uptake deficiency, even in the presence of residual proximal signaling [102]. Another cause is decreased GLUT4 mRNA levels, which reduce the total pool available for translocation, limit maximal translocation, and reduce glucose uptake capacity. Garvey et al. (1991) demonstrated a ~40% reduction in GLUT4 mRNA in adipocytes from patients with type 2 diabetes, even when muscle expression was normal [103]. This suggests tissue-specific regulation. Together, these early molecular defects in the insulin cascade, ranging from IRS-1 dysregulation to alterations of final vesicular trafficking effectors, lead to a marked reduction in GLUT4 density on the myocyte membrane, establishing the pathophysiological basis for fasting and postprandial hyperglycemia characteristic of insulin-resistant states [98,99].
Protectively, muscle contraction pathways remain largely functional: this is a crucial finding from a pathophysiological and therapeutic perspective. The Ca²⁺/CaMKII and p38 MAPK γ/δ mechanisms are preserved or even relatively potentiated in the muscle of individuals with T2DM [80,104]. This explains the clinical enigma of why patients with T2DM, highly resistant to insulin action, respond almost normally to the exercise stimulus, increasing glucose uptake. The emergency system, evolved to fuel working muscle, remains operational when the normal hormonal system fails.
7.1. Alterations in GSV Storage, Retention, and Fusion
Translocation of GLUT4, stored in intracellular GSVs under basal conditions, can be altered by defects in proteins of the TUG (Tether containing UBX domain for GLUT4) family, AS160 (Akt substrate of 160 kDa), or in the GAPDH protein, compromising the formation, stability, or intracellular retention of GLUT4 clusters, and as a consequence, GLUT4 is not efficiently retained and leaks into constitutive recycling pathways, reducing the pool specifically mobilizable by insulin. Yu et al. (2012) showed that the TUG protein mediates GLUT4 retention in a storage compartment distinct from the recycling endosome. Disruption of TUG leads to increased basal translocation and decreased insulin response (Yu et al., 2007).
Even when GSVs are translocated to the cell cortex, they require the SNARE machinery (e.g., VAMP2, Syntaxin4, SNAP23) to fuse. Altered expression or function of these components, or their regulators (such as Munc18c), will prevent GLUT4 from fusing with the membrane. Consequently, GSVs accumulated in the periphery cannot release their contents, a phenomenon sometimes termed "docking without fusion". The work of Kawanishi et al. (2000); in adipocytes from diabetic db/db models showed decreased expression of syntaxin 4 and SNAP23, correlated with impaired fusion of GLUT4-containing vesicles [105].
7.2. Alterations in Endocytosis and Recycling
We must consider that endocytosis and re-sorting of GLUT4 into GSVs are crucial steps in its recycling. Defects in endocytosis (e.g., due to alterations in clathrin, dynamin, or the internalization motif of GLUT4) can lead to loss of GLUT4 from the cell surface or trafficking to degradative compartments. This defect can cause both chronic loss of the transporter and an inability to replenish the GSV pool after stimulation. Blot & McGraw (2008) demonstrated that a single residue (Phe5) in the intracellular loop of GLUT4 is critical for its efficient internalization. Mutations in this motif disrupt the entire cycle and the insulin response [40]. Therefore, coordination in the cortical actin network is essential for the translocation and anchoring of GSVs near the plasma membrane. In states of insulin resistance, actin organization is altered, hindering final vesicle movement and fusion and contributing to the post-receptor defect. Omata et al. (2000) showed that insulin reorganizes actin in adipocytes and that pharmacological disruption of this cytoskeleton blocks GLUT4 translocation, independently of insulin signaling [106]. In summary, intrinsic defects in the GLUT4 cycle represent a fundamental cellular pathology that goes beyond mere insulin signaling and partly explains the irreversibility and progression of insulin resistance in key tissues.
8. Pharmacological Regulators
Knowledge of the mechanisms indicated above has driven the development of specific drugs that act as direct and indirect activators of GLUT4 translocation and glucose uptake. As direct activators (binding to the complex), we have AICAR, a prodrug that is intracellularly converted to ZMP, an AMP analog that activates AMPK in an LKB1-dependent manner. And A-769662 and 991, which bind to a distinct allosteric site at the α-β interface, stabilizing the phosphorylated form and inhibiting dephosphorylation. Their action can be independent of LKB1. As indirect activators (inducers of energy stress), we have metformin, a mild inhibitor of mitochondrial complex I. Its activation of AMPK is indirect, mediated by increased AMP/ADP and LKB1. Berberine and resveratrol are natural compounds with multiple mechanisms of action, including mitochondrial inhibition and modulation of SIRT1/AMPK. However, due to complex physiology, all drugs are non-specific and present various complications. For example, chronic and systemic AMPK activation can have adverse effects (e.g., cancer, autoimmune problems, cardiac hypertrophy) [107]. The future will lie in developing complex-specific activators (e.g., for muscle α1β1γ1) or tissue-targeted delivery systems.
8.1. Agonists of the Nuclear Receptor PPARγ
Thiazolidinediones (TZDs: rosiglitazone, pioglitazone) are a class of drugs that improve insulin sensitivity. Their primary mechanism is totally independent of acute insulin signaling pathways (PI3K/Akt) and AMPK, operating at the level of gene regulation [104]. TZDs are synthetic high-affinity ligands for the nuclear receptor PPARγ (peroxisome proliferator-activated receptor gamma), abundantly expressed in adipocytes. Upon ligand binding, PPARγ heterodimerizes with the retinoid X receptor (RXR). This complex binds to specific DNA sequences, called PPAR response elements (PPRE), located in the promoter region of the SLC2A4 gene. The ligand-bound PPARγ/RXR complex recruits coactivators (interestingly, including PGC-1α), which in turn recruit HATs and transcription machinery, inducing a massive and specific increase in GLUT4 transcription in adipose tissue. Implications: This effect is slow (requires days to manifest as increased protein) and is direct. This partly explains how TZDs improve glucose uptake into the bloodstream, although their adverse effects (weight gain, fluid retention, and possible cardiac risk) have limited their clinical use.
In pathological states (e.g., immunological conditions or certain cancers), chronic stress increases the concentrations of stress cytokines (e.g., TNF-α, ROS), and deacetylases and sirtuins can also activate AMPK pathways to increase energy availability. In this sense, the physiological role of Transforming Growth Factor-β-activated Kinase 1 (TAK1) is least clear and seems context-dependent. It may be important for AMPK activation in response to chronic stress [79]. But its activation is usually slower and indirect [80]. Besides calcium via CaMKKβ, other routes can modulate AMPK activity without directly altering adenylate charge, adding layers of regulation. Among them: a) Reactive oxygen species (ROS) can activate AMPK by oxidizing cysteine residues. For example, oxidation of Cys299 and Cys304 in the human α subunit induces conformational changes that favor Thr172 phosphorylation; b) some ROS promote LKB1 translocation to the cytosol, and oxidative stress can inhibit PP2A/PP2C; c) inhibitory phosphorylation by PKA is a crucial hormonal negative feedback mechanism. In response to catabolic hormones like glucagon and adrenaline, activated PKA phosphorylates AMPKα at Ser173 (adjacent to Thr172). This phosphorylation prevents subsequent phosphorylation at Thr172 by LKB1, ensuring that AMPK does not activate glucose uptake and fatty acid oxidation when the body needs to mobilize its energy reserves [85]; d) acetylation of lysines on the α and β subunits can affect AMPK activity and localization.
Sirtuins (SIRT1), NAD+-dependent deacetylases, can deacetylate and activate AMPK, establishing a molecular link between redox state (NAD+/NADH) and energy status [82]. Sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) are evolutionarily conserved energy sensors that form a reciprocal signaling loop linking a cell's redox state (NAD+/NADH ratio) to its energy status (AMP/ATP ratio) [66]. During metabolic challenges such as exercise or caloric restriction, AMPK acts as the primary sensor. Increased AMPK activity elevates the cellular NAD+/NADH ratio by stimulating nicotinamide phosphoribosyltransferase (NAMPT) expression, the rate-limiting enzyme in the NAD+ salvage pathway [108]. This increase in NAD+ bioavailability subsequently activates SIRT1, an NAD+-dependent deacetylase [109]. Once activated, SIRT1 deacetylates and modulates the activity of key downstream targets, most notably Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and FOXO transcription factors, to promote mitochondrial biogenesis, fatty acid oxidation, and oxidative stress resistance [66,109]. Therefore, the molecular link is established by AMPK modulating the NAD+ pool to control SIRT1 activity, rather than by SIRT1 directly deacetylating and activating AMPK.
In states of insulin resistance, obesity, and type 2 diabetes, decreased GLUT4 content, especially in adipose tissue, is frequently observed, aggravating metabolic dysfunction. Both chronic hyperinsulinemia and hyperglycemia can activate PKC and hexosamines, thereby inhibiting transcription factors. Paradoxically, while acute insulin does not significantly regulate GLUT4 transcription, chronic hyperinsulinemia (as observed in compensated insulin resistance) can lead to downregulation of GLUT4 expression. This can occur through persistent activation of pathways like mTOR/S6K1, which can generate negative feedback on insulin signaling and affect GLUT4 mRNA stability. Furthermore, macrophage infiltration and the secretion of pro-inflammatory cytokines, such as TNF-α and IL-6, in the adipose tissue of obese individuals activate kinases, including JNK and IKKβ/NF-κB. These pathways not only interfere with insulin signaling (phosphorylating IRS-1 at inhibitory sites) but can also directly repress SLC2A4 gene expression [86]. Under conditions of low insulin/Akt signaling, the transcription factor FOXO1 is not phosphorylated and therefore not exported from the nucleus to the cytoplasm. In the nucleus, FOXO1 can bind the SLC2A4 promoter to repress its transcription and activate gluconeogenic genes in the liver [107,110]. Finally, the glucotoxicity environment can alter transcription factor function and modify histones, contributing to a state of transcriptional repression
9. Lesser-Known Systems of GLUT4 Regulation
The GLUT4 regulatory system does not operate via separate compartments. In the healthy organism, and particularly in skeletal muscle, the distinct signaling pathways that regulate the synthesis, compartmentalization, and translocation of GLUT4 to the cytoplasmic membrane interact dynamically, generating integrated, additive, or synergistic responses, dependent on and independent of AMPK activation [111]. In metabolic disease, this network becomes selectively unbalanced.
In skeletal muscle, the effects of insulin and muscle contraction on glucose uptake are not only additive but also often synergistic. This interaction is explained by the convergence of their distinct signaling pathways onto a common set of molecular nodes, many aspects of which remain under active investigation [112]. The proteins TBC1D1 and TBC1D4 (also known as AS160) function as key platforms for signal integration. These Rab-GTPase-activating proteins (RabGAPs) are phosphorylated by multiple kinases, including Akt, CaMKII, AMPK, and p38 MAPK, at distinct residues. The current model suggests that the inhibition of their GAP activity, which permits GLUT4 vesicle translocation, is maximized when multiple sites are phosphorylated simultaneously, reflecting a unique phosphosignature for each stimulus or combination thereof [113]. For example, exercise can phosphorylate primary sites on these proteins, while subsequent insulin stimulation can add phosphorylation at other sites, stabilizing the inhibited state and further enhancing glucose uptake [111,114].
Supporting this model, research has demonstrated that a single 30-minute aerobic exercise session significantly increases the phosphorylation of AS160 and TBC1D1 in the skeletal muscle of healthy men [115]. Moreover, a 60-minute aerobic exercise session has been reported to increase phosphorylation of AS160 and TBC1D1 in healthy men [116]. This exercise-induced phosphorylation occurred at sites known to be targets of AMPK (e.g., AS160 Ser711, TBC1D1 Ser231) and was not accompanied by an increase in Akt phosphorylation, confirming its independence from the canonical insulin signaling pathway [115]. This exercise-specific modification of the signaling proteins is believed to prime the system.
Consequently, it is well established that an acute exercise session improves skeletal muscle insulin sensitivity for 24 to 48 hours afterward. This enhanced state persists well after the direct, contraction-stimulated increase in glucose uptake has returned to basal levels [117,118]. The mechanisms underlying this prolonged effect are a subject of intense research but are thought to involve sustained phosphorylation of key proteins, such as AS160, which enhances GLUT4 translocation to the membrane in response to subsequent insulin stimulation [119,120]. Proposed mechanisms include:
- -
- Increased PI3K/Akt activity: Prior exercise can prime the insulin pathway, possibly reducing inhibitory serine phosphorylation on IRS-1 or increasing expression of key signaling proteins.
- -
- Improved glycogen turnover: Post-exercise muscle glycogen depletion creates an internal demand for glucose that sensitizes the cell to insulin action.
- -
- Changes in GLUT4 distribution: Exercise can redistribute GLUT4 to a pool of vesicles more readily fusing or closer to the plasma membrane, from which insulin can recruit them more efficiently [121].
Beyond defects in signaling, the vesicular trafficking machinery itself may be altered; for example, reduction of the total GLUT4 pool, especially pronounced in adipose tissue. Alterations in SNARE fusion and reduced expression of Syntaxin 4 and Munc18c have been reported in muscle and adipocytes in some insulin resistance models, which could create a bottleneck in the final step of exocytosis [105]; dysfunction of Rab proteins or their effectors, a less studied pathway, with post-translational modifications (e.g., glycation, nitration) or alterations in the expression of specific Rabs could contribute to deficient trafficking; and finally, exacerbated constitutive endocytosis it is theoretically possible that internalization mediated by the FQQI motif is hyperactive in some states, although this is not well established.
Together, this selective disconnect between pathways (insulin broken, contraction intact) not only explains the pathophysiology of T2DM but also points the direction for rational therapeutic interventions: enhance the pathways that still work, i.e., those activated by exercise
In summary, when acute exercise becomes chronic, it increases SLC2A4 gene expression, which encodes GLUT4, and optimizes the actions of several pathways and proteins implicated in energy metabolism, such as the Krebs cycle, respiratory chain, PGC-1α (a master regulator of mitochondrial and metabolic biogenesis), MEF2, and HDAC. Besides, it improves the biogenesis and the trafficking machinery of GSV on the cytoskeleton. Chronic exercise also increases the expression of key cycle proteins, such as TUG (for retention), SNAREs (for fusion), and cytoskeletal components, thereby optimizing the entire system. By stimulating vesicular machinery, exercise favors a more efficient cycle of endocytosis, sorting, and re-storage of GLUT4, thereby avoiding its degradation or loss through constitutive pathways and reversing insulin resistance
10. Therapeutic Perspectives and Future Research Directions
The biochemical study of canonical and alternative pathways regulating GLUT4 and glucose uptake will help us understand the complex molecular mechanisms underlying insulin resistance and type 2 diabetes. Future treatments must leverage this knowledge to identify how the type and form of short- or long-term exercise could be effective in reducing weight and decreasing these diseases. pathway alone. Similarly, search for alternative pathways that will mimic exercise through robotics in sedentary individuals. In addition, this manuscript proposes developing alternative therapies that selectively activate key components of muscle contraction signaling pathways, thereby improving glucose uptake and insulin sensitivity independently of the insulin receptor.
10.1. Development Proposals
- -
- Develop specific p38 MAPK γ/δ activators: given the fundamental role of p38γ/δ in exercise-induced GLUT4 translocation and its preservation in T2DM, it is an attractive target. The challenge is achieving absolute pharmacological specificity for the α and β isoforms, which are associated with unwanted inflammatory and cellular stress responses.
- -
- Perform high-throughput screens to identify small molecules that bind to unique allosteric pockets of the γ/δ isoforms. Developing allosteric phosphatase inhibitors that specifically deactivate p38γ/δ (e.g., MKP-1) could also potentiate this pathway.
- -
- Chronic activation of p38 MAPK may be associated with muscle and cardiac hypertrophy, as well as cellular stress responses. Long-term toxicology studies and possibly intermittent dosing regimens are required.
- -
- Identify modulators of the calcium/CaMKII pathway: Targeting CaMKII directly is risky due to its central role in cardiac and neuronal excitability. Therefore, strategies must be subtler:
- -
- Identify and block the protein phosphatases that specifically dephosphorylate the CaMKII-phosphorylated residues of TBC1D1 (e.g., PP2A associated with a specific regulatory complex). This would prolong the contraction signal without globally elevating intracellular calcium.
- -
- Identifying mild agonists or allosteric modulators of ryanodine receptors (RyR) in skeletal muscle could sensitize Ca²⁺ release in response to depolarization, potentiating the endogenous signal without causing massive release.
- -
- Develop AMPK activators with high complexity and tissue specificity: The lesson from the pan-AMPK activator MK-8722 (effective for glucose but inducing cardiac hypertrophy) is clear: specificity is needed [107].
- -
- Develop complex-specific activators: Compounds like PF-06409577 prefer the α1β1γ1 complex, predominant in skeletal muscle and liver, over the α2β1γ1 complex, which is more important in the heart [110]. This selectivity could dissociate beneficial metabolic effects from adverse cardiac effects.
- -
- Develop tissue-activated prodrugs, molecules that are selectively activated by enzymes abundant in skeletal muscle, such as creatine kinase or myokinase. This would concentrate drug activity in the target tissue.
- -
- Develop modulators of upstream kinases: Instead of activating AMPK directly, tissue-specific LKB1 activators or modulators of the LKB1-STRAD-MO25 interaction could be developed to potentiate the response to natural energy stress.
- -
- Another alternative strategy is to target more distal components of the signaling cascade, where all pathways converge.
- -
- Develop stabilizers of the GTP-active form of Rabs: small molecules that bind to specific Rabs (Rab8A, Rab10) and stabilize their active (GTP-bound) conformation, mimicking the effect of inhibiting TBC1D1/D4.
- -
- Develop modulators or enhancers that facilitate the formation of the trans-SNARE complex (VAMP2-Syntaxin4-SNAP23) or stabilize the interaction with Munc18 c. This approach would be useful regardless of the activation pathway.
Besides, to correct more permanent defects or induce sustained adaptations, advanced epigenetic therapies offer revolutionary potential. For example, GLUT4 overexpression via viral vectors: preclinical studies with adeno-associated (AAV) vectors expressing GLUT4 under muscle-specific promoters (e.g., myosin light chain promoter) have demonstrated improved glucose tolerance and insulin sensitivity in diabetic mouse models [123]. Challenges include immunogenicity, efficient delivery to sufficient muscle mass, and long-term control of expression. Epigenetic Editing and Modulation: to imprint a metabolic memory similar to exercise without the need for continuous exercise. Develop targeted transcriptional activators, such as CRISPR/dCas9 systems coupled to transcription activation domains (e.g., dCas9-VPR) or enzymes that deposit histone activation marks (e.g., dCas9-p300), to specifically target the SLC2A4 gene promoter and increase its expression in a durable, regulable manner [124]. For example, develop drugs that inhibit specific histone deacetylases (HDACs). Since AMPK phosphorylates and inhibits HDAC5, selective HDAC5 inhibitors could be developed to alleviate chromatin repression of SLC2A4 and other metabolic genes, thereby replicating this effect.
Conclusion
The glucose transporter GLUT4 serves as a central node in metabolic physiology, whose complex regulation underscores its critical importance for energy homeostasis. This review has integrated current evidence, revealing an extraordinary system of sophistication and redundancy, synthesized in the following 5 points.
1. Architecture and Function: GLUT4 is an exquisitely designed molecular machine, whose alternating access transport mechanism is finely tuned. Its localization motifs (FQQI, C-terminal domain) determine its dynamic traffic between a specialized intracellular storage compartment (GSVs) and the plasma membrane.
2. Canonical Insulin Pathway: The PI3K/Akt/TBC1D4 axis represents the high-fidelity system for acute translocation in the fed state. Its activation triggers an orchestrated cascade that releases the molecular brake imposed by the GAPs TBC1D4/D1, allowing Rab GTPases to orchestrate the transport, anchoring, and fusion of GSVs.
3. Insulin-Independent Pathways: Muscle contraction activates parallel and robust mechanisms, led by Ca²⁺ signals (via CaMKII) and mechanical/metabolic stress (via p38 MAPK γ/δ). These pathways converge on the inhibition of the GAPs TBC1D1/D4 via distinct phosphorylation targets, ensuring glucose uptake in response to acute energy demand, independent of hormonal status.
4. AMPK as an Integrative Sensor: AMPK acts as a guardian of energy status, activated by both ATP deficit (via LKB1) and calcium (via CaMKKβ). It contributes to GLUT4 translocation and its long-term expression, but is not essential for the main insulin or contraction pathways.
5. Transcriptional Regulation: The expression of the SLC2A4 gene is controlled by long-term programs, with the exercise-induced PGC-1α/MEF2 axis being the most potent physiological regulator and the nuclear receptor PPARγ being the direct pharmacological target
Author Contributions
Conceptualization, A.R-J. and J.A.R-H.; methodology, J.G-A.; formal analysis, M.R-V.; investigation, E.G-R and V.M-B.; writing—original draft preparation, A.R-J.; writing—review and editing, R.P.H-T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
During the preparation of the figures, the author(s) used [Biorender, Notebook ver. Ultra, and Nanobanana 2.0. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAV | Adeno-associated vectors |
| ADP | Adenosine diphosphate |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| AP-1 / AP-2 | Adaptor protein complexes 1 and 2 |
| AS160 | Akt substrate of 160 kDa (also known as TBC1D4) |
| ATP | Adenosine triphosphate |
| CaM | Calmodulin |
| CaMK / CaMKII | Calcium/calmodulin-dependent protein kinase II |
| CaMKKβ | Calcium/calmodulin-dependent protein kinase kinase β |
| CBM | Carbohydrate-binding module |
| CBS | Cystathionine β-synthase |
| CCB | Cytochalasin B |
| CRISPR/dCas9 | Clustered Regularly Interspaced Short Palindromic Repeats / dead Cas9 |
| DHPR | Dihydropyridine receptors |
| dKO | Double knockout |
| EPAC | Exchange protein directly activated by cAMP |
| GAP | GTPase-activating protein |
| GLP-1 | Glucagon-like peptide-1 |
| GLUT1 / GLUT2 / GLUT3 / GLUT4 | Glucose transporter types 1, 2, 3, and 4 |
| GSIS | Glucose-stimulated insulin secretion |
| GSV | GLUT4 storage vesicle |
| HATs | Histone acetyltransferases |
| HDAC / HDAC4 / HDAC5 | Histone deacetylases |
| IKKβ | Inhibitor of nuclear factor-kappa B kinase subunit beta |
| IR | Insulin receptor |
| IRAP | Insulin-regulated aminopeptidase |
| IRS / IRS-1 | Insulin receptor substrate (1-4) |
| IRV | Insulin-responsive vesicle |
| JNK | c-Jun N-terminal kinase |
| KATP | ATP-sensitive potassium channels |
| LKB1 | Liver Kinase B1 |
| MAPK | Mitogen-activated protein kinase (specifically p38 MAPK ) |
| MEF2 / MEF2A | Myocyte enhancer factor 2 / 2A |
| MK2 | MAPK-activated protein kinase 2 |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| NAD+ / NADH | Nicotinamide adenine dinucleotide (oxidized and reduced forms) |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| nNOS | Neuronal nitric oxide synthase |
| PDK1 | 3-Phosphoinositide-dependent protein kinase-1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PH domain | Pleckstrin-homology domain |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PKA | Protein kinase A |
| PKB | Protein kinase B (also known as Akt) |
| PKCθ | Protein kinase C theta |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PPRE | PPAR response elements |
| PTB | Phosphotyrosine-binding |
| ROS | Reactive oxygen species |
| RXR | Retinoid X receptor |
| RyR | Ryanodine receptors |
| SIRT1 | Sirtuin 1 |
| SLC2A / SLC2A1-4 | Solute carrier family 2 (facilitative glucose transporter) members |
| SNAP23 / SNAP-25 | Synaptosome-associated proteins 23 and 25 |
| SNARE | Soluble NSF Attachment Protein Receptor |
| T2DM | Type 2 diabetes mellitus |
| TBC1D1 / TBC1D4 | TBC1 domain family members 1 and 4 |
| TGN | Trans-Golgi network |
| TM / TMD | Transmembrane / Transmembrane domain |
| TNF-α | Tumor necrosis factor alpha |
| TUG | Tether containing UBX domain for GLUT4 |
| VAMP2 | Vesicle-associated membrane protein 2 |
| VASP | Vasodilator-stimulated phosphoprotein |
References
- Spartano, N.L.; Sultana, N.; Lin, H.; Cheng, H.; Lu, S.; Fei, D.; Murabito, J.M.; Walker, M.E.; Wolpert, H.A.; Steenkamp, D.W. Defining Continuous Glucose Monitor Time in Range in a Large, Community-Based Cohort Without Diabetes. J. Clin. Endocrinol. Metab. 2025, 110, 1128–1134. [CrossRef]
- Charron, M.J.; Brosius, F.C.; Alper, S.L.; Lodish, H.F. A Glucose Transport Protein Expressed Predominately in Insulin-Responsive Tissues. Proc. Natl. Acad. Sci. 1989, 86, 2535–2539. [CrossRef]
- James, D.E.; Strube, M.; Muecdler, M. Molecular Cloning and Characterization of an Insulin-Regulatable Glucose Transporter. Nature 1989, 338, 83–87. [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of Glucose Transport by Insulin: Traffic Control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [CrossRef]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin Stimulates Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nature 2002, 415, 339–343. [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002, 8, 1288–1295. [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and Skeletal Muscle Glucose Uptake. Physiol. Rev. 2013, 93, 993–1017. [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of Glut1 mRNA by Hypoxia-Inducible Factor-1: INTERACTION BETWEEN H-Ras AND HYPOXIA *. J. Biol. Chem. 2001, 276, 9519–9525. [CrossRef]
- Dimitriadis, G.; Leighton, B.; Parry-Billings, M.; Sasson, S.; Young, M.; Krause, U.; Bevan, S.; Piva, T.; Wegener, G.; Newsholme, E.A. Effects of Glucocorticoid Excess on the Sensitivity of Glucose Transport and Metabolism to Insulin in Rat Skeletal Muscle. Biochem. J. 1997, 321, 707–712. [CrossRef]
- Yang; Zhi; Yang; Qin; Et., A. Molecular Identification of Glucose Transporter 4: The Responsiveness to Starvation, Glucose, Insulin and Glucagon on Glucose Transporter 4 in Common Carp (Cyprinus CarpioL.). J. Fish Biol. 2021. [CrossRef]
- Barth; Albuszies; Baumgart; Matejovic; Et., A. Glucose Metabolism and Catecholamines. Crit. Care Med. 2007. [CrossRef]
- Shulman, G.I. Cellular Mechanisms of Insulin Resistance. J. Clin. Invest. 2000, 106, 171–176. [CrossRef]
- Slot, J.W.; Geuze, H.J.; Gigengack, S.; Lienhard, G.E.; James, D.E. Immuno-Localization of the Insulin Regulatable Glucose Transporter in Brown Adipose Tissue of the Rat. J. Cell Biol. 1991, 113, 123–135. [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 Glucose Transporter. Cell Metab. 2007, 5, 237–252. [CrossRef]
- Kahn, B.B. Glucose Transport: Pivotal Step in Insulin Action. Diabetes 1996, 45, 1644–1654. [CrossRef]
- Kennedy, J.W.; Hirshman, M.F.; Gervino, E.V.; Ocel, J.V.; Forse, R.A.; Hoenig, S.J.; Aronson, D.; Goodyear, L.J.; Horton, E.S. Acute Exercise Induces GLUT4 Translocation in Skeletal Muscle of Normal Human Subjects and Subjects with Type 2 Diabetes. Diabetes 1999, 48, 1192–1197. [CrossRef]
- Sun, B.; Chen, H.; Xue, J.; Li, P.; Fu, X. The Role of GLUT2 in Glucose Metabolism in Multiple Organs and Tissues. Mol. Biol. Rep. 2023, 50, 6963–6974. [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose Transporters in Pancreatic Islets. Pflüg. Arch. - Eur. J. Physiol. 2020, 472, 1249–1272. [CrossRef]
- Takeda, J.; Kayano, T.; Fukomoto, H.; Bell, G.I. Organization of the Human GLUT2 (Pancreatic β-Cell and Hepatocyte) Glucose Transporter Gene. Diabetes 1993, 42, 773–777. [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) Is Not the Principal Glucose Transporter in Human Pancreatic Beta Cells: Implications for Understanding Genetic Association Signals at This Locus. Mol. Genet. Metab. 2011, 104, 648–653. [CrossRef]
- Ashcroft, F.M. KATP Channels and the Metabolic Regulation of Insulin Secretion in Health and Disease: The 2022 Banting Medal for Scientific Achievement Award Lecture. Diabetes 2023, 72, 693–702. [CrossRef]
- Mu-u-min, R.B.A.; Diane, A.; Allouch, A.; Al-Siddiqi, H.H. Ca2+-Mediated Signaling Pathways: A Promising Target for the Successful Generation of Mature and Functional Stem Cell-Derived Pancreatic Beta Cells In Vitro. Biomedicines 2023, 11. [CrossRef]
- Peng, X.; Wang, K.; Chen, L. Biphasic Glucose-Stimulated Insulin Secretion over Decades: A Journey from Measurements and Modeling to Mechanistic Insights. Life Metab. 2025, 4, loae038. [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) Family of Membrane Transporters. Mol. Aspects Med. 2013, 34, 121–138. [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal Structure of the Human Glucose Transporter GLUT1. Nature 2014, 510, 121–125. [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [CrossRef]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular Basis of Ligand Recognition and Transport by Glucose Transporters. Nature 2015, 526, 391–396. [CrossRef]
- Yu, C.; Cresswell, J.; Löffler, M.G.; Bogan, J.S. The Glut4 Regulating Protein Tug Is Essential for Highly Insulin Responsive Glucose Uptake in 3t3-L1 Adipocytes. J. Biol. Chem. 2007, 282, 7710–7722. [CrossRef]
- Stöckli, J.; Fazakerley, D.J.; James, D.E. GLUT4 Exocytosis. J. Cell Sci. 2011, 124, 4147–4159. [CrossRef]
- Yuan, Y.; Kong, F.; Xu, H.; Zhu, A.; Yan, N.; Yan, C. Cryo-EM Structure of Human Glucose Transporter GLUT4. Nat. Commun. 2022, 13, 2671. [CrossRef]
- Taylor, L.P.; Holman, G.D. Symmetrical Kinetic Parameters for 3-O-Methyl-d-Glucose Transport in Adipocytes in the Presence and in the Absence of Insulin. Biochim. Biophys. Acta BBA - Biomembr. 1981, 642, 325–335. [CrossRef]
- Sheena, A.; Mohan, S.S.; Haridas, N.P.A.; Anilkumar, G. Elucidation of the Glucose Transport Pathway in Glucose Transporter 4 via Steered Molecular Dynamics Simulations. PLOS ONE 2011, 6, 1–11. [CrossRef]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H.; et al. Mechanism of Inhibition of Human Glucose Transporter GLUT1 Is Conserved between Cytochalasin B and Phenylalanine Amides. Proc. Natl. Acad. Sci. 2016, 113, 4711–4716. [CrossRef]
- Drobiova, H.; Alhamar, G.; Ahmad, R.; Al-Mulla, F.; Madhoun, A.A. GLUT4 Trafficking and Storage Vesicles: Molecular Architecture, Regulatory Networks, and Their Disruption in Insulin Resistance. Int. J. Mol. Sci. 2025, 26. [CrossRef]
- Li, D.T.; Habtemichael, E.N.; Julca, O.; Sales, C.I.; Westergaard, X.O.; DeVries, S.G.; Ruiz, D.; Sayal, B.; Bogan, J.S. GLUT4 Storage Vesicles: Specialized Organelles for Regulated Trafficking. Yale J. Biol. Med. 2019, 92, 453–470.
- Kawaguchi, T.; Tamori, Y.; Kanda, H.; Yoshikawa, M.; Tateya, S.; Nishino, N.; Kasuga, M. The T-SNAREs Syntaxin4 and SNAP23 but Not v-SNARE VAMP2 Are Indispensable to Tether GLUT4 Vesicles at the Plasma Membrane in Adipocyte. Biochem. Biophys. Res. Commun. 2010, 391, 1336–1341. [CrossRef]
- Zhao, P.; Yang, L.; Lopez, J.A.; Fan, J.; Burchfield, J.G.; Bai, L.; Hong, W.; Xu, T.; James, D.E. Variations in the Requirement for V-SNAREs in GLUT4 Trafficking in Adipocytes. J. Cell Sci. 2009, 122, 3472–3480. [CrossRef]
- Martin, S.; Millar, C.A.; Lyttle, C.T.; Meerloo, T.; Marsh, B.J.; Gould, G.W.; James, D.E. Effects of Insulin on Intracellular GLUT4 Vesicles in Adipocytes: Evidence for a Secretory Mode of Regulation. J. Cell Sci. 2000, 113, 3427–3438. [CrossRef]
- Bryant, N.J.; Govers, R.; James, D.E. Regulated Transport of the Glucose Transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002, 3, 267–277. [CrossRef]
- Blot, V.; McGraw, T.E. Molecular Mechanisms Controlling GLUT4 Intracellular Retention. Mol. Biol. Cell 2008, 19, 3477–3487. [CrossRef]
- Bernhardt, U.; Carlotti, F.; Hoeben, R.C.; Joost, H.-G.; Al-Hasani, H. A Dual Role of the N-Terminal FQQI Motif in GLUT4 Trafficking. Biol. Chem. 2009, 390, 883–892. [CrossRef]
- Al-Hasani, H.; Kunamneni, R.K.; Dawson, K.; Hinck, C.S.; Müller-Wieland, D.; Cushman, S.W. Roles of the N- and C-Termini of GLUT4 in Endocytosis. J. Cell Sci. 2002, 115 Pt 1, 131–140.
- Blot, V.; McGraw, T.E. GLUT4 Is Internalized by a Cholesterol-dependent Nystatin-sensitive Mechanism Inhibited by Insulin. EMBO J. 2006, 25, 5648–5658. [CrossRef]
- Widberg, C.H.; Bryant, N.J.; Girotti, M.; Rea, S.; James, D.E. Tomosyn Interacts with the T-SNAREs Syntaxin4 and SNAP23 and Plays a Role in Insulin-Stimulated GLUT4 Translocation *. J. Biol. Chem. 2003, 278, 35093–35101. [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [CrossRef]
- Bai, L.; Wang, Y.; Fan, J.; Chen, Y.; Ji, W.; Qu, A.; Xu, P.; James, D.E.; Xu, T. Dissecting Multiple Steps of GLUT4 Trafficking and Identifying the Sites of Insulin Action. Cell Metab. 2007, 5, 47–57. [CrossRef]
- Scapin, G.; Dandey, V.P.; Zhang, Z.; Prosise, W.; Hruza, A.; Kelly, T.; Mayhood, T.; Strickland, C.; Potter, C.S.; Carragher, B. Structure of the Insulin Receptor-Insulin Complex by Single Particle CryoEM Analysis. Nature 2018, 556, 122–125. [CrossRef]
- Sun, X.J.; Pons, S.; Wang, L.M.; Zhang, Y.; Yenush, L.; Burks, D.; Myers, M.G.; Glasheen, E.; Copeland, N.G.; Jenkins, N.A.; et al. The IRS-2 Gene on Murine Chromosome 8 Encodes a Unique Signaling Adapter for Insulin and Cytokine Action. Mol. Endocrinol. 1997, 11, 251–262. [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical Nodes in Signalling Pathways: Insights into Insulin Action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [CrossRef]
- Sano, H.; Kane, S.; Sano, E.; Mı̂inea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated Phosphorylation of a Rab GTPase-Activating Protein Regulates GLUT4 Translocation *. J. Biol. Chem. 2003, 278, 14599–14602. [CrossRef]
- Mîinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peränen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt Substrate Regulating GLUT4 Translocation, Has a Functional Rab GTPase-Activating Protein Domain. Biochem. J. 2005, 391, 87–93. [CrossRef]
- Kane, S.; Sano, H.; Liu, S.C.H.; Asara, J.M.; Lane, W.S.; Garner, C.C.; Lienhard, G.E. A Method to Identify Serine Kinase Substrates: Akt Phosphorylates a Novel Adipocyte Protein with a Rab Gtpase-Activating Protein (Gap) Domain. J. Biol. Chem. 2002, 277, 22115–22118. [CrossRef]
- Ishikura; Klip Muscle Cells Engage Rab8A and Myosin Vb in Insulin-Dependent GLUT4 Translocation. Am. J. Physiol.-Cell Physiol. 2008. [CrossRef]
- Langlais, P.; Dillon, J.L.; Mengos, A.; Baluch, D.P.; Ardebili, R.; Miranda, D.N.; Xie, X.; Heckmann, B.L.; Liu, J.; Mandarino, L.J. Identification of a Role for CLASP2 in Insulin Action. J. Biol. Chem. 2012, 287, 39245–39253. [CrossRef]
- Inoue, M.; Chang, L.; Hwang, J.; Chiang, S.-H.; Saltiel, A.R. The Exocyst Complex Is Required for Targeting of Glut4 to the Plasma Membrane by Insulin. Nature 2003, 422, 629–633. [CrossRef]
- Sun, Y.; Bilan, P.J.; Liu, Z.; Klip, A. Rab8A and Rab13 Are Activated by Insulin and Regulate GLUT4 Translocation in Muscle Cells. Proc. Natl. Acad. Sci. 2010, 107, 19909–19914. [CrossRef]
- Kanda, H.; Tamori, Y.; Shinoda, H.; Yoshikawa, M.; Sakaue, M.; Udagawa, J.; Otani, H.; Tashiro, F.; Miyazaki, J.; Kasuga, M. Adipocytes from Munc18c-Null Mice Show Increased Sensitivity to Insulin-Stimulated GLUT4 Externalization. J. Clin. Invest. 2005, 115, 291–301. [CrossRef]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.-B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-Selective Targeting of the GLUT4 Gene Impairs Insulin Action in Muscle and Liver. Nature 2001, 409, 729–733. [CrossRef]
- Jørgensen, S.B.; Viollet, B.; Andreelli, F.; Frøsig, C.; Birk, J.B.; Schjerling, P.; Vaulont, S.; Richter, E.A.; Wojtaszewski, J.F.P. Knockout of the A2 but Not A1 5′-AMP-Activated Protein Kinase Isoform Abolishes 5-Aminoimidazole-4-Carboxamide-1-β-4-Ribofuranosidebut Not Contraction-Induced Glucose Uptake in Skeletal Muscle *. J. Biol. Chem. 2004, 279, 1070–1079. [CrossRef]
- Sakamoto, K.; McCarthy, A.; Smith, D.; Green, K.A.; Grahame Hardie, D.; Ashworth, A.; Alessi, D.R. Deficiency of LKB1 in Skeletal Muscle Prevents AMPK Activation and Glucose Uptake during Contraction. EMBO J. 2005, 24, 1810–1820. [CrossRef]
- Li; Yue, Y.; Hu, F.; Zhang, C.; Ma, X.; Li, N.; Qiu, L.; Fu, M.; Chen, L.; Yao, Z.; et al. Electrical Pulse Stimulation Induces GLUT4 Translocation in C(2)C(12) Myotubes That Depends on Rab8A, Rab13, and Rab14. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E478–E493. [CrossRef]
- Liu, S.; Zhang, J.; Qi, R.; Deng, B.; Ni, Y.; Zhang, C.; Niu, W. CaMKII and Kalirin, a Rac1-GEF, Regulate Akt Phosphorylation Involved in Contraction-Induced Glucose Uptake in Skeletal Muscle Cells. Biochem. Biophys. Res. Commun. 2022, 610, 170–175. [CrossRef]
- Pehmøller, C.; Treebak, J.T.; Birk, J.B.; Chen, S.; Mackintosh, C.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F.P. Genetic Disruption of AMPK Signaling Abolishes Both Contraction- and Insulin-Stimulated TBC1D1 Phosphorylation and 14-3-3 Binding in Mouse Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E665-675. [CrossRef]
- Vichaiwong, K.; Purohit, S.; An, D.; Toyoda, T.; Jessen, N.; Hirshman, M.F.; Goodyear, L.J. Contraction Regulates Site-Specific Phosphorylation of TBC1D1 in Skeletal Muscle. Biochem J 2010, 431, 311–320. [CrossRef]
- Chen; Liu, B.; Yao, X.; Yang, X.; Sun, J.; Yi, J.; Xue, F.; Zhang, J.; Shen, Y.; Chen, B.; et al. AMPK/SIRT1/PGC-1α Signaling Pathway: Molecular Mechanisms and Targeted Strategies From Energy Homeostasis Regulation to Disease Therapy. CNS Neurosci. Ther. 2025, 31, e70657. [CrossRef]
- Brault, J.J.; Pizzimenti, N.M.; Dentel, J.N.; Wiseman, R.W. Selective Inhibition of ATPase Activity during Contraction Alters the Activation of P38 MAP Kinase Isoforms in Skeletal Muscle. J. Cell. Biochem. 2013, 114.
- Ho, R.C.; Alcazar, O.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. P38gamma MAPK Regulation of Glucose Transporter Expression and Glucose Uptake in L6 Myotubes and Mouse Skeletal Muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R342-349. [CrossRef]
- Pogozelski, A.R.; Geng, T.; Li, P.; Yin, X.; Lira, V.A.; Zhang, M.; Chi, J.-T.; Yan, Z. P38γ Mitogen-Activated Protein Kinase Is a Key Regulator in Skeletal Muscle Metabolic Adaptation in Mice. PLOS ONE 2009, 4, 1–9. [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise Regulated Kinases and AMPK Substrates. Cell Metab 2015, 22, 922–935. [CrossRef]
- Treebak, J.T.; Pehmøller, C.; Kristensen, J.M.; Kjøbsted, R.; Birk, J.B.; Schjerling, P.; Richter, E.A.; Goodyear, L.J.; Wojtaszewski, J.F.P. Acute Exercise and Physiological Insulin Induce Distinct Phosphorylation Signatures on TBC1D1 and TBC1D4 Proteins in Human Skeletal Muscle. J Physiol 2014, 592, 351–375. [CrossRef]
- Kjøbsted, R.; Roll, J.L.W.; Jørgensen, N.O.; Birk, J.B.; Foretz, M.; Viollet, B.; Chadt, A.; Al-Hasani, H.; Wojtaszewski, J.F.P. AMPK and TBC1D1 Regulate Muscle Glucose Uptake After, but Not During, Exercise and Contraction. Diabetes 2019, 68, 1427–1440. [CrossRef]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative Stress Stimulates Skeletal Muscle Glucose Uptake through a Phosphatidylinositol 3-Kinase-Dependent Pathway. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E889–E897. [CrossRef]
- Merry, T.L.; Ristow, M. Nuclear Factor Erythroid-derived 2-like 2 (NFE2L2, Nrf2) Mediates Exercise-induced Mitochondrial Biogenesis and the Anti-oxidant Response in Mice. J. Physiol. 2016, 594, 5195–5207. [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK - a Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [CrossRef]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP Is a True Physiological Regulator of AMP-Activated Protein Kinase by Both Allosteric Activation and Enhancing Net Phosphorylation. Cell Metab. 2013, 18, 556–566. [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The Tumor Suppressor LKB1 Kinase Directly Activates AMP-Activated Kinase and Regulates Apoptosis in Response to Energy Stress. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 3329–3335. [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-Dependent Protein Kinase Kinase-β Is an Alternative Upstream Kinase for AMP-Activated Protein Kinase. Cell Metab. 2005, 2, 9–19. [CrossRef]
- Momcilovic, M.; Hong, S.-P.; Carlson, M. Mammalian TAK1 Activates Snf1 Protein Kinase in Yeast and Phosphorylates AMP-Activated Protein Kinase in Vitro. J. Biol. Chem. 2006, 281, 25336–25343. [CrossRef]
- Vichaiwong, K.; Purohit, S.; An, D.; Toyoda, T.; Jessen, N.; Hirshman, M.F.; Goodyear, L.J. Contraction Regulates Site-Specific Phosphorylation of TBC1D1 in Skeletal Muscle. Biochem. J. 2010, 431, 311–320. [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise and Myocyte Enhancer Factor 2 Regulation in Human Skeletal Muscle. Diabetes 2004, 53, 1208–1214. [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 2006, 27, 728–735. [CrossRef]
- McGee, S.L.; van Denderen, B.J.W.; Howlett, K.F.; Mollica, J.; Et., A. AMP-Activated Protein Kinase Regulates GLUT4 Transcription by Phosphorylating Histone Deacetylase 5. Diabetes 2008. [CrossRef]
- Myers, R.W.; Guan, H.-P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic Pan-AMPK Activator MK-8722 Improves Glucose Homeostasis but Induces Cardiac Hypertrophy. Science 2017, 357, 507–511. [CrossRef]
- Djouder, N.; Tuerk, R.D.; Suter, M.; Salvioni, P.; Thali, R.F.; Scholz, R.; Vaahtomeri, K.; Auchli, Y.; Rechsteiner, H.; Brunisholz, R.A.; et al. PKA Phosphorylates and Inactivates AMPKα to Promote Efficient Lipolysis. EMBO J. 2010, 29, 469–481. [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [CrossRef]
- Carrillo, E.D.; Hernández, D.I.; Clara, M.V.; Lezama, I.; García, M.C.; Sánchez, J.A. Exercise Increases MEF2A Abundance in Rat Cardiac Muscle by Downregulating microRNA-223-5p. Sci. Rep. 2023, 13, 14481. [CrossRef]
- Chavanelle, V.; Boisseau, N.; Otero, Y.F.; Combaret, L.; Dardevet, D.; Montaurier, C.; Delcros, G.; Peltier, S.L.; Sirvent, P. Effects of High-Intensity Interval Training and Moderate-Intensity Continuous Training on Glycaemic Control and Skeletal Muscle Mitochondrial Function in Db/Db Mice. Sci. Rep. 2017, 7, 204. [CrossRef]
- Feng, Y.; Zhang, J.; Tian, X.; Wu, J.; Lu, J.; Shi, R. Mechanical Stretch Activates Glycometabolism-Related Enzymes via Estrogen in C2C12 Myoblasts. J. Cell. Physiol. 2020, 235, 5702–5710. [CrossRef]
- Ray, A.; Wen, J.; Yammine, L.; Culver, J.; Parida, I.S.; Garren, J.; Xue, L.; Hales, K.; Xiang, Q.; Birnbaum, M.J.; et al. Regulated Dynamic Subcellular GLUT4 Localization Revealed by Proximal Proteome Mapping in Human Muscle Cells. J. Cell Sci. 2023, 136, jcs261454. [CrossRef]
- Benatti, F.B.; Miyake, C.N.H.; Dantas, W.S.; Zambelli, V.O.; Shinjo, S.K.; Pereira, R.M.R.; Silva, M.E.R.; Sá-Pinto, A.L.; Borba, E.; Bonfá, E.; et al. Exercise Increases Insulin Sensitivity and Skeletal Muscle AMPK Expression in Systemic Lupus Erythematosus: A Randomized Controlled Trial. Front. Immunol. 2018, Volume 9-2018. [CrossRef]
- Muñoz, V.R.; Botezelli, J.D.; Gaspar, R.C.; da Rocha, A.L.; Vieira, R.F.L.; Crisol, B.M.; Braga, R.R.; Severino, M.B.; Nakandakari, S.C.B.R.; Antunes, G.C.; et al. Effects of Short-Term Endurance and Strength Exercise in the Molecular Regulation of Skeletal Muscle in Hyperinsulinemic and Hyperglycemic Slc2a4+/− Mice. Cell Mol Life Sci 2023, 80, 122. [CrossRef]
- O’Gorman, D.J.; Karlsson, H.K.R.; McQuaid, S.; Yousif, O.; Rahman, Y.; Gasparro, D.; Glund, S.; Chibalin, A.V.; Zierath, J.R.; Nolan, J.J. Exercise Training Increases Insulin-Stimulated Glucose Disposal and GLUT4 (SLC2A4) Protein Content in Patients with Type 2 Diabetes. Diabetologia 2006, 49, 2983–2992. [CrossRef]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute Exercise Remodels Promoter Methylation in Human Skeletal Muscle. Cell Metab. 2012, 15, 405–411. [CrossRef]
- Flores-Opazo, M.; McGee, S.L.; Hargreaves, M. Exercise and GLUT4. Exerc. Sport Sci. Rev. 2020, 48, 110. [CrossRef]
- McGee, S.L. Exercise and MEF2–HDAC Interactions. Appl. Physiol. Nutr. Metab. 2007, 32, 852–856. [CrossRef]
- Röckl, K.S.C.; Witczak, C.A.; Goodyear, L.J. Diabetes, Mitocondrias y Ejercicio. Rev. Esp. Cardiol. Supl. 2008, 8, 27C-34C. [CrossRef]
- Sylow, L.; Tokarz, V.L.; Richter, E.A.; Klip, A. The Many Actions of Insulin in Skeletal Muscle, the Paramount Tissue Determining Glycemia. Cell Metab. 2021. [CrossRef]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The Aetiology and Molecular Landscape of Insulin Resistance. Nat. Rev. Mol. Cell Biol. 2021. [CrossRef]
- Samuel, V.T.; Shulman, G.I. Integrating Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [CrossRef]
- Ijuin, T.; Takenawa, T. Regulation of Insulin Signaling and Glucose Transporter 4 (GLUT4) Exocytosis by Phosphatidylinositol 3,4,5-Trisphosphate (PIP3) Phosphatase, Skeletal Muscle, and Kidney Enriched Inositol Polyphosphate Phosphatase (SKIP). J. Biol. Chem. 2012, 287, 6991–6999. [CrossRef]
- Bogan, J.S. Regulation of Glucose Transporter Translocation in Health and Diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [CrossRef]
- Garvey, W.T.; Maianu, L.; Huecksteadt, T.P.; Birnbaum, M.J.; Molina, J.M.; Ciaraldi, T.P. Pretranslational Suppression of a Glucose Transporter Protein Causes Insulin Resistance in Adipocytes from Patients with Non-Insulin-Dependent Diabetes Mellitus and Obesity. J. Clin. Invest. 1991, 87, 1072–1081. [CrossRef]
- Hawley, J.A.; Lessard, S.J. Exercise Training-Induced Improvements in Insulin Action. Acta Physiol. 2008, 192, 127–135. [CrossRef]
- Kawanishi, M.; Tamori, Y.; Okazawa, H.; Araki, S.; Shinoda, H.; Kasuga, M. Role of SNAP23 in Insulin-Induced Translocation of GLUT4 in 3T3-L1 Adipocytes: MEDIATION OF COMPLEX FORMATION BETWEEN SYNTAXIN4 AND VAMP2 *. J. Biol. Chem. 2000, 275, 8240–8247. [CrossRef]
- Omata, W.; Shibata, H.; Li, L.; Takata, K.; Kojima, I. Actin Filaments Play a Critical Role in Insulin-Induced Exocytotic Recruitment but Not in Endocytosis of GLUT4 in Isolated Rat Adipocytes. Biochem. J. 2000, 346, 321–328. [CrossRef]
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The Forkhead Transcription Factor Foxo1 (Fkhr) Confers Insulin Sensitivity onto Glucose-6-Phosphatase Expression. J. Clin. Invest. 2001, 108, 1359–1367. [CrossRef]
- Lee, Y.; Kim, E.-K. AMP-Activated Protein Kinase as a Key Molecular Link between Metabolism and Clockwork. Exp. Mol. Med. 2013, 45, e33–e33. [CrossRef]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab. 2010, 11, 213–219. [CrossRef]
- Cameron, K.O.; Kung, D.W.; Kalgutkar, A.S.; Kurumbail, R.G.; Et., A. Discovery and Preclinical Characterization of 6-Chloro-5-[4-(1-Hydroxycyclobutyl)Phenyl]-1H-Indole-3-Carboxylic Acid (PF-06409577), a Direct Activator of Adenosine Monophosphate-Activated Protein Kinase (AMPK), for the Potential Treatment of Diabetic Nephropathy. J. Med. Chem. 2016. [CrossRef]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [CrossRef]
- Cartee, G.D. Roles of TBC1D1 and TBC1D4 in Insulin- and Exercise-Stimulated Glucose Transport of Skeletal Muscle. Diabetologia 2015, 58, 19–30. [CrossRef]
- Cartee, G.D.; Funai, K. Exercise and Insulin: Convergence or Divergence at AS160 and TBC1D1? Exerc. Sport Sci. Rev. 2009, 37, 188–195. [CrossRef]
- Kjøbsted, R.; Roll, J.L.W.; Jørgensen, N.O.; Birk, J.B.; Foretz, M.; Viollet, B.; Chadt, A.; Al-Hasani, H.; Wojtaszewski, J.F.P. AMPK and TBC1D1 Regulate Muscle Glucose Uptake After, but Not During, Exercise and Contraction. Diabetes 2019, 68, 1427–1440. [CrossRef]
- Jessen, N.; An, D.; Lihn, A.S.; Nygren, J.; Hirshman, M.F.; Thorell, A.; Goodyear, L.J. Exercise Increases TBC1D1 Phosphorylation in Human Skeletal Muscle. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, E164–E171. [CrossRef]
- Vendelbo, M.H.; Møller, A.B.; Treebak, J.T.; Gormsen, L.C.; Goodyear, L.J.; Wojtaszewski, J.F.P.; Jørgensen, J.O.L.; Møller, N.; Jessen, N. Sustained AS160 and TBC1D1 Phosphorylations in Human Skeletal Muscle 30 Min after a Single Bout of Exercise. J. Appl. Physiol. 2014, 117, 289–296. [CrossRef]
- Cartee, G.D. Mechanisms for Greater Insulin-Stimulated Glucose Uptake in Normal and Insulin-Resistant Skeletal Muscle after Acute Exercise. Am. J. Physiol.-Endocrinol. Metab. 2015. [CrossRef]
- Stephens, F.B.; Tsintzas, K. Metabolic and Molecular Changes Associated with the Increased Skeletal Muscle Insulin Action 24-48 h after Exercise in Young and Old Humans. Biochem. Soc. Trans. 2018, 46, 111–118. [CrossRef]
- Cartee, G.D. Mechanisms for Greater Insulin-Stimulated Glucose Uptake in Normal and Insulin-Resistant Skeletal Muscle after Acute Exercise. Am. J. Physiol.-Endocrinol. Metab. 2015. [CrossRef]
- Mikines, K.J.; Sonne, B.; Farrell, P.A.; Tronier, B.; Galbo, H. Effect of Physical Exercise on Sensitivity and Responsiveness to Insulin in Humans. Am. J. Physiol.-Endocrinol. Metab. 1988, 254, E248–E259. [CrossRef]
- Wojtaszewski, J.F.; Higaki, Y.; Hirshman, M.F.; Michael, M.D.; Dufresne, S.D.; Kahn, C.R.; Goodyear, L.J. Exercise Modulates Postreceptor Insulin Signaling and Glucose Transport in Muscle-Specific Insulin Receptor Knockout Mice. J Clin Invest 1999, 104, 1257–1264. [CrossRef]
- Wojtaszewski, J.F.; Higaki, Y.; Hirshman, M.F.; Michael, M.D.; Dufresne, S.D.; Kahn, C.R.; Goodyear, L.J. Exercise Modulates Postreceptor Insulin Signaling and Glucose Transport in Muscle-Specific Insulin Receptor Knockout Mice. J. Clin. Invest. 1999, 104, 1257–1264. [CrossRef]
- Liu; Gibbs; McCoid; Milici; Et., A. Transgenic Mice Expressing the Human GLUT4/Muscle-Fat Facilitative Glucose Transporter Protein Exhibit Efficient Glycemic Control. Proc. Natl. Acad. Sci. 1993. [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly Specific Epigenome Editing by CRISPR/Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat Methods 2015, 12, 1143–1149. [CrossRef]
Figure 1.
Schematic of the beta cell. Insulin is synthesized in the endoplasmic reticulum as preproinsulin and converted to proinsulin. In the Golgi apparatus, it is cleaved into mature insulin and C-peptide. It is then packaged into secretory granules, where it is stored until glucose stimulates its release into the bloodstream. A failure in this process contributes to type 2 diabetes.
Figure 1.
Schematic of the beta cell. Insulin is synthesized in the endoplasmic reticulum as preproinsulin and converted to proinsulin. In the Golgi apparatus, it is cleaved into mature insulin and C-peptide. It is then packaged into secretory granules, where it is stored until glucose stimulates its release into the bloodstream. A failure in this process contributes to type 2 diabetes.

Figure 2.
Topological organization and intracellular regulatory regions of GLUT4.

Figure 3.
GLUT4-mediated glucose transport mechanism under the “alternating access” model.

Figure 4.
Representative scheme of GSV retention and release: The 5-stage journey of GLUT4.

Figure 5.
Molecular sequence of insulin-induced GLUT4 vesicular trafficking. Representative scheme of the coordinated events that regulate GLUT4 mobilization and recycling. (1) Release of storage vesicles (GSVs) from their intracellular retention state through activation of Rab proteins and dissociation of anchoring complexes; (2) organization of the fusion machinery at the plasma membrane, including t-SNARE proteins; (3) vesicular approach and initial assembly of the SNARE complex with VAMP2 involvement; (4) stabilization of docking in the cortical region; (5) vesicular fusion and insertion of GLUT4 into the plasma membrane; (6) facilitated glucose transport; (7) internalization of the transporter via clathrin-dependent endocytosis; and (8) cytoskeleton-associated intracellular recycling and redirection for subsequent reuse.
Figure 5.
Molecular sequence of insulin-induced GLUT4 vesicular trafficking. Representative scheme of the coordinated events that regulate GLUT4 mobilization and recycling. (1) Release of storage vesicles (GSVs) from their intracellular retention state through activation of Rab proteins and dissociation of anchoring complexes; (2) organization of the fusion machinery at the plasma membrane, including t-SNARE proteins; (3) vesicular approach and initial assembly of the SNARE complex with VAMP2 involvement; (4) stabilization of docking in the cortical region; (5) vesicular fusion and insertion of GLUT4 into the plasma membrane; (6) facilitated glucose transport; (7) internalization of the transporter via clathrin-dependent endocytosis; and (8) cytoskeleton-associated intracellular recycling and redirection for subsequent reuse.

Figure 6.
Structural organization of the AMPK heterotrimer.

Figure 7.
Convergence of exercise-induced signals on the TBC1D1/4–Rab–GLUT4 axis.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



