Submitted:
05 March 2026
Posted:
06 March 2026
You are already at the latest version
Abstract
Background. Quaternary phosphonium salts (QPSs) are extensively researched since represent new promising weapons to counteract critical superbugs, regardless their robust pattern of resistance. Methods. Here, dynamic light scattering analysis was carried out on QPSs 1, 3 and 4 recently reported and already found active against cancer cells, and phosphine 2 unveiling particles of 700-800 nm for 2, 3 and 4 and positive Zeta-potential (ζ-p ) for all (+4.2-+38.1 mV). 1, 3 and 4 plus 2, were microbiologically evaluated, assessing minimum inhibitory concentration values (MICs) (1-4), time-killing curves (1), and anti-biofilm capacity (1). Results. MICs on a total of 23 Gram-positive and Gram-negative clinically isolated superbugs, evidenced that, poorly soluble 2, 3 and 4 exhibited not reproducible MICs, while 1 provided interesting MICs, which made it worthy of further investigations. In fact, 1 was active against clinically relevant multidrug-resistant (MDR) Gram-positive species and not active against MDR Gram-negative species including Pseudomonas aeruginosa. Specifically, MICs = 16-32 µg/mL and 16-64 µg/mL were determined against methicillin-resistant Staphylococcus aureus (MRSA) and S. epidermidis (MRSE) respectively. MICs = 32-64 µg/mL were observed against teicoplanin- and vancomycin-resistant (VRE) Enterococcus faecalis and E. faecium and no activity against P. aeruginosa (> 128 µg/mL). Notably, time-kill experiments established that 1 was bactericidal against MRSA, while strongly inhibited (up to 100%) the formation of biofilm produced by the strongest biofilm-producers S. epidermidis and S. aureus isolates of our collection, at MICs and 2.5 × MIC concentrations, depending on isolates considered. Interestingly, if used against Staphylococci, and mainly MRSA, 1 was softly haemolytic. It was no cytotoxic against not tumorigenic human keratinocytes (HaCaT) and murine embryonic fibroblasts (3T3) in all cases. Structure-activity relationships have been studied, leading to outcomes which could be of great help for designing optimized new QPSs. Conclusions. Findings of this study overturn previous antimicrobial reports on compound 1, suggesting it as a new excellent weapon to counteract bacterial resistance and biofilm production by MRSA and MRSE superbugs, as well as thinkable for future in vivo experiments and clinical development.
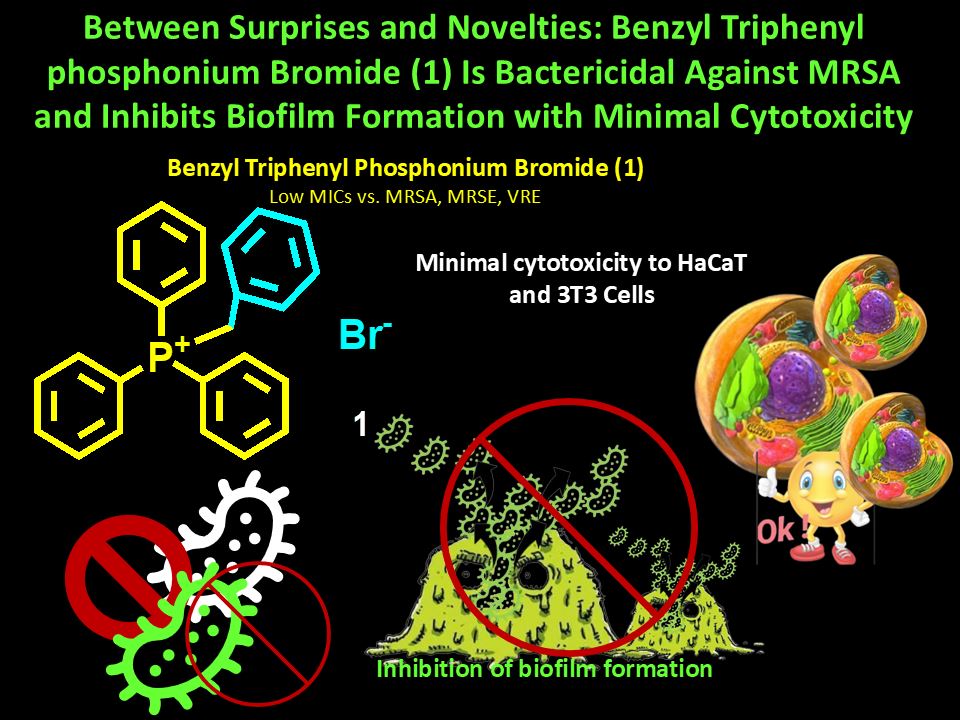
Keywords:
1. Introduction
1.1. Why Phosphonium Salts 1, 3 and 4, as well as Phosphine 2 Were Chosen for Antimicrobial Experiments in this Study?
2. Results and Discussion
2.1. Synthesis of Quaternary Benzyl Phosphonium Bromides 1, 3 and 4
2.2. Morphology Compounds 1-4 in Methanol and Water by Optical Microscopy
2.3. Dynamic Ligh Scattering (DLS) Analyses of 2, 3 and 4
2.4. Antibacterial Properties of QPSs 1-4
2.4.1. In Vitro Antibacterial Activity of Compounds 1-4 by Determining MIC Values (MICs)
2.4.2. Structure Activity Relationships (SAR)
2.4.3. Time-Killing Curves
2.4.4. Antibiofilm Capacity of 1
2.5. Cytotoxicity and Haemolytic Toxicity of Compound 1
2.5.1. Cytotoxicity of 1 Against Human Keratinocytes (HaCaT) and Murine Fibroblasts (3T3)
2.5.2. Haemolytic Effects of 1 Against Red Blood Cells (RBCs)
2.5.3. Selectivity of 1 for Bacteria
3. Materials and Methods
3.1. Compounds 1-4
3.3. Optical Microscopy
3.4. Dynamic Light Scattering Analysis (DLS)
3.5. Microbiologic Experiments
3.5.1. Clinically Relevant Superbugs Used in This Study
3.5.2. Determination of MICs
3.5.3. Time–Kill Curves
3.5.4. Detection of Biofilm Production Using the Microliter Plate Method
3.6. Cytotoxicity of Compound 1 on Eukaryotic Cells
3.7. Haemolytic Effects of Compound 1 on Red Blood Cells (RBCs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandrasekhar, D.; Joseph, C.M.; parambil, J.C.; Murali, S.; Yahiya, M.; K, S. Superbugs: An Invicible Threat in Post Antibiotic Era. Clin. Epidemiol. Glob. Health 2024, 28. [Google Scholar] [CrossRef]
- Rajendran, R. Superbug Infection. J. Drug Metab. Toxicol. 2018, 09. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. The Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus Aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, H.; Hu, G.; Liu, J.; Lian, S.; Pang, S.; Zhu, G.; Ding, X. Unmasking MRSA’s Armor: Molecular Mechanisms of Resistance and Pioneering Therapeutic Countermeasures. Microorganisms 2025, 13, 1928. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Approaches to Combating Methicillin-Resistant Staphylococcus Aureus (MRSA) Biofilm Infections. Expert Opin. Investig. Drugs 2024, 33, 1–3. [Google Scholar] [CrossRef]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiology and Molecular Biology Reviews 2014, 78, 510–543. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus Epidermidis — the “accidental” Pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-Negative Staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef]
- McCann, M.T.; Gilmore, B.F.; Gorman, S.P. Staphylococcus Epidermidis Device-Related Infections: Pathogenesis and Clinical Management. Journal of Pharmacy and Pharmacology 2008, 60, 1551–1571. [Google Scholar] [CrossRef]
- Trobos, M.; Firdaus, R.; Svensson Malchau, K.; Tillander, J.; Arnellos, D.; Rolfson, O.; Thomsen, P.; Lasa, I. Genomics of Staphylococcus Aureus and Staphylococcus Epidermidis from Periprosthetic Joint Infections and Correlation to Clinical Outcome. Microbiol. Spectr. 2022, 10. [Google Scholar] [CrossRef]
- Takahashi, C.; Sato, M.; Sato, C. Biofilm Formation of Staphylococcus Epidermidis Imaged Using Atmospheric Scanning Electron Microscopy. Anal. Bioanal. Chem. 2021, 413, 7549–7558. [Google Scholar] [CrossRef]
- Arias, C.A.; Contreras, G.A.; Murray, B.E. Management of Multidrug-Resistant Enterococcal Infections. Clinical Microbiology and Infection 2010, 16, 555–562. [Google Scholar] [CrossRef]
- Arias, C.A.; Murray, B.E. The Rise of the Enterococcus: Beyond Vancomycin Resistance. Nat. Rev. Microbiol. 2012, 10, 266–278. [Google Scholar] [CrossRef]
- Lee, T.; Pang, S.; Abraham, S.; Coombs, G.W. Antimicrobial-Resistant CC17 Enterococcus Faecium: The Past, the Present and the Future. J. Glob. Antimicrob. Resist. 2019, 16, 36–47. [Google Scholar] [CrossRef]
- Mohamed, J.A.; Huang, D.B. Biofilm Formation by Enterococci. J. Med. Microbiol. 2007, 56, 1581–1588. [Google Scholar] [CrossRef]
- Alfei, S.; Signorello, M.G.; Tinedi, S.; Khaledizadeh, E.; Giordani, P.; Reggio, C.; Marengo, B.; Domenicotti, C. Quaternary Phosphonium Salts Outperformed Vemurafenib (PLX) and Etoposide Against BRAF<Sup>V600D, V600E</Sup> PLX-Resistant, Melanoma and MDR Neuroblastoma, Exhibiting No/Low Toxicity on 3T3/HaCaT Cells 2026.
- Kodjo Amengor, C.D.; Amaning Danquah, C.; Adusei, E.B.A.; Kekessie, F.K.; Ofosu-Koranteng, F.; Peprah, P.; Harley, B.K.; Orman, E.; Adu, J.; Saaka, Y. Synthesized Phosphonium Compounds Demonstrate Resistant Modulatory and Antibiofilm Formation Activities against Some Pathogenic Bacteria. Heteroatom Chemistry 2022, 2022, 1–9. [Google Scholar] [CrossRef]
- Parchebafi, A.; Tamanaee, F.; Ehteram, H.; Ahmad, E.; Nikzad, H.; Haddad Kashani, H. The Dual Interaction of Antimicrobial Peptides on Bacteria and Cancer Cells; Mechanism of Action and Therapeutic Strategies of Nanostructures. Microb. Cell Fact. 2022, 21, 118. [Google Scholar] [CrossRef]
- Zając, M.; Kotyńska, J.; Zambrowski, G.; Breczko, J.; Deptuła, P.; Cieśluk, M.; Zambrzycka, M.; Święcicka, I.; Bucki, R.; Naumowicz, M. Exposure to Polystyrene Nanoparticles Leads to Changes in the Zeta Potential of Bacterial Cells. Sci. Rep. 2023, 13, 9552. [Google Scholar] [CrossRef]
- Vejzovic, D.; Piller, P.; Cordfunke, R.A.; Drijfhout, J.W.; Eisenberg, T.; Lohner, K.; Malanovic, N. Where Electrostatics Matter: Bacterial Surface Neutralization and Membrane Disruption by Antimicrobial Peptides SAAP-148 and OP-145. Biomolecules 2022, 12, 1252. [Google Scholar] [CrossRef]
- Halder, S.; Yadav, K.K.; Sarkar, R.; Mukherjee, S.; Saha, P.; Haldar, S.; Karmakar, S.; Sen, T. Alteration of Zeta Potential and Membrane Permeability in Bacteria: A Study with Cationic Agents. Springerplus 2015, 4, 672. [Google Scholar] [CrossRef]
- Khan, F.; Saha, P.; Bera, D.; Das, S. Structure Activity Relationship of Novel Antibacterial Phosphonium Ionic Liquids/Organic Salts in Dispersions and on Films: Potential Antifouling Coating Materials. Mater. Chem. Phys. 2023, 309, 128389. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers (Basel). 2020, 12, 1195. [Google Scholar] [CrossRef]
- Bacchetti, F.; Schito, A.M.; Milanese, M.; Castellaro, S.; Alfei, S. Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt. Int. J. Mol. Sci. 2024, 25, 2761. [Google Scholar] [CrossRef]
- Alfei, S.; Zuccari, G.; Bacchetti, F.; Torazza, C.; Milanese, M.; Siciliano, C.; Athanassopoulos, C.M.; Piatti, G.; Schito, A.M. Synthesized Bis-Triphenyl Phosphonium-Based Nano Vesicles Have Potent and Selective Antibacterial Effects on Several Clinically Relevant Superbugs. Nanomaterials 2024, 14, 1351. [Google Scholar] [CrossRef]
- Graikioti, D.; Athanassopoulos, C.M.; Schito, A.M.; Alfei, S. Synthesis and Characterization of Triphenyl Phosphonium-Modified Triterpenoids with Never Reported Antibacterial Effects Against Clinically Relevant Gram-Positive Superbugs 2025.
- Ermolaev, V.; Miluykov, V.; Rizvanov, I.; Krivolapov, D.; Zvereva, E.; Katsyuba, S.; Sinyashin, O.; Schmutzler, R. Phosphonium Ionic Liquids Based on Bulky Phosphines: Synthesis, Structure and Properties. Dalton Transactions 2010, 39, 5564. [Google Scholar] [CrossRef]
- Arkhipova, D.M.; Samigullina, A.I.; Minyaev, M.E.; Lyubina, A.P.; Voloshina, A.D.; Ermolaev, V. V. Synthesis, Crystal Structure, and Biological Activity of Menthol-Based Chiral Quaternary Phosphonium Salts (CQPSs). Struct. Chem. 2024, 35, 75–88. [Google Scholar] [CrossRef]
- Ermolaev, V. V.; Arkhipova, D.M.; Miluykov, V.A.; Lyubina, A.P.; Amerhanova, S.K.; Kulik, N. V.; Voloshina, A.D.; Ananikov, V.P. Sterically Hindered Quaternary Phosphonium Salts (QPSs): Antimicrobial Activity and Hemolytic and Cytotoxic Properties. Int. J. Mol. Sci. 2021, 23, 86. [Google Scholar] [CrossRef]
- Nunes, B.; Cagide, F.; Fernandes, C.; Borges, A.; Borges, F.; Simões, M. Efficacy of Novel Quaternary Ammonium and Phosphonium Salts Differing in Cation Type and Alkyl Chain Length against Antibiotic-Resistant Staphylococcus Aureus. Int. J. Mol. Sci. 2023, 25, 504. [Google Scholar] [CrossRef]
- Nunes, B.; Cagide, F.; Borges, F.; Simões, M. Antimicrobial Activity and Cytotoxicity of Novel Quaternary Ammonium and Phosphonium Salts. J. Mol. Liq. 2024, 401, 124616. [Google Scholar] [CrossRef]
- Banerjee, A.; Aremu, B.R.; Dehghandokht, S.; Salama, R.; Zhou, H.; Lackie, S.M.; Seifi, M.; Kennepohl, P.; Trant, J.F. Lethal Weapon IL: A Nano-Copper/Tetraalkylphosphonium Ionic Liquid Composite Material with Potent Antibacterial Activity. RSC Sustainability 2023, 1, 1783–1797. [Google Scholar] [CrossRef]
- Mukherjee, I.; Manna, K.; Dinda, G.; Ghosh, S.; Moulik, S.P. Shear- and Temperature-Dependent Viscosity Behavior of Two Phosphonium-Based Ionic Liquids and Surfactant Triton X-100 and Their Biocidal Activities. J. Chem. Eng. Data 2012, 57, 1376–1386. [Google Scholar] [CrossRef]
- Das, S.; Paul, A.; Bera, D.; Dey, A.; Roy, A.; Dutta, A.; Ganguly, D. Design, Development and Mechanistic Insights into the Enhanced Antibacterial Activity of Mono and Bis-Phosphonium Fluoresceinate Ionic Liquids. Mater. Today Commun. 2021, 28, 102672. [Google Scholar] [CrossRef]
- Metelytsia, L.O.; Hodyna, D.M.; Semenyuta, I. V.; Kovalishyn, V. V.; Rogalsky, S.P.; Derevianko, K.Y.; Brovarets, V.S.; Tetko, I. V. Theoretical and Experimental Studies of Phosphonium Ionic Liquids as Potential Antibacterials of MDR Acinetobacter Baumannii. Antibiotics 2022, 11, 491. [Google Scholar] [CrossRef]
- O’Toole, G.A.; Wathier, M.; Zegans, M.E.; Shanks, R.M.Q.; Kowalski, R.; Grinstaff, M.W. Diphosphonium Ionic Liquids as Broad-Spectrum Antimicrobial Agents. Cornea 2012, 31, 810–816. [Google Scholar] [CrossRef]
- Terekhova, N. V.; Khailova, L.S.; Rokitskaya, T.I.; Nazarov, P.A.; Islamov, D.R.; Usachev, K.S.; Tatarinov, D.A.; Mironov, V.F.; Kotova, E.A.; Antonenko, Y.N. Trialkyl(Vinyl)Phosphonium Chlorophenol Derivatives as Potent Mitochondrial Uncouplers and Antibacterial Agents. ACS Omega 2021, 6, 20676–20685. [Google Scholar] [CrossRef]
- Simões, M.; Pereira, A.R.; Simões, L.C.; Cagide, F.; Borges, F. Biofilm Control by Ionic Liquids. Drug Discov. Today 2021, 26, 1340–1346. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gilmore, B.F. The Antimicrobial Potential of Ionic Liquids: A Source of Chemical Diversity for Infection and Biofilm Control. Int. J. Antimicrob. Agents 2015, 46, 131–139. [Google Scholar] [CrossRef]
- Galkina, I.; Bakhtiyarova, Y.; Andriyashin, V.; Galkin, V.; Cherkasov, R. Synthesis and Antimicrobial Activities of Phosphonium Salts on Basis of Triphenylphosphine and 3,5-Di-Tert-Butyl-4-Hydroxybenzyl Bromide. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 15–18. [Google Scholar] [CrossRef]
- Terekhova, N. V.; Tatarinov, D.A.; Shaihutdinova, Z.M.; Pashirova, T.N.; Lyubina, A.P.; Voloshina, A.D.; Sapunova, A.S.; Zakharova, L.Ya.; Mironov, V.F. Design and Synthesis of Amphiphilic 2-Hydroxybenzylphosphonium Salts with Antimicrobial and Antitumor Dual Action. Bioorg. Med. Chem. Lett. 2020, 30, 127234. [Google Scholar] [CrossRef]
- Alfei, S.; Zuccari, G.; Athanassopoulos, C.M.; Domenicotti, C.; Marengo, B. Strongly ROS-Correlated, Time-Dependent, and Selective Antiproliferative Effects of Synthesized Nano Vesicles on BRAF Mutant Melanoma Cells and Their Hyaluronic Acid-Based Hydrogel Formulation. Int. J. Mol. Sci. 2024, 25, 10071. [Google Scholar] [CrossRef]
- Alfei, S.; Torazza, C.; Bacchetti, F.; Signorello, M.G.; Passalacqua, M.; Domenicotti, C.; Marengo, B. Tri-Phenyl-Phosphonium-Based Nano Vesicles: A New In Vitro Nanomolar-Active Weapon to Eradicate PLX-Resistant Melanoma Cells. Int. J. Mol. Sci. 2025, 26, 3227. [Google Scholar] [CrossRef]
- Alfei, S.; Giannoni, P.; Signorello, M.G.; Torazza, C.; Zuccari, G.; Athanassopoulos, C.M.; Domenicotti, C.; Marengo, B. The Remarkable and Selective In Vitro Cytotoxicity of Synthesized Bola-Amphiphilic Nanovesicles on Etoposide-Sensitive and -Resistant Neuroblastoma Cells. Nanomaterials 2024, 14, 1505. [Google Scholar] [CrossRef]
- Alfei, S.; Torazza, C.; Bacchetti, F.; Milanese, M.; Passalaqua, M.; Khaledizadeh, E.; Vernazza, S.; Domenicotti, C.; Marengo, B. TPP-Based Nanovesicles Kill MDR Neuroblastoma Cells and Induce Moderate ROS Increase, While Exert Low Toxicity To-Wards Primary Cell Cultures: An in Vitro Study. IJMS 2025, 26, 4991. [Google Scholar] [CrossRef]
- Cieniecka-Rosłonkiewicz, A.; Pernak, J.; Kubis-Feder, J.; Ramani, A.; Robertson, A.J.; Seddon, K.R. Synthesis, Anti-Microbial Activities and Anti-Electrostatic Properties of Phosphonium-Based Ionic Liquids. Green Chemistry 2005, 7, 855. [Google Scholar] [CrossRef]
- Milenković, M.R.; Živković-Radovanović, V.; Andjelković, L. Synthesis and Antimicrobial Activity of (3-Formyl-4-Hydroxybenzyl)Triphenylphosphonium Chloride Acylhydrazones. Russ. J. Gen. Chem. 2020, 90, 1716–1720. [Google Scholar] [CrossRef]
- Valenti, G.E.; Alfei, S.; Caviglia, D.; Domenicotti, C.; Marengo, B. Antimicrobial Peptides and Cationic Nanoparticles: A Broad-Spectrum Weapon to Fight Multi-Drug Resistance Not Only in Bacteria. Int. J. Mol. Sci. 2022, 23, 6108. [Google Scholar] [CrossRef]
- Zuccari, G.; Zorzoli, A.; Marimpietri, D.; Alfei, S. Development of Mixed Micelles for Enhancing Fenretinide Apparent Solubility and Anticancer Activity Against Neuroblastoma Cells. Curr. Drug Deliv. 2025, 22, 1017–1029. [Google Scholar] [CrossRef]
- EUCAST. European Committee on Antimicrobial Susceptibility Testing. Available online: Https://Www.Eucast.Org/Ast_of_bacteria/ (accessed on 20 January 2024).
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Alfei, S. Broad-Spectrum Bactericidal Activity of a Synthetic Random Copolymer Based on 2-Methoxy-6-(4-Vinylbenzyloxy)-Benzylammonium Hydrochloride. Int. J. Mol. Sci. 2021, 22, 5021. [Google Scholar] [CrossRef]
- Schito, A.M.; Schito, G.C.; Alfei, S. Synthesis and Antibacterial Activity of Cationic Amino Acid-Conjugated Dendrimers Loaded with a Mixture of Two Triterpenoid Acids. Polymers (Basel). 2021, 13, 521. [Google Scholar] [CrossRef]
- Schito, A.M.; Alfei, S. Antibacterial Activity of Non-Cytotoxic, Amino Acid-Modified Polycationic Dendrimers against Pseudomonas Aeruginosa and Other Non-Fermenting Gram-Negative Bacteria. Polymers (Basel). 2020, 12, 1818. [Google Scholar] [CrossRef]
- Crémet, L.; Corvec, S.; Batard, E.; Auger, M.; Lopez, I.; Pagniez, F.; Dauvergne, S.; Caroff, N. Comparison of Three Methods to Study Biofilm Formation by Clinical Strains of Escherichia Coli. Diagn. Microbiol. Infect. Dis. 2013, 75, 252–255. [Google Scholar] [CrossRef]
- Chandrasekhar, B.; Gor, R.; Ramalingam, S.; Thiagarajan, A.; Sohn, H.; Madhavan, T. Repurposing FDA-Approved Compounds to Target JAK2 for Colon Cancer Treatment. Discover Oncology 2024, 15, 226. [Google Scholar] [CrossRef]
- Cui, M.; Li, Z.; Tang, R.; Jia, H.; Liu, B. Novel (E)-5-Styryl-2,2′-Bithiophene Derivatives as Ligands for β-Amyloid Plaques. Eur. J. Med. Chem. 2011, 46, 2908–2916. [Google Scholar] [CrossRef]
- Ammer, J.; Nolte, C.; Karaghiosoff, K.; Thallmair, S.; Mayer, P.; Devivie-Riedle, R.; Mayr, H. Ion-Pairing of Phosphonium Salts in Solution: C-H…halogen and C-H…π Hydrogen Bonds. Chemistry - A European Journal 2013, 19. [Google Scholar] [CrossRef]
- Ceccacci, F.; Sennato, S.; Rossi, E.; Proroga, R.; Sarti, S.; Diociaiuti, M.; Casciardi, S.; Mussi, V.; Ciogli, A.; Bordi, F.; et al. Aggregation Behaviour of Triphenylphosphonium Bolaamphiphiles. J. Colloid Interface Sci. 2018, 531, 451–462. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Valenti, G.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef]
- Alfei, S.; Piatti, G.; Caviglia, D.; Schito, A. Synthesis, Characterization, and Bactericidal Activity of a 4-Ammoniumbuthylstyrene-Based Random Copolymer. Polymers (Basel). 2021, 13, 1140. [Google Scholar] [CrossRef]
- Godoy, C.A.; Balic, I.; Moreno, A.A.; Diaz, O.; Arenas Colarte, C.; Bruna Larenas, T.; Gamboa, A.; Caro Fuentes, N. Antimicrobial and Antibiofilm Activity of Chitosan Nanoparticles Against Staphylococcus Aureus Strains Isolated from Bovine Mastitis Milk. Pharmaceutics 2025, 17, 186. [Google Scholar] [CrossRef]
- Xu, W.; Lin, Z.; Cortez-Jugo, C.; Qiao, G.G.; Caruso, F. Antimicrobial Phenolic Materials: From Assembly to Function. Angewandte Chemie International Edition 2025, 64. [Google Scholar] [CrossRef]
- Chen, X.; Lan, W.; Xie, J. Natural Phenolic Compounds: Antimicrobial Properties, Antimicrobial Mechanisms, and Potential Utilization in the Preservation of Aquatic Products. Food Chem. 2024, 440, 138198. [Google Scholar] [CrossRef]
- Terekhova, N. V.; Tatarinov, D.A.; Shaihutdinova, Z.M.; Pashirova, T.N.; Lyubina, A.P.; Voloshina, A.D.; Sapunova, A.S.; Zakharova, L.Ya.; Mironov, V.F. Design and Synthesis of Amphiphilic 2-Hydroxybenzylphosphonium Salts with Antimicrobial and Antitumor Dual Action. Bioorg. Med. Chem. Lett. 2020, 30, 127234. [Google Scholar] [CrossRef]
- Li, Y.-T.; Huang, L.; Wen, Z.-M.; Zhu, M.-T.; Zheng, X.-T.; Zhang, Z.-H.; Wang, Z.; Ni, C.-L. Synthesis, Crystal Structure, Optical and Antimicrobial Properties of 2-Nitrobenzyl Triphenylphosphonium Tetrabromocobaltate(II). Journal of Structural Chemistry 2024, 65, 243–255. [Google Scholar] [CrossRef]
- Andreeva, O. V.; Voloshina, A.D.; Lyubina, A.P.; Garifullin, B.F.; Strobykina, I.Yu.; Belenok, M.G.; Babaeva, O.B.; Babaev, V.M.; Aznagulov, R.F.; Saifina, L.F.; et al. Antimicrobial Activity of Triphenylphosphonium (TPP) Conjugates of Alkynyl−substituted Nucleic Bases and Their Analogues. J. Antibiot. (Tokyo). 2025, 78, 731–756. [Google Scholar] [CrossRef]
- Andreeva, O. V.; Voloshina, A.D.; Lyubina, A.P.; Garifullin, B.F.; Sapunova, A.S.; Amerhanova, S.K.; Strobykina, I.Yu.; Belenok, M.G.; Babaeva, O.B.; Babaev, V.M.; et al. Triphenylphosphonium (TPP) Conjugates of 1,2,3-Triazolyl Nucleoside Analogues. Synthesis, Cytotoxicity and Antimicrobial Activity. Medicinal Chemistry Research 2025, 34, 367–391. [Google Scholar] [CrossRef]
- Galkina, I. V.; Andriyashin, V. V.; Romanov, S.R.; Egorova, S.N.; Vorob’eva, N. V.; Shulaeva, M.P.; Pozdeev, O.K.; Litvinov, I.A.; Bakhtiyarova, Y. V. Synthesis, Structure and Antimicrobial Activity of Sterically Hindered Bis-Phosphonium Derivatives of 2,6-Di-Tert-Butyl-4-Methylphenol. Mendeleev Communications 2023, 33, 635–637. [Google Scholar] [CrossRef]
- Miller, W.R.; Arias, C.A. ESKAPE Pathogens: Antimicrobial Resistance, Epidemiology, Clinical Impact and Therapeutics. Nat. Rev. Microbiol. 2024, 22, 598–616. [Google Scholar] [CrossRef]
- Kang, S.; Sunwoo, K.; Jung, Y.; Hur, J.K.; Park, K.-H.; Kim, J.S.; Kim, D. Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus Aureus (MRSA). Antibiotics 2020, 9, 758. [Google Scholar] [CrossRef]
- Knauf, G.A.; Cunningham, A.L.; Kazi, M.I.; Riddington, I.M.; Crofts, A.A.; Cattoir, V.; Trent, M.S.; Davies, B.W. Exploring the Antimicrobial Action of Quaternary Amines against Acinetobacter Baumannii. mBio 2018, 9. [Google Scholar] [CrossRef]
- Leggett, M.J.; Setlow, P.; Sattar, S.A.; Maillard, J.-Y. Assessing the Activity of Microbicides against Bacterial Spores: Knowledge and Pitfalls. J. Appl. Microbiol. 2016, 120, 1174–1180. [Google Scholar] [CrossRef]
- Maillard, J.-Y. Impact of Benzalkonium Chloride, Benzethonium Chloride and Chloroxylenol on Bacterial Antimicrobial Resistance. J. Appl. Microbiol. 2022, 133, 3322–3346. [Google Scholar] [CrossRef]
- Noel, D.J.; Keevil, C.W.; Wilks, S.A. Synergism versus Additivity: Defining the Interactions between Common Disinfectants. mBio 2021, 12. [Google Scholar] [CrossRef]
- Worthing, K.A.; Marcus, A.; Abraham, S.; Trott, D.J.; Norris, J.M. Qac Genes and Biocide Tolerance in Clinical Veterinary Methicillin-Resistant and Methicillin-Susceptible Staphylococcus Aureus and Staphylococcus Pseudintermedius. Vet. Microbiol. 2018, 216, 153–158. [Google Scholar] [CrossRef]
- Buzón-Durán, L.; Alonso-Calleja, C.; Riesco-Peláez, F.; Capita, R. Effect of Sub-Inhibitory Concentrations of Biocides on the Architecture and Viability of MRSA Biofilms. Food Microbiol. 2017, 65, 294–301. [Google Scholar] [CrossRef]
- Rahmi, K.A.; Purwono, P.B.; Rochmanti, M. Benzalkonium Chloride Effectiveness as a Disinfectant against Hospital-Associated Methicillin-Resistant Staphylococcus Aureus (HA-MRSA). Malays. J. Microbiol. 2019. [Google Scholar] [CrossRef]
- Rahmi, K.A.; Purwono, P.B.; Rochmanti, M. Benzalkonium Chloride Effectiveness as a Disinfectant against Hospital-Associated Methicillin-Resistant Staphylococcus Aureus (HA-MRSA). Malays. J. Microbiol. 2019. [Google Scholar] [CrossRef]
- Akimitsu, N.; Hamamoto, H.; Inoue, R.; Shoji, M.; Akamine, A.; Takemori, K.; Hamasaki, N.; Sekimizu, K. Increase in Resistance of Methicillin-Resistant Staphylococcus Aureus to β-Lactams Caused by Mutations Conferring Resistance to Benzalkonium Chloride, a Disinfectant Widely Used in Hospitals . Antimicrob. Agents Chemother. 1999, 43, 3042–3043. [Google Scholar] [CrossRef]
- Ferk, F.; Misik, M.; Hoelzl, C.; Uhl, M.; Fuerhacker, M.; Grillitsch, B.; Parzefall, W.; Nersesyan, A.; Micieta, K.; Grummt, T.; et al. Benzalkonium Chloride (BAC) and Dimethyldioctadecyl-Ammonium Bromide (DDAB), Two Common Quaternary Ammonium Compounds, Cause Genotoxic Effects in Mammalian and Plant Cells at Environmentally Relevant Concentrations. Mutagenesis 2007, 22, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Matt, C.; Ilic Balestri, L.J.; Skillinghaug, B.; Odell, L.R. Synthesis of Phosphonium Ylides. Comprehensive Organic Synthesis 2025, 601–648. [Google Scholar] [CrossRef]
- Emery - Pharma The Time-Kill Kinetic Essay. Available online: https://emerypharma.com/solutions/cell-microbiology-services/time-kill-kinetics-assay/ (accessed on 6 February 2026).
- Time Kill Assay: Principles, Methods, and Antimicrobial Applications. Available online: https://biologyinsights.com/time-kill-assay-principles-methods-and-antimicrobial-applications/ (accessed on 6 February 2026).
- Schito, A.M.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, G.C.; Alfei, S. Efficacy of Ursolic Acid-Enriched Water-Soluble and Not Cytotoxic Nanoparticles against Enterococci. Pharmaceutics 2021, 13, 1976. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D.; Zorzoli, A.; Marimpietri, D.; Spallarossa, A.; Lusardi, M.; Zuccari, G.; Schito, A.M. Potent and Broad-Spectrum Bactericidal Activity of a Nanotechnologically Manipulated Novel Pyrazole. Biomedicines 2022, 10, 907. [Google Scholar] [CrossRef]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Zorzoli, A.; Marimpietri, D.; Alfei, S. Bactericidal Activity of Non-Cytotoxic Cationic Nanoparticles against Clinically and Environmentally Relevant Pseudomonas Spp. Isolates. Pharmaceutics 2021, 13, 1411. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Bactericidal Activity of a Self-Biodegradable Lysine-Containing Dendrimer against Clinical Isolates of Acinetobacter Genus. Int. J. Mol. Sci. 2021, 22, 7274. [Google Scholar] [CrossRef]
- ASTM E2315-23Standard; Assessment of the Antimicrobial Activity Using a Time Kill Procedure.
- Baishya, H. Application of Mathematical Models in Drug Release Kinetics of Carbidopa and Levodopa ER Tablets. J. Dev. Drugs 2017, 06. [Google Scholar] [CrossRef]
- Alfei, S.; Grasso, F.; Orlandi, V.; Russo, E.; Boggia, R.; Zuccari, G. Cationic Polystyrene-Based Hydrogels as Efficient Adsorbents to Remove Methyl Orange and Fluorescein Dye Pollutants from Industrial Wastewater. Int. J. Mol. Sci. 2023, 24, 2948. [Google Scholar] [CrossRef]
- Alfei, S.; Orlandi, V.; Grasso, F.; Boggia, R.; Zuccari, G. Cationic Polystyrene-Based Hydrogels: Low-Cost and Regenerable Adsorbents to Electrostatically Remove Nitrites from Water. Toxics 2023, 11, 312. [Google Scholar] [CrossRef]
- Blondeau, J.; DeCory, H. In Vitro Time-Kill of Common Ocular Pathogens with Besifloxacin Alone and in Combination with Benzalkonium Chloride. Pharmaceuticals 2021, 14, 517. [Google Scholar] [CrossRef]
- Nordholt, N.; Kanaris, O.; Schmidt, S.B.I.; Schreiber, F. Persistence against Benzalkonium Chloride Promotes Rapid Evolution of Tolerance during Periodic Disinfection. Nat. Commun. 2021, 12, 6792. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Jayakumar, S.; Bihani, S.C.; Shetake, N.; Naidu, R.; Kutala, V.K.; Sarma, H.D.; Gupta, G.D.; Sandur, S.K.; Kumar, V. Pharmacological Characterization of a Structurally New Class of Antibacterial Compound, Triphenyl-Phosphonium Conjugated Diarylheptanoid: Antibacterial Activity and Molecular Mechanism. J. Biosci. 2020, 45, 147. [Google Scholar] [CrossRef] [PubMed]
- Saseendran Nair, S.; Anand, V.; De Silva, K.; Wiles, S.; Swift, S. The Antibacterial Potency and Antibacterial Mechanism of a Commercially Available Surface-Anchoring Quaternary Ammonium Salt (SAQAS)-Based Biocide in Vitro. J. Appl. Microbiol. 2022, 133, 2583–2598. [Google Scholar] [CrossRef]
- Xiao, Y.-H.; Chen, J.-H.; Fang, M.; Xing, X.-D.; Wang, H.; Wang, Y.-J.; Li, F. Antibacterial Effects of Three Experimental Quaternary Ammonium Salt (QAS) Monomers on Bacteria Associated with Oral Infections. J. Oral Sci. 2008, 50, 323–327. [Google Scholar] [CrossRef]
- Alfei, S.; Caviglia, D. Prevention and Eradication of Biofilm by Dendrimers: A Possibility Still Little Explored. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Malone, M.; Bjarnsholt, T.; McBain, A.J.; James, G.A.; Stoodley, P.; Leaper, D.; Tachi, M.; Schultz, G.; Swanson, T.; Wolcott, R.D. The Prevalence of Biofilms in Chronic Wounds: A Systematic Review and Meta-Analysis of Published Data. J. Wound Care 2017, 26, 20–25. [Google Scholar] [CrossRef]
- Fernández-Calderón, M.C.; Fernández-Babiano, I.; Navarro-Pérez, M.L.; Pazos-Pacheco, C.; Calvo-Cano, A. Biofilm Formation and Role of Other Pathogenic Factors in the Virulence of Staphylococcus Epidermidis Clinical Isolates. Front. Cell. Infect. Microbiol. 2025, 15. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Otto, M. Staphylococcus Epidermidis —Key to Understanding Biofilms, Commensalism, and More. J. Bacteriol. 2025, 207. [Google Scholar] [CrossRef]
- Jonblat, S.; As-sadi, F.; Zibara, K.; El Sabban, M.; Dermesrobian, V.; El Khoury, A.; Kallassy, M.; Chokr, A. Staphylococcus Epidermidis Biofilm Assembly and Self-Dispersion: Bacteria and Matrix Dynamics. International Microbiology 2023, 27, 831–844. [Google Scholar] [CrossRef]
- Sabaté Brescó, M.; Harris, L.G.; Thompson, K.; Stanic, B.; Morgenstern, M.; O’Mahony, L.; Richards, R.G.; Moriarty, T.F. Pathogenic Mechanisms and Host Interactions in Staphylococcus Epidermidis Device-Related Infection. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Heilmann, C. Molecular Basis of Biofilm Formation by Staphylococcus Epidermidis. In Medical Implications of Biofilms; Cambridge University Press, 2003; pp. 110–135. [Google Scholar]
- Percival, S.L.; Suleman, L.; Vuotto, C.; Donelli, G. Healthcare-Associated Infections, Medical Devices and Biofilms: Risk, Tolerance and Control. J. Med. Microbiol. 2015, 64, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival Mechanisms of Clinically Relevant Microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus Aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef]
- STEPANOVIĆ, S.; VUKOVIĆ, D.; HOLA, V.; DI BONAVENTURA, G.; DJUKIĆ, S.; ĆIRKOVIĆ, I.; RUZICKA, F. Quantification of Biofilm in Microtiter Plates: Overview of Testing Conditions and Practical Recommendations for Assessment of Biofilm Production by Staphylococci. APMIS 2007, 115, 891–899. [Google Scholar] [CrossRef]
- O’Toole, G.A. Microtiter Dish Biofilm Formation Assay. Journal of Visualized Experiments 2011. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Y. Structure–Activity Relationship of Cationic Surfactants as Antimicrobial Agents. Curr. Opin. Colloid Interface Sci. 2020, 45, 28–43. [Google Scholar] [CrossRef]
- Erick Ngehdzeka, C.; Menkem Elisabeth, Z. Bacterial Biofilm Eradication in Human Infections. In Recent Advances in Bacterial Biofilm Studies - Formation, Regulation, and Eradication in Human Infections; IntechOpen, 2024. [Google Scholar]
- Wang, Y.; Bian, Z.; Wang, Y. Biofilm Formation and Inhibition Mediated by Bacterial Quorum Sensing. Appl. Microbiol. Biotechnol. 2022, 106, 6365–6381. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Y.; Ge, Y.; Zhu, X.; Pan, J. Regulatory Mechanisms and Promising Applications of Quorum Sensing-Inhibiting Agents in Control of Bacterial Biofilm Formation. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Provencher, E.A.P.; Ehrig, M.R.; Cecere, A.G.; Cousins, S.C.; Maybin, M.A.; Meredith, T.C.; Miyashiro, T.I. Inhibition of Biofilm Formation by a Lipopolysaccharide-Associated Glycosyltransferase in the Bacterial Symbiont Vibrio Fischeri. Frontiers in Bacteriology 2023, 2. [Google Scholar] [CrossRef]
- Nguyen, A.N.X.; Thirapanmethee, K.; Audshasai, T.; Khuntayaporn, P.; Chomnawang, M.T. Insights into Molecular Mechanisms of Phytochemicals in Quorum Sensing Modulation for Bacterial Biofilm Control. Arch. Microbiol. 2024, 206, 459. [Google Scholar] [CrossRef]
- Juszczuk-Kubiak, E. Molecular Aspects of the Functioning of Pathogenic Bacteria Biofilm Based on Quorum Sensing (QS) Signal-Response System and Innovative Non-Antibiotic Strategies for Their Elimination. Int. J. Mol. Sci. 2024, 25, 2655. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Hu, X.; Zhang, X.; Wang, W.; Sun, J.; Su, Z.; Zhu, C. Strategies to Prevent, Curb and Eliminate Biofilm Formation Based on the Characteristics of Various Periods in One Biofilm Life Cycle. Front. Cell. Infect. Microbiol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, Y.; Lin, S.; Zhang, W.; Shu, G.; Lin, J.; Li, H.; Xu, F.; Tang, H.; Peng, G.; et al. Strategies for Interfering With Bacterial Early Stage Biofilms. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, K.K.; Goldmann, D.A.; Pier, G.B. Use of Confocal Microscopy To Analyze the Rate of Vancomycin Penetration through Staphylococcus Aureus Biofilms. Antimicrob. Agents Chemother. 2005, 49, 2467–2473. [Google Scholar] [CrossRef]
- Tian, L.; Shi, S.; Zhang, X.; Han, F.; Dong, H. Newest Perspectives of Glycopeptide Antibiotics: Biosynthetic Cascades, Novel Derivatives, and New Appealing Antimicrobial Applications. World J. Microbiol. Biotechnol. 2023, 39, 67. [Google Scholar] [CrossRef]
- Hu, T.; Wang, L. Vancomycin Resistance in Gram-Positive Infections: Evolutionary Strategies of Survival. Arch. Microbiol. 2026, 208, 148. [Google Scholar] [CrossRef]
- Grooters, K.E.; Ku, J.C.; Richter, D.M.; Krinock, M.J.; Minor, A.; Li, P.; Kim, A.; Sawyer, R.; Li, Y. Strategies for Combating Antibiotic Resistance in Bacterial Biofilms. Front. Cell. Infect. Microbiol. 2024, 14. [Google Scholar] [CrossRef]
- Rivani, E.; Arfijanto, M.V.; Widodo, A.D.W. Vancomycin for Methicillin-Resistant Staphylococcus Aureus Biofilm Eradication Is Associated with the Emergence of Heterogeneous Vancomycin Intermediate Staphylococcus Aureus. Int. J. Health Sci. (Qassim). 2022, 811–818. [Google Scholar] [CrossRef]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef]
- Singh, R.; Sahore, S.; Kaur, P.; Rani, A.; Ray, P. Penetration Barrier Contributes to Bacterial Biofilm-Associated Resistance against Only Select Antibiotics, and Exhibits Genus-, Strain- and Antibiotic-Specific Differences. Pathog. Dis. 2016, 74, ftw056. [Google Scholar] [CrossRef]
- Pedroni, M.A.; Ribeiro, V.S.T.; Cieslinski, J.; Lopes, A.P. de A.; Kraft, L.; Suss, P.H.; Tuon, F.F. Different Concentrations of Vancomycin with Gentamicin Loaded PMMA to Inhibit Biofilm Formation of Staphylococcus Aureus and Their Implications. Journal of Orthopaedic Science 2024, 29, 334–340. [Google Scholar] [CrossRef]
- Shiri, M.; Ashrafi, F. The Bactericidal and Antibiofilm Effects of New Liposomes Containing Vancomycin Formulation Against Clinical Biofilm Positive Staphylococcus Aureus Isolates. Appl. Biochem. Microbiol. 2023, 59, 824–832. [Google Scholar] [CrossRef]
- Huang, Z.; Li, Y.; Yin, W.; Raby, R.B.N.; Liang, H.; Yu, B. A Magnetic-Guided Nano-Antibacterial Platform for Alternating Magnetic Field Controlled Vancomycin Release in Staphylococcus Aureus Biofilm Eradication. Drug Deliv. Transl. Res. 2025, 15, 1249–1264. [Google Scholar] [CrossRef]
- Alharbi, O.; Alhazmi, K.; Gazzaz, M.; Almuhayya, S.; Aldehalan, F.; Sharif, A.; Redwan, B.; Alzain, M.; Alhazmi, W.; Altarawneh, H.; et al. A Review Vancomycin Role in Gram Positive Biofilm-Associated Infections: Challenges and Emerging Solutions. Ther. Clin. Risk Manag. 2025, Volume 21, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, O.; Alhazmi, K.; Gazzaz, M.; Almuhayya, S.; Aldehalan, F.; Sharif, A.; Redwan, B.; Alzain, M.; Alhazmi, W.; Altarawneh, H.; et al. A Review Vancomycin Role in Gram Positive Biofilm-Associated Infections: Challenges and Emerging Solutions. Ther. Clin. Risk Manag. 2025, Volume 21, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Sahore, S.; Kaur, P.; Rani, A.; Ray, P. Penetration Barrier Contributes to Bacterial Biofilm-Associated Resistance against Only Select Antibiotics, and Exhibits Genus-, Strain- and Antibiotic-Specific Differences. Pathog. Dis. 2016, 74, ftw056. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.B.; Yao, L.Q.; Guo, Z.Y.; Ma, Y.C.; Wang, F.; Yang, J.H. Enhancing Biofilm Disruption and Bactericidal Efficiency Using Vancomycin-Loaded Microbubbles in Sonodynamic Therapy. JAC. Antimicrob. Resist. 2025, 7. [Google Scholar] [CrossRef]
- Borges, N.H.; Suss, P.H.; Ortis, G.B.; Dantas, L.R.; Tuon, F.F. Synergistic Activity of Vancomycin and Gentamicin Against Staphylococcus Aureus Biofilms on Polyurethane Surface. Microorganisms 2025, 13, 1119. [Google Scholar] [CrossRef]





















| Sample | Z-Ave (nm), kcps | ζ-p (mV), kcps |
| 1 | N.D. | 38.10±6.15, 143.2.1 |
| 2 | 604.9±218.2, 12.7 | 4.19±3.26, 1.1 |
| 3 | 156.9±54.4, 14.2 | 15.7±5.07, 31.9 |
| 4 | 140.4±22.9, 24.2 | 13.4±7.63, 93.8 |
| Strains |
1 (433.3)1 |
2 (356.5) 1 |
3 (527.5) 1 |
4 (509.5) 1 |
V/T | OXA |
| MIC µg/mL | ||||||
| S. aureus ATCC 29213 MSSA | 16 | N.L. | N.L. | N.L. | 0.5(V) | 0.5 |
| S. aureus B MRSA | 8 | N.L. | N.L. | N.L. | 0.25(V) | 512 |
| S. epidermidis 22 MRSE | 16 | N.L. | N.L. | N.L. | 0.5(V) | 128 |
| E. faecalis 1VRE, * | 64 | N.L. | N.L. | N.L. | 256(V); 64(T) | N.R. |
| E. faecium 152 VRE, * | 64 | N.L. | N.L. | N.L. | 128(V); 64(T) | N.R. |
| P. aeruginosa 259 **, CF, CR | > 128 | N.L. | N.L. | N.L. | N.R. | 128 |
| E. coli **, *** | > 128 | N.L. | N.L. | N.L. | N.R. | 128 |
| K. pneumoniae **, *** | > 128 | N.L. | N.L. | N.L. | N.R. | 256 |
| Compounds | 1 (433.3) 1 | oxacillin | vancomycin/teicoplanin |
| Gram-positive Strains | MIC (µg/mL) | MIC (µg/mL) | MIC (µg/mL) |
| S. aureus 18 MRSA | 32 | 512 | 0.5(V) |
| S. aureus ATCC 29213 MSSA | 32 | 128 | 0.5(V) |
| S. aureus 189 MRSA | 16 | 0.5 | 0.5(V) |
| S. aureus B MRSA | 8 | 512 | 0.25(V) |
| S. epidermidis 22 MRSE | 16 | 128 | 0.5(V) |
| S. epidermidis 25 MRSE | 64 | 256 | 0.5(V) |
| S. epidermidis 64 MRSE | 64 | 128 | 0.5(V) |
| S. epidermidis 147 MRSE | 64 | 64 | 1(V) |
| E. faecalis 1 VRE, *, ** | 64 | N. R. | 256(V); 64(T) |
| E. faecalis 439 VRE, * | 64 | N. R. | 256(V); 64(T) |
| E. faecalis 365 VRE, *, ** | 64 | N. R. | 32(V) ;1(T) |
| E. faecalis 451 VRE, * | 64 | N. R. | 128(V); 32(T) |
| E. faecium 152 VRE, *, ** | 64 | N. R. | 128(V); 64(T) |
| E. faecium 183 VRE, *, ** | 64 | N. R. | 256(V); 64(T) |
| E. faecium 185 VRE, *, ** | 64 | N. R. | 256(V); 32(T) |
| E. faecium 364 VRE, * | 64 | N. R. | 64(V); 005(T) |
| Gram-negative Strains | MIC (µg/mL) | MIC (µg/mL) | MIC (µg/mL) |
| P. aeruginosa 259 ***, CF, CR | > 128 | > 128 | N. R. |
| P. aeruginosa 229 ***, CF | > 128 | > 128 | N. R. |
| P. aeruginosa 247 *** | > 128 | > 128 | N. R. |
| P. aeruginosa 256 *** | > 128 | > 128 | N. R. |
| P. aeruginosa 268 *** | > 128 | > 128 | N. R. |
| Kinetic Model | Control | 1 |
| PFO | 0.9616 | 0.9550 |
| PSO | 0.9925 | 0.9631 |
| Parameter | CTR * | CTR (EXP) | 1 | 1 (EXP) |
| GKe (Log10CFU/mL) | 11.34 (Ge) | 10.91 | 0.6668 (Ke) | 0.6990 |
| KPSO ** | 0.0706 | N.A. | 0.5089 | N.A. |
| Compounds | Mathematical Model | KPSO | Time (h) | Refs. |
| TPP- salts | PSO | 0.5681 | 24 | [32,72,96] |
| QASs | 714.9 | 8 | [97,98] | |
| P7 (QAPs) | 1.25 | 24 | [53] | |
| P5 (QAPs) | 3.07 | 24 | [62] | |
| 1 | 0.5089 | 24 | This work |
| OD (Optical Density) | Biofilm classification |
| < 0.1 | Not adhering (No producer) |
| 0.1 – 0.5 | Weak biofilm producer |
| 0.5 – 1.0 | Moderate biofilm producer |
| > 1.0 | Strong biofilm producer |
| Compound (MW) | Cells | IC50 (µg/mL) | HC50 (µg/mL) |
| 1 (433.3) | RBCs | - | 23.0±4.7 |
| HaCaT | 216.7±V.W. * | - | |
| 3T3 | 656.9± V.W. * | - |
| Compounds | 1 (433.3) 1 | SIs | ||
| Gram-positive Strains | MIC (µg/mL) | RBCs | HaCaT | 3T3 |
| S. aureus 18 MRSA | 32 | 0.72 | 6.77 | 20.53 |
| S. aureus ATCC 29213 | 32 | 0.72 | 6.77 | 20.53 |
| S. aureus 189 MRSA | 16 | 1.44 | 13.54 | 41.06 |
| S. aureus B MRSA | 8 | 2.88 | 27.09 | 82.11 |
| S. epidermidis 22 MRSE | 16 | 1.44 | 13.54 | 41.06 |
| S. epidermidis 25 MRSE | 64 | 0.36 | 3.39 | 10.26 |
| S. epidermidis 64 MRSE | 64 | 0.36 | 3.39 | 10.26 |
| S. epidermidis 147 MRSE | 64 | 0.36 | 3.39 | 10.26 |
| E. faecalis 1 VRE *, ** | 64 | 0.36 | 3.39 | 10.26 |
| E. faecalis 439 VRE * | 64 | 0.36 | 3.39 | 10.26 |
| E. faecalis 365 VRE *, ** | 64 | 0.36 | 3.39 | 10.26 |
| E. faecalis 451VRE * | 64 | 0.36 | 3.39 | 10.26 |
| E. faecium 152 VRE *, ** | 64 | 0.36 | 3.39 | 10.26 |
| E. faecium 183 VRE *, ** | 64 | 0.36 | 3.39 | 10.26 |
| E. faecium 185 VRE *, ** | 64 | 0.36 | 3.39 | 10.26 |
| E. faecium 364 VRE * | 64 | 0.36 | 3.39 | 10.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



