
Submitted:
01 March 2026
Posted:
03 March 2026
You are already at the latest version
Abstract
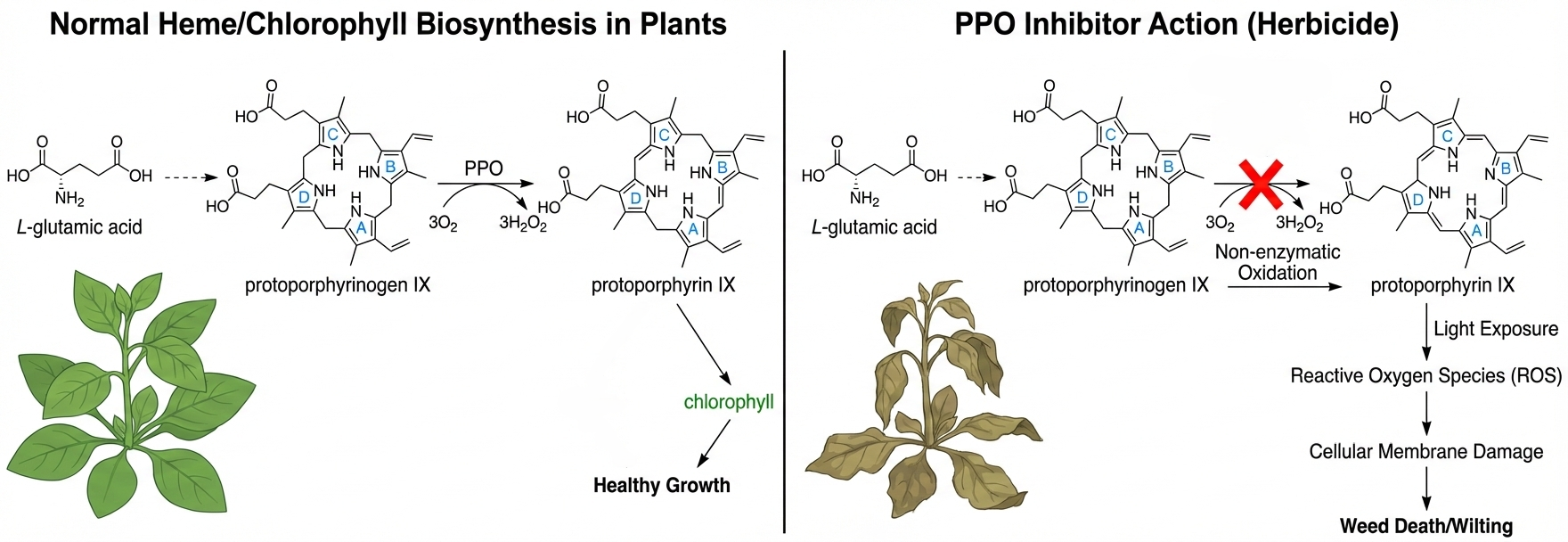
As the key enzyme catalyzing the final step in the biosynthesis of heme and chlorophyll, protoporphyrinogen oxidase (PPO) has become a crucial target for herbicide development. To date, more than 40 PPO-inhibiting herbicides have been developed, exhibiting multiple advantageous characteristics: they combine high efficacy with environmental friendliness, feature low effective concentrations, rapid action, long-lasting effects, and excellent control of both monocotyledonous and dicotyledonous weeds. In recent years, significant progress has been made in the structural biology of PPO—five crystal structures from tobacco, humans, and various bacteria have been resolved, most of which are presented as enzyme-inhibitor complexes. Although the development of such herbicides spans over five decades, novel PPO inhibitors still hold broad potential for innovation due to the resistance of early applied PPOs. This review systematically summarizes the three-dimensional structures of PPO from different sources, the interaction mechanisms between the enzyme and inhibitors, studies on quantitative structure-activity relationships of inhibitors, and outlines molecular design directions for the next generation of PPO inhibitors.
Keywords:
PPO inhibitors
; herbicide discovery
; molecular design
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



