
Submitted:
21 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
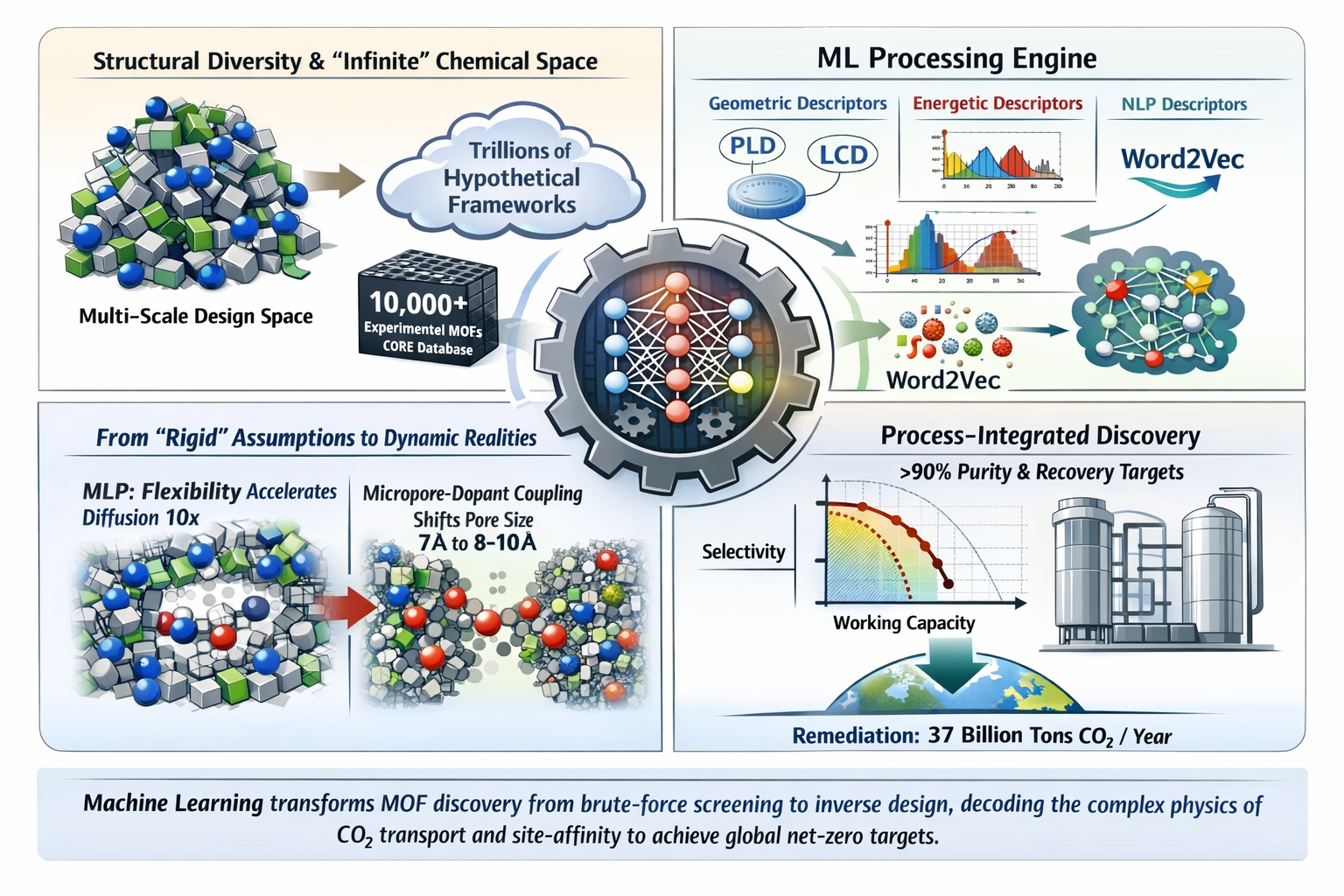
The accelerating climate crisis, driven by annual CO₂ emissions exceeding 37 billion metric tons, necessitates the rapid advancement of Carbon Capture and Storage (CCS) and Direct Air Capture (DAC) technologies. Metal–Organic Frameworks (MOFs) have emerged as highly promising sorbents due to their tunable pore architectures and exceptional surface areas; however, exploration of their vast chemical design space remains computationally prohibitive. This review systematically examines the expanding role of Machine Learning (ML) in accelerating CO₂ capture research within MOFs. Using a structured evaluation protocol, we assess state-of-the-art models across four key dimensions: predictive performance, descriptor physical relevance, mechanistic interpretability, and process-level applicability. Recent advances in Machine Learning Interatomic Potentials (MLPs) demonstrate that framework flexibility significantly influences adsorption thermodynamics and diffusivity, challenging conventional rigid-lattice assumptions. Generative approaches—including Deep Reinforcement Learning and transformer-based architectures—enable inverse design of high affinity frameworks, while physics-informed descriptor engineering improves predictive accuracy across pressure regimes (R² > 0.90). Importantly, the field is transitioning from isolated property prediction toward multiscale, process-integrated optimization, where ML models couple material features with industrial performance metrics such as CO₂ purity and recovery in pressure swing adsorption systems. Collectively, these developments indicate that future progress will depend on physics informed, interpretable architectures capable of bridging molecular-scale discovery with experimentally robust and water-stable materials suitable for industrial deployment.
Keywords:
machine learning
; MOFs
; CO2
; green house gases
; adsorption
; artificial intelligence
Introduction
The average global temperature has been rising steadily, with 2023 recorded as approximately 1.2 °C warmer than the preindustrial average. This warming is primarily driven by greenhouse gas emissions, with carbon dioxide (CO2) accounting for over 60% of the effect. Annual global CO2 emissions exceeded 37.15 billion tons in 2022, a trend that underscores the urgent need to achieve “net zero” targets by 2050. To manage these levels, Carbon Capture and Storage (CCS) and Direct Air Capture (DAC) have emerged as promising solutions to mitigate emissions from both industrial point sources and ambient air [1].
A key challenge for these technologies lies in identifying materials that exhibit strong CO2 affinity and high selectivity over other gases like nitrogen (N2) or atmospheric water vapor (H2O) [2]. Metal-Organic Frameworks (MOFs)—crystalline porous materials composed of metal clusters linked by organic ligands—have surfaced as leading candidates due to their ultrahigh surface areas, tunable pore sizes, and programmable functionalities. Unlike traditional adsorbents, MOFs can be precisely engineered with open metal sites (OMS) or specific chemical functional groups (e.g., diamines) to enhance CO2 binding efficacy [3].
However, the chemical design space for MOFs is nearly infinite, with over 100,000 synthesized structures and trillions more hypothesized in silico. Comprehensive evaluation of these materials using traditional laboratory synthesis or high-fidelity molecular simulations, such as Grand Canonical Monte Carlo (GCMC) [4] or Density Functional Theory (DFT) is computationally prohibitive and time intensive. This bottleneck necessitates a shift toward data-driven computational screening and generative design to accelerate the discovery of next-generation carbon capture materials. Machine Learning (ML) has become a transformative tool in the remediation of CO2 by offering a compromise between the high accuracy of ab initio methods and the speed of classical force fields. In the context of MOFs, ML applications serve four primary remediated functions:
ML models, such as Random Forests (RF) and Artificial Neural Networks (ANN), are used to rapidly predict CO2 working capacity and selectivity, enabling the screening of tens of thousands of materials in a fraction of the time required for traditional simulations [5].
Advanced Machine-Learning Interatomic Potentials (MLPs) allow researchers to model framework flexibility, revealing that structural vibrations can accelerate CO2 diffusivity by an order of magnitude compared to rigid models. Furthermore, the integration of artificial intelligence with high-throughput computational screening has further refined predictive modeling, guiding experimental efforts toward optimal materials and enhancing CO2 adsorption efficiency through the design of core-shell MOFs [6]
Techniques like Deep Reinforcement Learning (DRL) and Large Language Models are employed for the inverse design of MOFs, navigating complex subspaces to identify materials with extreme CO2 affinity that are rarely found in existing databases. ML is utilized to bridge the gap between material properties and industrial cycle performance, predicting process-level metrics such as CO2 purity, recovery, and energy productivity in pressure swing adsorption cycles [7].
By integrating explainable ML models with SHAP or PDP analysis, researchers can now quantify the influence of specific material features—such as Lewis acidity, pore geometry, and steric hindrance—to provide a theoretical basis for the next generation of CO2 capture experiments [8].
The reviews were written based on the following foundational criteria:
Each review identifies the specific properties predicted (e.g., uptake, selectivity, or TOF) and the performance metrics (e.g., R2 or RMSE) achieved against established “ground truth” labels.
Models were evaluated based on whether their inputs (descriptors) were derived from fundamental physics or chemical intuition, such as pore limiting diameter, metal partial charges, or energy-based radial distribution functions.
The reviews scrutinize how models were validated (e.g., k-fold cross-validation) and whether they demonstrated transferability to unseen material classes or external experimental data. Beyond reporting accuracy, each review highlights the new scientific insight provided by the model—such as the “bifurcation” of optimal pore sizes or the “coupling effect” between functional dopants and micropore volume.
Each analysis addresses the limitations of the work, such as the reliance on rigid-framework assumptions, the exclusion of open metal sites, or the failure to model chemical reactions in humid streams.
This systematic base ensures that the resulting review summary accurately reflects the model’s utility for industrial scalability and its role in advancing the field of computational materials science.
1. Physics-Informed Descriptor Engineering
1.1. Descriptor Engineering Strategies
The integration of spatially aware energy descriptors represents a vital advancement in capturing the complex potential energy surfaces (PES) of porous materials. By supplementing traditional geometric features and Henry’s constants with surface energy histograms and radial distribution functions (RDFs), researchers have achieved an R2>0.97 for nitrogen and R2>0.87 for carbon dioxide isotherms. This approach explicitly addresses the “intermediate pressure bottleneck”—the regime where both binding energy and spatial heterogeneity dictate uptake—by providing the ML model with a physically meaningful representation of surface shape.
Crucially, the use of XGBoost trained on over 10,000 structures reveals that while affinity distribution (f(E)) sets the energetic baseline, the spatial decay of interaction sites (captured via RDF) is the primary differentiator for capacity in frameworks with identical energy levels. However, the model’s modest performance in the CO2 chemisorption regime highlights the current ceiling of single-charge probes. To reach industrial-grade reliability, future physics-informed ML directions must incorporate dipole and quadrupole probes to account for the orientation-dependent packing and electrostatic multipoles of CO2, bridging the gap between simplified physisorption models and real-world selective separation [9].
By moving beyond standalone pore or doping engineering, researchers have identified a critical micropore-dopant coupling mode that determines CO2 transport in carbon-based adsorbents. Using a Random Forest architecture trained on multi-scale simulation data (R2=0.934), this study introduces Free volume (Vf) as a primary descriptor (accounting for 25% relative importance) to quantify the steric effects induced by surface functionalization. This approach reveals a significant thermodynamic shift: while basic dopants (e.g., NH2) utilize Lewis’s acid-base interactions to optimize adsorption at 7 Å, physisorption-dominant dopants (e.g., oxygen groups) occupy available nano space, necessitating an enlarged optimal pore size of 8–10 Å to maximize capacity. Experimental verification confirmed these findings, as adsorbents engineered with this specific coupling mode achieved a leading-level capacity of 4 mmol/g, a 130% improvement over non-optimized frameworks. Such results underscore the necessity of physics-informed descriptor engineering to resolve long-standing debates regarding the inhibitory vs. promotional effects of heteroatom doping in porous carbons [10].
The integration of advanced descriptor engineering—specifically the addition of over 700 “calculated” molecular features—has been shown to improve CO2 adsorption prediction accuracy by 15%–20% compared to traditional structural models. While the XGBoost framework achieves a high coefficient of determination (R2>0.94) at industrial pressures (2.5 bar), its performance sensitivity at low pressures (0.01 bar) highlights the challenges of capturing sparse gas-solid interactions. Crucially, the use of SHAP interpretability reveals a fundamental thermodynamic transition: the predictive weight of atomic mass and number (representing van der Waals forces) at low pressures is gradually superseded by charge distribution and electronegativity features (representing Coulomb forces) as pressure increases. However, the reliance on a hypothetical dataset (hMOF) and simulated GCMC labels presents a risk of overestimating real-world performance; without experimental validation, the model’s ability to account for structural defects or competitive adsorption in complex flue gases remains a critical gap for industrial scalability [11].
1.2. Hybrid Textural-Optimization and Outlier-Aware Predictive Modeling
The shift from standalone machine learning to hybridized optimization frameworks represents a critical advancement in overcoming the “black-box” limitations of traditional adsorption modeling. By integrating the Growth Optimization (GO) algorithm with Least Square Support Vector Machines (LSSVM), researchers achieved a remarkable R2 of 0.9798, effectively neutralizing the hyperparameter tuning errors that lead to underestimation in standalone models at high uptake regimes. A database-driven analysis of 475 experimental points reveals that SBET is the primary structural driver of capture, outperforming Langmuir surface area and pore volume in predictive sensitivity.
Critically, the use of Williams plot analysis confirms that 94.95% of the experimental data falls within the model’s applicable domain, providing a level of statistical robustness essential for rapid screening. However, the current model’s reliance on experimental textural measurements suggests a persistent bottleneck in purely computational discovery pipelines. To enhance industrial scalability, future physics-informed ML directions must bridge the gap between these high-accuracy experimental models and high-throughput theoretical proxies, ensuring that the “metal-affinity” and “pore-filling” dynamics identified here can be exploited in the de novo design of next-generation carbon capture materials [12].
1.3. Ensemble Interaction Mapping and Node-Affinity Hierarchies
The deployment of custom stacking ensemble architectures represents a significant leap in bridging the accuracy gap between individual machine learning models and complex experimental adsorption data. By synthesizing the predictive strengths of tree-based, kernel based, and neural network learners, researchers achieved a benchmark-surpassing R2 of 0.9833 for CO2 uptake across 1,212 experimental data points. This study moves beyond simple performance reporting to provide a multi-criteria interpretability framework, utilizing ablation studies and permutation importance to decouple the influence of thermodynamics from framework architecture.
Critically, the analysis identifies a “metal-affinity hierarchy,” where Copper and Magnesium centers exhibit a clear productivity advantage over traditional Zinc-based nodes, a finding validated by partial dependence plots that quantify the stepwise shift in CO2 binding efficacy. However, the 70% dominance of Zinc-based entries in current experimental repositories highlights a persistent data imbalance challenge that risks model overfitting toward well studied materials. To enhance industrial scalability, future physics-informed ML directions must incorporate stratified sampling and synthetic oversampling (e.g., SMOTE) to ensure that the discovery of high-performance MOFs extends into the underrepresented chemical subspaces of rare or complex metal centers [13]
While physics-informed descriptor engineering establishes the foundational language through which MOF structures are numerically represented, the true utility of these features emerges in large-scale predictive workflows. Once chemically meaningful descriptors are defined, they enable supervised learning models to rapidly estimate adsorption properties across vast chemical spaces. This progression from feature construction to high-throughput prediction marks the first major acceleration point in ML-driven CO2 remediation research
2. High-Throughput Screening and Universal Property Prediction
2.1. High-Throughput Screening and Discovery
The shift toward hybrid physics-ML models allows for the screening of composite materials with an efficiency that traditional molecular simulations cannot match. By training a stacked ensemble regression model on over 54,000 hybrid membranes, researchers achieved an exceptional predictive accuracy of R2 =0.96, demonstrating that transfer learning is a robust strategy for predicting “unseen” materials like 6FDA-DAM based on known polymer characteristics. Crucially, big data mining of this dataset unveiled that the bottleneck for elite performance transitions from the polymer matrix to the MOF filler; in top-performing membranes, PLD and LCD importance spikes to over 30%, suggesting that pore-size engineering is more critical than polymer selection once a high-permeability baseline is established.
However, constructive evaluation reveals a dependency on the Maxwell model assumption of ideal interfaces. In real-world industrial scalability, the presence of interfacial gaps and polymer chain rigidity often compromises the theoretical “Robeson limit” performance. Future physics-informed ML directions should integrate interfacial morphology descriptors to account for the complex bonding between organic linkers and polyimide matrices, ensuring that high-throughput screening leads to materials that maintain their separation efficacy under the mechanical stresses of flue gas streams [14].
The transition from simulation-heavy datasets to experimental-based ML models provides a more rigorous benchmark for real-world CO2 capture. By utilizing the CATBoost architecture, researchers have demonstrated that ensemble learning can navigate the heterogeneous landscape of 236 unique experimental MOFs with a 15% RMSE improvement over traditional gradient-boosting frameworks. While the model confirms that pressure and surface area remain the dominant thermodynamic drivers, the integration of SHAP interpretability uncovers a deeper layer of chemical control: localized atomic environments and electronic state distributions (e.g., PEOE_VSA7) are the true differentiators for optimizing adsorption.
However, this study also serves as a critical evaluation of generalization limits. The disparity between the model’s training performance (R2 =0.99) and its validation score (R2 =0.84) underscores a persistent overfitting challenge when training complex models on limited experimental data. Future efforts in physics-informed ML must move toward dataset diversification, incorporating a wider range of chemical functionalities to ensure that highthroughput screening of hypothetical frameworks can reliably translate to industrially scalable, high-stability sorbents [15].
2.2. Universal Property Prediction and Isotherm Generalization
The transition toward unified experimental datasets represents a critical advancement in zeolite-based carbon capture, moving beyond material-specific empirical models. By training a Gradient Boosted Trees (GBT) model on over 5,700 experimental data points, researchers have achieved a “universal” predictive capability that significantly outperforms traditional Langmuir isotherm fittings in both accuracy and generalization. This approach identifies the Si/Al ratio and cation composition as pivotal chemical determinants, allowing the model to generalize across diverse frameworks (e.g., FAU, ZSM-5, 13X) and extreme operational conditions up to 45bar.
Critically, the use of external validation on unseen datasets confirms the model’s robustness, effectively addressing the overfitting challenges common in literature-derived datasets. However, a constructive critique reveals that 32% of the training data lacked surface area reporting, and the dataset remains skewed toward low-uptake regimes (<2 mmol/g). To enhance industrial scalability, future physics-informed ML directions must prioritize the inclusion of high-uptake experimental benchmarks and standardized reporting of structural parameters to further refine model sensitivity in peak performance regimes [16]
2.3. Property-Driven ML Applications
This study is the application of ML to predict two specific properties—CO2 working capacity and CO2/N2 selectivity. Artificial Neural Networks (ANNs) provide a computationally efficient alternative to Graph Neural Networks for the rapid screening of Metal-Organic Frameworks, achieving high predictive accuracy (R2) for carbon capture metrics. By integrating industrial, field, and simulation data, this model quantifies a critical design insight: CO2 working capacity is predominantly driven by pore size and surface area, showing a weak negative correlation with increased chemical complexity. However, the model’s reliability is non-uniform; while CO2 capacity predictions are robust (MAE = 0.8 mmol/g), the CO2/N2 selectivity predictions exhibit substantial dispersion (MAE = 25), suggesting that intrinsic properties alone may not fully capture the competitive adsorption physics required for high-selectivity forecasting. Furthermore, without specific thermodynamic benchmarks (Fixed T, P) or external experimental validation, the model’s generalization capability to novel, uncharacterized structural fragments remain restricted. Future physics-informed directions must address the high uncertainty in selectivity modeling to ensure that rapid AI-based screening translates into dependable industrial carbon reduction technologies [17].
Although high-throughput screening significantly reduces computational burden, predictive accuracy alone does not guarantee scientific understanding. Black-box models risk obscuring the physicochemical principles governing adsorption behavior. Consequently, interpretability frameworks and thermodynamic mapping approaches are essential to extract mechanistic insights from trained models and ensure that predictions remain grounded in adsorption physics
3. Model Interpretability and Thermodynamic Mapping
3.1. Model Interpretability and Physical Insight
Machine-learning potentials (MLPs) trained on quantum chemistry data provide a high-fidelity alternative to the rigid-lattice assumptions typically used in molecular simulations of gas transport. By utilizing the DeepPot-SE model to capture the dynamic flexibility of MgMOF-74, researchers achieved exceptional predictive accuracy (R2 =0.9916) for system energies. This approach reveals that structural flexibility is a dominant factor in predicting CO2 diffusivity; rigid models significantly overestimate adsorption free-energy barriers, resulting in diffusion coefficients nearly ten times slower than those observed in flexible frameworks.
While this integration of thermodynamics and ML highlights the “hopping” mechanism between open metal sites, the model’s generalization capability remains a point of critical evaluation. Because the MLP was trained exclusively on simulated DFT-MD trajectories from a single 30ps window, its performance is intrinsically tied to the accuracy of the underlying semi-empirical lot. Furthermore, the lack of direct experimental validation for Mg-MOF-74 diffusivity underscores a broader need for standardized experimental benchmarks to verify the industrial scalability of such physics-informed ML models. Studies relying on limited simulated datasets, while useful for revealing physical trends like flexibility-enhanced diffusion, must be cautiously applied to real-world applications where complex gas mixtures and structural defects may alter transport kinetics [18]
3.2. Ensemble-Averaged Thermodynamics and Potential Energy Surface (PES) Mapping
The development of transferable machine learning force fields (MLFFs) represents a paradigm shift in the high-throughput screening of porous materials for direct air capture. By finetuning a foundation model (MACE-MP) with a specialized GoldDAC dataset, researchers have achieved ab initio quality thermodynamics at computational speeds comparable to classical methods. This approach reveals that standard UFF+DDEC models suffer from systemic inaccuracies in chemically complex hybrid environments—particularly in lanthanide-based frameworks, where H2O adsorption energies are overestimated by up to 17.8%. Furthermore, the move from single-point interaction energies to ensemble-averaged properties via the DAC-SIM package has identified 161 promising candidates, demonstrating that the “selective pocket” mechanism provided by parallel benzene rings (PAR) and uncoordinated nitrogen is essential for maintaining a high CO2 /H2O selectivity (KH ratio > 100). While these models successfully ground screening in real-world physics, the current reliance on rigid-framework assumptions remains a hurdle for accurately capturing the regeneration costs and chemisorption dynamics critical for large-scale industrial deployment [19]
The deployment of Machine-Learning Interatomic Potentials (MLPs) has enabled the exploration of gas transport in complex, functionalized frameworks at a scale and precision previously unattainable by classical MD (over 4,000 atoms on nanosecond timescales). By systematically analyzing the transition from Mg2(dobdc) to Mg2(dotpdc), this study quantifies that pore size enlargement alone can accelerate CO2 diffusivity by a factor of 4 to 6. Crucially, the model provides deep mechanistic clarity on the role of diamine functionalization: rather than increasing the population of high-affinity sites, these chemical modifiers redistribute the majority of CO2 (up to 77%) into intermediate interaction regions. Thermodynamic insights derived from interpretable models clarify equilibrium adsorption behavior; however, practical carbon capture systems are governed not only by equilibrium capacity but also by mass transport kinetics and multicomponent interactions. A comprehensive understanding therefore, requires extending ML frameworks beyond static adsorption metrics toward molecular diffusion, interfacial transport phenomena, and competitive gas separation mechanisms.
This integration of thermodynamics and ML reveals that the “cooperative insertion mechanism” is less about static binding and more about a dynamic redistribution that favors faster kinetics. However, while the study utilizes experimental NMR data for validation, its reliance on a purely computational MLP framework highlights the limitations in modeling chemical transitions to carbamates or carbonates in humid streams. To ensure industrial scalability, future physics-informed ML directions must bridge the gap between these transport models and the complex stability and regeneration cycles required in high-humidity flue gas environments [20]
4. Molecular Transport and Multicomponent Separation Mechanisms
4.1. Molecular-Level Transport and Adsorption Mechanisms
The implementation of fragment-based Neural Network Potentials (NNPs) addresses the long-standing computational bottleneck of modeling flexible frameworks with open metal sites (OMS) at ab initio accuracy. By utilizing an E (3)-equivariant architecture (NequIP) trained on a limited set of ~2,000 DFT conformers, this study achieved an exceptional force RMSE of 0.039 eV/Å, while maintaining high transferability with only 0.54% deviation in lattice parameters compared to SCXRD experimental benchmarks. Crucially, the integration of thermodynamics and ML through a hybrid MD/GCMC workflow reveals that structural dynamics are essential for realistic adsorption modeling: framework flexibility facilitates structural relaxation, which delocalizes CO2 molecules and optimizes the energy landscape at low pressures (0.1–1.0 bar).
While traditional rigid-lattice GCMC models provide a reasonable approximation at 298 K, they significantly underestimate uptake at higher temperatures by failing to accommodate the necessary structural adjustments between the guest and host. Despite the success of this active-learning-driven screening, a critical evaluation of the fragment-based approach suggests that while transferable, such models must be carefully validated against diverse chemical environments to ensure they do not “forget” complex interactions outside their initial training fragment. Future physics-informed ML directions should focus on utilizing these high-fidelity potentials to explore the cooperative insertion mechanisms and regeneration cycles required for large-scale industrial carbon capture [21]
4.2. Multicomponent Gas Separation and Structural Design Strategies
The transition from binary/ternary gas models to actual 6-component natural gas mixtures (N2,CO2,CH4,C2H6,C3H8,H2S) marks a critical step toward the industrial deployment of MOF-based carbon capture. By integrating GCMC simulations with a Random Forest architecture, this study successfully identified 10 elite MOF candidates from the 12,020-structure CoRE-MOF database, achieving a predictive accuracy of R2=0.922 for material renderability. A database-driven analysis of the results reveals a “volcanic” structure-property relationship, where optimal separation performance is constrained to a density window of 0.5–1.7 g/cm3; frameworks outside this range suffer from either kinetic exclusion or a loss of selectivity due to oversized pores.
The study defines a clear tripartite design strategy for next-generation adsorbents: (i) replacing metal nodes, such as substituting Cd with Mn, to quadruple working capacity; (ii) incorporating nitrogen-rich organic linkers (e.g., pyridine or azoles) to exploit electrostatic interactions; and (iii) regulating topological connectivity to optimize pore geometry, While the rigid-framework assumption successfully identified top-tier materials like XIGWUF and ETECOX, future physics-informed ML directions must move beyond static lattice models to incorporate the thermodynamic and kinetic impacts of framework flexibility in real-world, high-pressure natural gas streams [22].
While transport modeling elucidates how CO2 migrates through porous architectures, adsorption selectivity and catalytic activation are ultimately dictated by local chemical environments within the framework. Engineering Lewis acid–base interactions, electronic structure modulation, and synergistic site arrangements represents a complementary strategy to purely structural optimization, bridging adsorption thermodynamics with chemical reactivity
5. Active-Site and Electronic Structure Engineering
5.1. Lewis Acid-Base Site Engineering and Catalytic Kinetics
The integration of low-cost descriptors with explainable machine learning enables the rapid screening of catalytic frameworks without the prohibitive cost of density functional theory (DFT). By training a Random Forest architecture on 372 high-yield experimental data points, researchers achieved a remarkable 97% accuracy in predicting CO2 cycloaddition activity. This study moves beyond “black-box” predictions by utilizing SHAP and PDP analysis to quantify the “optimal Lewis acidity” of metal nodes; results show that a metal partial charge between 1.2 and 2.0 maximizes epoxide substrate activation while preventing active site deactivation through saturated coordination.
The successful experimental validation of MOF-76(Y)—which achieved a top-tier TOF of 64.72 h−1—confirms that ML can bridge the gap between hypothetical structure generation and real-world CO2 utilization. However, a critical evaluation suggests that the reliance on literature-derived datasets necessitates careful data cleaning (e.g., filtering for high yields) to ensure comparability across non-linear reaction profiles. Future physics-informed ML directions should focus on standardizing these experimental benchmarks to further improve the industrial scalability of screened catalysts for diverse catalytic reactions [23].
5.2. Electronic Property Modulation and Electrocatalytic Selectivity
The integration of DFT-driven discovery with Gradient Boosting Regression (GBR) architectures has successfully identified a elite class of 2D conjugated MOFs (2D c-MOFs) that surpass traditional Cu(211) benchmarks in CO2 electroreduction performance. By systematically modifying both the metal active sites and organic ligands (HDQ series), this research quantifies a fundamental stability hierarchy (TMN4>TMN2O2>TMO4) and uncovers catalysts with remarkably low limiting potentials, such as NiN4−HDQ (UL=−0.04V for CO).
A critical ML-based sensitivity analysis reveals that electron affinity (EA) and electronegativity (χ) are the dominant electronic “handles” for tuning activity, collectively accounting for over 34% of feature importance. While these 2D c-MOFs exhibit high thermal stability at 400 K and maintain pristine surfaces in aqueous environments, the study confirms that ligand-induced orbital overlap is the primary driver of intermediate adsorption strength. To ensure industrial scalability, future physics-informed ML directions must bridge these high-fidelity C1 models with multi-carbon (C2+) coupling kinetics, unlocking the full potential of these highly conductive, tunable frameworks for artificial carbon cycle applications [24]
5.3. Synergistic Site Engineering and Composite Pore Modulation
The integration of Convolutional Neural Networks (CNNs) with inception modules addresses the computational “time-trap” of simulating complex composite adsorbents, providing a robust framework for multi-criteria screening. By training on 700 GCMC-validated structures, this model achieved a high predictive fidelity (R2≈0.90) for both CO2 working capacity and selectivity, effectively navigating the inherent trade-offs between these two metrics. Crucially, big data analysis of the 1,631-composite pool reveals that while most ionic liquids enhance selectivity by physically blocking N2 transport, rare “synergistic” frameworks like IL@MARJAQ utilize the IL to generate entirely new potential energy minima for CO2.
This study also quantifies the non-linear impact of IL loading, demonstrating that “more is not always better”; for example, the selectivity of IL@GUBKUL can be tuned from 614 to over 7,000 simply by optimizing the number of inserted molecules. Such insights move the field away from trial-and-error post-synthetic modification toward precision loading strategies. However, the current model’s rigid-framework assumption and its tendency to underestimate uptake in frameworks with open metal sites (OMS) suggest that future physicsinformed ML directions must incorporate lattice dynamics to fully capture the process economics and regeneration efficiencies required for industrial flue gas separation [25]
Insights obtained from active-site engineering and electronic modulation naturally inform the next evolutionary step: inverse design. Rather than screening existing materials, generative machine learning models exploit learned structure–property relationships to propose entirely new MOF architectures optimized for multiple objectives. This shift from predictive modeling to autonomous material generation fundamentally redefines the discovery paradigm.
6. Multi-Objective Inverse Design and Generative MOF Discovery
6.1. Multi-Objective Inverse Design and Chemical Subspace Exploration
The transition from brute-force screening to Deep Reinforcement Learning (DRL) for inverse design addresses the challenge of navigating the nearly infinite chemical space of porous crystals. By integrating Transformer-based predictors into a reward-driven environment, researchers have successfully generated physisorbents for Direct Air Capture with Qst values exceeding 40 kJ/mol and CO2/H2O selectivities greater than 1. A database-driven analysis of these results reveals a critical trade-off in material “genes”: high affinity for CO2 is often driven by open metal sites (e.g., Mn-based N131 nodes) which concurrently attract water, whereas high selectivity is governed by specific Cu and Zn clusters that occupy a separate subspace in the chemical design landscape.
While the DRL approach demonstrates robust extrapolation capability—producing structures that compete with top-performing experimental MOFs like KAUST-7—its current reliance on classical force fields limits its ability to model chemisorptive interactions involving charge transfer. To improve industrial scalability, future physics-informed ML directions must incorporate DFT-derived charges and active learning loops to refine the predictors, ensuring that inverse-designed candidates can maintain efficacy in the humid, high-dilution environments characteristic of real-world atmospheric capture [26].
The shift from manual high-throughput screening to reinforcement learning-enhanced generative design marks a pivotal advancement in the discovery of materials tailored for specific carbon capture regimes. By leveraging the MOFGPT framework, researchers have demonstrated that while traditional supervised fine-tuning fails to generate a single chemically valid structure (0% validity), a reward-guided RL policy can achieve 100% validity even when targeting extreme CO2 adsorption performance (mean+2σ). This approach allows for the navigation of a nearly infinite chemical space by optimizing MOFid string sequences—encoding both SMILES-based chemistry and RCSR-based topologies— without the need for manual assembly of building blocks.
A database-driven analysis of the generated candidates confirms that the RL model captures intrinsic structure-property correlations; for example, high CO2 uptake is consistently linked to open Cu2+ paddle wheel units and nitrogen-rich linkers that enhance electrostatic interactions. Furthermore, with novelty rates consistently above 63%, the framework demonstrates an ability to explore underrepresented regions of the chemical landscape that traditional screening might overlook. However, the current reliance on rigid-lattice assumptions remains a notable limitation, as it excludes the dynamic flexibility often critical for CO2 transport kinetics. Future physics-informed ML directions should integrate 3D structural construction and DFT-based stability filters to bridge the gap between AI-designed strings and synthetically robust industrial sorbents [27]
Despite remarkable advances in generative discovery, material-level optimization alone does not guarantee industrial viability. Adsorption performance must ultimately be evaluated within realistic process environments, including pressure swing and temperature swing cycles. Integrating machine learning with process simulation therefore closes the loop between molecular design and system-scale performance, enabling translation from computational prediction to deployable carbon capture technologies.
7. Process-Level Integration and Industrial Translation
7.1. Process-Integrated Generative Design and Pore-Size Bifurcation
The development of a multiscale generative workflow marks a significant transition from simple property screening to process-level material design. By utilizing MOF-NET, an architecture grounded in NLP-style word embeddings, researchers have successfully navigated a landscape of trillions of hypothetical structures to identify candidates that “substantially outperform” benchmarks like 13X zeolite and CALF-20. A database-driven analysis of the top-performing materials reveals a structural dichotomy: optimal performance is achieved either through strict size exclusion (3–5 Å pores) or through the engineering of high-density binding pockets (6–30 Å pores) where CO2 molecules are stabilized by multiple oxygen-rich nodes.
Critically, this study identifies Cu-based nodes and fluorinated short-linkers as the “genetic markers” of elite adsorbents, providing a clear design guideline for experimentalists. While the best-in-class small-pore material (hjm + N387 + E44) demonstrates a significant productivity advantage over CALF-20 due to its superior N2 rejection, the study highlights a persistent innovation gap. Many computationally designed materials continue to face hurdles in synthesizability and water stability, emphasizing that the next frontier for physics-informed ML must be the integration of synthetic accessibility (SA) scores and lattice dynamics to ensure these “theoretical possibilities” can survive the transition from computer to the laboratory [28]
Conclusion:
The integration of Machine Learning (ML) into the discovery and optimization of Metal– Organic Frameworks (MOFs) has transformed the paradigm of carbon capture research. The field has progressed beyond large-scale brute-force screening toward data-driven strategies that integrate prediction, interpretation, and generative design within unified computational workflows. A central advance has been the recognition that framework flexibility plays a critical role in adsorption thermodynamics and transport kinetics. Machine-learning interatomic potentials and hybrid simulation frameworks have demonstrated that rigid-lattice approximations can underestimate diffusivity and misrepresent adsorption near open metal sites. These findings highlight the importance of dynamic structural effects in accurately modeling CO2 behavior within porous architectures. Simultaneously, inverse design approaches—enabled by reinforcement learning and transformer-based generative models— have shifted material discovery from passive screening to proactive exploration of chemical subspaces. When coupled with physically meaningful descriptors, these models provide insight into how pore geometry, functional group distribution, and confinement effects collectively influence adsorption performance. Despite rapid methodological progress, a gap persists between computational discovery and industrial implementation. Many models remain biased toward well-characterized chemistries and often neglect factors critical for deployment, including water stability, multicomponent gas competition, and synthetic feasibility. Bridging this gap will require the integration of stability-aware objectives, synthetic accessibility metrics, and process-level constraints into generative and predictive frameworks. Looking forward, the most promising direction lies in physics-informed, interpretable ML architectures that operate across molecular, material, and process scales. By coupling dynamic adsorption modeling with industrial performance evaluation, future research can accelerate the development of experimentally viable and scalable adsorbent platforms aligned with global net-zero objectives.
Comparative Overview of ML Models for CO2 and Gas Remediation:
| Study Focus | Primary ML Algorithm(s) | Key Descriptor(s) | Predictive Performance (R2) | Core Scientific Insight |
| Pore Energy Mapping | XGBoost | Energy-based RDFs & Surface Histograms | > 0.81 CO2 > 0.97 N2 | Spatially aware energy RDFs resolve the “intermediate pressure bottleneck” in isotherms. |
| Statistical Void Analysis | ERT / RF / XGBoost | Void Fraction Moments ( VFn) |
Up to 0.984 | The distribution of voids (VF2) is as critical as the total void fraction for uptake capacity. |
| Composite Modulation | CNN (Inception) | Geometric + Chemical (Ionic Liquids) | ≈ 0.90 | Ionic liquids can act as synergistic sites, creating new potential energy minima for CO2. |
| Kinetic Transport | DeepPot-SE (MLP) | Atomic coordinates (Flexible) | 0.9916 (Energy) | Framework flexibility accelerates CO2 diffusivity by 10x compared to rigid models. |
| Generative Design | MOFGPT (Transformer) | MOFid (NLP-based strings) | 35–100% Validity | Reinforcement learning effectively navigates the “extreme tail” of property distributions. |
| Process-Level Design | MOF-NET (ANN) | Word Embeddings of Building Blocks | Elite purity/recovery | Optimal design bifurcates into small-pore exclusion vs. large-pore binding. |
| Mixed Matrix Membranes | Stacking Ensemble | Polymer FFV + MOF PLD/LCD | 0.96 | A “10x permeability rule” exists where filler must exceed polymer permeability for gain. |
| Experimental Benchmarking | Stacking (RF/XGB/MLP) | Textural (BET) + Operational (P, T) | 0.9833 | Identified a metal-affinity hierarchy where Mg and Cu centers provide superior binding sites. |
| Hybrid Optimization | LSSVM-GO | Textural + Operational | 0.9798 | Growth Optimization (GO) significantly reduces prediction errors in high-uptake regimes. |
| Multicomponent Separation | Random Forest | Structural + Chemical Descriptors | 0.922 (R%) | MOF renderability is optimized within a specific density window of 0.5–1.7 g/cm3. |
| Electrocatalytic Selectivity | Gradient Boosting (GBR) | Electronic (EA, chi, d-band) | 0.9998 | Catalytic activity is primarily governed by electron affinity and electronegativity. |
| Universal Zeolite Prediction | GBT / RF / DL | Si/Al Ratio + Cation type | 0.936 | Provides a universal framework without case-specific parameter fitting required by Langmuir models. |
References
- Ozkan M., Akhavi A.-A., Coley W.C., Shang R., Ma Y., Progress in carbon dioxide capture materials for deep decarbonization, Chem 8 (1) (2022) 141–173. [CrossRef]
- Carrascal-Hernández, D.C.; Grande-Tovar, C.D.; Mendez-Lopez, M.; Insuasty, D.; García-Freites, S.; Sanjuan, M.; Márquez, E. CO2 Capture: A Comprehensive Review and Bibliometric Analysis of Scalable Materials and Sustainable Solutions. Molecules 2025, 30, 563. [CrossRef]
- Mahajan S., Lahtinen M., Recent progress in metal-organic frameworks (MOFs) for CO2 capture at different pressures, J. Environ. Chem. Eng. 10 (6) (2022) 108930. [CrossRef]
- Multi-Scale Computational Design of Metal–Organic Frameworks for Carbon Capture Using Machine Learning and Multi-Objective Optimization.
- Zijun Deng and Lev SarkisovChemistry of Materials 2024 36 (19), 9806-9821DOI: 10.1021/acs.chemmater.4c01969.
- Hussin F., Md Rahim S.A.N., Mohamed Hatta N.S., Aroua M.K., Mazari S.A., A systematic review of machine learning approaches in carbon capture applications, J. CO2 Util. 71 (2023) 102474. [CrossRef]
- Coudert F.-X., Recent advances in stimuli-responsive framework materials: Understanding their response and searching for materials with targeted behavior, Coord. Chem. Rev. 539 (2025) 216760. [CrossRef]
- Park H., Majumdar S., Zhang X., Kim J., Smit B., Inverse design of metal–organic frameworks for direct air capture of CO2 via deep reinforcement learning, Digit. Discov. 3 (4) (2024) 728–741. [CrossRef]
- Fathalian, Farnoush & Aarabi, Sepehr & Ghaemi, Ahad & Hemmati, Alireza. (2022). Intelligent prediction models based on machine learning for CO2 capture performance by graphene oxide-based adsorbents. Scientific Reports. 12. 21507. 10.1038/s41598-022-26138-6.
- Z. Deng, L. Sarkisov, Engineering machine learning features to predict adsorption of carbon dioxide and nitrogen in metal–organic frameworks, J. Phys. Chem. C (2024). [CrossRef]
- J. Zuo, F. Sun, Z. Qu, C. Yang, L. Xie, Y. Zhang, X. Li, J. Li, Unraveling the coupling effect of micropore confinement and functional sites of carbon-based adsorbents on flue gas CO2 adsorption: A machine learning study based on multi-scale simulations, Carbon Capture Sci. Technol. (2025). [CrossRef]
- Y. Teng, G. Shan, Interpretable machine learning for materials discovery: Predicting CO2 adsorption properties of metal–organic frameworks, APL Mater. 12 (2024) 081115. [CrossRef]
- P.O. Longe, S. Davoodi, M. Mehrad, D.A. Wood, Robust machine-learning model for prediction of carbon dioxide adsorption on metal–organic frameworks, J. Alloys Compd. (2024). [CrossRef]
- Z. Iyiola, E.T. Brantson, N.J. Okeke, K. Sanni, P. Longe, Carbon capture using metal organic frameworks (MOFs): Novel custom ensemble learning models for prediction of CO2 adsorption, Processes 13 (2025). [CrossRef]
- H. Wan, Y. Fang, M. Hu, S. Guo, Z. Sui, X. Huang, Z. Liu, Y. Zhao, H. Liang, Y. Wu, H. Gao, Z. Qiao, Interpretable machine-learning and big data mining to predict the CO2 separation in polymer–MOF mixed matrix membranes, Adv. Sci. (2024). [CrossRef]
- S. Achour, Z. Hosni, ML-driven models for predicting CO2 uptake in metal–organic frameworks (MOFs), Can. J. Chem. Eng. (2024). [CrossRef]
- E. Kirtil, Universal prediction of CO2 adsorption on zeolites using machine learning: A comparative analysis with Langmuir isotherm models, ChemEngineering 9 (2025) 80. [CrossRef]
- E.V. Kotov, J. Sravanthi, G. Logabiraman, H. Dhall, M. Chandna, P. Madan, V. Sharma, Carbon capture and storage optimization with machine learning using an ANN model, E3S Web Conf. 588 (2024) 01003. [CrossRef]
- B. Zheng, G.X. Gu, C. dos Santos, R.N.B. Ferreira, M. Steiner, B. Luan, Simulating CO2 diffusivity in rigid and flexible Mg-MOF-74 with machine-learning force fields, APL Mach. Learn. 2 (2024) 026115. [CrossRef]
- Y. Lim, H. Park, A. Walsh, J. Kim, Accelerating CO2 direct air capture screening for metal–organic frameworks with a transferable machine learning force field, ChemRxiv (2024). [CrossRef]
- J. Randrianandraina, C.S. Hong, J.-H. Lee, Unraveling the effects of pore size and diamine functionalization on CO2 diffusion in metal–organic frameworks using machine-learning interatomic potentials, ChemRxiv (2025). [CrossRef]
- Tayfuroglu, S. Keskin, Modeling CO2 adsorption in flexible MOFs with open metal sites via fragment-based neural network potentials, ChemRxiv (2025). [CrossRef]
- Y. Zhou, S. Ji, S. He, W. Fan, L. Zan, L. Zhou, X. Ji, G. He, Machine-learning-assisted high-throughput screening of metal–organic frameworks for CO2 separation from CO2-rich natural gas, Ind. Eng. Chem. Res. (2024). [CrossRef]
- X. Bai, Y. Li, Y. Xie, Q. Chen, X. Zhang, J.-R. Li, High-throughput screening of CO2 cycloaddition MOF catalyst with an explainable machine learning model, Green Energy Environ. (2024). [CrossRef]
- G. Xing, S. Liu, G. Sun, J.-Y. Liu, Modification of metals and ligands in two-dimensional conjugated metal–organic frameworks for CO2 electroreduction: A combined DFT and machine learning study, SSRN Electron. J. (2024). [CrossRef]
- M. Sheng, X. Zhang, H. Cheng, Z. Song, Z. Qi, Multi-criteria computational screening of [BMIM][DCA]@MOF composites for CO2 capture, Green Chem. Eng. (2024). [CrossRef]
- H. Park, B. Smit, S. Majumdar, X. Zhang, J. Kim, Inverse design of metal–organic frameworks for direct air capture of CO2 via deep reinforcement learning, Digit. Discov. (2024). [CrossRef]
- S. Badrinarayanan, R. Magar, A. Antony, R.S. Meda, A.B. Farimani, MOFGPT: Generative design of metal–organic frameworks using language models, J. Chem. Inf. Model. (2025). [CrossRef]
- Z. Deng, L. Sarkisov, Multi-scale computational design of metal–organic frameworks for carbon capture using machine learning and multi-objective optimization, Chem. Mater. (2024). [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



