Submitted:
23 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Diabetes Mellitus is a chronic metabolic disorder characterized by impaired insulin production and/or action, leading to persistent hyperglycemia and insulin resistance. It has been associated with several comorbidities, including cognitive dysfunction, affecting functions such as attention, memory, and processing speed. Mounting evidence indicates a complex relationship between type 2 Diabetes Mellitus (DM2) and neurodegenerative disorders such as mild cognitive impairment and Alzheimer’s disease (AD). Beyond the conventional hallmarks of each pathology, patients with DM2 face an increased risk of neuronal degeneration, while AD is characterized by a marked reduction in insulin receptor density. Although aging, neuroinflammation, and vascular dysfunction have been recognized as key risk factors in AD, the precise molecular mechanisms driving AD pathogenesis remain incompletely understood. Various studies have been conducted to identify reliable biomarkers that elucidate the connection between DM2 and AD, including insulin dysregulation, neuroinflammation, amyloid-β aggregation, and tau hyperphosphorylation. Investigation on these biomarkers is still ongoing and may serve not only as diagnostic tools but also as therapeutic targets. Here, we review the current evidence supporting a convergent biological framework between DM2 and AD. Clarifying these shared pathways may improve early detection and guide the development of targeted therapeutic strategies aimed at reducing neurodegeneration in metabolically vulnerable populations.
Keywords:
Alzheimer's disease
; amyloid-β
; biomarkers
; hyperglycemia
; neuroinflammation
; cognitive dysfunction
; type 2 diabetes mellitus
1. Introduction
Diabetes Mellitus (DM) is a metabolic disorder that accounts for 90% - 95% of all diabetes cases and is considered one of the most significant public health concerns (Burillo et al., 2021). The global prevalence of DM in 2019 was 9.3% (463 million people), and it is projected to rise to 10.2% (578 million people) by 2030 and 10.9% (700 million people) by 2045 (Takeishi et al., 2021). DM is characterized by disrupted insulin secretion and/or function, leading to persistent hyperglycemia and insulin resistance (IR) in peripheral tissues (Bakker et al., 2009; Saini, 2010).
In addition to its metabolic complications, DM has been increasingly associated with cognitive dysfunction, including impairments in executive functions, attention, memory, and processing speed (G. J. Biessels & Despa, 2018; Hameed et al., 2015; Sebastian et al., 2023). Recent studies have identified DM as a potential risk factor for cognitive decline, including mild cognitive impairment (MCI), vascular dementia, and particularly Alzheimer's disease (AD) (Hameed et al., 2015).
AD is the most prevalent form of dementia worldwide, affecting nearly 50 million people, and is marked by progressive memory loss and cognitive decline (Takeishi et al., 2021). At the molecular level, AD is characterized by extracellular amyloid-β (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, and activation of immune glial cells, mainly microglia (Calabrò et al., 2021; Lee et al., 2022).
Emerging evidence suggests that DM and AD share overlapping pathophysiological mechanisms, including insulin signaling dysfunction, Aβ accumulation, tau hyperphosphorylation, oxidative stress, vascular dysfunction, and neuroinflammation (Arvanitakis et al., 2004; Rasool et al., 2021). About 50-52% of patients with DM develop dementia as a late complication, with an increased risk of over 65% for AD (Arvanitakis et al., 2004; Barbagallo, 2014; Rasool et al., 2021). Understanding the intricate relationship between peripheral metabolic disorders and brain pathologies is critical to unraveling the molecular mechanisms that link DM -particularly DM2- and AD (Behl et al., 2022).
The relationship between metabolic dysfunction and cognitive impairment has gradually emerged over the past decades. In 1980, hypoglycemia was first observed to influence brain function (Adolfsson et al., 1980), and by 1994, researchers began to associate altered glucose metabolism with dementia (Razay & Wilcock, 1994). In 1996, further studies reported that DM2 patients and elevated blood glucose levels exhibited learning and memory deficits (Messier & Gagnon, 1996). Similarly, in AD patients, hyperglycemia was associated with neuronal damage due to Aβ accumulation (Messier & Gagnon, 1996). This led to the conceptualization of the term "brain diabetes" in 2005 (Li & Hölscher, 2007), to describe overlapping features between diabetes and AD, and to the identification of shared pathological features between DM2 and AD in subsequent years, such as the presence of fibrillar protein aggregates: amylin in pancreatic islets in DM2, and Aβ plaques and NFTs in AD patients (de la Monte & Wands, 2008; Götz et al., 2009).
Additional contributors to this intersection have been identified, including inflammation, oxidative stress, insulin receptor downregulation, and Aβ accumulation (Park, 2011; Saini, 2010). DM2 can impact brain function through various mechanisms, including glucose-induced toxicity, blood-brain barrier (BBB) disruption, increased production of reactive oxygen species (ROS), and IR in the brain (Behl et al., 2022; Garcia-Serrano & Duarte, 2020). Hyperglycemia and reduced brain glucose uptake also contribute to the formation of advanced glycation end products (AGEs), which are now considered a molecular bridge between DM2 and AD pathologies (Behl et al., 2022; Kong et al., 2020; Uribarri et al., 2020).
Patients with DM2 have a higher risk of neurodegeneration and AD development (Stanciu et al., 2020). In AD, insulin receptor density is reduced by 80%, and dysfunctional insulin signaling is believed to play a pivotal role in the development of AD (Hernández-Contreras et al., 2021). Therefore, DM2 patients not only have a higher predisposition to cognitive impairments but also an elevated risk of developing AD later in life (Pasquier et al., 2006; Verdelho et al., 2007). These cognitive impairments have also been replicated in animal models of diabetes, reinforcing the likelihood of a biological connection (Biju & Paulose, 1998; Winocur & Greenwood, 2005).
Additionally, the brains of AD patients exhibit features like those of insulin-deficient states, including reduced insulin and Insulin Growth Factor (IGF) signaling, as well as increased markers of oxidative and inflammatory stress (Craft, 2005, 2006; Rivera-Meza et al., 2017; Steen et al., 2005). Together, these findings support the notion that DM2 and AD are interrelated conditions, linked by common metabolic and neurobiological pathways.
This review aims to explore the neurobiological mechanisms linking DM2 to AD, identify additional risk factors contributing to this association, and analyze current advancements in shared biomarkers. By assessing these connections, we aim to provide a comprehensive understanding of the relationship between DM2 and AD from a neurobiological perspective and to suggest targeted therapeutic strategies (Figure 1).
3. Bridging the Gap: Exploring the Interplay Between Insulin Signaling, Diabetes, and Alzheimer's Disease.
Insulin is a hormone secreted by pancreatic β-cells permeable to the BBB and, therefore, able to enter the CNS via systemic circulation (Xourgia et al., 2019). In the brain, insulin acts as a potent neurotrophic factor, regulating metabolism, synaptic plasticity, and cognitive function through the activation of insulin receptors (Galea, 2021; Sima & Zhang, 2014; Sonar & Lal, 2018).
Insulin receptors are ubiquitously expressed in the brain, particularly in the hippocampus, cortex, hypothalamus, olfactory bulb, and pituitary, with a higher density in neurons compared to glia (J. Duarte, 2014; Kleinridders et al., 2014). Studies have shown that insulin receptors are densely expressed in the axons of hippocampal pyramidal neurons and are predominantly localized in brain regions essential for learning, memory, and cognitive functions. In addition to their structural presence, functional studies in healthy conditions provide strong evidence that insulin signaling is crucial for optimal cognitive performance (Freychet, 2000; McNay et al., 2010; Pomytkin et al., 2018). Specifically, when insulin receptors are blocked or inhibited within the hippocampus, there is a significant impairment in memory performance, indicating that endogenous insulin signaling is necessary for the maintenance of normal memory processes. This functional disruption emphasizes that insulin action within the hippocampus is not merely supportive but vital for proper cognitive function, with receptor blockade leading directly to deficits in spatial working memory and other hippocampal-dependent tasks (Freychet, 2000; McNay et al., 2010; Pomytkin et al., 2018).
In DM2 patients, a decrease in insulin receptor density and alterations in insulin signaling have been observed, particularly in the hippocampus, cortex, and choroid plexus (A. I. Duarte et al., 2012). Metabolic disturbances associated with diabetes, including hyperglycemia, hyperinsulinemia, and hypercholesterolemia, have been linked to brain atrophy and neuropathological features of AD (C.-C. Huang et al., 2014; Scheltens et al., 2021; Verdile et al., 2015). Furthermore, even in the absence of dementia, insulin resistance has been associated with cognitive impairment in aging (Burns et al., 2012; Gong et al., 2023).
Insulin regulates protein synthesis, post-translational modifications (PTMs), the assembly of cytoskeletal and adhesive proteins, and intracellular support molecules important for axonal integrity (Sima & Zhang, 2014; Yang et al., 2023). Dysregulation of these processes is associated with an increased risk of developing diabetes and can also lead to neuronal dysfunction, synaptic deficits, and increased vulnerability to neurodegeneration (J. M. N. Duarte, 2023; Yang et al., 2023).
Importantly, insulin regulates the metabolism of Aβ and tau, the basic components of amyloid plaques and NFTs, respectively (G. J. Biessels & Kappelle, 2005; Craft et al., 1998; Kim et al., 2013; Steen et al., 2005). Reduced insulin production, as observed in aging and AD, correlates with decreased insulin activity in regions such as the frontal cortex, hippocampus, and hypothalamus (De Felice et al., 2022).
Knopman, in 2014, studied glucose metabolism using positron emission tomography with fluorodeoxyglucose (PET-FDG) in cognitively normal APOE4 carriers, showing reduced uptake in various brain regions, including the temporal lobe (Knopman et al., 2014). Finally, the APOE3 allele is the most common isoform and tends to remain neutral regarding the disease, neither increasing nor decreasing the risk (Langella et al., 2023; Rebeck et al., 2002). Other studies confirm APOE ε4’s association with accelerated cognitive decline, especially when combined with different mutations, such as PSEN1 (Langella et al., 2023; Polsinelli et al., 2023) (Figure 4).
Interestingly, some studies suggest that APOE ε4 carriers with diabetes may experience distinct cognitive trajectories compared to non-carriers. While APOE ε4 primarily affects long-term memory, diabetes is more closely linked to deficits in working memory (Ravipati et al., 2022). This suggests that DM2 and APOE ε4 may influence cognition through partially independent but converging mechanisms.
The importance of considering APOE genotype and diabetes as potential risk factors for AD highlights the need for further research to elucidate how these factors contribute to the development and progression of the disease (Ravipati et al., 2022; Shinohara et al., 2021) (Figure 4). Insights gained from such studies could pave the way for targeted interventions and personalized treatment approaches in the field of AD research and management.
4. Therapeutic Opportunities and Pharmacological Interventions
Late-onset AD and DM2 share common features, including impaired insulin signaling, chronic inflammation, mitochondrial dysfunction, and glucose dysregulation, suggesting a link between the two conditions (Cholerton et al., 2013; Mancinetti et al., 2023). As mentioned, IR and impaired glucose metabolism may contribute to the development and progression of AD (Mancinetti et al., 2023). These shared mechanisms have raised interest in evaluating anti-diabetic drugs as potential therapies for AD and cognitive decline.
In experimental models, including rodent studies of ischemia and neurodegeneration, pioglitazone has been shown to exert vasculoprotective effects, such as reducing inflammatory responses in adipose tissue, decreasing urinary TGF-β1 excretion, and promoting endothelial progenitor cell mobilization, which enhances angiogenesis (Desouza & Shivaswamy, 2010). These effects are mediated through activation of PPAR gamma and beta/delta pathways, leading to reduced inflammation, improved endothelial function, and potential neuroprotective mechanisms. Clinically, in patients with DM2, pioglitazone's benefits include improved glycemic control, decreased inflammatory mediators like C-reactive protein and VEGF, and possible nephroprotective effects, as evidenced by reduced urinary TGF-β1 excretion. However, risks such as peripheral edema, weight gain, and hypoglycemia when combined with insulin or other hypoglycemic agents are notable considerations (Desouza & Shivaswamy, 2010). By inhibiting the reabsorption of glucose in the kidneys and promoting glucose excretion in the urine, SGLT2 inhibitors improve glycemic control while also modulating brain metabolism and neuroinflammatory pathways implicated in AD (Desouza & Shivaswamy, 2010; Dolan et al., 2010; Majid et al., 2025).
Recent population-based studies have compared the effects of SGLT2 inhibitors with other anti-diabetic drugs, such as dipeptidyl peptidase-4 (DPP4) inhibitors, indicating that SGLT2 inhibitors may be more effective in reducing the risk of dementia in DM2 patients (DeFronzo et al., 2013; Mancinetti et al., 2023). The neuroprotective effects of these drugs extend beyond their anti-diabetic properties, suggesting a direct impact on brain health and cognitive function (Cholerton et al., 2013).
Furthermore, SGLT2 inhibitors have been shown to prevent memory impairment in AD animal models, showing beneficial effects on neurogenesis, synaptic plasticity, and neurodegeneration (DeTure & Dickson, 2019). These findings suggest that targeting glucose metabolism through SGLT2 inhibition may offer novel therapeutic strategies for managing cognitive decline associated with AD (DeFronzo et al., 2013).
On the other hand, AD clinical presentations differ between early- and late-onset forms. Early-onset AD is associated with language, visuospatial, or executive function impairments, while late-onset AD patients exhibit the classic amnestic pattern of the disease (Palasí et al., 2015). Comorbidities such as diabetes, cardiovascular dysfunction, and obesity are more commonly observed in late-onset pathology, supporting the metabolic contribution to disease progression (Chen et al., 2017; Gerritsen et al., 2016).
Insulin plays a crucial role in brain health, influencing cerebral bioenergetics, synaptic viability, neurotransmitter turnover, and proteostasis (Kellar & Craft, 2020). Insulin signaling dysregulation in the brain can lead to neurodegeneration by impairing the clearance of Aβ and tau, promoting vascular damage, and increasing inflammation (Busiguina et al., 2000; Clark & Vissel, 2018; El Khoury et al., 2014).
Peripheral IR may result in reduced brain insulin concentrations, impairing insulin signaling within neurons (Könner et al., 2011). Notably, insulin directly interacts with Aβ, reducing its synaptotoxicity and facilitating clearance mechanisms (Craft et al., 2020). This interaction is critical because reduced insulin levels in the brain and cerebrospinal fluid (CSF) have been reported in AD patients (Craft et al., 2020; Kellar & Craft, 2020).
Emerging therapeutic strategies now aim to restore brain insulin function. Intranasal insulin administration has shown promise by enhancing cognitive performance in both preclinical and clinical studies (Craft et al., 2020). This approach increases brain insulin availability without affecting blood glucose, reducing the risk of hypoglycemia.
Other therapeutic targets include insulin-like growth factor-1 (IGF-1) and insulin receptor substrate-1 (IRS-1), which are frequently impaired in AD brains (Arvanitakis et al., 2020; Talbot et al., 2012). Postmortem studies of AD patients have revealed deficient insulin signaling in regions associated with cognition, such as the frontal cortex and hippocampus (Arvanitakis et al., 2020). These abnormalities are associated with both Aβ accumulation and tau hyperphosphorylation, underscoring the relevance of insulin-related pathways in AD pathogenesis (Craft et al., 2020).
In conclusion, therapies originally designed for DM2, such as SGLT2 inhibitors, may have potential in slowing or preventing cognitive decline associated with AD. These strategies target the intersection of metabolism, neuroinflammation, and neurodegeneration, offering a multi-modal approach to treating patients at the crossroads of diabetes and dementia. Further research is warranted to elucidate the specific mechanisms by which SGLT2 inhibitors exert their neuroprotective effects and to optimize their clinical use.
5. Biomarkers Related to Neurodegeneration in Diabetes Mellitus and Alzheimer's Disease
In recent years, the development of highly sensitive immunoassays has significantly advanced the early and less invasive detection of AD biomarkers in plasma and cerebrospinal fluid (Kivisäkk et al., 2024). Given the growing evidence that DM2 may contribute to the pathogenesis of AD, neurodegeneration has been considered a possible mechanism establishing a connection between DM2 and AD (Madhusudhanan et al., 2020).
Several converging mechanisms, such as vascular endothelial dysfunction, insulin dysregulation, dysglycemia, neuroinflammation, Aβ accumulation, and abnormal tau phosphorylation, have been identified as shared contributors in the pathogenesis of cognitive impairment associated with both DM2 subtypes and AD (Burillo et al., 2021; Sible et al., 2022; Trimm & Red-Horse, 2023; Tumminia et al., 2018; Willette et al., 2015) (Table 1).
AD is pathologically defined by extracellular Aβ plaques and intracellular NFTs composed of hyperphosphorylated tau (Zhang et al., 2021). In CSF, lower levels of Aβ42 and lower Aβ42:Aβ40 ratios have been related to AD (Andersson et al., 2023). This is due to the sequestration of Aβ42 into amyloid plaques, which induces elevated levels of total tau (t-tau) and tau phosphorylated at Thr 181 (p-tau181) in CSF (Lim et al., 2023). Additionally, increases in t-tau and p-tau181 in CSF tend to correlate with Aβ burden rather than with NFT load (Horie et al., 2023), suggesting that they may reflect early pathophysiological changes rather than late-stage neuronal loss (Barthélemy et al., 2023).
Recent advances in plasma biomarkers have demonstrated that phosphorylated tau species, particularly at Thr181 (p-tau181), Thr217 (p-tau217), and Thr231 (p-tau231), can predict AD pathology with high accuracy (Milà-Alomà et al., 2022). For example, p-tau231 in CSF is considered an early marker of Aβ pathology, and p-tau217 may offer better discrimination between AD patients and control groups, with better accuracy than proton emission tomography (Groot et al., 2022; Kimura et al., 2023; Milà-Alomà et al., 2022).
Tau hyperphosphorylation remains a central pathological mechanism in AD and may be exacerbated by chronic hyperglycemia, as seen in diabetes (R. Huang et al., 2020). Tau protein is essential for microtubule stability in neurons, but high glucose levels promote tau hyperphosphorylation in hippocampal neurons, leading to its detachment from microtubules, misfolding, and aggregation into NFTs, inducing cognitive dysfunction in diabetes (Barbier et al., 2019; Cheng et al., 2022; Ke et al., 2009; Tabeshmehr & Eftekharpour, 2023).
This pathological process disrupts axonal transport and cytoskeletal integrity, resulting in neuronal dysfunction, synaptic damage, and cell death (Cheng et al., 2022). Tau is also subject to proteolytic cleavage by endogenous enzymes, such as caspases and calpains, which enhances its aggregation propensity (Barthélemy et al., 2020; Mietelska-Porowska et al., 2014). These cleaved forms of tau further promote NFT formation and correlate with cognitive decline (Olesen & Quintanilla, 2023; Pérez et al., 2018).
Patients with DM2 often exhibit cognitive impairment, including deficits in verbal and visual memory, processing speed, and executive function (Y. Liu et al., 2024). Insulin has been implicated in the clearance of Aβ and the regulation of tau phosphorylation in the CNS (Mullins et al., 2017), with evidence supporting a protective role against Aβ synaptotoxicity by regulating Aβ elimination through the modulation of lipid metabolism, proteases, and insulin-degrading enzymes (Saraya et al., 2023).
C-peptide, traditionally considered a byproduct of insulin synthesis, has gained recognition as a bioactive molecule that may modulate insulin signaling, influencing diabetes (Wang et al., 2012). Emerging evidence suggests that treatment with a C-peptide receptor agonist, such as Glucagon-like peptide-1 receptor agonists (GLP1-RA), may offer therapeutic benefits by improving insulin signaling and reducing the incidence of dementia in type 2 diabetes (DM2) patients (García-Casares et al., 2023; Klausen et al., 2022).
On the other hand, a negative correlation has been observed between insulin signaling and tau phosphorylation (Gonçalves et al., 2019). Upon its binding, the insulin receptor activates different IRS, such as phosphorylated IRS-1, leading to PI3K activation, the phosphorylation of MAPK and protein kinase B (AKT) downstream, and inhibiting GSK-3β, a prominent tau kinase, white its inactivation by AKT signaling inhibits GSK-3β-mediated tau phosphorylation (Boucher et al., 2014; Gabbouj et al., 2019; Ye et al., 2017). When insulin signaling is impaired, GSK-3β remains active, resulting in tau hyperphosphorylation and NFT formation (Hobday & Parmar, 2021; Woodfield et al., 2022).
Hyperglycemia also exacerbates cognitive impairment through osmotic stress, oxidative damage, and inflammatory responses, particularly through the formation of advanced glycation end products (AGE), which subsequently leads to the production of ROS and the release of inflammatory cytokines such as IL-1β and IL-6 (C. Liu et al., 2015), causing systemic inflammation, microvascular dysfunction, and long-term neuronal injury (Ehtewish et al., 2022).
6. Conclusions and Perspectives
This comprehensive review highlights the intricate connections between insulin, neurodegeneration, and AD, shedding light on the multifaceted roles of insulin in maintaining cognitive function. Insulin receptors are ubiquitously expressed in brain regions critical for memory and learning, including hippocampus, cortex, hypothalamus, olfactory bulb, and pituitary (Mullins et al., 2017). Notably, the high density of insulin receptors in hippocampal neurons highlights insulin's crucial role in synaptic plasticity and memory consolidation (G.-J. Biessels et al., 1998; Malenka, 1994).
Both DM2 and AD exhibit reduced insulin levels and altered signaling, particularly in the hippocampus, cortex, and choroid plexus. These changes are associated with brain atrophy and cognitive decline, indicating a potential connection between metabolic disturbances and neurodegeneration.
Insulin plays a multifaceted role in several cellular processes relevant for neuronal survival, including cytoskeletal assembly, synaptic plasticity, energy metabolism, and the clearance of Aβ and phosphorylated tau (Mullins et al., 2017; Saraya et al., 2023). As insulin production declines with aging and disease progression, the loss of its neurotrophic and protective effects may directly contribute to AD pathology.
Genetic factors significantly contribute to the risk of AD. Variants of the APOE gene, particularly the ε4 allele, are strongly associated with increased AD risk and earlier onset. APOE4 influences lipid metabolism, neuroinflammation, and amyloid β (Aβ) aggregation. In contrast, the ε2 allele appears protective, while ε3 remains neutral (Raulin et al., 2022). Understanding these genetic influences not only clarifies the clinical variability seen in AD but also has the potential to guide the development of personalized interventions.
Biomarkers offer another promising avenue. Advances in immunoassays have enabled the measurement of p-tau isoforms in plasma and CSF, enhancing early and precise AD diagnosis. Key biomarkers, including insulin dysregulation, endothelial dysfunction, neuroinflammation, Aβ accumulation, and abnormal tau phosphorylation, serve as indicators of disease progression and therapeutic targets (Milà-Alomà et al., 2020).
Therapeutic interventions aimed at restoring insulin signaling and related molecules are under scrutiny. GLP1 receptor agonists and SGLT2 inhibitors show promise not only in controlling glycemia but also in reducing the incidence and progression of dementia in DM2 patients (Gonçalves et al., 2019). These agents may exert neuroprotective effects through multiple mechanisms, including reducing oxidative stress, enhancing neurogenesis, and improving mitochondrial function.
Hyperglycemia leads to neuronal damage through mechanisms such as osmotic imbalance, oxidative injury, and chronic inflammation. The accumulation of AGEs and release of cytokines like IL-1β and IL-6 further exacerbate microvascular damage and contribute to cognitive decline (Ehtewish et al., 2022; García-Casares et al., 2023; C. Liu et al., 2015; Luna et al., 2021). Insulin protective role in Aβ clearance and tau phosphorylation regulation highlights the potential of targeting insulin signaling pathways in both the prevention and treatment of AD.
Future research should focus on clarifying the molecular links between insulin resistance, genetic risk, and neurodegeneration, with the goal of identifying predictive biomarkers and tailoring individualized therapies. Moreover, non-invasive methods to improve brain insulin signaling, such as the use of intranasal insulin, should be further explored in clinical studies.
In conclusion, the overlap between DM2 and AD reflects a complex interplay between metabolism and neurodegeneration. A deeper understanding of insulin central role in these conditions opens new opportunities for early diagnosis, the development of biomarkers, and the design of targeted treatments aimed at delaying or preventing cognitive decline.
Author Contributions
G.I.G and F.C: conceived and designed the major ideas developed in the manuscript. G.I.G, F.C, F.C.O and A.F wrote the paper. A.F, C.B, F.A, C.R-L, L.M.R, M.B, F.C.O, F.C and GIG: reviewed the literature and designed the figures; wrote and edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) Grant 1250485 (to GIG).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
| Aβ | Amyloid-beta |
| AD | Alzheimer's Disease |
| AGEs | Advanced Glycation End Products |
| AKT | Protein Kinase B |
| APOE | Aprolipoprotein E |
| APP | Amyloid Precursor Protein |
| BBB | Blood-Brain Barrier |
| BMECs | Brain Microvascular Endothelial Cells |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| DM2 | Type 2 Diabetes Mellitus |
| GLP1-RA | Glucagon-Like Peptide-1 Receptor Agonist |
| GLUT1 and GLUT3 | Glucose Transporters |
| GSK-3β | Glycogen Synthase Kinase 3 beta |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| IR | Insulin resistance |
| MAPK | Mitogen-Activated Protein Kinase |
| NFT | Neurofibrillary Tangles |
| PET-FDG | Positron Emission Tomography with Fluorodeoxyglucose |
| PI3K | Phosphoinositide 3-Kinase |
| PS1/PS2 | Presenilin-1/Presenilin-2 |
| p-tau | Phosphorylated Tau Protein |
| PTM | Posttranslational Modifications |
| RAGEs | Receptor for AGEs |
| ROS | Reactive Oxygen Species |
| t-tau | Total Tau Protein |
| VEGF | Vascular Endothelial Growth Factor |
References
- Adolfsson, R.; Bucht, G.; Lithner, F.; Winblad, B. Hypoglycemia in Alzheimer’s disease. Acta Med Scand 1980, 208, 387–8. [Google Scholar] [CrossRef]
- Andersson, E.; Schultz, N.; Saito, T.; Saido, T.C.; Blennow, K.; Gouras, G.K.; Zetterberg, H.; Hansson, O. Cerebral Aβ deposition precedes reduced cerebrospinal fluid and serum Aβ42/Aβ40 ratios in the AppNL−F/NL−F knock-in mouse model of Alzheimer’s disease. Alzheimers Res Ther 2023, 15, 64. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wang, H.; Capuano, A.W.; Khan, A.; Taïb, B.; Anokye-Danso, F.; Schneider, J.A.; Bennett, D.A.; Ahima, R.S.; Arnold, S.E. Brain Insulin Signaling, Alzheimer Disease Pathology, and Cognitive Function. Ann Neurol 2020, 88, 513–525. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch Neurol 2004, 61, 661. [Google Scholar] [CrossRef] [PubMed]
- Bakker, W.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W.M. Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res 2009, 335, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M. Type 2 diabetes mellitus and Alzheimer’s disease. World J Diabetes 2014, 5, 889. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front Aging Neurosci 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, Tammie.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; Morris, J.C.; Karch, C.M.; Xiong, C.; Allegri, R.; Mendez, P.C.; Berman, S.B.; Ikeuchi, T.; Mori, H.; Shimada, H.; Shoji, M.; Suzuki, K.; Noble, J.; Farlow, M.; Chhatwal, J.; Graff-Radford, N.R.; Salloway, S.; Schofield, P.R.; Masters, C.L.; Martins, R.N.; O’Connor, A.; Fox, N.C.; Levin, J.; Jucker, M.; Gabelle, A.; Lehmann, S.; Sato, C.; Bateman, R.J.; McDade, E. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Saef, B.; Li, Y.; Gordon, B.A.; He, Y.; Horie, K.; Stomrud, E.; Salvadó, G.; Janelidze, S.; Sato, C.; Ovod, V.; Henson, R.L.; Fagan, A.M.; Benzinger, T.L.S.; Xiong, C.; Morris, J.C.; Hansson, O.; Bateman, R.J.; Schindler, S.E. CSF tau phosphorylation occupancies at T217 and T205 represent improved biomarkers of amyloid and tau pathology in Alzheimer’s disease. Nat Aging 2023, 3, 391–401. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 2015, 9. [Google Scholar] [CrossRef]
- Behl, T.; Arora, A.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S.; Mostafavi, E. Molecular and Biochemical Pathways Encompassing Diabetes Mellitus and Dementia. CNS Neurol Disord Drug Targets 2022, 21, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Biessels, G.-J.; Kamal, A.; Urban, I.J.A.; Spruijt, B.M.; Erkelens, D.W.; Gispen, W.H. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 1998, 800, 125–135. [Google Scholar] [CrossRef]
- Biessels, G.J.; Kappelle, L.J. Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans 2005, 33, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Biju, M.P.; Paulose, C.S. Brain glutamate dehydrogenase changes in streptozotocin diabetic rats as a function of age. Biochem Mol Biol Int 1998, 44, 1–7. [Google Scholar] [CrossRef]
- Bogush, M.; Heldt, N.A.; Persidsky, Y. Blood Brain Barrier Injury in Diabetes: Unrecognized Effects on Brain and Cognition. Journal of Neuroimmune Pharmacology 2017, 12, 593–601. [Google Scholar] [CrossRef]
- Bohórquez Moreno, C.E.; Barreto Vásquez, M.; Muvdi Muvdi, Y.P.; Rodríguez Sanjuán, A.; Badillo Viloria, M.A.; Martínez de la Rosa, W.Á.; Mendoza Sánchez, X. FACTORES MODIFICABLES Y RIESGO DE DIABETES MELLITUS TIPO 2 EN ADULTOS JÓVENES: UN ESTUDIO TRANSVERSAL. Ciencia y Enfermería 2020, 26. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb Perspect Biol 2014, 6, a009191–a009191. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Burns, J.M.; Honea, R.A.; Vidoni, E.D.; Hutfles, L.J.; Brooks, W.M.; Swerdlow, R.H. Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2012, 1822, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Busiguina, S.; Fernandez, A.M.; Barrios, V.; Clark, R.; Tolbert, D.L.; Berciano, J.; Torres-Aleman, I. Neurodegeneration Is Associated to Changes in Serum Insulin-like Growth Factors. Neurobiol Dis 2000, 7, 657–665. [Google Scholar] [CrossRef]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: a review. AIMS Neurosci 2021, 8, 86–132. [Google Scholar] [CrossRef]
- Chelombitko, M.A. Role of Reactive Oxygen Species in Inflammation: A Minireview. Moscow Univ Biol Sci Bull 2018, 73, 199–202. [Google Scholar] [CrossRef]
- Chen, Y.; Sillaire, A.R.; Dallongeville, J.; Skrobala, E.; Wallon, D.; Dubois, B.; Hannequin, D.; Pasquier, F. Low Prevalence and Clinical Effect of Vascular Risk Factors in Early-Onset Alzheimer’s Disease. Journal of Alzheimer’s Disease 2017, 60, 1045–1054. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, J.-R.; Jian, J.-M.; He, C.-Y.; Xu, M.-Y.; Zeng, G.-H.; Tan, C.-R.; Shen, Y.-Y.; Jin, W.-S.; Chen, D.-W.; Li, H.-Y.; Yi, X.; Zhang, Y.; Bu, X.-L.; Wang, Y.-J. Associations of plasma angiostatin and amyloid-β and tau levels in Alzheimer’s disease. Transl Psychiatry 2022, 12, 194. [Google Scholar] [CrossRef]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, and dementia. Eur J Pharmacol 2013, 719, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Chrem Mendez, P.; Surace, E.; Bérgamo, Y.; Calandri, I.; Vázquez, S.; Sevlever, G.; Allegri, R.F. Biomarkers for Alzheimer’s disease. Where we stand and where we are headed. Medicina (B Aires) 2019, 79, 546–551. [Google Scholar] [PubMed]
- Ciudin, A. Diabetes mellitus tipo 2 y enfermedad de Alzheimer: una relación para no olvidar. Endocrinología y Nutrición 2016, 63, 191–193. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, α-synuclein, amyloid-β and insulin resistance in neurodegenerative diseases. Br J Pharmacol 2018, 175, 3859–3875. [Google Scholar] [CrossRef] [PubMed]
- Costache, A.D.; Ignat, B.E.; Grosu, C.; Mastaleru, A.; Abdulan, I.; Oancea, A.; Roca, M.; Leon, M.M.; Badescu, M.C.; Luca, S.; Jigoranu, A.R.; Chetran, A.; Mitu, O.; Costache, I.I.; Mitu, F. Inflammatory Pathways in Overweight and Obese Persons as a Potential Mechanism for Cognitive Impairment and Earlier Onset Alzeihmer’s Dementia in the General Population: A Narrative Review. Biomedicines 2023, 11, 3233. [Google Scholar] [CrossRef]
- Craft, S. Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord 2006, 20, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Insulin resistance and cognitive impairment: a view through the prism of epidemiology. Arch Neurol 2005, 62, 1043–4. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology 1998, 50, 164–168. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.-K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; Gessert, D.; Aisen, P.S. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol 2020, 77, 1099. [Google Scholar] [CrossRef]
- Cukierman-Yaffe, T.; Kasher-Meron, M.; Fruchter, E.; Gerstein, H.C.; Afek, A.; Derazne, E.; Tzur, D.; Karasik, A.; Twig, G. Cognitive Performance at Late Adolescence and the Risk for Impaired Fasting Glucose Among Young Adults. J Clin Endocrinol Metab 2015, 100, 4409–4416. [Google Scholar] [CrossRef]
- De Felice, F.G.; Gonçalves, R.A.; Ferreira, S.T. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat Rev Neurosci 2022, 23, 215–230. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2008, 2, 1101–13. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; LaCreta, F.P.; Griffen, S.C. Characterization of Renal Glucose Reabsorption in Response to Dapagliflozin in Healthy Subjects and Subjects With Type 2 Diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2019, 11, 392–404. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Simon, D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 2012, 13, 583–594. [Google Scholar] [CrossRef]
- Desouza, C.V.; Shivaswamy, V. Pioglitazone in the Treatment of Type 2 Diabetes: Safety and Efficacy Review. Clin Med Insights Endocrinol Diabetes 2010, 3, CMED.S5372. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Dolan, H.; Crain, B.; Troncoso, J.; Resnick, S.M.; Zonderman, A.B.; Obrien, R.J. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of aging cohort. Ann Neurol 2010, 68, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.O.; Pagano, M.A.; Marschoff, E.R.; González, S.E.; Repetto, M.G.; Serra, J.A. Enfermedad de Alzheimer y deterioro cognitivo asociado a la diabetes mellitus de tipo 2: relaciones e hipótesis. Neurología 2014, 29, 567–572. [Google Scholar] [CrossRef]
- Donoso, S.A.; Behrens, P.M.I. Variabilidad y variantesde la enfermedad de Alzheimer. Rev Med Chil 2005, 133. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in Central Nervous System: More than Just a Peripheral Hormone. J Aging Res 2012, 2012, 1–21. [Google Scholar] [CrossRef]
- Duarte, J. Metabolic Alterations Associated to Brain Dysfunction in Diabetes. Aging Dis 2014. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.N. Loss of brain energy metabolism control as a driver for memory impairment upon insulin resistance. Biochem Soc Trans 2023, 51, 287–301. [Google Scholar] [CrossRef]
- Ehtewish, H.; Arredouani, A.; El-Agnaf, O. Diagnostic, Prognostic, and Mechanistic Biomarkers of Diabetes Mellitus-Associated Cognitive Decline. Int J Mol Sci 2022, 23, 6144. [Google Scholar] [CrossRef]
- El Khoury, N.B.; Gratuze, M.; Papon, M.-A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front Cell Neurosci 2014, 8. [Google Scholar] [CrossRef]
- Fava, S. Glycaemic Control: A Balancing Act or A Different Approach? Curr Diabetes Rev 2014, 10, 124–130. [Google Scholar] [CrossRef]
- Formiga, F.; Reñe, R.; Pérez-Maraver, M. Demencia y diabetes: ¿relación casual o causal? Med Clin (Barc) 2015, 144, 176–180. [Google Scholar] [CrossRef]
- Freychet, P. Insulin receptors and insulin actions in the nervous system. Diabetes Metab Res Rev 2000, 16, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; Natunen, T. Altered Insulin Signaling in Alzheimer’s Disease Brain – Special Emphasis on PI3K-Akt Pathway. Front Neurosci 2019, 13. [Google Scholar] [CrossRef]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell Mol Immunol 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- García-Casares, N.; González-González, G.; de la Cruz-Cosme, C.; Garzón-Maldonado, F.J.; de Rojas-Leal, C.; Ariza, M.J.; Narváez, M.; Barbancho, M.Á.; García-Arnés, J.A.; Tinahones, F.J. Effects of GLP-1 receptor agonists on neurological complications of diabetes. Rev Endocr Metab Disord 2023, 24, 655–672. [Google Scholar] [CrossRef]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn From Diet-Induced Diabetes Models? Front Neurosci 2020, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, A.A.J.; Bakker, C.; Verhey, F.R.J.; de Vugt, M.E.; Melis, R.J.F.; Koopmans, R.T.C.M.; Oosterveld, S.M.; Kessels, R.P.; Olde Rikkert, M.G.; Hamel, R.; Ramakers, I.H.; Aalten, P.; Sistermans, N.; Smits, L.L.; Pijnenburg, Y.A.; van der Flier, W.M. Prevalence of Comorbidity in Patients With Young-Onset Alzheimer Disease Compared With Late-Onset: A Comparative Cohort Study. J Am Med Dir Assoc 2016, 17, 318–323. [Google Scholar] [CrossRef]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The Link Between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front Cell Neurosci 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Luo, H.; Li, Z.; Feng, Y.; Liu, Z.; Chang, J. Metabolic Profile of Alzheimer’s Disease: Is 10-Hydroxy-2-decenoic Acid a Pertinent Metabolic Adjuster? Metabolites 2023, 13, 954. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M.; Lim, Y.-A. Common features between diabetes mellitus and Alzheimer’s disease. Cell Mol Life Sci 2009, 66, 1321–5. [Google Scholar] [CrossRef] [PubMed]
- Groot, C.; Cicognola, C.; Bali, D.; Triana-Baltzer, G.; Dage, J.L.; Pontecorvo, M.J.; Kolb, H.C.; Ossenkoppele, R.; Janelidze, S.; Hansson, O. Diagnostic and prognostic performance to detect Alzheimer’s disease and clinical progression of a novel assay for plasma p-tau217. Alzheimers Res Ther 2022, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Rodelo, C.; Roura-Guiberna, A.; Olivares-Reyes, J.A. Molecular Mechanisms of Insulin Resistance: An Update. Gac Med Mex 2017, 153, 214–228. [Google Scholar]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J Diabetes 2015, 6, 598. [Google Scholar] [CrossRef]
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med 2023, 55, 1–12. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; Ankarcrona, M. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proceedings of the National Academy of Sciences 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Hernández-Contreras, K.A.; Martínez-Díaz, J.A.; Hernández-Aguilar, M.E.; Herrera-Covarrubias, D.; Rojas-Durán, F.; Aranda Abreu, G.E. Mecanismos de asociación entre Enfermedad de Alzheimer y Diabetes Mellitus: La paradoja de la insulina. Archivos de Neurociencias 2021, 25. [Google Scholar] [CrossRef]
- Hobday, A.L.; Parmar, M.S. The Link Between Diabetes Mellitus and Tau Hyperphosphorylation: Implications for Risk of Alzheimer’s Disease; Cureus, 2021. [Google Scholar] [CrossRef]
- Horie, K.; Salvadó, G.; Barthélemy, N.R.; Janelidze, S.; Li, Y.; He, Y.; Saef, B.; Chen, C.D.; Jiang, H.; Strandberg, O.; Pichet Binette, A.; Palmqvist, S.; Sato, C.; Sachdev, P.; Koyama, A.; Gordon, B.A.; Benzinger, T.L.S.; Holtzman, D.M.; Morris, J.C.; Mattsson-Carlgren, N.; Stomrud, E.; Ossenkoppele, R.; Schindler, S.E.; Hansson, O.; Bateman, R.J. CSF MTBR-tau243 is a specific biomarker of tau tangle pathology in Alzheimer’s disease. Nat Med 2023, 29, 1954–1963. [Google Scholar] [CrossRef]
- Huang, C.-C.; Chung, C.-M.; Leu, H.-B.; Lin, L.-Y.; Chiu, C.-C.; Hsu, C.-Y.; Chiang, C.-H.; Huang, P.-H.; Chen, T.-J.; Lin, S.-J.; Chen, J.-W.; Chan, W.-L. Diabetes Mellitus and the Risk of Alzheimer’s Disease: A Nationwide Population-Based Study. PLoS One 2014, 9, e87095. [Google Scholar] [CrossRef]
- Huang, R.; Tian, S.; Zhang, H.; Zhu, W.; Wang, S. Chronic hyperglycemia induces tau hyperphosphorylation by downregulating OGT-involved O-GlcNAcylation in vivo and in vitro. Brain Res Bull 2020, 156, 76–85. [Google Scholar] [CrossRef]
- Jabeen, K.; Rehman, K.; Akash, M.S.H. Genetic mutations of APOEε4 carriers in cardiovascular patients lead to the development of insulin resistance and risk of Alzheimer’s disease. J Biochem Mol Toxicol 2022, 36. [Google Scholar] [CrossRef]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. Journal of Clinical Investigation 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Ke, Y.D.; Delerue, F.; Gladbach, A.; Götz, J.; Ittner, L.M. Experimental Diabetes Mellitus Exacerbates Tau Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. PLoS One 2009, 4, e7917. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol 2020, 19, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Backus, C.; Oh, S.; Feldman, E.L. Hyperglycemia-Induced Tau Cleavage in vitro and in vivo: A Possible Link Between Diabetes and Alzheimer’s Disease. Journal of Alzheimer’s Disease 2013, 34, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Aota, T.; Aso, Y.; Yabuuchi, K.; Sasaki, K.; Masuda, T.; Eguchi, A.; Maeda, Y.; Aoshima, K.; Matsubara, E. Predicting positron emission tomography brain amyloid positivity using interpretable machine learning models with wearable sensor data and lifestyle factors. Alzheimers Res Ther 2023, 15, 212. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Fatima, H.A.; Cahoon, D.S.; Otieno, B.; Chacko, L.; Minooei, F.; Demos, C.; Stengelin, M.; Sigal, G.; Wohlstadter, J.; Arnold, S.E. Clinical evaluation of a novel plasma pTau217 electrochemiluminescence immunoassay in Alzheimer’s disease. Sci Rep 2024, 14, 629. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.K.; Thomsen, M.; Wortwein, G.; Fink-Jensen, A. The role of glucagon-like peptide 1 (GLP-1) in addictive disorders. Br J Pharmacol 2022, 179, 625–641. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jack, C.R.; Wiste, H.J.; Lundt, E.S.; Weigand, S.D.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; Mielke, M.M.; Roberts, R.O.; Boeve, B.F.; Petersen, R.C. 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging 2014, 35, 2096–2106. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: the Receptor for Advanced Glycation End Products (RAGE). Front Aging Neurosci 2020, 12. [Google Scholar] [CrossRef]
- Könner, A.C.; Hess, S.; Tovar, S.; Mesaros, A.; Sánchez-Lasheras, C.; Evers, N.; Verhagen, L.A.W.; Brönneke, H.S.; Kleinridders, A.; Hampel, B.; Kloppenburg, P.; Brüning, J.C. Role for Insulin Signaling in Catecholaminergic Neurons in Control of Energy Homeostasis. Cell Metab 2011, 13, 720–728. [Google Scholar] [CrossRef]
- Lamport, D.J.; Dye, L.; Mansfield, M.W.; Lawton, C.L. Acute glycaemic load breakfast manipulations do not attenuate cognitive impairments in adults with type 2 diabetes. Clinical Nutrition 2013, 32, 265–272. [Google Scholar] [CrossRef]
- Lamport, D.J.; Lawton, C.L.; Mansfield, M.W.; Moulin, C.A.J.; Dye, L. Type 2 diabetes and impaired glucose tolerance are associated with word memory source monitoring recollection deficits but not simple recognition familiarity deficits following water, low glycaemic load, and high glycaemic load breakfasts. Physiol Behav 2014, 124, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Langella, S.; Barksdale, N.G.; Vasquez, D.; Aguillon, D.; Chen, Y.; Su, Y.; Acosta-Baena, N.; Acosta-Uribe, J.; Baena, A.Y.; Garcia-Ospina, G.; Giraldo-Chica, M.; Tirado, V.; Muñoz, C.; Ríos-Romenets, S.; Guzman-Martínez, C.; Oliveira, G.; Yang, H.-S.; Vila-Castelar, C.; Pruzin, J.J.; Ghisays, V.; Arboleda-Velasquez, J.F.; Kosik, K.S.; Reiman, E.M.; Lopera, F.; Quiroz, Y.T. Effect of apolipoprotein genotype and educational attainment on cognitive function in autosomal dominant Alzheimer’s disease. Nat Commun 2023, 14, 5120. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Choe, Y.M.; Suh, G.-H.; Choi, I.-G.; Lee, J.H.; Kim, H.S.; Hwang, J.; Yi, D.; Kim, J.W. A combination of midlife diabetes mellitus and the apolipoprotein E ε4 allele increase risk for cognitive decline. Front Aging Neurosci 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hölscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 2007, 56, 384–402. [Google Scholar] [CrossRef]
- Lim, Y.Y.; Yassi, N.; Bransby, L.; Ayton, S.; Buckley, R.F.; Eratne, D.; Velakoulis, D.; Li, Q.-X.; Fowler, C.; Masters, C.L.; Maruff, P. CSF Aβ42 and tau biomarkers in cognitively unimpaired Aβ- middle-aged and older APOE ε4 carriers. Neurobiol Aging 2023, 129, 209–218. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Z.; Song, Y.; Wu, D.; Zheng, X.; Li, P.; Jin, J.; Xu, N.; Li, L. Effects of Berberine on Amelioration of Hyperglycemia and Oxidative Stress in High Glucose and High Fat Diet-Induced Diabetic Hamsters In Vivo. Biomed Res Int 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chu, B.; Jin, S.; Li, M.; Xu, Y.; Yang, H.; Feng, Z.; Bi, J.; Wang, P. Vascular endothelial growth factor alleviates mitochondrial dysfunction and suppression of mitochondrial biogenesis in models of Alzheimer’s disease. International Journal of Neuroscience 2021, 131, 154–162. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, Y.; Du, W.; Gao, B.; Gao, J.; Hu, S.; Song, Q.; Wang, W.; Miao, Y. White matter microstructure alterations in type 2 diabetes mellitus and its correlation with cerebral small vessel disease and cognitive performance. Sci Rep 2024, 14, 270. [Google Scholar] [CrossRef]
- Luna, R.; Talanki Manjunatha, R.; Bollu, B.; Jhaveri, S.; Avanthika, C.; Reddy, N.; Saha, T.; Gandhi, F. A Comprehensive Review of Neuronal Changes in Diabetics. Cureus. [CrossRef] [PubMed]
- Madhusudhanan, J.; Suresh, G.; Devanathan, V. Neurodegeneration in type 2 diabetes: Alzheimer’s as a case study. Brain Behav 2021, 2020, 10. [Google Scholar] [CrossRef]
- Majid, H.; Islam, S.U.; Kohli, S.; Nidhi. Neuroinflammation and metabolic dysregulation as predictors of cognitive impairment, depression, and quality of life in type 2 diabetes mellitus patients on SGLT2 inhibitors and sulfonylureas. Inflammopharmacology 2025. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538. [Google Scholar] [CrossRef]
- Mancinetti, F.; Xenos, D.; De Fano, M.; Mazzieri, A.; Porcellati, F.; Boccardi, V.; Mecocci, P. Diabetes-Alzheimer’s connection in older age: SGLT2 inhibitors as promising modulators of disease pathways. Ageing Res Rev 2023, 90, 102018. [Google Scholar] [CrossRef]
- Martínez, S.; Ochoa, B.; Pérez, M.R.; Torrico, F.; García, I.; Garcia, C.C. Polimorfismos del gen de la apolipoproteína E en adultos mayores de 60 años con disminución de la memoria cognitiva y enfermedad de Alzheimer en diferentes poblaciones venezolanas. Biomédica 2022, 42, 116–129. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem 2010, 93, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Gagnon, M. Glucose regulation and cognitive functions: relation to Alzheimer’s disease and diabetes. Behavioural Brain Research 1996, 75, 1–11. [Google Scholar] [CrossRef]
- Mestizo Gutiérrez, S.L.; Herrera Rivero, M.; Cruz Ramírez, N.; Hernández, E.; Aranda-Abreu, G.E. Decision trees for the analysis of genes involved in Alzheimer׳s disease pathology. J Theor Biol 2014, 357, 21–25. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int J Mol Sci 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; Day, T.A.; González-Escalante, A.; Sánchez-Benavides, G.; Minguillon, C.; Fauria, K.; Molinuevo, J.L.; Dage, J.L.; Zetterberg, H.; Gispert, J.D.; Suárez-Calvet, M.; Blennow, K. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat Med 2022. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Salvadó, G.; Gispert, J.D.; Vilor-Tejedor, N.; Grau-Rivera, O.; Sala-Vila, A.; Sánchez-Benavides, G.; Arenaza-Urquijo, E.M.; Crous-Bou, M.; González-de-Echávarri, J.M.; Minguillon, C.; Fauria, K.; Simon, M.; Kollmorgen, G.; Zetterberg, H.; Blennow, K.; Suárez-Calvet, M.; Molinuevo, J.L. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer’s continuum. Alzheimer’s & Dementia 2020, 16, 1358–1371. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front Aging Neurosci 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Shimozawa, M.; Bereczki, E.; Li, X.; Liu, J.; Jiang, R.; Giraud, R.; Leal, N.S.; Pinho, C.M.; Berger, E.; Falk, V.L.; Dentoni, G.; Ankarcrona, M.; Nilsson, P. Mitochondrial hypermetabolism precedes impaired autophagy and synaptic disorganization in App knock-in Alzheimer mouse models. Mol Psychiatry 2023, 28, 3966–3981. [Google Scholar] [CrossRef]
- Olesen, M.A.; Quintanilla, R.A. Pathological Impact of Tau Proteolytical Process on Neuronal and Mitochondrial Function: a Crucial Role in Alzheimer’s Disease. Mol Neurobiol 2023, 60, 5691–5707. [Google Scholar] [CrossRef]
- Palasí, A.; Gutiérrez-Iglesias, B.; Alegret, M.; Pujadas, F.; Olabarrieta, M.; Liébana, D.; Quintana, M.; Álvarez-Sabín, J.; Boada, M. Differentiated clinical presentation of early and late-onset Alzheimer’s disease: is 65 years of age providing a reliable threshold? J Neurol 2015, 262, 1238–1246. [Google Scholar] [CrossRef]
- Park, S.A. A common pathogenic mechanism linking type-2 diabetes and Alzheimer’s disease: evidence from animal models. J Clin Neurol 2011, 7, 10–8. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, F.; Boulogne, A.; Leys, D.; Fontaine, P. Diabetes mellitus and dementia. Diabetes Metab 2006, 32, 403–14. [Google Scholar] [CrossRef]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front Neurosci 2018, 12. [Google Scholar] [CrossRef]
- Petermann, F.; Troncoso-Pantoja, C.; Martínez, M.A.; Leiva, A.M.; Ramírez-Campillo, R.; Poblete-Valderrama, F.; Garrido-Méndez, A.; Díaz-Martínez, X.; Ulloa, N.; Concha, Y.; Celis-Morales, C. Asociación entre diabetes mellitus tipo 2018, 2, historia familiar de diabetes y deterioro cognitivo en adultos mayores chilenos. Rev Med Chil 146, 872–881. [CrossRef] [PubMed]
- Polsinelli, A.J.; Logan, P.E.; Lane, K.A.; Manchella, M.K.; Nemes, S.; Sanjay, A.B.; Gao, S.; Apostolova, L.G. APOE ε4 carrier status and sex differentiate rates of cognitive decline in early- and late-onset Alzheimer’s disease. Alzheimer’s & Dementia 2023, 19, 1983–1993. [Google Scholar] [CrossRef]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci Ther 2018, 24, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Pooja Naik, L.C. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J Pharmacovigil 2014, 02. [Google Scholar] [CrossRef]
- Ramírez Rincón, A.; Saldarriaga Betancur, S.; García Ramos, A.F.; González Arango, J.; Estupiñán Vargas, V. Tratamiento farmacológico del paciente que vive con diabetes mellitus tipo 2. CES Medicina 2022, 36, 81–105. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Waquar, S.; Zaheer, A.; Asif, M.; Iqbal, Z.; Gauthaman, K.; Kamal, M.A.; Pushparaj, P.N. Cellular and Molecular Mechanisms of Dementia: Decoding the Causal link of Diabetes Mellitus in Alzheimer’s Disease. CNS Neurol Disord Drug Targets 2021, 20, 602–612. [Google Scholar] [CrossRef]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol Neurodegener 2022, 17, 72. [Google Scholar] [CrossRef]
- Ravipati, K.; Chen, Y.; Manns, J.R. Reassessing Diabetes and APOE Genotype as Potential Interacting Risk Factors for Alzheimer’s Disease. Am J Alzheimers Dis Other Demen 2022, 37, 153331752110709. [Google Scholar] [CrossRef]
- Razay, G.; Wilcock, G.K. Hyperinsulinaemia and Alzheimer’s disease. Age Ageing 1994, 23, 396–9. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W.; Kindy, M.; LaDu, M.J. Apolipoprotein E and Alzheimer’s disease: The protective effects of ApoE2 and E3. Journal of Alzheimer’s Disease 2002, 4, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Meza, M.; Muñoz, D.; Jerez, E.; Quintanilla, M.E.; Salinas-Luypaert, C.; Fernandez, K.; Karahanian, E. Fenofibrate Administration Reduces Alcohol and Saccharin Intake in Rats: Possible Effects at Peripheral and Central Levels. Front Behav Neurosci 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Montoliu-Gaya, L.; Villegas, S. The Role of Apolipoprotein E Isoforms in Alzheimer’s Disease. Journal of Alzheimer’s Disease 2019, 68, 459–471. [Google Scholar] [CrossRef]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef]
- Sánchez-Zúñiga, M. de J.; Carrillo-Esper, R.; Sánchez-Pérez, H.; González-Chávez, A.; Elizondo-Argueta, S. Circuito insulinérgico cerebral. De las bases a su impacto en la clínica. Cir Cir 2020, 88. [Google Scholar] [CrossRef]
- Saraya, A.W.; Tunvirachaisakul, C.; Sonpee, C.; Katasrila, P.; Sathaporn, T.; Tepmongkol, S.; Tangwongchai, S. Serum proinsulin levels as peripheral blood biomarkers in patients with cognitive impairment. Sci Rep 2023, 13, 22436. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. The Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Sebastian, M.J.; Khan, S.K.; Pappachan, J.M.; Jeeyavudeen, M.S. Diabetes and cognitive function: An evidence-based current perspective. World J Diabetes 2023, 14, 92–109. [Google Scholar] [CrossRef]
- Shinohara, M.; Suzuki, K.; Bu, G.; Sato, N. Interaction Between APOE Genotype and Diabetes in Longevity. Journal of Alzheimer’s Disease 2021, 82, 719–726. [Google Scholar] [CrossRef]
- Sible, I.J.; Yew, B.; Jang, J.Y.; Alitin, J.P.M.; Li, Y.; Gaubert, A.; Nguyen, A.; Dutt, S.; Blanken, A.E.; Ho, J.K.; Marshall, A.J.; Kapoor, A.; Shenasa, F.; Rodgers, K.E.; Sturm, V.E.; Head, E.; Martini, A.; Nation, D.A. Blood pressure variability and plasma Alzheimer’s disease biomarkers in older adults. Sci Rep 2022, 12, 17197. [Google Scholar] [CrossRef] [PubMed]
- Sienes Bailo, P.; Llorente Martín, E.; Calmarza, P.; Montolio Breva, S.; Bravo Gómez, A.; Pozo Giráldez, A.; Sánchez-Pascuala Callau, J.J.; Vaquer Santamaría, J.M.; Dayaldasani Khialani, A.; Cerdá Micó, C.; Camps Andreu, J.; Sáez Tormo, G.; Fort Gallifa, I. Implicación del estrés oxidativo en las enfermedades neurodegenerativas y posibles terapias antioxidantes. Advances in Laboratory Medicine / Avances en Medicina de Laboratorio 2022, 3, 351–360. [Google Scholar] [CrossRef]
- Sima, A.A.F.; Zhang, W. Mechanisms of diabetic neuropathy; 2014; pp. 429–442. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 2010, 6, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.J. Introducing Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, an open access journal of the Alzheimer’s Association. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2015, 1, 1–4. [Google Scholar] [CrossRef]
- Sonar, S.A.; Lal, G. Blood–brain barrier and its function during inflammation and autoimmunity. J Leukoc Biol 2018, 103, 839–853. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Cobzaru, A.; Paduraru, L.; Bulea, D. Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. J Clin Med 2020, 9, 1713. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? Journal of Alzheimer’s Disease 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Tabeshmehr, P.; Eftekharpour, E. Tau; One Protein, So Many Diseases. Biology (Basel) 2023, 12, 244. [Google Scholar] [CrossRef]
- Takeishi, J.; Tatewaki, Y.; Nakase, T.; Takano, Y.; Tomita, N.; Yamamoto, S.; Mutoh, T.; Taki, Y. Alzheimer’s Disease and Type 2 Diabetes Mellitus: The Use of MCT Oil and a Ketogenic Diet. Int J Mol Sci 2021, 22, 12310. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; Arvanitakis, Z.; Schneider, J.A.; Wolf, B.A.; Bennett, D.A.; Trojanowski, J.Q.; Arnold, S.E. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. Journal of Clinical Investigation 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Tobon-Velasco, J.; Cuevas, E.; Torres-Ramos, M. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol Disord Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Trimm, E.; Red-Horse, K. Vascular endothelial cell development and diversity. Nat Rev Cardiol 2023, 20, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int J Mol Sci 2018, 19, 3306. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Buchman, A.S.; Cai, W.; Haroutunian, V.; Beeri, M.S. Human brain and serum advanced glycation end products are highly correlated: Preliminary results of their role in Alzheimer’s disease and type 2 diabetes. Alzheimer’s & Dementia 2020, 16. [Google Scholar] [CrossRef]
- Vanhandsaeme, G.; Benhalima, K. The long-term metabolic and neurocognitive risks in offspring of women with type 1 diabetes mellitus. Acta Diabetol 2021, 58, 845–858. [Google Scholar] [CrossRef]
- Verdelho, A.; Madureira, S.; Ferro, J.M.; Basile, A.-M.; Chabriat, H.; Erkinjuntti, T.; Fazekas, F.; Hennerici, M.; O’Brien, J.; Pantoni, L.; Salvadori, E.; Scheltens, P.; Visser, M.C.; Wahlund, L.-O.; Waldemar, G.; Wallin, A.; Inzitari, D.; LADIS Study. Differential impact of cerebral white matter changes, diabetes, hypertension and stroke on cognitive performance among non-disabled elderly. The LADIS study. J Neurol Neurosurg Psychiatry 2007, 78, 1325–30. [Google Scholar] [CrossRef]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis 2015, 84, 22–38. [Google Scholar] [CrossRef]
- von Bernhardi, R. La Barrera Hemato-Encefálica en la patología del Sistema Nervioso Central: su importancia en la Respuesta Inflamatoria. Rev Chil Neuropsiquiatr 2004, 42. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Zheng, Y.; Hou, J.; Dou, Y.; Zhang, S.; Luo, X.; Cai, X. The Role of Insulin C-Peptide in the Coevolution Analyses of the Insulin Signaling Pathway: A Hint for Its Functions. PLoS One 2012, 7, e52847. [Google Scholar] [CrossRef]
- Watson, D.; Castaño, E.; Kokjohn, T.A.; Kuo, Y.-M.; Lyubchenko, Y.; Pinsky, D.; Connolly, E.S.; Esh, C.; Luehrs, D.C.; Stine, W.B.; Rowse, L.M.; Emmerling, M.R.; Roher, A.E. Physicochemical characteristics of soluble oligomeric A β and their pathologic role in Alzheimer’s disease. Neurol Res 2005, 27, 869–881. [Google Scholar] [CrossRef]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D.; Alzheimer’s Disease Neuroimaging Initiative. Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 2015, 64, 1933–40. [Google Scholar] [CrossRef]
- Winocur, G.; Greenwood, C.E. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging 2005, 26 1, 46–9. [Google Scholar] [CrossRef]
- Woodfield, A.; Gonzales, T.; Helmerhorst, E.; Laws, S.; Newsholme, P.; Porter, T.; Verdile, G. Current Insights on the Use of Insulin and the Potential Use of Insulin Mimetics in Targeting Insulin Signalling in Alzheimer’s Disease. Int J Mol Sci 2022, 23, 15811. [Google Scholar] [CrossRef] [PubMed]
- Xourgia, E.; Papazafiropoulou, A.; Melidonis, A. Antidiabetic treatment on memory and spatial learning: From the pancreas to the neuron. World J Diabetes 2019, 10, 169–180. [Google Scholar] [CrossRef]
- Yaffe, K. The Metabolic Syndrome, Inflammation, and Risk of Cognitive Decline. JAMA 2004, 292, 2237. [Google Scholar] [CrossRef]
- Yang, C.; Wei, M.; Zhao, Y.; Yang, Z.; Song, M.; Mi, J.; Yang, X.; Tian, G. Regulation of insulin secretion by the post-translational modifications. Front Cell Dev Biol 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Maji, S.; Sanghera, N.; Gopalasingam, P.; Gorbunov, E.; Tarasov, S.; Epstein, O.; Klein-Seetharaman, J. Structure and dynamics of the insulin receptor: implications for receptor activation and drug discovery. Drug Discov Today 2017, 22, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Tragodara, D.; Roque, H.; Parodi, J.F. Relación entre trastornos neurocognitivos,Diabetes Mellitus Tipo 2 y otros factores en adultos mayores del Centro Médico Naval del Perú, entre los años 2010 a 2015. Rev Neuropsiquiatr 2020, 83, 87–96. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int J Biol Sci 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
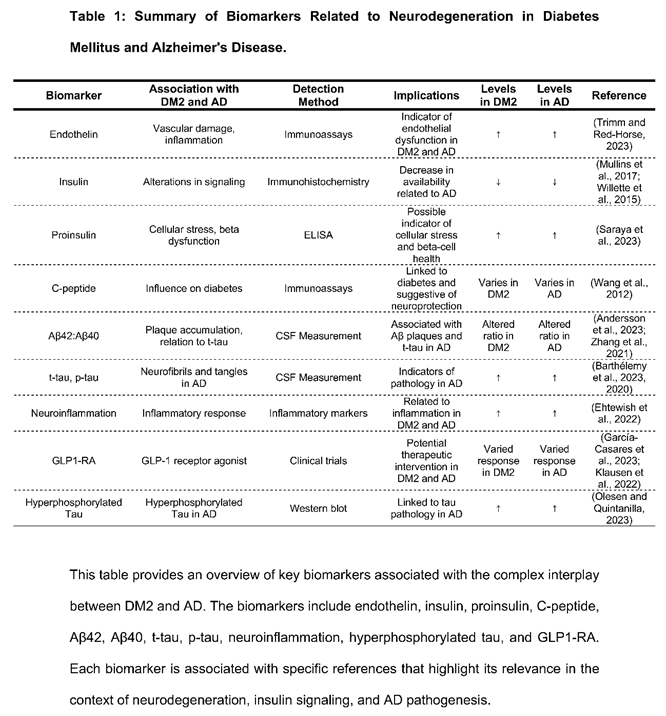
Figure 1.
Mechanistic links between type 2 diabetes mellitus (DM2) and Alzheimer's disease. Chronic hyperglycemia and insulin resistance in DM2 result in altered insulin receptor signaling. This altered insulin signaling compromises neuronal energy homeostasis and synaptic plasticity. Concurrently, hyperglycemia increases oxidative stress through the overproduction of ROS, leading to mitochondrial dysfunction and endothelial damage that contribute to the disruption of the blood-brain barrier. Barrier impairment/permeabilization facilitates infiltration of peripheral immune cells and amplifies central neuroinflammatory responses, mediated by the activation of microglia and astrocytes. Chronic inflammation further promotes the accumulation of Aβ and tau hyperphosphorylation, leading to the formation of neurofibrillary tangles. Together, these pathological processes converge to accelerate neuronal loss, cortical and hippocampal atrophy, and cognitive decline. Created in https://BioRender.com.
Figure 1.
Mechanistic links between type 2 diabetes mellitus (DM2) and Alzheimer's disease. Chronic hyperglycemia and insulin resistance in DM2 result in altered insulin receptor signaling. This altered insulin signaling compromises neuronal energy homeostasis and synaptic plasticity. Concurrently, hyperglycemia increases oxidative stress through the overproduction of ROS, leading to mitochondrial dysfunction and endothelial damage that contribute to the disruption of the blood-brain barrier. Barrier impairment/permeabilization facilitates infiltration of peripheral immune cells and amplifies central neuroinflammatory responses, mediated by the activation of microglia and astrocytes. Chronic inflammation further promotes the accumulation of Aβ and tau hyperphosphorylation, leading to the formation of neurofibrillary tangles. Together, these pathological processes converge to accelerate neuronal loss, cortical and hippocampal atrophy, and cognitive decline. Created in https://BioRender.com.
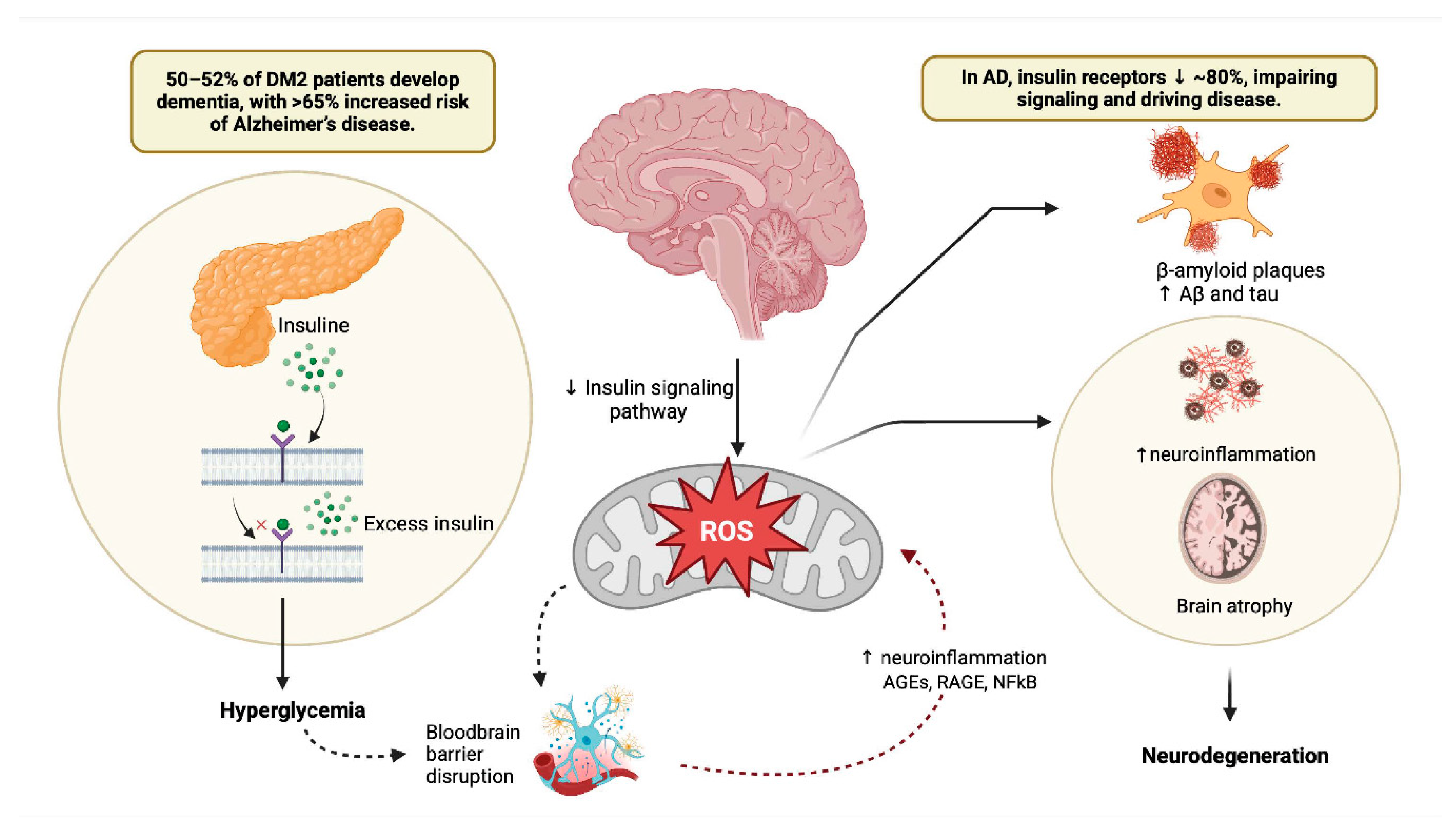
Figure 2.
Blood-Brain Barrier Dysfunction in Diabetes Mellitus and Alzheimer's Disease: Mechanisms and Therapeutic Targets. This figure illustrates the intricate relationship between DM2 and AD through the lens of BBB dysfunction. The BBB, composed of endothelial cells, astrocytes, neurons, and pericytes, acts as a critical defense mechanism for the brain against neurotoxic compounds. The figure outlines the breakdown of the BBB in AD, emphasizing the role of impaired tight junctions and adherents’ junctions in brain microvascular endothelial cells. Furthermore, the figure delves into the similarities between DM2 and neurodegenerative diseases, such as vascular dementia and AD, showcasing how hyperglycemia, oxidative stress, and chronic inflammation contribute to BBB impairment in DM2. Overall, this comprehensive figure provides insights into the multifaceted mechanisms of BBB dysfunction in the context of DM2 and AD, offering potential avenues for therapeutic interventions. Created in https://BioRender.com.
Figure 2.
Blood-Brain Barrier Dysfunction in Diabetes Mellitus and Alzheimer's Disease: Mechanisms and Therapeutic Targets. This figure illustrates the intricate relationship between DM2 and AD through the lens of BBB dysfunction. The BBB, composed of endothelial cells, astrocytes, neurons, and pericytes, acts as a critical defense mechanism for the brain against neurotoxic compounds. The figure outlines the breakdown of the BBB in AD, emphasizing the role of impaired tight junctions and adherents’ junctions in brain microvascular endothelial cells. Furthermore, the figure delves into the similarities between DM2 and neurodegenerative diseases, such as vascular dementia and AD, showcasing how hyperglycemia, oxidative stress, and chronic inflammation contribute to BBB impairment in DM2. Overall, this comprehensive figure provides insights into the multifaceted mechanisms of BBB dysfunction in the context of DM2 and AD, offering potential avenues for therapeutic interventions. Created in https://BioRender.com.
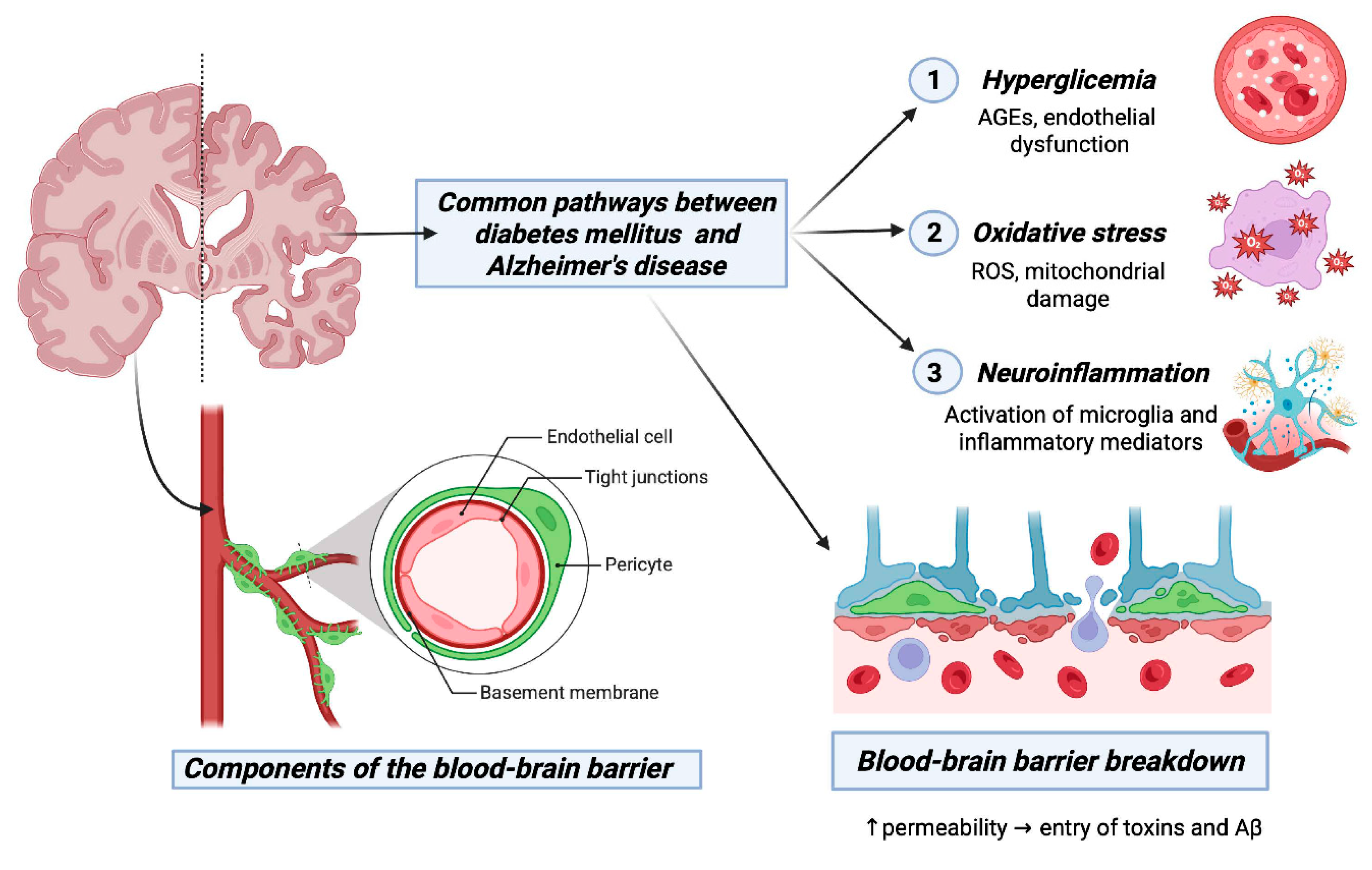
Figure 3.
Interconnected Mechanisms Linking Diabetes Mellitus Type 2 and Alzheimer's Disease. The figure illustrates the interconnected mechanisms that establish a link between DM2 and AD. It focuses on shared elements such as insulin resistance, chronic inflammation, and common brain proteins. The impact of insulin on cognitive processes, Aβ peptide levels, and tau protein hyperphosphorylation is highlighted. Additionally, it shows mechanisms like neuroinflammation, Aβ accumulation, and tau phosphorylation, emphasizing blood biomarkers for AD diagnosis. In the amyloidogenic pathway, amyloid precursor protein is first cleaved by β-secretase and subsequently cleaved by γ-secretase to generate Aβ peptides. The accumulation and aggregation of Aβ peptides results in neurotoxic amyloid plaques. This figure provides a visually comprehensive overview of the complex interplay between DM2 and AD, unraveling the interconnected mechanisms that contribute to their shared pathophysiology and mutual promotion of cognitive dysfunction. Created in https://BioRender.com.
Figure 3.
Interconnected Mechanisms Linking Diabetes Mellitus Type 2 and Alzheimer's Disease. The figure illustrates the interconnected mechanisms that establish a link between DM2 and AD. It focuses on shared elements such as insulin resistance, chronic inflammation, and common brain proteins. The impact of insulin on cognitive processes, Aβ peptide levels, and tau protein hyperphosphorylation is highlighted. Additionally, it shows mechanisms like neuroinflammation, Aβ accumulation, and tau phosphorylation, emphasizing blood biomarkers for AD diagnosis. In the amyloidogenic pathway, amyloid precursor protein is first cleaved by β-secretase and subsequently cleaved by γ-secretase to generate Aβ peptides. The accumulation and aggregation of Aβ peptides results in neurotoxic amyloid plaques. This figure provides a visually comprehensive overview of the complex interplay between DM2 and AD, unraveling the interconnected mechanisms that contribute to their shared pathophysiology and mutual promotion of cognitive dysfunction. Created in https://BioRender.com.
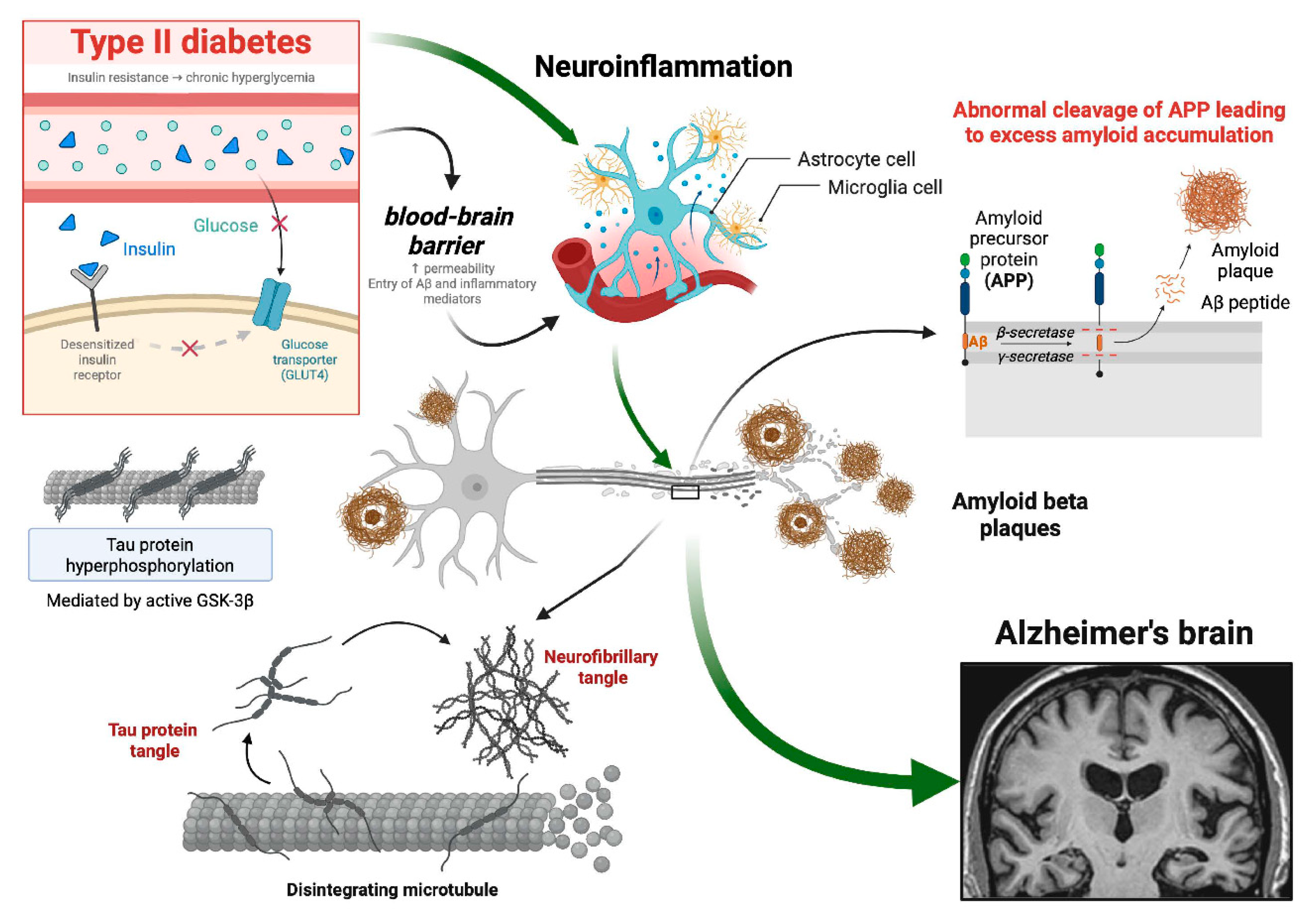
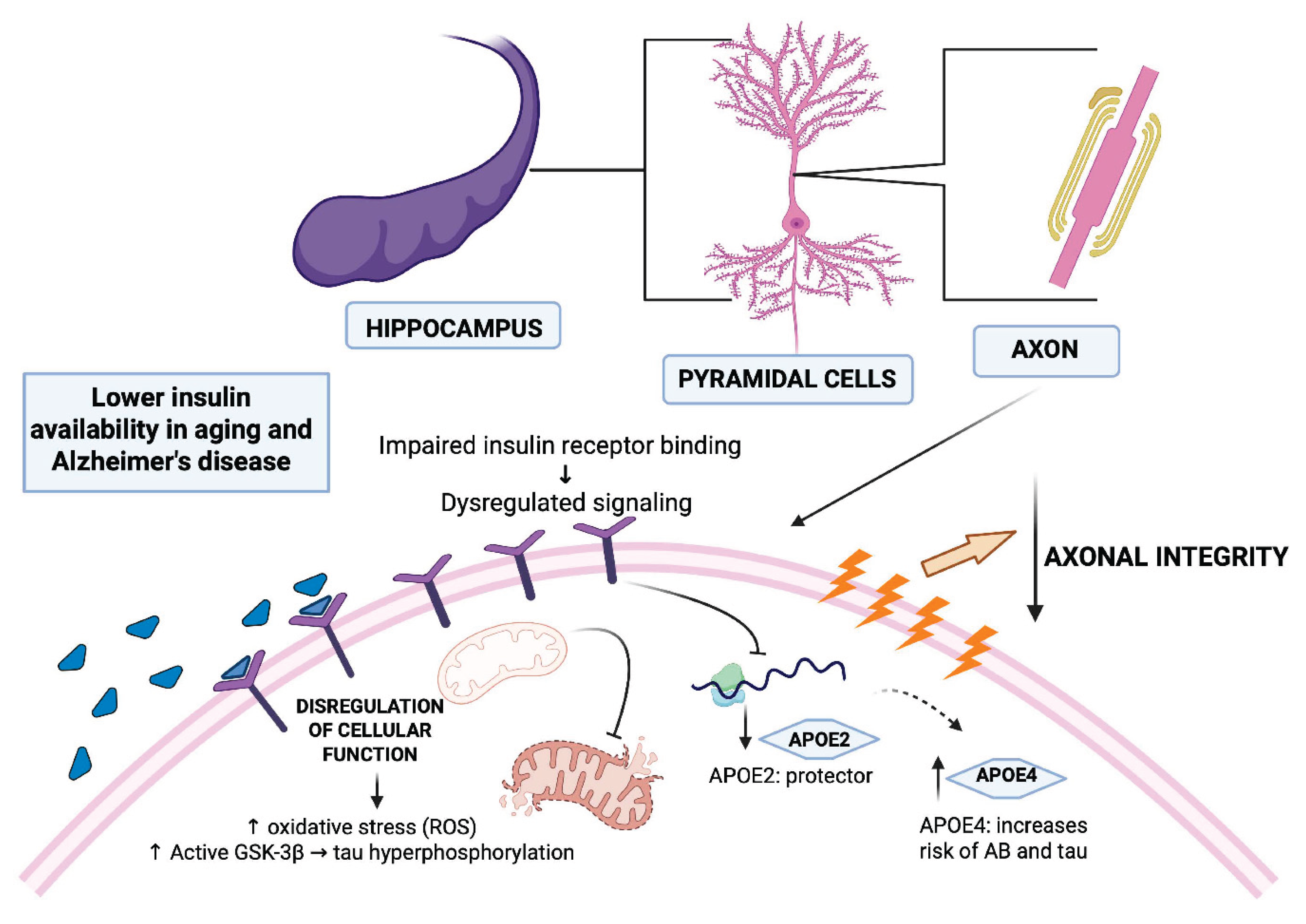
Figure 4.
Interplay of Insulin, APOE Genotypes, and Alzheimer's Disease Onset. This figure illustrates the intricate relationships between insulin, APOE genotypes, and AD. Insulin, a crucial trophic factor in the CNS, is transported across the BBB and interacts with insulin receptors, which are widely expressed in the hippocampus. The figure highlights how alterations in insulin levels and signaling contribute to neurodegeneration and cognitive impairment in the context of DM2 and aging. It visualizes how alterations in insulin levels and signaling, particularly in brain regions like the hippocampus, contribute to neurodegeneration in the context of diabetes mellitus and aging. The figure also highlights the impact of APOE genotypes on AD risk, emphasizing the neuroprotective role of APOE2, and the increased risk associated with APOE4. Created in https://BioRender.com.
Figure 4.
Interplay of Insulin, APOE Genotypes, and Alzheimer's Disease Onset. This figure illustrates the intricate relationships between insulin, APOE genotypes, and AD. Insulin, a crucial trophic factor in the CNS, is transported across the BBB and interacts with insulin receptors, which are widely expressed in the hippocampus. The figure highlights how alterations in insulin levels and signaling contribute to neurodegeneration and cognitive impairment in the context of DM2 and aging. It visualizes how alterations in insulin levels and signaling, particularly in brain regions like the hippocampus, contribute to neurodegeneration in the context of diabetes mellitus and aging. The figure also highlights the impact of APOE genotypes on AD risk, emphasizing the neuroprotective role of APOE2, and the increased risk associated with APOE4. Created in https://BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



