Submitted:
26 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Therapeutic stagnation in Alzheimer’s disease (AD) highlights the role of non-genetic mechanisms like metabolic dysregulation. We propose a paradigm: exercise reprograms phosphoglycerate dehydrogenase (PHGDH) from a pathogenic nuclear transcription factor to a protective cytoplasmic metabolic enzyme through brain-periphery crosstalk. Under AD-associated stress, PHGDH translocate to the nucleus, where its helix-turn-helix (HHTH) domain activates pro-inflammatory genes (e.g., IKKα, HMGB1), suppresses autophagy, and accelerates Aβ deposition. Centrally, exercise-induced myokine irisin suppresses PHGDH nuclear translocation via AMPK/PGC-1α signaling, preserving its metabolic role. Peripherally, exercise inhibits PHGDH in hepatic Kupffer cells, reducing systemic IL-1β release and neuroinflammation. Notably, the HHTH-targeting inhibitor NCT-503 reduces Aβ plaques by 44–62% in APP-KI mice, validating this strategy. This framework positions exercise as a precision intervention, enabling novel diagnostics (e.g., PET probes for PHGDH compartmentalization) and synergistic “exercise-pharmacotherapy.”
Keywords:
neurodegenerative diseases
; PHGDH
; exercise
1. Introduction
PHGDH: Redefining a Metabolic Enzyme’s Paradigm in Neurodegeneration
Alzheimer’s disease (AD), the most prevalent neurodegenerative disorder, is characterized by progressive cognitive decline, extracellular amyloid-β (Aβ) plaques, and neurofibrillary tangles composed of hyperphosphorylated tau. With global aging accelerating, AD currently affects over 55 million people worldwide, a figure projected to triple by 2050, imposing an estimated economic burden of $1.8 trillion [1]. Despite decades of research, existing therapeutics (e.g., acetylcholinesterase inhibitors and anti-Aβ antibodies) offer merely symptomatic relief without halting disease progression. This therapeutic impasse stems from AD’s multifactorial pathogenesis, where genetic risk factors (e.g., APOE4) explain only 30–50% of sporadic AD (sAD) cases, highlighting the critical role of non-genetic mechanisms such as metabolic dysregulation and neuroinflammation [2,3].
Phosphoglycerate dehydrogenase (PHGDH), the rate-limiting enzyme in de novo serine biosynthesis, has emerged as a critical node in Alzheimer’s disease (AD) pathogenesis. Longitudinal multi-cohort analyses reveal that PHGDH expression is elevated in both brain tissue and blood of sAD patients, correlating with Braak staging and cognitive decline even in asymptomatic individuals, positioning it as a predictive biomarker for early diagnosis [4]. Beyond its canonical metabolic role, PHGDH exhibits a non-canonical function as a transcriptional regulator in astrocytes. Under glucose deprivation—a hallmark of AD brains—PHGDH undergoes p38 MAPK-mediated nuclear translocation. Therein, its helix-helix-turn-helix (HHTH) domain binds promoters of pro-inflammatory genes (IKKα, HMGB1), acting independently of its enzymatic activity. This binding triggers NF-κB/mTOR activation and suppresses autophagy, accelerating Aβ deposition in 3xTg-AD mice and human brain organoids (BOs) [3]. Crucially, enzyme-dead mutants (PHGDH-ED) retain this pathogenic function, confirming the dissociation of metabolic and transcriptional roles [2].
Therapeutically, the BBB-penetrant small-molecule inhibitor NCT-503 disrupts PHGDH’s DNA-binding capacity by targeting the HHTH domain. In APP-KI mice, NCT-503 reduces hippocampal Aβ plaques by 44–62% and rescues spatial memory deficits without perturbing serine metabolism, underscoring the primacy of transcriptional inhibition over metabolic modulation [2]. These findings caution against serine supplementation—an approach under clinical evaluation—as elevated PHGDH in AD likely augments endogenous serine production, rendering exogenous intake ineffective or potentially detrimental [4].
Epidemiological studies consistently demonstrate that physical activity reduces AD incidence by 45% and decelerates cognitive decline in prodromal stages, with aerobic exercise (AE) identified as the most effective modality for enhancing global cognition in elderly AD patients [5,6,7]. Single-nucleus RNA sequencing reveals that exercise restores neuroprotective astrocyte subpopulations (e.g., CDH4+ cells) in AD mice, enhancing neurovascular coupling and Aβ phagocytosis, while simultaneously augmenting disease-associated microglial gene expression (e.g., TREM2) to promote amyloid clearance [8,9]. Notably, exercise-induced reactivation of the metabolic gene ATPIF1 promotes neuronal survival and synaptic plasticity, accounting for 64% of transcriptional repair in oligodendrocyte precursor cells [8,9]. Clinically, moderate-intensity continuous training (e.g., 4,000 steps/day) increases hippocampal volume by 2%, effectively reversing age-related atrophy and correlating with a 25–50% risk reduction for AD conversion [6,7].
For patients with advanced frailty, pharmacological mimetics targeting exercise-activated pathways (e.g., NCT-503 inhibiting PHGDH transcriptional activity) reduce Aβ plaques by 44–62% and improve spatial memory, offering potential “exercise-in-a-pill” strategies [2].
2. The Metabolic and Transcriptional Duality of PHGDH in Astrocytes: A Bifunctional Regulator in Neurodegenerative Pathogenesis
Phosphoglycerate dehydrogenase (PHGDH), the gatekeeper of the serine biosynthesis pathway, is canonically defined by its catalytic conversion of 3-phosphoglycerate (3-PG) to 3-phosphohydroxypyruvate (3-PHP), a process integral to one-carbon metabolism and nucleotide biosynthesis [10]. This traditional view is now being challenged by the revelation of a non-canonical, moonlighting function for PHGDH as a transcriptional regulator, a role that is especially pronounced in astrocytes—the principal glial population of the central nervous system (CNS) [2]. This functional duality situates PHGDH at the confluence of metabolic reprogramming and epigenetic regulation, positioning it as a pivotal player in the pathogenesis of neurodegenerative diseases, notably Alzheimer’s disease (AD) [10,11]. In the context of AD, astrocytes—long relegated to the status of metabolic support cells for neurons—undergo a pathological reprogramming characterized by aberrant PHGDH expression. Strikingly, the magnitude of this dysregulation scales with Braak staging and parallels cognitive deterioration [11].
2.1. Metabolic Functions: Serine Synthesis and Beyond
In astrocytes, phosphoglycerate dehydrogenase (PHGDH) is the master regulator of de novo serine synthesis, yielding D-serine—an essential co-agonist of NMDA receptors for synaptic plasticity (Figure 1). Genetic ablation of PHGDH in murine models depletes D-serine pools, thereby impairing long-term potentiation (LTP) and spatial memory [14]. Beyond its role in neurotransmission, PHGDH-derived serine fuels the folate cycle, which produces S-adenosylmethionine (SAM). SAM serves as the universal methyl donor for reactions governing histone modifications (e.g., H3K27me3) and DNA stability in neurons [12].
Under conditions of glucose deprivation—a hallmark of Alzheimer’s disease (AD) brains—PHGDH undergoes dynamic subcellular redistribution. Specifically, p38 MAPK phosphorylates PHGDH at Ser371, triggering its nuclear translocation (Figure 1) [10]. Concurrently, AMPK-mediated phosphorylation at Ser55 reprograms PHGDH’s catalytic activity to utilize alternative substrates like malate. This metabolic shift generates NADH to sustain redox homeostasis [10]. Such metabolic plasticity enables astrocytes to maintain the NAD+/NADH balance during energetic stress, a process elegantly visualized using nuclear-targeted SoNar/Frex biosensors [10].
2.2. Transcriptional Regulation: The Non-Canonical Paradigm
In astrocytes within the Alzheimer’s disease (AD) brain, PHGDH assumes a novel, non-canonical role in the nucleus, functioning independently of its well-characterized metabolic activity. Utilizing its helix-helix-turn-helix (HHTH) domain, PHGDH acts as a DNA-binding protein, as demonstrated by chromatin immunoprecipitation sequencing (ChIP-seq) which identified its occupancy at promoter regions. This binding directly drives the expression of key pro-inflammatory genes, IKKα and HMGB1 (Figure 1) [10]. The downstream effects are profoundly pathogenic: IKKα potentiates NF-κB signaling, while HMGB1 disrupts the autophagic removal of amyloid-beta (Aβ), establishing a self-perpetuating cycle that amplifies amyloid deposition [11].
This dual functionality is unequivocally demonstrated by the finding that enzyme-dead PHGDH mutants (e.g., PHGDH-ED) retain their transcriptional capabilities, confirming a clear dissociation between its metabolic and gene-regulatory functions (Figure 1) [10]. The HHTH domain (residues 210–310) is the critical mediator of this activity, as its deletion via mutagenesis prevents PHGDH from activating IKKα and HMGB1, even when the protein is overexpressed to pathological levels [10]. Furthermore, the structural homology of this domain to established transcription factors provides the molecular basis for its ability to engage in sequence-specific DNA recognition, specifically targeting CpG-rich sites near neuroinflammatory gene promoters [10].
2.3. Pathogenic Implications in Neurodegenerative Diseases
Elevated RNA and protein levels of PHGDH in astrocytes have been consistently demonstrated in multi-cohort analyses of late-onset Alzheimer’s disease (LOAD), with these levels positively correlating with amyloid-beta (Aβ) burden and Braak staging [11]. This relationship is further supported by functional studies in 3xTg-AD mice, where astrocyte-specific overexpression of PHGDH resulted in a 2.3-fold increase in Aβ plaques. In contrast, PHGDH knockdown led to a substantial reduction of 60–70% in plaque formation [11]. At the mechanistic level, the PHGDH-IKKα-HMGB1 axis contributes to Aβ accumulation by suppressing autophagy and enhancing the activity of β-secretase (BACE1) [11].
The function of nuclear PHGDH extends to the integration of metabolic stress with immune responses. Under conditions of low glycolytic flux, such as the depletion of 3-phosphoglycerate (3-PGA), PHGDH promotes the assembly of a PHGDH-AXIN-HIPK2 complex. This complex facilitates the phosphorylation of p53 at Serine 46, initiating pro-apoptotic pathways in astrocytes under duress [16]. On the other hand, through its role in producing S-adenosylmethionine (SAM), PHGDH fuels the trimethylation of H3K27 (H3K27me3) at the promoters of key neuroprotective genes like BDNF, leading to their transcriptional silencing and a subsequent increase in neuronal vulnerability [11,12].
3. Multi-Organ PHGDH Network Remodeling Underlies Systemic Benefits of Exercise Training
3.1. Exercise-Induced Irisin Modulates the Brain Metabolic Landscape via AMPK/PGC-1α-Mediated Suppression of PHGDH Nuclear Translocation
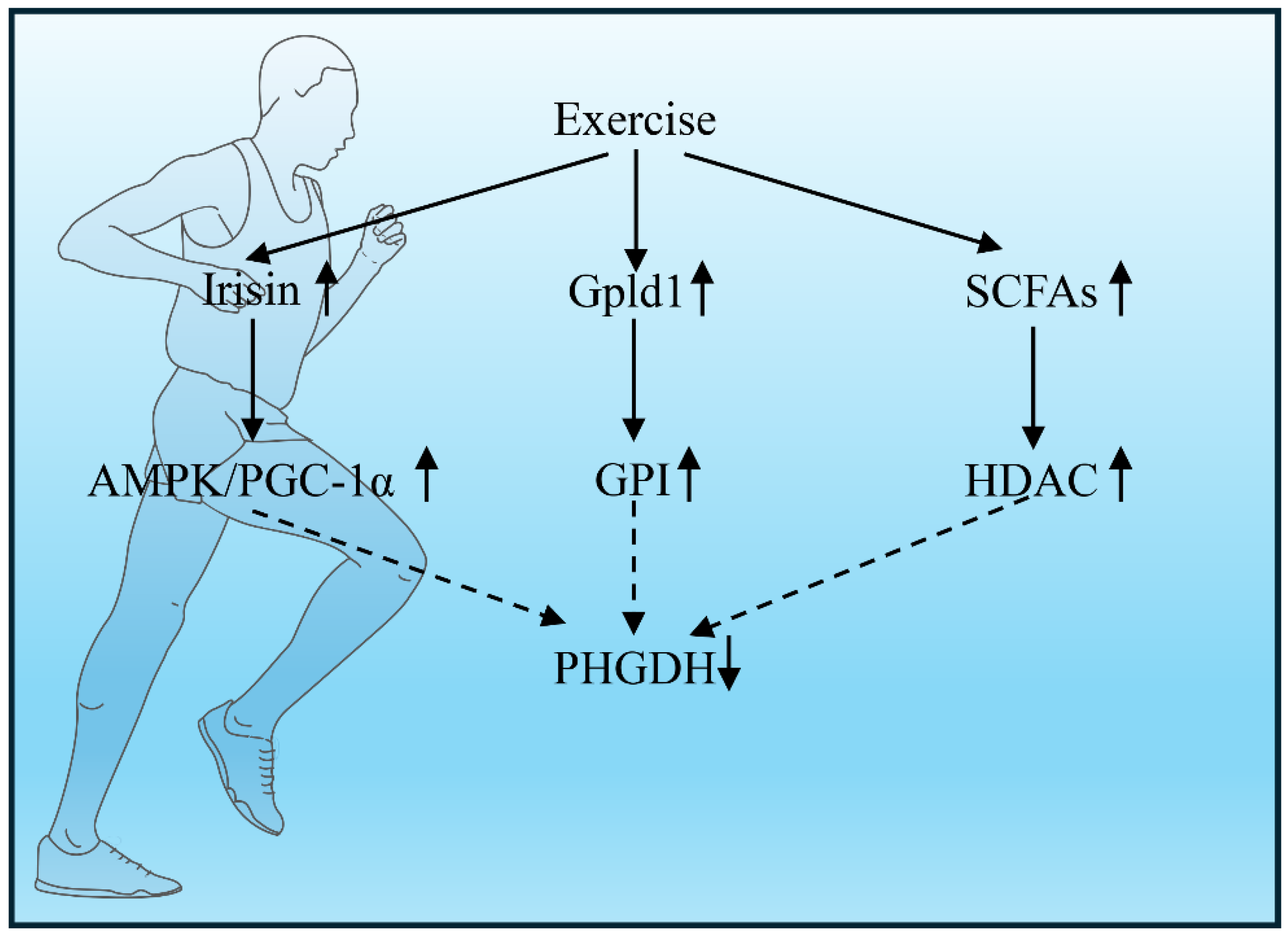
Physical exercise emerges as a potent non-pharmacological intervention that consistently demonstrates neuroprotective benefits across clinical and preclinical studies. Intriguingly, the muscle-brain endocrine axis has been implicated as a critical mediator of exercise-induced neuroprotection, with the myokine irisin serving as a key signal transducer. This review posits a novel hypothesis: Exercise-derived irisin activates the AMPK/PGC-1α axis to suppress nuclear translocation of phosphoglycerate dehydrogenase (PHGDH), thereby reprogramming brain metabolism and attenuating neurodegenerative pathology [13,14].
Skeletal muscle functions as a dynamic endocrine organ during physical activity, secreting myokines that communicate with distant organs, including the brain. Irisin, a cleaved fragment (∼12 kDa) of the membrane protein fibronectin type III domain-containing protein 5 (FNDC5), is robustly upregulated by exercise via the PGC-1α-dependent pathway in contracting muscle. Upon release into circulation, irisin crosses the blood-brain barrier (BBB) via saturable transport mechanisms, achieving physiologically relevant concentrations in the hippocampus, cortex, and cerebrospinal fluid. Crucially, irisin deficiency exacerbates synaptic loss and cognitive impairment in AD models, whereas its administration rescues neurogenesis deficits [14]. This establishes irisin as a principal exercise-derived mediator of central nervous system (CNS) adaptation.
Irisin initiates a neuroprotective signaling cascade by binding to integrin αV/β5 receptors on the surface of neurons and glial cells. This interaction triggers the activation of AMP-dependent protein kinase (AMPK), a master cellular energy sensor that is phosphorylated at Thr172 in response to irisin-induced shifts in the cellular AMP:ATP ratio. Once active, AMPK directly phosphorylates peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) at Ser538, which enhances its stability as a transcriptional coactivator and promotes its retention within the nucleus [15]. In turn, PGC-1α orchestrates a transcriptional program that favors mitochondrial biogenesis—mediated by NRF1 and TFAM—and bolstered antioxidant defenses through SOD2 and CAT. Notably, activation of the AMPK/PGC-1α axis has demonstrated neuroprotective effects in ischemic stroke models, correlating with improved mitochondrial membrane potential and enhanced ATP generation [15].
Phosphoglycerate dehydrogenase (PHGDH), which catalyzes the rate-limiting step of de novo serine biosynthesis, diverts glycolytic flux toward amino acid production and one-carbon metabolism. Under pathological stress conditions, such as exposure to amyloid-beta (Aβ) oligomers, PHGDH undergoes translocation to the nucleus. Within the nucleus, it locally generates serine to support histone methylation (specifically H3K27me3), leading to the repression of genes essential for neurogenesis. This elevation of nuclear PHGDH correlates with impaired hippocampal neurogenesis and deficits in memory consolidation in models of Alzheimer’s disease (AD). We hypothesize that exercise-induced irisin signaling suppresses this aberrant nuclear localization. To mechanistically validate this axis, we will investigate whether exercise intervention fails to suppress PHGDH nuclear translocation in Fndc5 murine models, thereby exacerbating tau hyperphosphorylation through the amplification of this pathological cascade.
3.2. Exercise-Mediated Suppression of Hepatic Macrophage PHGDH Attenuates Neuroinflammation via the Liver-Brain Immune Axis
Neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), are characterized by progressive neuronal loss and neuroinflammation driven by microglial activation and cytokine dysregulation (e.g., IL-1β, TNF-α). While central mechanisms have been extensively studied, recent evidence highlights the liver as a critical peripheral regulator of brain immunity via the “liver-brain immune axis” [16,17]. This axis orchestrates systemic metabolic and immune homeostasis, wherein hepatic macrophages (Kupffer cells) act as key sentinels. Intriguingly, physical exercise—a non-pharmacological intervention—modulates this axis by suppressing phosphoglycerate dehydrogenase (PHGDH) in hepatic macrophages, thereby reducing IL-1β production and mitigating neuroinflammation [2,17].
PHGDH catalyzes the rate-limiting step in de novo serine synthesis, a pathway linked to inflammatory responses. In aged or metabolically stressed livers, PHGDH is upregulated in Kupffer cells, promoting serine flux toward IL-1β biosynthesis via the NLRP3 inflammasome. Serine-derived metabolites fuel mitochondrial respiration, enhancing ROS generation and caspase-1 activation, which cleaves pro-IL-1β into its active form [2]. Elevated IL-1β enters systemic circulation, crosses the compromised blood-brain barrier (BBB) in neurodegeneration, and activates microglia, exacerbating Aβ/tau pathology in AD and α-synuclein aggregation in PD [18]. Critically, PHGDH inhibition reduces IL-1β by >40% in murine models, confirming its pro-inflammatory role [2].
Exercise induces multi-organized crosstalk that converges on hepatic PHGDH regulation (Figure 2):
- AMPK/SIRT1 Activation: Exercise initiates a powerful metabolic signal by releasing myokines (IL-6, irisin) from skeletal muscle that activate hepatic AMPK and SIRT1. These enzymes orchestrate a two-pronged attack on PHGDH: AMPK flags PHGDH for destruction by phosphorylating it at Ser49, while SIRT1 suppresses PHGDH gene transcription by deacetylating NF-κB [16,19].
- Gpld1-Mediated Signaling: The liver counters inflammation through the exercise-induced hepatokine Gpld1. Gpld1 disrupts inflammatory signaling in Kupffer cells by cleaving GPI-anchored proteins, which directly halts PHGDH-driven serine synthesis and blocks IL-1β maturation. This pathway is so crucial that its absence in Gpld1-deficient mice renders the anti-inflammatory effects of exercise completely ineffective [16,19].
- Gut-Liver Axis Modulation: Exercise also reshapes the gut ecosystem, fostering beneficial bacteria like Bifidobacterium. These microbes generate short-chain fatty acids, notably butyrate, which act as epigenetic regulators. In Kupffer cells, butyrate inhibits histone deacetylases, effectively silencing the expression of key inflammatory genes, PHGDH and IL1B [16].
4. Precision Exercise Intervention: Diagnostic and Therapeutic Innovations Targeting PHGDH
Addressing the critical unmet need for novel therapeutics in neurodegenerative disorders, we propose to establish a comprehensive experimental framework for PHGDH-targeted precision exercise interventions. This initiative is founded on three highly innovative and synergistic components, each supported by a rigorous and systematic experimental design.
Aim 1: Pioneering a Diagnostic PET Probe for PHGDH Subcellular Dynamics. Our first objective is to develop a revolutionary molecular imaging tool: a PET probe engineered with the HHTH structural domain to quantify the PHGDH nuclear-to-cytoplasmic ratio (N/C ratio). This technology will, for the first time, allow for in vivo, high-resolution spatiotemporal mapping of PHGDH compartmentalization. By providing a quantitative biomarker of PHGDH activation states, this probe will transform early disease detection and enable the precise stratification of patients for personalized therapeutic interventions.
Aim 2: Validating a Synergistic Exercise-Pharmacotherapy Strategy. We will investigate the synergistic potential of combining aerobic exercise with pharmacotherapy, specifically testing the hypothesis that exercise enhances the blood-brain barrier permeability and central nervous system (CNS) bioavailability of the PHGDH inhibitor NCT-503. To validate this premise, we will execute a systematic experimental program encompassing: (1) the development and characterization of transgenic animal models with robust, disease-relevant phenotypes; (2) detailed pharmacokinetic and pharmacodynamic (PK/PD) profiling of NCT-503 under controlled exercise conditions; and (3) the application of multidimensional behavioral batteries to assess functional outcomes.
Aim 3: Translational Optimization of Combinatorial Parameters. The final phase of our research will be to translate these findings into an optimized therapeutic protocol. We will systematically determine the ideal combinatorial parameters, including the most effective dosing regimens for NCT-503 and the optimal intensity, duration, and timing of exercise interventions. This will establish a comprehensive, clinically actionable framework for PHGDH-targeted precision exercise therapy.
5. Precision Exercise Targeting PHGDH in Neurodegenerative Diseases: Challenges, and Future Experimental Designs
To further explore the therapeutic potential of gene-exercise crosstalk, we will employ AAV-mediated delivery of a constitutively active PHGDH-S553D gene variant, engineered to mimic exercise-induced phosphorylation. This innovative strategy requires a sophisticated experimental framework that incorporates: (1) quantitative evaluation of transduction efficiency using advanced molecular techniques, (2) longitudinal therapeutic monitoring through multimodal imaging and functional assays, and (3) rigorous safety profiling to assess potential off-target effects. Through an interdisciplinary collaboration integrating biomedical engineering, molecular neuroscience, and clinical neurology, we anticipate achieving transformative breakthroughs in mitigating the progression of motor neuron degeneration.
A central objective of our future experimental design is to elucidate a fundamental question: how does exercise specifically regulate the HHTH domain of PHGDH without affecting its canonical enzymatic activity? To comprehensively dissect this complex mechanism, we have devised two complementary experimental strategies.
First, we will leverage cutting-edge spatial omics technologies, integrating single cell sequencing with subcellular proteomics, to map the comprehensive landscape of the PHGDH interactome following exercise. This approach will enable us to identify key proteins that dynamically interact with the PHGDH HHTH domain across different cell types and subcellular compartments, thereby revealing the specific regulatory mechanisms engaged during physical activity.
Second, we will construct organoid models, specifically by introducing a PHGDH-luc reporter gene into late-onset Alzheimer’s disease brain organoids (LOAD-BOs), to simulate and study the regulatory effects of exercise on PHGDH. This platform will serve to screen pharmacological compounds capable of mimicking the beneficial effects of exercise. Such screening will not only validate the generalizability of our mechanistic findings but also identify promising therapeutic targets and lead compounds for future drug development. We are confident that the synergistic combination of these two strategies will open new avenues for elucidating the molecular mechanisms by which exercise regulates PHGDH.
6. Conclusions
In conclusion, this review fundamentally reframes our understanding of how physical exercise confers neuroprotection, identifying PHGDH as a pivotal metabolic switch whose spatial function is reprogrammed through brain-periphery crosstalk. We have elucidated a dual regulatory mechanism: centrally, exercise-induced irisin activates the AMPK/PGC-1α axis in astrocytes, suppressing the pathological nuclear translocation of PHGDH and thereby preserving its beneficial metabolic role while inhibiting its non-canonical function as a pro-inflammatory transcription factor. Peripherally, exercise modulates the liver-brain immune axis by suppressing PHGDH activity in hepatic Kupffer cells, which curtails the systemic release of IL-1β and mitigates neuroinflammation.
These findings collectively transcend the conventional view of PHGDH as merely a metabolic enzyme, establishing it as a bifunctional regulator linking metabolic stress directly to gene expression and immune dysregulation in neurodegeneration. This paradigm shift not only provides a unified mechanistic explanation for the well-documented neuroprotective effects of exercise but also opens new avenues for therapeutic intervention. The demonstrated efficacy of selectively targeting PHGDH’s non-canonical HHTH domain with inhibitors like NCT-503, which reduces amyloid pathology without disrupting serine metabolism, validates the promise of precision medicine strategies that decouple these dual functions.
Looking ahead, our proposed framework—encompassing the development of a novel PET probe to quantify PHGDH subcellular dynamics and the validation of synergistic exercise-pharmacotherapy regimens-charts a clear course for translational research. By establishing the principles of PHGDH spatial regulation, this work lays the groundwork for a new era of “precision exercise medicine,” offering tangible hope for altering the trajectory of Alzheimer’s disease and other neurodegenerative disorders.
Author Contributions
Wen Guo and Liang Guo conceived and designed the “Exercise-CTSS-AD Axis” hypothesis and framework. Dong Yang performed the literature search, data synthesis, and figure creation, and drafted the initial manuscript. Wen Guo and Liang Guo critically reviewed and edited the manuscript for important intellectual content. All authors read and approved of the final manuscript. Wen Guo, Liang Guo, and Bihan Wang are the co-corresponding authors.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- You, Y.; Shou, X.; Zhang, X.; Fan, S.; Chai, R.; Xue, W.; Hu, Y.; He, Q. Psycho-Cardiological Disease: A Bibliometric Review From 2001 to 2021. Front Cardiovasc Med 2022, 9, 890329. [Google Scholar] [CrossRef]
- Chen, J.; Hadi, F.; Wen, X.; Zhao, W.; Xu, M.; Xue, S.; Lin, P.; Calandrelli, R.; Richard, J.L.C.; Song, Z.; et al. Transcriptional regulation by PHGDH drives amyloid pathology in Alzheimer’s disease. Cell 2025, 188, 3513–3529.e3526. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Zhang, C.S.; Xiong, J.; Cai, D.Q.; Wang, C.Z.; Wang, Y.; Liu, Y.H.; Wang, Y.; Li, Y.; Wu, J.; et al. Low glucose metabolite 3-phosphoglycerate switches PHGDH from serine synthesis to p53 activation to control cell fate. Cell Res 2023, 33, 835–850. [Google Scholar] [CrossRef]
- Chen, X.; Calandrelli, R.; Girardini, J.; Yan, Z.; Tan, Z.; Xu, X.; Hiniker, A.; Zhong, S. PHGDH expression increases with progression of Alzheimer’s disease pathology and symptoms. Cell Metab 2022, 34, 651–653. [Google Scholar] [CrossRef]
- Hu, F.; Peng, J.; Wang, W.; Shen, L.; Jia, M. Comparing the impact of various exercise modalities on old adults with Alzheimer’s disease: A Bayesian network meta-analysis. Complement Ther Clin Pract 2025, 59, 101968. [Google Scholar] [CrossRef]
- De la Rosa, A.; Olaso-Gonzalez, G.; Arc-Chagnaud, C.; Millan, F.; Salvador-Pascual, A.; García-Lucerga, C.; Blasco-Lafarga, C.; Garcia-Dominguez, E.; Carretero, A.; Correas, A.G.; et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J Sport Health Sci 2020, 9, 394–404. [Google Scholar] [CrossRef]
- Raji, C.A.; Meysami, S.; Hashemi, S.; Garg, S.; Akbari, N.; Ahmed, G.; Chodakiewitz, Y.G.; Nguyen, T.D.; Niotis, K.; Merrill, D.A.; et al. Exercise-Related Physical Activity Relates to Brain Volumes in 10,125 Individuals. J Alzheimers Dis 2024, 97, 829–839. [Google Scholar] [CrossRef]
- da Rocha, J.F.; Lance, M.L.; Luo, R.; Schlachter, P.; Moreira, L.; Iqbal, M.A.; Kuhn, P.; Gardner, R.S.; Valaris, S.; Islam, M.R.; et al. Protective exercise responses in the dentate gyrus of Alzheimer’s disease mouse model revealed with single-nucleus RNA-sequencing. Nat Neurosci 2025, 28, 1546–1561. [Google Scholar] [CrossRef]
- Madhu, L.N.; Somayaji, Y.; Shetty, A.K. Promise of irisin to attenuate cognitive dysfunction in aging and Alzheimer’s disease. Ageing Res Rev 2022, 78, 101637. [Google Scholar] [CrossRef]
- Ma, C.; Zheng, K.; Jiang, K.; Zhao, Q.; Sha, N.; Wang, W.; Yan, M.; Chen, T.; Zhao, Y.; Jiang, Y. The alternative activity of nuclear PHGDH contributes to tumour growth under nutrient stress. Nat Metab 2021, 3, 1357–1371. [Google Scholar] [CrossRef]
- Shan, X.; Hu, P.; Ni, L.; Shen, L.; Zhang, Y.; Ji, Z.; Cui, Y.; Guo, M.; Wang, H.; Ran, L.; et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1-p38 axis. Cell Mol Immunol 2022, 19, 1263–1278. [Google Scholar] [CrossRef]
- Cai, Z.; Li, W.; Hager, S.; Wilson, J.L.; Afjehi-Sadat, L.; Heiss, E.H.; Weichhart, T.; Heffeter, P.; Weckwerth, W. Targeting PHGDH reverses the immunosuppressive phenotype of tumor-associated macrophages through α-ketoglutarate and mTORC1 signaling. Cell Mol Immunol 2024, 21, 448–465. [Google Scholar] [CrossRef]
- Kong, J.; Xie, Y.; Fan, R.; Wang, Q.; Luo, Y.; Dong, P. Exercise orchestrates systemic metabolic and neuroimmune homeostasis via the brain-muscle-liver axis to slow down aging and neurodegeneration: a narrative review. Eur J Med Res 2025, 30, 475. [Google Scholar] [CrossRef]
- Zhao, R. Can exercise benefits be harnessed with drugs? A new way to combat neurodegenerative diseases by boosting neurogenesis. Transl Neurodegener 2024, 13, 36. [Google Scholar] [CrossRef]
- Wu, X.; Li, C.; Ke, C.; Huang, C.; Pan, B.; Wan, C. The activation of AMPK/PGC-1α/GLUT4 signaling pathway through early exercise improves mitochondrial function and mitigates ischemic brain damage. Neuroreport 2024, 35, 648–656. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 2020, 369, 167–173. [Google Scholar] [CrossRef]
- Bono, F.; Tomasoni, Z.; Mutti, V.; Sbrini, G.; Kumar, R.; Longhena, F.; Fiorentini, C.; Missale, C. G Protein-Dependent Activation of the PKA-Erk1/2 Pathway by the Striatal Dopamine D1/D3 Receptor Heteromer Involves Beta-Arrestin and the Tyrosine Phosphatase Shp-2. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Hynes, T.J.; Chernoff, C.S.; Hrelja, K.M.; Tse, M.T.L.; Avramidis, D.K.; Lysenko-Martin, M.R.; Calderhead, L.; Kaur, S.; Floresco, S.B.; Winstanley, C.A. Win-Paired Cues Modulate the Effect of Dopamine Neuron Sensitization on Decision Making and Cocaine Self-administration: Divergent Effects Across Sex. Biol Psychiatry 2024, 95, 220–230. [Google Scholar] [CrossRef]
- Zhou, F.; Wei, L.; Wang, Y.; Chen, W. Aerobic exercise modulates the striatal Erk/MAPK signaling pathway and improves LID in a mouse model of Parkinson’s disease. Brain Res Bull 2024, 209, 110906. [Google Scholar] [CrossRef]
Figure 1.
Duality of PHGDH Functions in Astrocytes. Canonical metabolic function of PHGDH: Under healthy physiological conditions, PHGDH is primarily located in the cytoplasm, where it serves as the rate-limiting enzyme of the serine synthesis pathway. This pathway generates D-serine, a co-agonist of NMDA receptors that supports synaptic plasticity and cognitive function. Concurrently, serine metabolism fuels the production of S-adenosylmethionine (SAM), a methyl donor for epigenetic modifications (e.g., histone methylation) that maintains neuronal homeostasis. Pathological mode of PHGDH (Non-canonical transcriptional function): Under pathological conditions such as Alzheimer’s disease (AD), particularly during glucose deprivation, PHGDH is phosphorylated by p38 MAPK and translocases from the cytoplasm to the nucleus. Within the nucleus, its HHTH domain acts as a transcription factor, directly binding to and activating the promoters of pro-inflammatory genes, such as IKKα and HMGB1. This function is independent of its enzymatic activity, leading to the inhibition of autophagy via the NF-κB pathway, accelerated amyloid-beta (Aβ) deposition, and ultimately, neurodegeneration.
Figure 1.
Duality of PHGDH Functions in Astrocytes. Canonical metabolic function of PHGDH: Under healthy physiological conditions, PHGDH is primarily located in the cytoplasm, where it serves as the rate-limiting enzyme of the serine synthesis pathway. This pathway generates D-serine, a co-agonist of NMDA receptors that supports synaptic plasticity and cognitive function. Concurrently, serine metabolism fuels the production of S-adenosylmethionine (SAM), a methyl donor for epigenetic modifications (e.g., histone methylation) that maintains neuronal homeostasis. Pathological mode of PHGDH (Non-canonical transcriptional function): Under pathological conditions such as Alzheimer’s disease (AD), particularly during glucose deprivation, PHGDH is phosphorylated by p38 MAPK and translocases from the cytoplasm to the nucleus. Within the nucleus, its HHTH domain acts as a transcription factor, directly binding to and activating the promoters of pro-inflammatory genes, such as IKKα and HMGB1. This function is independent of its enzymatic activity, leading to the inhibition of autophagy via the NF-κB pathway, accelerated amyloid-beta (Aβ) deposition, and ultimately, neurodegeneration.
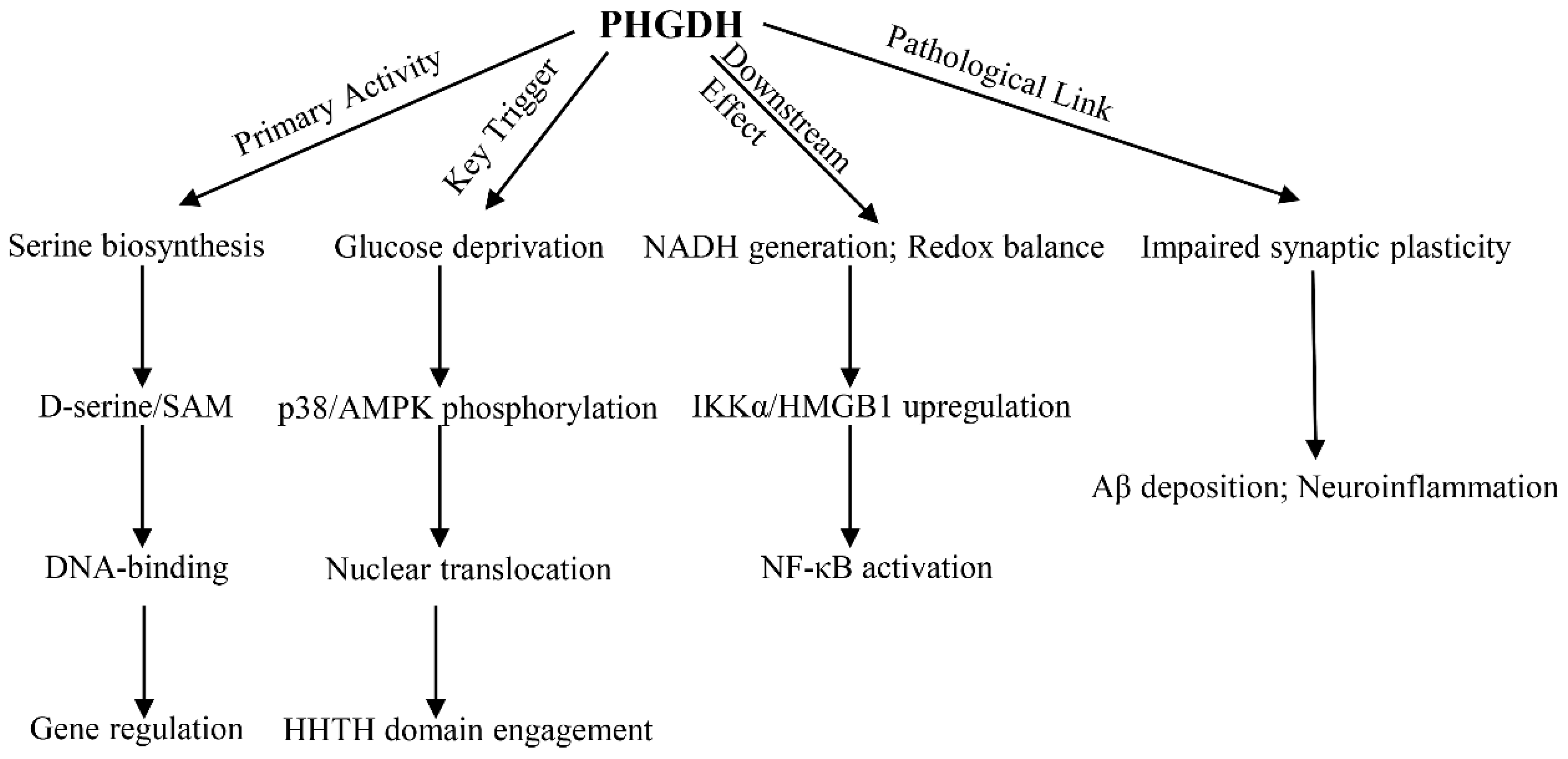
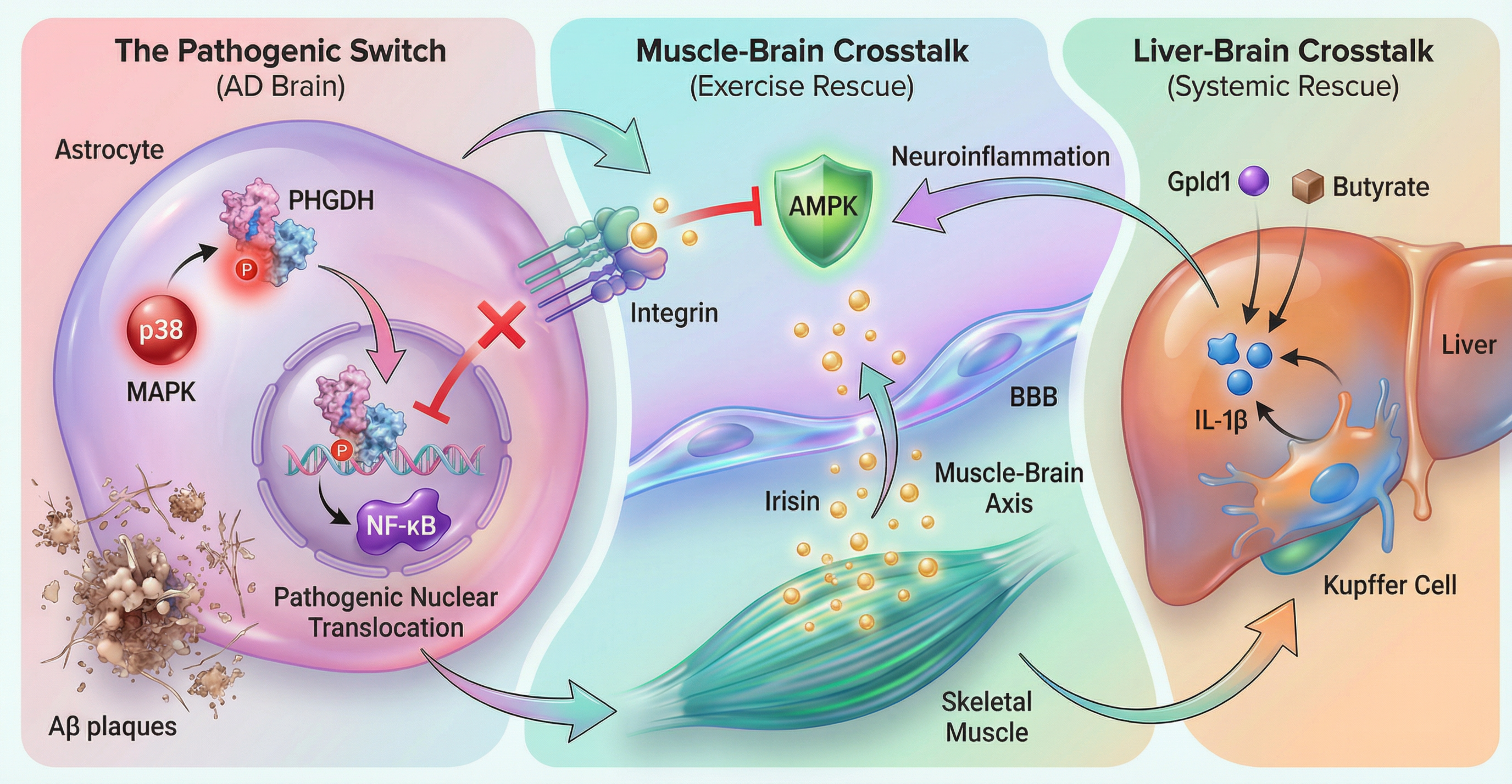
Figure 2.
Molecular Pathways Linking Exercise to Hepatic PHGDH-IL-1β Suppression. Central pathway (Intra-brain): Exercise induces the release of the myokine irisin from skeletal muscle. Irisin crosses the blood-brain barrier and activates the AMPK/PGC-1α signaling pathway in the brain (primarily in astrocytes), thereby inhibiting PHGDH nuclear translocation and retaining it in the cytoplasm to perform its beneficial metabolic functions. Peripheral pathway (Liver-brain axis): Exercise modulates hepatic immunity by suppressing PHGDH in liver macrophages (Kupffer cells), reducing the systemic release of the pro-inflammatory cytokine IL-1β, which in turn mitigates neuroinflammation in the brain.
Figure 2.
Molecular Pathways Linking Exercise to Hepatic PHGDH-IL-1β Suppression. Central pathway (Intra-brain): Exercise induces the release of the myokine irisin from skeletal muscle. Irisin crosses the blood-brain barrier and activates the AMPK/PGC-1α signaling pathway in the brain (primarily in astrocytes), thereby inhibiting PHGDH nuclear translocation and retaining it in the cytoplasm to perform its beneficial metabolic functions. Peripheral pathway (Liver-brain axis): Exercise modulates hepatic immunity by suppressing PHGDH in liver macrophages (Kupffer cells), reducing the systemic release of the pro-inflammatory cytokine IL-1β, which in turn mitigates neuroinflammation in the brain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



