
Submitted:
23 February 2026
Posted:
25 February 2026
You are already at the latest version
Abstract
Burkholderia pseudomallei, the causative agent of melioidosis, presents significant challenges in both treatment and environmental decontamination. Bacteriophages, or phages, are increasingly being explored as potential diagnostic, therapeutic and biocontrol agents against this bacterial pathogen. Our recent investigation has shown that most B. pseudomallei genomes contained pro-phage(s) associated with specific tRNA gene loci, prompting us to explore these detectable pro-phages as sources of temperate phages, for further applications. Transcriptomic profiling of B. pseudomallei Bp82, a model strain that possesses three different prophages, revealed high expression levels of the integrase and certain transcriptional regulatory genes within its prophages during normal exponential growth. Using one of its temperate phages, namely φBP82.2, a P2-like phage, as a model, we investigated the lysogenic-lytic control mechanisms. Mutagenesis of the integrase gene, phiBP82.2_gp51, did not improve killing activity compared to the wildtype phage. In contrast, deletion of phiBP82.2_gp38, a putative transcriptional regulatory gene, and two-downstream hypothetical protein genes, phiBP82.2_gp36 and phiBP82.2_gp37, resulted in significant lytic improvement. We conclude that these genes play a crucial role in the lysogenic-lytic switch of φBP82.2, suggesting a new avenue for engineering temperate phages for future applications.
Keywords:
Burkholderia pseudomallei
; melioidosis
; prophage
; phage engineering
1. Introduction
Burkholderia pseudomallei is a Gram-negative saprophytic bacterium found in soil and fresh water [1,2,3]. It poses a significant challenge to tropical and sub-tropical regions worldwide, serving as the causative agent of melioidosis in both humans and animals [4,5,6,7]. Melioidosis is considered a difficult-to-treat disease due to the bacterium's natural multidrug resistance and its ability to survive intracellularly [8]. Thus, effective antibiotic treatments for melioidosis necessitate a prolonged course, often extending up to 20 weeks and incurring high costs. A select few antibiotics have demonstrated efficacy against B. pseudomallei involving an acute phase with parenteral ceftazidime, amoxicillin–clavulanic acid, or meropenem, followed by an eradication phase with oral trimethoprim–sulfamethoxazole [9,10,11,12]. While these antibiotics have proven against B. pseudomallei effectively, the prolonged treatment duration raises concerns about antibiotic resistance [8,13,14]. Moreover, the widespread presence of B. pseudomallei in the environments also complicates efforts to decontaminate affected areas [15]. These dual problems complicate efforts to address melioidosis in many countries, raising serious public health concerns.
In response to these challenges, researchers are currently exploring bacteriophage (phage) therapy as a promising alternative, offering potential for diagnostic, therapeutic, and environmental biocontrol applications [16,17,18,19,20]. However, phages are highly host-specific, and the environmental bacteria commonly develop resistance to phages. Finding the right phage(s) to target most B. pseudomallei strains may be difficult, which hinders the progress of phage research for this bacterium. Although phages infecting B. pseudomallei were first reported as early as 1956 [21] , only a few lytic phages, such as ST79 and vB_BpP _ HN01 have been recently characterized [17,19]. GenBank database indicates that the majority of known B. pseudomallei phages are temperate, which are associated with the prophage regions of the bacterial genomes, where the phage persists in the bacterial chromosome after its DNA enters the bacterium [22]. Temperate phages usually contain a site-specific recombination (SSR) sequence, also known as a phage attachment site (attP) in their genomes. This specific DNA sequence is used to recombine its genome with a specific homologous sequence on the bacterial chromosome known as attB site [23]. After integration, a prophage or prophage island is formed, and a short direct repeat sequence, identical to attP or attB, is present at the 3’end region of the prophage [24]. Terms attL and attR are used to describe the homologous sequences flanking the left and right ends of a prophage, respectively. The prophage is also replicated when the host cell divides and is believed to play an important role in microbial evolution [22,23,24].
Our group has reported that most prophage regions are associated with genomic islands in B. pseudomallei where the foreign genetic materials recombined into the bacterial chromosome which may allow the bacteria to survive in various environmental conditions [25]. We also observed that the SSR events frequently occurred in highly conserved regions of the bacterial chromosome such as at genes that encode tRNAs, where prophages are located downstream of the genes flanked by the SSR sequences. We also observed that most of the B. pseudomallei strains contain at least one lysogenic phage [22]. Based on these observations, we used bioinformatic tools to identify the prophage regions of multiple B. pseudomallei strains. This was done by tracking a short direct repeat sequence of tRNA genes in B. pseudomallei genomes obtained from GenBank. This strategy allowed us to easily predict intact prophages and the potential temperate phages that infect B. pseudomallei. However, the success in isolating these temperate phages from B. pseudomallei raised an important question of whether we could utilize these phages to infect other B. pseudomallei strains. We have learned that their killing efficacy is relatively poor, largely due to their temperate nature. Thus, understanding the regulatory mechanisms governing the lysogenic life cycle of these phages is essential. Such knowledge may allow us to engineer these phages to more effectively kill B. pseudomallei, making them more suitable for downstream applications. These temperate phages could then serve as a substantial resource for future applications if their lysogenic to lytic phenotypes can be specifically modified.
In brief, this investigation was developed based on the observation that certain prophage genes such as phage integrase and repressor genes were always expressed during the exponential growth phase, while their structural protein genes were mostly silent during the normal growth of B. pseudomallei. This led to the discovery of the lysogenic-lytic control mechanisms in a prophage known to be associated with phage φBP82.2, a P2 - like phage in B. pseudomallei Bp82.
2. Results
2.1. Genomic Analysis of B. pseudomallei Prophages
We previously observed that most prophages in B. pseudomallei are associated with at least eight tRNA gene locations. These prophages serve as sources for two major temperate phage groups, myophages and siphophages [22]. Notably, comparative genomic analysis of available Burkholderia myophages genomes reveals a high degree of conservation across species. These phages are classified as P2-like phages and belong to the Peduoviridae family, which includes the well-characterized Enterobacteria phage P2. Notably, the structural genes encoding capsid, tail, and lysis proteins are highly conserved within this group. However, variability was observed in the enzymatic gene regions (comprised of non-structural genes), particularly within one-third of the phage genome if these phages had different integration sites e.g., different tRNA genes, or different genomic islands (GIs). As shown in Figure 1A, φBP82.2 was associated with tRNA-Arginine (anticodon CCG) and φBP82.3 with tRNA-Phenylalanine (anticodon GAA), while φPK23 associated with a prophage within genomic island 15 (GI 15), a non-tRNA gene location (Figure 1A; [22,26]). In contrast, Burkholderia siphophages exhibit greater diversity and possess genomes approximately 1.5 times larger than those of Burkholderia myophages. Comparative analysis of prophage regions reveals some structural gene variations among siphophages when located at different genomic sites; for example, φBt-TXDOH which is associated with tRNA-Serine (anticodon GGA) gene on chromosome 2 of B. thailandensis TXDOH, versus φBP82.1, associated with tRNA-Proline (anticodon UGG) gene on chromosome 1 of B. pseudomallei Bp82 (Figure 1B). Nevertheless, when siphophages are integrated at the same tRNA gene locus, their genomes remain conserved, similar to the pattern observed in myophages.
Another intriguing finding from our prophage genomic analysis is the strong correlation between phage-encoded integrase genes and tRNA - SSR sequences. For prophage regions linked to a particular tRNA - SSR site, such as prophages integrated at the tRNA-Phe locus in B. pseudomallei strains 52237, K96243, and MSHR2543 (Figure 1A), or at the tRNA-Pro locus in strains 1026b, Burk178-Type2, and MSHR5848 (Figure 1B), the corresponding phages consistently encode the same integrase gene sequence. In addition, the non-structural protein-coding regions, which typically comprise approximately one-third of the phage genome, are largely conserved among prophages integrated at the same SSR locus. Notably, comparison of intact phage genomes with their integrated prophage counterparts, such as φBP82.3 versus prophages at tRNA-Phe gene locus, and φBP82.1 relative to prophages at tRNA-Pro, reveals that the phage genomes are split at the SSR junction upon integration. This genomic rearrangement results in repositioning of the integrase gene to either the first or last open reading frame within the prophage region following chromosomal integration.
2.2. Isolation of a Lysogenic Clone of B. pseudomallei 576mn Following φBP82.2 Infection
To investigate whether the temperate phages exhibit specificity for particular phage integration site known as the attB sequence of bacterial genome, we used a temperate phage φBP82.2 from B. pseudomallei Bp82, an attenuated derivative of B. pseudomallei 1026b to infect its susceptible host B. pseudomallei 576mn as previously described [22]. Bp82 carries three functional prophages on chromosome 1 (Figure 2A [22]). Following infection, a lysogen was isolated from a resulting bacterial colony grown inside the plaque of φBP82.2 and subjected to whole-genome sequencing. The analysis showed that φBP82.2 integrated into the tRNA-Arg (CCG) gene of strain 576mn, precisely at the same site as in Bp82 genome (Figure 2B). Notably, the phage integration attB site, corresponds to a 45-bp 3’end sequence of the tRNA-Arg gene. As observed previously, the phage genome containing an attP sequence, was recombined with this attB sequence through homologous recombination, resulting in the integrase gene being repositioned as the first gene in the integrated prophage downstream of tRNA-Arg gene. Furthermore, the prophage region was flanked by an identical sequence attP/attB (also known as attL and attR) right at the end of the prophage. Since the integrase gene is located next to the attB site, it could play a major role in mediating site-specific recombination at the attB site during phage transduction, and a reverse recombination for phage excision to enter the lytic cycle. Genomes of B. pseudomallei 576mn and its lysogenic derivative strain 576mn-phiBP82.2 are available through GenBank with accession numbers: JBQYPK000000000 and JBQYPL000000000, respectively.

2.3. Expression Profile of the Prophages in B. pseudomallei Bp82 During Normal Exponential Growth
To identify prophage genes involved in bacterial lysogenic life cycle, we conducted RNAseq analysis during the normal exponential growth of B. pseudomallei Bp82. Expression profiles of the prophage genes were generated by mapping sequencing reads to each phage genome for a simplified visualization (Figure 3). Applying a 3000 read coverage cut-off window view, we observed that both prophages sourced of the myophages, φBP82.2 and φBP82.3, displayed higher read coverages compared to the siphophage, φBP82.1. This finding correlates with the higher plaque-forming activity of φBP82.2 and φBP82.3 than that of φBP82.1 on the B. pseudomallei 576mn host (Data not shown). Interestingly, structural protein genes encoding tail, capsid, and endolysin genes were transcriptionally silent across all three phages, indicating a dormant state consistent with lysogeny. Both myophages exhibited nearly identical gene expression profiles within the non-structural part of their genome (Figure 3B and Figure 3C). This similarity persisted despite gene variations due to the different tRNA-SSR location of each prophage. Intriguingly, both prophages associated with φBP82.2 and φBP82.3 showed a consistent pattern of gene expression, with high expression of the integrase gene. Additionally, a cluster of repressor genes upstream of the integrase also exhibited high expression in both prophages. Furthermore, a set of hypothetical genes downstream of the integrase, with unknown functions, displayed high expression. In contrast, integrase gene in prophage associated with φBP82.1 was not expressed, while genes gp45-52, predicted to be acted as the repressors were expressed (Figure 3A).
2.4. Deletion of the Integrase Gene gp51 in the φBP82.2 Prophage Resulted in the Loss of Phage Excision
To investigate the role of an integrase gene in φBP82.2 prophage, we used the lysogenic B. pseudomallei 576mn-φBP82.2 as a model strain. This is because B. pseudomallei 576mn does not contain any intact prophages in its genome which eliminates potential confounding factors of integrase genes from other prophages. We observed that this lysogenic strain exhibited immunity against φBP82.2 infection, while it was susceptible to φBP82.1 and φBP82.3 (Figure 4A).

To assess the function of the integrase gene in φBP82.2 prophage, we engineered a suicide plasmid vector, pExKm5 [27], to target the integrase gene (int, gp51) present in the integrated φBP82.2 prophage in the lysogenic B. pseudomallei 576mn-phiBP82.2. Upon transforming the plasmid into the bacteria, we successfully isolated merodiploid colonies and counterselection resulting in a marker-less mutagenesis of the gp51 gene. The resulting integrase deletion mutant was confirmed by PCR, designated as B. pseudomallei 576mn-phiBP82.2Δint. Sequencing of the mutant phage, φBP82.2Δint, confirmed the deletion of the integrase gene (Figure 4D, and GenBank accession no. PX400618). We did not observe any phage released from this mutant when it was spontaneously induced in liquid culture and propagated with the wildtype 576mn (Figure 4B). To test if this was due to the deletion of phage integrase gene, we then conducted a trans-complementation of the integrase gene controlling by the lac promoter on a recombinant plasmid pBIC (pBRR1K::phiBP82.2_gp51) into the mutant strain. After six hours of IPTG induction, the supernatant was collected for spontaneous phage analysis. As expected, we successfully observed plaque formations on the double agar overlay culture of 576mn (data not shown). The phage produced from 576mn-phiBP82.2Δint mutant with int-trans complementation were used in the phage-host challenge test for the comparison of the killing efficacy with the wildtype φBP82.2. Surprisingly, the killing efficacy of this mutant phage did not improve compared to the wildtype φBP82.2 (Figure 4C). This suggests that the integrase does not play a major role in the switch to the phage lytic life cycle.
2.5. Repressor Gene Involvement in the Lysogenic-Lytic Switch
To further identify the genes responsible for maintaining lysogeny, we hypothesized these genes would be highly expressed during the normal growth of bacteria to suppress the excision of phages. Based on the RNAseq data, we observed a group of highly expressed genes corresponding to gp36, gp37, and gp38 in the φBP82.2 prophage, which were located near the integrase gene. Although prophage φBP82.3 contains a different set of genes at the same genomic location when aligned with φBP82.2, genes in this location were also highly expressed despite limited sequence similarity. To investigate the function of these genes in the model φBP82.2 prophage, we proceeded with mutagenesis of genes gp36, gp37, and gp38 genes in the lysogenic B. pseudomallei 576mn-phiBP82.2 using the same approach described above. We successfully resolved the merodiploid following counterselection of the gp36 deletion mutant by plating on medium containing 15% sucrose. In contrast, no bacterial colonies were recovered after counterselection of the gp37 or gp38 deletion mutants. We therefore hypothesized that deletion of these two genes could trigger activation of the lytic life cycle, resulting in host cell lethality.
To assess this, we attempted to resolve the merodiploid by cultivating the suspected clones in LB broth containing 15% sucrose instead of LB agar, with the expectation of recovering mutant phages that could be released into the liquid phase following host cell death. As expected, we successfully recovered the mutant phages φBP82.2Δgp37 (gp37 deletion), and φBP82.2Δgp38 (gp38 deletion). Using the same approach, we successfully created a triple gene deletion mutant, φBP82.2Δgp37-38 Δint. The triple genes mutant phage was confirmed by PCR (Figure 5B). A phage-host challenge test of these mutant phages demonstrated a fascinating improvement in the killing efficacy against B. pseudomallei 576mn host (Figure 5A). The highest concentrations of the phages also improved about a 2-log difference (Figure 5D), suggesting that the repressor genes play a crucial role in lytic switch of these Burkholderia P2-like prophages. However, we also performed a comparison of the killing efficacy of the engineered phages with φPK23V1, one of the most B. pseudomallei-specific lytic-like phages previously described [26]. The φPK23V1 consistently exhibited superior killing efficacy and higher phage titer compared with the engineered phages (Figure 5C, D).
3. Discussion
In the era of advanced genetic modification, the potential of engineered bacteriophages has garnered attention as an alternative agent against antibiotic-resistant bacteria, driven by the intentional design to fix some weaknesses of phages for specific purposes. Two notable examples of successful engineered phages were Mycobacteriophage ZoeJ and BPs, which have been effectively utilized to treat multidrug-resistant Mycobacterium abscessus in a cystic fibrosis patient [28,29,30]. These temperate phages underwent genetic engineering, involving the deletion of immunity genes and the attP site sequence, ultimately transforming the temperate phages into virulent lytic forms. In B. pseudomallei, we previously identified a widespread prevalence of prophages in the population, with approximately fifty percent of bacterial genomes containing at least one prophage [22]. Thus, we were curious to explore the possibility of modifying these phages to enhance their lytic activity, aiming to improve the killing efficiency of these phages for downstream applications. Our work has illustrated the process from the initial discovery of the temperate phages and used one of them as a model to engineer for practical use. Here are the main points to discuss:
3.1. Temperate Phages are Common in B. pseudomallei and May Contribute to Host Survival
We have recently demonstrated that the majority of reported bacteriophages infecting B. pseudomallei are P2-like myophages belonging to the family Peduoviridae, followed by siphophages. Consistent with this trend, the attenuated strain Bp82 harbors two myophages, φBP82.2 and φBP82.3, and one siphophage, φBP82.1, integrated within its genome, providing a useful model for studying phage biology in this bacterium [22]. One likely advantage of lysogeny is the protection it confers against superinfection by related phages. In support of this, φBP82.2 failed to infect both the 576mn-phiBP82.2 lysogen and the parental Bp82 strain, demonstrating classical superinfection immunity (Figure 4A). These findings suggest that temperate phages may enhance bacterial survival by preventing infection from closely related phages in the environment.
3.2. The Integrase Gene is Required for Phage Excision but Does Not Enhance Lytic Activity
We previously identified the unique phage integration sequences, including a site-specific recombination sequences (SSRs) [22]. These elements were conserved when prophages occupied the same genomic loci. Notably, the integrase gene exhibited high expression levels during the exponential bacterial growth in liquid medium, leading us to hypothesize that it may play a crucial role in regulating the dormant prophage stage. To test this hypothesis, we deleted the integrase gene (gp51) in the prophage of the lysogenic 576mn-phiBP82.2 strain. Unexpectedly, the resulting mutant strain was defective in spontaneous phage induction. However, complementation of the integrase gene successfully restored spontaneous induction, leading to the release of the φBP82.2int mutant and demonstrating that the integrase gene is required for the phage excision. Despite its essential role in phage excision, deletion of the integrase gene did not enhance phage-mediated killing. This observation suggests that the primary regulatory mechanisms governing the lytic switch are not directly controlled by the integrase. Consistent with our finding, Shitrit and colleagues demonstrated that both the integrase gene and the phage integration site are essential for integration, yet deletion of these genes did not increase the lytic activity of the engineered phage [31]. Together, these results indicate that although integrase function is critical for prophage excision and integration, it does not represent a limiting factor for enhancing lytic activity in this system.
3.3. The Deletion of Repressor Genes Activates the Lysogenic - Lytic Switch
P2-like phages are widely distributed among Gram-negative bacteria, with Enterobacteria phage P2 (GenBank accession no. NC_001895.1) serving as a well-studied prototype. In our comparative analysis, we performed BLASTp analysis to assess the protein-level similarity between P2 phage and our φBP82.2 phage (Supplemental Figure 1). This analysis revealed limited similarity among structural protein genes, whereas nonstructural or accessory protein genes showed little to no detectable homology. Despite the low sequence conservation, overall gene orientation was largely conserved across the two genomes. Notably, the highly expressed φBP82.2-associated prophage genes gp36, gp37, and gp38, located on the reverse strand, were transcribed in the opposite orientation relative to genes involved in the lytic cycle. We hypothesized that these genes may function as regulators of the lytic-lysogenic decision, analogous to the C gene in the P2 phage [32]. Consistent with this hypothesis, deletion of gp36, gp37, or gp38 in φBP82.2 successfully triggered activation of the lytic life cycle. A similar phenomenon has been well documented in phage lambda, where deletion of the transcriptional repressor results in obligately lytic phages, underscoring the conserved role of repressor proteins in maintaining lysogeny across diverse temperate phage systems [33].
Supporting this model, Yao and colleagues reported that mutations near promoter regions of similarly oriented reverse-strand genes resulted in strict virulence in Burkholderia phage Milagro, a P2-like phage infecting B. cenocepacia [34]. The authors proposed that face-to-face promoters (PL and PR) in this genomic region compete with one another, leading to antagonistic regulation of lytic genes encoded on the forward strand. A comparable regulatory architecture exists in P2 phage, where the C and cox genes are arranged in opposite orientations, binding of the C protein to the Pe promoter inhibits cox expression, thereby promoting lysogeny. Consistent with this model, transcriptional profiling of φBP82.2 prophage during the exponential growth phase, together with promoter prediction using BPROM (SoftBerry Inc., Mount Kisco, NY, USA), identified a σ70-dependent promoter upstream of gp38. In contrast, promoter elements on the forward strand remain poorly defined. Furthermore, the precise inhibitory mechanisms mediated by the gp37-gp38 proteins remain to be elucidated and warrant further investigation.
4. Materials and Methods
4.1. Bacterial Culture Condition
Burkholderia spp. and Escherichia coli strains were cultured at 37°C with aeration in LB media. The attenuated strains B. pseudomallei Bp82 [35] and 576mn [36], the biosafe non-select agent strains, were additionally supplemented with 80 mg/L of adenine. Bacteriophages were propagated on bacterial host soft agar by combining 100 μL of log phase 576mn culture with 4.5 mL of molten LB adenine soft agar (0.35% w/v) in a sterile 15 mL tube. The resulting mixture was poured onto an LB agar plate supplemented with 80 mg/L of adenine. Once the agar solidified, one hundred microliters of the phage solution was dropped onto the bacterial soft agar plate, gently rotated the plate to spread the phage solution throughout plate surface, then allowed to dry for 10 minutes. Following drying, the plate was inverted and incubated at 37°C overnight, and subsequently, plaque formations were observed. Bacterial growth curves and host challenge tests were conducted by combining 100 ml of fresh 576mn culture at 0.1 optical density 600 nm (O.D. 600) assuming 108 cfu/mL of the bacterial host with 100 mL of 105 pfu/ml phage in 500 ml LB adenine media in a 48-well flat-bottomed plate with a lid. The cultures were shaken using linear shaking mode at 493 cpm in a BioTek Synergy HTX plate reader. This process was performed in triplicate at 37 °C. The O.D. 600 nm was measured every 10 minutes for a duration of 24 hrs. It is important to note that a low multiplicity of infections (~MOI 0.001) was used due to the limited production of the temperate phages.
4.2. Prophage Genome Analysis
We conducted pangenome analysis using the local NCBI BLASTn (v 2.2.18) [37] command line to acquire the coordinate sequences at the locations of the prophages associated with tRNA genes, as detailed in our previous publication [22]. The coordinates encompassing the start and end bases of each prophage region, along with the chromosome strand information, were compiled into a table. This table was then utilized to generate BED files. Subsequently, the BED files were employed to extract a multiFASTA file for each prophage genome using the BEDtools command [38]. Also, the coordinates were used to download the GenBank file of each prophage region from the GenBank database directly. To perform whole phage genome alignments, rigorous comparisons were done in the Artemis Comparison Tool [39] and Easyfig 2.2.2 OSX [40] to compare the conserved regions of the prophages.
4.3. Phage Induction and Lysogen Isolation
The Bp82.2 phage was induced from B. pseudomallei strain Bp82, following the procedure outlined in [22]. The phage solution was spotted on top of the surface of a 576mn LB adenine soft agar plate and incubated overnight. The presence of a clear zone, with colonies growing inside the clear zone, was observed on the next day. A sterile inoculating loop was used to stab the clear zone, and was streaked onto a new LB adenine plate, then incubated for 48 hours. For the isolation of lysogen colonies, each single colony was assigned a number and individually stabbed onto a 576mn LB adenine soft agar plate, followed by an overnight incubation. The presence of a clear zone ring around the stabbed colony indicated that it was a lysogen colony. Subsequently, these lysogen colonies were purified three times and confirmed for the integrase gene of Bp82.2 through colony PCR.
4.4. RNA Sequencing
A 3 mL volume of fresh Bp82 culture with an O.D. 600 of 0.1 was incubated at 37°C with aeration for 3 hours, using triplicate tubes. Subsequently, a 1 mL aliquot of each culture was centrifuged at 16,000 x g for 1 minute to obtain a pellet in each sample. RNA extraction from each bacterial pellet was carried out using the Direct-zolTM RNA Miniprep kit (Zymo Research) following the manufacturer's protocol. For RNA sequencing method, the samples underwent DNAse treatment with Invitrogen DNAse (RNAse free). Library preparation was executed using Illumina's Stranded Total RNA Prep Ligation with Ribo-Zero Plus kit and 10-bp unique dual indices (UDI). Sequencing was performed on a NovaSeq X Plus, generating paired end 150bp reads. Demultiplexing, quality control, and adapter trimming were executed using bcl-convert (v4.1.5). Subsequently, reads were trimmed with Trimmomatic [41] and mapped onto B. pseudomallei 1026b, φBP82.1, φBP82.2, and φBP82.3 genome using BWA-MEM [42]. The resulting BAM files were visualized on Artemis [43].
4.5. Phage Integrase Gene Mutagenesis
A plasmid pExKm5 was provided as a gift by Dr. Herbert P Schweizer at the University of Florida. To mutate the phage integrase (int) gene or phiBP82.2_gp51, around 900 base pairs upstream and downstream of the gene were amplified using the primers listed on Supplemental Table 1. The PCR fragments were gel purified using Zymoclean Gel DNA Recovery Kits. To construct pExKm5-Bp82.2@int plasmid, we modified a method from López CM et al, 2009 [27]. Briefly, the pExKm5 was digested by EcoRI-HF and NotI-HF (New England Biolabs, NEB) and gel purified as the previous PCR products. The digested plasmid and the PCR fragments were assembled using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, NEB) using manufacturer's protocol. The assembled plasmid was then transformed into E. coli DH5a competent cells on an LB plate supplemented with 50ug/mL of kanamycin (Km) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). White colonies were selected and used for plasmid amplification. B. pseudomallei 576mn-phiBP82.2 lysogen electrocompetent cells were made in house by washing three times with 300mM sucrose as described by Choi KH et al in 2005 [44]. Approximately 100-200 ng of purified plasmid DNA was then electroporated into the 100 µl competent cells using a 0.2 mm cuvette (Bio-Rad) at 2500 V 200@. The transformed cells were recovered in LB broth with shaking at 200-250 rpm for two hours. Then the mixture was plated on LB plates containing 250 μg/mL Km and 50 μg/ml of 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (X-Gluc) for 48 hrs. A blue Kmr colony was picked up and the merodiploid was solved by streaking with the presence of 15% sucrose in YT agar supplemented with 0.8 ug/mL adenine for 48 hours. White colonies were then tested by PCR to confirm the mutant lysogen using upstream forward and downstream reverse primers.
4.6. Construction of Replicative Plasmid for Integrase Supplementation
Plasmid pBbB1k-GFP (addgene#35342) [45] were used as a plasmid backbone to construct pBBR1K-Bp82.2integrase-chromoprotein (pBIC). The 660-bp chromoprotein gene was amplified from plasmid pSB1C3 (tsPurple chromoprotein; addgene#117848) [46] with primers listed on Supplemental Table 1, using Q5 High-Fidelity DNA Polymerase (New England Biolabs, NEB) at annealing temperature 55 oC for 45 seconds and extension at 72 oC for 1 minute. An integrase gene was amplified from phiBP82.2_gp51 gene using primers listed on Supplemental Table 1, at annealing temperature 60 oC for 45 seconds and extension at 72 oC for 90 seconds. The PCR products were gel purified using Zymoclean Gel DNA Recovery Kits. To construct a pBIC plasmid, the pBbB1k-GFP plasmid was digested with NdeI and BamHI-HF (New England Biolabs, NEB), then assembled with the purified phiBP82.2_gp51 DNA and chromoprotein PCR fragments with NEBuilder HiFi DNA Assembly Master Mix. The pBIC was then transformed into competent cells of E. coli DH5α for amplification. The successful transformants were identified by the appearance of pink-purple colonies. For integrase complementation to induce the mutant phage, the 576mn-phiBP82.2△int lysogen were transformed with pBIC plasmid using electroporation protocol described above. The recovery cells were plated on LB adenine supplemented with 250 µg/mL Km. Plasmid presence in a clone was confirmed by PCR using primers targeting the phiBP82.2_gp51 integrase gene. To induce the expression of the integrase to promote spontaneous phage production, the confirmed clone was cultured in LB adenine broth containing 250 µg/mL Km until reaching a log phase and then induced with 0.5 M Isopropyl ß-D-1-thiogalactopyranoside (IPTG) for 6 hours. One milliliter of the culture was then centrifuged at 16,000 x g for 1 minute, and the supernatant was then filtered on a 0.2 µm filter membrane. A volume of 100 µL of the supernatant was plated on top of the 576mn LB adenine soft agar plate. The mutant phages were isolated from plaques after overnight incubation.
4.7. Phage Repressor Gene Mutagenesis
To mutate the phage repressor genes, we used the same technique as described above with all the primers listed in Supplemental Table 1. However, resolution of the merodiploid resulted in host cell lethality with no mutant clone observed on sucrose-containing LB plate. Therefore, the merodiploid was resolved in liquid media as follows. A blue colony was inoculated in 3 mL of LB-adenine-Km250-X-gluc media overnight. The culture was centrifuged at 4000 x g for 10 minutes. The supernatant was discarded, and the pellet was resuspended in 3 mL of LB broth. This washing step was repeated three times to remove the residual antibiotic. The pellet was then resuspended in 3 mL of LB–adenine broth containing 15% sucrose and incubated at 37 oC for 4 hours. After incubation, 1 mL of the culture was aliquoted and centrifuged for phage isolation as described above, and then the filtered supernatant was used to perform phage isolation.
5. Conclusions
Our study established φBP82.2, a temperate phage from B. pseudomallei Bp82, as a tractable model for phage engineering. We demonstrated distinct functional roles for the integrase and the hypothetical repressor genes, with the latter acting as key regulators of the lysogenic-lytic switch in this P2-like phage. Deletion of the integrase gene impaired phage excision, whereas deletion of these hypothetical repressor genes enhanced lytic activity. We believe that these engineered phage derivatives provide a framework for converting prophages into functional tools and represent a valuable resource for future phage biology and engineering studies.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Genomic comparison of Enterobacter phage P2 and Burkholderia phage φBP82.2; and Supplemental Table 1: List of plasmids and PCR primers used in this study.
Author Contributions
Conceptualization, P.K. and A.T.; methodology, P.K., S.M.D., N. S., Y. P.; validation, A.T. and Y.P. ; formal analysis, P.K., and A.T; investigation, P.K.; resources, H.P.S., A.M., A.T; data curation, A.T.; writing—original draft preparation, P.K. and N.S.; writing—review and editing, S.M.D., A.T.; supervision, S.M.D. and A.T.; project administration, A.T.; funding acquisition, H.P.S., A.M., A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the Defense Threat Reduction Agency (DTRA) Grant no. HDTRA1-21-1-0029.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- White, N.J., Melioidosis. Lancet, 2003. 361(9370): p. 1715-22.
- Wiersinga, W.J., et al., Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol, 2006. 4(4): p. 272-82. [CrossRef]
- Pongmala, K., et al., Distribution of Burkholderia pseudomallei within a 300-cm deep soil profile: implications for environmental sampling. Sci Rep, 2022. 12(1): p. 8674. [CrossRef]
- Limmathurotsakul, D., et al., Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat Microbiol, 2016. 1(1). [CrossRef]
- Elschner, M.C., et al., Isolation of the highly pathogenic and zoonotic agent Burkholderia pseudomallei from a pet green Iguana in Prague, Czech Republic. BMC Vet Res, 2014. 10: p. 283. [CrossRef]
- Limmathurotsakul, D., et al., Melioidosis in animals, Thailand, 2006-2010. Emerg Infect Dis, 2012. 18(2): p. 325-7. [CrossRef]
- Wiersinga, W.J., et al., Melioidosis. Nat Rev Dis Primers, 2018. 4: p. 17107.
- Schweizer, H.P., Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Future Microbiol, 2012. 7(12): p. 1389-99. [CrossRef]
- Crowe, A., et al., Current antimicrobial susceptibility of first-episode melioidosis Burkholderia pseudomallei isolates from the Northern Territory, Australia. Int J Antimicrob Agents, 2014. 44(2): p. 160-2. [CrossRef]
- Dance, D., Treatment and prophylaxis of melioidosis. Int J Antimicrob Agents, 2014. 43(4): p. 310-8.
- Dance, D.A., et al., Trimethoprim/sulfamethoxazole resistance in Burkholderia pseudomallei. Int J Antimicrob Agents, 2014. 44(4): p. 368-9. [CrossRef]
- Sullivan, R.P., et al., 2020 Review and revision of the 2015 Darwin melioidosis treatment guideline; paradigm drift not shift. PLoS Negl Trop Dis, 2020. 14(9): p. e0008659. [CrossRef]
- Bugrysheva, J.V., et al., Antibiotic Resistance Markers in Burkholderia pseudomallei Strain Bp1651 Identified by Genome Sequence Analysis. Antimicrob Agents Chemother, 2017. 61(6). [CrossRef]
- Sarovich, D.S., et al., Development of ceftazidime resistance in an acute Burkholderia pseudomallei infection. Infect Drug Resist, 2012. 5: p. 129-32. [CrossRef]
- Sommanustweechai, A., et al., Environmental management procedures following fatal melioidosis in a captive chimpanzee (Pan troglodytes). J Zoo Wildl Med, 2013. 44(2): p. 475-9. [CrossRef]
- Lim, Y.M., et al., Effective Therapeutic Options for Melioidosis: Antibiotics versus Phage Therapy. Pathogens, 2022. 12(1). [CrossRef]
- Wang, Y., et al., A novel lytic phage potentially effective for phage therapy against Burkholderia pseudomallei in the tropics. Infect Dis Poverty, 2022. 11(1): p. 87. [CrossRef]
- Gatedee, J., et al., Isolation and characterization of a novel podovirus which infects burkholderia pseudomallei. Virol J, 2011. 8: p. 366. [CrossRef]
- Yordpratum, U., et al., Novel lytic bacteriopphages from soil that lyse Burkholderia pseudomallei. FEMS Microbiol Lett, 2011. 314(1): p. 81-88. [CrossRef]
- Guang-Han, O., et al., Experimental Phage Therapy for Burkholderia pseudomallei Infection. PLoS One, 2016. 11(7): p. e0158213. [CrossRef]
- Leclerc, H. and P. Sureau, [Research on bacteriophages of the Whitmore bacillus in stagnant waters of Hanoi]. Bull Soc Pathol Exot Filiales, 1956. 49(5): p. 874-82.
- Khrongsee, P., et al., A comprehensive study of prophage islands in Burkholderia pseudomallei complex. Frontiers in Bacteriology, 2024. 3: p. 1339809. [CrossRef]
- DeShazer, D., Genomic diversity of Burkholderia pseudomallei clinical isolates: subtractive hybridization reveals a Burkholderia mallei-specific prophage in B. pseudomallei 1026b. J Bacteriol, 2004. 186(12): p. 3938-50. [CrossRef]
- Woods, D.E., et al., Burkholderia thailandensis E125 harbors a temperate bacteriophage specific for Burkholderia mallei. J Bacteriol, 2002. 184(14): p. 4003-17. [CrossRef]
- Tuanyok, A., et al., Genomic islands from five strains of Burkholderia pseudomallei. BMC Genomics, 2008. 9: p. 566. [CrossRef]
- Khrongsee P, K.J., Alami-Rose M, Subramaniam K, Waltzek TB, Schweizer HP, Tuanyok A, Exploring Burkholderia pseudomallei-specific bacteriophages: overcoming O-antigen specificity and adaptive mutation in phage tail fiber. Front. Bacteriol, 2024. 3:1433593 .
- Lopez, C.M., et al., Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl. Environ. Microbiol., 2009. 75: p. 6496-6503. [CrossRef]
- Dedrick, R.M., et al., Mycobacteriophage ZoeJ: A broad host-range close relative of mycobacteriophage TM4. Tuberculosis (Edinb), 2019. 115: p. 14-23. [CrossRef]
- Dedrick, R.M., et al., Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med, 2019. 25(5): p. 730-733. [CrossRef]
- Broussard, G.W., et al., Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell, 2013. 49(2): p. 237-48. [CrossRef]
- Shitrit, D., et al., Genetic engineering of marine cyanophages reveals integration but not lysogeny in T7-like cyanophages. Isme j, 2022. 16(2): p. 488-499. [CrossRef]
- Christie, G.E. and R. Calendar, Bacteriophage P2. Bacteriophage, 2016. 6(1): p. e1145782.
- Guarente, L., et al., Mutant lambda phage repressor with a specific defect in its positive control function. Proc Natl Acad Sci U S A, 1982. 79(7): p. 2236-9. [CrossRef]
- Yao, G., et al., Phage Milagro: a platform for engineering a broad host range virulent phage for Burkholderia. J Virol, 2023. 97(11): p. e0085023. [CrossRef]
- Propst, K.L., et al., A Burkholderia pseudomallei delta-purM mutant is a avirulent in immune competent and immune deficient animals: candidate strain for exclusion from select agent lists. Infection and Immunity, 2010. 78: p. 3136-3143.
- Norris, M.H., et al., An avirulent Burkholderia pseudomallei ∆purM strain with atypical type B LPS: expansion of the toolkit for biosafe studies of melioidosis. BMC Microbiol, 2017. 17(1): p. 132. [CrossRef]
- Camacho, C., et al., BLAST+: architecture and applications. BMC Bioinformatics, 2009. 10(1): p. 421.
- Quinlan, A.R. and I.M. Hall, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 2010. 26(6): p. 841-2. [CrossRef]
- Carver, T.J., et al., ACT: the Artemis Comparison Tool. Bioinformatics, 2005. 21(16): p. 3422-3. [CrossRef]
- Sullivan, M.J., N.K. Petty, and S.A. Beatson, Easyfig: a genome comparison visualizer. Bioinformatics, 2011. 27(7): p. 1009-10. [CrossRef]
- Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20. [CrossRef]
- Li, H., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013(preprint arXiv:1303.3997).
- Carver, T., et al., Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics, 2012. 28(4): p. 464-9. [CrossRef]
- Choi, K.H., A. Kumar, and H.P. Schweizer, A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods, 2006. 64(3): p. 391-7. [CrossRef]
- Lee, T.S., et al., BglBrick vectors and datasheets: A synthetic biology platform for gene expression. J Biol Eng, 2011. 5: p. 12. [CrossRef]
- Liljeruhm, J., et al., Engineering a palette of eukaryotic chromoproteins for bacterial synthetic biology. J Biol Eng, 2018. 12: p. 8. [CrossRef]
Figure 1.
Genomic comparison of temperate phage genomes or prophage DNA sequences associated with B. pseudomallei and B. thailandensis. Phage structural and late genes are color-coded in red, brown, and green, whereas the integrase gene is in purple. A) Myophages, φPK23, φBP82.2, and φBP82.3, are known to be associated with genomic island 15 (GI15), tRNA-Arginine (anticodon CCG) gene, and tRNA-Phenylalanine (anticodon GAA) gene, respectively. B) Siphophages, φBt-TXDOH and φBP82.1, are known to be associated with tRNA-Serine (anticodon GGA) gene and tRNA-Proline (anticodon UGG) gene, respectively. Phages or prophages sharing the same site-specific recombination (SSR) sites exhibit highly similar gene content across their genomes.
Figure 1.
Genomic comparison of temperate phage genomes or prophage DNA sequences associated with B. pseudomallei and B. thailandensis. Phage structural and late genes are color-coded in red, brown, and green, whereas the integrase gene is in purple. A) Myophages, φPK23, φBP82.2, and φBP82.3, are known to be associated with genomic island 15 (GI15), tRNA-Arginine (anticodon CCG) gene, and tRNA-Phenylalanine (anticodon GAA) gene, respectively. B) Siphophages, φBt-TXDOH and φBP82.1, are known to be associated with tRNA-Serine (anticodon GGA) gene and tRNA-Proline (anticodon UGG) gene, respectively. Phages or prophages sharing the same site-specific recombination (SSR) sites exhibit highly similar gene content across their genomes.
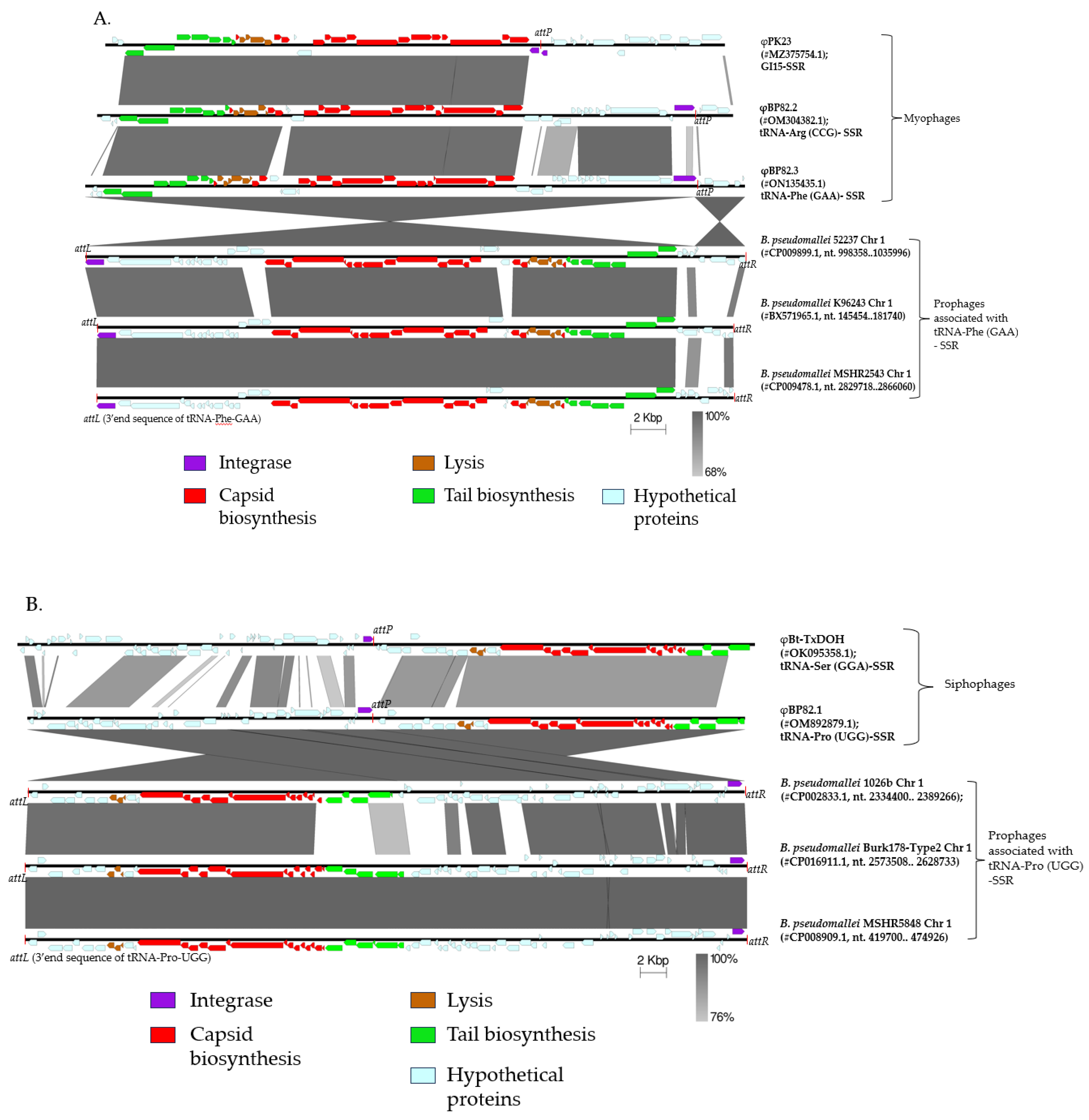
Figure 2.
Three prophages in B. pseudomallei 1026b genome, and the integration of a model phage φBP82.2 from its attenuated derivative strain Bp82 into the chromosome of B. pseudomallei 576mn, a susceptible host strain; A: showing the genomic locations of these prophages on chromosome 1 of B. pseudomallei 1026b, and B: depicting the prophage formation in B. pseudomallei 576mn following φBP82.2 infection. In panel B, the genomic comparison showed that φBP82.2 genome was integrated into the 3’end sequence of tRNA-Arg (anticodon CCG) gene through the site-specific recombination. The resulting prophage in 576mn was flanked by a short 45-bp repeat, identical to the 3’end sequence of tRNA-Arg gene, at both ends, termed attL and attR. This prophage in 576mn is identical to the prophage associated with the tRNA-Arg gene on chromosome 1 of B. pseudomallei 1026b.
Figure 2.
Three prophages in B. pseudomallei 1026b genome, and the integration of a model phage φBP82.2 from its attenuated derivative strain Bp82 into the chromosome of B. pseudomallei 576mn, a susceptible host strain; A: showing the genomic locations of these prophages on chromosome 1 of B. pseudomallei 1026b, and B: depicting the prophage formation in B. pseudomallei 576mn following φBP82.2 infection. In panel B, the genomic comparison showed that φBP82.2 genome was integrated into the 3’end sequence of tRNA-Arg (anticodon CCG) gene through the site-specific recombination. The resulting prophage in 576mn was flanked by a short 45-bp repeat, identical to the 3’end sequence of tRNA-Arg gene, at both ends, termed attL and attR. This prophage in 576mn is identical to the prophage associated with the tRNA-Arg gene on chromosome 1 of B. pseudomallei 1026b.
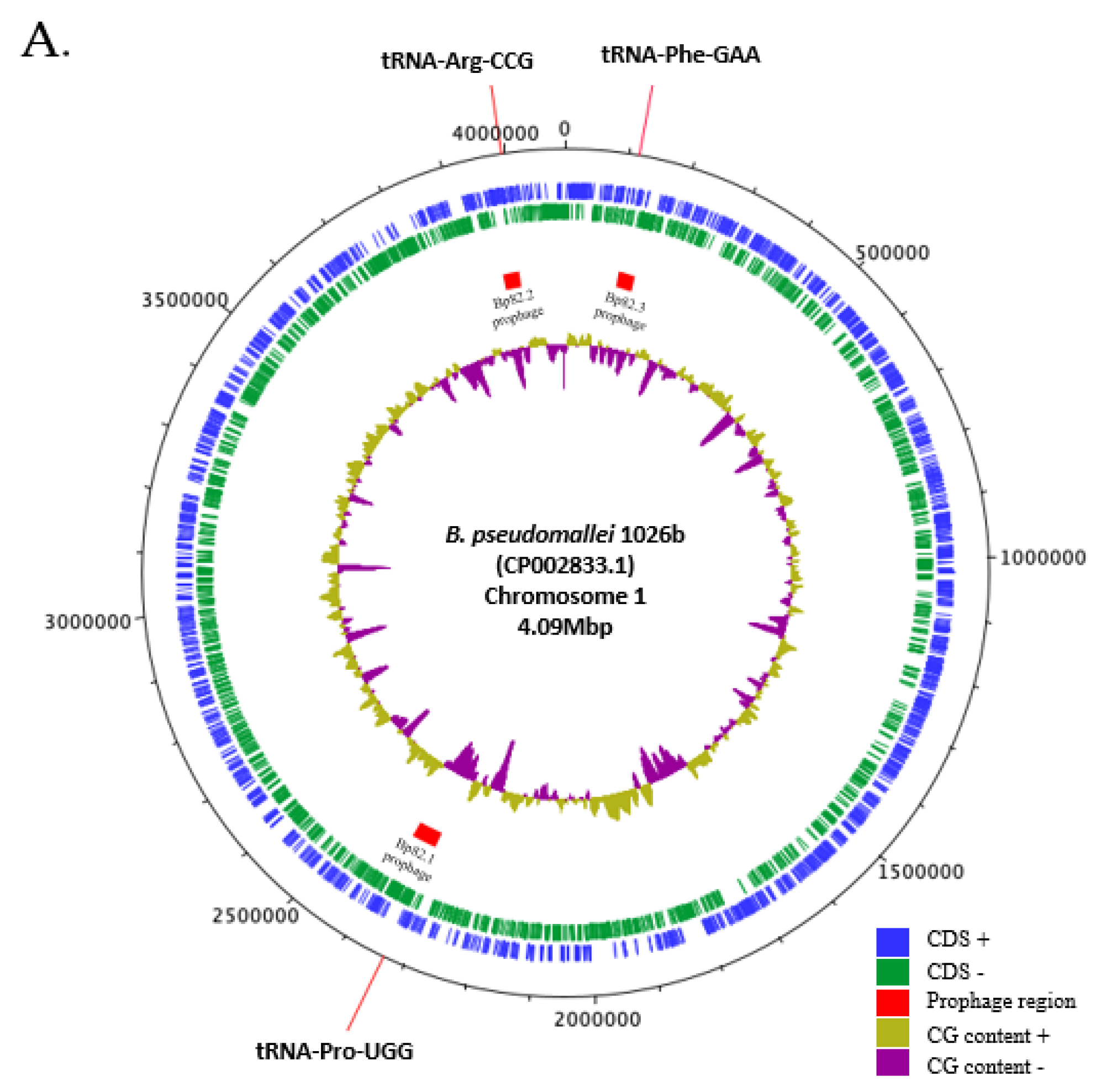
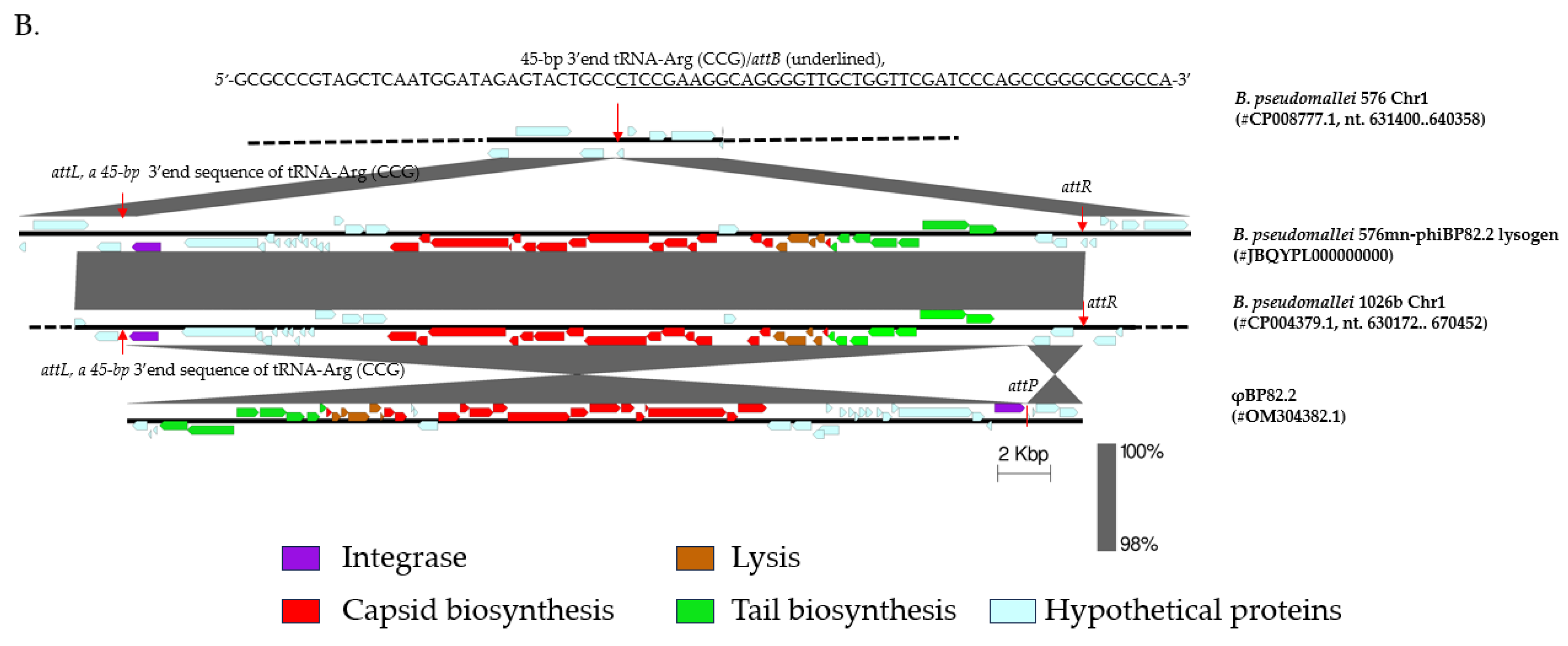
Figure 3.
Mapping of transcriptional sequencing reads against three temperate phage genomes of B. pseudomallei Bp82 to simplify the visualization of the expression of prophage genes. The RNA sequenced reads (in blue) were mapped to each temperate phage: A) φBP82.1, B) φBP82.2, and C) φBP82.3. Overall, most of the structural protein genes (color coded in red, green and brown,) of all three prophages were silent, while integrase genes (purple) and some hypothetical protein genes (cyan) were highly expressed in prophages associated with both myophages φBP82.2 and φBP82.3. We noted that the integrase gene in φBP82.1 prophage was not expressed. Interestingly, genes gp35-38 in φBP82.2 predicted as transcriptional repressor gene cassette were also highly expressed.
Figure 3.
Mapping of transcriptional sequencing reads against three temperate phage genomes of B. pseudomallei Bp82 to simplify the visualization of the expression of prophage genes. The RNA sequenced reads (in blue) were mapped to each temperate phage: A) φBP82.1, B) φBP82.2, and C) φBP82.3. Overall, most of the structural protein genes (color coded in red, green and brown,) of all three prophages were silent, while integrase genes (purple) and some hypothetical protein genes (cyan) were highly expressed in prophages associated with both myophages φBP82.2 and φBP82.3. We noted that the integrase gene in φBP82.1 prophage was not expressed. Interestingly, genes gp35-38 in φBP82.2 predicted as transcriptional repressor gene cassette were also highly expressed.
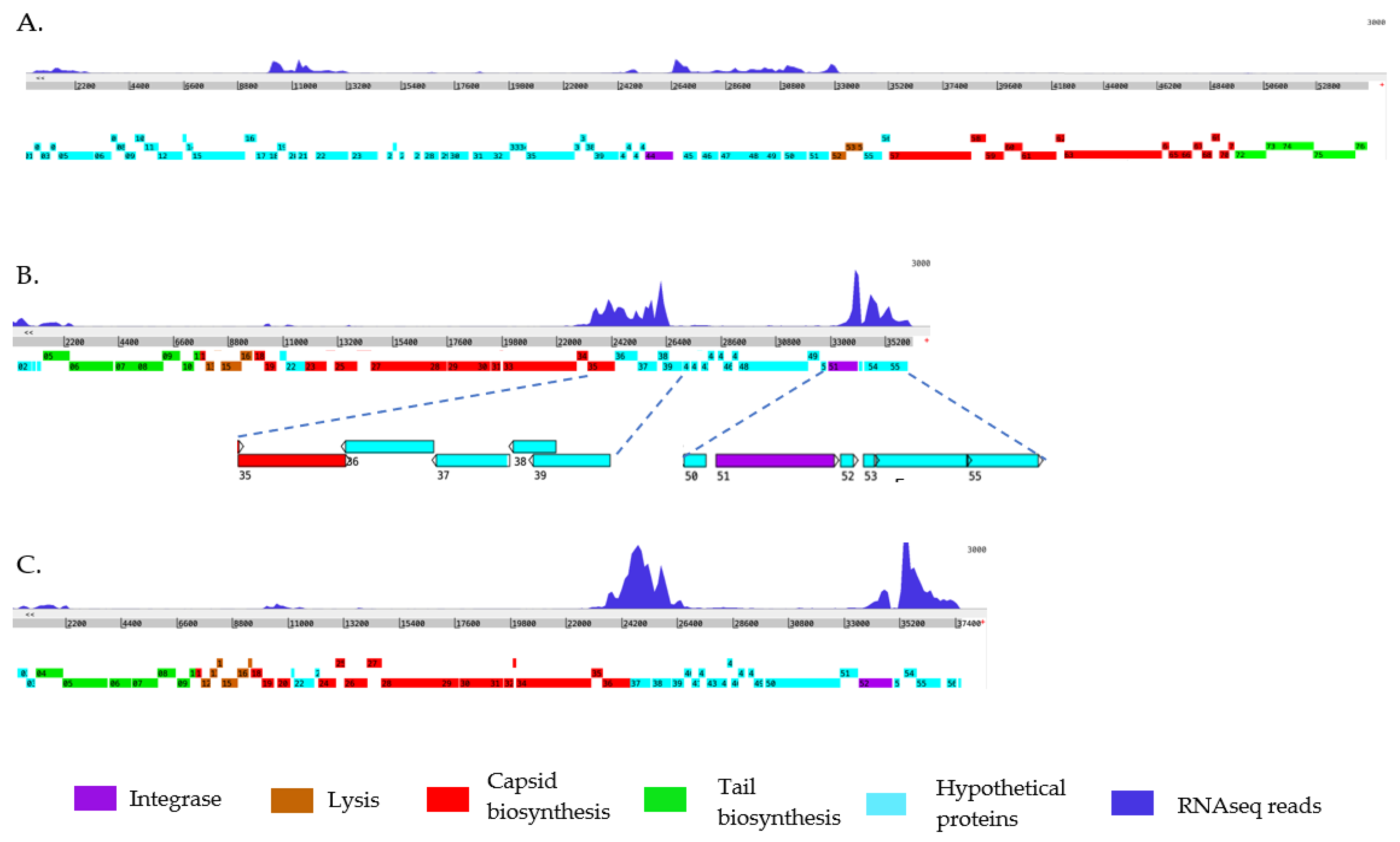
Figure 4.
Phenotypic characterization of B. pseudomallei 576mn-phiBP82.2 lysogen resulting from φBP82.2 integration: A), spot test results of φBP82.1, φBP82.2, φBP82.3, and φPK23 (control) against B. pseudomallei strains Bp82 (source strain), 576mn (a susceptible host), and 576mn-phiBP82.2 lysogen; B), plaques observed from 576mn-phiBP82.2 lysogen on 576mn lawn following spontaneous induction, while no plaques observed from its integrase gene (gp51, int) deleted mutant; C), killing efficacy of the φBP82.2Δint mutant (resulted from int-trans complementation), against 576mn with no improvement when compared with the wildtype phage φBP82.2. The experiment was performed in triplicates; D), whole genome sequencing of the φBP82.2 Δint confirmed the absence of its integrase gene.
Figure 4.
Phenotypic characterization of B. pseudomallei 576mn-phiBP82.2 lysogen resulting from φBP82.2 integration: A), spot test results of φBP82.1, φBP82.2, φBP82.3, and φPK23 (control) against B. pseudomallei strains Bp82 (source strain), 576mn (a susceptible host), and 576mn-phiBP82.2 lysogen; B), plaques observed from 576mn-phiBP82.2 lysogen on 576mn lawn following spontaneous induction, while no plaques observed from its integrase gene (gp51, int) deleted mutant; C), killing efficacy of the φBP82.2Δint mutant (resulted from int-trans complementation), against 576mn with no improvement when compared with the wildtype phage φBP82.2. The experiment was performed in triplicates; D), whole genome sequencing of the φBP82.2 Δint confirmed the absence of its integrase gene.
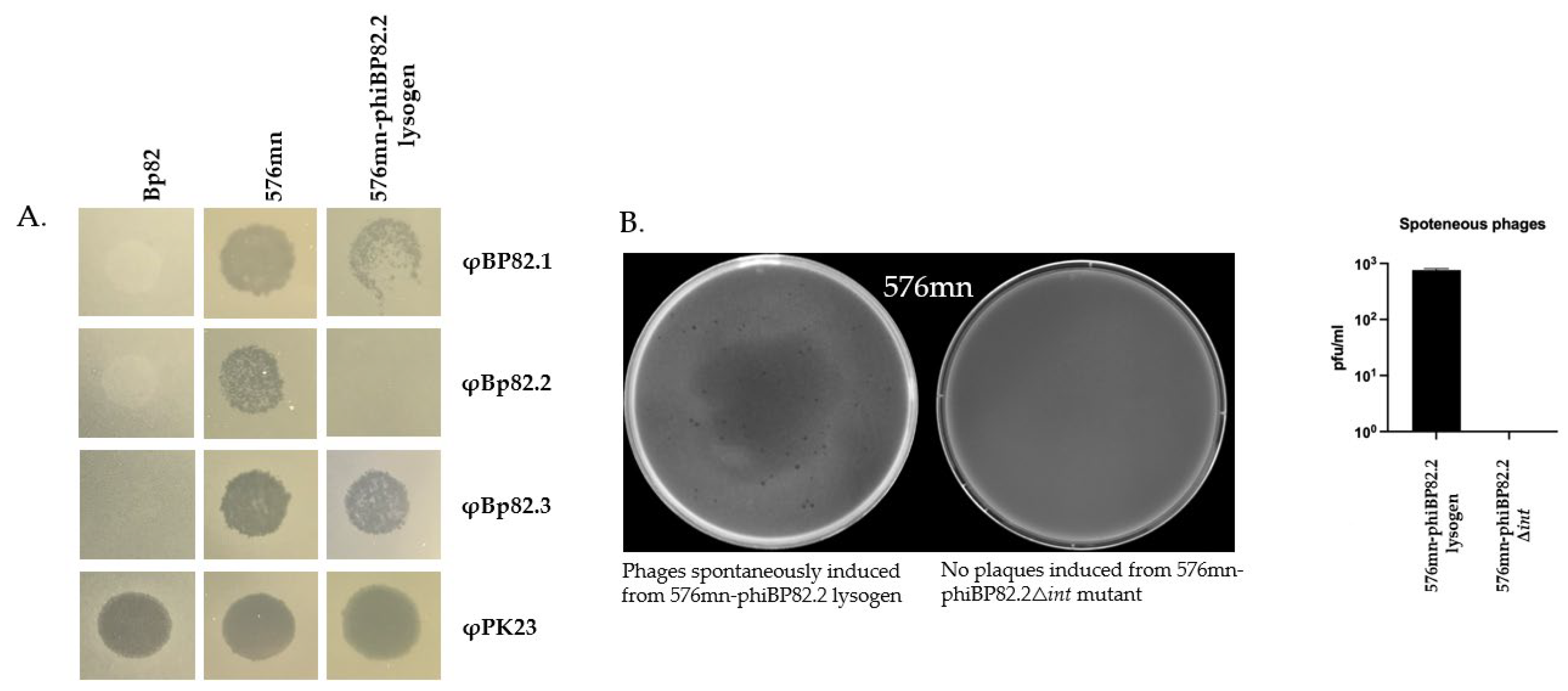
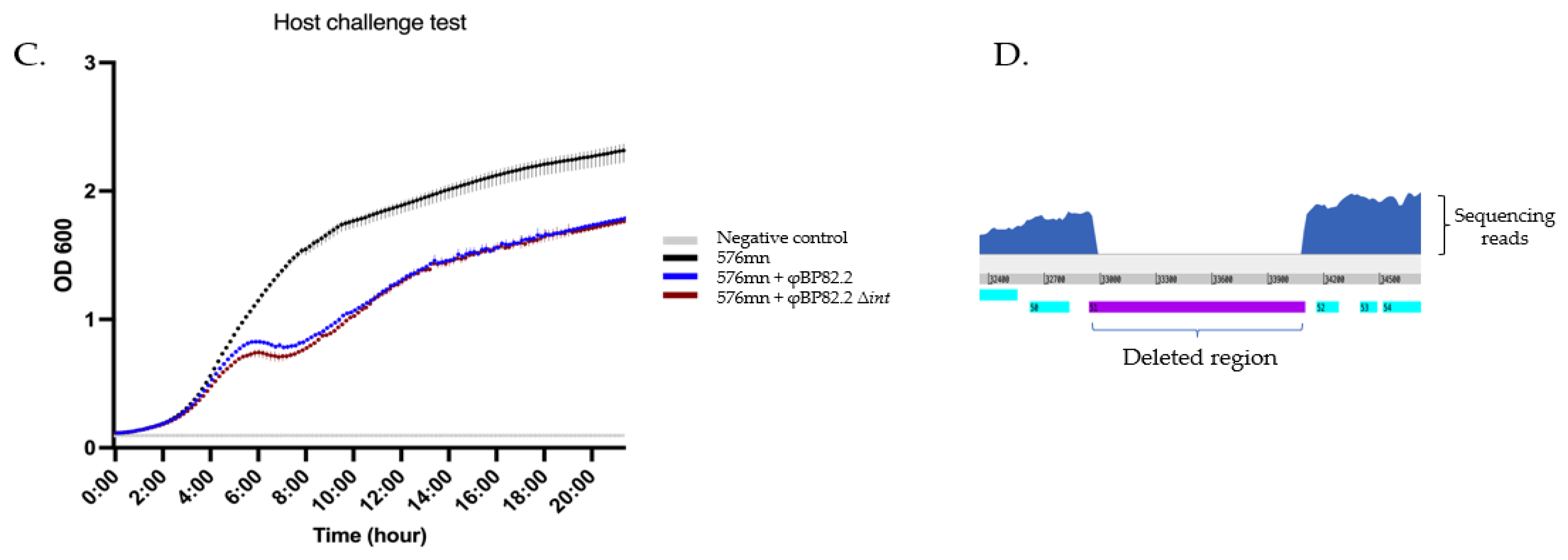
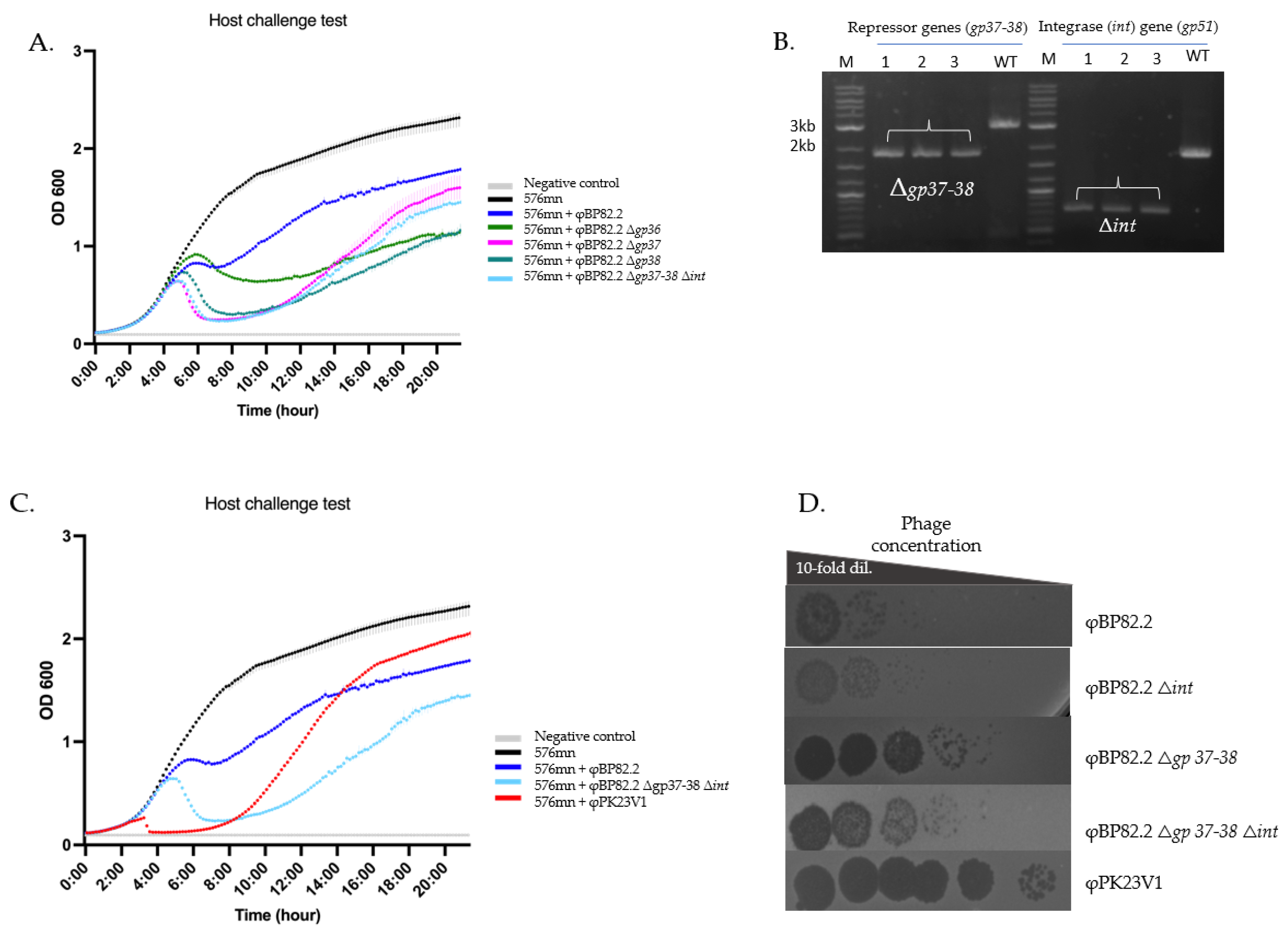
Figure 5.
Enhanced killing activity of mutant phages. This figure illustrates the phenotypic changes associated with repressor gene mutant phages. (A) Killing curve of the mutant phages, demonstrating that the deletion of gp36, gp37 and gp38 genes in φBp82.2 effectively enhanced phage lytic activity. We also evaluated a triple mutant, φBp82.2 △gp37-38△int, which showed a similar killing activity compared with the φBp82.2△gp 37 mutant. (B) PCR confirmed deletion of the hypothetical repressor genes gp37-38 and the integrase gene gp51. (C) Comparison of the killing activity of φBp82.2△gp 37-38△int mutant and φPK23V1, a highly B. pseudomallei-specific lytic-like phage. The mutant phage exhibited slower killing but better control of bacterial growth compared with φPK23V1. D) Spot test resulted from 10-fold serial dilution of phage preparations. Phage stocks were serially diluted 10-fold and 5 μL of each dilution was spotted onto bacterial lawns to estimate relative phage titers. Deletion of the integrase gene did not result in increased phage production, as indicated by similar clearing patterns across dilutions. In contrast, the gp37-38 gene-deleted mutant phages showed an approximately 2-log increase in their titters compared with the wildtype phage. Despite this increase, phage titers of the gp37-38 mutants remained lower than those of the φPK23V1 control.
Figure 5.
Enhanced killing activity of mutant phages. This figure illustrates the phenotypic changes associated with repressor gene mutant phages. (A) Killing curve of the mutant phages, demonstrating that the deletion of gp36, gp37 and gp38 genes in φBp82.2 effectively enhanced phage lytic activity. We also evaluated a triple mutant, φBp82.2 △gp37-38△int, which showed a similar killing activity compared with the φBp82.2△gp 37 mutant. (B) PCR confirmed deletion of the hypothetical repressor genes gp37-38 and the integrase gene gp51. (C) Comparison of the killing activity of φBp82.2△gp 37-38△int mutant and φPK23V1, a highly B. pseudomallei-specific lytic-like phage. The mutant phage exhibited slower killing but better control of bacterial growth compared with φPK23V1. D) Spot test resulted from 10-fold serial dilution of phage preparations. Phage stocks were serially diluted 10-fold and 5 μL of each dilution was spotted onto bacterial lawns to estimate relative phage titers. Deletion of the integrase gene did not result in increased phage production, as indicated by similar clearing patterns across dilutions. In contrast, the gp37-38 gene-deleted mutant phages showed an approximately 2-log increase in their titters compared with the wildtype phage. Despite this increase, phage titers of the gp37-38 mutants remained lower than those of the φPK23V1 control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



