
Submitted:
22 February 2026
Posted:
25 February 2026
You are already at the latest version
Abstract
Advances in technology have provided a better understanding of the genetic basis of neurodegenerative disorders and their underlying molecular pathophysiology. However, treating these disorders with conventional strategies is a major challenge. The approval of gene-targeted therapy for spinal muscular atrophy (SMA) has laid the foundation for developing therapies for other neurodegenerative disorders. Highly personalized gene therapy trials have been reported. As intensive research and efforts to advance gene-targeted therapies continue, this review provides an overview of viral and non-viral vectors and delivery methods, as well as treatment strategies, including gene addition, replacement, editing, silencing, and splice modulation. Gene-targeted approaches and clinical trials for SMA and amyotrophic lateral sclerosis (ALS) have demonstrated success, and additional studies are in progress. The design of efficient clinical trials which facilitate successful translation into clinical practice is of critical importance. Key considerations include the selection of appropriate disease models, understanding the natural history of the disease, and establishing well-defined outcome measures to assess prognosis of the disease and therapeutic efficacy. Finally, the precision of CRISPR-gene editing may facilitate the development of new therapies.
Keywords:
gene therapy
; spinal muscular atrophy
; amyotrophic lateral sclerosis
; clinical trial readiness
1. Introduction
Neurodegenerative diseases (NDDs) refer to a group of disorders that cause progressive and irreversible loss of neurons in the nervous system. They affect millions of people worldwide and are considered one of the leading causes of death and disability [1]. The complexity of the nervous system and lack of relevant disease models have limited our understanding of the underlying disease pathophysiology in NDDs. Treating these disorders has always been a major challenge with conventional therapies due to the heterogeneous and complex nature of the nervous system, its slow regeneration capacity, need for repeated dosing and poor accessibility of the tissue due to the blood brain barrier. As a result, there is no cure or disease-modifying therapy for many of these disorders. Nonetheless, advances in technology have provided a better understanding of the disease mechanism and subsequent advancement of gene-targeted therapies for several NDDs.
Gene therapy is defined by FDA as a technique that modifies a person’s genes to treat or cure disease by replacing a disease-causing gene with a healthy copy of the gene, inactivating a disease-causing gene that is not functioning properly or introducing a new or modified gene into the body to help treat a disease [2]. Gene-targeted therapy studies have gained more interest due to advances in gene delivery and tools for gene manipulation, which has resulted in an acceleration of the current pipeline in recent years (Figure 1). To date several gene therapy products have been approved, mostly for cancer. The pace of development for NDDs has increased, especially after the approval of the first gene-targeted therapy for spinal muscular atrophy (SMA). Encouraging advances in personalized gene therapy studies have already been reported. Considering this progress, this review will discuss gene-targeted therapy strategies for NDDs, focusing on their clinical applications in SMA and amyotrophic lateral sclerosis (ALS), and the importance of clinical trial readiness.
2. Delivery Methods, Vectors, and Strategies for Gene-Targeted Therapies
In a broad sense, two therapeutic approaches can be implemented to introduce genetic material into the cells: in vivo and ex vivo (Figure 2). During in vivo gene therapy, genetic material is modified inside the body. With ex vivo gene therapy, cells are harvested from a patient (autologous) or a donor (allogenic) and then reintroduced to the patient after gene modification in culture. This approach is mostly implemented in blood-related disorders, and is not suitable for many cell types that cannot be isolated from the body or that are not able to survive ex vivo for a long time [3,4]. Gene therapy’s progress to clinical reality has been accomplished by addressing vector choice and design, route of delivery, dosing and immune response of the patient and scalability. More research is needed to optimize the technology and extend its use for various monogenic or multifactorial disorders.
Delivery of therapeutic nucleic acid to the target site requires vectors to overcome a series of obstacles including of the rapid clearance of genetic material by systemic endonucleases, the lack of tissue-specific distribution and low efficiency of cellular uptake [5]. Immune system activation is another reason to choose a suitable delivery vector approach. Viral and non-viral vectors are used to deliver genetic material to the target cells or tissue. Each vector has advantages and disadvantages, and several points needs to be considered when choosing a suitable gene delivery vector including; 1) type and packing size of the genetic material, 2) the expression efficiency of the gene, 3) duration of therapy, 4) cytotoxicity and immunogenicity of the vector, 5) feasibility of vector production, 6) route of administration and targeted cell type, and 7) previous infection with the same virus chosen as vector [6,7,8].
The majority of clinical trials (70%) have been conducted with viral vectors [6]. Viruses have long been used as vectors for gene delivery, aiming to treat or prevent neurodegenerative, muscular, cardiovascular, ophthalmological, and hematological diseases as well as cancer [9]. Adenovirus, adeno-associated virus (AAV), and retroviruses, including lentiviruses are the most widely used vectors in clinical trials. Viral vectors are composed of three basic elements, including the transgene itself, the regulatory sequences that provide the expression and stability of the transgene such as the promoter, enhancer and poly-A regions, and the surrounding capsid [10]. Structural differences provide the opportunity for their preferred use in specific applications. Adenovirus (Ad) has a double-stranded linear DNA genome, containing early and late phase genes within “inverted terminal repeats” (ITRs), and is surrounded by a capsid. Different vectors are designed by deleting some or most of the genomic sequences to permit space for the transgene insertion. For instance, helper-dependent Ad vectors only contain ITRs and cis-packaging signals, therefore they can accommodate 36 kb of exogenous DNA sequences [11]. Ad vectors have broad tropism and high transduction efficiency that enable infection of both dividing and quiescent cells. However, the presence of neutralizing antibodies from previous infections and strong induction of immune response by Ads reduces their transduction efficiencies. To improve their clinical use, adenovirus serotypes can be genetically manipulated to reduce immunogenicity and increase transduction efficiency [10]. In 1965, a defective virus was identified in Ad preparations, called adeno-associated viruses (AAVs) [12]. Today, AAVs are the main viral vectors preferred for in vivo gene therapy applications due to their broad tropisms, low immunogenicity, non-pathogenicity, rare genome integration potential and episomally durable transgene expression [9,13,14]. AAVs have a 4.7 kb single stranded linear DNA genome, containing ITRs at both ends. Between ITRs, there are cap and rep genes for both replication and capsid synthesis. Vectors are designed by replacing both cap and rep genes with the transgene and tissue/cell specific or ubiquitous promoter, enhancer, 3’UTR region including regulatory elements and a poly-A tail [16]. There are at least twelve natural AAV serotypes and hundreds of variants have been identified [9,13,17]. A specific AAV serotype can be selected to bind the desired target receptor and infect the cells of interest [9,17]. For example, AAV9, a clinically approved serotype vector delivering the SMN1 gene for spinal muscular atrophy (SMA) treatment, can infect any tissue including brain by binding galactose as the primary receptor and laminin receptor 1 (LamR) and AAV receptor, as co-receptors [9]. After binding to their receptors, vectors are internalized via endocytosis and migrate through the trans-Golgi network towards the nucleus, and escape endosomal vesicle entry before nuclear import. The capsid releases its single-stranded genome (ssDNA) in the nucleus and replication occurs to generate double-stranded DNA (dsDNA) that remains mostly episomal [15]. Nonetheless, a better understanding of the mechanisms of intracellular trafficking of different AAV serotypes would help to develop more specific and potent vectors [16]. In this regard, self-complementary AAVs (scAAVs) were generated by mutating one of the ITRs to by-pass the necessity of ssDNA to dsDNA conversion. As a result, transduction efficiency is increased, although packaging capacity of the scAAVs is reduced to <2.5 kb from 4.7 kb [17]. scAAVs have successfully been translated into the clinic, as demonstrated by the use of scAAV9 vector in the treatment of SMA, which is the first neurological disease approved for AAV-based gene therapy. New vectors are designed to increase cell/tissue-type specificity, transduction efficiency, and reduce immune response via modification of either capsid or other elements, including the transgene cassette, promoter, enhancer or regulatory sequences [18].
In addition to AAVs, retroviruses are also used as vectors in the clinic for the treatment of different diseases. Retroviruses are enveloped viruses, having a single stranded RNA genome. Lentiviruses belong to the retroviridae family of retroviruses, capable of transducing both dividing and post-mitotic cells, including neurons. Lentiviruses have structural (gag, pol and env), regulatory (tat and rev), and auxiliary (Vpu, Vpr, Vip, Nef) genes in regions between long terminal repeats (LTRs) [10]. Vectors generated from lentiviruses lack required elements for viral replication and gene expression; therefore, they can deliver exogenous DNA almost 10 kb in size [19]. Vector tropism can be altered by modifying envelope proteins. Due to their host genome-integrating ability, lentiviral and retroviral vectors are preferred for ex vivo gene therapy to provide long-term transgene expression, especially in hematopoietic stem cells and T cells. Currently, these vectors are used in CAR-T cell therapies for the treatment of blood cancers [9]. Lentiviral vectors prefer transcriptionally active sites to integrate, therefore increasing the risk of insertional mutagenesis. To overcome this issue, non-integrating lentiviral vectors have also been developed [9].
Although recent advancements in viral vector design and manufacturing have improved, there are several issues to be addressed starting with cytotoxicity and immunogenicity. Insertional mutagenesis is a phenomenon that raises concerns of malignancy which can result from oncogene activation or tumor suppressor gene disruption during viral vector integration into the host genome. Non-viral vectors, therefore, have gained significant attention recently, after being ignored for years since they have poor delivery efficiency and transient transgene expression. Non-viral vectors have reduced cytotoxicity, immunogenicity and mutagenesis risk, flexible insert size of DNA, cost-effectiveness, ease of largescale production, and have become an attractive area of gene therapy research [20,21].
The non-viral vectors are classified as naked DNA, nanoparticle based and chemical-based vectors. Small nucleic acid molecules (antisense oligonucleotides-ASOs, DNAi, RNAi, miRNAs mimics, aptamers and CpG oligodeoxynucleotides) and large nucleic acid molecules (plasmid DNA and mRNA) can be transferred with non-viral vectors by chemical and physical methods [20,22]. Chemical methods use synthetic or natural biodegradable particles such as inorganic particles, lipids, peptides and polymers to transfer nucleic acids to the cell. This delivery method is less destructive and can be used in vivo; however, it is less efficient than physical methods [23]. Physical methods include a variety of methods to introduce therapeutic nucleic acids into the cell. While needle injections and ballistic DNA injections introduce genetic materials directly into the cell or tissue, electrical pulse (electroporation), sound wave (sonoporation), hydrodynamics (hydroporation), laser pulse (photoporation) and magnetic fields (magnetofection) are used to permeabilize the cell membrane to allow nucleic acid entry [20,22]. Although high transfection efficiency can be achieved with physical methods, it has some drawbacks including high cytotoxicity and difficulty of in vivo application. There are some challenges to overcome including gene transfer efficiency, extracellular stability, internalization, intracellular trafficking, nuclear entry, specificity, gene expression duration, and safety [20,24]. In order to achieve efficient and safer gene targeted therapies, both viral and non-viral vector design and delivery methods need further investigation and improvement.
3. Gene-Targeted Therapy Approaches for Neurodegenerative Diseases
Depending on the causative defect, gene-targeted therapy strategies can include gene addition, gene replacement, gene editing, gene silencing and splice modulation therapies (Figure 2). These strategies can only be implemented in somatic cells as germline gene studies are not approved.
3.1. Gene Addition and Replacement
Understanding the genetic basis of diseases and the underlying molecular pathophysiology has facilitated the development of novel gene addition therapies. Gene addition strategies include delivery of a functional copy of a faulty gene that could restore gene function in the context of monogenic disorders. In this case it is usually described as gene replacement. Furthermore, it is plausible to introduce a new gene targeting molecular mechanisms driving neurodegeneration or neuronal survival to compensate for the defective gene or to improve the disease phenotype. This approach is also called gene overexpression and could be applied for monogenic diseases or disorders with complex etiology. Exogenous genetic material could be delivered locally or systemically via viral or non-viral vectors. The advantage of the gene addition approach is that the therapeutic protein can be continuously expressed in nondividing cells, potentially enabling one-time therapy. Nonetheless, there are several challenges for gene addition therapies to overcome, including but not limited to effective and safe delivery methods, off-target effects, cost and accessibility issues.
3.2. Gene Editing
strategies allow for specific changes in the nucleotide sequence of the genome with the help of programmable nucleases [25]. Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are the earliest gene editors preceding the CRISPR-Cas systems [26,27]. Two elements give ZFNs and TALENs the ability to modify specific regions of the genome: endonuclease Fok I and specially designed amino acids that guide the nuclease to the desired DNA domain. When the targeted DNA sequence is cleaved by Fok I nuclease, the endogenous dsDNA break repair mechanisms are activated, enabling gene disruption or precise alterations on this particular site [28,29]. Instead of synthetic peptides, the CRISPR-Cas system directs the nuclease domain to the targeted region by guide RNA sequences [30]. The CRISPR-Cas system is a natural adaptive immune system mechanism developed against bacteriophage infections and mobile genetic elements in bacteria and archaea [30,31,32]. The discovery of the potential of this system as a genome editing tool has resulted in unprecedented avenues for many research areas from disease modeling to gene therapy [30,33]. In principle, the guide RNA (gRNA) directs the Cas nuclease to the target genomic region, where the Cas protein introduces a double-stranded break in the DNA sequence complementary to the gRNA. Double strand DNA (dsDNA) break repair activates either non-homologous end-joining (NHEJ) or homologous recombination (HR) repair mechanisms [34]. The error prone NHEJ pathway causes frameshift or premature stop codon formation with insertions/deletions that occur during repair, providing a mechanism to generate loss of function models of the target gene. In the HR mechanism, it is possible to repair the DNA precisely when an exogenous template sequence is introduced. In this way it is possible to perform genome editing during repair by giving a donor plasmid containing the desired nucleotide sequence (such as insertion, deletion, point mutation) to the cell. Over time, different versions of the CRISPR technique have emerged through the creation of Cas9 protein variants to overcome the limitations of dsDNA break dependance, efficiency and specificity issues. A catalytically inactive Cas9 protein (deadCas9 or dCas9) can be generated by engineering Cas9 with nuclease-inactivating D10A and H840A mutations. Not able to create dsDNA breaks, dCas9 can be directed to the specific target sites to regulate gene expression at the transcriptional level enabling CRISPR interference and CRISPR activation [37,38]. Retaining only the D10A mutation, researchers generated the Cas9 nickase (nCas9) enzyme and fused the protein with a deaminase enzyme to create CRISPR base editors [39]. nCas9 is directed to the genomic target by a single gRNA (sgRNA) and when nCas9-sgRNA-DNA complex is formed, a strech of single strand DNA (ssDNA) becomes a substrate for a deaminase reaction and base conversion [33]. The resulting mismatch and the nick introduced in the non-edited strand activate the cell’s DNA repair machinery, which corrects the non-edited strand using the edited strand as a template with enhanced efficiency and precision [35,36,40]. Prime editing is a more recent technology that was developed by fusing nCas9 (H840A) to a reverse transcriptase [41,42]. Prime editing gRNA (pegRNA) comprises of a spacer sequence to direct the nCas9 to the target strand, a reverse trancriptase template encoding desired edits to the genome and a primer binding site. When nCas9-pegRNA complex binds to the target region, nCas9 creates a nick on the non—target strand and the exposed 3’ -hyroxyl group anneals to the primer binding site on the 3’ end of pegRNA. Subsequently, reverse transcriptase fused to nCas9 transcribes the reverse transcriptase template on pegRNA. Unedited 5’ flap is removed and edited 3’ flap hybridizes with the unedited strand resulting in heteroduplex formation. DNA mismatch repair then resolves the heteroduplex using the edited strand as a template, allowing incorporation of the desired edits into the genome. While base editors can perform a limited number of modifications to the genome, prime editing can introduce various type of modifications including base substitutions, insertions and deletions. Developments in base and prime editing technologies continue to improve the efficiency and precision of the editing technologies and hold therapeutic promises for several disorders [43].
3.3. Gene Silencing
is used for knockdown of a gene that is not functioning properly. Knockdown of gene expression can be achieved through oligonucleotide-based therapeutics, including gapmer ASOs, small interfering RNA (siRNA) or microRNA (miRNA) molecules. In recent years substantial progress has been made in development of ASOs. ASOs are synthetic single strand oligonucleotides that target mRNA molecules by sequence specific Watson-Crick base-pairing to cause either splice modulation or transcript knockdown [44]. Knocking down the mRNA transcripts can be accomplished by gapmers. Gapmers are typically 15-22 base pair long single stranded ASOs that have a core DNA sequence complementary to target RNA (pre-mRNAs or mRNAs) and flanking sugar modified RNA nucleotides at each end [45]. Gapmers are internalized by endocytosis and hybridize with RNA molecules in the nucleus or cytoplasm [46]. Formation of a DNA-RNA heteroduplex activates endonuclease ribonuclease RNaseH1 and induces cleavage of target RNA degradation, and release of the gapmer to bind to another target RNA [45,47,48]. siRNAs (20-25 base pairs) or miRNA mimics (~22 base pairs) are synthetic non-coding RNA molecules can be used to regulate gene expression post-transcriptionally by inducing RNA interference. After introducing synthetic double-stranded siRNA and miRNA molecules, they are incorporated into the RNA-induced silencing complex (RISC). The sense strand of siRNA is degraded by argonaute 2 protein, while with miRNA it is unwound and released from the complex. The antisense strand guides the RISC complex to the complementary target mRNA, leading to either cleavage of target mRNA or interruption of translation. An siRNA perfectly matches to one specific target mRNA and causes mRNA degradation, while a miRNA can target multiple genes at the same time and can be perfectly or partially complementary to the target mRNA. The degree of the miRNA complementarity to its target mRNA will determine the fate of the mRNA. Gapmer ASOs, siRNA and miRNA-mediated gene knockdown approaches can be considered as a therapy for gain of function mutations if the resulting loss of function due to therapy causes no additional harm to the patient. This approach could also be useful for disorders caused by a dominant negative mechanism of action. Dominant-negative effect occurs when the mutant gene product interferes with the wild type gene product. Knock down of the mutant gene expression could be a strategy to eliminate the dominant negative effect [49]. ASOs can be administered naked, whereas siRNAs and miRNAs require viral vectors or non-viral carriers for delivery [50,51,52]. To treat neurodegenerative disorders these oligonucleotides should reach the central nervous system (CNS), but they cannot cross the blood brain barrier due to their size and negative charge. Although intrathecal or intracerebroventricular administration has been done successfully, several approaches are currently being developed to achieve effective and noninvasive delivery of oligonucleotides to the CNS. ASOs have been subjected to combinations of chemical modifications on the backbone (phosphorothioate bond) or sugar residues (e.g. 2′ -O-methyl, 2′-O-methoxyethyl, 2′-fluoro, locked nucleic acid and constrained ethyl modifications) to confer resistance to nucleases and to increase stability, binding affinity to RNA, tissue uptake, potency, solubility, and reduced toxicity [53,54,55,56]. siRNAs and miRNAs can have comparable chemical modifications to ASOs including modifications that would help to ensure correct loading on to RISC, and efficient strand selection to unwind and discard the sense strand [57,58]. Recently developed divalent siRNAs, which are composed of two fully chemically modified, phosphorothioate-containing siRNAs connected by a linker, have shown promise for treating neurological disorders due to their potency, enhanced distribution in brain regions and long-lasting gene silencing effects with minimal toxicity [59]. Ongoing research in oligonucleotide chemistry and delivery methods holds promise to overcome the current limitations of oligonucleotide therapies.
3.4. Splice Modulating
In addition to their degrading mechanisms, ASOs can act as steric blockers to upregulate or downregulate gene expression. Splice modulating ASOs can alter pre-mRNA splicing by binding to splice sites, enhancers, or silencer sequences and result in exon skipping or inclusion. In addition, they can alter mRNA stability by changing polyadenylation site selection and inhibit translation by sterically blocking ribosomal subunits and RNA binding proteins. Also, they can increase the translation of a protein from its main open reading frame (ORF) by blocking the upstream open reading frame (uORF) within the 5’UTR [53,60,61]. These mechanisms allow splice modulation therapies to restore the reading frame to increase protein levels or modify the transcript to synthesize a partially functional protein where the disease mechanism is loss of function [49]. By masking splicing regulatory elements from the spliceosome machinery, they can also disrupt reading frames to decrease protein levels when the disease mechanism is from a gain of function. In the case of haploinsufficiency or dominant negative effect, upregulating the expression of the wild type allele is also possible [60]. Additional new ASO strategies will be developed as our understanding of ASO mechanisms continues to expand. Understanding disease mechanisms is crucial for developing an ASO strategy to use for treatment [49].
4. Current Gene-Targeted Therapies in Neurodegenerative Disorders: SMA and ALS
4.1. Spinal Muscular Atrophy (SMA)
Spinal muscular atrophy (SMA) is a rare inherited disease primarily affecting children. Survival of motor neuron 1 (SMN1) gene mutations, especially deletions, are responsible for SMA. Almost 95% of the patients have homozygous deletions in exon 7 or both exons 7 and 8; however, the clinical phenotype is substantially different among patients who have the same mutation. Historically, SMA is classified into 5 groups according to age of disease onset and achieved motor functions. Type 0 is the most severe form of the disease, which begins prenatally and survival is generally few days to weeks. Alternatively, symptoms can occur after the age of 30 in type 4 patients with a normal life expectancy [62]. SMN1 is not the sole gene that encodes survival motor neuron (SMN) protein in the human genome. The nearly identical copy of SMN1, namely SMN2, exists in multiple copies ranging from 1 to 6. However, due to C to T transition, splicing of SMN2-encoded mRNAs is defective, and almost 90% of transcripts lack exon 7. Therefore, a minor fraction of the transcripts are available for translation of full-length and functional SMN protein. The increased copy number of SMN2 allows for the synthesis of more full-length SMN protein; therefore, higher SMN2 copy numbers are associated with milder phenotypes. However, due to the inequivalence of SMN2 copies, clinical severity does not absolutely correlate with SMN2 copy numbers [63]. SMN2 is the primary target of two of the three approved treatments for SMA.
Deficiency of SMN causes several cellular perturbations that lead to the progressive degeneration of alpha motor neurons in the spinal cord and results in symmetrical muscle weakness and atrophy. Moreover, non-neuronal tissues, including skeletal and cardiac muscle and kidney, have also been reported to be affected by the loss of function, and SMA is considered a multisystem disease [64,65,66]. SMN functions in different cellular processes, including snRNA biogenesis, endocytosis, translation, and cytoskeleton regulation; therefore, loss of SMN impairs multiple molecular mechanisms that are currently under investigation [67,68,69,70,71,72]. Restoration of SMN-related perturbations by enhancing SMN protein level have been extensively studied in both pre-clinical and clinical studies [73,74,75]. At the end of these efforts, the first success came from studies with an ASO, namely Nusinersen (Spinraza®). Nusinersen was approved in 2016 by the FDA and then by the European Medicines Agency (EMA) and is now currently available for the treatment of all types of SMA. Nusinersen is delivered intrathecally to reach motor neurons, which are the most sensitive cell types affected by SMN deficiency. The ASO targets SMN2-encoded pre-mRNAs, aiming to restore the inclusion of exon 7. Nusinersen is an 18-mer long oligonucleotide, which has backbone modifications of 2‘-O-2-methoxyethyl phosphorothioate to provide protection from nucleases [76]. The ASO enters the nucleus and binds to the intronic splice silencer N1 (ISS-N1) sequence, located in the immediate downstream 5’ splice site of the intron 7 SMN2-encoded mRNA. Thereby, it prevents the binding of heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and leads to the inclusion of exon 7 (Figure 3). To date, nusinersen has been used in more than 14,000 SMA patients to enhance cellular SMN protein levels [76,77,78,79].
In 2019, a second therapeutic approach also gained approval from the FDA, namely onasemnogene abeparvovec (Zolgensma®), and more than 4000 SMA patients have received this therapy [80]. Onasemnogene abeparvovec is a gene replacement therapy, aiming to enhance SMN protein level by AAV9-mediated SMN1 gene delivery. SMN cDNA, together with the human cytomegalovirus enhancer and chicken beta actin promoter, is packed into a non-replicating self-complementary adeno-associated virus serotype 9 (scAAV9) capsid. The journey of AAV vectors starts with cellular internalization via receptor-mediated endocytosis. After release from the endosome, it passes through the nuclear pores and is uncoated. In the nucleus, single-stranded DNA forms a double strand via complementarity at inverted terminal repeats, thus allowing for transcription to occur. mRNAs leave the nucleus, and the target protein is translated in the cytoplasm [9,75]. Onasemnogene abeparvovec can increase the level of functional SMN protein in both neuronal and peripheral tissues due to its distribution by intravenous administration [81].
To correct SMN2 splicing, small molecules have also been studied extensively. The approval for SMA therapy came with risdiplam (Evrysdi®) in 2020, and its tablet form was subsequently approved in 2025 [73,82]. Several analogues have been shown to modify SMN2 splicing in pre-clinical studies, in fact, the mechanism of action has been demonstrated for its analogues but not for risdiplam itself at the time. It has been reported that the SMN-C3 analogue interacts with the AG-rich motif within exon 7, while analogue SMN-C5 stabilizes U1 snRNP on the 5′ splice site. A recent in vitro study with risdiplam demonstrated the effects of an AC-containing motif in exon 7 on exon inclusion and proposed a mechanism for its mechanism of action [83]. The advantage of risdiplam over nusinersen is oral delivery, thereby reaching not only nervous system but also peripheral tissues. Off-target effects are the major drawbacks that have been recently reported in vitro [76,83,84,85]. Considering their complex mechanisms of action and off-target effects, additional studies are needed to develop more specific splicing-modulator molecules. Effects of risdiplam on patients previously treated with the aforementioned therapies was recently reported [86]. The effects of risdiplam in combination with onasemnogene abeparvovec, as well as switching from nusinersen to risdiplam have been investigated [87,88].
Translational medicine has helped to develop therapies for SMA, starting from bench to the bedside. However, this is not the end, since further clinical and molecular investigations are needed to understand the effects of these therapies on cellular mechanisms and implications for improving patient health. Clinical studies of ASOs (NCT05067790 for a higher dose of nusinersen on patients who were treated with risdiplam; NCT05575011 for another ASO, BIIB115) and gene therapy (NCT05335876 for OAV101) approaches are ongoing for SMA [89].
4.2. ALS
ALS is a fatal neurodegenerative disorder that results from progressive motor neuron degeneration in the brain, brainstem, and spinal cord [90]. Resulting muscle weakness and paralysis lead to respiratory failure and death within 2-4 years from diagnosis [91,92]. The worldwide incidence of ALS is 2/100,000, and the mean age of adult-onset ALS varies between 40-63 years [93,94,95].
ALS is classified either as familial (fALS; 10% of the cases) or sporadic (sALS; 90% of the cases); however, this classification overlooks the complex genetics that underlie ALS pathophysiology [96]. More than 40 genes have been associated with the disease and known gene mutations can explain 70% of the fALS and 15% of the sporadic cases [96,97,98]. The four most common ALS-related genes are superoxide dismutase 1 (SOD1; MIM147450), fused in sarcoma (FUS; MIM137070), TAR-DNA binding protein (TARDBP; MIM605078), and chromosome 9 open reading frame 72 (C9orf72; MIM614260). These four genes account for 40-55% of fALS and 5% of sporadic cases and have different frequency, inheritance pattern, and penetrance. The disease pathophysiology is complicated and incompletely understood [96,99,100]. Consequently, ALS treatment has been limited to three drugs on the market: Riluzole (1996 EMA and 1995 FDA approval), Edaravone (2017 FDA approval) and Relyvrio (2023 FDA approval). These drugs do not have specific targets, their exact mechanism of action is not fully understood, and they have a modest effect on the disease phenotype. Tofersen (Qalsody®) is the first precision medicine approved for SOD1 ALS by the FDA in 2023. This highlights the urgent need to develop more targeted and effective therapies, leveraging advances in genetic testing and gene-based treatments.
As gene targeted therapies are challenging for various neurological disorders, the field of ALS therapeutics has its own challenges due to the complex etiology of the disease and the need for effective delivery to both the cerebral cortex and the anterior horn cells of the spinal cord [50]. Since toxic gain of function is the predominant mechanism in ALS, several approaches reaching clinical trials have been implemented to silence gene expression and treat the genetic cause of the disease. Current clinical studies of the gene targeted therapies in ALS are summarized in Table 1.
4.2.1. SOD1
More than 200 mutations have been reported in SOD1 gene since its discovery in 1993 as the first gene associated with fALS [99]. SOD1 mutations account for 2% of all ALS cases (10-14% of fALS and 1-2% of sALS in European ancestries) [99,101]. The SOD1 gene encodes the superoxide dismutase enzyme, which is a metalloprotease that metabolizes superoxide radicals to molecular oxygen and hydrogen peroxide, thus providing a defense against oxygen toxicity [102,103,104]. Although the underlying disease pathology is not fully understood, the predominant mechanism is the gain of function with rare loss of function studies also reported [105]. Lowering the concentration of misfolded and aggregated mutant SOD1 protein offers a potential strategy for therapy, with gene editing, RNAi, and ASO based approaches implemented to silence mutant SOD1 protein expression [106,107]. ASO 333611 is the first promising candidate to treat SOD1-ALS and was able to decrease both SOD1 mRNA and protein levels in rat and extended survival 37% after disease onset [108]. These encouraging results have led to a groundbreaking first-in-human clinical trial (NCT01041222) showing for the first time the feasibility of intrathecal administration of CNS targeted ASOs to treat genetic forms of ALS [109]. Advancements in ASO technology have facilitated the development of a more potent ASO called Tofersen (BIIB067) [110]. Tofersen (Qalsody®) is the first FDA approved precision medicine for a genetic form of ALS (Figure 4). The molecular formula of Tofersen is C230H317N72O123P19S15 which contain 20-bases with an RNA–DNA–RNA (5–10–5) gapmer mixed backbone containing oligonucleotide that has a molecular weight of 7127.86 atomic mass units [111]. After the entry to the motor neurons and astrocytes, Tofersen forms a DNA:RNA hybrid inside cytoplasm which is recognized and cleaved by the enzyme RNaseH1. In rodents and non-human primates Tofersen was able to decrease SOD1 mRNA and protein levels, significantly extending survival, and decrease serum and cerebrospinal fluid (CSF) plasma neurofilament light chain (NfL) which are biomarkers of axonal injury and neurodegeneration. Tofersen enabled reversal of neurodegeneration even after disease onset [110]. ALS patients with SOD1 mutations received ascending doses of Tofersen by intrathecal administration over a period of 12 weeks in phase I/II [112]. The safety and efficacy of Tofersen was then evaluated in a phase 3, randomized, double-blind, placebo-controlled VALOR trial (NCT02623699) and a long-term extension study (NCT03070119, completed) [113]. Proceedure associated adverse events were common and neurological adverse events were observed in approximately 7% of patients. The primary endpoint of the trial was change from baseline to week 28 in the ALSFRS-R total score in the faster-progression subgroup. Although Tofersen did not meet the clinical end points, the drug was able to reduce CSF SOD1 protein levels and plasma concentrations of NfL in patients [113,114]. The drug was approved by the FDA in April 2023 and an expanded access program is ongoing (NCT04972487).
Another approach to lower SOD1 protein involves RNAi molecules. ARO-SOD1 (siRNA), RAG-17 (siRNA) and AMT-162 (a.k.a. APB-102; adeno-associated virus rh10 containing an anti- SOD1 microRNA) have recently been developed to treat adult patients with SOD1 mutations [107,115,116,117]. The preclinical studies in rodents and non-human primates have shown improved efficacy, long duration of action, and potency, as they delayed disease progression, preserved motor function, and extended survival. A study in two SOD1-ALS patients with A4V and D90A mutations showed the safety of intrathecal infusion of adeno-associated virus rh10 containing an anti- SOD1 microRNA as a potential treatment. This proof-of-concept study lead the way for phase I/II clinical trial of AMT-162 (NCT06100276), which recently enrolled 20 SOD1-ALS patients. A first-in-human trial of RAG-17 (NCT05903690) showed safety and efficacy in SOD-1 ALS patients [118]. CSF SOD1 and plasma NfL levels were significantly decreased. Moreover, the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) score was improved for all patients, and forced vital capacity was stabilized. These results show promise for treating SOD1-ALS patients with RAG-17 (NCT06556394).
4.2.2. FUS
Discovered in 2009, FUS gene mutations are known to cause a rare early onset, rapidly progressive form of ALS (ALS6) that is responsible for 4% of familial and 2% of sporadic cases [119,120,121,122]. The vast majority of the mutations are inherited in an autosomal dominant fashion and mostly impact the C-terminal nuclear localization signal (NLS) region of the protein [123]. As a result, FUS protein is mislocalized to the cytoplasm and forms cytoplasmic inclusions. Consequently, underlying disease pathology is suggested to be both loss of function in the nucleus and gain of toxic function in the cytoplasm. A potential approach for therapy involves reducing levels of mutant FUS protein [124,125]. Jacifusen (also referred to as ION363 or UlefnersenTM), is an ASO molecule that targets FUS gene and reduces FUS protein levels [125]. Jacifusen was first introduced to a 25-year-old patient (JH) with a FUS P525L mutation whose identical twin sister passed away from the same mutation [125,126]. Although Jacifusen was well-tolerated and slowed the rate of ALS-FRS-R score decline, she passed away almost a year after starting treatment. The autopsy examination has revealed that jacifusen has a broad distribution throughout the CNS, even 2 months after the last therapeutic infusion, and dramatically decreased mutant FUS expression and FUS-positive aggregates. Following the first in-human study, results from the 11 patients who were enrolled in an investigator-initiated treatment program have provided evidence of the safety and therapeutic potential of jacifusen [127]. Serial intrathecal administration of the drug decreased FUS mRNA and protein levels and reduced CSF NfL concentration by up to 82·8%. One patient showed exceptional functional recovery in fine motor, gross motor, and respiratory domains. The results of the ongoing phase 1-3 clinical trial (Fusion, NCT04768972) will evaluate the clinical efficacy of jacifusen in FUS-ALS patients worldwide.
4.2.3. C9orf72
Autosomal dominantly inherited mutations in C9orf72 were discovered later than other ALS related genes and represents the most common genetic cause of ALS [128,129]. Hexanucleotide repeat (GGGGCC) expansion in the first intron or the promotor region of C9orf72 is responsible for 40% of fALS and 7% of sALS cases [128,130,131]. Healthy individuals carry up to eight repeats, and 24-29 repeats is considered as risk factor. Thirty repeats is considered as a pathogenic threshold and patients can have up to thousands of repeats. [132,133,134]. C9orf72 pathology is suggested to be caused by both loss and gain of protein function. Loss of function may occur due to haploinsufficiency resulting from decreased C9orf72 gene expression and disrupted vesicular trafficking [135,136]. Gain of function toxicity may occur by two mechanisms: 1. Mutant mRNA generation of nuclear foci and sequestration of essential RNA-binding proteins, which may impair their function [114]. 2. Sense and antisense strands of C9orf72 RNA are translated by repeat-associated non-ATG translation and generate toxic aggregates of dipeptide repeat proteins (sense: polyGR, polyGA, polyGP; antisense: polyGP, polyPR, polyPA) [138,139].
Gene targeted therapy strategies to treat C9orf72-ALS include the targeted reduction of hexanucleotide repeat-containing C9orf72 transcripts and dipeptide repeat proteins. The gene has three transcript variants called V1 (short protein isoform), V2 and V3 (full-length protein) [140]. Hexanucleotide repeats are located between non-coding exons 1a and 1b and exon two has the translational start site (ATG). V1 and V3 transcripts have exons starting from exon 1a and the hexanucleotide repeat sequence, but the V2 transcript includes exons starting from exon 1b, which resides downstream of the hexanucleotide repeat. One therapeutic strategy under investigation involves ASOs that selectively reduce the V1 and V3 transcripts without impacting transcript V2. A proof-of-concept study has shown that Afinersen (ASO 5-2) was able to degrade both V1 and V3 transcripts by RNase-H1-mediated degradation in C9orf72 disease models, including patient-derived cells, mice, sheep, and monkeys. Encouraging results have led to the treatment of a single patient. The intrathecally injected Afinersen was well tolerated, the ALSFRS score improved, and dipeptide repeat proteins were reduced 80% in CSF [140]. A larger clinical trial is needed to evaluate the efficacy of Afinersen.
BIIB078 (IONIS-C9Rx/Tadnersen) is another investigational ASO that selectively targets exon 1a-initiated transcripts V1 and V3 without altering V2 transcript levels. When the ASO was administered to mice expressing C9orf72 RNAs with up to 450 GGGGCC repeats or with one or both C9orf72 alleles inactivated, it reduced RNA foci, poly-glycine-proline and poly-glycine-alanine dipeptide-repeat proteins and improved behavioral deficits [141]. In a phase I study (NCT04288856) of adult patients with C9orf72-associated ALS, administration of BIIB078 did not show benefit on clinical outcomes and did not reduce NfL concentrations in CSF or plasma [142]. The trial was terminated by the sponsor, and potential challenges have been identified, including the heterogeneity of pathobiology in C9orf72-ALS, limitations of outcome measures, and the need for further understanding of pathogenic effects of antisense oligonucleotides. The growing body of studies and trials will help lay the foundation for the development of effective treatments in the future.
4.2.4. TARDBP
Another common gene mutated in ALS is TARDBP. Since its discovery in 2006, more than 50 mutations have been reported and are responsible for 3.3% of fALS (autosomal dominant inheritance) and 0.5% of sALS [143]. TAR DNA-binding protein 43 (TDP-43) is a ubiquitously expressed and tightly regulated DNA/RNA-binding protein encoded by the TARDBP gene. It has roles in several cellular mechanisms, including RNA biogenesis, processing, and translation [144,145]. TDP-43 is predominantly found in the nucleus, but in 97% of ALS cases with or without TARDBP mutations, the protein mislocalizes to the cytoplasm and forms aggregates in the brain and spinal cord of ALS patients as a pathological hallmark of the disease [143,145,146,147]. TDP-43-related pathology is suggested to be caused by both loss of TDP-43 function in the nucleus and gain of function in the cytoplasm [148]. Different strategies have been developed to target pathological forms of TDP-43 and to restore its homeostasis [149]. A recent study in ALS/FTD mice showed the potential of a gapmer-type ASO against human TDP-43 as a possible disease-modifying therapy [150]. Alternatively, indirect methods to lower toxic TDP-43 aggregates have been investigated, such as ASO-mediated knockdown of other non-TARDBP ALS-related genes [148]. Two promising target genes are ataxin-2 and stathmin-2. An increase in CAG repeats (27-33 repeats) in the ataxin-2 gene has been reported to increase ALS disease risk by 11-fold [142]. ASO (BIIB105/ION541) or AAV-based delivery of RNAi (miRNA) targeting ataxin-2 in TDP-43 mice has shown promising results. These strategies were able to decrease TDP-43 aggregation, improve motor function, and markedly extend survival [151,152]. A phase I/II study (NCT04494256) with the ataxin-2 targeting ASO drug BIIB105 was ongoing but recently terminated since treatment did not reduce plasma NfL or impact clinical outcomes [153].
Stathmin-2 (STMN2) is a tubulin-binding protein that is required for axon outgrowth, maintenance, and regeneration [154,155,156]. TDP-43 regulates pre-mRNA processing and is required for functional full-length stathmin-2 protein synthesis [157]. In healthy individuals, TDP-43 sterically blocks a cryptic splice site that resides in the first intron of the STMN2 gene. When nuclear TDP-43 is lost in ALS patients, the cryptic splice site usage results in inclusion of exon2a which introduces an early stop codon and premature polyadenylation signal. This results in truncated protein synthesis and stathmin-2 loss of function. The ASO drug QRL-201 is developed to target the regulatory elements that TDP-43 regulates in STMN2 pre-mRNA processing, restore functional stathmin-2 protein, and rescue TDP-43-related axonal regeneration [153]. A phase I study of QRL-201 (NCT05633459) is ongoing [158].
5. Clinical Trial Readiness and Future Considerations
The last decade has witnessed the potential of gene targeted therapies for several disorders and clinical trials are now designed in a personalized way. A recent study highlighted that gene targeted therapies possess a higher rate of clinical development success compared to other therapeutic modalities [159]. While we currently have improved tools and technology to develop these therapies, improving clinical trial readiness is necessary for future success of the trials for various neurodegenerative disorders. In this sense, several considerations should be addressed, including but not limited to the role of disease models, knowing the natural history of the disease, and the need for well-established outcome measures.
Disease models are important resources for studying disease pathophysiology and therapeutics development. Better understanding of disease and early diagnosis is vital, as experience has shown the importance of early therapeutic intervention in SMA before significant motor neuron loss occurs [160]. Animal models have been used as the gold standard in preclinical trials, though they have limitations. These model systems may not fully recapitulate the disease phenotype due to the complexity of human neuroanatomy, and evaluation of safety could be difficult. Interspecies differences are also a concern for gene targeted therapies, as species-specific gene editing efficacy in mice compared to primates has been reported [161]. In this sense, humanized mouse models and large non-human primates are pharmacologically more relevant disease models for dosing, efficacy, and safety studies [162,163]. However, the cost, time requirement, and inefficiency of genetic manipulations of large animals limit their widespread use. Another limitation is the diverse genetic and ethnic backgrounds of patients.
Patient-derived cell models have been used as a valuable resource to study disease mechanisms with the advantage of having the genetic background of the patient. In diseases with high genetic heterogeneity, such as ALS, the specific genes and mutations causing disease can vary. As a result, generalizing findings from a single animal model may lead to oversight of certain gene and mutation-specific phenotypes. Fibroblast cells have been used for studying disease mechanisms since they share similar gene expression profiles with neuronal cells. However, they are not the primary cell types affected in neurological disorders, which may limit the observations in a cell type-specific manner [164]. Induced pluripotent stem cells (iPSCs) are invaluable resources as they have the potential to differentiate into disease-relevant neuronal and glial cell types [165,166]. Generation of isogenic cell lines by correcting the patient’s mutation would eliminate the genetic background differences and help to elucidate the molecular mechanisms caused by a specific mutation. The Answer ALS platform provides a large set of data from patient and control iPSC-derived motor neurons and resources for studying gender involvement, dysregulated genes, and disrupted pathways in disease pathology [99,167]. Combining gene editing technologies such as CRISPR with stem cell technologies including organoids and organs-on-a-chip allows for the generation of disease models that can provide new insights into disease mechanisms and allow for drug testing in a human-relevant context [168]. A recent study in an SMA spinal cord organoid (SCO) model was generated using patient-derived iPSCs and isogenic controls. This study reported early neurodevelopmental defects in the disease pathogenesis, which might support even earlier therapeutic intervention in patients [169]. Early treatment of the SCO model with an SMN-restoring ASO rescued morphological and functional deficits as well as SMN-dependent splicing defects [170]. Neuromuscular organoids, 3D human cortical organoid slice culture models, and presymptomatic cerebral organoids were derived from iPSCs of C9orf72 ALS patients and were able to recapitulate spinal neuromuscular pathologies, early changes in 3D human brain tissue organization, synaptic structure and function, and mature astroglial/neuronal phenotypes [171,172,173]. Although each model has its own limitations, these tools provide opportunities to deepen our understanding of disease mechanisms and enable the development of effective treatment approaches for neurodegenerative disorders.
Besides the need for disease-relevant models, the natural history of disease is crucial for facilitating clinical trial readiness. The natural history of a disease is traditionally defined as the course a disease takes in the absence of intervention in individuals with the disease, from the disease’s onset until either the disease’s resolution or the individual’s death [174]. Natural history studies provide detailed information on initial disease symptoms, disease progression, and severity. This knowledge is valuable for identifying patient subpopulations within a heterogeneous cohort and can help recognize subgroups that may benefit from a specific treatment. In addition, natural history studies allow for the development of clinical outcome assessments for identifying disease features, monitoring and managing the clinical status of the disease, and testing drug efficacy in clinical trials [175,176]. Patient-reported outcome measurements are also important to identify the importance and prevalence of key symptoms and factors linked to disease severity [175]. Natural history studies allow for the collection of blood, tissue, or bodily fluid specimens and imaging (e.g., magnetic resonance imaging of brain and skeletal muscle), which will help in developing biomarkers for diagnostic, prognostic, or predictive determinations [176,177,178]. These biomarkers can serve as endpoints in clinical trials when they are validated. In recent years, NfL and phosphorylated neurofilament heavy chain (pNfH) proteins have been identified as biomarkers for neuron degeneration and drug response for several neurodegenerative disorders. Combining molecular and non-molecular biomarkers (electrophysiological or imaging) can also be very informative [179,180]. As a final point it is important to emphasize the critical role of patient registries in establishing outcome and endpoint measures in addition to supporting patient recruitment for clinical trials.
Looking forward, advancements in precision medicine and emerging technologies will offer promising gene-targeted therapy opportunities for various disorders. Genetic screening will play a crucial role in addressing the genetic heterogeneity of diseases such as ALS, enabling the development of tailored therapies for specific mutations [181]. Gene editing technologies, particularly CRISPR-based approaches, will remain integral in the development of disease models and precise therapies for various disorders. Casgevy (NCT03745287) and Lyfgenia (NCT04293185) are the first examples of approved CRISPR-Cas9 gene editing therapies for sickle cell disease and beta thalassemia, showing the substantial potential of these approaches. Gene editing approaches are already being explored in SMA and ALS, demonstrating the potential of these therapies to offer effective treatments. Adenosine base editors have demonstrated the potential to correct C to T transition in SMN2, allowing exon 7 retention and increasing SMN levels in patient-derived fibroblasts and in mice, with no off-target effects. This correction resulted in improved motor function and extended lifespan in animal models [182,183]. Base editing of SMN2 allows for the endogenous regulation of the SMN protein, which could eliminate potential toxicity due to the constitutive expression driven by onasemnogene abeparvovec-xioi (Zolgensma). RNA editing is also receiving attention due to safety concerns associated with editing DNA. Recent studies suggest this approach should be considered for ALS. CRISPR-Cas13d variants have been shown to significantly reduce C9orf72 sense and antisense repeat transcripts in patient-derived iPSC-neuron lines and different C9orf72 mouse models without affecting V2 transcript levels [184,185]. In conclusion, while challenges and safety concerns remain, the continued progress in gene-targeted therapies offers unprecedented potential for treating neurodegenerative diseases with greater precision and efficacy.
Author Contributions
Conceptualization: AYK, GBA, HEY, CG; Writing-original draft preparation; AYK, GBA, HEY, CG; Visualization: AYK, GBA; Writing-review and editing: AYK, GBA, HEY, CG.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the National Institutes of Health (NIH). The contributions of the NIH author(s) are considered Works of the United States Government. The findings and conclusions presented in this paper are those of the author(s) and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAV | Adeno-associated virus |
| AAV9 | Adeno-associated virus serotype 9 |
| Ad | Adenovirus |
| ALS | Amyotrophic lateral sclerosis |
| ALSFRS-R | Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised |
| ASO | Antisense oligonucleotide |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CAR-T | Chimeric antigen receptor T-cell |
| Cas9 | CRISPR-associated protein 9 |
| Cas13d | CRISPR-associated protein 13d |
| CNS | Central nervous system |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CSF | Cerebrospinal fluid |
| DNA | Deoxyribonucleic acid |
| dsDNA | Double-stranded DNA |
| EMA | European Medicines Agency |
| fALS | Familial amyotrophic lateral sclerosis |
| FDA | Food and Drug Administration |
| FTD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| gRNA | Guide RNA |
| HR | Homologous recombination |
| iPSC | Induced pluripotent stem cell |
| ITR | Inverted terminal repeat |
| LTR | Long terminal repeat |
| miRNA | MicroRNA |
| mRNA | Messenger ribonucleic acid |
| NDDs | Neurodegenerative diseases |
| NfL | Neurofilament light chain |
| NHEJ | Non-homologous end joining |
| RNA | Ribonucleic acid |
| RNAi | RNA interference |
| RNaseH1 | Ribonuclease H1 |
| sALS | Sporadic amyotrophic lateral sclerosis |
| scAAV | Self-complementary adeno-associated virus |
| sgRNA | Single guide RNA |
| siRNA | Small interfering RNA |
| SMA | Spinal muscular atrophy |
| SMN | Survival motor neuron |
| SMN1 | Survival motor neuron 1 |
| SMN2 | Survival motor neuron 2 |
| SOD1 | Superoxide dismutase 1 |
| STMN2 | Stathmin-2 |
| TALEN | Transcription activator-like effector nuclease |
| TARDBP | TAR DNA-binding protein |
| TDP-43 | TAR DNA-binding protein 43 |
| UTR | Untranslated region |
| ZFNs | Zinc finger nucleases |
References
- Arabi F, Mansouri V, Ahmadbeigi N. Gene therapy clinical trials, where do we go? An overview. Biomed Pharmacother. 2022;153:113324. [CrossRef]
- What is gene therapy? [Internet]. Silver Spring (MD): FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy (accessed on 27 October 2025).
- Razi Soofiyani S, Baradaran B, Lotfipour F, Kazemi T, Mohammadnejad L. Gene therapy, early promises, subsequent problems, and recent breakthroughs. Adv Pharm Bull. 2013;3(2):249-55. [CrossRef]
- Seow Y, Wood MJ. Biological gene delivery vehicles: beyond viral vectors. Mol Ther. 2009;17(5):767-77. [CrossRef]
- Alhakamy NA, Curiel DT, Berkland CJ. The era of gene therapy: From preclinical development to clinical application. Drug Discov Today. 2021;26(7):1602-19. [CrossRef]
- Shin MH, He Y, Marrogi E, Piperdi S, Ren L, Khanna C, et al. A RUNX2-Mediated Epigenetic Regulation of the Survival of p53 Defective Cancer Cells. PLoS Genet. 2016;12(2):e1005884. [CrossRef]
- Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73-80. [CrossRef]
- Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci USA. 2016;113(11):2868-73. [CrossRef]
- Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21(4):255-72; [CrossRef]
- Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021;6(1):53. [CrossRef]
- Alba R, Bosch A, Chillon M. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther. 2005;12 Suppl 1:S18-27. [CrossRef]
- Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science. 1965;149(3685):754-6. [CrossRef]
- Wang JH, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 2024;9(1):78. [CrossRef]
- Penaud-Budloo M, Le Guiner C, Nowrouzi A, Toromanoff A, Cherel Y, Chenuaud P, et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J Virol. 2008;82(16):7875-85. [CrossRef]
- Riyad JM, Weber T. Intracellular trafficking of adeno-associated virus (AAV) vectors: challenges and future directions. Gene Ther. 2021;28(12):683-96. [CrossRef]
- Ling Q, Herstine JA, Bradbury A, Gray SJ. AAV-based in vivo gene therapy for neurological disorders. Nat Rev Drug Discov. 2023;22(10):789-806. [CrossRef]
- Pupo A, Fernández A, Low SH, François A, Suárez-Amarán L, Samulski RJ. AAV vectors: The Rubik's cube of human gene therapy. Mol Ther. 2022;30(12):3515–3541. [CrossRef]
- Suarez-Amaran L, Song L, Tretiakova AP, Mikhail SA, Samulski RJ. AAV vector development, back to the future. Mol Ther. 2025;33(5):1903–1936. [CrossRef]
- van Haasteren J, Li J, Scheideler OJ, Murthy N, Schaffer DV. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat Biotechnol. 2020;38(7):845-55. [CrossRef]
- Ramamoorth M, Narvekar A. Non viral vectors in gene therapy- an overview. J Clin Diagn Res. 2015;9(1):GE01-6. [CrossRef]
- Picanco-Castro V, Pereira CG, Covas DT, Porto GS, Athanassiadou A, Figueiredo ML. Emerging patent landscape for non-viral vectors used for gene therapy. Nat Biotechnol. 2020;38(2):151-7. [CrossRef]
- Shahryari A, Burtscher I, Nazari Z, Lickert H. Engineering Gene Therapy: Advances and Barriers. Advanced Therapeutics. 2021;4(9). [CrossRef]
- Zhou H, He Y, Xiong W, Jing S, Duan X, Huang Z, et al. MSC based gene delivery methods and strategies improve the therapeutic efficacy of neurological diseases. Bioact Mater. 2023;23:409-37. [CrossRef]
- Zu H, Gao D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021;23(4):78. [CrossRef]
- Giovannelli I, Higginbottom A, Kirby J, Azzouz M, Shaw PJ. Prospects for gene replacement therapies in amyotrophic lateral sclerosis. Nat Rev Neurol. 2023;19(1):39-52. [CrossRef]
- Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K, et al. Gene Therapy Leaves a Vicious Cycle. Front Oncol. 2019;9(927):297. [CrossRef]
- Poletto E, Baldo G, Gomez-Ospina N. Genome Editing for Mucopolysaccharidoses. Int J Mol Sci. 2020;21(2):500. [CrossRef]
- Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021;24(3):297-311. [CrossRef]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321-34. [CrossRef]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816-21. [CrossRef]
- Mojica FJM D-VJ, García-Martínez Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174-82. [CrossRef]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709-12. https://doi.org.10.1126/science.113814.
- Wang JY, Doudna JA. CRISPR technology: A decade of genome editing is only the beginning. Science. 2023;379(6629):eadd8643. [CrossRef]
- Jiang F, Doudna JA. CRISPR–Cas9 Structures and Mechanisms. Annual Review of Biophysics. 2017;46(Volume 46, 2017):505-29. [CrossRef]
- Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. 2023;24(3):161-77. [CrossRef]
- Newby GA, Liu DR. In vivo somatic cell base editing and prime editing. Mol Ther. 2021;29(11):3107-24. [CrossRef]
- Casas-Mollano JA, Zinselmeier MH, Erickson SE, Smanski MJ. CRISPR-Cas Activators for Engineering Gene Expression in Higher Eukaryotes. CRISPR J. 2020;3(5):350-64. [CrossRef]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173-83. [CrossRef]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420-4. [CrossRef]
- Kang Y, Chu C, Wang F, Niu Y. CRISPR/Cas9-mediated genome editing in nonhuman primates. Dis Model Mech. 2019;12:dmm039982. [CrossRef]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149-157. [CrossRef]
- Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. 2023;24(3):161–177. [CrossRef]
- Zhao Z, Shang P, Mohanraju P, Geijsen N. Prime editing: advances and therapeutic applications. Trends Biotechnol. 2023;41(8):1000-1012. [CrossRef]
- Schoch KM, Miller TM. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron. 2017;94(6):1056-70. [CrossRef]
- Bennett CF, Kordasiewicz HB, Cleveland DW. Antisense Drugs Make Sense for Neurological Diseases. Annu Rev Pharmacol Toxicol. 2021;61:831-52. [CrossRef]
- Marrosu E, Ala P, Muntoni F, Zhou H. Gapmer Antisense Oligonucleotides Suppress the Mutant Allele of COL6A3 and Restore Functional Protein in Ullrich Muscular Dystrophy. Mol Ther Nucleic Acids. 2017;8:416-27. [CrossRef]
- Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279(17):17181-9. [CrossRef]
- Lima WF, De Hoyos CL, Liang XH, Crooke ST. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res. 2016;44(7):3351-63. [CrossRef]
- Lauffer MC, van Roon-Mom W, Aartsma-Rus A, Collaborative N. Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun Med (Lond). 2024;4(1):6. [CrossRef]
- Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021;29(12):3345-58. [CrossRef]
- Brillante S, Volpe M, Indrieri A. Advances in MicroRNA Therapeutics: From Preclinical to Clinical Studies. Hum Gene Ther. 2024;35(17-18):628-648. [CrossRef]
- Borel F, Kay MA, Mueller C. Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol Ther. 2014;22(4):692–701. [CrossRef]
- Bennett CF. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev Med. 2019;70:307-21. [CrossRef]
- Leung AK, Tam YY, Cullis PR. Lipid nanoparticles for short interfering RNA delivery. Adv Genet. 2014;88:71-110. [CrossRef]
- Dahlman JE, Kauffman KJ, Langer R, Anderson DG. Nanotechnology for in vivo targeted siRNA delivery. Adv Genet. 2014;88:37-69. [CrossRef]
- Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46-51. [CrossRef]
- Garreau M, Weidner J, Hamilton R, Kolosionek E, Toki N, Stavenhagen K, et al. Chemical modification patterns for microRNA therapeutic mimics: a structure-activity relationship (SAR) case-study on miR-200c. Nucleic Acids Research. 2024;52(6):2792-2807. [CrossRef]
- Wang P, Zhou Y, Richards AM. Effective tools for RNA-derived therapeutics: siRNA interference or miRNA mimicry. Theranostics. 2021 ;11(18):8771-8796. [CrossRef]
- Alterman JF, Godinho BMDC, Hassler MR, Ferguson CM, Echeverria D, Sapp E, et al. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat Biotechnol. 2019;37(8):884-894. [CrossRef]
- Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9-21. [CrossRef]
- Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44(14):6549-63. [CrossRef]
- Wirth B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021;44(4):306-22. [CrossRef]
- Yeo CJJ, Tizzano EF, Darras BT. Challenges and opportunities in spinal muscular atrophy therapeutics. Lancet Neurol. 2024;23(2):205–218. [CrossRef]
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19(20):4059-71. [CrossRef]
- Allardyce H, Kuhn D, Hernandez-Gerez E, Hensel N, Huang YT, Faller K, et al. Renal pathology in a mouse model of severe Spinal Muscular Atrophy is associated with downregulation of Glial Cell-Line Derived Neurotrophic Factor (GDNF). Hum Mol Genet. 2020;29(14):2365-78. [CrossRef]
- Somers E, Lees RD, Hoban K, Sleigh JN, Zhou H, Muntoni F, et al. Vascular Defects and Spinal Cord Hypoxia in Spinal Muscular Atrophy. Ann Neurol. 2016;79(2):217-30. [CrossRef]
- Hensel N, Claus P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist. 2018;24(1):54-72. [CrossRef]
- Kaymaz AY, Bal SK, Bora G, Talim B, Ozon A, Alikasifoglu A, et al. Alterations in insulin-like growth factor system in spinal muscular atrophy. Muscle Nerve. 2022;66(5):631-8. [CrossRef]
- Bora G, Hensel N, Rademacher S, Koyunoğlu D, Sunguroğlu M, Aksu-Mengeş E, et al. Microtubule-associated protein 1B dysregulates microtubule dynamics and neuronal mitochondrial transport in spinal muscular atrophy. Hum Mol Genet. 2021;29(24):3935-44. [CrossRef]
- Chaytow H, Huang YT, Gillingwater TH, Faller KME. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci. 2018;75(21):3877-94. [CrossRef]
- Tapken I, Schweitzer T, Paganin M, Schüning T, Detering NT, Sharma G, Niesert M, Saffari A, Kuhn D, Glynn A, Cieri F, Santonicola P, Cannet C, Gerstner F, Faller KME, Huang YT, Kothary R, Gillingwater TH, Di Schiavi E, Simon CM, Claus P. The systemic complexity of a monogenic disease: the molecular network of spinal muscular atrophy. Brain. 2025;148(2):580–596. [CrossRef]
- Özer PZ, Koyunoğlu D, Son ÇD, Erdem-Yurter H, Bora G. SMN loss dysregulates microtubule-associated proteins in spinal muscular atrophy model. Mol Cell Neurosci. 2022;120:103725. [CrossRef]
- Dayangaç-Erden D, Bora G, Ayhan P, Kocaefe C, Dalkara S, Yelekçi K, et al. Histone deacetylase inhibition activity and molecular docking of (e )-resveratrol: its therapeutic potential in spinal muscular atrophy. Chem Biol Drug Des. 2009;73(3):355-64. [CrossRef]
- Chaytow H, Faller KME, Huang YT, Gillingwater TH. Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine. Cell Rep Med. 2021;2(7):100346. [CrossRef]
- Mercuri E, Pera MC, Scoto M, Finkel R, Muntoni F. Spinal muscular atrophy - insights and challenges in the treatment era. Nat Rev Neurol. 2020;16(12):706-15. [CrossRef]
- Jablonka S, Hennlein L, Sendtner M. Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders. Neurol Res Pract. 2022;4(1):2. [CrossRef]
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333-46. [CrossRef]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834-48. [CrossRef]
- Biogen. Spinraza® (nusinersen) [Internet]. Cambridge (MA): Biogen; c2024 Available online: https://wwwspinrazacom/ (accessed on 13 May 2025).
- Zolgensma®. Bannockburn (IL): Novartis; c2024. Available online: https://www.zolgensma.com/ (14 May 2025).
- Thomsen G, Burghes AHM, Hsieh C, Do J, Chu BTT, Perry S, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med. 2021;27(10):1701-11. [CrossRef]
- Roche. FDA approves Roche’s Evrysdi tablet as first and only tablet for Spinal Muscular Atrophy (SMA). 2025 Feb 12. https://www.roche.com/media/releases/med-cor-2025-02-12.
- Ottesen EW, Singh NN, Luo D, Kaas B, Gillette BJ, Seo J, et al. Diverse targets of SMN2-directed splicing-modulating small molecule therapeutics for spinal muscular atrophy. Nucleic Acids Res. 2023;51(12):5948-80. [CrossRef]
- Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet. 2020;21:231-61. [CrossRef]
- Singh RN, Ottesen EW, Singh NN. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci Insights. 2020;15:2633105520973985. [CrossRef]
- Chiriboga CA, Bruno C, Duong T, Fischer D, Mercuri E, Kirschner J, Kostera-Pruszczyk A, Jaber B, Gorni K, Kletzl H, Carruthers I, Martin C, Scalco RS, Fontoura P, Muntoni F, JEWELFISH Study Group. JEWELFISH: 24-month results from an open-label study in non-treatment-naïve patients with SMA receiving treatment with risdiplam. J Neurol. 2024;271(8):4871–4884. [CrossRef]
- Bekircan-Kurt CE, Subramanian S, Chagat S, Mackenzie SJ, Iammarino M, Reash N, Richardson C, Tsao CY, Noritz G, Gushue C, Kotha K, Paul G, Shell R, Alfano LN, Lowes LP, Connolly AM, Waldrop MA. Transitioning from nusinersen to risdiplam for spinal muscular atrophy in clinical practice: A single-center experience. Muscle Nerve. 2025;71(3):414–421. [CrossRef]
- Oechsel KF, Cartwright MS. Combination therapy with onasemnogene and risdiplam in spinal muscular atrophy type 1. Muscle Nerve. 2021;64(4):487–490. [CrossRef]
- Cure SMA. SMA drug pipeline [Internet]. Chicago (IL): Cure SMA. Available online: https://www.curesma.org/sma-drug-pipeline/ (accessed on 13 May 2025).
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [CrossRef]
- Van Damme P, Robberecht W, Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. 2017;10(5):537-49. [CrossRef]
- Shatunov A, Al-Chalabi A. The genetic architecture of ALS. Neurobiol Dis. 2021;147:105156. [CrossRef]
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118-30. [CrossRef]
- Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. 2020;267(4):944-53. [CrossRef]
- Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385-90. [CrossRef]
- Ruffo P, Traynor BJ, Conforti FL. Advancements in genetic research and RNA therapy strategies for amyotrophic lateral sclerosis (ALS): current progress and future prospects. J Neurol. 2025;272(3):233. [CrossRef]
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. Lancet. 2022;400(10360):1363-80. [CrossRef]
- Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17(1):94-102. [CrossRef]
- Akcimen F, Lopez ER, Landers JE, Nath A, Chio A, Chia R, et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet. 2023;24(9):642-58. [CrossRef]
- Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17-23. [CrossRef]
- Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. 2018;89(8):817-27. [CrossRef]
- Sherman L, Dafni N, Lieman-Hurwitz J, Groner Y. Nucleotide sequence and expression of human chromosome 21-encoded superoxide dismutase mRNA. Proc Natl Acad Sci USA. 1983;80(18):5465-9. [CrossRef]
- Niwa J, Yamada S, Ishigaki S, Sone J, Takahashi M, Katsuno M, et al. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J Biol Chem. 2007;282(38):28087-95. [CrossRef]
- Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247(1):1-11. [CrossRef]
- Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136(Pt 8):2342-58. [CrossRef]
- Lim CKW, Gapinske M, Brooks AK, Woods WS, Powell JE, Zeballos CM, et al. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol Ther. 2020;28(4):1177-89. [CrossRef]
- Duan W, Guo M, Yi L, Liu Y, Li Z, Ma Y, et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020;27(3-4):157-69. [CrossRef]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290-6. [CrossRef]
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435-42. [CrossRef]
- McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558-67. [CrossRef]
- Jin J, Zhong XB. ASO drug Qalsody (tofersen) targets amyotrophic lateral sclerosis. Trends Pharmacol Sci. 2023;44(12):1043-4. [CrossRef]
- Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109-19. [CrossRef]
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-110. [CrossRef]
- Blair HA. Tofersen: First Approval. Drugs. 2023;83:1039–1043. [CrossRef]
- Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabrera GC, Song L, et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum Gene Ther. 2016;27(1):19-31. [CrossRef]
- Arrowhead Pharmaceuticals. Poster for Arrowhead Pharmaceuticals [Internet]. Preclinical profile of ARO-SOD1, an siRNA therapy for SOD1-ALS. Available online: https://ir.arrowheadpharma.com/static-files/804aa94c-7323-4917-a04f-bd0812d2e2fb (accessed on 5 June 2025).
- Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med. 2020;383(2):151–158. [CrossRef]
- Ye J, Jiang L, Wang L, Pan Y, Wang X, Qu H, et al. RAG-17, a Novel siRNA Therapy for SOD1-ALS: Safety and Preliminary Efficacy from a First-in-human Trial (S5.003). Neurology. 2025;104(7_Supplement_1). [CrossRef]
- Assoni AF, Foijer F, Zatz M. Amyotrophic Lateral Sclerosis, FUS and Protein Synthesis Defects. Stem Cell Rev Rep. 2023;19(3):625-38. [CrossRef]
- Tarlarini C, Lunetta C, Mosca L, Avemaria F, Riva N, Mantero V, et al. Novel FUS mutations identified through molecular screening in a large cohort of familial and sporadic amyotrophic lateral sclerosis. Eur J Neurol. 2015;22(11):1474-81. [CrossRef]
- Akiyama T, Warita H, Kato M, Nishiyama A, Izumi R, Ikeda C, et al. Genotype-phenotype relationships in familial amyotrophic lateral sclerosis with FUS/TLS mutations in Japan. Muscle Nerve. 2016;54(3):398-404. [CrossRef]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208-11. [CrossRef]
- Xiao X, Li M, Ye Z, He X, Wei J, Zha Y. FUS gene mutation in amyotrophic lateral sclerosis: a new case report and systematic review. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(1-2):1-15. [CrossRef]
- Reber S, Stettler J, Filosa G, Colombo M, Jutzi D, Lenzken SC, et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016;35(14):1504-21. [CrossRef]
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104-16. [CrossRef]
- Conte A, Lattante S, Zollino M, Marangi G, Luigetti M, Del Grande A, et al. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul Disord. 2012;22(1):73-5. [CrossRef]
- Shneider NA, Harms MB, Korobeynikov VA, Rifai OM, Hoover BN, Harrington EA, et al. Antisense oligonucleotide jacifusen for FUS-ALS: an investigator-initiated, multicentre, open-label case series. Lancet. 2025;405(10494):2075-86. [CrossRef]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257-68. [CrossRef]
- Smeyers J, Banchi EG, Latouche M. C9ORF72: What It Is, What It Does, and Why It Matters. Front Cell Neurosci. 2021;15:661447. [CrossRef]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245-56. [CrossRef]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323-30. [CrossRef]
- Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev. 2017;44:117-24. [CrossRef]
- Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24-30 repeats are associated with ALS. Acta Neuropathol Commun. 2019;7(1):115. [CrossRef]
- Sellier C, Corcia P, Vourc'h P, Dupuis L. C9ORF72 hexanucleotide repeat expansion: From ALS and FTD to a broader pathogenic role? Rev Neurol (Paris). 2024;180(5):417-428. [CrossRef]
- Braems E, Swinnen B, Van Den Bosch L. C9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 2020;140(5):625-43. [CrossRef]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–325. Swinnen B, Robberecht W, Van Den Bosch L. RNA toxicity in non-coding repeat expansion disorders. Embo j. 2020;39(1):e101112. https://doi.org/10.15252/embj.2018101112. [CrossRef]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639-46. [CrossRef]
- Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530-9. [CrossRef]
- Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117-24. [CrossRef]
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90(3):535-50. [CrossRef]
- van den Berg LH, Rothstein JD, Shaw PJ, Babu S, Benatar M, Bucelli RC, et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: a phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 2024: 23(9):901-912. [CrossRef]
- Meijboom KE, Brown RH. Approaches to Gene Modulation Therapy for ALS. Neurotherapeutics. 2022;19(4):1159-79. [CrossRef]
- Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138 Suppl 1:95-111. [CrossRef]
- Guo L, Shorter J. Biology and Pathobiology of TDP-43 and Emergent Therapeutic Strategies. Cold Spring Harb Perspect Med. 2017;7(9). [CrossRef]
- Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, Magistrello M, et al. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010;20(2):351-60. [CrossRef]
- Suk TR, Rousseaux MWC. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15(1):45. [CrossRef]
- Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19(4):671-83. [CrossRef]
- Babazadeh A, Rayner SL, Lee A, Chung RS. TDP-43 as a therapeutic target in neurodegenerative diseases: Focusing on motor neuron disease and frontotemporal dementia. Ageing Res Rev. 2023;92:102085. [CrossRef]
- Takeuchi T, Maeta K, Ding X, Oe Y, Takeda A, Inoue M, et al. Sustained therapeutic benefits by transient reduction of TDP-43 using ENA-modified antisense oligonucleotides in ALS/FTD mice. Mol Ther Nucleic Acids. 2023;31:353-66. [CrossRef]
- Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367-71. [CrossRef]
- Amado DA, Robbins AB, Smith AR, Whiteman KR, Chillon Bosch G, Chen Y, et al. AAV-based delivery of RNAi targeting Ataxin-2 improves survival, strength, and pathology in mouse models of rapidly and slowly progressive sporadic ALS. BioRxiv. 2024. [CrossRef]
- Biogen and Ionis Pharmaceuticals. Biogen and Ionis announce topline Phase 1/2 study results of investigational drug in amyotrophic lateral sclerosis. Biogen. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-12-study-results (accessed on 16 May 2024 ).
- Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180-90. [CrossRef]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167-79. [CrossRef]
- Shin JE, Geisler S, DiAntonio A. Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol. 2014;252:1-11. [CrossRef]
- Baughn MW, Melamed Z, Lopez-Erauskin J, Beccari MS, Ling K, Zuberi A, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science. 2023;379(6637):1140-9. [CrossRef]
- Genge A, Salmon K, Polzer J, Martinez C, Boggs B, Eon V, et al. QRL-201-01—A Multi-center, Randomized, Double-blind, Placebo-controlled Multiple Ascending Dose Study to Evaluate the Safety and Tolerability of QRL-201 in Amyotrophic Lateral Sclerosis (P6-11.010). Neurology. 2024;102(7_supplement_1). [CrossRef]
- Phares S, Phillip K, Trusheim M. Clinical development success rates for durable cell and gene therapies. Nat Rev Drug Discov. 2025;24(5):329-30. [CrossRef]
- Sumner CJ, Crawford TO. Early treatment is a lifeline for infants with SMA. Nat Med. 2022;28(7):1348-9. [CrossRef]
- Rothgangl T, Dennis MK, Lin PJC, Oka R, Witzigmann D, Villiger L, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat Biotechnol. 2021;39(8):949-57. [CrossRef]
- Janssen P, Isa T, Lanciego J, Leech K, Logothetis N, Poo MM, et al. Visualizing advances in the future of primate neuroscience research. Curr Res Neurobiol. 2023;4:100064. [CrossRef]
- Westerling-Bui AD, Fast EM, Soare TW, Venkatachalan S, DeRan M, Fanelli AB, et al. Transplanted organoids empower human preclinical assessment of drug candidate for the clinic. Sci Adv. 2022;8(27):eabj5633. [CrossRef]
- Evangelisti C, Ramadan S, Orlacchio A, Panza E. Experimental Cell Models for Investigating Neurodegenerative Diseases. Int J Mol Sci. 2024;25(17). [CrossRef]
- Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, Ward ME. Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr Protoc Cell Biol. 2018;79(1):e51. [CrossRef]
- Giacomelli E, Vahsen BF, Calder EL, Xu Y, Scaber J, Gray E, et al. Human stem cell models of neurodegeneration: From basic science of amyotrophic lateral sclerosis to clinical translation. Cell Stem Cell. 2022;29(1):11-35. [CrossRef]
- Answer ALS. Answer ALS [Internet]. New Orleans (LA): Answer ALS; Available online: https://www.answerals.org (accessed on 27 May 2025).
- Okano H, Morimoto S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell. 2022;29(2):189-208. [CrossRef]
- Grass T, Dokuzluoglu Z, Buchner F, Rosignol I, Thomas J, Caldarelli A, et al. Isogenic patient-derived organoids reveal early neurodevelopmental defects in spinal muscular atrophy initiation. Cell Rep Med. 2024;5(8):101659. [CrossRef]
- Faravelli I, Rinchetti P, Tambalo M, Simutin I, Mapelli L, Mancinelli S, et al. Targeted Antisense Oligonucleotide Treatment Rescues Developmental Alterations in Spinal Muscular Atrophy Organoids. bioRxiv 2025. [CrossRef]
- Szebenyi K, Wenger LMD, Sun Y, Dunn AWE, Limegrover CA, Gibbons GM, et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat Neurosci. 2021;24(11):1542-54. [CrossRef]
- Gao C, Shi Q, Pan X, Chen J, Zhang Y, Lang J, et al. Neuromuscular organoids model spinal neuromuscular pathologies in C9orf72 amyotrophic lateral sclerosis. Cell Rep. 2024;43(3):113892. [CrossRef]
- van der Geest AT, Jakobs CE, Ljubikj T, Huffels CFM, Canizares Luna M, Vieira de Sa R, et al. Molecular pathology, developmental changes and synaptic dysfunction in (pre-) symptomatic human C9ORF72-ALS/FTD cerebral organoids. Acta Neuropathol Commun. 2024;12(1):152. [CrossRef]
- U.S. Food and Drug Administration. Rare diseases: natural history studies for drug development. Draft guidance for industry. FDA-2019-D-0481. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rare-diseases-natural-history-studies-drug-development (Accessed on 25 October 2025).
- Alqahtani A, Kokkinis A, Zizzi C, Dilek N, Fischbeck KH, Heatwole CR, et al. Patient-Reported Impact of Symptoms in Spinal and Bulbar Muscular Atrophy. Neurol Clin Pract. 2023;13(6):e200213. [CrossRef]
- Grunseich C, Patankar A, Amaya J, Watts JA, Li D, Ramirez P, et al. Clinical and Molecular Aspects of Senataxin Mutations in Amyotrophic Lateral Sclerosis 4. Ann Neurol. 2020;87(4):547-55. [CrossRef]
- FDA-NIH Biomarker Working Group, 2017, BEST (Biomarkers, EndpointS, and other Tools) Resource, Silver Spring, Maryland: Food and Drug Administration, and Bethesda, Maryland:National Institutes of Health Available online https://www.ncbi.nlm.nih.gov/books/NBK326791/pdf/Bookshelf_NBK326791.pdf (accessed on October 2025).
- Klickovic U, Zampedri L, Sinclair CDJ, Wastling SJ, Trimmel K, Howard RS, et al. Skeletal muscle MRI differentiates SBMA and ALS and correlates with disease severity. Neurology. 2019;93(9):e895-e907. [CrossRef]
- Tebbenkamp AT, Huggett SB, Lombardi V, Zampedri L, AlQahtani A, Kokkinis A, et al. Protein biomarker signature in patients with spinal and bulbar muscular atrophy. JCI Insight. 2024;9(13). [CrossRef]
- Xing X, Liu X, Li X, Li M, Wu X, Huang X, et al. Insights into spinal muscular atrophy from molecular biomarkers. Neural Regen Res. 2025;20(7):1849-63. [CrossRef]
- Ruffo P, Traynor BJ, Conforti FL. Advancements in genetic research and RNA therapy strategies for amyotrophic lateral sclerosis (ALS): current progress and future prospects. J Neurol. 2025;272(3):233. [CrossRef]
- Arbab M, Matuszek Z, Kray KM, Du A, Newby GA, Blatnik AJ, et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science. 2023;380(6642):eadg6518. [CrossRef]
- Alves CRR, Ha LL, Yaworski R, Sutton ER, Lazzarotto CR, Christie KA, et al. Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy. Nat Biomed Eng. 2024;8(2):118-31. [CrossRef]
- Kempthorne L, Vaizoglu D, Cammack AJ, Carcolé M, Roberts MJ, Mikheenko A, et al. Dual-targeting CRISPR-CasRx reduces C9orf72 ALS/FTD sense and antisense repeat RNAs in vitro and in vivo. Nat Commun. 2025;16(1):459. [CrossRef]
- McCallister TX, Lim CKW, Singh M, Zhang S, Ahsan NS, Terpstra WM, et al. A high-fidelity CRISPR-Cas13 system improves abnormalities associated with C9ORF72-linked ALS/FTD. Nat Commun. 2025;16(1):460. [CrossRef]
Figure 1.
Gene therapy clinical trials and approved drugs. (a) Timeline of gene or cell therapy clinical trials that have been conducted in years between 1991-2025. Data is retrieved from World Health Organization International Clinical Trials Registry Platform search portal (https://trialsearch.who.int/Default.aspx). (b) Timeline of gene therapy drugs, approved for clinical use. Note that withdrawn drugs were also included to show advancements over the years. Lentiviruses are not depicted independently of retroviruses. Created with BioRender.com.
Figure 1.
Gene therapy clinical trials and approved drugs. (a) Timeline of gene or cell therapy clinical trials that have been conducted in years between 1991-2025. Data is retrieved from World Health Organization International Clinical Trials Registry Platform search portal (https://trialsearch.who.int/Default.aspx). (b) Timeline of gene therapy drugs, approved for clinical use. Note that withdrawn drugs were also included to show advancements over the years. Lentiviruses are not depicted independently of retroviruses. Created with BioRender.com.
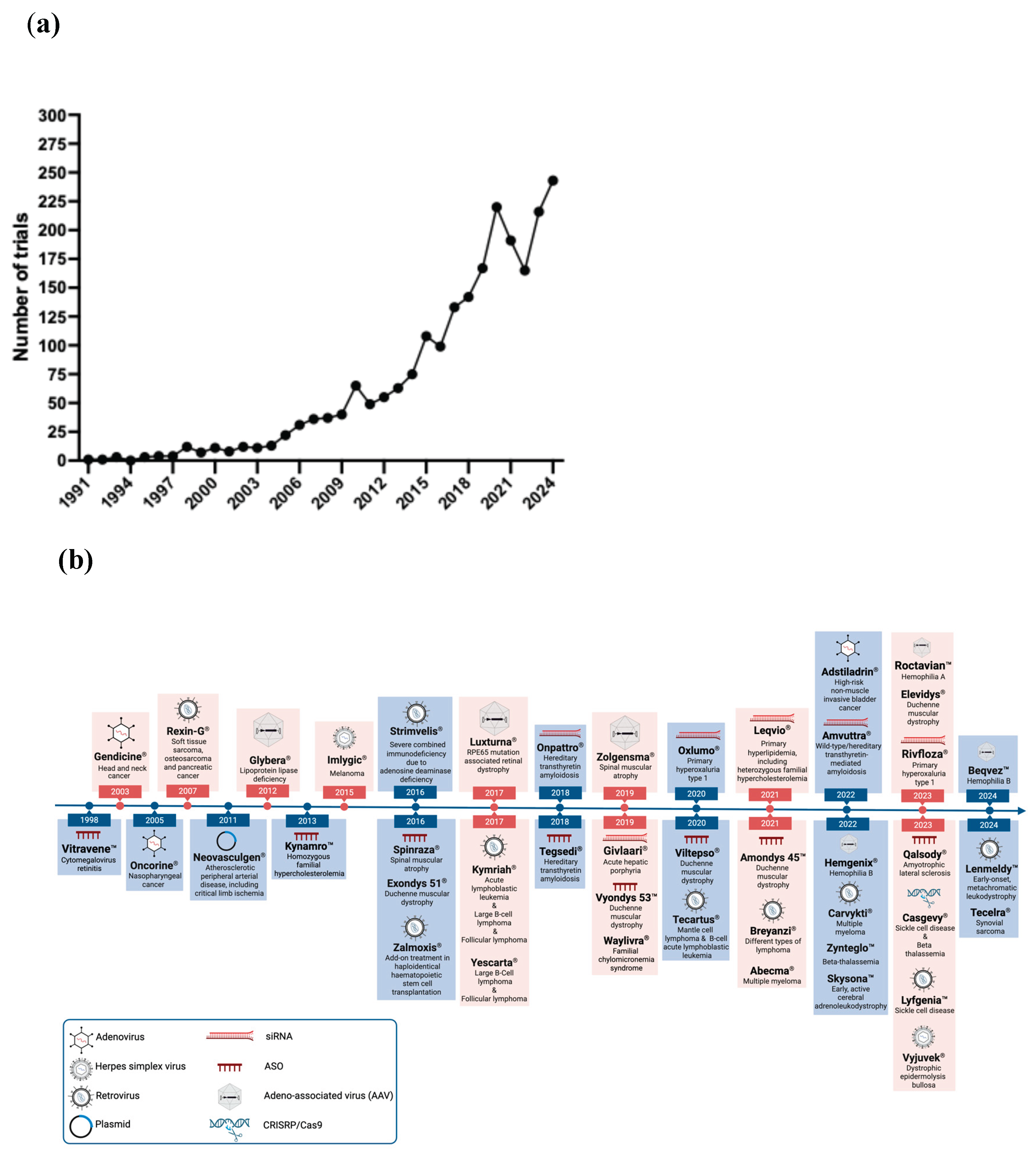
Figure 2.
Vectors, delivery methods and gene-therapy strategies. Created with BioRender.com.
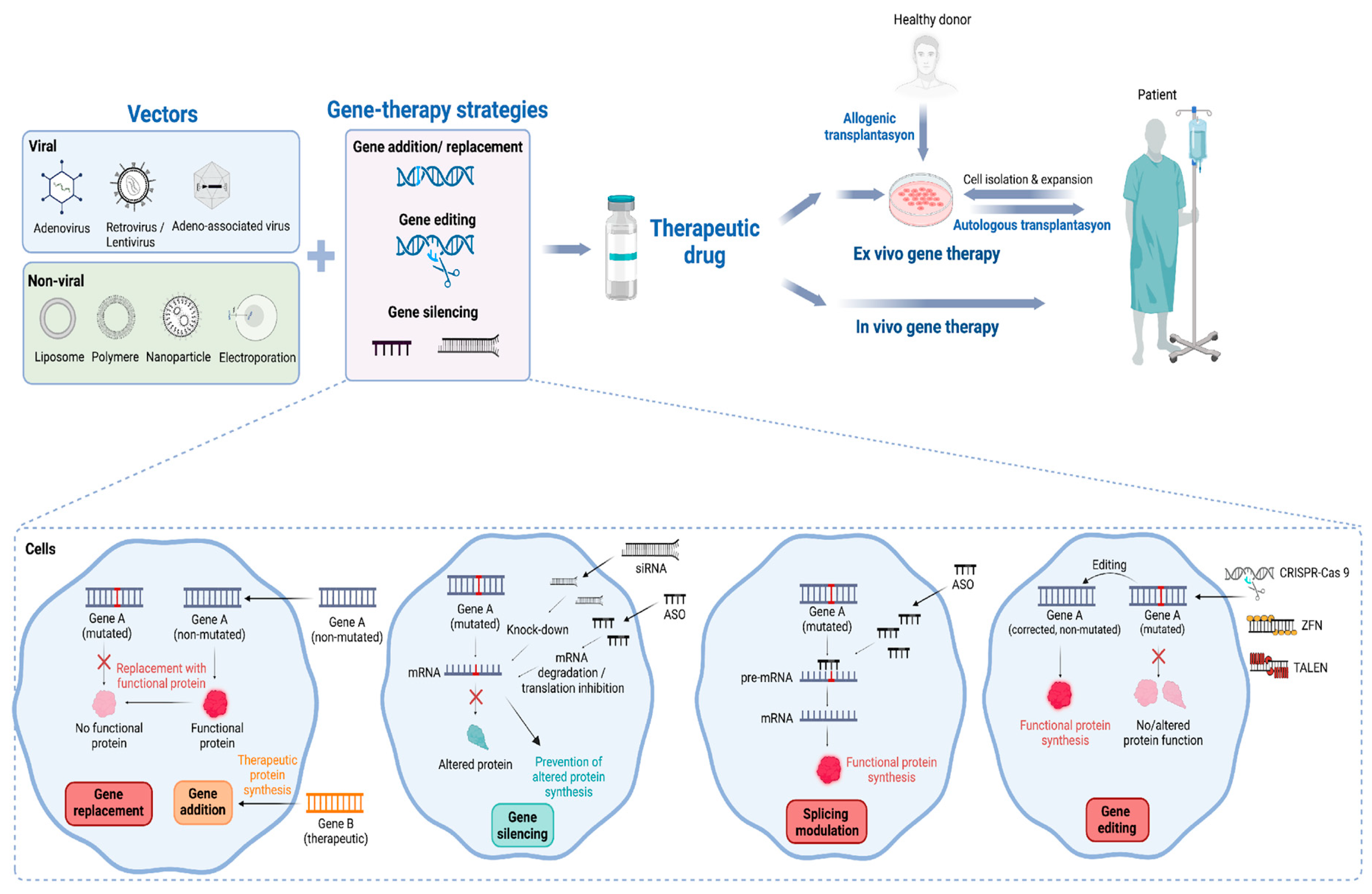
Figure 3.
Mechanisms of clinically approved therapies for spinal muscular atrophy. Spinraza induces exon 7 inclusion in SMN2-encoded mRNA by preventing the binding of hnRNP A1. Zolgensma enables the synthesis of functional SMN protein through SMN1 gene replacement using the scAAV9 vector. Created with BioRender.com.
Figure 3.
Mechanisms of clinically approved therapies for spinal muscular atrophy. Spinraza induces exon 7 inclusion in SMN2-encoded mRNA by preventing the binding of hnRNP A1. Zolgensma enables the synthesis of functional SMN protein through SMN1 gene replacement using the scAAV9 vector. Created with BioRender.com.
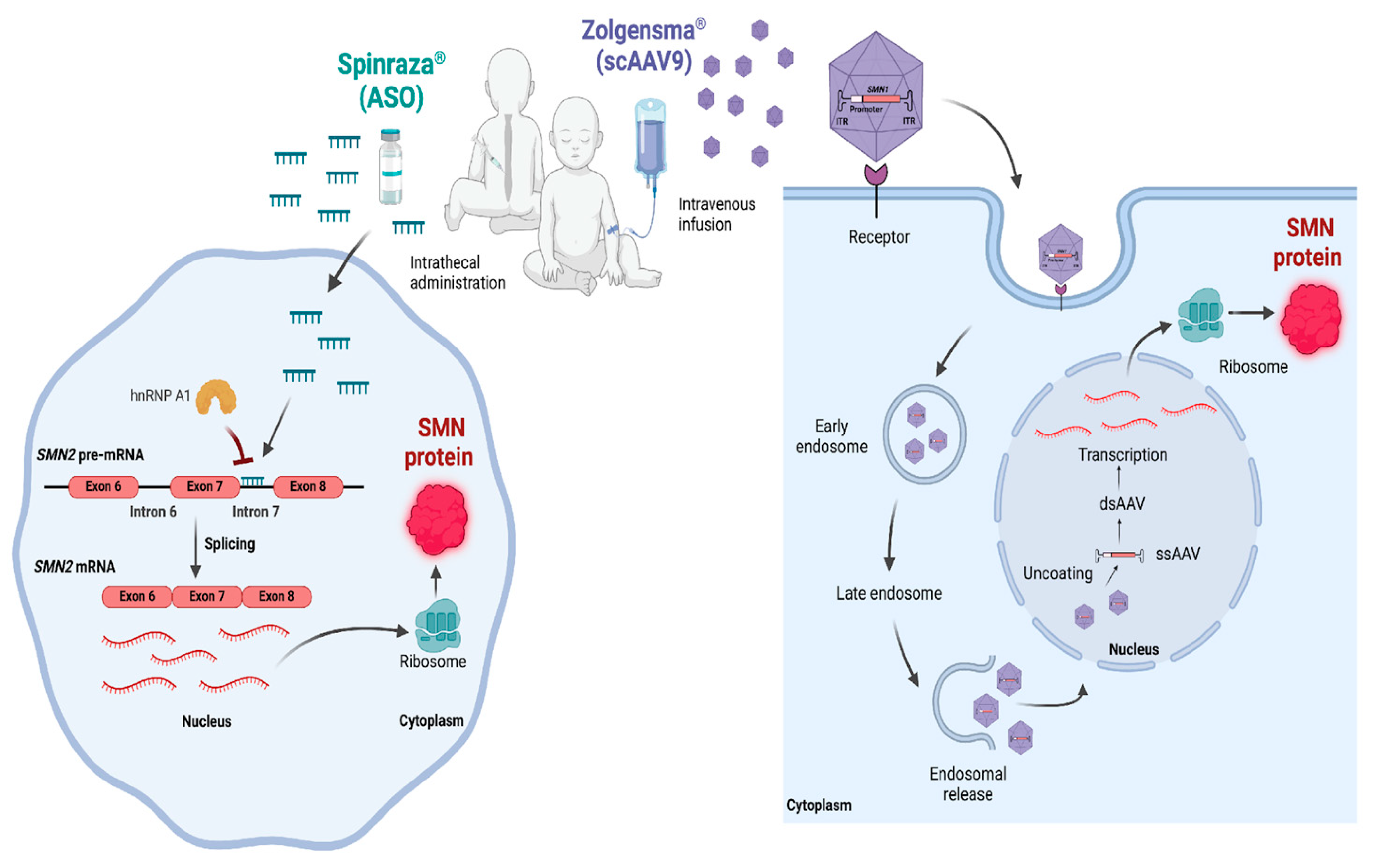
Figure 4.
Mechanisms of clinically approved therapy for ALS. Qalsody (Tofersen) prevents the aggregation of mutant SOD1 protein via inducing mRNA degradation. Created with BioRender.com.
Figure 4.
Mechanisms of clinically approved therapy for ALS. Qalsody (Tofersen) prevents the aggregation of mutant SOD1 protein via inducing mRNA degradation. Created with BioRender.com.
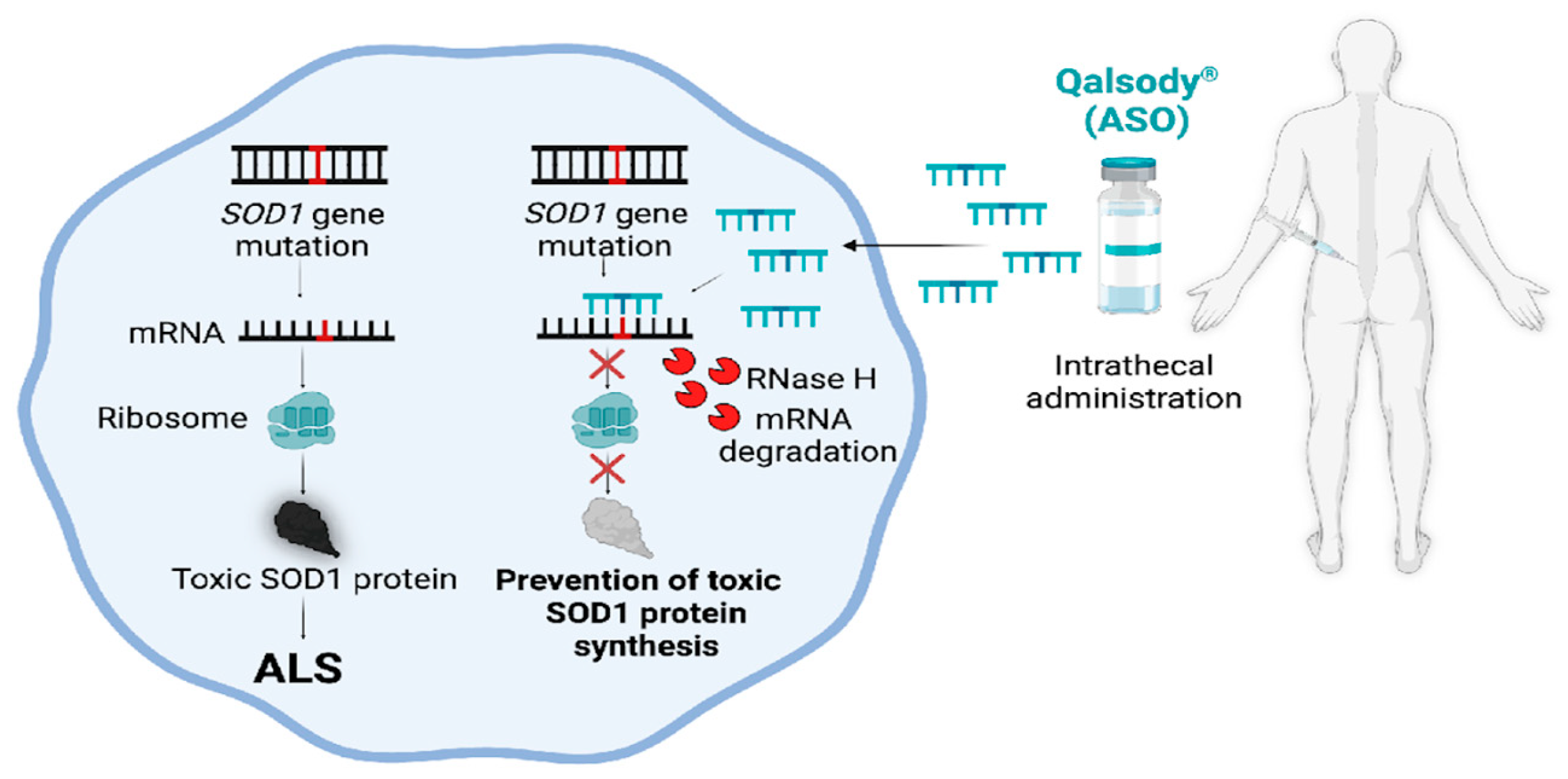

Table 1.
Current ALS gene-targeted therapies in clinical trials.
| National Clinical Trial Identifier | Target | Interventions | Modality | Phases | Study Status |
|---|---|---|---|---|---|
| NCT03764488 NCT02623699 NCT03070119 NCT04856982 |
SOD1 | BIIB067 (Tofersen) | ASO | III | Approved for marketing |
| NCT05903690 NCT05903690 |
SOD1 | RAG-17 | RNAi | I | Completed Recruiting |
| NCT06100276 | SOD1 | AMT-162 (APB-102) |
AAV-RNAi (miRNA) | I/II | Active, not recruiting |
| NCT04768972 | FUS | ION363 (Jacifusen) | ASO | III | Active, not recruiting |
| NCT04632225 NCT05176093 NCT02039401 |
HGF addition | Engensis (VM202) | Non-viral plasmid gene delivery | II | Completed |
| NCT05633459 | STMN2 | QRL-201 | ASO | I | Active, not recruiting |
*ASO, antisense oligonucleotide; RNAi, RNA interference; NA, not applicable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



