
Submitted:
10 February 2026
Posted:
12 February 2026
You are already at the latest version
Abstract
Thermochemical energy storage (TCES) using metal oxides offers a promising path for applications requiring high-temperature heat and long duration storage. Among numerous mixed metal oxides that have been studied for TCES, magnesium manganese oxide (MgMnO) has emerged as a promising candidate. A significant challenge of using this material in shaped form is crack formation during manufacturing and redox cycling. To develop mitigation strategies, we have first used a novel additive manufacturing process for manufacturing rods and plates of MgMnO that allows to rapid material mixing and produce a variety of shapes with relative ease. Graphite powder was utilized as pore-forming material to prevent the material from cracking and also to increase the porosity, thereby improving the thermal storage. After cycling, samples demonstrated high energy density that remained consistent. The introduction of graphite powder appeared to significantly reduce cracks, and yielded rods with over 17.24% higher energy density, and improved cyclability.
Keywords:
thermochemical energy storage
; metal oxide
; magnesium manganese oxide
; experimental setup
1. TOC Graphic
The growing global demand for clean and dispatchable energy has led to the development of thermal energy storage technologies to bridge the gap between intermittent energy supply and variable end-use demand. Thermochemical energy storage (TCES) systems face challenges in ensuring efficient heat and mass transfer within reactors, managing pressure drops, and developing cost-effective, durable materials. Sintering caused by coarsening at high temperatures during the reaction is a main limitation of several TCES materials. A wide array of candidate materials have been explored for TCES, spanning metal chlorides (e.g., CaCl2·H2O), zeolites, silica gels, metal hydrides, carbonates (e.g., CaCO3/CaO), hydroxides, ammonia–water chemisorption pairs, organics, and redox-active metal oxides [1]. Each material group has its limitations. Metal chlorides and hydrides often exhibit very high reaction enthalpies but may suffer from corrosivity, hygroscopicity, or challenging regeneration conditions. Carbonates offer robust cycling at high temperatures but require substantial thermal input for calcination. Sorption materials like zeolites and silica gels combine moderate densities with rapid kinetics but are limited by relatively low reaction enthalpies and complex reactor designs [2]. Ammonia–water pairs deliver favorable economics in closed systems but involve handling risks. Metal oxides, in contrast, strike a balance of high energy density, thermal stability, environmental friendliness, and relatively low material costs, making them among the most promising for high-temperature, grid-scale applications [3].
Magnesium manganese oxide has emerged as particularly attractive candidate among mixed metal oxides. This material forms a non-stoichiometric spinel structure (Mg0.5Mn0.5)3– O4 that undergoes highly endothermic oxygen-release (charging) at around 1500 °C and exothermic re-oxidation (discharging) at nearly 1000 °C via the following simplified reaction:
where x is the molar ratio of Mg to Mn, and y1 and y2 are the excess oxygen in the material at the oxidized and reduced states, respectively. Mixed oxides also improve reactive stability over pure manganese oxide, delaying reduction onset and enabling lower-temperature operation [4]. Additionally, magnesium acts as an anti-sintering agent. Recent experimental investigations have focused on optimizing manganese to magnesium ratios, redox kinetics, and material cyclability. King et al. [4] prepared solid-state-synthesized powders with Mn:Mg molar ratio of 2:3, 1:1, and 2:1 and identified the 1:1 molar ratio as providing the highest chemical plus sensible gravimetric energy density (1,029±57 kJ/kg at 1500 °C/0.01 atm reduction, 1000 °C oxidation) and peak volumetric density around this ratio. Randhir et al. [5] conducted tube furnace redox cycling of 10 g batches (4-hour reduction at 1500 °C in Ar, 1-hour oxidation at 1000–1500 °C in Ar/O2 mixtures) with on-line mass spectrometry, demonstrating reproducible oxygen exchange profiles and enabling kinetic modeling under varying oxygen pressure and temperature conditions [6]. More recent sol–gel syntheses and doping studies (e.g., Fe, Co, Cu, Zn, Ni at 0–20 mol%) have shown that modest Fe or Cu incorporation can further enhance kinetics, reduce hysteresis, and improve cyclic stability, pointing toward tunable formulations for specific operating windows [7]. Taken together, these efforts underscore magnesium manganese oxide as a leading TCES material class for high-temperature, grid-scale storage, combining high energy densities, favorable thermodynamics, and robust cycling behavior.
Functionality of MgMnO at different temperatures, both for oxidation and reduction reactions, has been investigated by Randhir et al. [5], which showed that the energy density will increase as the reduction temperature rises, while storage efficiency decreases with temperature rise. The optimum temperatures to reach 850 kJ/kg energy density have been reported as 1500 and 1000 °C for reduction and oxidation, respectively. Packed bed form of MgMnO pellets has also been constructed and tested under realistic thermodynamic conditions, performing well with good energy density and cyclability, and no major problems during the experiments (Rahmatian et al. [8]). Thermal conductivity of the packed bed MgMnO was determined as well using the sheathed hot-wire method [9]. The range was reported to be 0.5-1.81 W/m °C between 306 and 1393 °C. In addition, moving-bed reactors of MgMnO were studied by Schimmels [10] and Hayes et al. [11]. A vertical reactor was designed and constructed that employed MgMnO particle flow to carry out the redox reactions. An air counterflow through the particles was applied to cool the outlet particles and preheat the inlet particles. This approach enabled continuous energy storage and maintained the inlet and outlet particles at room temperature. King et al. [4] investigated the oxygen pressure during reduction. Lowering the oxygen pressure significantly improved the energy density up to 55% depending on the molar ratio. They also performed doping MgMnO with cobalt, iron, zinc, and nickel oxides, none of which showed any improvement in reactivity of the material. However, Hashimoto et al. [7] found that doping it with 0.5%/g iron increases the reduction rate, but higher amounts were shown to be counterproductive. Rahman et al. [12] experimented a packed bed reactor with BaO2 pellets that were mixed with 15 wt% MgO to reach more stability which also resulted a volumetric energy density of around 627.9 MJ/m3. Ortiz-Ulloa et al. [13] investigated the effect of particle size on a moving bed reactor and found that oxidation is controlled by reaction kinetics in particles smaller than 6 mm, whereas diffusion limits the process for larger particles. Major challenges observed using flowing pelletized material include low radial heat diffusion through the material [14], which limits scalability. In addition, ensuring flowability of the particles requires gas bypasses and sophisticated L-valves for particulate flow control.
In this research, we have tried to shape MgMnO in forms such as plates and rods, suitable for developing a fixed bed thermal battery. The thermal storage material is placed and fixed inside the reactor. During reduction a sweeping gas passes through the material, while air is flowed during oxidation. A major challenge faced with such fixed bed system is cracking or deformation of the material, either during manufacturing or cycling [15].
MgO and MnO powders obtained from Sigma Aldrich with particle sizes of 250 m and 45 m respectively were mixed with 1:1 molar ratio. They were then ball milled inside a plastic jar with ceramic pellets using a roller mixer. In order to investigate the effect of particle sizes, mixed powders were prepared with and without the ball milling process. Moreover, different weight percentages of graphite as well as different particle sizes of graphite were used to prepare different samples (See Table 1). The graphite powders obtained from Asbury Carbons were mixed with MgMnO powder before the printing process.
In order to print the samples, a 3D printing system, so-called scalable and expeditious additive manufacturing (SEAM), was utilized [16,17]. SEAM integrates the advantages of binder jet printing and stereolithography by UV-curing a metal-powder mixed with UV-curable resin suspension, yielding smooth surfaces, high resolution, and low residual stress while maintaining throughput suitable for high-volume production. This process is widely used in the ceramic industry, and it has been developed for metals by Nguyen et al. [18]. Because it is powder-bed-based, surrounding powder provides inherent support for overhangs, eliminating dedicated support structures during printing. In this system, CPS 3010 UV-curable resin is first mixed with the material. The suspension can be shaped by curing the polymer using UV light. This UV curing step is integrated into the powder bed process, so each layer solidifies under UV light before the print bed moves down for the next layer. The system uses a powder bed setup where the print bed moves down by one layer each time until the part is finished. The thickness of each layer is 150 m. A top-down deposition unit replaces the usual supply bed and also mixes the material. During each cycle, the mixer deposits just enough suspension for one layer and keeps it well stirred, then a spreading blade spreads that layer onto the print bed. Next, a digital DLP projector shines the precise pattern with UV light for that layer onto the bed, after which the bed lowers by 150 m. Curing tests confirmed that a 150 µm layer is fully cured with 55 seconds of DLP exposure, balancing complete curing with high printing speed. These steps repeat continuously until the entire 3D part is built (See Figure 1).
After the printing process, the polymer is combusted out of the sample. The graphite needs to be oxidized out to leave behind a porous structure. The sample thus undergoes a process called debinding, where it is heated to 800 °C for 8 hours. During this process, the polymer is eliminated but the graphite is oxidized out to leave behind a porous structure. Doing so, MgMnO particles also form a "necking" structure, avoiding collapse after binder and graphite are burned out. The sample is still fragile at this stage. It then goes through the sintering process to fuse the particles and create the desired metal oxide. For this purpose, the sample is heated to 1500 °C for 20 hours, after which a rigid rod with a porous structure is obtained, consisting of only MgMnO. When building the sample without graphite, minor cracks were observable on the surface of the sample after sintering. These cracks were eliminated when graphite was added during printing, which suggests that pore forming of graphite also has the advantage of significantly reducing crack formation. Figure 2 shows the sample with 5 wt% graphite at different stages. The top green rod is the sample after printing, and the white rod in the middle and the black rod at the bottom are the samples after debinding and sintering, respectively.
Since the rods showed an increase in the chemical energy density with good cycling stability, which will be discussed later, a mold was designed to make additional rods rapidly. At this stage, polyvinyl alcohol (PVA) was utilized instead of UV-cured polymer to mold the MgMnO. It is first mixed well with MgMnO powder, and then poured into the mold. When the mold is fastened, it is kept in room temperature for 12 hours to become solid. After molding, the same debinding and sintering processes were done. Samples which were produced by this method also showed no cracks on their surface when graphite powder was used. In addition, the same graphite percentage resulted in the same oxygen absorption capacity compared to the samples that were 3D printed, confirming that molding has the same output for same mixture of material. As a result, the manufacturing method does not have an impact on the results.
For cycling the samples through reduction and oxidation reactions, a Sentrotech STT-1700-2-6 horizontal tube furnace with a 1700 °C maximum temperature and a power of 4 kW was used. The sample is put inside the tube, and both ends of the tube are fastened with flanges. Based on the reaction, nitrogen for reduction and air for oxidation could be inserted into the tube through flanges. An Alicat MCR gas flow controller was used to keep the entering gas flow to the tube at 1 SLPM. Moreover, an AMI Model 70 oxygen analyzer was used to measure the oxygen percentage of the output gas from the tube, both in oxidation and reduction reactions. All data from measuring devices were transferred to a Labjack T7-pro data acquisition system that was connected to a PC. Signals taken from the flow controller and oxygen analyzer were sampled continuously at 1 Hz and logged with Labview. In order to perform the reduction reaction, the tube furnace was set to reach 1500 °C with a 5 °C/min ramp rate and with the nitrogen flow present inside the tube. For the oxidation reaction, the furnace was set to reach 1000 °C with the same ramp rate and with air flow present inside the tube. After each reduction and oxidation, which form one cycle, the amount of oxygen being absorbed or released by the sample can be measured. These measured values match since they correspond the same amount of oxygen being released or absorbed. They were measured by a Mettler Toledo XS105 scale with an accuracy of 0.01 mg. This amount is directly related to the chemical energy density of the sample. The following equation is used to estimate the gravimetric energy storage density ():
where is the difference in enthalpy of formation per mole of excess oxygen between reduced and oxidized MgMnO sample, nO2 is the amount of oxygen stored (oxidation) or released (reduction) by the sample in moles. These two terms together form the chemical energy density of the material. is the sensible heat change between the reduced and oxidized state.
Figure 3 shows the percentage of oxygen in the output gas from the tube furnace, both for reduction and oxidation reactions. As the temperature rises, the sample releases oxygen during phase change, resulting in a small increase in the measured oxygen percentage for a short period during the reduction process. During oxidation, the sample absorbs oxygen as the temperature rises, which causes a temporary decrease in oxygen measured. These time periods in redox reaction depend on the sample mass and vary with the sample dimensions. The oxygen measured during reduction has two peaks. The first peak corresponds to a surface layer of MgMnO that reaches the required reduction temperature. The second peak is interpreted as corresponding to the release of oxygen associated with the diffusion of heat inside the sample [21]. This likely indicates that the temperature ramp rate of 5 °C/min is higher than the sample temperature due to delay in heat diffusion. Complete reduction is nonetheless achieved as the sample is held at 1500 °C after no more oxygen appears to be released by the sample. The oxidation graph, however, has only one peak, corresponding to internal oxidation which releases heat internally. To ensure that the experiment was repeatable, each sample was cycled five times. The maximum difference in the amount of oxygen absorbed between all five cycles was 0.6 cm3/g.
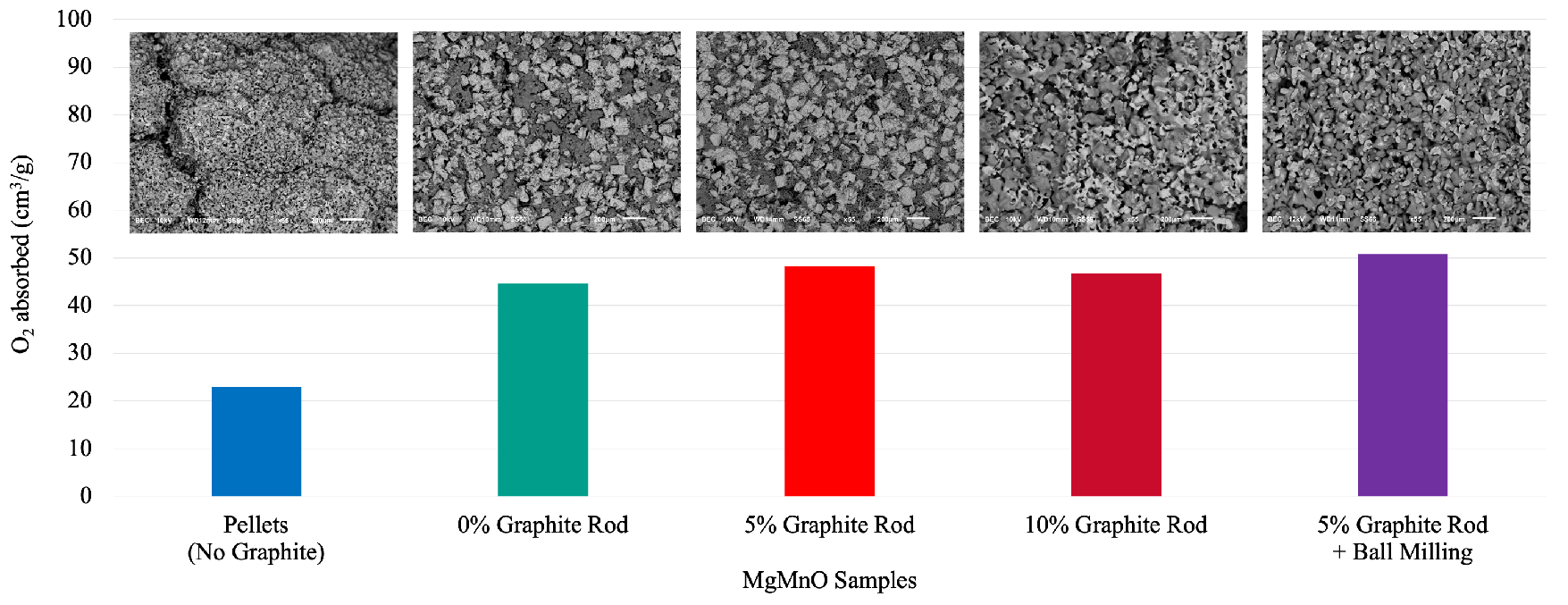
To determine the morphological changes of the material during reduction/oxidation, the SEM images of the samples are presented in Figure 4. The images on the top row show the samples at reduced state, and the bottom images show the samples at oxidized state. Figures belong to the pellet obtained following [10], rod with no graphite, rod with 5 wt% graphite, rod with 10 wt% graphite, and rod with 5 wt% graphite that is made with ball-milled MgMnO. The pore-forming effect of graphite is observable especially in the oxidized phase. Also, the effect of ball-milling on the structure uniformity is visible in the image. The sizes and the shapes of the dark spots which are the pores of the samples vary from left to right, representing the difference in pore forming by varying graphite amount and integration of the ball milling process.
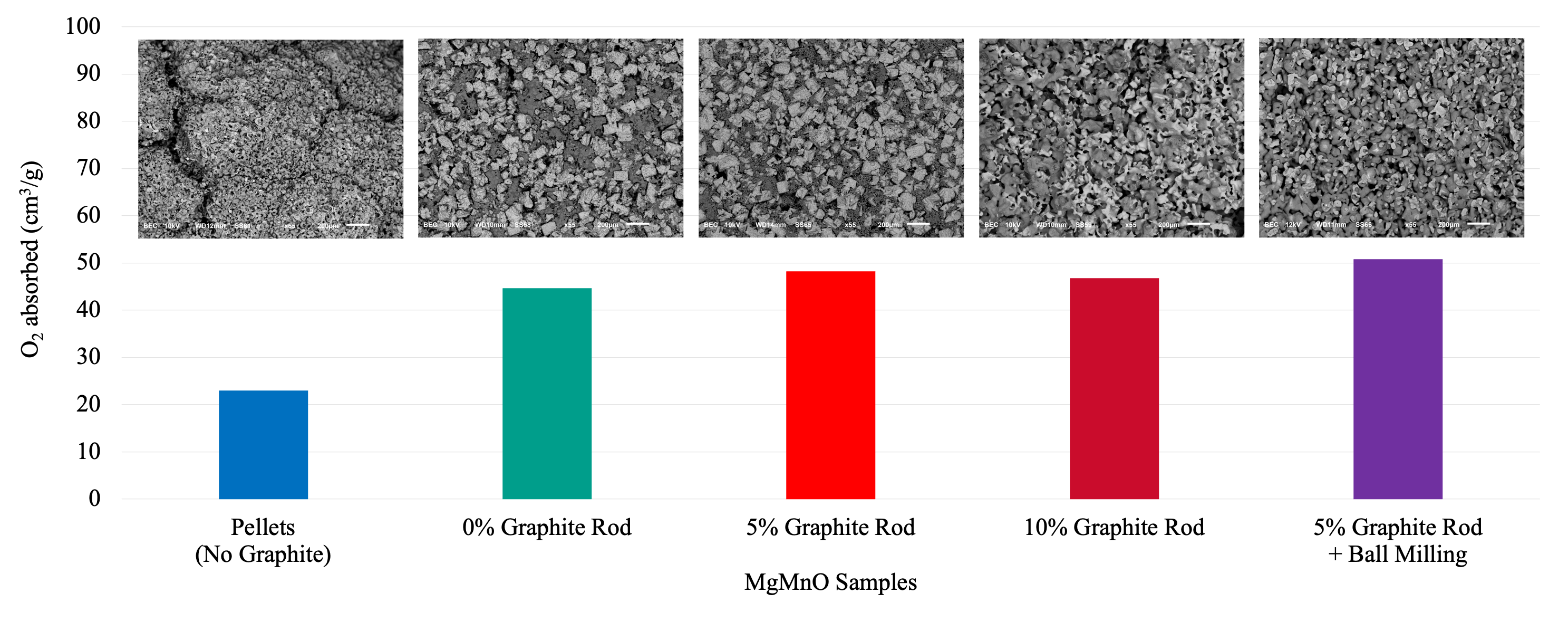
The amount of oxygen being absorbed/released by various samples is shown in Figure 5. Compared to pelletized MgMnO, the rod-shaped sample with no graphite shows a higher capacity for oxygen absorption. Although this sample shows an increase in chemical energy density, minor cracks were observed on its surface. By adding 5 wt% graphite during production, an increase in oxygen absorption capacity was achieved compared to the rod with no graphite. In addition, it resolved the crack issue. No cracks were observed on the samples with graphite. As the graphite burns out during the debinding process, the porous structure is formed. This porosity helps to inhibit crack propagation during sintering and redox processes. In addition, it improves gas flow through the sample. Increasing the amount of graphite to 10 wt% resulted in a negative effect compared to the sample with 5 wt% graphite, which indicates that the optimum amount of graphite is somewhere between 0 and 10 wt%. The sample made with ball-milled MgMnO and 5 wt% graphite showed even higher oxygen absorption capacity, illustrating the effect of ball-milling to reach a uniform microstructure from solid state processing. The uniform microstructure was observable in the SEM images that were discussed in the previous section.
To investigate the possibility of manufacturing channels, two hollow samples were constructed and tested as well. The cross section of these two samples corresponded to a hollow square with two different wall thicknesses, 5.42 and 2.8 mm. Although they differed in cross section geometry, no significant difference was observed in their chemical energy density compared to conventional rods. This suggests that thickness of 5.42 mm is not yet limiting gas transport. The effect of more complex geometries has not been investigated in this study, and will be carried out in future works.
To test the higher amounts of graphite on the pressed sample, three other samples with 25, 40, and 60 wt% graphite were molded and produced. Redox experiment result is presented in Figure 6 (right), which shows the negative impact of high graphite percentage. At higher amounts of graphite, the pore-forming process adversely affects the oxygen absorption capacity. The effect of graphite particle size was also investigated. Keeping the graphite amount constant at 5 wt%, four different particle size ranges were studied. One sample with the particle size between 150 to 850 m, one with 44, and two others between 7 to 11 and 3 to 7 m. Figure 6 (left) shows the effect of different particle sizes of graphite. The sample with particle size between 7-11 m show the best result.
The cross-section of the rod affects both the oxygen absorption capacity and reaction time. Three different samples with the same length and height, but different width were prepared and tested. As the cross-section shrinks, the oxygen absorption capacity increases, and the reaction time shortens. This is explained by the diffusion of oxygen. The higher the cross-section, the lower the oxygen diffusion, which results in a lower reaction rate. A linear correlation was observed between oxygen absorption capacity and cross-section, and also reaction time with cross section. Based on different applications (e.g. long time heat release, quick and high heat release, etc) the material can be optimized for the best efficiency. Figure 7 shows these relations together.
Accurate enthalpy of formation values are needed to estimate the energy storage capacity of samples as processed. To verify the reported enthalpy of formation values, the enthalpy of dissolution is measured below using acid solution calorimetry [22]. A setup was prepared including a mechanical stirrer, PFA-coated thermocouple, insulation, and data acquisition to measure the enthalpy of dissolution. Hydrochloric acid was used as the solution inside a Dewar flask, and MgO, MnO, Mg, and Mn powders were weighted and poured into the acid to dissolve in order to measure the temperature rise separately. This measurement was repeated three times with different amounts, and a linear trend was obtained for each material. The calorimetry was also done for MgMnO both after reduction and after oxidation, where the samples were ground using mortar and pestle under acetone to avoid oxidation. After the powder was dried, the same approach was applied on them. The relation between enthalpy of dissolution, temperature rise, and material weight is as follows.
where is the enthalpy of dissolution, T is the temperature rise, m is the mass, M is the molecular weight, and C is the calibration constant of the setup and is calculated by measuring the enthalpy of dissolution of the known materials. The value was calculated to be 1.514 ±0.394 kJ/°C for the setup. After measuring the enthalpy of dissolution of different compounds, the enthalpy of formation of MgMnO at reduced and oxidized phases was calculated using the derived relations between the enthalpy of formation for different reactions. Using the difference of enthalpy of formation of MgMnO at reduced and oxidized phases as well as the amount of absorbed oxygen, the chemical energy density can be calculated.
Figure 8 shows the chemical energy density for different cases. It appears that shaping MgMnO to rods leads to improved energy density compared to pelletized form. It appears that thin rods and plates of MgMnO can be utilized to manufacture a thermal battery.
In summary, a new shaping method for porous MgMnO was introduced to reduce cracking. It also enhanced its chemical energy density. The 3D printing and molding of MgMnO in different dimensions and geometries were successfully done, and cycling of the material was done on the rod shaped samples. Graphite powder was utilized to eliminate the cracks on the sample and to enhance the chemical energy density by pore forming during production of the material. The following results were obtained during the experiment:
- Rod-shaped MgMnO samples yielded up to 17.24% higher chemical energy density than packed bed forms for both additive manufacturing and molding approaches.
- Using graphite powder for pore forming during production appears to eliminate the formation of cracks.
- Porosity at micro-scale appeared to affect the oxygen absorption and increase the chemical energy density as a result. Both particle size and the weight percent of graphite influence the pore-forming of MgMnO. The porous samples that were ball-milled during production, yielded up to 8.67% higher energy density than conventional rods.
- As the cross-section of the MgMnO rod decreases at constant lengths, the reaction time shortens, and oxygen absorption increases. Thus, smaller cross-section results in more chemical energy density due to higher heat diffusion through the material.
Numerical simulation of more complex geometries, utilizing water as sweeping gas, and also microwave heating of the sample instead of conventional heating method are scheduled as future work to optimize MgMnO for industrial consumption in TCES industry.
Author Contributions
Farshid Kassaei, Philipp Schimmels, and Zhiyuan Qu: Literature search, Experimental setup, Data analysis, Visualization, Writing, and Original draft preparation. André Bénard, Haseung Chung, and James Klausner: Supervision, Review, and Editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, S.; Zhou, C.; Doroodchi, E.; Nellore, R.; Moghtaderi, B. A review on high-temperature thermochemical energy storage based on metal oxides redox cycle. Energy conversion and management 2018, 168, 421–453. [Google Scholar] [CrossRef]
- Vasta, S.; Brancato, V.; La Rosa, D.; Palomba, V.; Restuccia, G.; Sapienza, A.; Frazzica, A. Adsorption heat storage: State-of-the-art and future perspectives. Nanomaterials 2018, 8, 522. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Wu, H.B.; Xie, Y.; Lou, X.W. Mixed transition-metal oxides: design, synthesis, and energy-related applications. Angewandte Chemie International Edition 2014, 53, 1488–1504. [Google Scholar] [CrossRef] [PubMed]
- King, K.; Randhir, K.; Petrasch, J.; Klausner, J. Enhancing thermochemical energy storage density of magnesium-manganese oxides. Energy Storage 2019, 1, e83. [Google Scholar] [CrossRef]
- Randhir, K.; King, K.; Rhodes, N.; Li, L.; Hahn, D.; Mei, R.; AuYeung, N.; Klausner, J. Magnesium-manganese oxides for high temperature thermochemical energy storage. Journal of Energy Storage 2019, 21, 599–610. [Google Scholar] [CrossRef]
- Randhir, K.; King, K.; Petrasch, J.; Klausner, J. Oxidation Kinetics of Magnesium-Manganese Oxides for High-Temperature Thermochemical Energy Storage. Energy Technology 2020, 8, 2000063. [Google Scholar] [CrossRef]
- Hashimoto, J.; Bayon, A.; Tamburro, O.; Muhich, C.L. Thermodynamic and structural effects of Fe doping in magnesium manganese oxides for thermochemical energy storage. Energy & Fuels 2023, 37, 4692–4700. [Google Scholar] [CrossRef]
- Rahmatian, N.; Bo, A.; Randhir, K.; Klausner, J.; Petrasch, J. Bench-scale demonstration of thermochemical energy storage using the Magnesium-Manganese-Oxide redox system. Journal of Energy Storage 2022, 45, 103682. [Google Scholar] [CrossRef]
- Hayes, M.; Masoomi, F.; Schimmels, P.; Randhir, K.; Klausner, J.; Petrasch, J. Ultra-high temperature thermal conductivity measurements of a reactive magnesium manganese oxide porous bed using a transient hot wire method. Journal of Heat Transfer 2021, 143, 104502. [Google Scholar] [CrossRef]
- Schimmels, P. Development of a Thermo-Chemical Moving Bed Reduction Reactor for Charging a Pelletized Material Capable of Short-and Long-Term Energy Storage; Michigan State University, 2024. [Google Scholar]
- Hayes, M.; Korba, D.; Schimmels, P.; Klausner, J.; Petrasch, J.; AuYeung, N.; Li, L.; Randhir, K. Experimental demonstration of high-temperature (> 1000° C) heat extraction from a moving-bed oxidation reactor for thermochemical energy storage. Applied Energy 2023, 349, 121625. [Google Scholar] [CrossRef]
- Rahman, M.; Korba, D.; Zhao, J.; AuYeung, N.; Li, L. Thermochemical energy storage in a lab-scale packed-bed reactor using MgO supported BaO2/BaO redox system. Journal of Energy Storage 2025, 133, 117917. [Google Scholar] [CrossRef]
- Ortiz-Ulloa, J.; Rahman, M.; Randhir, K.; Li, L.; AuYeung, N. Design and Scale-Up of Direct-Contact Continuous Oxidation Reactors for High-Temperature Thermochemical Storage Using Concentrated Solar Energy. Energy & Fuels 2025. [Google Scholar]
- Schimmels, P.; Hayes, M.; Randhir, K.; Petrasch, J.; Klausner, J.F. Enhancing the chemical energy flux in a high-temperature tubular counterflow solid fuel synthesis reactor using a bypass. Industrial & Engineering Chemistry Research 2023, 62, 14671–14678. [Google Scholar] [CrossRef]
- Han, X.; Wang, L.; Ling, H.; Ge, Z.; Lin, X.; Dai, X.; Chen, H. Critical review of thermochemical energy storage systems based on cobalt, manganese, and copper oxides. Renewable and sustainable energy reviews 2022, 158, 112076. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Suen, H.; Poudel, B.; Qu, Z.; Ahmad, M.U.; Kwon, P.; Benard, A.; Chung, H. From Photopolymerization of Metal Suspension to Practical and Economical Additive Manufacturing of Haynes 214 Alloy for High Temperature Application. Proceedings of the International Manufacturing Science and Engineering Conference 2022, Vol. 86601, V001T02A006. [Google Scholar]
- Qu, Z.; Song, G.; Olortegui-Revoredo, J.; Kwon, P.; Chung, H. Dispersant-Induced Enhancement of Rheological Properties in Metal–Photopolymer Mixtures for 3D Printing. Journal of Manufacturing and Materials Processing 2025, 9, 244. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Suen, H.; Poudel, B.; Kwon, P.; Chung, H. Development of an innovative, high speed, large-scaled, and affordable metal additive manufacturing process. CIRP Annals 2020, 69, 177–180. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Poudel, B.; Qu, Z.; Kwon, P.; Chung, H. Photopolymerization of Stainless Steel 420 Metal Suspension: Printing System and Process Development of Additive Manufacturing Technology toward High-Volume Production. Journal of Manufacturing and Materials Processing 2024, 8, 191. [Google Scholar] [CrossRef]
- Panda, S.K.; Jung, I.H. Critical evaluation and thermodynamic modeling of the Mg–Mn–O (MgO–MnO–MnO 2) system. Journal of the American Ceramic Society 2014, 97, 3328–3340. [Google Scholar] [CrossRef]
- Schmalzried, H. Oxide solid solutions and its internal reduction reactions. Berichte der Bunsengesellschaft für physikalische Chemie 1984, 88, 1186–1191. [Google Scholar] [CrossRef]
- King, K.; Petrasch, J. Measurement of the thermochemical energy discharge process in reactive packed beds of magnesium-manganese oxide. PhD thesis, Michigan State University, 2021. [Google Scholar]
Figure 1.
The SEAM system is shown with the components used to 3D print the material [19].
Figure 1.
The SEAM system is shown with the components used to 3D print the material [19].

Figure 2.
Rod sample at different stages; 3D printed (top), debinded (middle), sintered (bottom). The change in appearance from debinding to sintering is likely due to the formation of a cubic spinel structure with MnO2 at high temperature [20].
Figure 2.
Rod sample at different stages; 3D printed (top), debinded (middle), sintered (bottom). The change in appearance from debinding to sintering is likely due to the formation of a cubic spinel structure with MnO2 at high temperature [20].

Figure 3.
Oxygen percentage and furnace temperature graph during reduction (top) and oxidation (bottom). Reduction is inferred by the release of oxygen between 800 and 1500 degrees, while oxidation occurs apparently between 600 and 1000 degrees.
Figure 3.
Oxygen percentage and furnace temperature graph during reduction (top) and oxidation (bottom). Reduction is inferred by the release of oxygen between 800 and 1500 degrees, while oxidation occurs apparently between 600 and 1000 degrees.
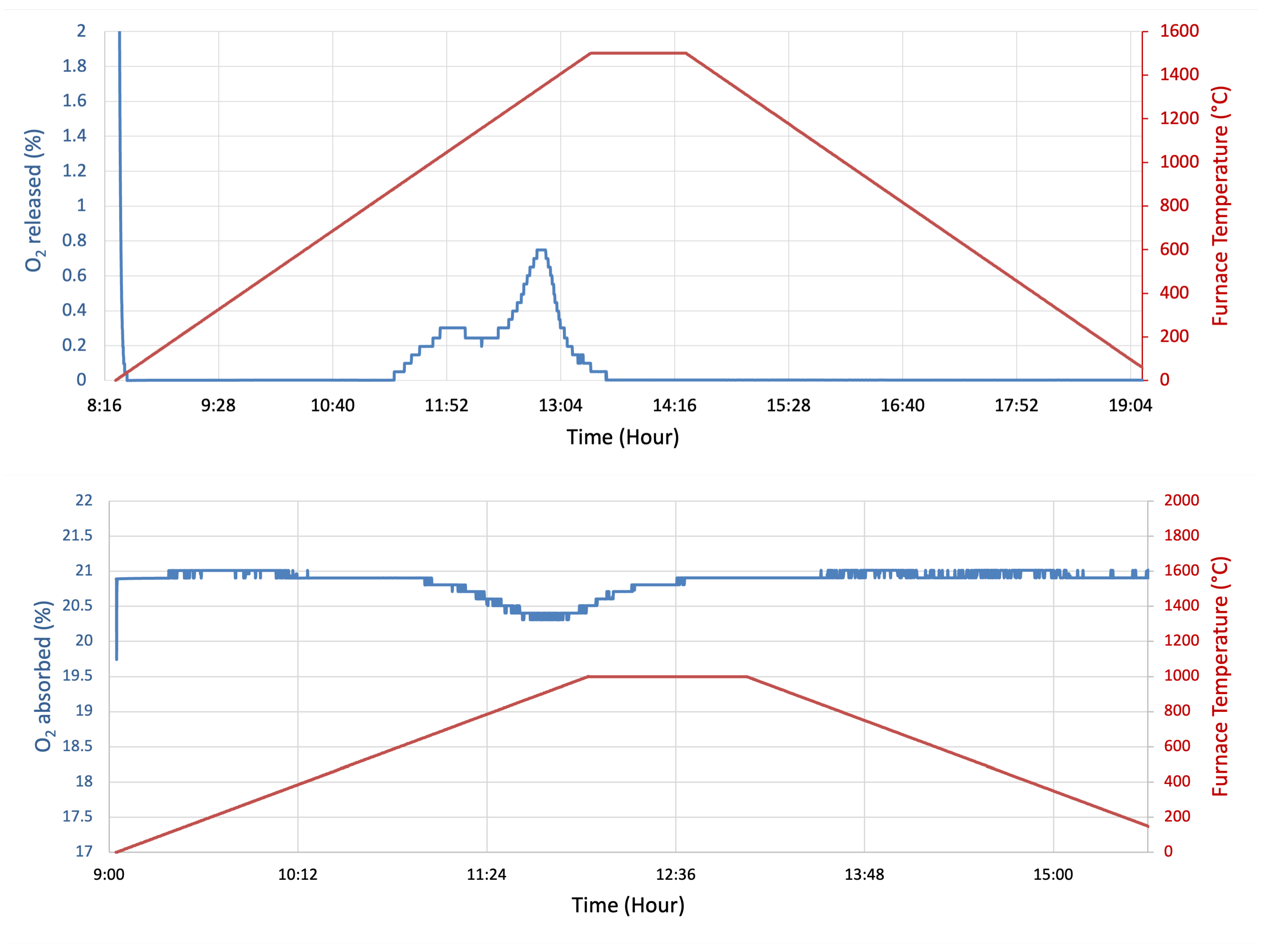
Figure 4.
SEM images at 55x magnification and scale of 200 m. The top row shows the samples at reduced phase, while bottom shows oxidized samples. The images represent pellet, rod with no graphite, rod with 5 wt% graphite, rod with 10 wt% graphite, rod with ball milling and 5 wt% graphite, respectively from left to right.
Figure 4.
SEM images at 55x magnification and scale of 200 m. The top row shows the samples at reduced phase, while bottom shows oxidized samples. The images represent pellet, rod with no graphite, rod with 5 wt% graphite, rod with 10 wt% graphite, rod with ball milling and 5 wt% graphite, respectively from left to right.
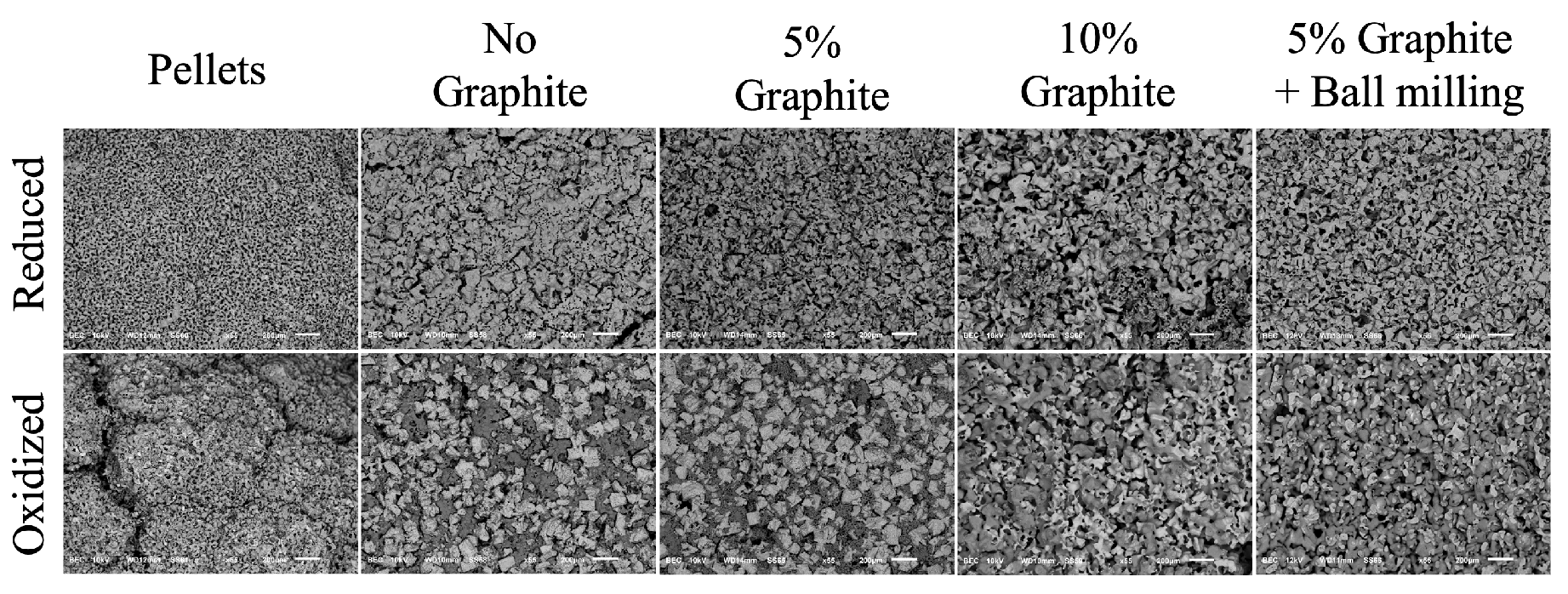
Figure 5.
Comparison of oxygen absorption between rods produced with different pore structures and pelletized MgMnO. The rod with 5 wt% graphite and ball milling process has the highest oxygen absorption due to higher and more uniform porosity.
Figure 5.
Comparison of oxygen absorption between rods produced with different pore structures and pelletized MgMnO. The rod with 5 wt% graphite and ball milling process has the highest oxygen absorption due to higher and more uniform porosity.
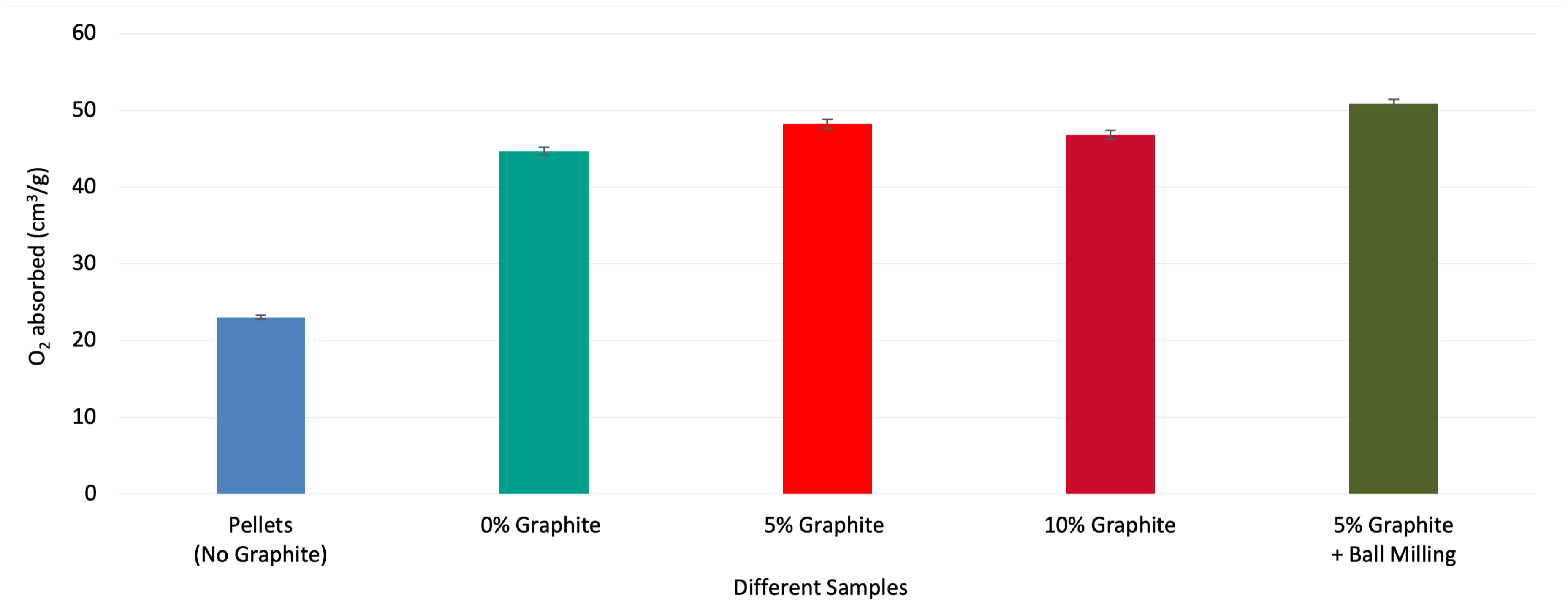
Figure 6.
Effect of different graphite particle sizes at constant 5wt% (left) and effect of higher graphite weight percentages at constant 44 m particle size (right). Increasing the amount of graphite higher than 5 wt% negatively affects the oxygen absorption. Moreover, the 7-11 m graphite particle size at constant 5 wt% results in slightly higher oxygen absorption.
Figure 6.
Effect of different graphite particle sizes at constant 5wt% (left) and effect of higher graphite weight percentages at constant 44 m particle size (right). Increasing the amount of graphite higher than 5 wt% negatively affects the oxygen absorption. Moreover, the 7-11 m graphite particle size at constant 5 wt% results in slightly higher oxygen absorption.
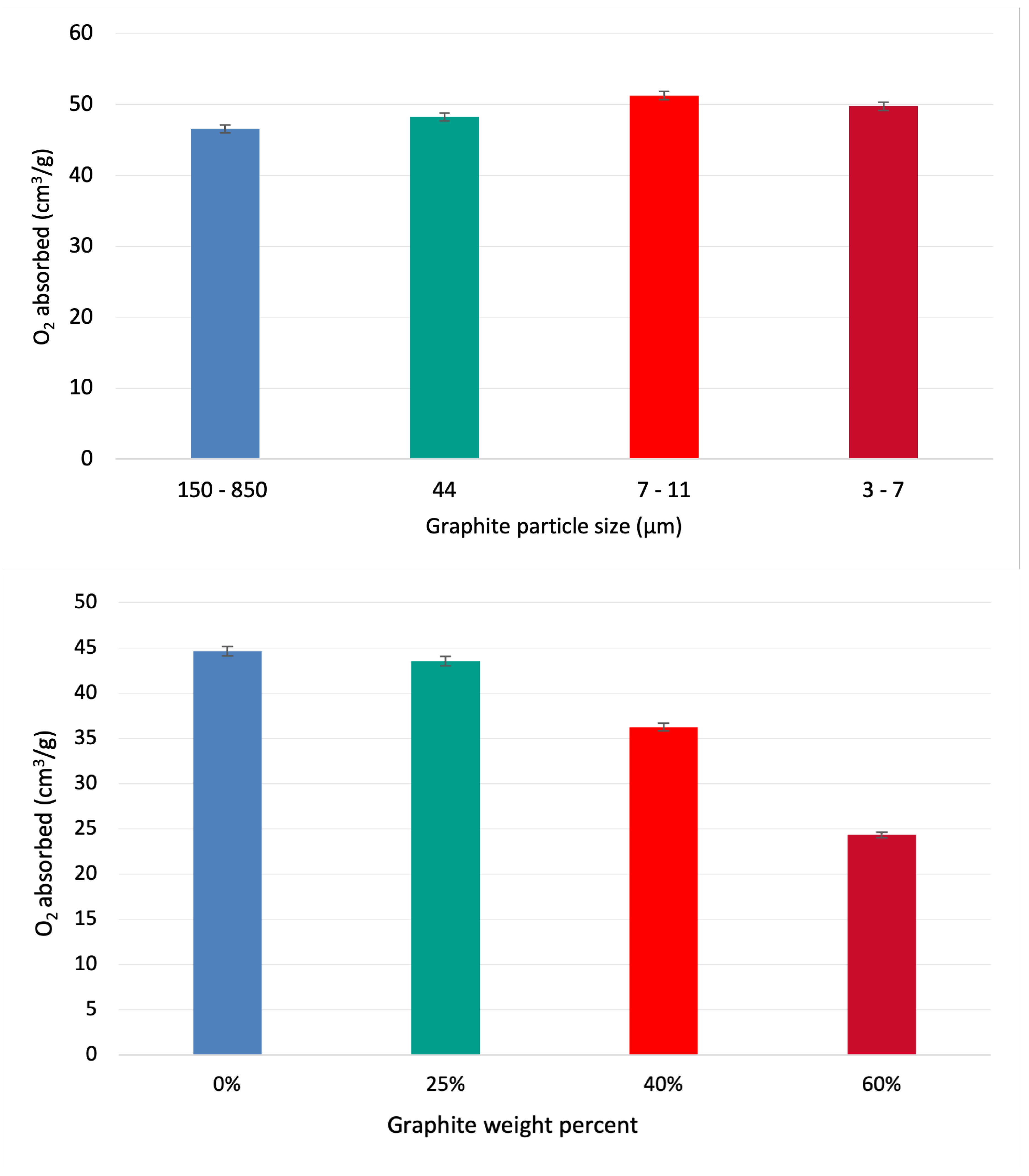
Figure 7.
Relation between cross-section, oxygen absorption capacity, and reaction time. As the cross-section increases, the reaction time will increase, while the oxygen absorption will decrease.
Figure 7.
Relation between cross-section, oxygen absorption capacity, and reaction time. As the cross-section increases, the reaction time will increase, while the oxygen absorption will decrease.
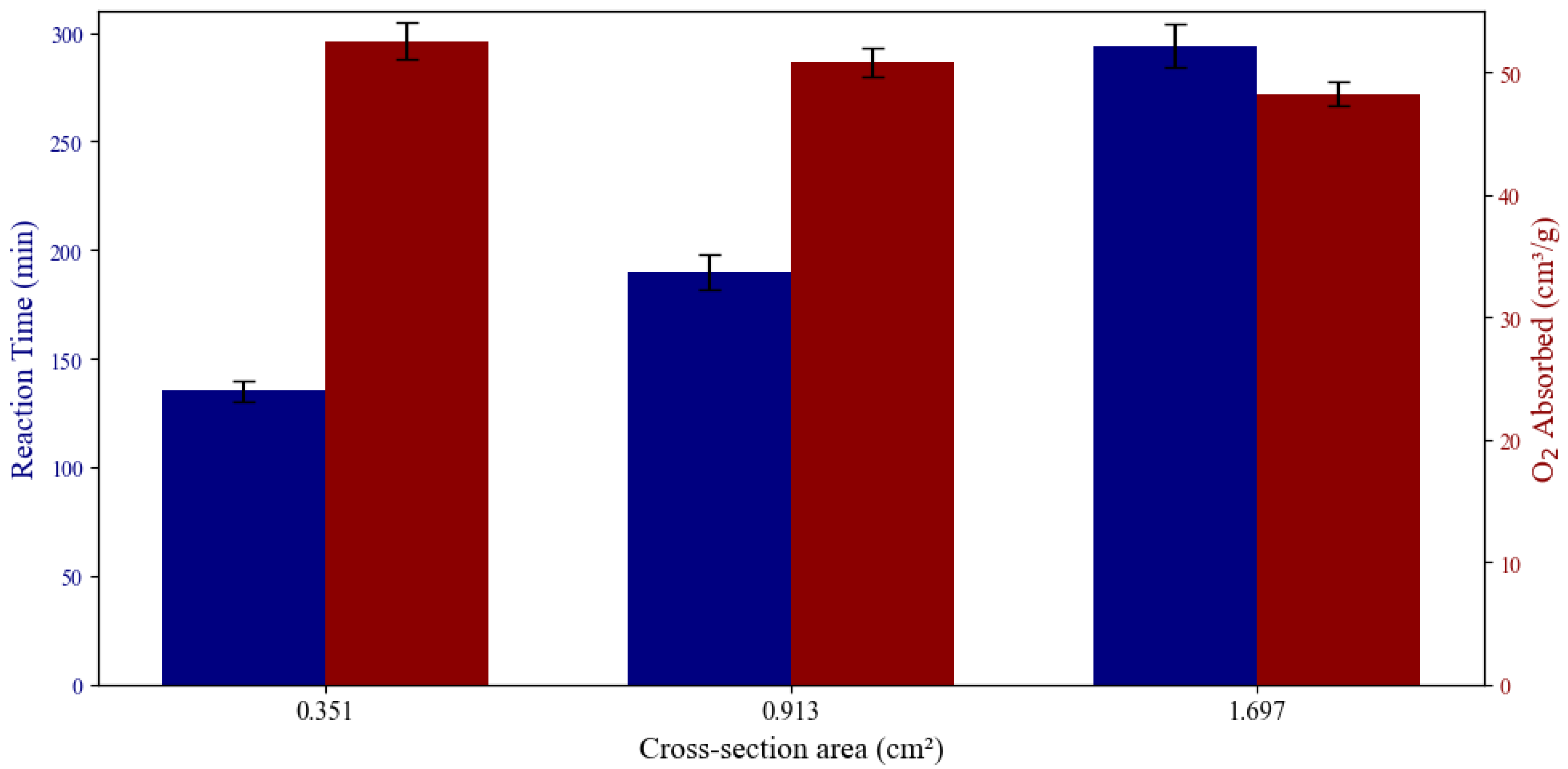
Figure 8.
Comparison of energy density for different MgMnO rods and pellets. The rod with 5 wt% graphite and ball milling showed the highest energy density compared to other samples.
Figure 8.
Comparison of energy density for different MgMnO rods and pellets. The rod with 5 wt% graphite and ball milling showed the highest energy density compared to other samples.
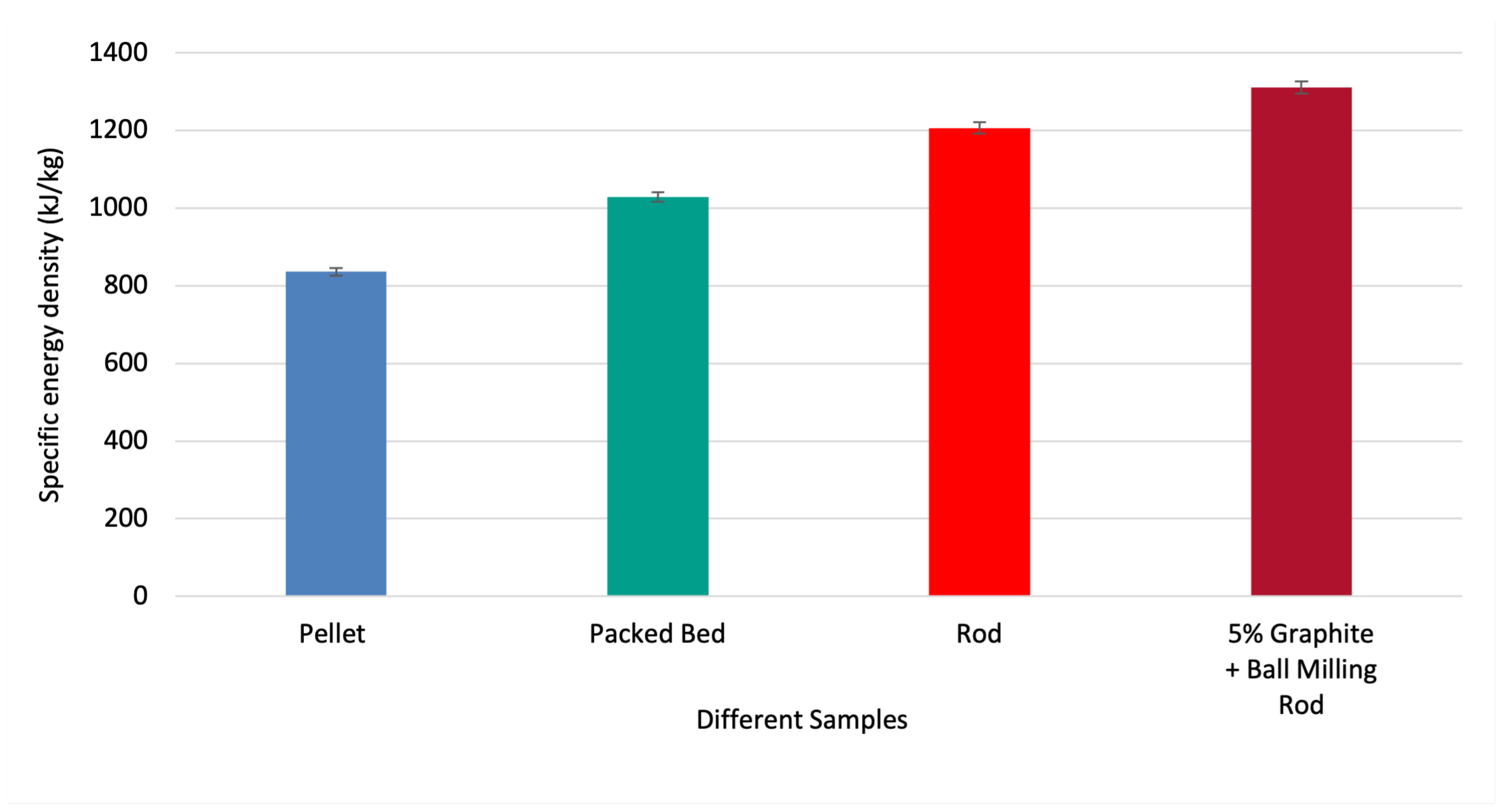

Table 1.
Graphite weight percentage and particle size for each sample.
| Sample number | Graphite content (% weight) | Graphite particle size (m) |
|---|---|---|
| 1 | 0 | – |
| 2 | 5 | 44 |
| 3 | 10 | 44 |
| 4 | 25 | 44 |
| 5 | 40 | 44 |
| 6 | 60 | 44 |
| 7 | 5 | 3–7 |
| 8 | 5 | 7–11 |
| 9 | 5 | 150–850 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



