Submitted:
08 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Direct air capture (DAC) of CO2 via temperature-swing adsorption (TSA) can support sustainable carbon dioxide removal, but only if sorbents regenerate with low energy demand and maintain performance under humid ambient air. Here, we evaluate three commercial molecular sieves (JLPM3, 13X and 4A) in packed-bed tests using humid ambient air. We compared 40 g samples as received with 200 g samples conditioned for 12 days at 100 °C to emulate prolonged exposure to regeneration temperature (the cumulative effect of many heating/desorption cycles); all cycle-stabilized uptake values are reported from the conditioned materials. JLPM3 delivered the highest stabilized CO2 uptake (0.24 ± 0.01 mmol·g-1), consistent with a combined physisorption/chemisorption mechanism. Its higher total porosity and smaller mesopores promoted rapid mass transfer and site accessibility, while slightly greater micropore area and volume than 13X supported its marginally higher capacity. Evidence of partial structural degradation under mechanical and thermal stress indicates that minimizing strain during cycling will be important for scale-up and for reducing sorbent replacement. Conditioning at 100 °C activated additional chemisorption sites across all sieves but reduced physisorption capacity. Importantly, a ~100 °C desorption step fully regenerated physisorbed CO2 while purging moisture from zeolite pores, indicating that low-temperature TSA (compatible with low-grade or waste heat) can replace harsher 300 °C regeneration and lower energy demand. CO2–H2O competition experiments confirmed substantial site occupancy by water vapor, which limits capture under humid conditions and motivates water-management strategies. Overall, maximizing DAC performance requires tailoring pore structure and operating conditions while preserving sorbent integrity; JLPM3 emerges as a promising candidate for more energy- and resource-efficient DAC.
Keywords:
direct air capture
; carbon dioxide removal
; negative emissions
; sustainable adsorption
; temperature swing adsorption
; low-temperature regeneration
; humidity
; water management
; zeolite molecular sieves
; sorbent durability
1. Introduction
The ongoing rise in atmospheric carbon dioxide (CO₂) concentrations is one of the most pressing challenges of our time, with levels exceeding 420 ppm as of 2024, an alarming increase from pre-industrial ~280 ppm [1,2]. This rapid accumulation, driven by fossil fuel combustion, deforestation, and industrial processes, has already led to profound climatic changes, including rising global temperatures, more extreme weather events, and widespread ecological disruption [3]. While deep emissions cuts via renewable energy, efficiency improvements, and ecosystem stewardship are essential, mitigation alone will not suffice. Even if emissions ceased immediately, excess atmospheric CO₂ would continue to warm the climate for centuries, underscoring the need for carbon dioxide removal (CDR) alongside rapid decarbonization [4].
From a sustainability standpoint, DAC must deliver net-negative CO₂ when energy, water, and material inputs are considered. In adsorption-based DAC, regeneration heat and moisture management strongly influence operating energy, lifecycle emissions, and cost. Likewise, sorbent lifetime and mechanical robustness affect resource demand and waste generation, making cyclic stability a key sustainability metric for scalable deployment.
Within the CDR portfolio, Direct Air Capture (DAC) offers a scalable, location-flexible route to remove CO₂ directly from ambient air and to address residual emissions from hard-to-abate sectors [5,6]. Although capturing CO₂ at ~420 ppm is technically demanding, DAC can be paired with permanent geological storage (CCS) or with utilization pathways (CCU) to enable negative emissions while supporting a range of end-uses [7]. Achieving practical DAC at scale hinges on sorbent materials that couple high capacity and selectivity with stability and regenerability under realistic operating conditions, including humidity [9]. Zeolitic molecular sieves are leading physisorbents for temperature-swing adsorption (TSA) because their framework chemistry, pore structure, and surface properties can be tuned for CO₂ capture [6,8]. However, real systems operate with humid ambient air and undergo many thermal cycles near typical regeneration temperatures, factors that can shift the balance between physisorption and chemisorption and adversely impact long-term sorbent performance. In particular, moisture in ambient air competitively occupies adsorption sites in hydrophilic zeolites, drastically curtailing CO₂ uptake and increasing regeneration energy requirements [9]. Repeated heating and pressurization cycles can also induce structural and capacity degradation in zeolites over time [10]. Accordingly, this study focuses on three commercial zeolites (13X, 4A, and JLPM3) to: (i) benchmark CO₂ capture from humid ambient air in a packed-bed TSA process; (ii) quantify CO₂–H₂O competitive adsorption under varying conditions; (iii) determine the regeneration conditions needed to fully desorb physisorbed CO₂ while purging accumulated moisture; (iv) assess how prolonged exposure at 100 °C, used to emulate the cumulative effect of many desorption/heating cycles, alters adsorption mechanisms and mechanical robustness; and (v) relate performance differences to textural attributes (porosity, mesopore size, and micropore volume).
To meet these objectives, we conducted two complementary experimental phases. In Phase 1, ambient air was fed through a packed bed containing 40 g of each sieve (no pre-treatment) at 2.94 L/min (3.09 NL/min) under ambient conditions. Breakthrough curves established CO₂ uptake; desorption was quantified during regeneration up to 310–315 °C; and concurrent water capture was tracked to elucidate hygroscopic behavior. In Phase 2, the materials underwent 12 days of pre-conditioning at 100 °C prior to testing, after which one TSA cycle was performed with a 200 g bed using humid ambient air feed. This deliberate, long-duration conditioning was designed to simulate the multiple desorption heating cycles encountered in real DAC/TSA operation at typical regeneration temperatures. All cycle-stabilized uptake values reported refer to these 12-day/100 °C-conditioned samples.
By linking humidity effects, regeneration temperature, and prolonged thermal exposure to pore architecture and mechanical integrity, this work provides actionable guidance to improve the energy efficiency, durability, and overall sustainability of zeolite-based DAC and to translate laboratory metrics into durable, real-world operation.
2. Experimental
2.1. Experimental Setup and Procedure
The experimental setup (Figure 1a) consists of a fixed-bed reactor made of a quartz tube with an internal diameter of 90 mm and a length of 800 mm. The ends of the quartz tube are sealed using vacuum flanges; one flange is equipped with a single valve, while the other has two valves for gas flow control. The quartz tube is oriented vertically (Figure 1b), and a metallic filter with 1 mm perforations is placed inside to support the molecular sieves.
The molecular sieves used in the experiments include 13X, 4A, and JLPM3, with particle sizes ranging from 1.6 to 2.5 mm.
Humid ambient air is pumped into the reactor from the bottom using a micro-diaphragm air pump that operates continuously at a flow rate of 2,94 L/min (3,09 NL/min). The air passes through the packed bed of molecular sieves, and a portion of the exiting air is directed to an online NDIR CO2 gas analyzer. The analyzer allows continuous CO2 concentration measurements in two ranges: 0–980 mg/m³ and 0–19,600 mg/m³, with an error margin of 2% in each range.
For the desorption process, the same setup is used, but the fixed-bed reactor is placed horizontally inside a tubular furnace (Figure 1.c) capable of reaching temperatures up to 1200°C.
2.2. Data Analysis
During the adsorption process, graphs like the one shown in Figure 2 are obtained. In these graphs, represents the inlet concentration of CO2. This value is determined by passing air through the reactor without the molecular sieves and averaging the measured concentrations. To calculate the total amount of CO2 adsorbed (), the following equation is used (Equation (1)):
The equation to calculate the total amount of CO2 adsorbed includes several key components. The volumetric flow rate of air, , represents the amount of air passing through the reactor and is expressed in units such as liters per minute (L/min) or cubic meters per second (m³/s). In this study, is set at 2,94 L/min (3,09 NL/min), equivalent to 0,000049 m³/s (0,0000515 Nm³/s). The inlet CO2 concentration, , reflects the amount of CO2 in the air entering the reactor, measured in milligrams per cubic meter (mg/m³) or parts per million (ppm). This provides the baseline for the CO2 available for adsorption. The outlet CO2 concentration, , corresponds to the CO2 content measured in the analyzer after passing through the packed bed of molecular sieves, indicating the CO2 that remains unadsorbed. The time at which the adsorption process reaches saturation, , represents the point at which the adsorbent material can no longer effectively capture CO2, serving as the upper limit of integration.
For the graphs obtained during the desorption process, a similar integration is performed to calculate the total amount of CO2 desorbed (). However, in this case, the integration is applied to the expression () from to the time when the desired temperature is reached. The outlet CO2 concentration () is adjusted by subtracting the inlet CO2 concentration () because during desorption, the CO2 released from the molecular sieves due to heating is mixed with the air being pumped through the system. This results in an additional being recorded by the analyzer, which must be accounted for to accurately quantify the CO2 desorbed.
The input concentration of H2O () is calculated theoretically using the temperature and relative humidity (RH) recorded during the experiments. These values were obtained from the meteorological station located in Los Rodeos, San Cristóbal de La Laguna, Santa Cruz de Tenerife. The calculation is based on the following equation:
Here, represents the water vapor concentration, expressed in units of g/L. The term refers to the percentage of water vapor present in the air relative to its maximum capacity at a given temperature, while denotes the saturation vapor pressure of water at that temperature, obtainable from empirical tables or formulas. The molar mass of water, (18.016 g/mol), converts the result into mass-based units. The universal gas constant, (0.0821 L⋅atm/(mol⋅K)), ensures proper scaling of the ideal gas law, and , the temperature in Kelvin, accounts for the thermal effects on air’s capacity to hold moisture. Dividing by 100 adjusts the relative humidity from a percentage to a fractional value for consistency in the calculation. Finally, the obtained value is multiplied by 106 to convert units to mg/m3.
2.3. Characterization Techniques
2.3.1. Nitrogen Adsorption
The samples were degassed at 200 °C for 16 hours under vacuum to remove moisture and other physisorbed species, ensuring that subsequent N₂ uptake reflects the accessible pore volume. Adsorption–desorption isotherms were then measured on a Micromeritics ASAP 2420 system with MicroActive 5.02 software. Liquid nitrogen cooling maintained a bath temperature of 77.3 K, and high-purity N₂ gas was used as the adsorptive. Data points were collected over a relative pressure range of p/p0≈0.001–0.987p/p_0 ≈ 0.001–0.987p/p0≈0.001–0.987 with a fixed 20 s equilibration interval at each step.
The instrument software generated, for each sample: single-point BET surface area, multi-point BET surface area, t-plot micropore area and volume, BJH adsorption/desorption pore-size distribution, and Horváth–Kawazoe micropore width. These textural parameters were used to correlate structural properties with adsorption performance.
2.3.2. Mercury Porosimetry
Mercury intrusion–extrusion measurements were performed with a high-pressure porosimeter (Micromeritics AutoPore IV) controlled by MicroActive software. Powder samples were loaded into a 3-bulb, 0.412-stem glass penetrometer (volume = 3.1276 mL). Intrusion was recorded from vacuum up to ~60,000 psia. Calculations assumed a mercury contact angle of 141°. The resulting mercury intrusion data provided macropore size distributions and bulk density measurements, supplementing the nitrogen physisorption data to give a full picture of the pore architecture in each material.
2.3.3. X-Ray Diffraction (XRD)
Powder specimens (10 mm track length) were examined on a θ–2θ diffractometer operated in Bragg–Brentano geometry. Cu K-α radiation was used (λ = 1.54060 Å; generator setting 45 kV / 40 mA). Key instrument parameters were: divergence slit = 0.1799°, incident-beam Soller slit = 0.040°, 10 mm beam mask, goniometer radius = 240 mm and specimen spinning “Yes”. Patterns were collected continuously from 5.013 to 79.97° 2θ with a step of 0.026° 2θ and 56.865 s counting time per step. Peak lists produced by the control software were matched against ICDD PDF-4+ entries, giving semi-quantitative phase abundances by the reference-intensity-ratio (RIR) routine included in the same package.
2.3.4. X-Ray Fluorescence (XRF)
Elemental concentrations were obtained from wavelength-dispersive XRF measurements. The instrument acquired two excitation ranges automatically: Range 3 at 20 kV and Range 2 at 40 kV (ranges shown in the report for every oxide). The three samples return analytical totals of 99.99–100.02 wt %, confirming measurement completeness within the stated precision.
3. CO2/H2O Adsorption-Desorption Performance
3.1. 40 g Packed Bed of Molecular Sieves JLPM3, 13X and 4A (No Pre-treatment)
Four TSA cycles were carried out on separate days in a fixed-bed reactor packed with 40 g of JLPM3, 13X and 4A molecular sieves. The first adsorption cycle (cycle 0) served as a pretreatment to activate the zeolites. Adsorption was performed under ambient conditions, while desorption was induced by a temperature swing from ambient () up to 310–315 °C. The packed beds had a radius () of 90 mm and lengths () of 0.8 cm, 0.8 cm, and 0.7 cm, respectively.
Four TSA cycles were performed on separate days with 40 g beds of JLPM3, 13X, and 4A using humid ambient air as feed (Section 2.1). Adsorption proceeded at ambient conditions; desorption used a temperature swing to 310–315 °C. The corresponding breakthrough and temperature-programmed desorption (TPD) profiles are shown in Figure 3, Figure 4 and Figure 5, and cycle-wise uptakes in Table 1, Table 2 and Table 3.
Typically, zeolites are considered physical adsorbents, but formation of chemisorbed species (like carbonates or bicarbonates) on zeolite cation sites is known under certain conditions [7]. For example, Amrit Kumar et al. (2015) observed that some physisorbents can bind CO2 strongly enough to require elevated temperatures for release, hinting at chemisorptive interactions in MOFs/zeolites during DAC [15]. For instance: “Similar dual desorption behavior has been reported for CO2 on Na-X zeolites, where a low-temperature desorption corresponds to weak physisorption and a high-temperature feature is attributed to CO2 binding as bicarbonate/carbonate on cation sites” [7]. In all three sieves, the CO₂ TPD curves show a reproducible bi-modal shape: a low-temperature peak (≈75–82 °C) followed by a broader peak at ≈214–220 °C (Figure 3b, Figure 4b and Figure 5b). We attribute the first to readily reversible physisorption in micropores and the second to stronger binding (e.g., bicarbonate/carbonate-like species on cationic/defect sites) that requires higher temperature to desorb, consistent with reports on FAU/LTA zeolites that separate weakly vs. strongly bound CO₂ in TPD features [6,7]. A fraction of stronger sites can also be introduced or amplified by composition/defects or metal modification (e.g., Fe@13X), which shifts part of the desorption to higher T [14].
As heat is applied from ambient up to 310–315 °C, a first peak appears between 75 °C and 82 °C (77.9 ± 1.2 °C for JLPM3; 75.3 ± 2.8 °C for 13X; 81.9 ± 0.6 °C for 4A), signaling the release of weakly adsorbed CO₂ (physisorption or surface interactions). Following this peak, the CO₂ concentration falls sharply (flush-out of residual gas) until roughly 156–168 °C. Then, a second, broader peak emerges at 214–220 °C (214.1 ± 7.3 °C for JLPM3; 216.6 ± 4.5 °C for 13X; 219.6 ± 5.1 °C for 4A), attributed to the desorption of more strongly bound CO₂ (chemisorption or internal zeolite interactions). A small, marginal rise near 300 °C is observed in all cases, likely due to combustion of trace organics rather than continued desorption.
Integrating the breakthrough curves, the initial CO₂ uptakes rank JLPM3 > 13X > 4A (Table 1, Table 2 and Table 3). JLPM3 shows the steepest front, indicative of faster kinetics, consistent with its higher porosity and smaller mesopores that shorten diffusion paths to CO₂-selective micropores (Section 4). Recent studies confirm that appropriately engineered zeolites can capture CO₂ at air-level concentrations with fast kinetics and modest heats of adsorption, particularly MOR-type frameworks under dry or controlled-humidity conditions [16]. These results align with our ranking (JLPM3 ≥ 13X ≫ 4A) and the observed sensitivity to moisture. All three materials, however, display substantial co-adsorption of H₂O; the measured H₂O/CO₂ selectivity increases across cycles (Figure 3, Figure 4 and Figure 5), reflecting the strong hydrophilicity of low-Si/Al zeolites and site blocking by water under ambient humidity [6,11]. The particularly low CO₂ capacity of 4A is consistent with its small LTA windows that limit CO₂ access and its strong affinity for H₂O [6,11,20]. Notably, while competitive adsorption by water is generally dominant in hydrophilic zeolites, specific K-MER sites have been shown to allow CO₂ to displace pre-adsorbed H₂O at 30 °C, highlighting framework/cation site effects under humid DAC conditions [17].
Quantitatively, the fraction of CO₂ released via the first mechanism (“low temperature” peak) varies by sorbent and cycle. For JLPM3 it remains around 80 % across cycles (with no statistically significant change) whereas 13X rises from 43 % in cycle 0 (preloaded and burning off organics) to about 85 % thereafter, and 4A from 55 % up to ~80–82 %. Weight measurements (Table 1, Table 2 and Table 3) confirm a large drop after the first cycle (40.01 g → 37.49 g for 13X; 40.01 g → 37.85 g for 4A), then only minor losses in subsequent cycles, corroborating removal of pre-existing CO₂/organics and stabilization of the material. Across all sorbents, after pre-treatment (after cycle 0), adsorption/desorption capacities (and match within margin of error each cycle, aside from cycle 0, but this is because during the desorption in cycle 0 there was still CO2 adsorbed in the zeolites and also part of the excess of CO2 comes from organic matter being burnt, demonstrating full reversibility once pre-treatment effects are accounted for.
Except for the first cycle, the integrated CO₂ desorbed () matches the CO₂ adsorbed () within the combined experimental uncertainty of the flow, baseline subtraction, and analyzer calibration. The apparent > observed in cycle 0 arises from (i) pre-loaded CO₂ in the as-received zeolites (CO₂ weakly bound and/or bicarbonate/carbonate species accumulated) that is released upon the first heat-up, and (ii) oxidation of trace organics on the pellets/binder, evidenced by the small shoulder near ~300 °C, which contributes additional CO₂ not associated with the immediately preceding adsorption step. After this conditioning cycle, baseline-corrected desorption profiles close the CO₂ mass balance in every cycle, confirming reversible uptake once pre-treatment effects are removed.
Despite this reversibility, total CO₂ uptake and release steadily decline with each TSA cycle (Figure 3a/3c, 4a/4c, 5a/5c), indicative of thermal degradation at 310–315 °C. This is consistent with thermally induced degradation of hydrophilic zeolites under repeated TSA exposure in humid feeds [10]. As high-quality CO₂ sites/accessibility decline, the H₂O/CO₂ selectivity rises because polar H₂O increasingly dominates competitive adsorption [6,11]. JLPM3 shows the greatest performance drop and physical fragmentation, producing dust, whereas 13X retains its structure best, and 4A lies in between. By cycle 3, JLPM3 and 13X converge to similar uptake levels, underscoring how JLPM3’s structural breakdown drives its loss. When comparing all three sorbents (Figure 6), JLPM3 initially delivers the highest CO₂ uptake, followed by 13X and then 4A, but all three diminish over cycles. Water uptake likewise converges, and the H₂O/CO₂ selectivity ratio increases cycle-by-cycle: as the sorbents’ electromagnetic adsorption strength weakens (affecting non-polar CO₂ more than polar H₂O), given that they preferentially capture water. These trends highlight that while the dual-peak desorption signature and reversible adsorption mechanisms persist, high-temperature cycling inexorably erodes sorbent capacity and selectivity, especially for JLPM3, suggesting a trade-off between initial performance and thermal durability.
The humidity penalty observed here and in literature [11,16,17,18] underscores the need for water management (e.g., layered beds or pre-drying) to enable moderate-temperature TSA (≈100–120 °C) with zeolites [11]. Framework topology and extra-framework cations critically tune DAC performance at 400 ppm, especially in LTA/4A where cation charge/size governs low-ppm uptake and moisture sensitivity [18]. These comparisons support our mechanistic interpretation and guide process/material strategies to mitigate water competition and thermal stress in DAC cycles.
3.2. Comparison of Molecular Sieves JLPM3, 13X and 4A (No-Pretreatment)
Figure 6 consolidates the cycle-wise CO₂ uptake, H₂O uptake, and H₂O/CO₂ selectivity for the three sieves (see Table 1, Table 2 and Table 3 for values and uncertainties). Trends are discussed in terms of framework topology, extra-framework cations, and competitive adsorption with water, which dominate DAC performance at ~400 ppm CO₂.
CO₂ uptake (Figure 6a): In the first cycle, the ranking is JLPM3 > 13X > 4A. JLPM3’s advantage is consistent with its higher accessible porosity and shorter diffusion paths to CO₂-selective micropores (Section 4), which sharpen the breakthrough front and improve bed utilization. 13X (FAU) provides large pore windows (~0.74 nm) and a high density of Na⁺ sites that polarize CO₂ effectively, explaining its robust, though lower, uptake. By contrast, 4A (LTA) has ~0.40–0.42 nm apertures that restrict CO₂ access and are readily occupied by H₂O under ambient humidity, depressing its effective capacity [6,11,16]. These observations align with DAC-specific studies showing zeolites can capture air-level CO₂ with fast kinetics under dry/controlled-humidity feeds (e.g., MOR- and FAU-type) and that framework/cation pairing strongly governs low-ppm performance [6,12,16].
H₂O uptake (Figure 6b): All three sieves co-adsorb substantial water, with 4A and 13X exhibiting the strongest moisture loading, consistent with their low Si/Al and hydrophilicity [6,11]. Preferential H₂O occupation of cationic sites reduces the number of electrostatically favorable CO₂ sites and lengthens mass-transfer paths, explaining earlier CO₂ breakthrough and lower integrated uptake relative to dry conditions [6,11]. While water typically “wins” on hydrophilic zeolites, site-specific exceptions exist (e.g., K-MER), where CO₂ can displace pre-adsorbed H₂O at 30 °C; this underscores that cation identity and local ring geometry can flip CO₂/H₂O selectivity under certain conditions [17].
H2O/CO2 (Figure 6c) increases with cycle number for all sieves. Two effects compound: (i) progressive loss of high-quality CO₂ sites and/or pore accessibility due to thermal/mechanical stress at 310–315 °C in humid feeds, well documented for 4A/13X-class materials [10], and (ii) H₂O “hogging” of the remaining cationic sites as the CO₂-affine population shrinks [6,11]. The net result is a rising water fraction in the working capacity even as both absolute uptakes decline.
The monotonic decrease in CO₂ capacity across cycles, the visible fragmentation in JLPM3, and the relative robustness of 13X are consistent with literature on thermo-humid cyclic degradation of hydrophilic zeolites [10]. By the last cycle, JLPM3 and 13X converge to similar CO₂ uptakes, implicating mechanical attrition and/or partial amorphization in JLPM3 as the cause of its steeper decline (see Section 4). 4A remains transport-limited and water-poisoned throughout, consistent with LTA access constraints and strong cation–H₂O interactions [6,11,16]. The pronounced cation sensitivity of LTA-type zeolites at 400 ppm reported elsewhere further explains 4A’s low working capacity under humid air [18].
Mechanistic link to desorption profiles. The bi-modal CO₂-TPD response discussed in Section 3.1, low-T, readily reversible physisorption followed by a higher-T contribution from stronger binding on cationic/defect sites, rationalizes the selective loss of CO₂ capacity under harsh cycling. Introducing or exposing a stronger-site fraction (e.g., via composition/defects or metal incorporation) shifts part of the desorption to higher temperature, as seen for Fe-modified FAU (Fe@13X) [14].
These results point to two levers for DAC with zeolites: (1) Water management, e.g., upstream drying or layered beds with a desiccant pre-layer [11]. (2) Framework/cation tuning, especially in LTA/FAU, controls the CO₂/H₂O balance at 400 ppm; cations with appropriate charge density and placement can improve low-ppm CO₂ uptake yet should avoid excessive water affinity [6,16,18]. These principles are consistent with the stronger initial performance of FAU-type materials here and with recent demonstrations of low-ppm CO₂ capture on optimized zeolites [12,16].
3.3. Effect of Prolonged Thermal Conditioning (12 Days at 100 ºC) on CO2 Uptake and Mechanism
A 12-day conditioning at 100 °C was applied to 200 g beds of JLPM3, 13X, and 4A to emulate cumulative desorption exposure and obtain cycle-stabilized behavior. After this treatment, one TSA cycle was performed under humid ambient air. The results (Figure 7a–f; Table 4) show (i) similar total CO₂ uptake for JLPM3 and 13X relative to their first cycles in section 3.1, (ii) a redistribution of CO₂ between weak and strong binding (a larger high-T desorption contribution), and (iii) persistent H₂O co-adsorption that continues to limit working capacity under humidity.
From Figure 7.b, 7.d and 7.f, the desorption profiles of the three molecular sieves as a function of temperature exhibit a distinct pattern: an initial peak (), followed by a relative minimum (), and finally, another peak (). These temperature points, detailed in Table 4, correspond to different desorption mechanisms. At , the zeolites release all CO2 stored via physisorption. The intermediate point marks the onset of desorption for CO2 bound by stronger interactions, while indicates the complete release of all stored CO2.
In the pre-treated 200 g beds, the CO₂-TPD traces display a low-temperature release centered near 95–105 °C (JLPM3 103.1 °C; 13X 103.7 °C; 4A 92.3 °C), followed by an intermediate minimum (~137–153 °C) and a pronounced high-temperature tail/peak extending to ~235–275 °C (Table 4; Figure 7b,d,f). We attribute the first feature to readily reversible physisorption in micropores and the higher-T release to stronger binding (bicarbonate/carbonate-like species on cationic/defect sites) that requires additional thermal input, consistent with established assignments for FAU/LTA zeolites and other physisorbents showing dual-mode CO₂ retention [6,7,15]. The growth of the high-T fraction after conditioning indicates that mild, extended heating can activate or expose stronger sites (e.g., by cation re-distribution or defect formation) [6,15], a trend aligned with observations that metal-modified FAU (e.g., Fe@13X) holds a portion of CO₂ more strongly and desorbs at higher T [14].
The total CO2 and H2O uptakes in Table 4 were calculated using the post-desorption weights of the molecular sieves, representing the fully regenerated samples. Table 4 shows that the weight of the molecular sieves after desorption is significantly lower than before adsorption, confirming the presence of CO2 and H2O stored in the materials even after 12 days at 100°C. Post-conditioning CO₂ uptakes are 0.24 ± 0.01 mmol g⁻¹ (JLPM3), 0.20 ± 0.01 mmol g⁻¹ (13X), 0.14 ± 0.01 mmol g⁻¹ (4A) (Table 4), preserving the rank JLPM3 > 13X > 4A seen in 40 g tests. This hierarchy matches expectations from framework access and micropore volume (FAU ≫ LTA) and the kinetic advantage of hierarchical/mesoporous architectures [6,12,16]. Nevertheless, H₂O uptake remains substantial (2.51, 2.14, and 1.89 mmol g⁻¹ for JLPM3, 13X, and 4A), keeping H₂O/CO₂ selectivity ≈ 10–14 (Table 4). This confirms that under humid feeds, water preferentially occupies cationic sites, depressing CO₂ working capacity unless moisture is managed [6,9,11]. While water usually predominates on hydrophilic, low-Si/Al zeolites, site-specific motifs (e.g., K-MER) can allow CO₂ to displace pre-adsorbed H₂O at 30 °C, underscoring the role of cation identity and ring geometry in tuning competition [17].
Figure 7.a,c,e provide insights into the adsorption behavior after extended pre-conditioning. For JLPM3 (Figure 7.a), the CO2 concentration surpasses the inlet concentration () before stabilizing at a higher level. This phenomenon may result from the heat released during adsorption, which partially desorbs CO2, and from competition between H2O and CO2 for adsorption sites due to JLPM3's strong electromagnetic interactions. For 13X (Figure 7.c), the CO2 concentration initially exceeds before oscillating around this value, suggesting lower heat being emitted during adsorption and fast cooling of the sorbent, which gives a rapid equilibration. In contrast, the behavior of 4A (Figure 7.e) reflects a significant loss of adsorption capacity following thermal pre-treatment, with a markedly anomalous adsorption curve compared to JLPM3, 13X, and the results from Section 3.1 for the 40g packed bed of molecular sieves 4A.
Prolonged exposure at 100 °C under humid air likely dehydrates residual hydroxyls, drives limited cation migration, and stabilizes defect-adjacent sites, modestly increasing the population of stronger CO₂-binding sites while slightly reducing purely physisorptive capacity [6,7,15]. This dual effect explains the larger high-T desorption contribution in Figure 7, and the observation that JLPM3 and 13X have similar capacities relative to their unconditioned cycle 1 (section 3.1), whereas 4A remains transport-limited by LTA apertures under humid air [6,11,16,18]. The trend is consistent with literature where framework/cation tuning at DAC concentrations adjusts the balance between weak and strong binding, particularly for LTA/FAU families [6,18,19].
4. Textural Properties and Characterization
4.1. Nitrogen Adsorption
Table 5 summarizes BET areas and pore volumes derived from N₂ sorption at 77 K. As measured, 4A shows negligible measured micropore area/volume, while JLPM3 and 13X exhibit large micropore areas (≈710 and ≈673 m² g⁻¹) and micropore volumes (≈0.267 and ≈0.253 cm³ g⁻¹), respectively. The disparity primarily reflects aperture accessibility at 77 K: LTA (4A) eight-ring windows (~0.40–0.42 nm) impose diffusion limitations for N₂ at 77 K, so conventional N₂ sorption “sees” little internal surface, whereas FAU (13X/JLPM3) 12-ring windows (~0.74 nm) are accessible, yielding the much larger measured microporous surface [6]. Consequently, measured N₂-microporosity correlates with the higher DAC CO₂ uptake observed for FAU vs LTA in Section 3.1, Section 3.2 and Section 3.3. This trend is consistent with zeolite structure, property relationships discussed in [6] and with the hierarchical-transport benefits reported for small-mesopore chabazite analogues [12].
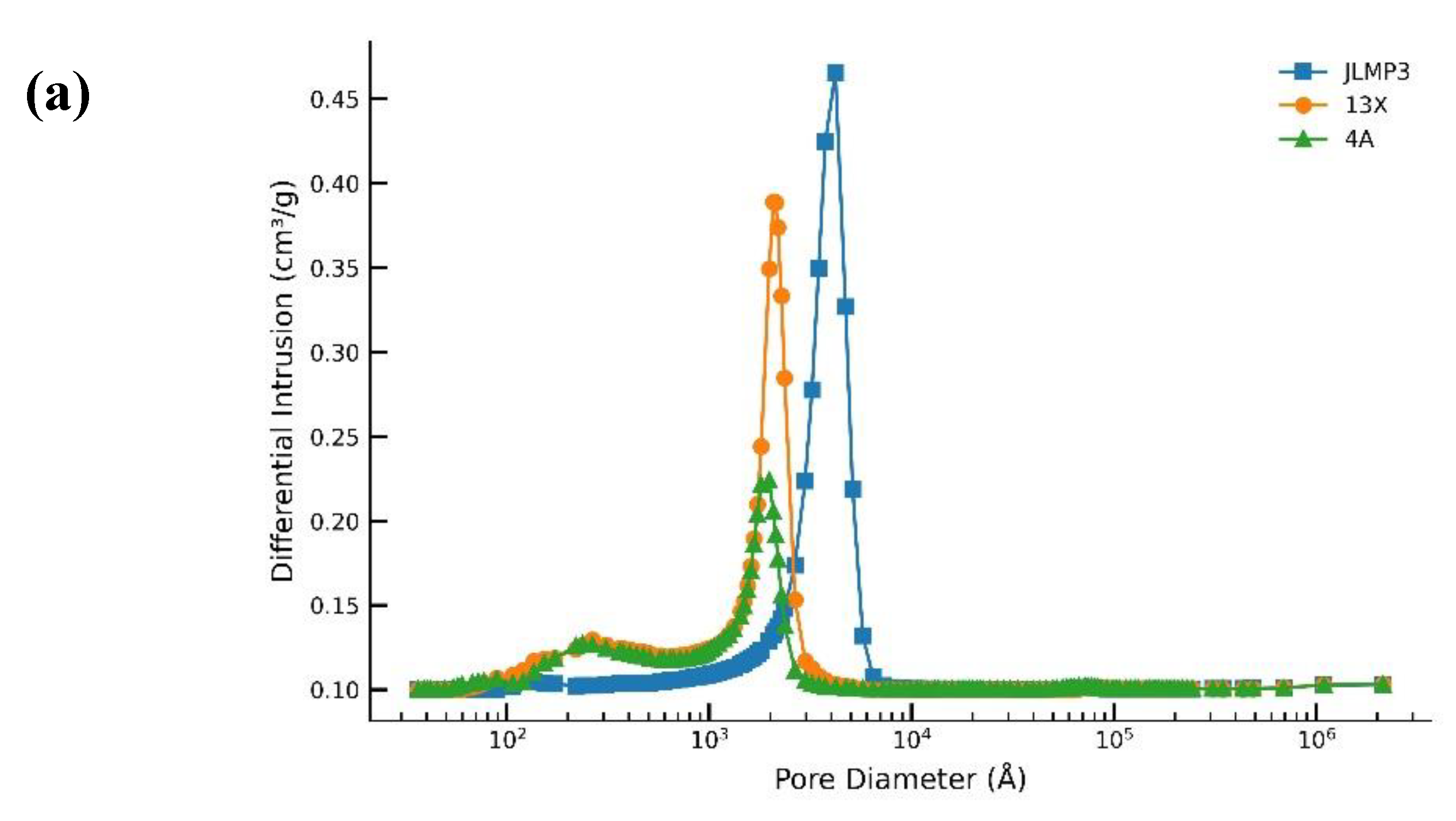
The HK micropore width for JLPM3 and 13X (~0.75 nm) indicates channels readily accessible to CO₂, while the BJH mesopore peaks (JLPM3 ≈7.6 nm; 13X ≈13.3 nm) suggest shorter diffusion paths in JLPM3 than in 13X, which helps explain JLPM3’s sharper breakthrough fronts and slightly higher working capacities under humid air (Section 3.1 and Section 3.2) [6,12,16]. In contrast, 4A’s low measured microporosity at 77 K aligns with its low working capacity at ~400 ppm in humid feeds due to combined access limitations and water competition [6,11,18].
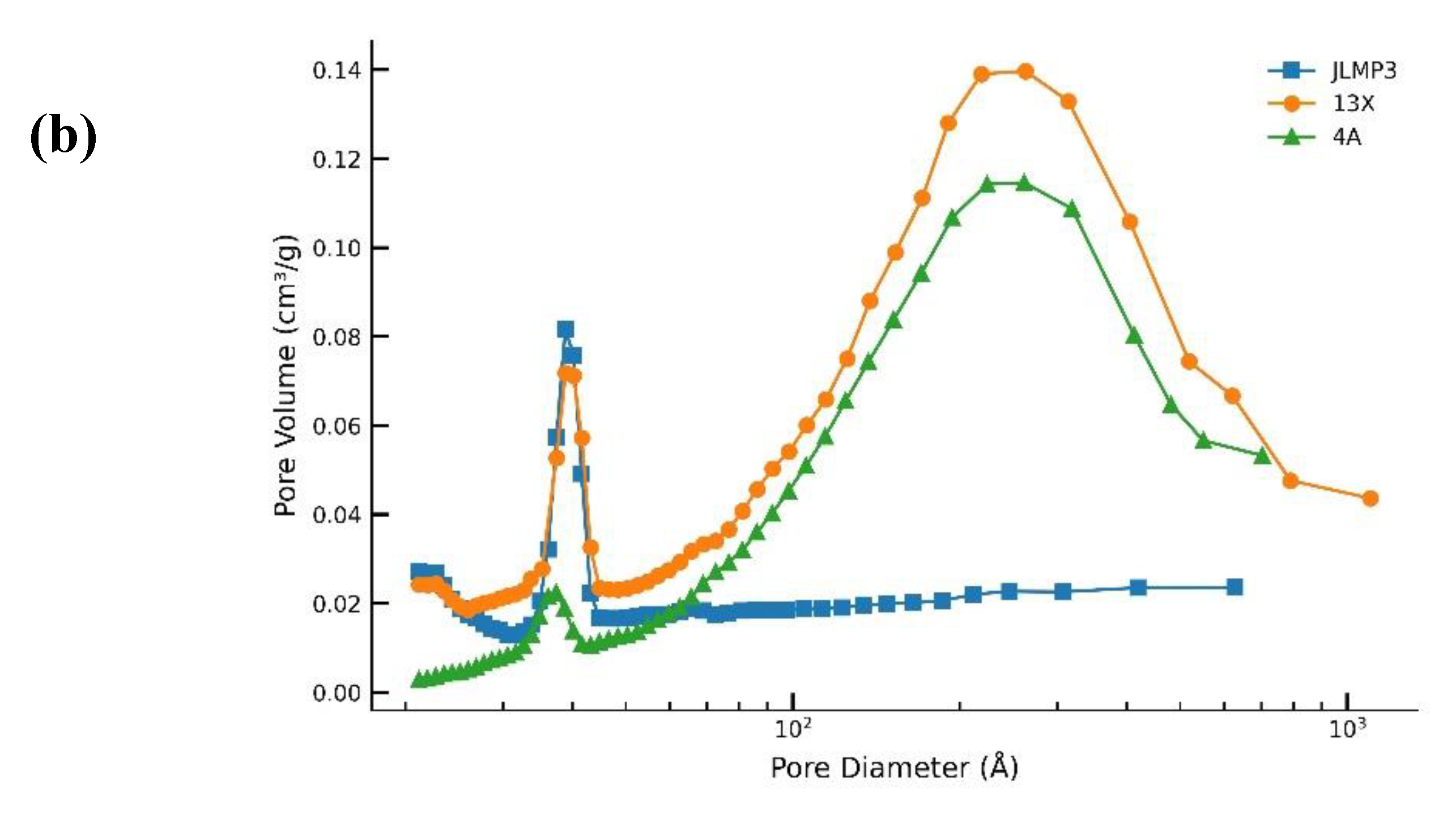
4.2. Mercury Porosimetry
Table 6 shows all three samples have similar bulk porosity (≈24–26 vol %), but distinct meso/macropore architectures. JLPM3 exhibits a narrower BJH mesopore distribution (~7–8 nm) than 13X and 4A (≈13–16 nm), which is consistent with shorter intraparticle paths to CO₂-selective micropores. Numerous studies on hierarchical zeolites (e.g., SSZ-13 with 5–8 nm mesopores) report accelerated uptake kinetics relative to purely microporous analogues, because a mesopore network reduces diffusion lengths into the micropore network [12]. These textural features rationalize JLPM3’s faster kinetics and higher initial utilization vs 13X/4A under humid ambient air, while also explaining why larger mesopores alone (13X, 4A) do not compensate for insufficient accessible micropore volume (notably in 4A) [6,12].
Figure 8.
(a) Mercury porosimetry and (b) N2 adsorption pore size distribution for molecular sieves JLPM3, 13X and 4A.
Figure 8.
(a) Mercury porosimetry and (b) N2 adsorption pore size distribution for molecular sieves JLPM3, 13X and 4A.
4.3. X-Ray Diffraction (XRD)
Figure 9 confirms the expected FAU topology for 13X and JLPM3 and LTA topology for 4A. Because reference-intensity-ratio (RIR) matches are semi-quantitative, the phase percentages should be interpreted cautiously; minor reflections may also include binder/amorphous components and carbonate/hydroxide species introduced during handling. The key point for DAC is structural: FAU frameworks (JLPM3, 13X) provide 12-ring access to large supercages, whereas LTA (4A) provides 8-ring access to α-cages, driving the accessibility differences discussed above [6]. The trace Fe indicated by XRF (Section 4.4) is consistent with small iron-bearing domains or extra-framework cations that can slightly modify local electrostatics, a factor known to influence low-ppm CO₂ binding on FAU-type materials [6,19].
4.4. X-Ray Fluorescence (XRF)
Table 7 shows all three are low-silica Na-zeolites (Si/Al ≈1.15–1.40), implying a high density of extra-framework cations that enhance adsorption of polarizable CO₂ via electrostatic/quadrupolar interactions [6]. The Si/Al trend helps explain strong CO₂ affinity in JLPM3 and 13X provided access is not rate-limiting, while 4A, despite similar Si/Al, remains transport-limited by its narrow windows under humid DAC conditions [6,18]. Minor levels of divalent/other cations (e.g., Mg²⁺) may adjust local fields or create defect-adjacent sites, but the dominant effects observed here are framework access (FAU vs LTA) and water competition (Section 3.1, Section 3.2 and Section 3.3) [6,11,18].
5. Conclusions
In conclusion, this comparative study has demonstrated distinct performance trade-offs among zeolite sorbents for direct air capture under humid TSA conditions. JLPM3 exhibited the highest cycle-stabilized CO₂ uptake (~0.24 mmol·g⁻¹) owing to a synergistic physisorption–chemisorption capacity and a more accessible pore architecture. However, JLPM3’s initial advantage was tempered by a pronounced capacity decline over repeated cycles due to mechanical and thermal strain, leading its uptake to converge toward that of the more robust 13X by the final cycles. Zeolite 13X achieved a slightly lower CO₂ capacity (~0.20 mmol·g⁻¹) but maintained superior structural integrity, showing minimal degradation across cycles. In contrast, 4A consistently displayed the smallest CO₂ uptake (~0.14 mmol·g⁻¹) because its narrow LTA-type pore windows and high intrinsic affinity for H₂O severely limited CO₂ access. All three sorbents showed substantial co-adsorption of water, which competed with CO₂ for adsorption sites and exacerbated performance differences.
Mechanistically, the sorbents were found to capture CO₂ via dual binding modes. We observed a readily reversible physisorbed component and a strongly bound chemisorbed component in all materials. The presence of a high-temperature desorption tail (extending up to ~250 °C in TPD profiles) confirms that a fraction of CO₂ forms stronger bonds (likely carbonate or bicarbonate species on cationic or defect sites) requiring higher thermal input to desorb. Mild thermal preconditioning (100 °C for 12 days) accentuated this effect by activating additional strong-binding sites (increasing the chemisorbed fraction) at the expense of some physisorptive capacity. Notably, a regeneration temperature of ~100 °C was sufficient to fully desorb the physisorbed CO₂ and to purge accumulated water from the zeolites. This indicates that more extreme heating (~300 °C) is unnecessary for routine CO₂ recovery and only serves to impose avoidable thermal stress. Humidity proved to be a critical factor: water vapor was shown to strongly compete for cationic adsorption sites, driving H₂O/CO₂ selectivity to ~10–14 in these low-Si/Al zeolites. Consequently, under humid ambient air, a significant portion of each sieve’s capacity was occupied by H₂O, directly limiting the available sites for CO₂ uptake unless moisture is rigorously managed.
From a practical standpoint, our findings underscore the importance of coupling water management strategies with gentle regeneration conditions to sustain DAC performance. Co-adsorbed water must be minimized, for example, by incorporating upstream dehumidification or layered bed configurations with a dedicated desiccant pre-layer – to prevent site blockage and preserve CO₂ capacity. Implementing such water-management measures enables effective CO₂ capture at moderate desorption temperatures (~100–120 °C), instead of having to heat adsorbent beds to ~310 °C. Indeed, maintaining milder TSA conditions was sufficient in this work to restore the sieves’ working capacity on each cycle while avoiding the structural degradation observed at higher temperatures. This gentle regeneration not only reduces energy requirements but also helps preserve sorbent integrity over extended operation.
For future development, sorbent tuning and hybrid bed configurations emerge as promising pathways to enhance performance. Tailoring the zeolite framework and cation composition can modulate the balance between CO₂ affinity and H₂O selectivity. For instance, adjusting Si/Al ratios or exchanging extra-framework cations may reduce excessive water binding while maintaining strong CO₂ adsorption at 400 ppm. Such compositional modifications, alongside creating hierarchical porosity, could produce materials that combine the high initial capacity of JLPM3 with the long-term stability of 13X. Equally important is improving the mechanical robustness of candidate sorbents: the partial attrition observed for JLPM3 indicates that strengthening particle morphology or binder formulation will be critical for durable cyclic use. Additionally, hybrid adsorption system designs (e.g. layered beds or sorbent blends) should be explored to leverage the complementary strengths of different materials – for example, using a moisture-tolerant layer to safeguard a CO₂-selective layer. Overall, the insights gained in this study provide a foundation for optimizing zeolitic sorbents and process conditions. By managing water uptake and limiting thermal stress, and by tuning sorbent properties to favor dual-mode CO₂ binding without incurring fragility, next-generation DAC systems can achieve more efficient and stable CO₂ capture from humid air in practical applications.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.de.
References
- IPCC. Climate Change 2023: Synthesis Report. Contribution of WGI, WGII and WGIII to the Sixth Assessment Report [Core Writing Team; Lee, H., Romero, J., Eds.; IPCC: Geneva, Switzerland, 2023. [Google Scholar] [CrossRef]
- Keith, D.; Holmes, G.; Angelo, D.; Heidel, K. A Process for Capturing CO₂ from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef]
- Anderson, K.; Peters, G. The Trouble with Negative Emissions. Science 2016, 354(6309), 182–183. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.; Psarras, P. C.; Liguori, S. Assessment of Reasonable Opportunities for Direct Air Capture. Environmental Research Letters 2017, 12(6), 065001. [Google Scholar] [CrossRef]
- Sahu, T.; Ghuman, K. K.; O’Brien, P. G. A Review of Materials Used for Carbon Dioxide Capture. In Progress in Sustainable Development; Elsevier, 2023; pp. 205–232. [Google Scholar] [CrossRef]
- Boer, D. G.; Langerak, J.; Pescarmona, P. P. Zeolites as Selective Adsorbents for CO₂ Separation. ACS Applied Energy Materials 2023, 6(5), 2634–2656. [Google Scholar] [CrossRef]
- Grün, R.; Hashim, A. S.; Grau Turuelo, C.; Breitkopf, C. Insights into CO₂ Diffusion on Zeolite 13X via Frequency Response Technique. Chemistry – Methods 2024, 4(9), e202400006. [Google Scholar] [CrossRef]
- Fu, D.; Davis, M. E. Carbon Dioxide Capture with Zeotype Materials. Chemical Society Reviews 2022, 51(22), 9340–9370. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Rim, G.; Kong, F.; Lively, R. P.; Jones, C. W. Cold-Temperature Capture of Carbon Dioxide with Water Coproduction from Air Using Commercial Zeolites. Industrial & Engineering Chemistry Research 2022, 61(36), 13624–13634. [Google Scholar] [CrossRef]
- Jacobs, J. H.; Deering, C. E.; Sui, R. H.; Lesage, K. L.; Marriott, R. A. Degradation of Desiccants in Temperature Swing Adsorption Processes: The Temperature Dependent Degradation of Zeolites 4A, 13X and Silica Gels. Chemical Engineering Journal 2024, 451, 139049. [Google Scholar] [CrossRef]
- Fu, D.; Davis, M. E. Toward the Feasible Direct Air Capture of Carbon Dioxide with Molecular Sieves by Water Management. Cell Reports Physical Science 2023, 4(5), 101389. [Google Scholar] [CrossRef]
- Hillen, L.; Degirmenci, V. Hierarchical Mesoporous SSZ-13 Chabazite Zeolites for Carbon Dioxide Capture. Catalysts 2021, 11(11), 1355. [Google Scholar] [CrossRef]
- Aquino, T.; Estevam, S.; Olivo Viola, V.; Melo, C.; Zancan, F.; Vasconcelos, L.; Riella, H.; Pires, M.; Morales-Ospino, R.; Torres, A.; Bastos-Neto, M.; Cavalcante, C., Jr. CO₂ Adsorption Capacity of Zeolites Synthesized from Coal Fly Ashes. Fuel 2020, 276, 118143. [Google Scholar] [CrossRef]
- Xiang, X.; Guo, T.; Yin, Y.; Gao, Z.; Wang, Y.; Wang, R.; An, M.; Guo, Q.; Hu, X. High Adsorption Capacity Fe@13X Zeolite for Direct Air CO₂ Capture. Industrial & Engineering Chemistry Research 2023, 62(12), 5420–5429. [Google Scholar] [CrossRef]
- Kumar, A.; Madden, D. G.; Lusi, M.; Chen, K-J.; Daniels, E. A.; Curtin, T.; Perry, J. J.; Zaworotko, M. J. Direct Air Capture of CO₂ by Physisorbent Materials. Angew. Chem. Int. Ed. 2015, 54(48), 14372–14377. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Park, Y.; Davis, M. E. Confinement Effects Facilitate Low-Concentration Carbon Dioxide Capture with Zeolites. Proc. Natl. Acad. Sci. USA 2022, 119(39), e2211544119. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Xie, D.; Zones, S. I.; Katz, A. CO₂ Desorbs Water from K-MER Zeolite under Equilibrium Control. J. Am. Chem. Soc. 2024, 146, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Tian, Y.; Ou, S. Y.; Gu, Q.; et al. Direct Air Capture of CO₂ by Metal Cation-Exchanged LTA Zeolites: Effect of the Charge-to-Size Ratio of Cations. AIChE J. 2023, 69(8), e18139. [Google Scholar] [CrossRef]
Figure 1.
(a) Schematic diagram of the experimental setup for CO2 analysis using a fixed bed reactor. (b) Experimental setup for adsorption. (c) Experimental setup for temperature swing desorption.
Figure 1.
(a) Schematic diagram of the experimental setup for CO2 analysis using a fixed bed reactor. (b) Experimental setup for adsorption. (c) Experimental setup for temperature swing desorption.

Figure 2.
Experimental breakthrough curve for CO2 adsorption.
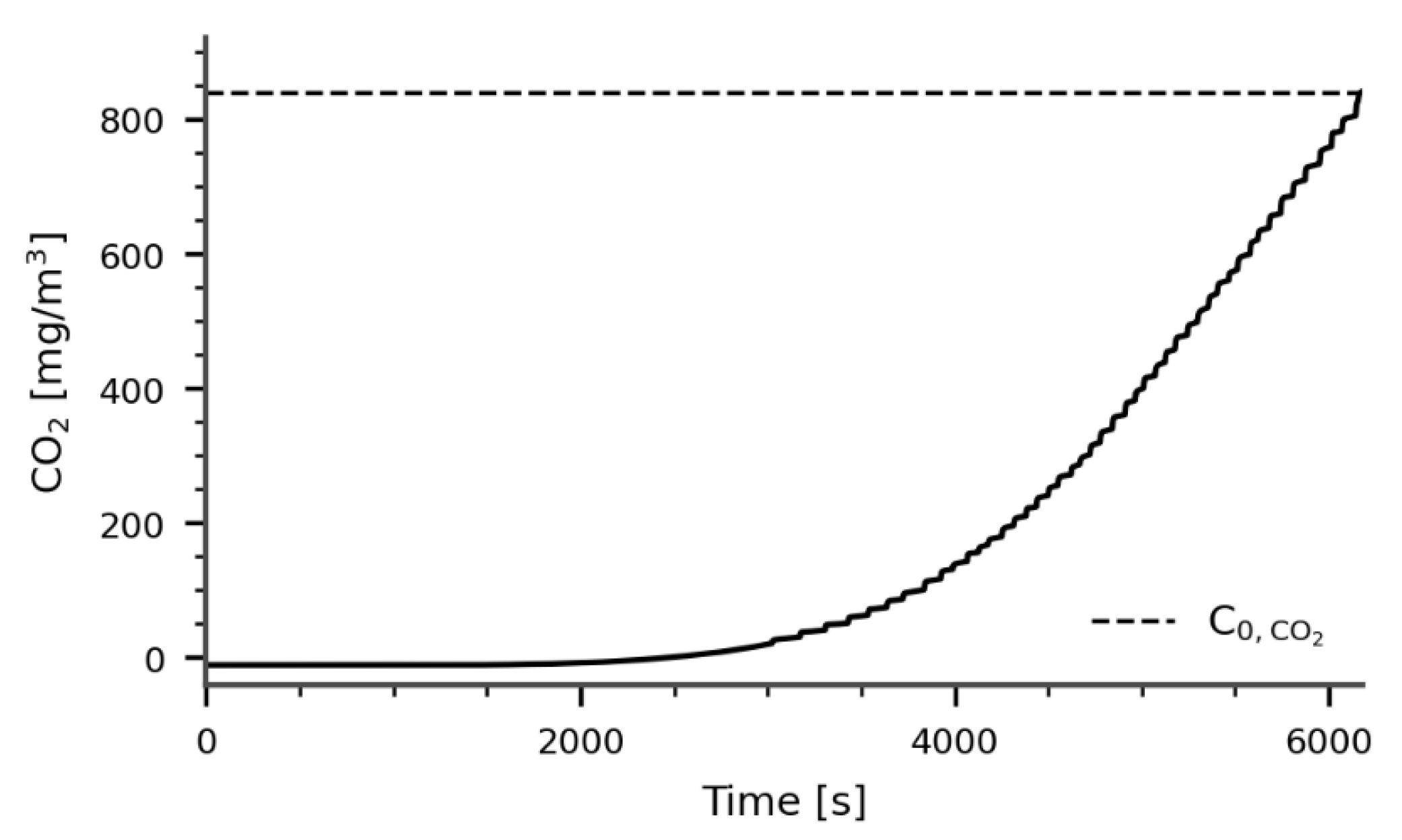
Figure 3.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves JLPM3. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves JLPM3. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves JLPM3.
Figure 3.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves JLPM3. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves JLPM3. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves JLPM3.
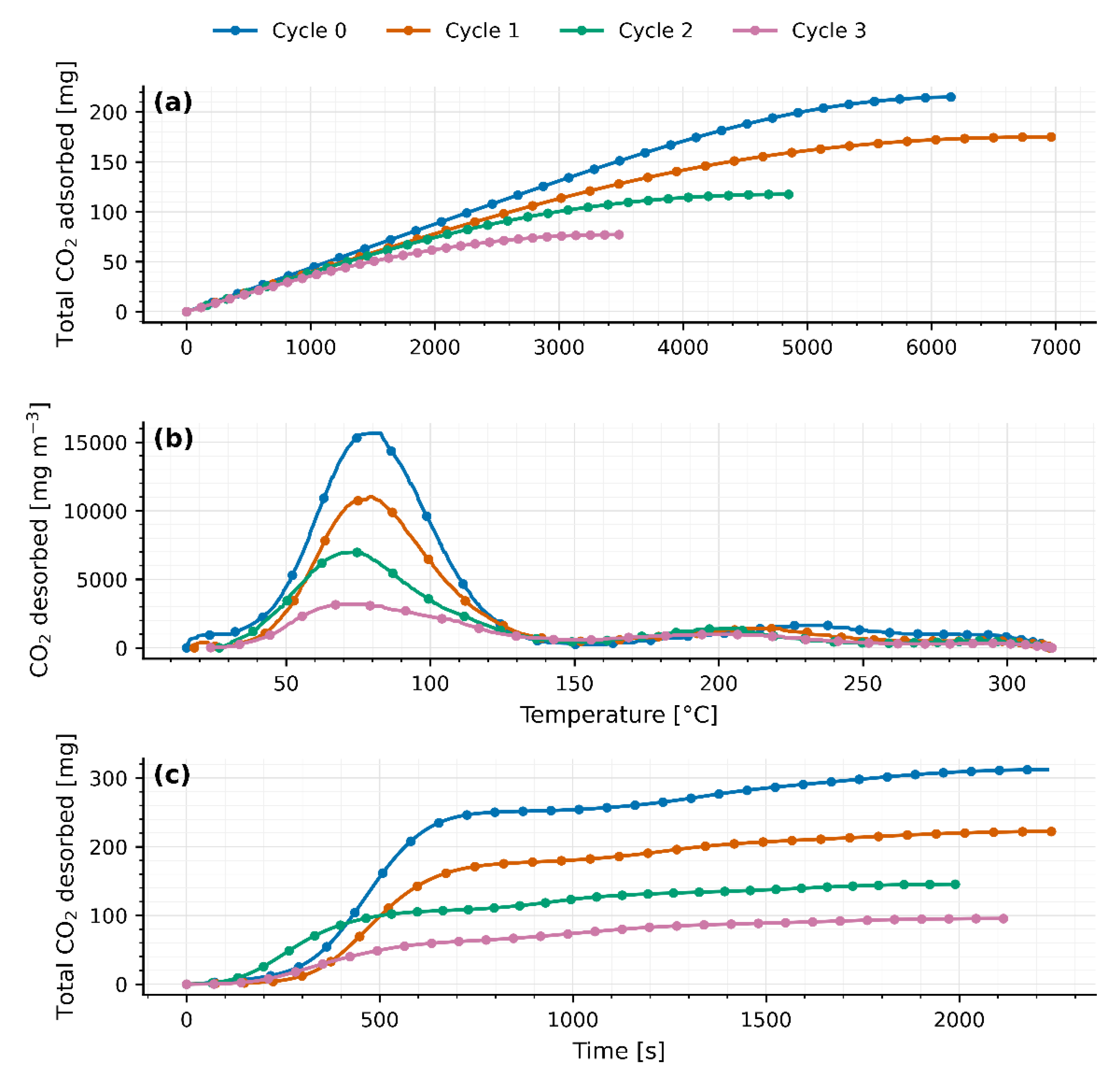
Figure 4.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves 13X. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves 13X. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves 13X.
Figure 4.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves 13X. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves 13X. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves 13X.
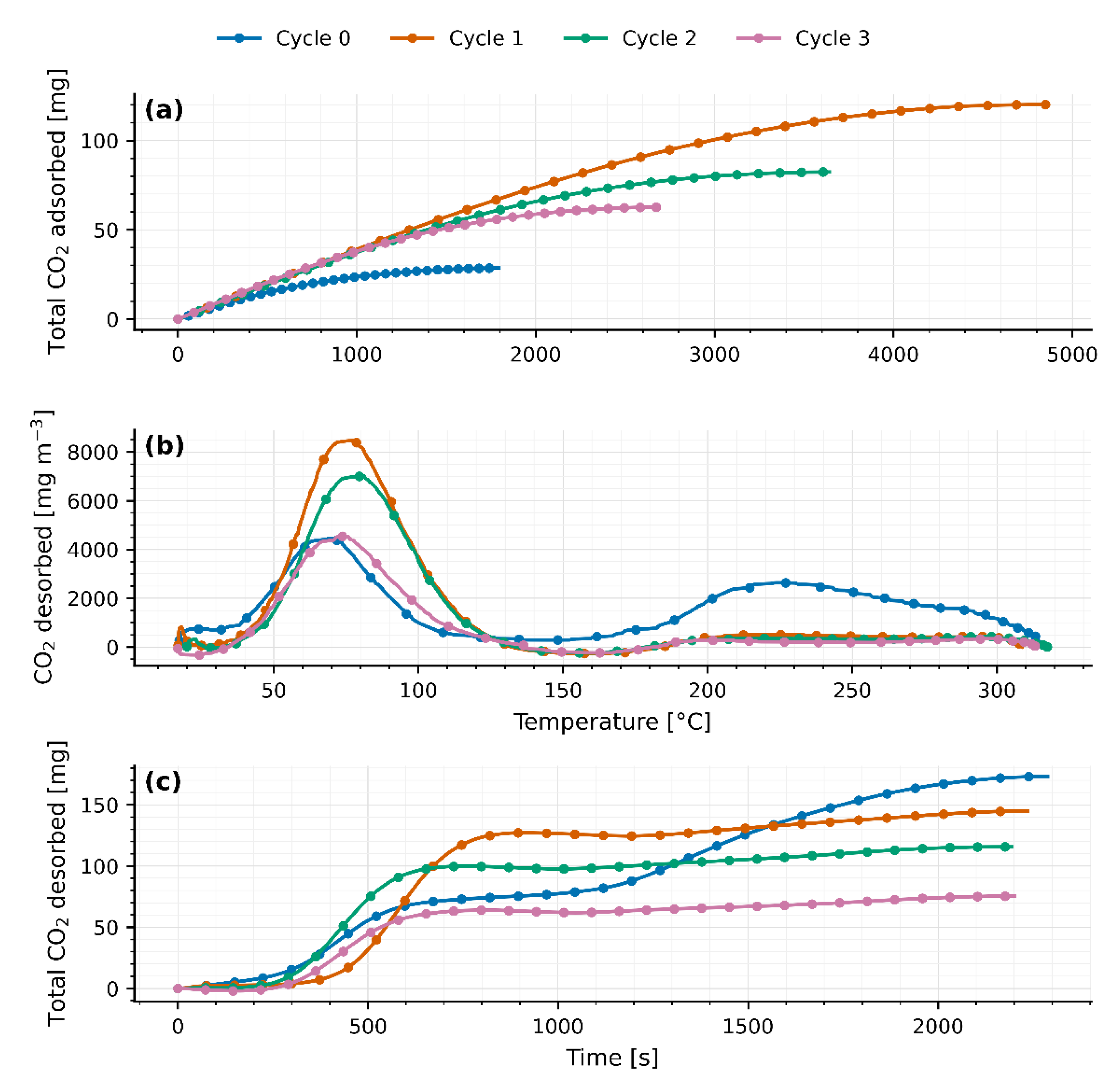
Figure 5.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves 4A. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves 4A. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves 4A.
Figure 5.
(a) Total CO2 adsorbed for each cycle as a function of time for 40g packed bed of molecular sieves 4A. (b) CO2 desorption on each cycle as a function of temperature for 40g packed bed of molecular sieves 4A. (c) Total CO2 desorbed for each cycle as a function of time for 40g packed bed of molecular sieves 4A.
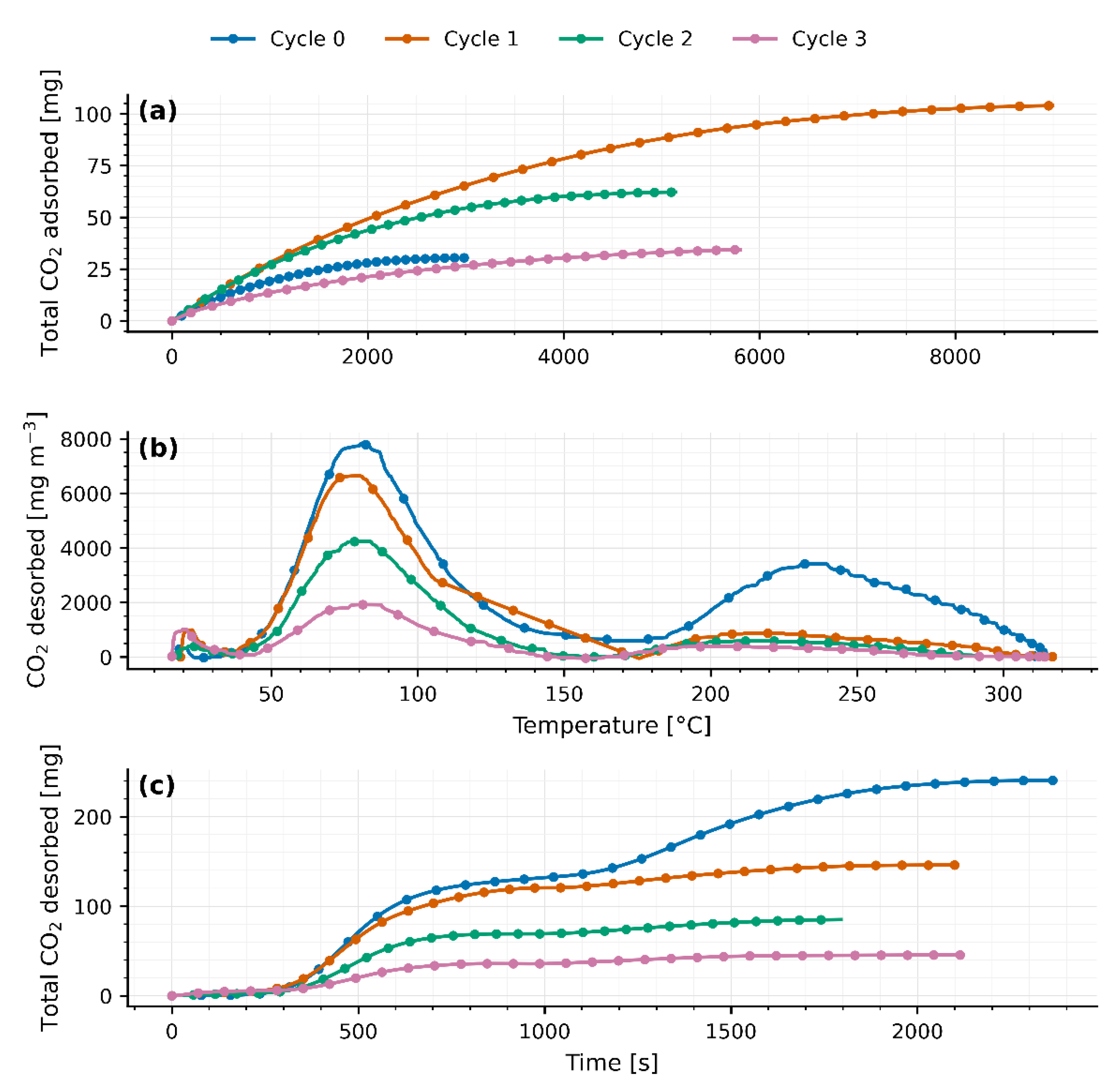
Figure 6.
(a) CO2 uptake comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle. (b) H2O uptake comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle. (c) H2O/CO2 selectivity comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle.
Figure 6.
(a) CO2 uptake comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle. (b) H2O uptake comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle. (c) H2O/CO2 selectivity comparison for 40g packed beds of molecular sieves JLPM3, 13X and 4A for each cycle.
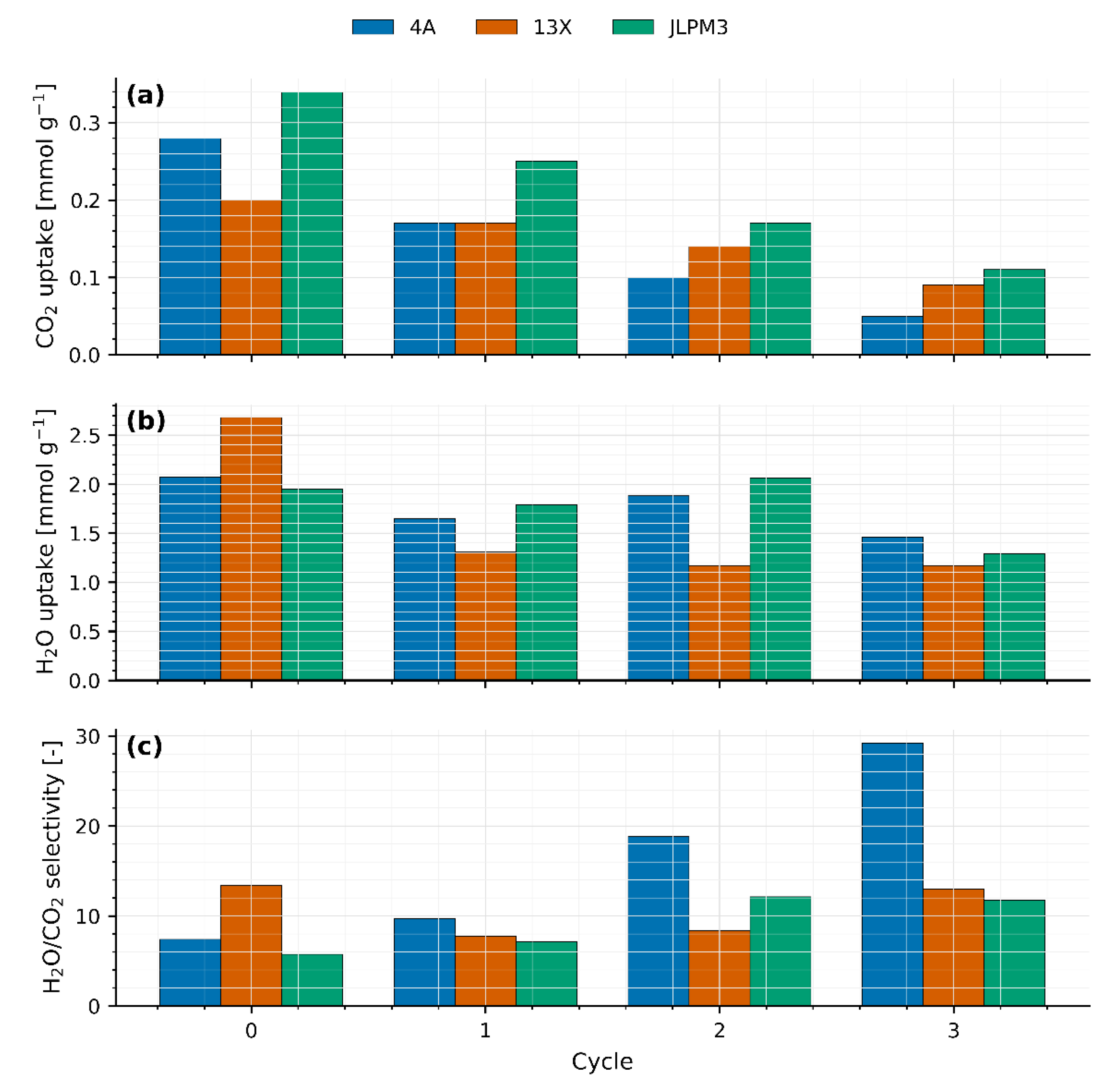
Figure 7.
(a) Breakthrough curve for CO2 adsorption on 200g of molecular sieves 4A with thermal pre-treatment. (b) CO2 desorption as a function of temperature of 200g molecular sieves 4A with thermal pre-treatment. (c) Breakthrough curve for CO2 adsorption on 200g of molecular sieves 13X with thermal pre-treatment. (d) CO2 desorption as a function of temperature on 200g of molecular sieves 13X with thermal pre-treatment. (e) Breakthrough curve for CO2 adsorption on 200g of molecular sieves JLPM3 with thermal pre-treatment. (f) CO2 desorption as a function of temperature on 200g of molecular sieves JLPM3 with thermal pre-treatment.
Figure 7.
(a) Breakthrough curve for CO2 adsorption on 200g of molecular sieves 4A with thermal pre-treatment. (b) CO2 desorption as a function of temperature of 200g molecular sieves 4A with thermal pre-treatment. (c) Breakthrough curve for CO2 adsorption on 200g of molecular sieves 13X with thermal pre-treatment. (d) CO2 desorption as a function of temperature on 200g of molecular sieves 13X with thermal pre-treatment. (e) Breakthrough curve for CO2 adsorption on 200g of molecular sieves JLPM3 with thermal pre-treatment. (f) CO2 desorption as a function of temperature on 200g of molecular sieves JLPM3 with thermal pre-treatment.
Figure 9.
(a) XRD spectra of molecular sieve JLPM3. (b) XRD spectra of molecular sieve 13X. (c) XRD spectra of molecular sieve 4A.
Figure 9.
(a) XRD spectra of molecular sieve JLPM3. (b) XRD spectra of molecular sieve 13X. (c) XRD spectra of molecular sieve 4A.
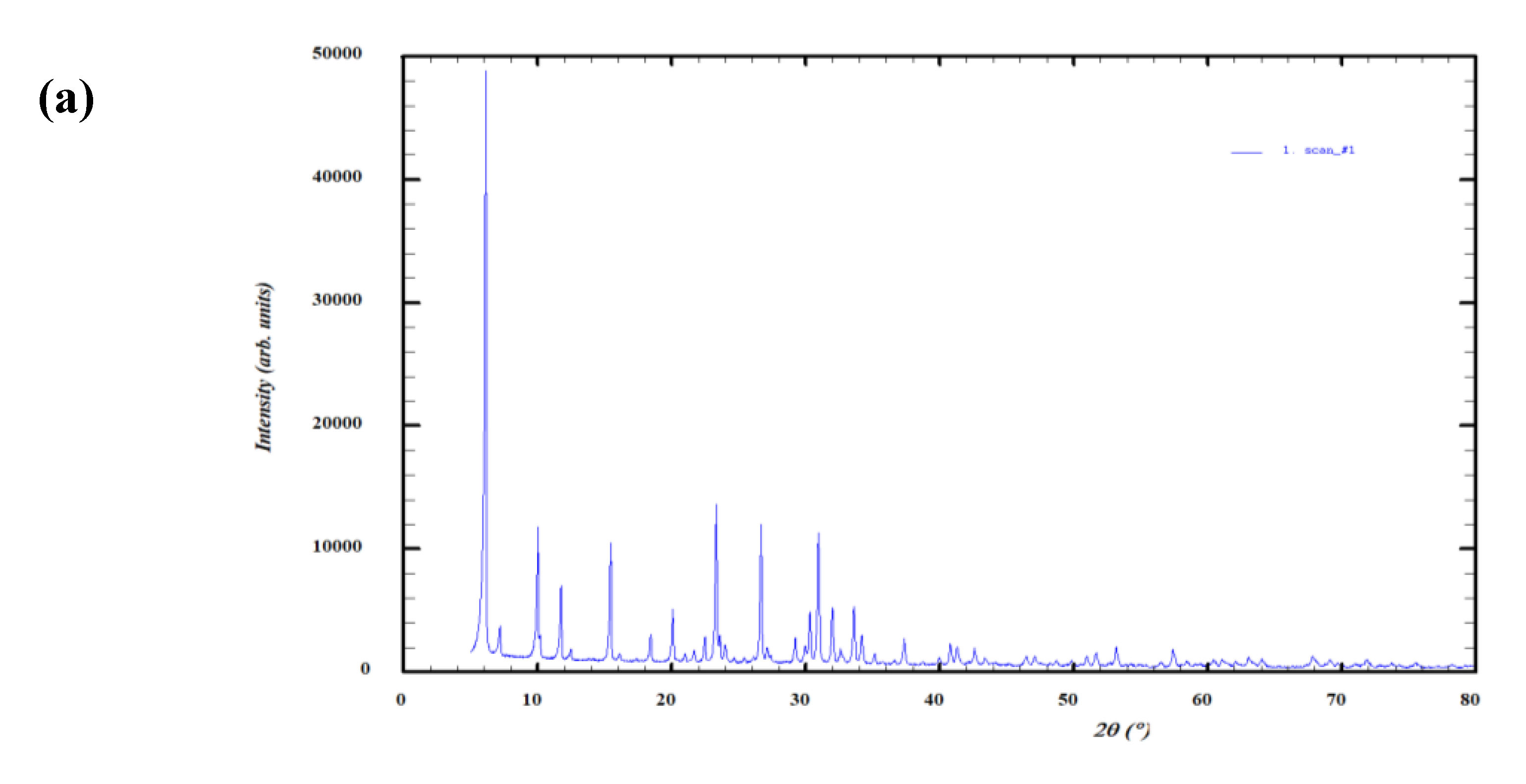
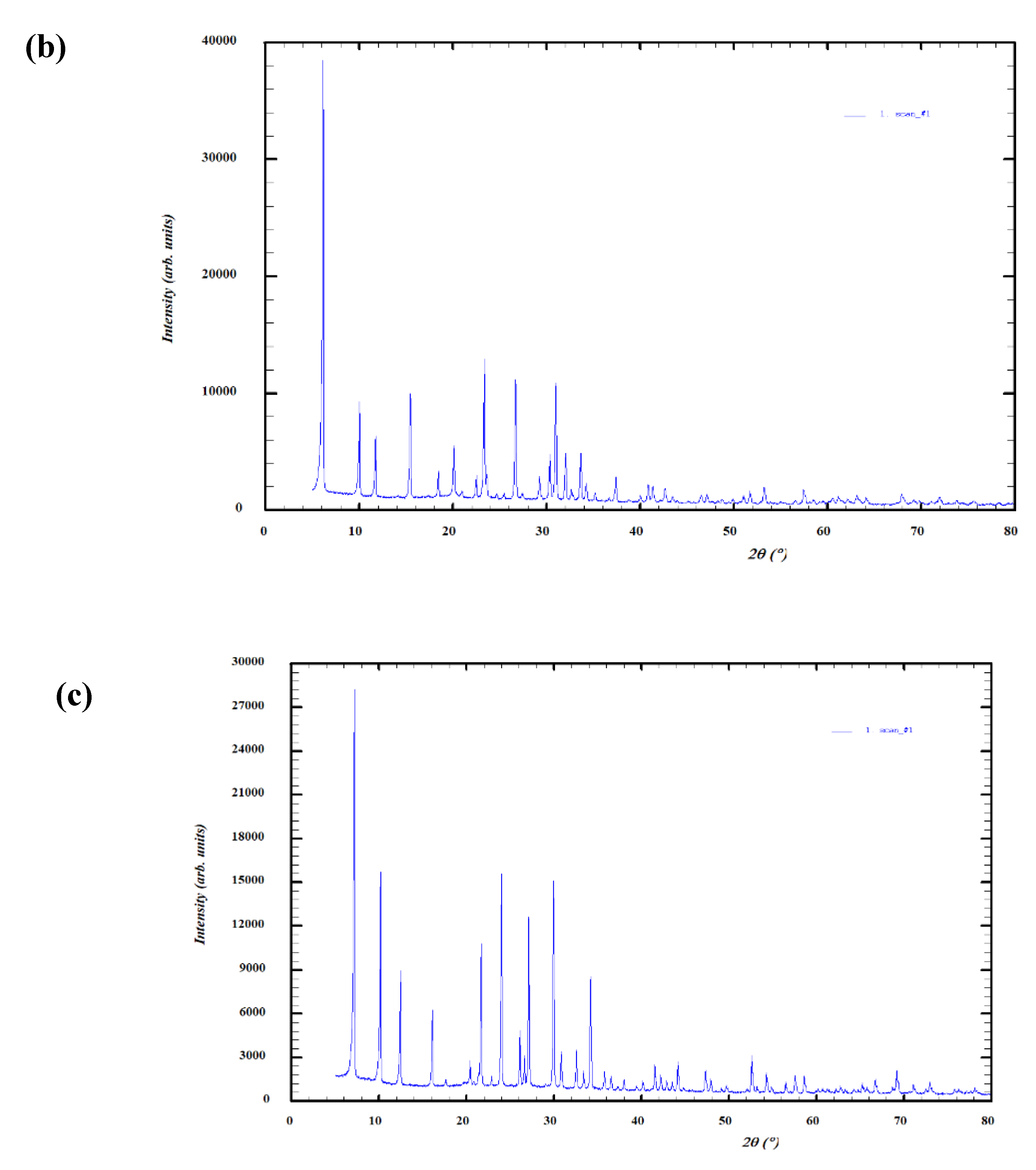

Table 1.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves JLPM3.
Table 1.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves JLPM3.
| Cycles | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| [mg/m3] | 840.85 ± 19.60 | 750.81 ± 19.60 | 744.99 ± 19.60 | 736.61 ± 19.60 |
| [mg/m3] | 11,662.40 | 5164.10 | 8014.29 | 8364.19 |
| Weight before adsorption [g] | 40.01 | 39.91 | 39.13 | 38.73 |
| Weight after adsorption [g] | 44.53 | 43.24 | 43.30 | 41.60 |
| [mg] | 214.91 ± 12.47 | 175.01 ± 14.08 | 117.48 ± 9.79 | 77.07 ± 7.08 |
| [mg] | 312.27 ± 46.54 | 222.13 ± 43.15 | 145.47 ± 38.75 | 95.48 ± 37.80 |
| [mg] | 4308 ± 46.54 | 3888 ± 43.15 | 4425 ± 38.75 | 2775 ± 37.80 |
| CO2 uptake [mmol/g] | 0.34 ± 0.05 | 0.25 ± 0.05 | 0.17 ± 0.04 | 0.11 ± 0.04 |
| H2O uptake [mmol/g] | 1.95 ± 0.02 | 1.79 ± 0.02 | 2.06 ± 0.02 | 1.29 ± 0.02 |
| H2O/CO2 Selectivity | 5.74 ± 0.85 | 7.16 ± 1.43 | 12.12 ± 2.85 | 11.73 ± 4.27 |
Table 2.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves 13X.
Table 2.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves 13X.
| Cycles | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| [mg/m3] | 784.68 ± 19.60 | 770.93 ± 19.60 | 770.29 ± 19.60 | 832.49 ± 19.60 |
| [mg/m3] | 12,878.63 | 6540.08 | 9520.27 | 14,366.6 |
| Weight before adsorption [g] | 40.01 | 37.49 | 37.07 | 36.88 |
| Weight after adsorption [g] | 43.25 | 39.91 | 39.38 | 39.36 |
| [mg] | 28.54 ± 3.62 | 120.10 ± 9.82 | 82.35 ± 7.35 | 62.64 ± 5.40 |
| [mg] | 173.07 ± 47.28 | 144.87 ± 36.18 | 115.68 ± 34.06 | 75.45 ± 35.51 |
| [mg] | 5587 ± 47.28 | 2695 ± 36.18 | 2384 ± 34.06 | 2405 ± 35.51 |
| CO2 uptake [mmol/g] | 0.20 ± 0.06 | 0.17 ± 0.04 | 0.14 ± 0.04 | 0.09 ± 0.04 |
| H2O uptake [mmol/g] | 2.68 ± 0.02 | 1.31 ± 0.02 | 1.17 ± 0.02 | 1.17 ± 0.02 |
| H2O/CO2 Selectivity | 13.40 ± 4.02 | 7.71 ± 1.82 | 8.36 ± 2.39 | 13.00 ± 5.78 |
Table 3.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves 4A.
Table 3.
Adsorption and desorption results for CO2 and H2O over 4 cycles for 40g packed bed of molecular sieves 4A.
| Cycles | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| [mg/m3] | 735.32 ± 19.60 | 728.58 ± 19.60 | 771.57 ± 19.60 | 723.62 ± 19.60 |
| [mg/m3] | 8991.64 | 5376.07 | 11,925.4 | 12,158.3 |
| Weight before adsorption [g] | 40.01 | 37.85 | 37.51 | 37.52 |
| Weight after adsorption [g] | 42.48 | 41.10 | 41.53 | 40.60 |
| [mg] | 30.46 ± 6.03 | 104.06 ± 18.17 | 62.24 ± 10.38 | 34.42 ± 11.72 |
| [mg] | 240.57 ± 44.13 | 146.02 ± 35.86 | 85.02 ± 28.80 | 45.46 ± 24.47 |
| [mg] | 4389 ± 44.13 | 3444 ± 35.86 | 3925 ± 28.80 | 3035 ± 24.47 |
| CO2 uptake [mmol/g] | 0.28 ± 0.05 | 0.17 ± 0.04 | 0.10 ± 0.03 | 0.05 ± 0.03 |
| H2O uptake [mmol/g] | 2.07 ± 0.02 | 1.65 ± 0.02 | 1.88 ± 0.01 | 1.46 ± 0.01 |
| H2O/CO2 Selectivity | 7.39 ± 1.32 | 9.71 ± 2.29 | 18.80 ± 5.64 | 29.20 ± 17.52 |
Table 4.
Adsorption and desorption results for CO2 and H2O over one TSA cycle with 12 days thermal pre-treatment at 100°C for molecular sieves JPM3, 13X and 4A.
Table 4.
Adsorption and desorption results for CO2 and H2O over one TSA cycle with 12 days thermal pre-treatment at 100°C for molecular sieves JPM3, 13X and 4A.
| Molecular Sieve | JLPM3 | 13X | 4A |
|---|---|---|---|
| Bed length () [cm] | 4.0 | 4.0 | 3.5 |
| [mg/m3] | 718.27 ± 19.60 | 745.33 ± 19.60 | 732.91 ± 19.60 |
| [mg/m3] |
8726.44 |
3382.61 |
9072.79 |
| Weight Before Adsorption [g] | 200.01 | 200.01 | 200.01 |
| Weight After Adsorption [g] | 214.22 | 207.39 | 208.83 |
| Weight After Desorption [g] | 187.14 | 184.6 | 188.44 |
| [mg] | 408.40 ± 24.95 | 128.95 ± 8.89 | 22.57 ± 19.62 |
| [mg] | 1009.93 ± 45.35 | 857.48 ± 43.45 | 588.55 ± 37.76 |
| [mg] | 26,070 ± 43.35 | 21,933 ± 43.45 | 19,802 ± 37.76 |
| CO2 uptake [mmol/g] | 0.24 ± 0.01 | 0.20 ± 0.01 | 0.14 ± 0.01 |
| H2Ouptake [mmol/g] | 2.51 ± 0.00 | 2.14 ± 0.00 | 1.89 ± 0.00 |
| H2O/CO2 Selectivity | 10.46 ± 0.44 | 10.70 ± 0.53 | 13.50 ± 0.96 |
| [ºC] | 103.1 | 103.7 | 92.3 |
| [ºC] | 152.6 | 136.9 | 126.6 |
| [ºC] | 275.3 | 273.2 | 236.6 |
Table 5.
BET/BJH data for molecular sieves JLPM3, 13X and 4A.
| Molecular Sieve | BET Surface Area (m2 g-1) | Micropore Area (m2 g-1) | External Surface Area (m2 g-1) | Total Pore Volume (cm3 g-1) | Micropore Volume (cm3 g-1) | Median Pore Width (nm) | BJH Average Mesopore Width (nm) |
|---|---|---|---|---|---|---|---|
| JLPM3 | 727.901 | 710.285 | 17.616 | 0.301 (at p/po = 0.977) | 0.267 | 0.758 | 7.569 (des.) |
| 13X | 710.088 | 672.618 | 37.470 | 0.359 (at p/po = 0.975) | 0.253 | 0.765 | 13.353 (des.) |
| 4A | 24.087 | 2.809 | 21.278 | 0.086 (at p/po = 0.976) | 0.001 | 41.560 | 15.699 (des.) |
Table 6.
Mercury intrusion porosimetry data for molecular sieves JLPM3, 13X and 4A.
| Molecular Sieve | Total Intrusion Volume at 59.862,34 Psia (mL g-1) | Total Pore Area at 59.862,34 Psia (m2 g-1) | Bulk Density at 1,02 Psia (g mL-1) | Apparent (Skeletal) Density at 59.862,34 Psia (g mL-1) | Porosity (%) |
|---|---|---|---|---|---|
| JLPM3 | 0.225 | 4.875 | 1.162 | 1.575 | 26.190 |
| 13X | 0.229 | 16.305 | 1.114 | 1.495 | 25.491 |
| 4A | 0.176 | 14.703 | 1.349 | 1.768 | 23.721 |
Table 7.
Elemental composition of molecular sieve JLPM3, 13X and 4A.
| Composition (wt.%) | JLPM3 | 13X | 4A |
|---|---|---|---|
| Na2O | 17.69 | 16.63 | 15.26 |
| MgO | 0.14 | 2.68 | 2.43 |
| SiO2 | 45.99 | 48.34 | 45.71 |
| Al2O3 | 33.82 | 29.33 | 32.62 |
| K2O | 0.21 | 0.24 | 0.35 |
| Fe2O3 | 0.60 | 1.13 | 1.44 |
| CaO | 0.97 | 0.83 | 1.15 |
| Other* | 1.58 | 0.82 | 1.04 |
| Si/Al** | 1.15 | 1.40 | 1.19 |
*“Other” includes P2O5, SO3, Cl, TiO2, MnO, SrO, ZrO2 and (where present) V2O5, ZnO, Rb2O; each individual component is ≤ 0.37 wt. %. ** Si/Al = Silicon/Aluminium.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



