Submitted:
07 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Skin inflammation is characterized by oxidative stress, excessive keratinocyte activa-tion, and the overproduction of pro-inflammatory cytokines. In a previous study, we demonstrated that the hydroalcoholic extract from Posidonia oceanica leaves (POE) mit-igates psoriasis-like skin inflammation in a mouse model. In the present study, we in-vestigated the cellular mechanisms underlying these effects in human HaCaT keratinocytes. Non-cytotoxic lipopolysaccharide (LPS) stimulation reproduced key in-flammatory features, including impaired cell proliferation, increased production of ROS and NO, and the upregulation of IL-1β, IL-6, TNF-α and CXCL8/IL-8. Co-treatment with POE significantly attenuated these alterations by restoring cell pro-liferation, suppressing oxidative stress, particularly NOS2/NO, and normalizing both cytokine expression and release. POE alone did not affect cell viability or inflammatory markers, confirming its favorable safety profile. However, POE alone induced a mild pro-apoptotic response, which may contribute to overcoming the apoptosis re-sistance typically observed in psoriatic keratinocytes. These in vitro findings are con-sistent with our previous in vivo results and demonstrate that POE exerts antioxidant and anti-inflammatory activities. Overall, these results support the POE as a promising marine-derived candidate for complementary strategies in the management of psoria-sis-associated inflammatory skin disorders.
Keywords:
Posidonia oceanica
; keratinocytes
; skin inflammation
; pro-inflammatory cytokines
; antioxidants
; marine natural products
; complementary medicine
; lipopolysaccharide
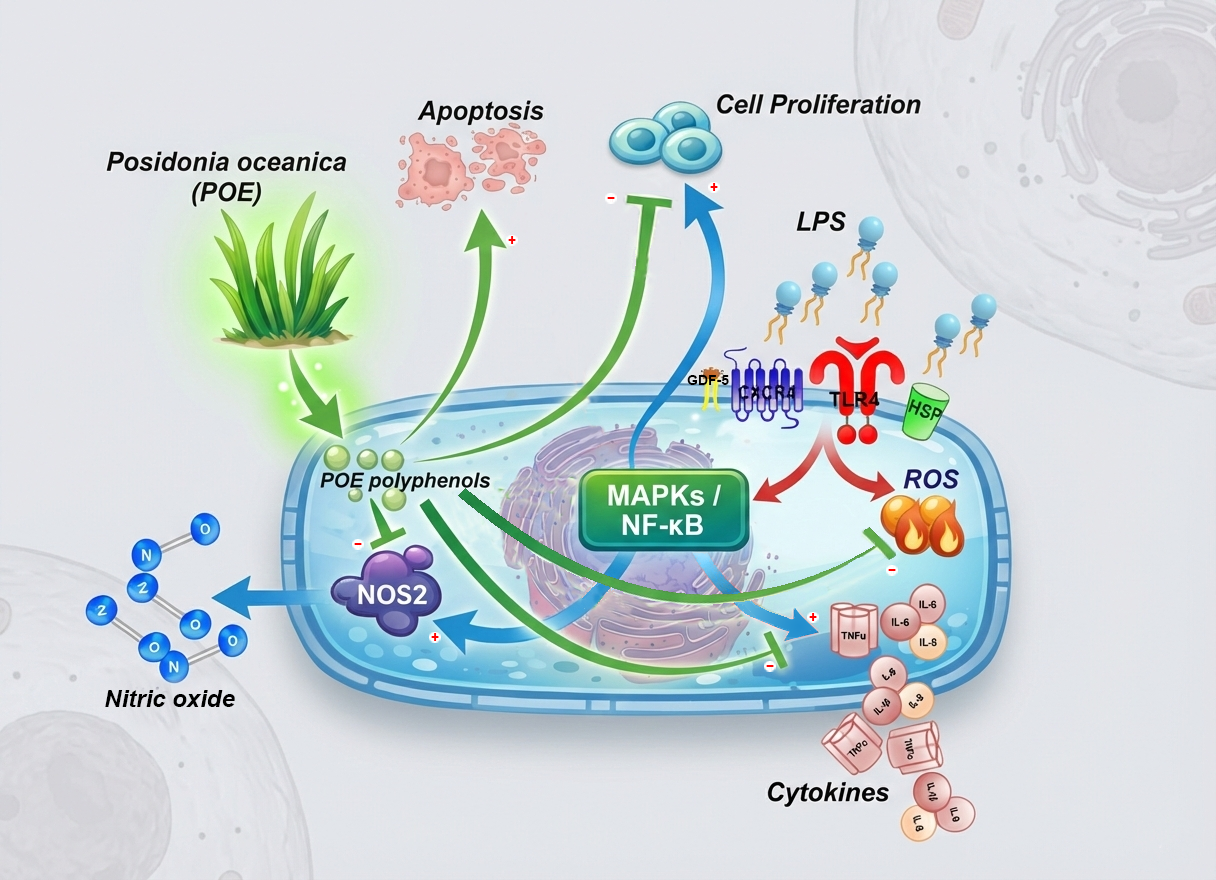
1. Introduction
Skin inflammation is a hallmark of numerous dermatological disorders, including atopic dermatitis, contact dermatitis, psoriasis, and other inflammatory skin conditions. These diseases are characterized by oxidative stress, dysregulated keratinocyte activity, and excessive production of pro-inflammatory cytokines, collectively contributing to epidermal barrier dysfunction and chronic inflammation [1,2]. Once considered inert structural cells, epidermal keratinocytes are now recognized as key initiators and amplifiers of skin inflammation. They actively contribute to the inflammatory milieu by secreting cytokines such as IL-1, IL-6, TNF-α, and the chemokine CXCL8/IL-8 [3,4].
In psoriasis, the inflammatory response is largely mediated by lymphocytes, particularly Th17 cells, which are activated by exogenous and endogenous stimuli and secrete cytokines including IL-17, IFN-γ, and IL-22. These cytokines drive keratinocyte hyperproliferation, promote anti-apoptotic signaling, and induce aberrant differentiation, leading to psoriatic plaque formation [5,6]. Increasing evidence indicates that innate immune activation precedes adaptive responses in psoriasis. Stimulation of Toll-like receptors (TLRs) by damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) represents an early initiating event, triggering innate immune responses in keratinocytes and dendritic cells (DCs). Upon DAMP or PAMP recognition, keratinocytes produce a wide array of pro-inflammatory cytokines, including IFN-β, IL-1β, IL-36, TNF-α, IL-6, and CXCL10, promoting the differentiation and recruitment of inflammatory T-cell subsets [1,7]. Th17 lymphocyte stimulation of keratinocytes further amplifies inflammation via enhanced secretion of TNF-α, IL-1, IL-6, and CXCL8/IL-8, sustaining the chronic inflammatory response characteristic of psoriasis [8].
Current therapies for inflammatory skin disorders, including psoriasis, often alleviate symptoms but may cause side effects or have limited efficacy, highlighting the need for safe and effective complementary or alternative approaches [9,10,11].
Marine-derived natural products have emerged as promising sources of bioactive compounds with antioxidant and anti-inflammatory properties. Posidonia oceanica (L.) Delile, a seagrass endemic to the Mediterranean Sea, is rich in polyphenols and other bioactive molecules with potent antioxidant activity. Previous studies in our laboratory demonstrated that a hydroalcoholic extract from P. oceanica leaves (POE) exhibits significant anti-inflammatory and antioxidant effects in various experimental models [12]. In our prior in vivo study using a mouse model of imiquimod-induced psoriasis-like skin inflammation, POE attenuated both clinical and histological features of psoriasis and significantly reduced expression of psoriatic pro-inflammatory cytokines, including IL-17A, IL-17F, IL-23, TNF-α, IFN-γ, and IL-2, as well as the inflammation-associated adipokine lipocalin-2 (LCN-2) [13]. These findings support the therapeutic potential of POE and provide a rationale for assessing its direct effects on keratinocytes, the key initiators of psoriatic inflammation.
In vitro models are essential for dissecting molecular mechanisms underlying keratinocyte-mediated inflammatory responses. Human immortalized keratinocytes, such as HaCaT cells, retain many biological characteristics of normal human keratinocytes [14,15] and respond to pro-inflammatory stimuli by producing cytokines and reactive oxygen species (ROS) [16,17,18]. Although HaCaT cells do not fully recapitulate all aspects of epidermal differentiation in vivo, they are stable and reproducible, making them a reliable system to study keratinocyte-mediated inflammatory mechanisms (Appendix A, Table A1).
Stimulation of HaCaT cells with lipopolysaccharide (LPS), a bacterial endotoxin, is widely used as an in vitro model of psoriasis (Appendix A, Table A1). LPS induces secretion of pro-inflammatory cytokines, including IL-1β, IL-6, TNF-α, and CXCL8/IL-8 [19,20], and upregulates inducible nitric oxide synthase 2 (NOS2), resulting in increased nitric oxide (NO) production [21]. NOS2 and NO are critical in the pathogenesis of psoriasis and other inflammatory skin disorders [22]. LPS binds to TLR4, which can also be activated by DAMPs [7], leading to chronic inflammation, oxidative stress, and epidermal barrier disruption. TLR4 signals via both MyD88-dependent and TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathways: the MyD88 pathway triggers rapid NF-κB and MAPK activation and pro-inflammatory cytokine expression, while the TRIF pathway mediates delayed NF-κB activation, amplifying inflammation [23,24]. Consistently, our previous studies showed that POE counteracts LPS-induced NF-κB activation in murine RAW264.7 macrophages by modulating ERK1/2 and Akt signaling cascades [25], highlighting its potential to interfere with LPS-driven inflammation.
Based on these premises, the present study aimed to elucidate the cellular mechanisms underlying the protective and anti-inflammatory effects of POE in keratinocytes, the primary initiators and amplifiers of psoriatic inflammation. Using LPS-stimulated HaCaT cells, we evaluated the effects of POE on cell proliferation, apoptosis, ROS production, transcriptional and secretory profiles of key psoriatic cytokines (IL-1β, IL-6, TNF-α, IL-8), and NOS2/NO expression. This approach allowed us to assess the anti-inflammatory potential of POE in keratinocyte-mediated psoriasis-relevant responses and provided the opportunity to explore its potential effects on keratinocyte proliferation and apoptosis.
2. Results
2.1. Determination of Safe Working Concentrations of POE and LPS
To identify the non-toxic threshold doses of POE and LPS for subsequent assays, HaCaT cell metabolic activity was assessed using the MTT assay, a colorimetric method that quantifies mitochondrial function. As shown in Figure 1, exposure to POE at concentrations up to 6.5 µg/mL (expressed as polyphenol equivalents; Figure 1a) or to LPS up to 2.5 µg/mL (Figure 1b) did not significantly affect metabolic activity compared with untreated controls. In contrast, higher concentrations of POE, such as 26 µg/mL polyphenol equivalents, induced a modest but significant reduction in metabolic activity (78.19 ± 3.10%, p < 0.05). Similarly, LPS at 5 and 10 µg/mL exerted marked cytotoxic effects, significantly reducing metabolic activity to 77.98 ± 3.94% and 66.29 ± 2.93%, respectively. Exposure to the vehicle DMSO did not significantly affect metabolic activity (Figure 1c).
Based on these findings, subsequent experiments were conducted using POE at 6.5 µg/mL polyphenol equivalents (dissolved in 14 mM DMSO as vehicle), 2.5 µg/mL LPS, and 14 mM DMSO alone as a vehicle control.
2.2. POE Mitigates the Stimulatory Effect of LPS on HaCaT Cell Proliferation
Cell proliferation was assessed using the green fluorescent nucleic acid–binding CyQUANT dye, which quantifies cell number based on DNA content. As shown in Figure 2, treatment with LPS (2.5 µg/mL) significantly increased HaCaT cell proliferation, resulting in an approximately 1.4-fold increase compared with control cells (Figure 2b; fold change vs. control: 1.35 ± 0.03, p < 0.05). In contrast, exposure to POE alone (6.5 µg/mL polyphenol equivalents) did not significantly affect cell proliferation (Figure 2b; fold change vs. control: 0.95 ± 0.08, p > 0.05). Treatment with DMSO (14 mM), used as the vehicle for POE, had no effect on HaCaT cell proliferation. Instead, the POE co-treatment in LPS-stimulated cells significantly attenuated the LPS-induced proliferative response, restoring cell proliferation to levels comparable to those of untreated control cells (Figure 2b; fold change vs. control: 0.89 ± 0.07), indicating that POE attenuates the proliferative effect of LPS on HaCaT cells.
2.3. Analyses of Apoptosis During LPS and POE Stimulation
Apoptosis was evaluated by flow cytometry using Annexin V/7-AAD staining. Representative Annexin V/7-AAD dot plots are shown in Figure 3a–f, while quantitative analyses of necrosis and apoptosis (early + late apoptosis) are summarized in Figure 3g. Untreated control cells exhibited low basal levels of both necrosis and apoptosis (Figure 3b and Figure 3g; necrosis: 2.6 ± 0.42%, apoptosis: 4.8 ± 0.21%).
Similar values were observed in HaCaT cells exposed to LPS (2.5 µg/mL) (Figure 3e and Figure 3g; necrosis: 2.8 ± 0.14%, apoptosis: 5.7 ± 0.01%), indicating that LPS at this concentration did not induce apoptotic or necrotic effects.
In contrast, exposure of HaCaT cells to POE (6.5 µg/mL, expressed as polyphenol equivalents) resulted in a moderate but significant increase in apoptosis compared with control cells (Figure 3d and Figure 3g; necrosis: 1.3 ± 0.07%, apoptosis: 7.75 ± 0.40%, p < 0.05). This pro-apoptotic effect was markedly lower than that observed in positive control cells treated with H₂O₂ (Figure 3a and Figure 3g; necrosis: 3.20 ± 0.28%, apoptosis: 19.95 ± 0.35%, p < 0.05).
2.4. POE Reduces the ROS Increase Triggered by LPS Stimulation
Evaluation of ROS levels using the 2′,7′-dichlorodihydrofluorescein diacetate (H₂DCF-DA) assay revealed that LPS stimulation (2.5 µg/mL) of HaCaT cells induced a significant but moderate increase in ROS compared with control cells (Figure 4b; fold change vs. control: 1.91 ± 0.09, p < 0.05), which was lower than the strong increase triggered by H₂O₂ (200 µM), used as a positive control (Figure 4b; fold change vs. control: 2.6 ± 0.07, p < 0.05). Treatment with POE alone (6.5 µg/mL polyphenol equivalents) or with the vehicle DMSO (14 mM) did not significantly affect ROS levels compared with control cells.
However, co-treatment with POE significantly attenuated the LPS-induced ROS increase, although a modest elevation remained (Figure 4b; fold change vs. control: 1.24 ± 0.09, p < 0.05).
2.5. POE reduces the LPS-Induced Nitric Oxide Secretion
Nitric oxide (NO) release following LPS stimulation (2.5 µg/mL) of HaCaT cells was quantified by measuring the accumulation of nitrite (NO₂⁻), a stable and quantifiable metabolite of NO, using the Griess colorimetric assay. LPS stimulation markedly increased NO₂⁻ levels compared with unstimulated control cells (Figure 5a; 29.20 ± 1.41 nmol/10⁶ cells vs. 2.00 ± 1.27 nmol/10⁶ cells in controls, p < 0.05), corresponding to an approximately fourteen-fold increase (Figure 5b; 14.60 ± 0.70-fold, p < 0.05).
Co-treatment of LPS-stimulated HaCaT cells with POE (6.5 µg/mL polyphenol equivalents) significantly reduced LPS-induced NO₂⁻ secretion (Figure 5a; 3.10 ± 0.40 nmol/10⁶ cells; Figure 5b; fold increase 1.5 ± 0.2, p < 0.05 vs. LPS-stimulated cells), restoring levels to values comparable with control cells.
Treatment with POE alone or with the vehicle DMSO (14 mM, corresponding to the vehicle concentration for POE) did not significantly affect nitrite levels compared with untreated control cells.
Overall, these findings demonstrate that POE inhibits LPS-induced NO production in HaCaT cells, further supporting its anti-inflammatory activity in this keratinocyte model.
2.6. POE Reduces LPS-Induced Cytokine Secretion in HaCaT Cells
The secretion of inflammatory cytokines following LPS stimulation (2.5 µg/mL) of HaCaT cells was quantified by ELISA for IL-1β, IL-6, IL-8, and TNF-α in the culture medium. LPS treatment markedly increased the secretion of IL-1β compared with unstimulated control cells (Figure 6a; 31.12 ± 1.72 pg/10⁶ cells vs. 14.03 ± 0.41 pg/10⁶ cells in controls, p < 0.05) and IL-6 (Figure 6c; 15.77 ± 0.90 pg/10⁶ cells vs. 8.06 ± 0.58 pg/10⁶ cells in controls, p < 0.05), with both cytokines showing approximately two-fold increases relative to controls (Figure 6b, 2.21 ± 0.12-fold; Figure 6d, 1.95 ± 0.11-fold). LPS also enhanced the secretion of IL-8 (Figure 6e; 8.70 ± 0.33 pg/10⁶ cells vs. 5.32 ± 1.00 pg/10⁶ cells in controls, p < 0.05) and TNF-α (Figure 6g; 6.88 ± 0.27 pg/10⁶ cells vs. 3.87 ± 0.45 pg/10⁶ cells in controls, p < 0.05), although these increases were more modest and did not reach the two-fold induction observed for IL-1β and IL-6 (Figure 6f, 1.60 ± 0.06-fold; Figure 6h, 1.77 ± 0.07-fold).
Exposure of LPS-stimulated cells to POE (6.5 µg/mL polyphenol equivalents) significantly attenuated the elevations of both IL-1β and IL-6 (Figure 6a, 20.96 ± 1.51 pg/10⁶ cells; Figure 6c, 10.89 ± 1.34 pg/10⁶ cells, p < 0.05), as well as those of IL-8 and TNF-α (Figure 6e, 7.07 ± 0.33 pg/10⁶ cells; Figure 6g, 5.65 ± 0.27 pg/10⁶ cells, p < 0.05), resulting in cytokine levels only slightly above those of untreated cells (Figure 6b, 1.49 ± 0.10-fold; Figure 6d, 1.35 ± 0.16-fold; Figure 6f, 1.28 ± 0.05-fold; Figure 6h, 1.46 ± 0.07-fold).
Treatment with POE alone, or with the vehicle DMSO (14 mM, the concentration used for POE), did not significantly affect cytokine secretion compared with control cells.
Overall, these findings indicate that POE effectively mitigates the LPS-induced inflammatory cytokine response in HaCaT cells.
2.7. POE Suppresses Cytokine and Nitric Oxide Synthase 2 Expression Induced by LPS Stimulation
The gene expression levels of IL-1β, IL-6, IL-8, TNF-α and nitric oxide synthase 2 (NOS2) were quantified by qRT-PCR. As shown in Figure 7, stimulation of HaCaT cells with LPS (2.5 µg/mL) markedly upregulated the transcription of pro-inflammatory cytokines and NOS2. Specifically, IL-1β and IL-6 mRNA levels nearly doubled compared with untreated control cells (Figure 7a, 1.95 ± 0.09-fold; Figure 7b, 1.91 ± 0.11-fold, respectively; p < 0.05). Similarly, NOS2 mRNA expression was approximately two-fold higher than in unstimulated cells (Figure 7e, 2.12 ± 0.19-fold; p < 0.05).
More moderate but statistically significant increases were also observed for IL-8 and TNF-α (Figure 7c, 1.38 ± 0.01-fold; Figure 7d, 1.44 ± 0.09-fold, respectively; p < 0.05), confirming that LPS activates a pro-inflammatory transcriptional program in HaCaT keratinocytes.
Exposure of LPS-stimulated HaCaT cells to POE (6.5 µg/mL polyphenol equivalents) restored the expression of all measured cytokines and NOS2 to levels comparable to those of untreated controls, demonstrating that POE effectively counteracts LPS-induced cytokine overexpression and exerts a clear anti-inflammatory effect at the transcriptional level.
Treatment with POE alone or with the vehicle DMSO (14 mM, the concentration used for POE) did not significantly affect cytokine or NOS2 expression compared with control cells. These findings are consistent with the results obtained from the analyses of cytokine and NOS2 secretion.
3. Discussion
Several chronic inflammatory skin disorders, including atopic dermatitis, hidradenitis suppurativa, and psoriasis, are driven not only by dysregulated adaptive immune responses but also by the aberrant activation of epidermal keratinocytes. In response to danger signals, keratinocytes initiate both protective and immunopathological reactions that critically contribute to disease onset and persistence [5,26,27,28].
Keratinocytes express a broad repertoire of TLRs, enabling them to recognize PAMPs derived from microbial structures, such as LPS, peptidoglycan, and flagellin, as well as endogenous danger-associated molecular patterns DAMPs, including host DNA released from dying cells, mitochondrial DNA, serum amyloid A, and amyloid β [29,30].
Engagement of TLRs triggers intracellular signaling cascades that converge on the transcription factor NF-κB, promoting the expression of cytokines, chemokines, and co-stimulatory molecules involved in immune and inflammatory responses [31,32]. Through these mechanisms, keratinocytes not only mount innate immune defenses but also orchestrate immune cell recruitment and activation, thereby shaping both acute and chronic inflammatory responses.
Although the psoriatic skin microbiota is predominantly associated with Gram-positive bacteria, particularly members of the Firmicutes and Actinobacteria phyla [33], recent evidence indicates that lesional areas of inverse psoriasis are characterized by a significantly higher abundance of Gram-negative bacteria compared with both psoriasis vulgaris and healthy skin [34]. These findings support a potential role for Gram-negative bacteria in sustaining specific subtypes of psoriatic inflammation and provide a rationale for investigating Gram-negative–derived inflammatory stimuli in psoriasis-associated models.
A commonly employed in vitro model to study psoriasis-related inflammatory mechanisms consists of stimulating human immortalized HaCaT keratinocytes with LPS, a prototypical PAMP derived from the outer membrane of Gram-negative bacteria. Canonical LPS-induced inflammatory signaling involves its binding to the serum LPS-binding protein (LBP), which facilitates the transfer of LPS to membrane-bound or soluble CD14. The resulting LPS–CD14 complex subsequently interacts with the receptor complex composed of TLR4 and myeloid differentiation factor 2 (MD-2) [35]. Indeed, TLR4 does not directly bind LPS; rather, the presence of the adaptor protein MD-2 is essential for ligand recognition. Formation of the activated LPS/MD-2/TLR4 heterodimer initiates intracellular signal transduction cascades that drive the inflammatory response [21,36].
While HaCaT keratinocytes express CD14 and TLR4, they lack MD-2 [37], leading some studies to favor alternative in vitro approaches to model psoriasis-like keratinocyte activation, such as the stimulation with the so-called M5 cytokine cocktail, composed of IL-17A, IL-22, oncostatin M, IL-1α, and TNF-α, which more closely recapitulates the cytokine milieu characteristic of psoriatic lesions [38].
Nevertheless, LPS remains a suitable stimulus for inducing inflammatory responses in HaCaT cells. In the absence of MD-2, LPS can signal through heterogeneous receptor complexes that include heat shock proteins 70 and 90 (HSP70 and HSP90), chemokine receptor 4 (CXCR4), and growth differentiation factor 5 (GDF5), thereby triggering pro-inflammatory signaling pathways in keratinocytes [39,40]. These alternative mechanisms support the continued use of LPS-stimulated HaCaT cells as a simpler in vitro model to investigate innate immune–driven inflammatory responses in keratinocytes. Unlike the M5 cytokine cocktail, which reproduces the inflammatory milieu resulting from the subsequent activation of adaptive immune cells, LPS stimulation relies on a single exogenous trigger that primarily induces early, keratinocyte-intrinsic activation of inflammatory pathways.
In a previous study [41], we demonstrated that oral administration of the hydroalcoholic extract from P. oceanica leaves (POE) significantly attenuated the clinical and histological features of imiquimod-induced psoriasis-like skin inflammation in mice, reducing the expression of key psoriatic pro-inflammatory cytokines and lipocalin-2. Building on these findings, the present study investigates the effects of POE at the cellular level, focusing on keratinocytes as primary responders in psoriasis. Accordingly, we assessed the ability of POE to counteract LPS-induced inflammatory, oxidative, and apoptotic responses in LPS-stimulated HaCaT cells.
Our results show that non-cytotoxic concentrations of LPS and POE exert biologically relevant effects on HaCaT keratinocytes. LPS stimulation (2.5 µg/mL) promoted keratinocyte hyperproliferation without affecting mitochondrial metabolic activity or inducing apoptosis, whereas POE exerted anti-proliferative effects and induced a mild, non-cytotoxic increase in apoptosis. The lack of a pro-apoptotic effect of LPS observed under our experimental conditions is consistent with previous studies reporting that low LPS concentrations do not induce apoptosis in HaCaT keratinocytes [42,43]. This finding could be explained by the dose-dependent nature of LPS signaling. At low, non-toxic concentrations, LPS might preferentially function as an inflammatory and mitogenic stimulus rather than triggering programmed cell death. Through interactions with its ligands, LPS initiates downstream signaling cascades involving NF-κB, MAPKs, and PI3K/Akt [44,45]. These pathways are well established in promoting cell survival, proliferation, and resistance to apoptosis in epithelial cells, including keratinocytes [46,47]. Consequently, the hyperproliferative response induced by LPS under these conditions might reflect the activation of pro-survival and pro-proliferative signaling pathways rather than an intrinsic inability of HaCaT cells to undergo apoptosis. This cellular response closely resembles a key feature of psoriatic epidermis, namely keratinocyte hyperproliferation, thereby supporting the use of LPS-stimulated HaCaT cells as a suitable in vitro model for mimicking psoriasis-associated alterations in keratinocyte homeostasis.
In contrast, POE treatment induced a modest but statistically significant increase in apoptosis (~8%), without affecting cell proliferation or metabolic activity. This POE-induced mild pro-apoptotic effect, which was not altered by co-treatment with LPS, may be particularly relevant in psoriatic phenotype, where chronic inflammation sustains keratinocyte resistance to apoptosis. In this context, POE may help restore a more balanced proliferation-apoptosis turnover [48].
Taken together, the anti-proliferative and mild apoptosis-modulating effects of POE observed in this study support its potential protective role in psoriatic inflammation. These findings are consistent with our previous in vivo evidence showing that POE alleviates imiquimod-induced psoriatic inflammation, as well as with other reports highlighting the therapeutic potential of natural compounds rich in polyphenols and flavonoids in counteracting key pathological features of psoriatic lesions [41,49,50].
Furthermore, we found that LPS stimulation induced moderate increases in intracellular ROS levels, together with a comparable activation of both transcription and secretion of the pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α. In addition, LPS stimulation resulted in a moderate upregulation of NOS2 expression, accompanied by a corresponding increase in the production of its downstream effector, nitric oxide (NO).
These results are in line with previous studies reporting that LPS stimulation in keratinocytes promotes oxidative stress, NO production, and NF-κB–dependent inflammatory cytokine expression [5,51,52,53,54,55]. In particular, increased NOS2 expression and NO production have been described in cultured normal human keratinocytes following stimulation with IL-8 and TNF-α, and the simultaneous expression of NOS2 and the IL-8 receptor has been observed in psoriatic epidermal keratinocytes [56,57]. Moreover, NO at moderate concentrations (<100 µM) has been shown to promote HaCaT cell proliferation, whereas higher concentrations induce cell-cycle arrest [58].
Therefore, in our experimental model, LPS may contribute to a moderate induction of NOS2 expression and NO production through a limited enhancement of IL-8 and TNF-α secretion (less than two-fold vs. control). Accordingly, the proliferative effect observed following LPS stimulation may be mediated by a moderate release of NO driven by cytokine-dependent, but not excessive, NOS2 induction. The moderate increase in ROS levels observed in our study during LPS stimulation at non-cytotoxic concentrations may contribute to a proliferative response in HaCaT cells, in the absence of pro-apoptotic signaling, which is typically associated with excessive ROS accumulation. This interpretation is consistent with previous studies reporting that low ROS levels promote cell proliferation, whereas high ROS levels trigger apoptosis and cytotoxic effects [59,60,61].
LPS-induced expression of pro-inflammatory cytokines was completely normalized by POE, as the mRNA levels of IL-1β, IL-6, IL-8, and TNF-α were restored to values comparable to those observed in untreated cells.
A parallel, although slightly less pronounced, effect was observed at the level of cytokine secretion. While POE markedly reduced LPS-induced cytokine release, it did not fully restore basal secretion levels. The partial persistence of low-level cytokine secretion is consistent with the well-established discordance between mRNA levels and protein secretion, a phenomenon driven by post-transcriptional regulation or differences in half-lives [62].
Inhibition of IL-1β and IL-6 by POE, both at the gene expression level and at the level of cytokine secretion, may interfere with early keratinocyte-derived signals that drive the initiation and maintenance of psoriatic inflammation, since keratinocyte-derived IL-1β and IL-6 are responsible for the recruitment and differentiation of pathogenic Th17 cell subsets [55,63]. Moreover, the inhibition of IL-8 and TNF-α at both the transcriptional and protein secretion levels may explain the suppression of LPS-stimulated keratinocyte proliferation through the down-regulation of NOS2 gene expression and the consequent reduction in NO release, as discussed above. Indeed, co-treatment with POE during LPS exposure normalized NOS2 expression and NO secretion, restoring levels to those of control cells.
The observed anti-inflammatory effects of POE in keratinocytes are consistent with our previous in vivo findings in an imiquimod-induced mouse model of psoriasis-like skin inflammation, in which POE significantly reduced clinical severity and downregulated Th17- and Th1-related cytokines, as well as inflammatory mediators such as LCN2 [41].
Although intracellular signaling pathways were not directly examined in this study, previous work from our group demonstrated that POE exerts anti-inflammatory activity in LPS-stimulated RAW264.7 murine macrophages by inhibiting NF-κB activation and modulating ERK1/2 and Akt signaling [25]. Therefore, it is plausible that similar mechanisms operate in keratinocytes, where NF-κB plays a central role in inflammation and apoptosis resistance [64,65,66]. In this context, future studies from our group will be specifically aimed at dissecting the intracellular signaling pathways underlying the anti-inflammatory effects of POE in the HaCaT inflammatory model, with particular emphasis on elucidating the context-dependent and paradoxical roles of NF-κB signaling in psoriasis pathogenesis [65,66].
In conclusion, this study demonstrates that P. oceanica leaf extract (POE) exerts potent antioxidant and anti-inflammatory effects in human keratinocytes exposed to inflammatory stimuli. By reducing oxidative stress, suppressing pro-inflammatory cytokine expression and release, POE effectively counteracts LPS-induced keratinocyte activation. Besides, the mild pro-apoptotic effect of POE may be beneficial in restoring the balance of proliferation and apoptosis, which is known to be impaired in psoriatic keratinocytes characterized by resistance to apoptosis. These findings further support the therapeutic potential of POE as a marine-derived bioactive phytocomplex and provide a strong rationale for its development as a complementary strategy for the management of psoriasis and other inflammatory skin disorders.
4. Materials and Methods
4.1. Chemicals and Reagents
Unless otherwise indicated, all chemicals and reagents were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
4.2. Cell Line and Culture Conditions
In this study, the HaCaT cell line, consisting of immortalized human keratinocytes (Cytion, formerly Cell Line Service, catalog no. 300493), was used. Cells were maintained at 37 °C in a humidified atmosphere with 5% CO₂, using high-glucose Dulbecco’s modified Eagle’s medium (DMEM) as the standard culture medium (Gibco DMEM High Glucose, Cat. No. 11965092, Thermo Fisher Scientific, Italy), containing 25 mM D-glucose, 4 mM L-glutamine, 44.0 mM NaHCO₃, and 1.8 mM calcium chloride, and supplemented with an additional 1 mM sodium pyruvate (Gibco, Cat. No. 11360070, Thermo Fisher Scientific, Italy) and 10% low-endotoxin, low-hemoglobin, heat-inactivated FBS (56 °C, 30 min) (Gibco Fetal Bovine Serum, Premium Plus, Cat. No. A5669701, Thermo Fisher Scientific, Italy).
Once cultures reached approximately 80% confluence (subconfluent cultures), cells were detached using TrypLE™ Express (Gibco TrypLE™ Express Enzyme 1×, no phenol red, Cat. No. 12604039, Thermo Fisher Scientific, Italy), collected, centrifuged for 10 min at 1000 rpm, and resuspended in complete medium. Cells were subsequently subcultured by seeding into new culture plates at a density of 18 × 10³ cells/cm². All experiments were performed using cells between passages 10 and 25. On the day prior to each experimental procedure, the culture medium was replaced with DMEM containing 1% FBS (serum-deprived medium).
4.3. Hydroalcoholic Extract of P. oceanica
The hydroalcoholic extract from P. oceanica leaves (POE) was prepared as previously described [13]. Briefly, POE obtained from 4 g of dried P. oceanica leaves yielded 52 mg/mL of dry extract after resuspension in 1000 µL of DMSO. Since POE is mainly composed of polyphenols (approximately 88%), as previously reported [67], the concentrations of POE used in all subsequent experimental procedures were expressed as polyphenol equivalents. This approach allows the biological activity to be related specifically to the phenolic fraction and ensures consistency with previously published studies on P. oceanica extracts.
The total polyphenol content of POE, quantified using the Folin–Ciocalteu method [68], was 6.5 ± 1.29 mg/mL, expressed as gallic acid equivalents. Antioxidant activity, measured by the FRAP assay [69], and free radical–scavenging capacity, assessed by the DPPH assay [70], were 0.98 ± 0.2 mg/mL and 1.3 ± 0.1 mg/mL of ascorbic acid equivalents, respectively.
4.4. Identification of Non-Toxic Doses of LPS and POE
To identify the non-toxic threshold doses of LPS and POE, defined as the highest concentrations that did not significantly reduce metabolic activity, the effects of these treatments were evaluated using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [72]. The MTT assay measures the reduction of the water-soluble yellow tetrazolium salt to insoluble purple formazan crystals by metabolically active cells, providing a quantitative readout of cellular metabolic activity. Non-toxic concentrations were defined as those that did not significantly alter metabolic activity, indicating preservation of viable cells and absence of cytotoxicity.
Briefly, subconfluent HaCaT cells were seeded into 96-well plates at a density of 1 × 10⁴ cells/well and cultured overnight in serum-deprived medium prior to treatment. The medium was then replaced with serum-deprived medium containing either two-fold serial dilutions of LPS (ranging from 10 to 0.625 µg/mL; eBioscience™ Lipopolysaccharide 500× solution, Escherichia coli serotype O26:B6, Cat. No. 00-4976-93, Thermo Fisher Scientific, Italy), two-fold serial dilutions of POE expressed as polyphenol equivalents (ranging from 26 ± 4.8 to 3.25 ± 0.6 µg/mL), or the corresponding DMSO concentrations used for POE to control for solvent-related effects (ranging from 0 to 56 mM). Untreated cells were used as controls.
After 12 h of incubation, the medium was removed, and cell monolayers were washed with phosphate-buffered saline (PBS). Subsequently, 100 µL/well of MTT solution (0.5 mg/mL) was added, and plates were incubated for 1 h at 37 °C in the dark. Cells were then washed with PBS and lysed with 100 µL/well of dimethyl sulfoxide (DMSO). The absorbance of the solubilized formazan was measured at 595 nm using a microplate reader (Bio-Rad Model 550 Microplate Reader, Bio-Rad Laboratories, Inc., CA, USA).
Metabolic activity was expressed as a percentage of untreated control cells. All experiments were performed in six replicate wells.
4.5. Cell Proliferation Assay
Once the minimal non-toxic concentrations of LPS and POE were identified, these doses were tested for their effects on cell proliferation using the CyQUANT® Cell Proliferation Assay (Invitrogen, CyQUANT™ Cell Proliferation Assays, Cat. No. C35011, Thermo Fisher Inc., Italy), according to the manufacturer’s instructions. This assay is based on the binding of the fluorometric CyQUANT GR dye to cellular DNA, providing a quantitative measure of DNA content and, therefore, allowing evaluation of cell proliferation and DNA replication [73].
Briefly, HaCaT cells were detached from subconfluent cultures and seeded into 96-well plates at a density of 1 × 10⁴ cells/well in serum-deprived medium. After overnight incubation, the medium was replaced with serum-deprived medium alone (control, non-supplemented) or supplemented with LPS (at its minimal non-toxic concentration), POE (at its minimal non-toxic concentration), or LPS + POE (both agents at their minimal non-toxic concentrations). Additional wells containing DMSO at concentrations corresponding to the POE treatment were included to control for solvent-related effects.
After 12 h of incubation, 100 µL of CyQUANT® 2X detection reagent were added to each well containing 100 µL of culture medium, and plates were incubated for 60 min at 37 °C. Fluorescence was then measured at an excitation wavelength of 485 nm and an emission wavelength of 538 nm using a BioTek Synergy 1H fluorimeter (Agilent Technologies, Santa Clara, CA, USA). Data were expressed either as fluorescence intensity or as fold change relative to control, and all experiments were performed in six replicate wells.
4.6. Flow Cytometric Analysis of Apoptosis
Apoptotic and necrotic cell populations were evaluated by flow cytometry to assess the impact of treatments on HaCaT keratinocyte viability. HaCaT cells (5 × 105) were cultured overnight in serum-deprived medium and subsequently incubated for 12 h with the indicated concentrations of LPS, POE, or their combination (LPS + POE). As a positive control, cells were treated with 200 µM H2O2 for 2 h. Following treatment, cells were detached using TrypLE™ Express (Gibco TrypLE™ Express Enzyme 1×, no phenol red; Cat. No. 12604039, Thermo Fisher Scientific, Italy) and collected for analysis.
Cells were harvested along with the supernatants, centrifuged at 1,000 × g, and resuspended in 500 µL of 1× Annexin Binding Buffer (BD Pharmingen™ Annexin V Binding Buffer, 10× concentrate, Cat. No. 556454, BD Biosciences, USA). The cell suspension was then incubated with 5 µL of Annexin V-BV421 (BD Horizon™ BV421 Annexin V, Cat. No. 563973, BD Biosciences, USA) and 5 µL of 7-Amino-Actinomycin D (7-AAD) (BD Pharmingen™ 7-AAD, Cat. No. 559925, BD Biosciences, USA) at room temperature for 30 min in the dark.
Cell suspensions from quadruplicate samples for each experimental condition were analyzed by flow cytometry using a BD FACSCanto™ II flow cytometer (BD Biosciences, USA). Data were processed with BD FACSDiva™ Software (BD Biosciences, USA), and a minimum of 10,000 events was collected per sample.
4.7. ROS Detection
To determine total ROS production in HaCaT cells following exposure to LPS, POE, or both, intracellular ROS levels were measured using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H₂DCF-DA) [74]. Briefly, HaCaT cells were seeded into 96-well plates at a density of 1 × 10⁴ cells/well and incubated overnight in serum-deprived medium. Cell monolayers were then incubated for 12 h in serum-deprived medium either alone (control) or supplemented with the indicated minimal non-toxic concentrations of LPS, POE, or both. In addition, cells were stimulated with H₂O₂ (200 µM) for 2 h as a positive control.
At the end of the incubation, the medium was removed, monolayers were gently washed with PBS, and 100 µL/well of 10 µM H₂DCF-DA in PBS was added. Cells were incubated for 1 h at 37 °C in the dark. Monolayers were then washed with PBS, and fluorescence was immediately measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm using a BioTek Synergy 1H fluorimeter (Agilent Technologies, Santa Clara, CA, USA).
ROS levels were normalized to cell metabolic integrity, as evaluated in replicate wells by the MTT assay, and data were expressed either as normalized fluorescence (arbitrary units) relative to MTT absorbance at 595 nm, or as fold change versus control. All experiments were performed in six replicate wells.
4.8. Assessment of Nitric Oxide Production
To evaluate the effect of POE on NO release during LPS stimulation, the accumulation of NO₂⁻ in the culture medium was measured. Nitrite is a stable end product of NO, which is highly reactive and rapidly converted in the presence of oxygen and water. Therefore, nitrite provides an indirect estimation of NO production and can be detected and quantified photometrically using the Griess colorimetric reaction [75].
Briefly, subconfluent HaCaT cells were detached as described above, seeded into 24-well plates at a density of 2 × 10⁵ cells/well, and incubated overnight in serum-deprived medium. The medium was then replaced with 200 µL per well of serum-deprived DMEM without phenol red (Gibco DMEM High Glucose, no phenol red, Cat. No. 31053028, Thermo Fisher Scientific, Italy), either alone (control) or supplemented with the indicated minimal non-toxic concentrations of LPS, POE, or their combination (LPS + POE). After 12 h of incubation, culture medium was collected and centrifuged at 500 × g for 5 min at 4 °C to remove cellular debris, and cells from each well were detached and counted using a hemocytometer.
Aliquots of 50 µL of the clarified supernatants were mixed with an equal volume of Griess reagent and incubated at room temperature for 15 min. Nitrite concentrations were determined in nmol/mL using a standard curve prepared with sodium nitrite in the range of 0–65 nmol/mL, by measuring absorbance at 540 nm with a microplate reader (Model 550 Microplate Reader, Bio-Rad, USA).
To normalize nitrite secretion to cell number, nitrite concentrations (nmol/mL) were converted to the total amount of nitrite per well and subsequently normalized to one million cells using the following formula:
Nitrite per 1,000,000 cells (nmol/10⁶ cells) = (Nitrite concentration in the supernatant (nmol/mL) × Supernatant volume (mL) ÷ Number of cells) × 1,000,000
Normalized values were used to compare nitrite secretion across experimental conditions.
4.9. Assessment of Cytokine Secretion by ELISA
To assess the effect of POE on IL-1β, IL-6, IL-8, and TNF-α secretion by HaCaT cells during LPS stimulation, cytokine levels were quantified by ELISA. Subconfluent HaCaT cells were detached as described above, seeded into 6-well plates at a density of 5 × 10⁵ cells/well, and incubated overnight in serum-deprived medium. The medium was then replaced with 2 mL per well of serum-deprived medium either alone (control) or supplemented with the indicated minimal non-toxic concentrations of LPS, POE, or their combination (LPS + POE). After 12 h of incubation, culture media were collected and centrifuged at 500 × g for 5 min at 4 °C to remove cellular debris, and cells from each well were detached and counted using a hemocytometer.
Aliquots of 50 µL of the clarified supernatants were subsequently analyzed in quadruplicate using the following ELISA kits, according to the manufacturers’ instructions: IL-1β, Human IL-1β High Sensitivity ELISA Kit, Invitrogen (Cat. No. BMS224-2HS; analytical sensitivity 0.05 pg/mL; assay range 0.16–10.0 pg/mL); IL-6, Human IL-6 High Sensitivity ELISA Kit, Invitrogen (Cat. No. BMS213-2HS; analytical sensitivity 0.03 pg/mL; assay range 0.08–5.0 pg/mL); IL-8, Human IL-8 Ultrasensitive ELISA Kit, Invitrogen (Cat. No. KHC0084-2; analytical sensitivity 0.19 ng/mL; assay range 0.31–20 ng/mL); TNF-α, Human TNF-α Ultrasensitive ELISA Kit, Invitrogen (Cat. No. KHC3014; analytical sensitivity <0.09 pg/mL; assay range 0.5–32 pg/mL).
Cytokine concentrations were calculated in pg/mL from standard curves by measuring absorbance at 450 nm using a microplate reader (Model 550 Microplate Reader, Bio-Rad, USA). To normalize cytokine secretion to cell number, cytokine concentrations (pg/mL) were converted to the total amount of cytokine per well and subsequently normalized to one million cells using the following formula:
Cytokine per 1,000,000 cells (pg/10⁶ cells) = (Cytokine concentration in the supernatant (pg/mL) × Supernatant volume (mL) ÷ Number of cells) × 1,000,000
Normalized values were used to compare cytokine secretion across experimental conditions.
4.10. Cytokine and Nitric Oxide Synthase 2 Expression by qRT-PCR
To assess the effect of POE on IL-1β, IL-6, IL-8, TNF-α, and NOS2 expression during LPS stimulation of HaCaT cells, qRT-PCR was performed. HaCaT cells were grown in serum-deprived medium, and after overnight incubation, the medium was replaced with serum-deprived medium either alone (control) or supplemented with the indicated minimal non-toxic concentrations of LPS, POE, or their combination (LPS + POE), as described above. After an additional 12 h of incubation, total RNA was extracted from 2 × 10⁶ cells using the RNeasy® Mini Kit (Cat. No. 74106, Qiagen, Milan, Italy) according to the manufacturer’s instructions. RNA concentration and purity were assessed by measuring absorbance at 260 nm and the 260/280 nm ratio, respectively.
A total of 1 µg of RNA was reverse-transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Cat. No. 205311, Qiagen). qRT-PCR reactions were performed in triplicate for each target gene and for the housekeeping gene using a BIO-RAD C1000 Touch Thermal Cycler with CFX96 Real-Time System. Each reaction contained 50 ng of cDNA and PowerSYBR® Green PCR Master Mix (Cat. No. A46109, Thermo Fisher, Italy). Primers (Table 1) were commercially sourced from Origene Technologies Inc. (OriGene Technologies GmbH, Herford, Germany).
Fluorescence was measured as the cycle threshold (Ct). GAPDH served as the reference gene, and relative mRNA expression levels were calculated using the 2⁻ΔΔCt method. PCR cycling conditions were as follows: 50 °C for 2 min (reverse transcription), 95 °C for 10 min (initial denaturation), followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min.
4.11. Statistical Analysis
All data are expressed as mean ± standard deviation (SD), and the number of samples analyzed for each group is indicated in the figure legends. Statistical differences among treatment groups were assessed using one-way ANOVA, followed by Tukey’s post hoc test for multiple comparisons. Analyses were performed using PSPP (version 2.0.0 64bit, GNU Project, Free Software Foundation). Differences were considered statistically significant at p < 0.05.
Author Contributions
Conceptualization, E.B.; methodology, E.B., D.D., M.V., M.L., N.S.; investigation, E.B., M.L., N.S.; writing—original draft preparation, E.B.; writing—review and editing, E.B., D.D., M.V., M.C., A.V., E.A.; supervision, E.B.; funding acquisition, E.B., D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by University of Florence (Fondi di Ateneo 2024 and 2025 to E.B. and D.D.).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We give our special thanks for providing us with Posidonia oceanica leaves to the Applied Ecology and Marine Biology Interuniversity Center “G. Bacci” (CIBM), Livorno, Italy.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Atopic dermatitis |
| Akt | Protein kinase B, Ak is the mouse strain and T is transforming ability |
| ANOVA | Analysis of variance |
| CD14 | Cluster of differentiation 14 |
| cDNA | Complementary DNA |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| CXCL8/IL-8 | C-X-C motif chemokine ligand 8 / Interleukin-8 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DAMPs | Damage-associated molecular patterns |
| DMSO | Dimethyl sulfoxide |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| ELISA | Enzyme-linked immunosorbent assay |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| FBS | Fetal bovine serum |
| FRAP | Ferric reducing antioxidant power |
| GDF5 | Growth/Differentiation Factor 5 |
| H₂O₂ | Hydrogen peroxide |
| H₂DCF-DA | 2′,7′-Dichlorodihydrofluorescein diacetate |
| HaCaT | Human adult (keratinocytes) propagated under low Ca2+ and elevated Temperature |
| HSP70 | Heat shock protein 70 |
| HSP90 | Heat shock protein 90 |
| IFN-β | Interferon beta |
| IFN-γ | Interferon gamma |
| IL-1α | Interleukin-1 alpha |
| IL-1β | Interleukin-1 beta |
| IL-2 | Interleukin-2 |
| IL-6 | Interleukin-6 |
| IL-8 / CXCL8 | Interleukin-8 / C-X-C motif chemokine ligand 8 |
| IL-17A | Interleukin-17A |
| IL-17F | Interleukin-17F |
| IL-22 | Interleukin-22 |
| IL-23 | Interleukin-23 |
| IMQ | Imiquimod |
| LBP | LPS-binding protein |
| LCN-2 | Lipocalin-2 |
| LPS | Lipopolysaccharide |
| MAPKs | Mitogen-activated protein kinases |
| MD-2 | Myeloid differentiation factor 2 |
| MyD88 | Myeloid differentiation primary response 88 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NO₂⁻ | Nitrite |
| NOS2 (iNOS) | Inducible nitric oxide synthase 2 |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PI3K | Phosphoinositide 3-kinase |
| POE | Posidonia oceanica extract |
| P. oceanica | Posidonia oceanica |
| PAMPs | Pathogen-associated molecular patterns |
| RAW264.7 | Raschke William 264.7 Murine macrophage cell line |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| Th1 | Type 1 T helper lymphocytes |
| Th17 | Type 17 T helper lymphocytes |
| TNF-α | Tumor necrosis factor alpha |
| TLRs | Toll-like receptors |
| TLR4 | Toll-like receptor 4 |
| UPLC | Ultra-performance liquid chromatography |
| Rpm | Revolutions per minute |
Appendix A
Appendix A.1

Table A1.
Main references from the last 5 years using LPS-stimulated HaCaT cells as inflammatory and psoriatic models.
Table A1.
Main references from the last 5 years using LPS-stimulated HaCaT cells as inflammatory and psoriatic models.
| Year | Reference | Main effect of LPS stimulation |
| 2025 | Plata-Babula, A.; Kulej, W.; Ordon, P.; Gajdeczka, J.; Stefaniak, M.; Chwalba, A.; Gościniewicz, P.; Kulpok, T.; Grabarek, BO. Modulation of Nuclear Factor Kappa B Signaling and microRNA Profiles by Adalimumab in LPS-Stimulated Keratinocytes. DOI: 10.3390/ijms262010035 |
The effect of Adalimumab, a monoclonal antibody that inhibits tumor necrosis factor-alpha (TNF-α) used in psoriasis therapy, was evaluated on LPS-induced NF-κB–driven inflammation in HaCaT cells. |
| 2024 | Chen, L.; Liu, C.; Xiang, X.; Qiu, W.; Guo, K. miR-155 promotes an inflammatory response in HaCaT cells via the IRF2BP2/KLF2/NF-κB pathway in psoriasis. DOI: 10.3892/ijmm.2024.5415 | LPS induces inflammatory phenotype (↑IL-1β, ↑IL-6, ↑NF-κB). |
| 2023 | Kim, H.J.; Kim, S.Y.; Bae, H.J.; Choi, Y.Y.; An, J.Y.; Cho, Y.E.; Cho, S.Y.; Lee, S.J.; Lee, S.; Sin, M.; Yun, Y.M.; Lee, J.R.; Park, S.J. Anti-Inflammatory Effects of the LK5 Herbal Complex on LPS- and IL-4/IL-13-Stimulated HaCaT Cells and a DNCB-Induced Animal Model of Atopic Dermatitis in BALB/c Mice. DOI: 10.3390/pharmaceutics16010040 |
LPS induces keratinocyte inflammation for anti-inflammatory evaluation. |
| 2023 | Li, S.; Li, G.; Li, X.; Wu, F.; Li, L. Etanercept ameliorates psoriasis progression through regulating high mobility group box 1 pathway. DOI: 10.1111/srt.13329 | LPS triggers pro-inflammatory activation to test Etanercept effects. |
| 2023 | Wang, X.; Yao, Y.; Li, Y.; Guo, S.; Li, Y.; Zhang, G. Experimental study on the effect of luteolin on the proliferation, apoptosis and expression of inflammation-related mediators in lipopolysaccharide-induced keratinocytes. DOI: 10.1177/03946320231169175 | LPS induces inflammation, apoptosis, and proliferation changes in HaCaT. |
| 2023 | Kim, S.-Y.; Hong, M.; Deepa, P.; Sowndhararajan, K.; Park, S.J.; Park, S.; Kim, S. Carthamus tinctorius Suppresses LPS-Induced Anti-Inflammatory Responses by Inhibiting the MAPKs/NF-κB Signaling Pathway in HaCaT Cells. DOI: 10.3390/scipharm91010014 | LPS (1 μg/mL) stimulates NO, IL-6, IL-1β, NF-κB/MAPK activation. |
| 2022 | Zhang, Y.; Liu, P.; You, S.; Zhao, D.; An, Q.; Wang, D.; Zhang, J.; Li, M.; Wang, C. Anti-Inflammatory Effects of Opuntia Milpa Alta Polysaccharides Fermented by Lactic Acid Bacteria in Human Keratinocyte HaCaT Cells. DOI: 10.1002/cbdv.202100923 |
LPS triggers inflammatory phenotype to test polysaccharide effects. |
| 2021 | Takuathung, MN.; Potikanond, S.; Sookkhee, S.; Mungkornasawakul, P.; Jearanaikulvanich, T.; Chinda, K.; Wikan, N.; Nimlamool, W. Anti-psoriatic and anti-inflammatory effects of Kaempferia parviflora in keratinocytes and macrophage cells. DOI: 10.1016/j.biopha.2021.112229 |
LPS induces inflammatory stress in HaCaT cells. |
| 2021 | Yang X.; Dang X.; Zhang X.; Zhao S. Liquiritin reduces lipopolysaccharide-aroused HaCaT cell inflammation damage via regulation of microRNA-31/MyD88. DOI: 10.1016/j.intimp.2021.108283 |
LPS arouses inflammatory damage (↓viability, ↑IL-6/TNF-α/COX-2). |
| 2020 | Thatikonda, S.; Pooladanda, V.; Sigalapalli, DK.; Godugu, C. Piperlongumine regulates epigenetic modulation and alleviates psoriasis-like skin inflammation via inhibition of hyperproliferation and inflammation. DOI: 10.1038/s41419-019-2212-y |
LPS induces inflammation e hyperproliferation in HaCaT cells. |
References
- Shirley, S.N.; Watson, A.E.; Yusuf, N. Pathogenesis of Inflammation in Skin Disease: From Molecular Mechanisms to Pathology. Int J Mol Sci. 2024, 25:10152. [CrossRef]
- Hänel, K.H.; Cornelissen, C.; Lüscher, B.; Baron, J.M. Cytokines and the skin barrier. Int. J. Mol. Sci. 2013, 14, 6720–6745. [CrossRef]
- Luger, T.A.; Schwarz, T. Evidence for an epidermal cytokine network. J. Invest. Dermatol. 1990, 95, 100S–104S. [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [CrossRef]
- Sieminska, I.; Pieniawska, M.; Grzywa, T.M. The Immunology of Psoriasis—Current Concepts in Pathogenesis. Clin. Rev. Allergy Immunol. 2024, 66, 164–191. [CrossRef]
- Chen, C.H.; Wu, N.L.; Tsai, T.F. How Cells Die in Psoriasis? Int. J. Mol. Sci. 2025, 26, 3747. [CrossRef]
- Sun, L.; Liu, W.; Zhang, L.J. The Role of Toll-Like Receptors in Skin Host Defense, Psoriasis, and Atopic Dermatitis. J. Immunol. Res. 2019, 2019, 1824624. [CrossRef]
- Matsushima, Y.; Hwang, S.T.; Simon, S.I. Mechanobiology of neutrophil inflammasome signaling in psoriasis. Curr. Opin. Immunol. 2025, 97, 102680. [CrossRef]
- Ufomadu, P.; Gill, B.J.; Orengo, I.; Rosen, T.; Shimizu, I. The Efficacy of Complementary and Alternative Medicines in Medical Dermatology: A Comprehensive Review. J. Clin. Aesthet. Dermatol. 2025, 18, E61–E79. https://pmc.ncbi.nlm.nih.gov/articles/PMC12189221/.
- Landis, E.T.; Davis, S.A.; Feldman, S.R.; Taylor, S. Complementary and alternative medicine use in dermatology in the United States. J. Altern. Complement Med. 2014, 20, 392–398. [CrossRef]
- Chalupczak, N.V.; Lio, P.A. Complementary and Alternative Therapies for Psoriasis. Arch. Dermatol. Res. 2024, 316, 531. [CrossRef]
- Vasarri, M.; De Biasi, A.M.; Barletta, E.; Pretti, C.; Degl’Innocenti, D. An Overview of New Insights into the Benefits of the Seagrass Posidonia oceanica for Human Health. Mar. Drugs 2021, 19, 476. [CrossRef]
- Micheli, L.; Vasarri, M.; Degl’Innocenti, D.; Di Cesare Mannelli, L.; Ghelardini, C.; Emiliano, A.; Verdelli, A.; Caproni, M.; Barletta, E. Posidonia oceanica (L.) Delile Is a Promising Marine Source Able to Alleviate Imiquimod-Induced Psoriatic Skin Inflammation. Mar. Drugs 2024, 22, 300. [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [CrossRef]
- Wilson, V.G. Growth and differentiation of HaCaT keratinocytes. Methods Mol. Biol. 2014, 1195, 33–41. [CrossRef]
- Kim, D.H.; Byamba, D.; Wu, W.H.; Kim, T.G.; Lee, M.G. Different characteristics of reactive oxygen species production by human keratinocyte cell line cells in response to allergens and irritants. Exp. Dermatol. 2012, 21, 99–103. [CrossRef]
- Mohamadzadeh, M.; Müller, M.; Hultsch, T.; Enk, A.; Saloga, J.; Knop, J. Enhanced expression of IL-8 in normal human keratinocytes and human keratinocyte cell line HaCaT in vitro after stimulation with contact sensitizers, tolerogens and irritants. Exp. Dermatol. 1994, 3, 298–303. [CrossRef]
- Wang, B.; Ruiz, N.; Pentland, A.; Caparon, M. Keratinocyte proinflammatory responses to adherent and nonadherent group A streptococci. Infect. Immun. 1997, 65, 2119–2126. [CrossRef]
- Lu, K.Y.; Cheng, L.C.; Hung, Z.C.; Chen, Z.Y.; Wang, C.W.; Hou, H.H. The Ethyl Acetate Extract of Caulerpa microphysa Promotes Collagen Homeostasis and Inhibits Inflammation in the Skin. Curr. Issues Mol. Biol. 2024, 46, 2701–2712. [CrossRef]
- Curzytek, K.; Maes, M.; Kubera, M. Immune-Regulatory and Molecular Effects of Antidepressants on the Inflamed Human Keratinocyte HaCaT Cell Line. Neurotox. Res. 2021, 39, 1211–1226. [CrossRef]
- An, J.Y.; Kim, S.Y.; Kim, H.J.; Bae, H.J.; Lee, H.D.; Choi, Y.Y.; Cho, Y.E.; Cho, S.Y.; Lee, S.J.; Lee, S.; Park, S.J. Geraniin from the methanol extract of Pilea mongolica suppresses LPS-induced inflammatory responses by inhibiting IRAK4/MAPKs/NF-κB/AP-1 pathway in HaCaT cells. Int. Immunopharmacol. 2024, 140, 112767. [CrossRef]
- Cals-Grierson, M.M.; Ormerod, A.D. Nitric oxide function in the skin. Nitric Oxide 2004, 10, 179–193. [CrossRef]
- Luo, R.; Yao, Y.; Chen, Z.; Sun, X. An examination of the LPS-TLR4 immune response through the analysis of molecular structures and protein-protein interactions. Cell Commun. Signal. 2025, 23, 142. [CrossRef]
- Kulms, D.; Schwarz, T. NF-kappaB and cytokines. Vitam. Horm. 2006, 74, 283–300. [CrossRef]
- Vasarri, M.; Leri, M.; Barletta, E.; Ramazzotti, M.; Marzocchini, R.; Degl’Innocenti, D. Anti-inflammatory properties of the marine plant Posidonia oceanica (L.) Delile. J. Ethnopharmacol. 2020, 247, 112252. [CrossRef]
- Burke, O.M.; Frerichs, V.R.; Garcia, D.F.; Stone, R.C.; Lev-Tov, H.; Czarnowicki, T.; Keane, R.W.; Ojeh, N.; Marjanovic, J.; Pastar, I.; Tomic-Canic, M.; de Rivero Vaccari, J.P.; Sawaya, A.P. The impact of innate immunity and epigenetics in the pathogenesis of hidradenitis suppurativa. Front. Immunol., 2025, 16, 1593253. [CrossRef]
- Ortiz-Lopez, L.I.; Choudhary, V.; Bollag, W.B. Updated perspectives on keratinocytes and psoriasis: keratinocytes are more than innocent bystanders. Psoriasis (Auckl.), 2022, 12, 73–87. [CrossRef]
- Chieosilaatham, P.; Kiatsurayanon, C.; Umehara, Y.; Trujillo-Paez, J.V.; Peng, G.; Yue, H.; Nguyen, L.T.H.; Niyonsaba, F. Keratinocytes: innate immune cells in atopic dermatitis. Clin. Exp. Immunol., 2021, 204, 296–309. [CrossRef]
- Morizane, S.; Mukai, T.; Sunagawa, K.; Tachibana, K.; Kawakami, Y.; Ouchida, M. Input/output cytokines in epidermal keratinocytes and the involvement in inflammatory skin diseases. Front. Immunol., 2023, 14, 1239598. [CrossRef]
- Miller, L.S. Toll-like receptors in skin. Adv. Dermatol., 2008, 24, 71–87. [CrossRef]
- Mao, H.; Zhao, X.; Sun, S.C. NF-κB in inflammation and cancer. Cell Mol. Immunol., 2025, 22, 811–839. [CrossRef]
- Salauddin, M.; Bhattacharyya, D.; Samanta, I.; Saha, S.; Xue, M.; Hossain, M.G.; Zheng, C. Role of TLRs as signaling cascades to combat infectious diseases: a review. Cell Mol. Life Sci., 2025, 82, 122. [CrossRef]
- Zhou, S.; Yao, Z. Roles of Infection in Psoriasis. Int. J. Mol. Sci., 2022, 23, 6955. [CrossRef]
- Ustuner, P.; Ceylan, A.N. Microbiological analysis of patients with inverse and plaque-type psoriasis. BMC Microbiol., 2025, 6, [Epub ahead of print]. [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci., 2021, 78, 1233–1261. [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines (Basel), 2017, 5, 34. [CrossRef]
- Köllisch, G.; Kalali, B.N.; Voelcker, V.; Wallich, R.; Behrendt, H.; Ring, J.; Bauer, S.; Jakob, T.; Mempel, M.; Ollert, M. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology, 2005, 114, 531–541. [CrossRef]
- Zheng, H.; Gu, L.; Wang, Z.; Zhou, H.; Zhang, C.; Teng, X.; Hu, Z.; Wei, X.; Liu, X.; Zeng, F.; Zhao, Q.; Hao, Y.; Hu, Y.; Wang, X.; Hu, J.; Yu, J.; Wu, W.; Zhou, Y.; Cui, K.; Huang, N.; Li, J. Establishing transcription profile of psoriasiform cutaneous in vitro using HaCaT cells stimulated with combination of cytokines. J. Vis. Exp., 2021, (169), [e61537]. [CrossRef]
- Triantafilou, M.; Triantafilou, K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol., 2002, 23, 301–304. [CrossRef]
- Triantafilou, M.; Lepper, P.M.; Briault, C.D.; Ahmed, M.A.; Dmochowski, J.M.; Schumann, C.; Triantafilou, K. Chemokine receptor 4 (CXCR4) is part of the lipopolysaccharide “sensing apparatus”. Eur. J. Immunol., 2008, 38, 192–203. [CrossRef]
- Micheli, L.; Vasarri, M.; Degl’Innocenti, D.; Di Cesare Mannelli, L.; Ghelardini, C.; Emiliano, A.; Verdelli, A.; Caproni, M.; Barletta, E. Posidonia oceanica (L.) Delile is a promising marine source able to alleviate imiquimod-induced psoriatic skin inflammation. Mar. Drugs, 2024, 22, 300. [CrossRef]
- Wang, J.; Luo, H.; Xiao, Y.; Wang, L. miR-125b inhibits keratinocyte proliferation and promotes keratinocyte apoptosis in oral lichen planus by targeting MMP-2 expression through PI3K/Akt/mTOR pathway. Biomed. Pharmacother., 2016, 80, 373–380. [CrossRef]
- Sangaran, P.G.; Ibrahim, Z.A.; Chik, Z.; Mohamed, Z.; Ahmadiani, A. Lipopolysaccharide pre-conditioning attenuates pro-inflammatory responses and promotes cytoprotective effect in differentiated PC12 cell lines via pre-activation of Toll-like receptor-4 signaling pathway leading to the inhibition of caspase-3/nuclear factor-κappa B pathway. Front. Cell Neurosci., 2021, 14, 598453. [CrossRef]
- Arya, P.; Sharma, V.; Singh, P.; Thapliyal, S.; Sharma, M. Bacterial endotoxin-lipopolysaccharide role in inflammatory diseases: an overview. Iran J. Basic Med. Sci., 2025, 28, 553–564. [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther., 2017, 2, 17023. [CrossRef]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J. Clin. Invest., 2005, 115, 2625–2632. [CrossRef]
- Liu, D.; Zhong, Z.; Karin, M. NF-κB: A Double-Edged Sword Controlling Inflammation. Biomedicines, 2022, 10, 1250. [CrossRef]
- Bhat, I.A.; Bhat, A.M.; Tasduq, A.S. Apoptosis-Mechanisms, Regulation in Pathology, and Therapeutic. In , Biochemistrym, Cell Death Regulation in Pathology, Carafa, V., Ed; IntechOpen Limited: London, UK, 2025; [CrossRef]
- Mustafa, A.M.; Atwa, A.M.; Elgindy, A.M.; Alkabbani, M.A.; Ibrahim, K.M.; Esmail, M.M.; El-Shiekh, R.A.; Mohamed, E.M.; Kamel, K.M. Targeting psoriatic inflammation with natural compounds: mechanistic insights and therapeutic promise. Inflammopharmacology, 2025, 33, 3843–3870. [CrossRef]
- Patel, P.; Garala, K.; Bagada, A.; Singh, S.; Prajapati, B.G.; Kapoor, D. Phyto-pharmaceuticals as a safe and potential alternative in management of psoriasis: a review. Z. Naturforsch. C J. Biosci., 2024, 80, 409–430. [CrossRef]
- Marrassini, C.; Arcos, M.L.B.; Cogoi, L.; Alonso, M.R.; Anesini, C. Anti-Inflammatory Effect of Urtica urens on LPS-Challenged Keratinocytes. J. Pharmacopuncture, 2025, 28, 311–320. [CrossRef]
- An, J.Y.; Kim, S.Y.; Kim, H.J.; Bae, H.J.; Lee, H.D.; Choi, Y.Y.; Cho, Y.E.; Cho, S.Y.; Lee, S.J.; Lee, S.; Park, S.J. Geraniin from the methanol extract of Pilea mongolica suppresses LPS-induced inflammatory responses by inhibiting IRAK4/MAPKs/NF-κB/AP-1 pathway in HaCaT cells. Int. Immunopharmacol., 2024, 140, 112767. [CrossRef]
- Jin, C.; Cha, Y.J.; Kim, H.E.; Jin, Y.; Zhang, X.; Le, L.T.T.; Giang, N.N.; Choi, K.; Lee, Y.H.; Kim, W.H.; Choi, J.H.; Chien, P.N.; Yeong, H.C. A Natural Composite Extract Restores Skin Barrier Function by Modulating Inflammatory, Hydration, and Redox Pathways. J. Cosmet. Dermatol., 2025, 24, e70613. [CrossRef]
- Wang, X.; Yao, Y.; Li, Y.; Guo, S.; Li, Y.; Zhang, G. Experimental study on the effect of luteolin on the proliferation, apoptosis and expression of inflammation-related mediators in lipopolysaccharide-induced keratinocytes. Int. J. Immunopathol. Pharmacol., 2023, 37, 3946320231169175. [CrossRef]
- Klimitz, F.J.; Shen, Y.; Repetto, F.; Brown, S.; Knoedler, L.; Ko, C.J.; Abu Hussein, N.; Crisler, W.J.; Adams, T.; Kaminski, N.; Lian, C.G.; Murphy, G.F.; Hsia, H.C.; Pomahac, B.; Kauke-Navarro, M. Keratinocytes as active regulators of cutaneous and mucosal immunity: a systematic review across inflammatory epithelial disorders. Front. Immunol., 2025, 16, 1694066. [CrossRef]
- Bruch-Gerharz, D.; Fehsel, K.; Suschek, C.; Michel, G.; Ruzicka, T.; Kolb-Bachofen, V. A proinflammatory activity of interleukin 8 in human skin: expression of the inducible nitric oxide synthase in psoriatic lesions and cultured keratinocytes. J. Exp. Med. 1996, 184, 2007–2012. [CrossRef]
- Sirsjö, A.; Karlsson, M.; Gidlöf, A.; Rollman, O.; Törmä, H. Increased expression of inducible nitric oxide synthase in psoriatic skin and cytokine-stimulated cultured keratinocytes. Br. J. Dermatol., 1996, 134, 643–648. [CrossRef]
- Frank, S.; Kämpfer, H.; Podda, M.; Kaufmann, R.; Pfeilschifter, J. Identification of copper/zinc superoxide dismutase as a nitric oxide-regulated gene in human (HaCaT) keratinocytes: implications for keratinocyte proliferation. Biochem. J., 2000, 346, 719–728. [CrossRef]
- Khorsandi, K.; Hosseinzadeh, R.; Esfahani, H.; Zandsalimi, K.; Shahidi, F.K.; Abrahamse, H. Accelerating skin regeneration and wound healing by controlled ROS from photodynamic treatment. Inflamm. Regen., 2022, 42, 40. [CrossRef]
- Bito, T.; Nishigori, C. Impact of reactive oxygen species on keratinocyte signaling pathways. J. Dermatol. Sci., 2012, 68, 3–8. [CrossRef]
- Venza, I.; Venza, M.; Visalli, M.; Lentini, G.; Teti, D.; d’Alcontres, F.S. ROS as Regulators of Cellular Processes in Melanoma. Oxid. Med. Cell Longev., 2021, 2021, 1208690. [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol., 2003, 4, 117. [CrossRef]
- Maddur, M.S.; Miossec, P.; Kaveri, S.V.; Bayry, J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am. J. Pathol., 2012, 181, 8–18. [CrossRef]
- Qin, J.Z.; Bacon, P.; Chaturvedi, V.; Nickoloff, B.J. Role of NF-kappaB activity in apoptotic response of keratinocytes mediated by interferon-gamma, tumor necrosis factor-alpha, and tumor-necrosis-factor-related apoptosis-inducing ligand. J. Invest. Dermatol., 2001, 117, 898–907. [CrossRef]
- Zhang, W.; Zhu, G.; Sun, H.; Jiang, C. NLRC3 affects the development of psoriasis by modulating the NF-κB signaling pathway mediated inflammatory response through its interaction with TRAF6. Immunol. Lett., 2025, 272, 106949. [CrossRef]
- Guo, J.; Zhang, H.; Lin, W.; Lu, L.; Su, J.; Chen, X. Signaling pathways and targeted therapies for psoriasis. Signal Transduct. Target Ther., 2023, 8, 437. Erratum in: Signal Transduct. Target Ther., 2024, 9, 25. https://doi.org/10.1038/s41392-023-01733-9. [CrossRef]
- Barletta, E.; Ramazzotti, M.; Fratianni, F.; Pessani, D.; Degl’Innocenti, D. Hydrophilic extract from Posidonia oceanica inhibits activity and expression of gelatinases and prevents HT1080 human fibrosarcoma cell line invasion. Cell Adh. Migr., 2015, 9, 422–431. [CrossRef]
- Medina, M.B. Simple and rapid method for the analysis of phenolic compounds in beverages and grains. J. Agric. Food Chem., 2011, 59, 1565–1571. [CrossRef]
- Pulido, R.; Bravo, L.; Saura-Calixto, F. Antioxidant activity of dietary polyphenols as determined by a modified ferric reducing/antioxidant power assay. J. Agric. Food Chem., 2000, 48, 3396–3402. [CrossRef]
- Fukumoto, L.R.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem., 2000, 48, 3597–3604. [CrossRef]
- Leri, M.; Ramazzotti, M.; Vasarri, M.; Peri, S.; Barletta, E.; Pretti, C.; Degl’Innocenti, D. Bioactive Compounds from Posidonia oceanica (L.) Delile Impair Malignant Cell Migration through Autophagy Modulation. Mar. Drugs, 2018, 16, 137. [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods, 1983, 65, 55–63. [CrossRef]
- Jones, L.J.; Gray, M.; Yue, S.T.; Haugland, R.P.; Singer, V.L. Sensitive determination of cell number using the CyQUANT cell proliferation assay. J. Immunol. Methods, 2001, 254, 85–98. [CrossRef]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol., 1992, 5, 227-31. [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem., 1982, 126, 131-8. [CrossRef]
Figure 1.
HaCaT cells were treated for 12 h with increasing concentrations of POE, LPS, or DMSO. (a) HaCaT cells treated with increasing concentrations of POE (0–26 µg/mL, expressed as polyphenol equivalents); (b) HaCaT cells treated with increasing concentrations of LPS (0–10 µg/mL); (c) HaCaT cells treated with increasing concentrations of DMSO (0–56 mM; vehicle control). Metabolic activity was assessed using the MTT assay and expressed as a percentage of untreated cells. Data are presented as mean ± SD (n = 6). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
Figure 1.
HaCaT cells were treated for 12 h with increasing concentrations of POE, LPS, or DMSO. (a) HaCaT cells treated with increasing concentrations of POE (0–26 µg/mL, expressed as polyphenol equivalents); (b) HaCaT cells treated with increasing concentrations of LPS (0–10 µg/mL); (c) HaCaT cells treated with increasing concentrations of DMSO (0–56 mM; vehicle control). Metabolic activity was assessed using the MTT assay and expressed as a percentage of untreated cells. Data are presented as mean ± SD (n = 6). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
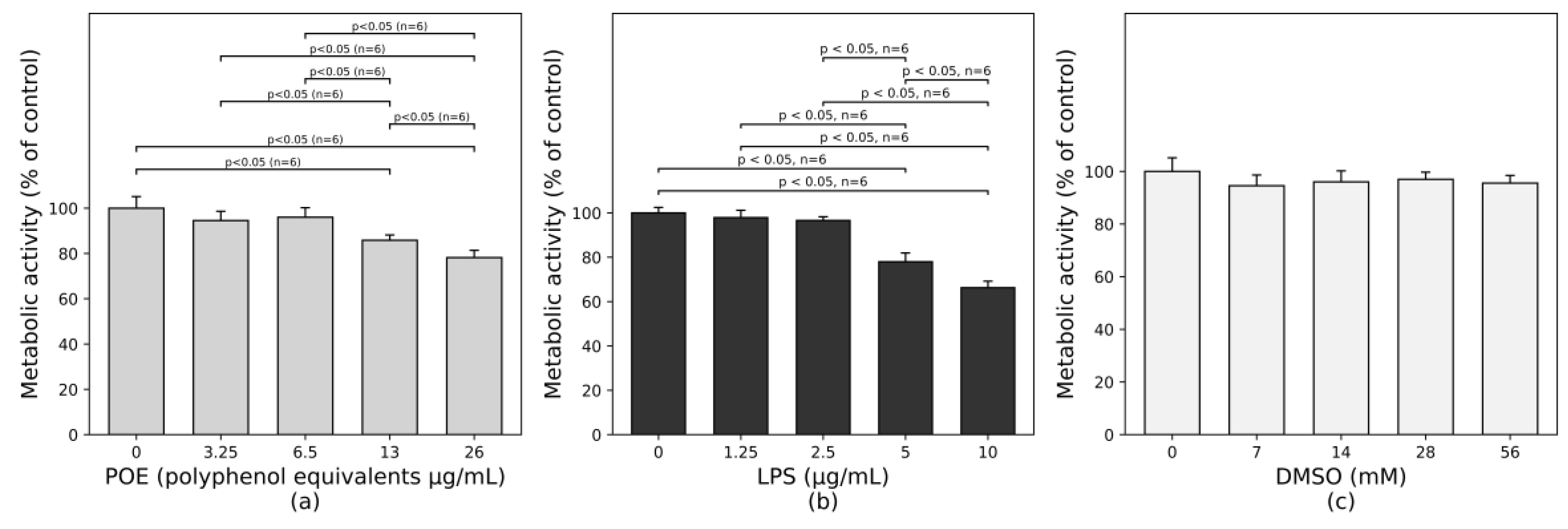
Figure 2.
Cell proliferation was assessed using the CyQUANT GR DNA-binding fluorescent dye. HaCaT cells were cultured under control conditions or treated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle for POE), or a combination of LPS + POE. (a) Cell proliferation expressed as arbitrary fluorescence units; (b) Cell proliferation expressed as fold change relative to control cells. Data are presented as mean ± SD (n = 6). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
Figure 2.
Cell proliferation was assessed using the CyQUANT GR DNA-binding fluorescent dye. HaCaT cells were cultured under control conditions or treated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle for POE), or a combination of LPS + POE. (a) Cell proliferation expressed as arbitrary fluorescence units; (b) Cell proliferation expressed as fold change relative to control cells. Data are presented as mean ± SD (n = 6). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
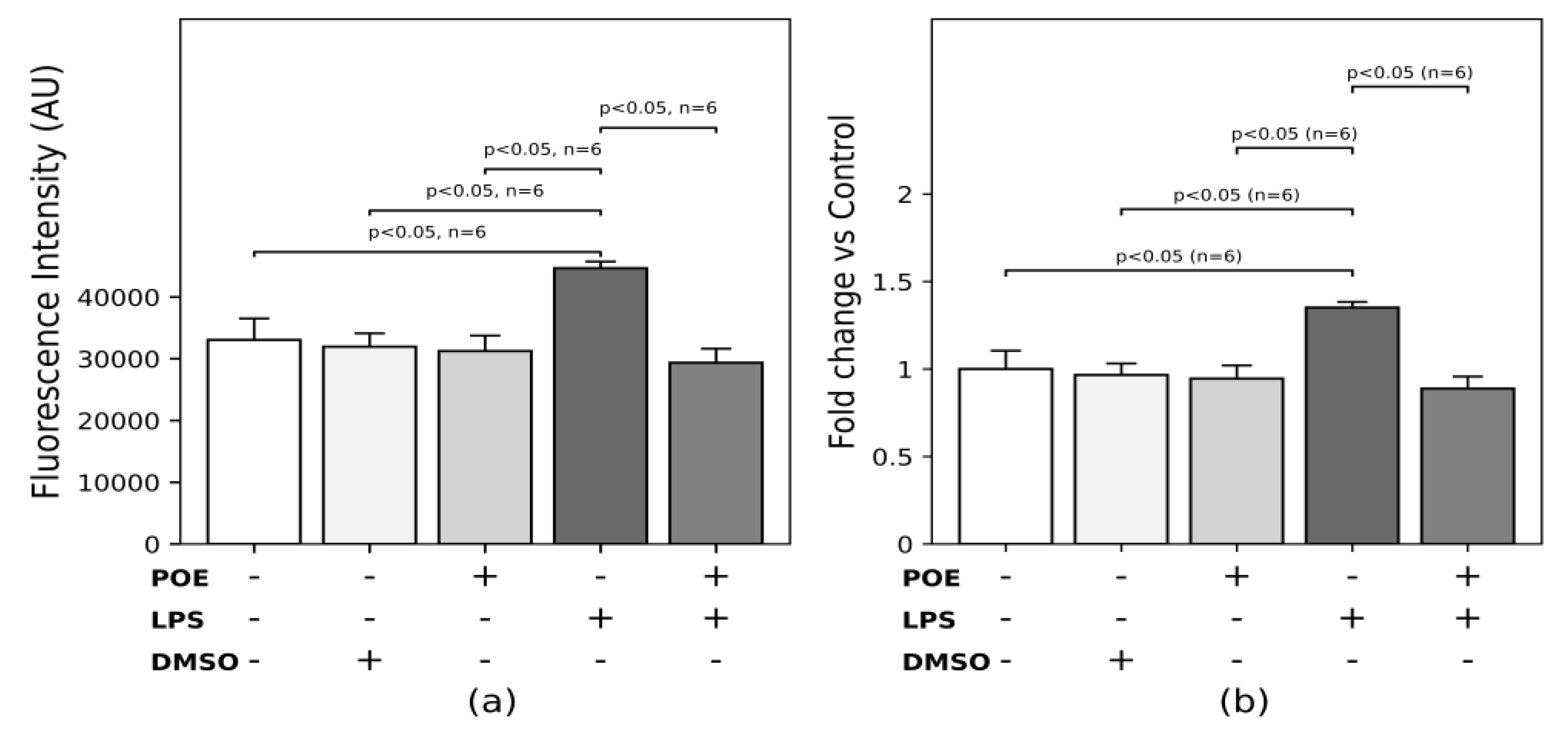
Figure 3.
Analysis of apoptosis and necrosis in HaCaT cells following LPS and POE stimulation. Apoptosis and necrosis were assessed by flow cytometry using Annexin V/7-AAD staining. Representative dot plots are shown for (a) H₂O₂-treated cells (positive control); (b) untreated control cells; (c) DMSO-treated cells (14 mM, vehicle for POE); (d) POE-treated cells (6.5 µg/mL, expressed as polyphenol equivalents); (e) LPS-treated cells (2.5 µg/mL); (f) cells co-stimulated with LPS and POE. Quadrant analysis distinguishes live, early apoptotic, late apoptotic, and necrotic populations. Data are representative of four independent experiments. (g) quantitative analysis of the percentage of live, necrotic, early apoptotic, and late apoptotic cells. The first six groups represent live cells, the next six represent necrotic cells, and the last six represent apoptotic cells, displayed as stacked bars separating early (E) and late (L) apoptosis. Treatments are H₂O₂, Control, DMSO, POE, LPS, and LPS + POE. Data are presented as mean ± SD (n = 4). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only significant differences are indicated; comparisons without p-values were not significant.
Figure 3.
Analysis of apoptosis and necrosis in HaCaT cells following LPS and POE stimulation. Apoptosis and necrosis were assessed by flow cytometry using Annexin V/7-AAD staining. Representative dot plots are shown for (a) H₂O₂-treated cells (positive control); (b) untreated control cells; (c) DMSO-treated cells (14 mM, vehicle for POE); (d) POE-treated cells (6.5 µg/mL, expressed as polyphenol equivalents); (e) LPS-treated cells (2.5 µg/mL); (f) cells co-stimulated with LPS and POE. Quadrant analysis distinguishes live, early apoptotic, late apoptotic, and necrotic populations. Data are representative of four independent experiments. (g) quantitative analysis of the percentage of live, necrotic, early apoptotic, and late apoptotic cells. The first six groups represent live cells, the next six represent necrotic cells, and the last six represent apoptotic cells, displayed as stacked bars separating early (E) and late (L) apoptosis. Treatments are H₂O₂, Control, DMSO, POE, LPS, and LPS + POE. Data are presented as mean ± SD (n = 4). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only significant differences are indicated; comparisons without p-values were not significant.
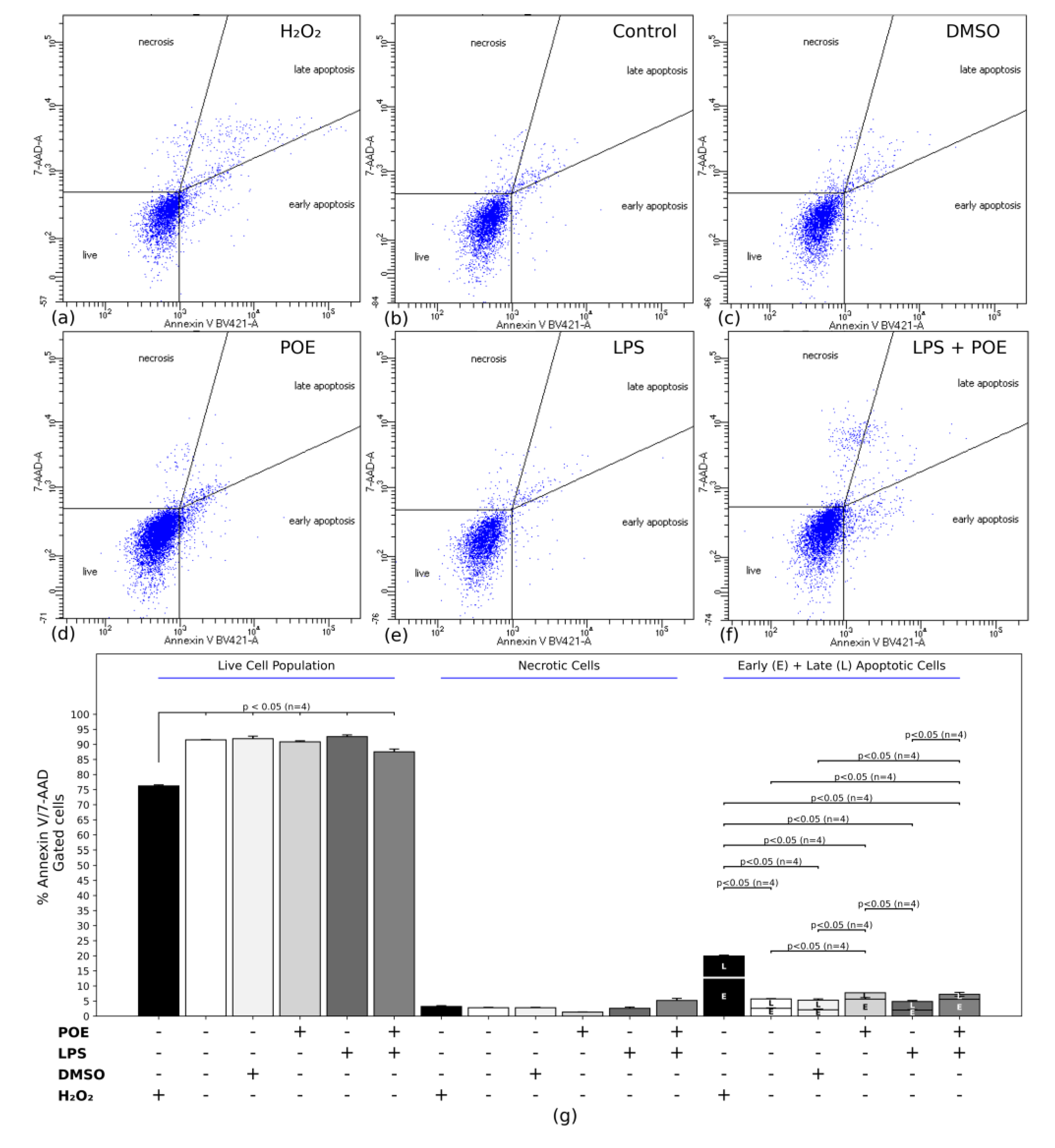
Figure 4.
Detection of ROS levels in HaCaT cells by H₂DCF-DA assay. HaCaT cells were treated for 12 h with H₂O₂ (200 µM, positive control), LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM, vehicle for POE), or a combination of LPS + POE. (a) ROS production expressed as normalized fluorescence (DCF-DA/MTT, arbitrary units); (b) ROS production expressed as fold change relative to untreated control cells. Data are presented as mean ± SD (n = 6). Statistical significance was assessed by one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
Figure 4.
Detection of ROS levels in HaCaT cells by H₂DCF-DA assay. HaCaT cells were treated for 12 h with H₂O₂ (200 µM, positive control), LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM, vehicle for POE), or a combination of LPS + POE. (a) ROS production expressed as normalized fluorescence (DCF-DA/MTT, arbitrary units); (b) ROS production expressed as fold change relative to untreated control cells. Data are presented as mean ± SD (n = 6). Statistical significance was assessed by one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
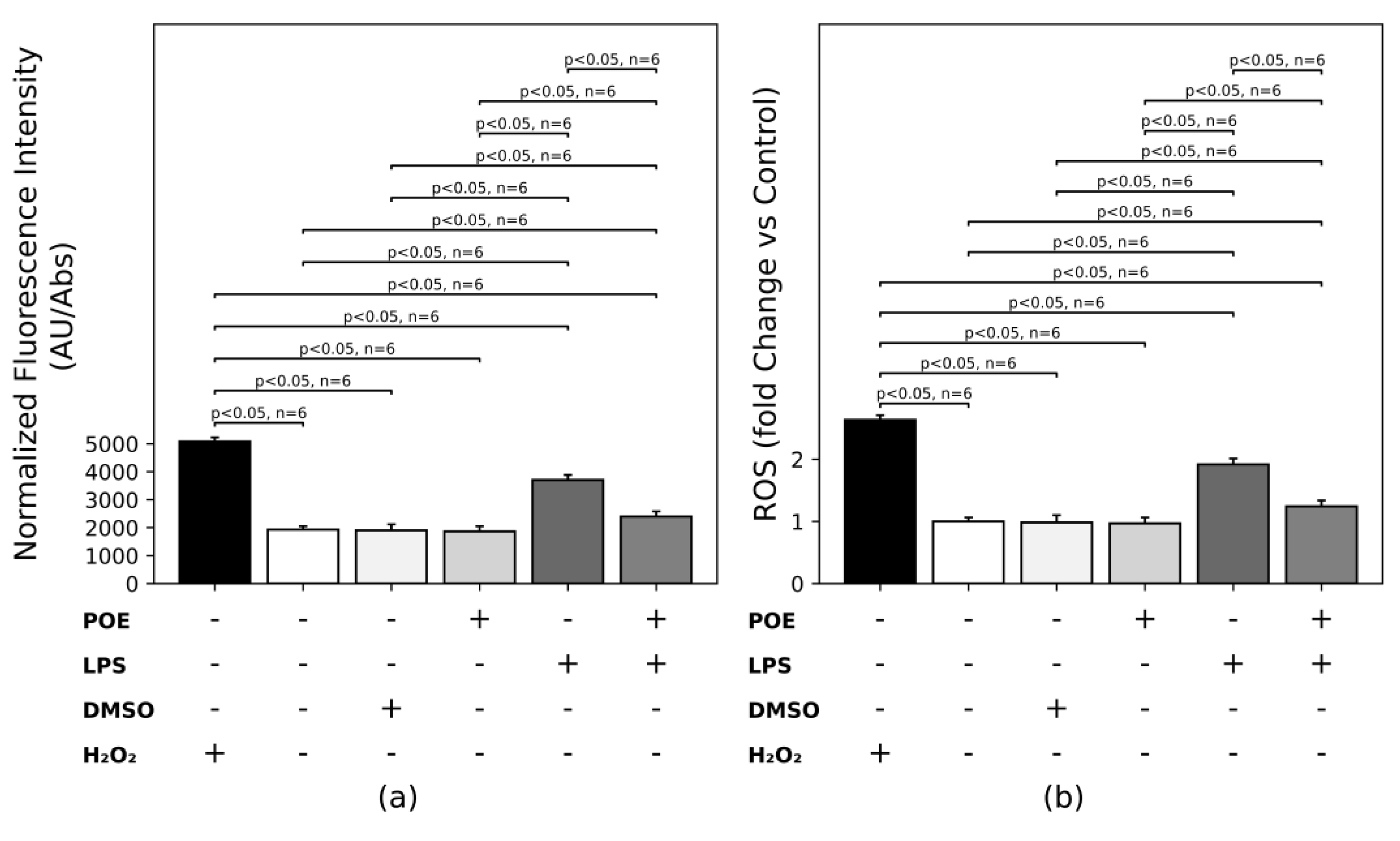
Figure 5.
Effects of POE on nitric oxide (NO) secretion. HaCaT cells were stimulated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle for POE), or a combination of LPS + POE. Nitrite (NO₂⁻), the stable metabolite of NO, was quantified in the culture medium using the Griess colorimetric assay and expressed as nmol/10⁶ cells. (a) Nitrite (NO₂⁻) levels; (b) Nitrite (NO₂⁻) fold change relative to untreated control cells. Data are presented as mean ± SD (n = 2). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
Figure 5.
Effects of POE on nitric oxide (NO) secretion. HaCaT cells were stimulated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle for POE), or a combination of LPS + POE. Nitrite (NO₂⁻), the stable metabolite of NO, was quantified in the culture medium using the Griess colorimetric assay and expressed as nmol/10⁶ cells. (a) Nitrite (NO₂⁻) levels; (b) Nitrite (NO₂⁻) fold change relative to untreated control cells. Data are presented as mean ± SD (n = 2). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; comparisons without p-values were not significant.
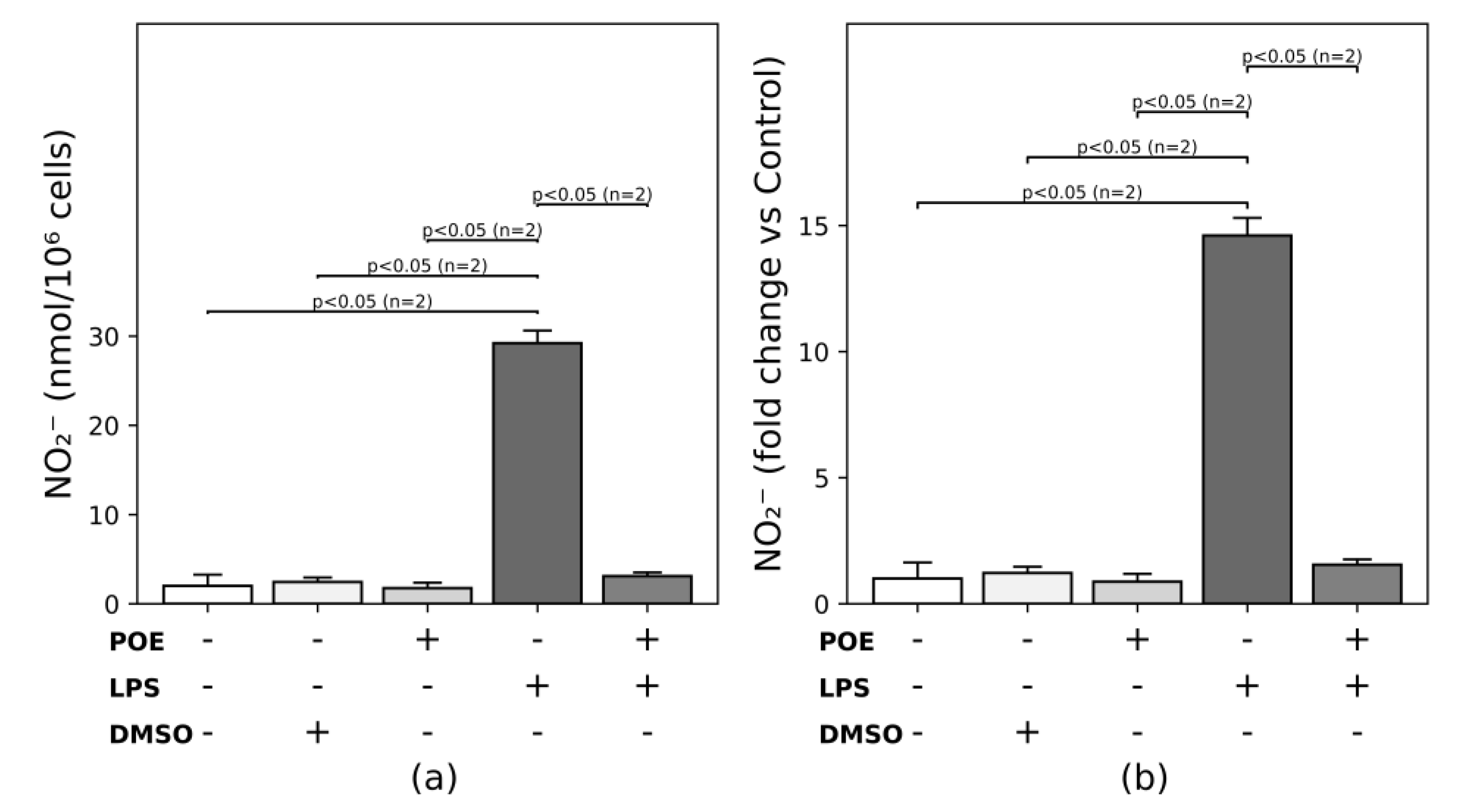
Figure 6.
Effects of POE on cytokine secretion during LPS stimulation of HaCaT cells. HaCaT cells were stimulated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle control), or a combination of LPS + POE. Levels of IL-1β, IL-6, IL-8, and TNF-α in the culture medium were quantified by ELISA and are shown as absolute concentrations (pg/10⁶ cells) and as fold changes relative to untreated controls. (a) IL-1β concentration; (b) IL-1β fold change relative to untreated control; (c) IL-6 concentration; (d) IL-6 fold change relative to untreated control; (e) IL-8 concentration; (f) IL-8 fold change relative to untreated control; (g) TNF-α concentration; (h) TNF-α fold change relative to untreated control. Data represent mean ± SD (n = 4). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; the absence of p-values denotes non-significant comparisons.
Figure 6.
Effects of POE on cytokine secretion during LPS stimulation of HaCaT cells. HaCaT cells were stimulated for 12 h with LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle control), or a combination of LPS + POE. Levels of IL-1β, IL-6, IL-8, and TNF-α in the culture medium were quantified by ELISA and are shown as absolute concentrations (pg/10⁶ cells) and as fold changes relative to untreated controls. (a) IL-1β concentration; (b) IL-1β fold change relative to untreated control; (c) IL-6 concentration; (d) IL-6 fold change relative to untreated control; (e) IL-8 concentration; (f) IL-8 fold change relative to untreated control; (g) TNF-α concentration; (h) TNF-α fold change relative to untreated control. Data represent mean ± SD (n = 4). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; the absence of p-values denotes non-significant comparisons.
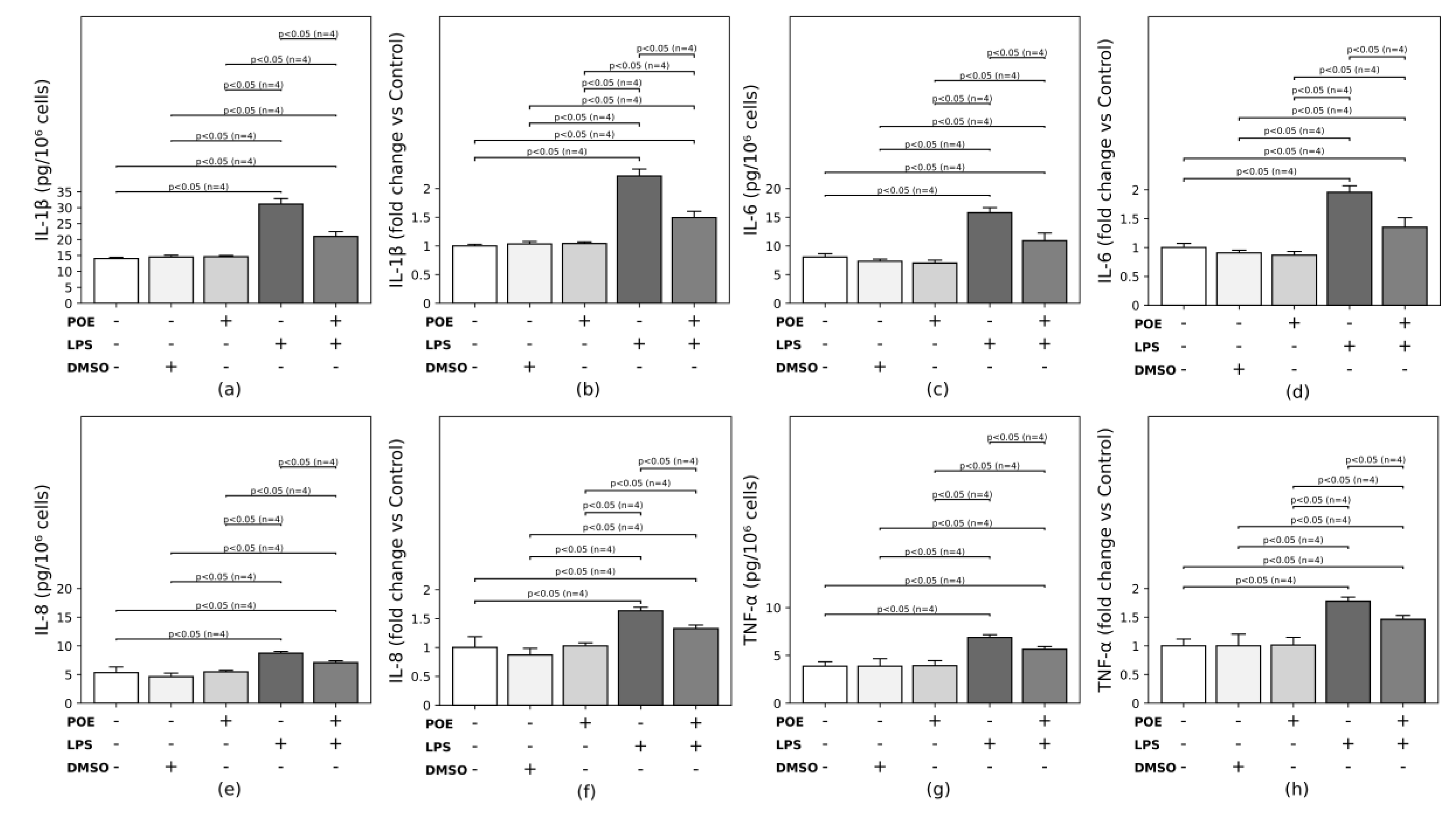
Figure 7.
HaCaT cells were treated for 12 h with one of the following conditions: LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle control), or a combination of LPS + POE. mRNA expression levels of IL-1β (a), IL-6 (b), IL-8 (c), TNF-α (d) and nitric oxide synthase 2 (NOS2); (e) were quantified by qRT-PCR and expressed as fold change relative to untreated control cells using the 2^–ΔΔCt method. Data are presented as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; the absence of p-values denotes non-significant comparisons.
Figure 7.
HaCaT cells were treated for 12 h with one of the following conditions: LPS (2.5 µg/mL), POE (6.5 µg/mL polyphenol equivalents), DMSO (14 mM; vehicle control), or a combination of LPS + POE. mRNA expression levels of IL-1β (a), IL-6 (b), IL-8 (c), TNF-α (d) and nitric oxide synthase 2 (NOS2); (e) were quantified by qRT-PCR and expressed as fold change relative to untreated control cells using the 2^–ΔΔCt method. Data are presented as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Differences were considered statistically significant at p < 0.05. Only statistically significant differences are indicated; the absence of p-values denotes non-significant comparisons.
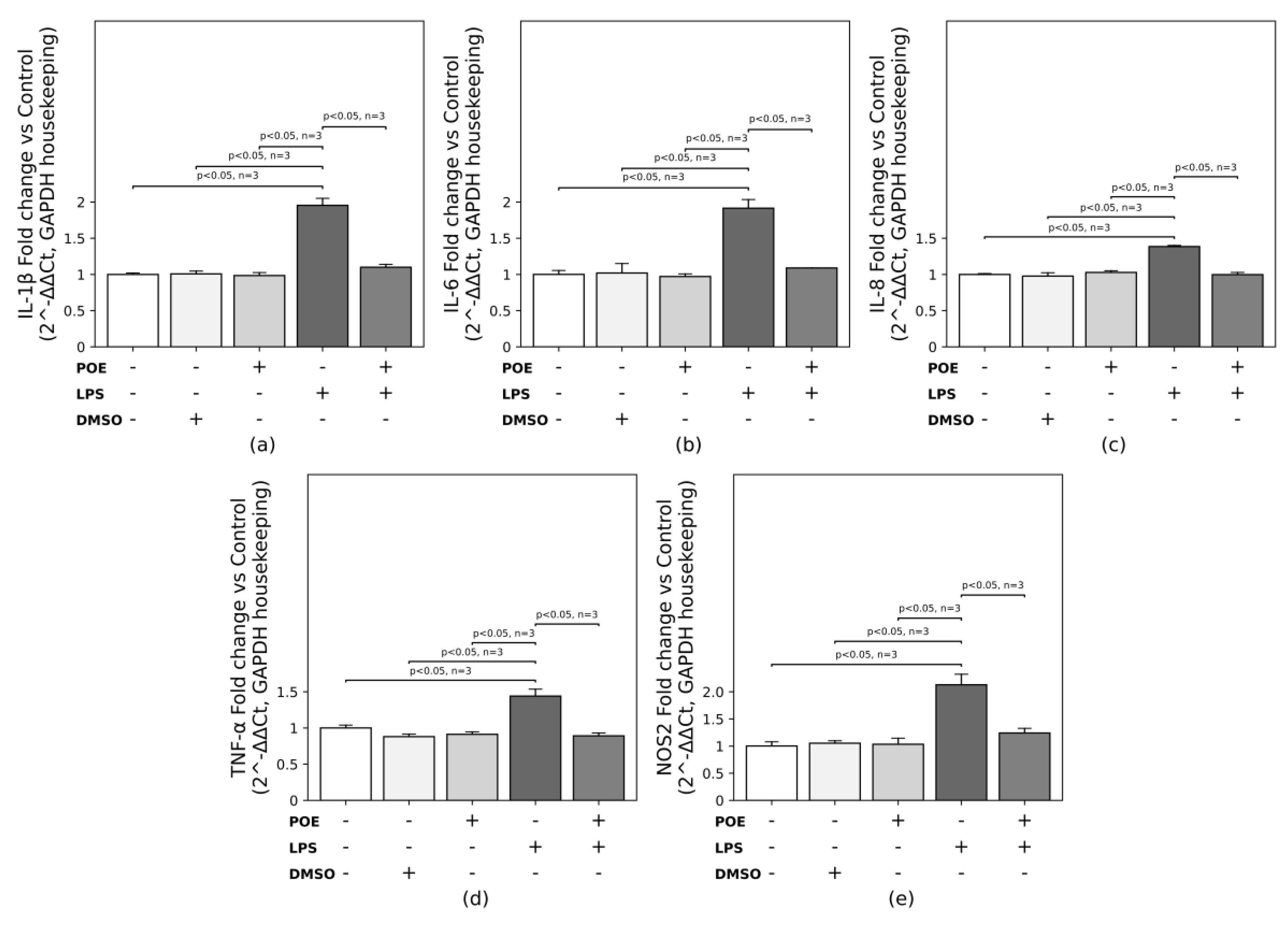
Figure 8.
Polyphenolic composition of Posidonia oceanica leaf extract. POE, analyzed by UPLC, contains a well-defined phenolic fraction representing approximately 88% of the extract. (+)-Catechin is the predominant component (84.76%), while gallic acid, chlorogenic acid, (−)-epicatechin, and ferulic acid are present in smaller amounts. Undefined polyphenols account for 11.11%. Percentages indicate the proportion of each compound within the extract.
Figure 8.
Polyphenolic composition of Posidonia oceanica leaf extract. POE, analyzed by UPLC, contains a well-defined phenolic fraction representing approximately 88% of the extract. (+)-Catechin is the predominant component (84.76%), while gallic acid, chlorogenic acid, (−)-epicatechin, and ferulic acid are present in smaller amounts. Undefined polyphenols account for 11.11%. Percentages indicate the proportion of each compound within the extract.
Table 1.
Primer Sequences and Target Genes for qRT-PCR.
| Gene product | Direction | Sequence (5’→3’) | Product size | Cat. No. |
| IL-1β | F | CCACAGACCTTCCAGGAGAATG | 131 bp | HP200544 |
| R | GTGCAGTTCAGTGATCGTACAGG | |||
| IL-6 | F | AGACAGCCACTCACCTCTTCAG | 132 bp | HP200567 |
| R | TTCTGCCAGTGCCTCTTTGCTG | |||
| IL-8 | F | GAGAGTGATTGAGAGTGGACCAC | 112 bp | HP200551 |
| R | CACAACCCTCTGCACCCAGTTT | |||
| TNF-α | F | CTCTTCTGCCTGCTGCACTTTG | 135 bp | HP200561 |
| R | ATGGGCTACAGGCTTGTCACTC | |||
| NOS2 | F | GCTCTACACCTCCAATGTGACC | 136 bp | HP200591 |
| R | CTGCCGAGATTTGAGCCTCATG | |||
| GAPDH | F | GTCTCCTCTGACTTCAACAGCG | 131 bp | HP205798 |
| R | ACCACCCTGTTGCTGTAGCCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



