Submitted:
06 February 2026
Posted:
09 February 2026
Read the latest preprint version here
Abstract
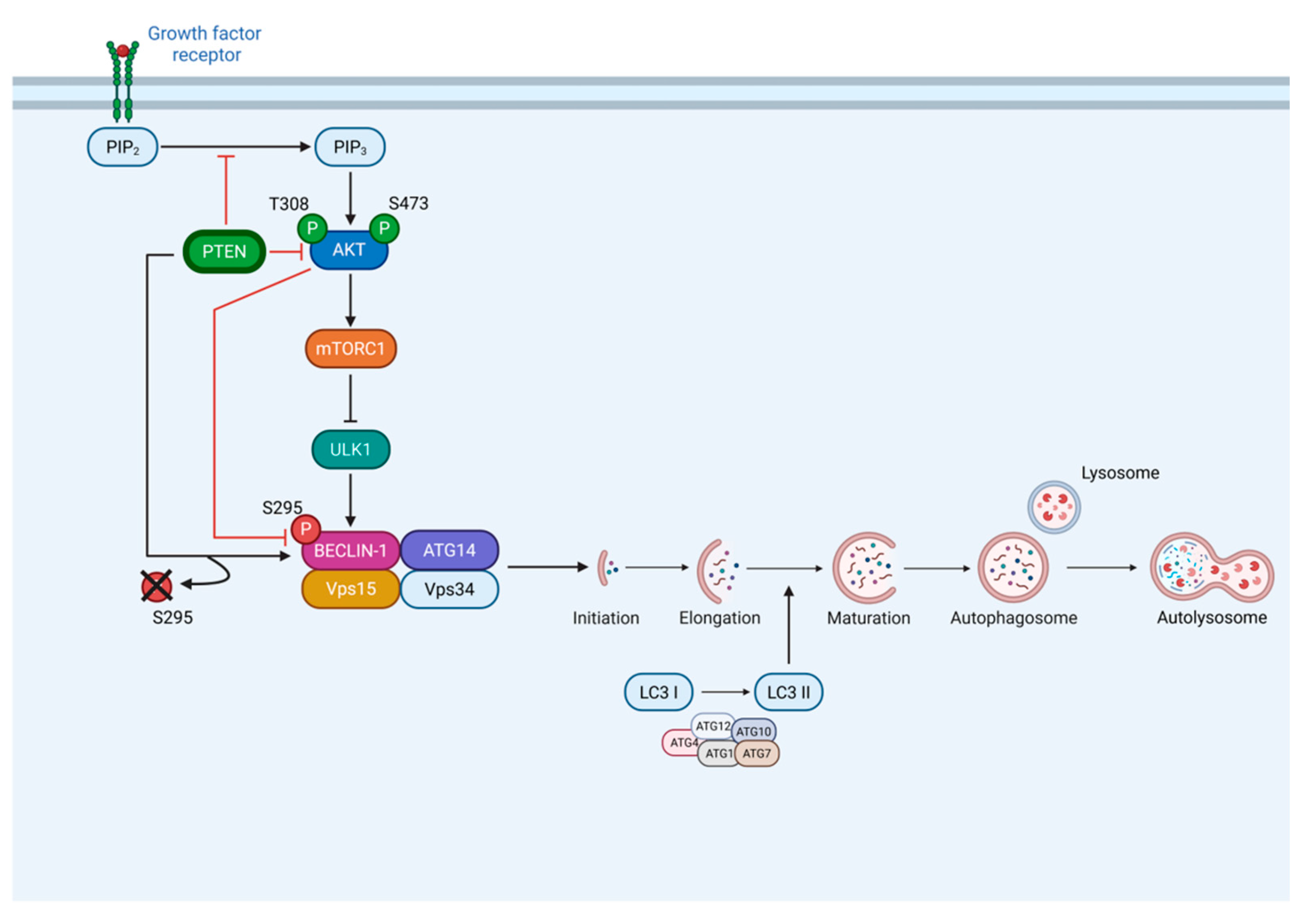
PTEN is a well-established tumor suppressor that plays a central role in the regulation of cell growth, metabolism, and survival. As a protein-lipid dual phosphatase, PTEN negatively regulates the PI3K/AKT signaling pathway, which in turn modulates autophagy, a conserved catabolic process that allows cells to degrade and recycle intracellular components, through the downstream effector mTORC1. While this represents the canonical mechanism by which PTEN influences autophagy, here we show that PTEN also regulates autophagy through an alternative, AKT-independent pathway. Specifically, through genetic manipulations of PTEN expression in thyroid cancer cells, we identify BECLIN-1 as a direct target of PTEN protein phosphatase activity. PTEN physically associates with BECLIN-1 under both basal and nutrient-deprived conditions, promoting its dephosphorylation at serine 295, thus relieving AKT inhibition resulting in autophagy activation. This regulatory event correlates with increased autophagic flux under starvation, as reflected by enhanced LC3 I to LC3 II conversion. Importantly, BECLIN-1 dephosphorylation is mediated by PTEN protein phosphatase activity and does not require its lipid phosphatase function. Furthermore, bioinformatic analyses reveal that high PTEN expression, together with enhanced autophagic activity (MAP1LC3B), is associated with improved clinical outcome in cancer patients. These findings uncover a direct, AKT-independent mechanism by which PTEN controls autophagy by modulating BECLIN-1 phosphorylation status. Together, our results provide novel insight into how PTEN coordinates cellular adaptation to metabolic stress and highlight an additional pathway through which PTEN regulates the autophagic machinery in cancer cells.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Transient Transfection
2.3. Antibodies
2.4. Western Blotting Analysis
2.5. Immunofluorescence
2.6. Co-Immunoprecipitation
2.7. Statistical Analysis
2.8. Protein-Protein Interaction Prediction
2.9. Bioinformatic Analysis
3. Results
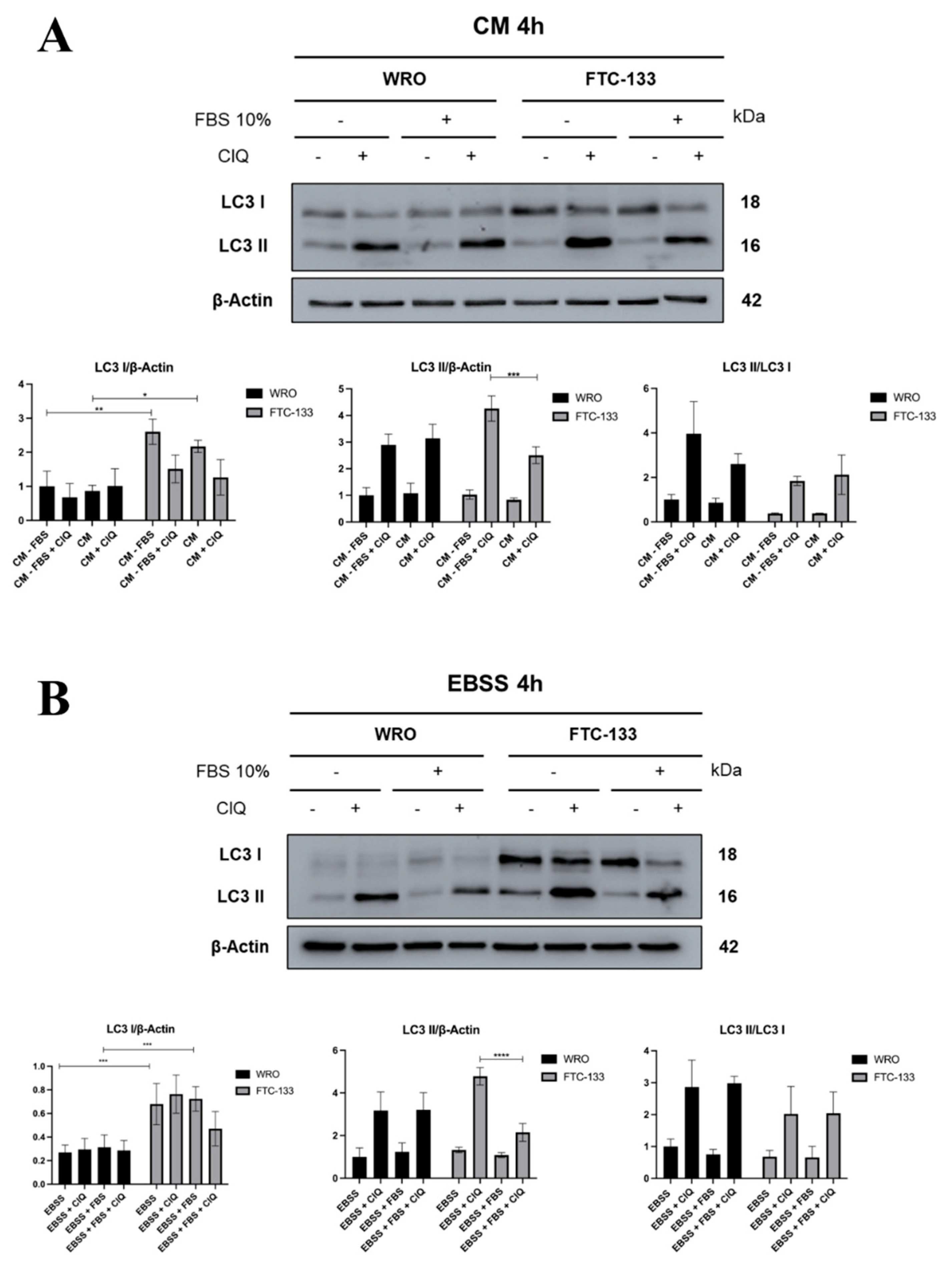
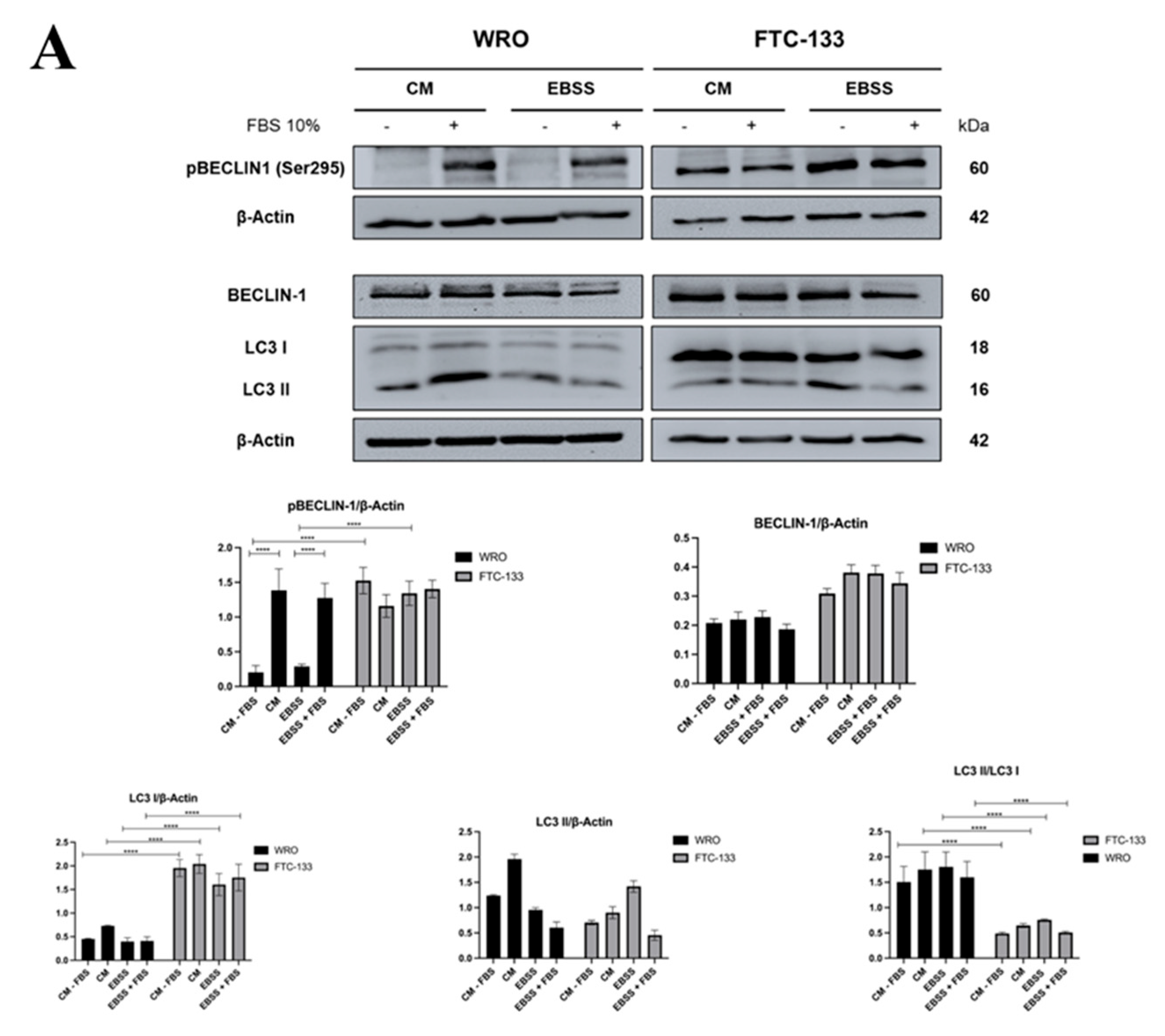
3.1. Serum inhibition of Autophagosome Formation is more Effective in PTEN-deficient than in PTEN-proficient Thyroid Cancer Cells
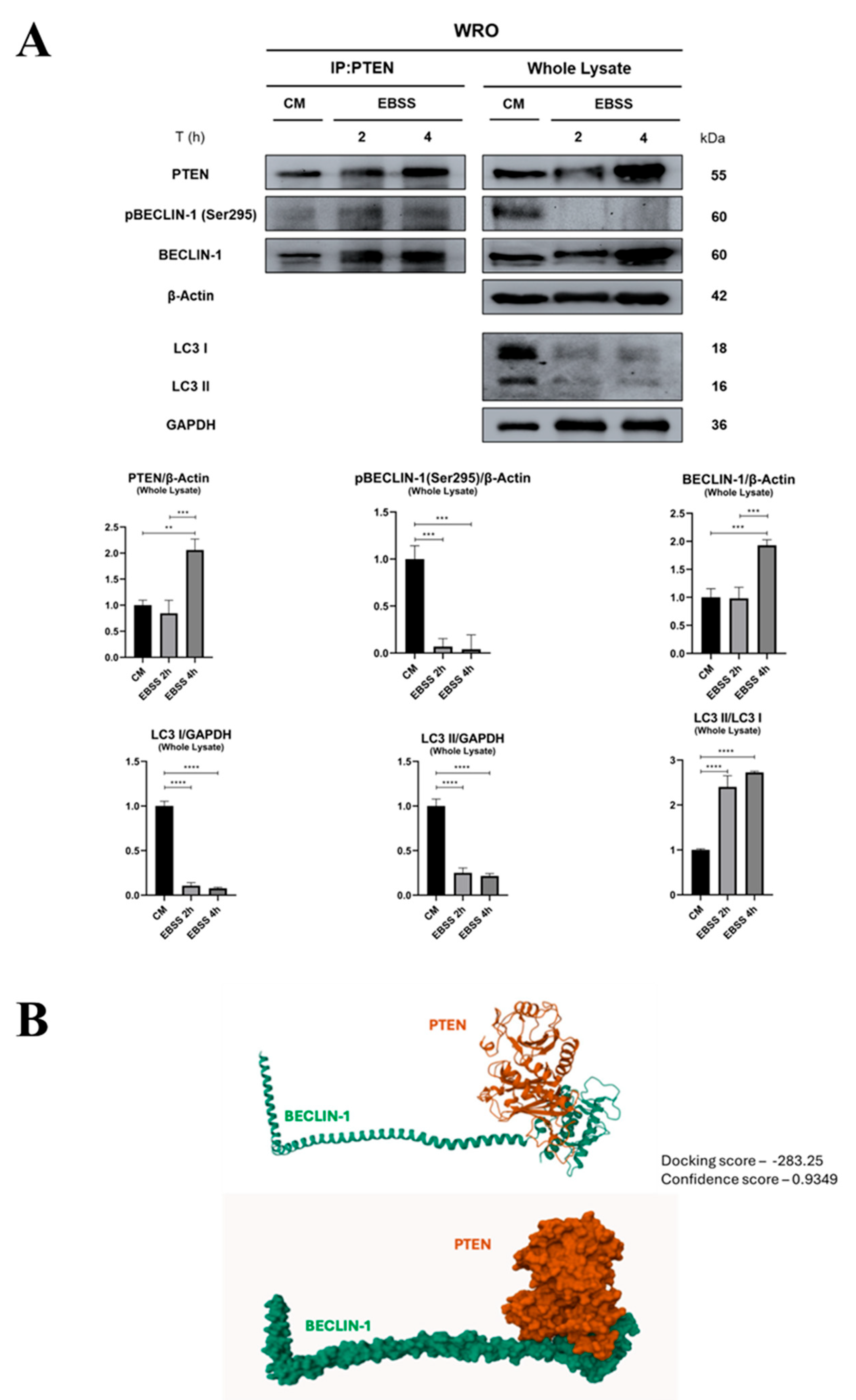
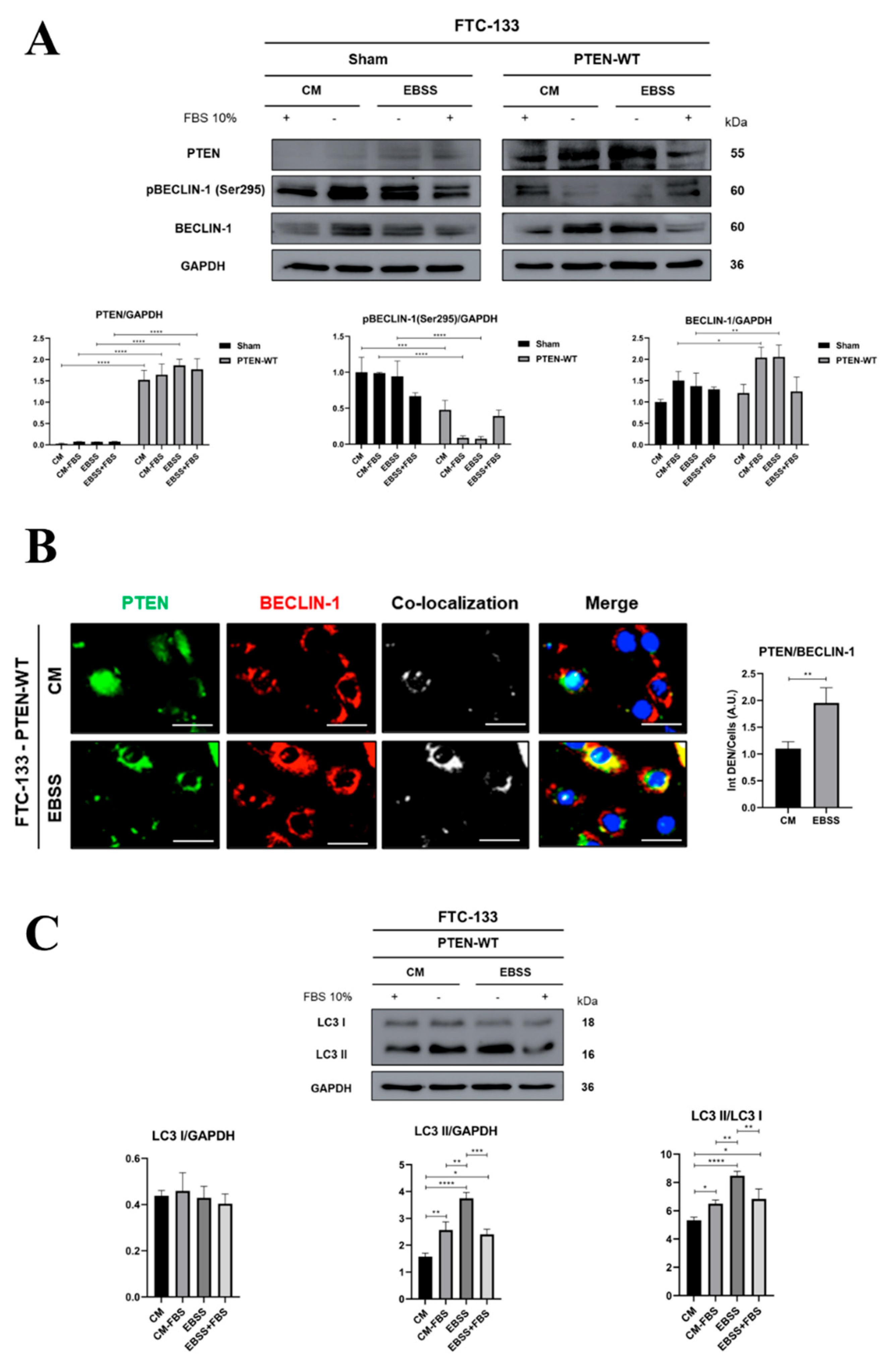
3.2. PTEN Interacts with BECLIN-1 and Promotes Ser295 Dephosphorylation
3.3. PTEN Protein Phosphatase Domain Is Mandatory for BECLIN-1 Dephosphorylation and Formation of the Autophagy Interactome
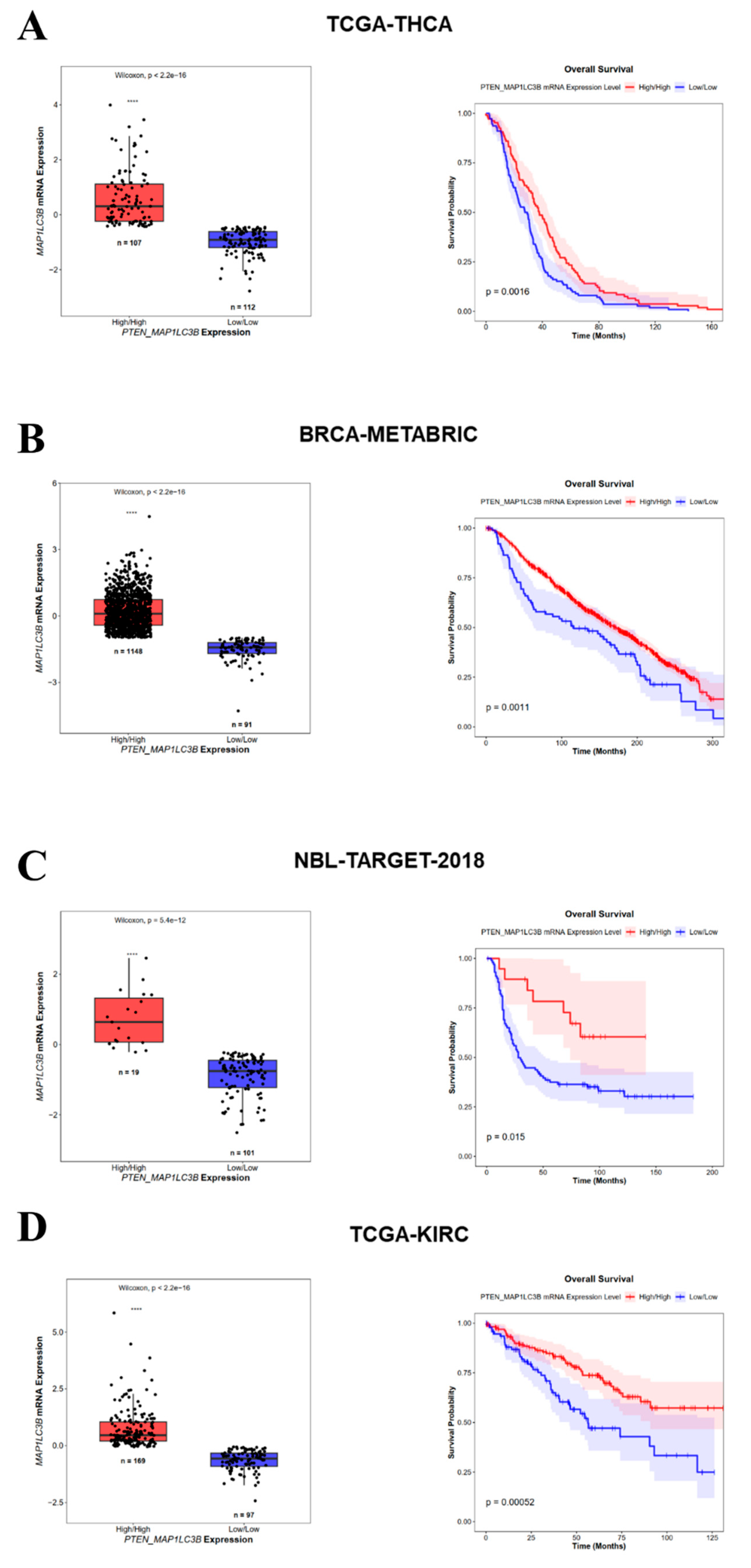
3.4. High PTEN and MAP1LC3B Expression Correlates with a Better Prognosis in Cancer Patients
4. Discussion
5. Conclusions
Abbreviation
| CM | Control Media |
| ClQ | Chloroquine |
| EBSS | Earle’s Balanced Salt Solution |
| FBS | Fetal Bovine Serum |
| Ser295 | Serine 295 |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niu, X; You, Q; Hou, K; Tian, Y; Wei, P; Zhu, Y; Gao, B; Ashrafizadeh, M; Aref, AR; Kalbasi, A; Cañadas, I; Sethi, G; Tergaonkar, V; Wang, L; Lin, Y; Kang, D; Klionsky, DJ. Autophagy in cancer development, immune evasion, and drug resistance. Drug Resist Updat. 2025, 78, 101170. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018, 20(5), 521–527. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Virgilio, L; Silva-Lucero, MD; Flores-Morelos, DS; Gallardo-Nieto, J; Lopez-Toledo, G; Abarca-Fernandez, AM; Zacapala-Gómez, AE; Luna-Muñoz, J; Montiel-Sosa, F; Soto-Rojas, LO; Pacheco-Herrero, M; Cardenas-Aguayo, MD. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11(15), 2262. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kang, R; Zeh, HJ; Lotze, MT; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011, 18(4), 571–80. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tran, S; Fairlie, WD; Lee, EF. BECLIN1: Protein Structure, Function and Regulation. Cells 2021, 10(6), 1522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Itakura, E; Kishi, C; Inoue, K; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008, 19(12), 5360–72. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maheshwari, C; Castiglioni, A; Walusimbi, U; Vidoni, C; Ferraresi, A; Dhanasekaran, DN; Isidoro, C. The Biological Role and Clinical Significance of BECLIN-1 in Cancer. Int J Mol Sci 2025, 26(19), 9380. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salwa, A; Ferraresi, A; Secomandi, E; Vallino, L; Moia, R; Patriarca, A; Garavaglia, B; Gaidano, G; Isidoro, C. High BECN1 Expression Negatively Correlates with BCL2 Expression and Predicts Better Prognosis in Diffuse Large B-Cell Lymphoma: Role of Autophagy. Cells 2023, 12(15), 1924. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, T; Li, D; He, Z; Feng, S; Zhao, S. Prognostic and clinicopathological significance of Beclin-1 in non-small-cell lung cancer: a meta-analysis. Onco Targets Ther 2018, 11, 4167–4175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, HC; Zhao, S; Xue, H; Zhao, EH; Jiang, HM; Hao, CL. The Roles of Beclin 1 Expression in Gastric Cancer: A Marker for Carcinogenesis, Aggressive Behaviors and Favorable Prognosis, and a Target of Gene Therapy. Front Oncol. 2020, 10, 613679. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, Z; Han, X; Ou, D; Liu, T; Li, Z; Jiang, G; Liu, J; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol 2020, 104(2), 575–587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, HP; Jiang, RY; Zhu, JY; Sun, KN; Huang, Y; Zhou, HH; Zheng, YB; Wang, XJ. PI3K/AKT/mTOR signaling pathway: an important driver and therapeutic target in triple-negative breast cancer. Breast Cancer 2024, 31(4), 539–551. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miricescu, D; Totan, A; Stanescu-Spinu, II; Badoiu, SC; Stefani, C; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int J Mol Sci. 2020, 22(1), 173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pareek, G; Kundu, M. Physiological functions of ULK1/2. J Mol Biol. 2024, 436(15), 168472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Melick, CH; Jewell, JL. Regulation of mTORC1 by Upstream Stimuli. Genes (Basel) 2020, 11(9), 989. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, RC; Wei, Y; An, Z; Zou, Z; Xiao, G; Bhagat, G; White, M; Reichelt, J; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338(6109), 956–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Papa, A; Pandolfi, PP. The PTEN⁻PI3K Axis in Cancer. Biomolecules 2019, 9(4), 153. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Phadngam, S; Castiglioni, A; Ferraresi, A; Morani, F; Follo, C; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7(51), 84999–85020. [Google Scholar] [CrossRef]
- Morani, F; Phadngam, S; Follo, C; Titone, R; Aimaretti, G; Galetto, A; Alabiso, O; Isidoro, C. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol. 2014, 53(2), 247–58. [Google Scholar] [CrossRef] [PubMed]
- Alobid, S. Targeting the PI3K/AKT/mTOR signaling pathway in prostate cancer: Molecular dysregulation, therapeutic advances, and future directions. Saudi Pharm J 2026, 34(1), 2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, Y; Wang, J; Chandarlapaty, S; Cross, J; Thompson, C; Rosen, N; Jiang, X. PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol. 2014, 21(6), 522–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Feng, J; Liang, J; Li, J; Li, Y; Liang, H; Zhao, X; McNutt, MA; Yin, Y. PTEN Controls the DNA Replication Process through MCM2 in Response to Replicative Stress. Cell Rep. 2015, 13(7), 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y; Tao, H; He, J; Huang, SY. The HDOCK server for integrated protein-protein docking. Nat Protoc 2020, 15(5), 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Morani, F; Phadngam, S; Follo, C; Titone, R; Thongrakard, V; Galetto, A; Alabiso, O; Isidoro, C. PTEN deficiency and mutant p53 confer glucose-addiction to thyroid cancer cells: impact of glucose depletion on cell proliferation, cell survival, autophagy and cell migration. Genes Cancer 2014, 5(7-8), 226–39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weng, LP; Gimm, O; Kum, JB; Smith, WM; Zhou, XP; Wynford-Thomas, D; Leone, G; Eng, C. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum Mol Genet. 2001, 10(3), 251–8. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C; Ferraresi, A; Seca, C; Secomandi, E; Isidoro, C. Methods for Monitoring Macroautophagy in Pancreatic Cancer Cells. Methods Mol Biol. 2019, 1882, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Levy, JMM; Towers, CG; Thorburn, A. Targeting autophagy in cancer. Nat Rev Cancer 2017, 17(9), 528–542. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Russell, RC; Yuan, HX; Guan, KL. Autophagy regulation by nutrient signaling. Cell Res 2014, 24(1), 42–57. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, M; Zhang, K; Zhang, Z; Zeng, X; Huang, Z; Qin, P; Xie, Z; Cai, X; Ashrafizadeh, M; Tian, Y; Wei, R. PI3K/AKT/mTOR Axis in Cancer: From Pathogenesis to Treatment. MedComm (2020) 2025, 6(8), e70295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- He, Y; Sun, MM; Zhang, GG; Yang, J; Chen, KS; Xu, WW; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther 2021, 6(1), 425. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, R; Chen, Y; Liu, G; Li, C; Song, Y; Cao, Z; Li, W; Hu, J; Lu, C; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11(9), 797. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vega-Rubin-de-Celis, S; Peña-Llopis, S; Konda, M; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13(3), 464–472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, J; Kundu, M; Viollet, B; Guan, KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011, 13(2), 132–41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lyu, J; Yu, X; He, L; Cheng, T; Zhou, J; Cheng, C; Chen, Z; Cheng, G; Qiu, Z; Zhou, W. The protein phosphatase activity of PTEN is essential for regulating neural stem cell differentiation. Mol Brain 2015, 8, 26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, A; Zhu, Y; Chen, W; Merlino, G; Yu, Y. PTEN Dual Lipid- and Protein-Phosphatase Function in Tumor Progression. Cancers (Basel) 2022, 14(15), 3666. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, X; Bok, I; Jasani, N; Wang, K; Chadourne, M; Mecozzi, N; Deng, O; Welsh, EA; Kinose, F; Rix, U; Karreth, FA. PTEN Lipid Phosphatase Activity Suppresses Melanoma Formation by Opposing an AKT/mTOR/FRA1 Signaling Axis. Cancer Res. 2024, 84(3), 388–404. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xiao, J; Hu, CP; He, BX; Chen, X; Lu, XX; Xie, MX; Li, W; He, SY; You, SJ; Chen, Q. PTEN expression is a prognostic marker for patients with non-small cell lung cancer: a systematic review and meta-analysis of the literature. Oncotarget 2016, 7(36), 57832–57840. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qiu, ZX; Zhao, S; Li, L; Li, WM. Loss of Expression of PTEN is Associated with Worse Prognosis in Patients with Cancer. Asian Pac J Cancer Prev. 2015, 16(11), 4691–8. [Google Scholar] [CrossRef] [PubMed]
- da Costa, AA; D’Almeida Costa, F; Ribeiro, AR; Guimarães, AP; Chinen, LT; Lopes, CA; de Lima, VC. Low PTEN expression is associated with worse overall survival in head and neck squamous cell carcinoma patients treated with chemotherapy and cetuximab. Int J Clin Oncol 2015, 20(2), 282–9. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



