Submitted:
05 February 2026
Posted:
06 February 2026
You are already at the latest version
Abstract
The longstanding dream of resurrecting dinosaurs faces formidable obstacles. DNA decays rapidly, making direct recovery impossible. Even if successful in their aims, and modern “synthetic” methods achieve a complete and functioning non-avian genome, they cannot recreate specific dinosaur species because at least some of the crucial genetic information is completely absent from living relatives. Alternatively, it is suggested here that microbial vectors once mediated horizontal gene transfer (HGT) between dinosaurs and the ancestors of extant species. While HGT appears rare, the ecological continuity between dinosaurs, microbes, and coexisting plants offers a faint but plausible route for fragments of dinosaur DNA to survived in extant species. Building on advances in paleogenomics, synthetic biology, and comparative genomics, we outline a multi-step strategy to search for, validate, and functionally test dinosaurian “genetic shrapnel” preserved in the “dark genome” of candidate organisms – particularly that of gymnosperms. If such material could be recovered, it might provide missing pieces needed to reconstruct extinct dinosaur species. Even if the goal of dinosaur de-extinction remains forever beyond reach, pursuing this line of research could yield transformative insights in evolutionary biology, synthetic biology, and conservation science — and offer a compelling narrative to engage the public.
Keywords:
horizontal gene transfer
; gymnosperm
; genomics
; synthetic biology
; transposable elements
Introduction
The prospect of dinosaur de-extinction has long fascinated scientists and the public alike—a fascination famously amplified by Michael Crichton’s Jurassic Park. Central to that story is the so-called “Mosquito Model”: the idea that dinosaur DNA might be recovered from the blood of ancient mosquitoes preserved in fossilized amber. Although conceptually compelling at the time, this approach is now widely regarded as scientifically implausible (Penney et al., 2013; Peris et al., 2020). DNA degrades far too rapidly due to chemical processes such as oxidation and has a half-life of just over 500 years (Allentoft et al., 2012). Moreover, fossilized amber is not airtight, which means there is, in practice, much opportunity for environmental factors to accelerate this degradation (Penney et al., 2013). Despite early enthusiasm in the 1990s and occasional reports of ancient DNA recovery from amber, later attempts failed to replicate those results (Peris et al., 2020; Penney et al., 2013). As such, the Mosquito Model has not brought us any closer to resurrecting dinosaurs in the decades since Jurassic Park first popularized the idea. Today, it survives more as a kind of scientific “fossil”—preserved not in amber, but in the collective imagination.
Two alternative approaches to the dream of dinosaur de-extinction have emerged with more scientifically credible foundations. One is the so-called “Dino-Chicken” project, which builds on the evolutionary continuity between non-avian theropod dinosaurs and modern birds. Birds are not merely descended from dinosaurs—they are dinosaurs. In fact, the genome of a modern chicken is likely closer to that of Tyrannosaurus rex than T. rex’s genome was to that of many other well-known but phylogenetically more distantly related dinosaurs, such as Allosaurus, Brontosaurus, or Stegosaurus. The Dino-Chicken approach rests on the idea that modifying gene expression in modern birds could reawaken dormant developmental pathways, producing phenotypes that resemble their non-avian ancestors (Horner & Gorman, 2009). Evolution often proceeds not by erasing old genetic instructions, but by overwriting or layering new adaptations over older ones—more like a palimpsest than a clean slate (Dawkins, 2024). This helps explain the appearance of atavisms: ancestral traits that occasionally re-emerge, such as chickens developing teeth or horses growing extra toes (Carroll, 2005; Gould, 1983). These phenomena suggest that the genetic instructions for long-lost traits may still be present, albeit silenced. On this view, the chicken genome is akin to a modern software program—for example, Microsoft Word Version 7—that retains legacy code from earlier versions, like Microsoft Word Version 3 (Dennett, 2017). By reactivating these latent instructions, researchers might engineer a bird with a long tail, toothed snout, and clawed forelimbs—features more typical of non-avian theropods than modern birds (Horner, 2009). While not identical to any specific dinosaur that has existed before, such a creature could be morphologically and functionally reminiscent of one. Evidence from experimental developmental biology supports this view, suggesting that bird genome retains many features inherited from its non-avian ancestors (Bhullar et al., 2015).
The second major approach to reconstructing dinosaur genomes draws on comparative genomics, which seeks to infer hypothetical ancestral genomes by analyzing the DNA of living relatives. This strategy—hereinafter referred to as the “synthetic approach”—focuses on sequencing the genomes of modern archosaurs, namely birds and crocodilians, and applying phylogenetic algorithms to reconstruct ancestral sequences (Griffin, Hassan, & Trifonov, 2019; O’Connor et al., 2018). These computational tools analyze genetic similarities and differences across species to estimate the genomic composition of their common ancestors. Such algorithms can identify conserved regions, estimate mutation rates, and model how genes diverged over time. The resulting phylogenetic trees not only map evolutionary relationships but also serve as scaffolds for assembling synthetic reconstructions of ancestral genomes (O’Connor et al., 2018). By comparing the genomes of birds and crocodilians, researchers can retroactively approximate segments of extinct dinosaur genomes (Griffin, Hassan, & Trifonov, 2019; Royal Veterinary College, 2024).
Additionally, the synthetic approach may be supplemented by insights from developmental biology and palaeontology. Fossil evidence can offer detailed anatomical and physiological information—such as tail length, limb proportions, or skull shape—that provide constraints on what genes must have been doing, even if their exact sequences are lost. In principle, if researchers develop a deep enough understanding of how genes regulate specific traits, it may be possible to functionally substitute missing genomic segments with sequences that encode the same developmental outcome, even if the original genetic "syntax" remains unknown. For instance, if fossil data suggest that Tyrannosaurus had a tail of a particular length, and if the genetic control of tail development is well understood, then new instructions could potentially be inserted into a synthetic genome to recapitulate this trait.
The three approaches discussed summarized in Figure 1 below. Despite their scientific merit, both the Dino-Chicken and synthetic approaches face inherent limitations that transcend current technological constraints. Even in the best-case scenario, the Dino-Chicken project could only reconstruct dinosaurs that are direct ancestors of modern birds. Crucial genetic components may be irretrievable from bird genomes due to mutations, deletions, or replacement by genes specific to avian adaptations. Similarly, while the synthetic approach may allow researchers to approximate a generalized non-avian dinosaur genome, it cannot recreate any specific species. Genes unique to Triceratops, Ankylosaurus, Deinonychus, and other extinct taxa would remain inaccessible—unless they were independently recovered from another source.
These limitations do not diminish the scientific value of either approach in advancing our understanding of developmental biology and evolutionary transitions. However, they mean these avenues offer little comfort to those hoping for true dinosaur de-extinction — or even a more plausible premise for dinosaur-themed films. The aim of this article is to explore a third, speculative possibility that may offer a new frontier. Namely, it is conceivable that fragments of dinosaur DNA endure within the cells of modern-day plants and fungi. If so, these overlooked reservoirs might provide a path toward recovering ancient genetic material.
The Genetic Shrapnel Approach
Consider an analogy from forensic ballistics—the science of reconstructing crime scenes from projectile fragments and impact patterns. Forensic experts infer a projectile’s type, trajectory, weapon of origin, firing distance, and number of shooters by analysing even the smallest surviving traces. In a similar way, dinosaurs—the dominant terrestrial vertebrates for nearly 180 million years—have left behind an abundance of indirect evidence. Although fossilization requires exceptional conditions, the sheer timespan and ecological dominance of dinosaurs have produced a diverse trace record: bones, footprints, coprolites, gastroliths, burrows, nests, eggshells, and feeding marks on other organisms. Palaeontologists, like forensic investigators, use these clues to reconstruct the morphology and behaviour of extinct species. If we set aside the fading hope of intact dinosaur DNA preserved in amber—and exclude birds as their direct descendants—we are left with a provocative question: could remnants of dinosaur DNA persist elsewhere in the biosphere? Like shrapnel embedded in unsuspected places, might genetic fragments survive today in unexpected forms?
One possibility is that fragments of dinosaur DNA were copied into the genomes of other organisms during the Mesozoic through horizontal gene transfer (HGT). HGT refers to the movement of genetic material between organisms by means other than parent-to-offspring inheritance. It often occurs via transposable elements (TEs), or “jumping genes”—mobile DNA sequences that can replicate and insert themselves throughout a genome and occasionally leap across species boundaries (Dunning Hotopp, 2011; Feschotte & Pritham, 2007; Richardson et al., 2015). In humans, for example, TEs make up roughly 50% of the genome, most of which are non-coding, but some are thought to have originated from ancient cross-species transfers embedded in what is sometimes called the “dark genome” (Dunning Hotopp, 2011; Richardson et al., 2015). We propose that similar mechanisms may have ferried dinosaur DNA into the genomes of other organisms – where it could persist in their descendants today, buried like genetic shrapnel in non-coding regions of extant taxa. This bypasses the problem of dinosaur DNA degrading, because the organisms that contain this genetic shrapnel have been actively duplicating it along with the rest of the genetic information that it has been embedded among.
It must be acknowledged that HGT from animals into other organisms is considered rare, and it appears that only a small fraction of well-characterized modern genomes to contain genes known to have arrived via this route (Crisp et al., 2015). However, a more optimistic perspective emerges when one considers the ecological context, evolutionary timescales, and the role of microbial intermediaries, to suggest that HGT from dinosaurs might have occurred with sufficient frequency to preserve meaningful genetic fragments—potentially enough to contribute to a reconstructed dinosaurian genome. The same line of reasoning also points to which types of organisms are most promising for detecting such ancient transfers.
HGT involving large animals is best documented in association with microorganisms, particularly bacteria and viruses. This is due to several biological features of microbes: they replicate rapidly, possess frequent genetic exchange mechanisms, and can integrate foreign DNA into their genomes. For instance, viruses can incorporate host genetic material during infection and later transfer it to other organisms, while bacteria can acquire DNA through transformation, transduction, or conjugation (Dunning Hotopp, 2011). Indeed, microbial genomes have been found to contain fragments of animal DNA, often embedded within transposable elements or associated with viral integration events (Boto, 2014). These findings raise the intriguing possibility that modern-day bacteria might harbor remnants of dinosaur DNA—molecular shrapnel preserved from ancient interactions. However, while microbes may have played a key role in facilitating gene transfer, they are unlikely themselves to serve as reliable long-term reservoirs of paleogenetic material. Bacterial genomes are small and highly dynamic, evolving quickly and subject to strong selection against non-functional DNA. As a result, any dinosaur-derived sequences that were once present are likely to have been degraded, lost, or mutated beyond recognition over the intervening 65 million years. Thus, while microbial vectors may have mediated ancient genetic transfers, they are unlikely to yield recoverable sequences suitable for genome reconstruction.
In contrast to bacteria and viruses, fungi may offer a more promising vehicle for preserving dinosaur DNA beyond the Cretaceous–Tertiary (K–T) extinction. Like bacteria, fungi engage in horizontal gene transfer (Richards et al., 2011) but possess far larger genomes that evolve more slowly (Narango-Ortiz & Gabaldón, 2019), increasing the likelihood of long-term DNA retention. There is some evidence of genetic material in fungi of animal origin (Alexander et al., 2016). Several fungal lineages that exist today were widespread during the age of dinosaurs, making ancient host–parasite interactions plausible (Redecker, Kodner, & Graham, 2000). Additionally, it seems reasonable to suppose that fungi’s capacity to infiltrate diverse host tissues could have facilitated acquisition of a broader array of host genes than would be likely of other parasites. Fungal genomes remain comparatively underexplored relative to those of bacteria, animals, and plants, and perhaps targeted genomic mining of fungi could reveal preserved dinosaur DNA fragments, although these sequences are likely to be fragmented and heavily modified by random mutational drift. We therefore advocate for targeted efforts to identify fungal lineages that likely interacted with dinosaurs and to survey their genomes for preserved genetic fragments. All that said, most fungi remain constrained by relatively modest genome sizes, meaning any retained dinosaur DNA is likely to be highly fragmented or heavily modified due to accumulated mutations. While fungi present a more favourable source than bacteria or viruses for discovering dinosaur genetic shrapnel, expectations for recovering complete or near-complete dinosaur genomes from fungal sources must remain tempered. Nonetheless, the potential insights gained from even partial sequences may warrant focused exploration.
Another candidate group are ticks because of their prolonged and intimate contact with host animals. Relative to microbial parasites, ticks contain a lot of genetic information, with most of this being a “dark genome”. The “dark genome” refers to the large portion of an organism’s genome that is not functionally active and tends to be where transposable elements reside (Feschotte & Pritham, 2007). There is reason to suspect that these regions, in ticks, contain genetic material that originated from host species, such as mammals and birds (Guilia-Nuss et al., 2016). At least six major extant genera of ticks share a common ancestor dating back to well before the extinction of dinosaurs (Klompen et al., 1996), meaning that potentially hundreds of species could trace their roots to lineages that independently engaged in HGT with dinosaurs. There is some direct evidence that ticks parasitized dinosaurs while not necessarily specializing on dinosaurs and so did not go extinct with them (Peñalver et al., 2017). A particularly interesting example of a tick that probably fed on dinosaur blood is Nuttallielliella namaqua. This is referred to as a “living fossil” because both molecular studies and morphological analysis of its fossilized ancestors suggest it has persisted virtually unchanged for hundreds of millions of years (Mans et al., 2011). These slow rates of evolutionary change mean that genes acquired through HGT from dinosaurs may still be present and hidden in their dark genome. In short, given ticks are notable because they have relatively large genomes for parasitic organisms – much of which is “dark” — and may be receptive to HGT from their host organisms, they could be especially worth searching to determine if they have been repositories for dinosaur genetic shrapnel.
The largest dark genomes of all are found in yet another major group of organisms with which dinosaurs had intimate and sustained interactions: plants—particularly gymnosperms such as conifers, cycads, and ginkgo. Unlike microorganisms, these plants possess vast, inherently slowly evolving genomes. A crucial reason their genomes are so large is because they have especially enormous dark genomes rich in transposable elements, many times larger than the largest of known terrestrial animal genomes (Leitch & Leitch, 2012). Gymnosperms dominated the ecosystems dinosaurs inhabited. Their ecological prominence alongside dinosaurs, combined with the presence of myriad ancient microbial symbionts they also surely interacted with, creates fertile ground for horizontal gene transfer bridging dinosaurs and plants via microbial intermediaries such as soil bacteria, gut microbes, and parasitic fungi. Moreover, hundreds of extant lineages of gymnosperms were already separated from common ancestors well before the extinction of dinosaurs (Biswas & Johri, 2013). This means that each of these lineages could be searched to collectively provide hundreds of samples potentially carrying dinosaur genomic fragments from ancient times to the present.
Given this ecological and genomic continuity, we propose a novel and potentially viable strategy for reconstructing dinosaur genomes, one that involves searching for genetic fragments — what we term “genetic shrapnel”—that may have been horizontally transferred into the germlines of organisms surviving the K-T extinction (the basic idea is illustrated in Figure 2). These fragments could have been transposed via microbial intermediaries such as viruses, bacteria, or fungi into the genomes of ancestral gymnosperms or other long-lived lineages. If present in sufficient quantity and quality, these genetic remnants could help fill gaps in dinosaur genomes, thereby complementing reconstructions based on comparative genomics of birds and crocodilians (i.e., what we summarized as the “synthetic approach” above).
We propose the following stepwise strategy to pursue this line of inquiry:
- Perform comprehensive genome sequencing of extant gymnosperm genera—particularly conifers—as well as relatively primitive (plesiomorphic) fungal and tick lineages.
- Screen the “dark genome” of these organisms—regions enriched with transposable elements and non-coding DNA—for sequences that appear to originate from non-plant sources.
- Cross-reference candidate sequences against known animal genomes and databases of horizontally transferred genes, focusing on segments plausibly derived from vertebrates.
- Apply molecular dating methods to estimate the timing of HGT events and evaluate whether these events plausibly occurred during the Mesozoic era.
- Use phylogenetic and comparative analyses to determine if these sequences correspond with known archosaur genomic features and to assess their likelihood of being dinosaurian in origin.
While speculative, this approach offers a novel and potentially transformative avenue for paleogenomic research. By leveraging the vast, slowly evolving genomes of gymnosperms and the ecological as well as microbial continuity spanning from the Mesozoic to the present, it explores the possibility that fragments of extinct dinosaur genomes may still be hiding in plain sight.
Finding a Dinosaur in a Haystack
Stephen Jay Gould’s essay “Dinosaur in a Haystack” exemplifies how scientific paradigms can shift through improved data sampling and theoretical reframing (Gould, 1995). Early palaeontologists interpreted the fossil record as showing a gradual dinosaur decline prior to the K–T extinction. However, the asteroid impact hypothesis prompted more systematic examination of fossil layers, revealing that dinosaurs remained diverse and abundant until the abrupt extinction event. This shift—from selective sampling to comprehensive scrutiny—offers a valuable methodological lesson for investigating rare biological phenomena. By analogy, HGT between animals and plants has long been considered vanishingly rare, based on limited data, theoretical barriers to cross-kingdom transfer, and perhaps underappreciation of the potential for HGT mediated by microbial intermediaries such as parasites (Bock, 2010; Crisp et al., 2015 Gilbert et al., 2010; Kambayashi et al., 2022). There is some clear evidence of HGT in the genomes of gymnosperms (Bergthorsson et al., 2004) although none, to our knowledge, thought to have originated in animals. However, a more systematic and targeted search may uncover unexpected genetic remnants of dinosaurs.
Another reason to suspect that HGT from dinosaurs to other organisms may have been more extensive than previously thought is that HGT events were likely more frequent during the Mesozoic than in subsequent eras. This period was generally characterized by warm, and humid environments with high levels of CO2, resulting in highly productive terrestrial ecosystems. Warmth and moisture increased microbial and parasite diversity and density, thereby enhancing opportunities for genetic exchange via HGT (González Villa & Vinas, 2019; Wani et al., 2022). Such high ecosystem productivity, combined with vast gymnosperm vegetation, supported dense and diverse microbial communities in soil and sediments — conditions known to foster horizontal gene transfer through physical proximity, biofilms, and abundant extracellular DNA. These relatively stable conditions were frequently punctuated by dramatic disturbances such as volcanic activity and continental drift, which would have disrupted habitats. Such challenges are also thought to elevate rates of HGT (González Villa & Vinas, 2019) and so may have been at levels during the Mesozoic that are beyond what prevail in more recent history.
Several considerations further justify optimism about this strategy’s prospects. First, detailed knowledge of modern taxa can help guide extensive searches through the vast “haystack” of plant and fungal genomes to identify likely dinosaur DNA. For example, regions adjacent to sequences that overlap with or are compatible with bird and synthetic archosaur genomes—but are conspicuously missing from these references—may represent uniquely dinosaurian genes. By “daisy-chaining” these overlapping fragments to progressively reconstruct a more complete core dinosaur genome, researchers could continually improve the sensitivity and accuracy of subsequent searches in a virtuous cycle.
Secondly, to further shrink the gap that remains to be filled by our genetic shrapnel approach, it should be noted that, although birds have evolved rapidly since their split with the ancestors of crocodilians, crocodilians have undergone relatively little genetic change (Green et al., 2014), helping to provide a potentially robust genomic template for archosaurs. Moreover, molecular evidence confirms Tyrannosaurus Rex and birds are more related to each other than birds are to any non-Avian animal today (Asara et al., 2007), and it is reasonable to suppose they would have shared approximately 80 per cent of the same genetic information needed to produce a viable organism1. So, if some of this difference can be made up for by the synthetic approach (i.e., using a generic archosaurian template, infer functionally equivalent genetic information for helping to control some phenotypic differences such as in growth rates or integumentary structures), the truly irreplaceable portion of the genome may be reduced further still.
Thirdly, if genetic information was transferred from dinosaurs to plants, this transfer may not represent random sampling. Genes located in genomic “hotspots” — regions that evolve rapidly and frequently underlie species-specific adaptations (Edsinger et al., 2024) —may be especially prone to be involved in HGT from host to microorganisms. This is because these genetic regions are more likely to be copied by microorganisms if those regions are especially highly expressed (Feschotte & Pritham, 2007) and structurally dynamic (Crisp et al. 2015). So, if these hotspots disproportionately contained traits that distinguished different dinosaur genera from one another and from their modern relatives, then the genetic shrapnel preserved and transferred according to our model may represent precisely those fragments most valuable for reconstructing distinctive dinosaur features.
Another consideration worth emphasizing is the vast biological throughput of dinosaurs. Well-known species like Tyrannosaurus rex existed for millions of years and were abundant across wide geographic ranges. It is estimated that approximately 2.5 billion adult T. rex individuals have ever lived (Marshall, Ward, & Smith, 2021) and would have widely roamed environments dominated by conifers, cycads, and fungi. Thus, even if the per-event probability of HGT into gymnosperm or fungal genomes was minuscule, this immense biological volume could have yielded meaningful quantities of genetic fragments. Moreover, this estimate likely underrepresents the potential for genetic recovery. Dinosaurs form a clade, meaning genomic information recovered from one lineage can inform missing segments in related lineages through comparative genomics (O’Connor et al., 2018). For example, while T. rex existed for roughly 2 million years, the broader Tyrannosauridae family persisted for around 7 million years (Brusatte & Carr, 2016). Genetic data missing from T. rex may be partially recoverable from other tyrannosaurids and theropods, with overlap proportional to their relatedness. Similarly, Brontosaurus lived for perhaps 2 million years, but the broader group of longed necked dinosaurs they were an example of (sauropods) thrived for roughly 70 million years and represent a sister clade to the theropod clade that gave rise to other groups like Tyrannosauridae and birds. Thus, although, say, Brontosaurus material would be less relevant to T. rex, it would still probably contribute information that would otherwise be missing and go some way in contributing to the reconstruction of sauropod genomes, and so forth. In other words, overlapping sequences among more abundant and longer-lasting relatives reduce the burden of recovering unique DNA for any particular dinosaur species that was around for less long, and so had less opportunity to leave behind much genetic shrapnel.
In short, genetic material from widespread and long-lived dinosaur clades likely contains overlapping sequences useful for reconstructing rarer varieties, thereby improving the prospects for comprehensive genome recovery. Far from replacing the Dino-Chicken and synthetic approaches, the genetic shrapnel strategy is predicated on their continued advancement. These existing approaches help minimize the genetic information that must be recovered as shrapnel and guide searches to be more targeted and effective. A maximally developed, “core archosaurian genome,” derived from extant birds and crocodilians, is fundamental.
To move from theoretical plausibility to any chance of real-life progress, a systematic and multi-pronged methodological approach is required. Figure 3 recapitulates the recommendations made above on how researchers might begin to identify, validate, and interpret potential dinosaur genetic fragments embedded in extant genomes while extending the synthetic approach towards resurrecting dinosaurian biology.
As mentioned above, the first step must be to cast a wide net and sequence as many candidate organisms as possible that might harbour dinosaur shrapnel, revolutions in Next Generation Sequencing technology allow the entire genomes of species to be sequenced at rates that were unimaginable when the mosquito model was first contemplated, and make it possible to sequence the entire genomes of gymnosperms, arachnid parasites such as ticks, and fungi. These could then systematically scan their genomes for regions exhibiting hallmarks of HGT events. These candidate sequences should then be filtered based on molecular markers indicating likely animal origin. If substantial amounts of such genetic material are found, they can be organized according to their resemblance to regions likely encoding proteins or serving regulatory roles, with comparisons made to the known or inferred “core archosaurian” genome. Molecular methods could be employed to date HGT events, helping to determine whether genes originated from the same or related genera. This temporal information would guide hypotheses about functional relationships between regions and their genomic organization. Finally, these hypotheses could undergo iterative generate-and-test cycles using gene-editing technologies combined with synthetic biology approaches, aiming to establish how these fragments might be workably incorporated into a more complete archosaurian genome and one that may progressively resemble that of a real dinosaur as this process proceeds.
Recent advances in large-scale genome analysis methods have dramatically improved our ability to explore vast genetic datasets for hidden gene clusters, evolutionary trends, and genetic sequences that have a history of being transferred horizontally between species. Tools like GATOR-GC (Genomic Assessment Tool for Orthologous Regions and Gene Clusters), for example, exemplify this progress. GATOR-GC enables researchers to efficiently examine millions of gene clusters across hundreds of genomes. This approach promises to combine information about evolutionary conservation and the surrounding genomic context at scales not imagined by approaches based on earlier methods (Cediel-Becerra et al., 2025).
To dig deeper into genetic information, scientists are now using advanced methods that look not just at DNA, but also at how genes are actively working and interacting within organisms. For example, transcriptomics studies RNA—the molecules that carry messages from DNA to make proteins—to understand which genes are switched on and how they function. Meanwhile, metabolomics examines small molecules called metabolites that play key roles in the body’s chemistry. Combining these approaches creates a powerful way to explore biological systems much more thoroughly than simply looking at DNA sequences alone (Go et al., 2024; Prabahar, 2022). On top of this, cutting-edge computer techniques may be useful for boosting our computational capacities to help detect faint signals in genetic data that point to ancient gene transfers between species (Abby et al., 2014) like “network-based analysis” (Popa, Landan, & Dagan, 2011) and “machine learning” (Perkel, 2025)— perhaps even when those gene sequences have changed a lot over millions of years.
At this point, it is worth noting that long mutational histories, gene conversion, recombination, and differing selective pressures can obscure the sorts of genetic signals needed to estimate precisely when genes may have first entered an organism via HGT from the time of the dinosaurs, and could have easily had effects that violate assumptions usually used in traditional molecular clock techniques used to estimate when genetic events occurred. A variety of techniques have evolved to help address these challenges. An example is the use of “relaxed clock models” that allow for the possibility of mutation rates varying across lineages (Bromham et al., 2020; Drummond & Rambaut, 2007; Ho & Duchêne, 2014). Statistical techniques are being developed to detect and account for rate variation and recombination (Rasmussen & Kellis, 2012). Reliable molecular dating of ancient HGT events could combine several approaches to reconcile speculative gene and species phylogenetic trees with information from the fossil record (Szöllősi et al., 2015), relaxed clock models applied to suspected transferred genes (Drummond & Rambaut, 2007; Ho & Duchêne, 2014), and integration of genomic and ecological data to constrain timing despite mutational complexities (Rasmussen & Kellis, 2012). By using some combination of methods like these, we may be able to approximate the ages of specific HGT events with improving accuracy as data and models improve.
In sum, by using these advanced tools, our search for dinosaur genetic “shrapnel” hidden inside the genomes of plants like conifers and fungi is at the forefront of modern genome research. These innovations make it increasingly possible to find, confirm, and understand ancient genetic fragments that might still exist within today’s living organisms.
Bully for Brontosaurus
Even if the ultimate and unquestionably laudable goal of resurrecting a dinosaur remains elusive, pursuing this research trajectory promises transformative benefits across multiple scientific fields and society at large. Efforts to identify ancient genetic fragments embedded in modern genomes could pioneer novel techniques in the emerging field of genomic archaeology. By combining comparative genomics, machine learning, and synthetic biology, these methods could be extended to other extinct lineages, greatly deepening our understanding of evolutionary history and the persistence of genomes over deep time.
This research could also significantly advance our understanding of HGT in multicellular eukaryotes. While HGT is well documented in microbes, its role in plants and animals remains poorly understood. Demonstrating that ancient animal genes are preserved through HGT may challenge and potentially reshape evolutionary theory and open new avenues for studying gene flow across kingdoms. Reconstructing extinct traits from partial genetic information would push the frontiers of synthetic biology, driving the development of novel tools for gene synthesis, regulatory network modelling, and developmental pathway engineering. These technologies could have wide-ranging applications in medicine, agriculture, and bioengineering. Furthermore, attempts to reverse-engineer dinosaur traits from genomic fragments could provide valuable insights into the genetic foundations of form and function. Such work could lead to new models in evolutionary developmental biology (evo-devo), especially concerning how complex traits emerge, diversify, and are shaped by phylogenetic constraints.
One major ethical argument against de-extinction efforts is that resources would be better spent on conserving existing species. A more nuanced version of this argument suggests that if people believe de-extinction is possible, they may feel less urgency to prevent extinctions in the first place—potentially increasing overall extinction rates. This is analogous to the paradoxical phenomenon of risk compensation observed in public health: for example, advances in AIDS treatment sometimes lead to riskier behaviour because individuals perceive the consequences of infection as less severe (Hedlund, 2000; Peltzman, 1975). Whether such an effect would occur with de-extinction is uncertain and depends on many factors.
A related objection is that investment in de-extinction research could crowd out funding for conservation efforts. However, this assumption relies on the premise of a fixed budget and that de-extinction and conservation are direct economic substitutes. In reality, public enthusiasm for resurrecting iconic species like dinosaurs is likely far greater than for many lesser known but endangered animals. Importantly, the groups motivated by these causes may also differ significantly.
A more plausible scenario is that interest in dinosaur de-extinction could actually increase overall funding for biological research, drawing government, philanthropic, and private investments beyond what is currently allocated to conservation. This influx of resources might not divert funds from conservation but instead create spillover benefits and synergies between the two fields. In this way, de-extinction research and conservation are better viewed as economic complements rather than substitutes.
More broadly, the pursuit of dinosaur gene recovery captures the public imagination and provides a compelling narrative to engage people with science. Such an inspiring goal — like landmark endeavours like the space race and the Human Genome Project — can spark widespread curiosity and promote scientific literacy. Just as the mosquito model once inspired a generation, this new, scientifically grounded approach has the potential to galvanize interest and support for genetics, palaeontology, and biotechnology today.
Summary and Conclusions
Dinosaur genes may have been channelled by fungi or microbial vectors in a way that resulted in fragments of dinosaur DNA becoming embedded into the genomes of plants and fungi, thereby preserving molecular echoes of these long-extinct animals. While this hypothetical scenario may seem improbable, whether sufficient, if any, dinosaur DNA could be recovered in this way remains an open empirical question. By applying systematic search strategies — guided by knowledge of phylogenetic relationships and targeting the “dark genomes” of living species where such shrapnel is most likely to reside (we have suggested gymnosperms) — the recovery of meaningful dinosaur genetic remnants could shift from speculation toward strategic plausibility and practical possibility. Even if full de-extinction remains out of reach, pursuing this research could yield profound scientific rewards, such as galvanizing public support for advances in molecular and developmental biology and deepening our understanding of horizontal gene transfer in complex multicellular organisms. If nothing else, if this article can provide some comfort to those who dream of dinosaurs one day again walking the earth, then it has achieved its purpose.
Statements and Declarations: I received no funding for the production of this manuscript and have no conflicts of interest to declare.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.
References
- Abby, S.S.; Tannier, E.; Gouy, M.; Daubin, V. Detecting lateral gene transfers by statistical reconciliation of phylogenetic forests. BMC Bioinform. 2014, 11, 324–324. [CrossRef]
- Alexander, W.G.; Wisecaver, J.H.; Rokas, A.; Hittinger, C.T. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc. Natl. Acad. Sci. 2016, 113, 4116–4121. [CrossRef]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, M.T.P.; Willerslev, E.; et al. The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils. Proc. R. Soc. B: Biol. Sci. 2012, 279, 4724–4733. [CrossRef]
- Asara, J.M.; Schweitzer, M.H.; Freimark, L.M.; Phillips, M.; Cantley, L.C. Protein Sequences from Mastodon and Tyrannosaurus Rex Revealed by Mass Spectrometry. Science 2007, 316, 280–285. [CrossRef]
- Bergthorsson, U.; Richardson, A.O.; Young, G.J.; Goertzen, L.R.; Palmer, J.D. Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm Amborella. Proc. Natl. Acad. Sci. 2004, 101, 17747–17752. [CrossRef]
- Bhullar, B.-A.S.; Morris, Z.S.; Sefton, E.M.; Tok, A.; Tokita, M.; Namkoong, B.; Camacho, J.; Burnham, D.A.; Abzhanov, A. A molecular mechanism for the origin of a key evolutionary innovation, the bird beak and palate, revealed by an integrative approach to major transitions in vertebrate history. Evolution 2015, 69, 1665–1677. [CrossRef]
- Biswas, C., & Johri, B. M. (2013). The gymnosperms. Springer Science & Business Media.
- Bock, R. The give-and-take of DNA: horizontal gene transfer in plants. Trends Plant Sci. 2010, 15, 11–22. [CrossRef]
- Boothby, T.C.; Tenlen, J.R.; Smith, F.W.; Wang, J.R.; Patanella, K.A.; Nishimura, E.O.; Tintori, S.C.; Li, Q.; Jones, C.D.; Yandell, M.; et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proc. Natl. Acad. Sci. 2015, 112, 15976–15981. [CrossRef]
- Boto, L. Horizontal gene transfer in evolution: facts and challenges. Proc. R. Soc. B: Biol. Sci. 2014, 277, 819–827. [CrossRef]
- Bromham, L.; Duchêne, S.; Hua, X.; Ritchie, A.M.; Duchêne, D.A.; Ho, S.Y.W. Bayesian molecular dating: opening up the black box. Biol. Rev. 2020, 93, 1165–1191. [CrossRef]
- Brusatte, S.L.; Carr, T.D. The phylogeny and evolutionary history of tyrannosauroid dinosaurs. Sci. Rep. 2016, 6, 20252. [CrossRef]
- Carroll, S. B. (2005). Endless forms most beautiful. W. W. Norton & Company.
- Cediel-Becerra, J.D.D.; Cumsille, A.; Guerra, S.; Ding, Y.; de Crécy-Lagard, V.; Chevrette, M.G. Targeted genome mining with GATOR-GC maps the evolutionary landscape of biosynthetic diversity. Nucleic Acids Res. 2025, 53. [CrossRef]
- Crisp, A.; Boschetti, C.; Perry, M.; Tunnacliffe, A.; Micklem, G. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 2015, 16, 50–50. [CrossRef]
- Dawkins, R. (2024). The Genetic Book of the Dead: A Darwinian Reverie. Yale University Press.
- Dennett, D. C. (2017). From bacteria to Bach and back: The evolution of minds. W. W. Norton & Company.
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [CrossRef]
- Hotopp, J.C.D. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011, 27, 157–163. [CrossRef]
- Edsinger, E.; Moroz, L.L. Genomic hotspots: localized chromosome gene expansions identify lineage-specific innovations as targets for functional biodiversity and predictions of stress resilience. Front. Mar. Sci. 2024, 11, 1434130. [CrossRef]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the Evolution of Eukaryotic Genomes. Annu. Rev. Genet. 2007, 41, 331–368. [CrossRef]
- Gilbert, C., Schaack, S., Pace II, J. K., Brindley, P. J., & Feschotte, C. (2010). A role for host–parasite interactions in the horizontal transfer of transposons across phyla. Nature, 464(7293), 1347-1350.
- Go, D.; Yeon, G.-H.; Park, S.J.; Lee, Y.; Koh, H.G.; Koo, H.; Kim, K.H.; Jin, Y.-S.; Sung, B.H.; Kim, J. Integration of metabolomics and other omics: from microbes to microbiome. Appl. Microbiol. Biotechnol. 2024, 108, 1–7. [CrossRef]
- González Villa, T., & Vinas, M. (2019). Horizontal gene transfer: breaking borders between living kingdoms. Springer.
- Gould, S. J. (1983). Hen’s teeth and horse’s toes: Further reflections in natural history. New York, NY: W. W. Norton & Company.
- Gould, S. J. (1995). Dinosaur in a Haystack: Reflections in Natural History. Harvard University Press.
- Green, R.E.; Braun, E.L.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.W.; John, J.A.S.; Capella-Gutiérrez, S.; Castoe, T.A.; et al. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 2014, 346, 1254449. [CrossRef]
- Gulia-Nuss, M.; Nuss, A.B.; Meyer, J.M.; Sonenshine, D.E.; Roe, R.M.; Waterhouse, R.M.; Sattelle, D.B.; de la Fuente, J.; Ribeiro, J.M.; Megy, K.; et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 2016, 7, 1–13. [CrossRef]
- Griffin, D. K., Hassan, S., & Trifonov, V. (2019). Time lapse: A glimpse into prehistoric genomics. Genes, 10(6), 464.
- Hedlund, J. Risky business: safety regulations, risk compensation, and individual behavior. Inj. Prev. 2000, 6, 82–89. [CrossRef]
- Ho, S.Y.W.; Duchêne, S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014, 23, 5947–5965. [CrossRef]
- Horner, J., & Gorman, J. (2009). How to build a dinosaur: The new science of reverse evolution. Penguin.
- Kambayashi, C.; Kakehashi, R.; Sato, Y.; Mizuno, H.; Tanabe, H.; Rakotoarison, A.; Künzel, S.; Furuno, N.; Ohshima, K.; Kumazawa, Y.; et al. Geography-Dependent Horizontal Gene Transfer from Vertebrate Predators to Their Prey. Mol. Biol. Evol. 2022, 39. [CrossRef]
- Klompen, J. S. H., Black, W., Keirans, J. E., & Oliver Jr, J. H. (1996). Evolution of ticks. Annual review of entomology, 41(1), 141-161.
- Leitch, I.J.; Leitch, A.R. (2012). Genome size diversity and evolution in land plants. In Plant genome diversity Volume 2: Physical structure, behaviour and evolution of plant genomes (pp. 307-322). Vienna: Springer Vienna.
- Mans, B.J.; de Klerk, D.; Pienaar, R.; Latif, A.A. Nuttalliella namaqua: A Living Fossil and Closest Relative to the Ancestral Tick Lineage: Implications for the Evolution of Blood-Feeding in Ticks. PLOS ONE 2011, 6, e23675. [CrossRef]
- Marshall, C. R., Ward, P. D., & Smith, F. A. (2021). Absolute abundance and preservation rate of Tyrannosaurus rex. Science, 372(6539), 284–287.
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: major ecological adaptations and evolutionary transitions. Biol. Rev. 2019, 94, 1443–1476. [CrossRef]
- National Human Genome Research Institute. (2010.). Why mouse matters. Retrieved August 21, 2025, from https://www.genome.gov/10001345/importance-of-mouse-genome.
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [CrossRef]
- O’cOnnor, R.E.; Romanov, M.N.; Kiazim, L.G.; Barrett, P.M.; Farré, M.; Damas, J.; Ferguson-Smith, M.; Valenzuela, N.; Larkin, D.M.; Griffin, D.K. Reconstruction of the diapsid ancestral genome permits chromosome evolution tracing in avian and non-avian dinosaurs. Nat. Commun. 2018, 9, 1–9. [CrossRef]
- Peñalver, E.; Arillo, A.; Delclòs, X.; Peris, D.; Grimaldi, D.A.; Anderson, S.R.; Nascimbene, P.C.; la Fuente, R.P.-D. Ticks parasitised feathered dinosaurs as revealed by Cretaceous amber assemblages. Nat. Commun. 2017, 8, 1924–1924. [CrossRef]
- Penney, D.; Wadsworth, C.; Fox, G.; Kennedy, S.L.; Preziosi, R.F.; A Brown, T. Absence of Ancient DNA in Sub-Fossil Insect Inclusions Preserved in ‘Anthropocene’ Colombian Copal. PLOS ONE 2013, 8, e73150. [CrossRef]
- Peris, D.; Janssen, K.; Barthel, H.J.; Bierbaum, G.; Delclòs, X.; Peñalver, E.; Solórzano-Kraemer, M.M.; Jordal, B.H.; Rust, J. DNA from resin-embedded organisms: Past, present and future. PLOS ONE 2020, 15, e0239521. [CrossRef]
- Perkel, J.M. Beyond AlphaFold: how AI is decoding the grammar of the genome. Nature 2025, 644, 829–832. [CrossRef]
- Prabahar, A. (2022). Integration of Transcriptomics Data and Metabolomic Data Using Biomedical Literature Mining and Pathway Analysis. In Biomedical Text Mining (pp. 301-316). New York, NY: Springer US.
- Peltzman, S. The Effects of Automobile Safety Regulation. J. Politi- Econ. 1975, 83, 677–725. [CrossRef]
- Rasmussen, M.D.; Kellis, M. Unified modeling of gene duplication, loss, and coalescence using a locus tree. Genome Res. 2012, 22, 755–765. [CrossRef]
- Redecker, D.; Kodner, R.; Graham, L.E. Glomalean Fungi from the Ordovician. Science 2000, 289, 1920–1921. [CrossRef]
- Richards, T.A.; Leonard, G.; Soanes, D.M.; Talbot, N.J. Gene transfer into the fungi. Fungal Biol. Rev. 2011, 25, 98–110. [CrossRef]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V.; Chandler, M.; Gellert, M.; Lambowitz, A.M.; Rice, P.A.; et al.
- Royal Veterinary College (RVC). (2024). Scientists use modern species to discover genome structure of dinosaurs. [Press release]. https://www.rvc.ac.uk/news-and-events/rvc-news/scientists-use-modern-species-to-discover-genome-structure-of-dinosaurs.
- Szöllősi, G.J.; Tannier, E.; Daubin, V.; Boussau, B. The Inference of Gene Trees with Species Trees. Syst. Biol. 2015, 64, e42–e62. [CrossRef]
- Wani, A.K.; Akhtar, N.; Sher, F.; Navarrete, A.A.; Américo-Pinheiro, J.H.P. Microbial adaptation to different environmental conditions: molecular perspective of evolved genetic and cellular systems. Arch. Microbiol. 2022, 204, 1–16. [CrossRef]
| 1 | Tyrannosaurus and birds share a common ancestor approximately 150-160 mya. This means the lineage giving rise to modern birds has had, say 150 million years to evolve differences from the common ancestor, while the Tyrannosaurus lineage had 85 million years (because it went extinct 65 mya). Thus, some 210 million years of evolutionary change separates Tyrannosaurus Rex from chickens. Humans and mice share a common ancestor around 90 mya and so have had about 180 million years to evolve differences. If we take it that approximately 85 per cent of expressed DNA in human and mice are identical (National Human Genome Research Institute, 2010), then 80 per cent does not seem an outlandish estimate. |
Figure 1.
Three Approaches to Dinosaur Paleogenomics. Venn diagram showing rough overlap of genetic identity between three archosaurians: crocodiles, chickens, and triceratops. The mosquito model hoped to clone dinosaurs from near complete genetic information preserved in fossilized mosquitos. The “dino-chicken” approach seeks to make heterochronic adjustments to the development of chickens to exploit genetic material inherited from their mid-Jurassic ancestors (genetic material shaded in light grey). Some of this genetic information and other material overlapping between crocodilians and both extinct dinosaurs and birds to reconstruct a basic archosaurian genetic template (probably not a functional animal).
Figure 1.
Three Approaches to Dinosaur Paleogenomics. Venn diagram showing rough overlap of genetic identity between three archosaurians: crocodiles, chickens, and triceratops. The mosquito model hoped to clone dinosaurs from near complete genetic information preserved in fossilized mosquitos. The “dino-chicken” approach seeks to make heterochronic adjustments to the development of chickens to exploit genetic material inherited from their mid-Jurassic ancestors (genetic material shaded in light grey). Some of this genetic information and other material overlapping between crocodilians and both extinct dinosaurs and birds to reconstruct a basic archosaurian genetic template (probably not a functional animal).
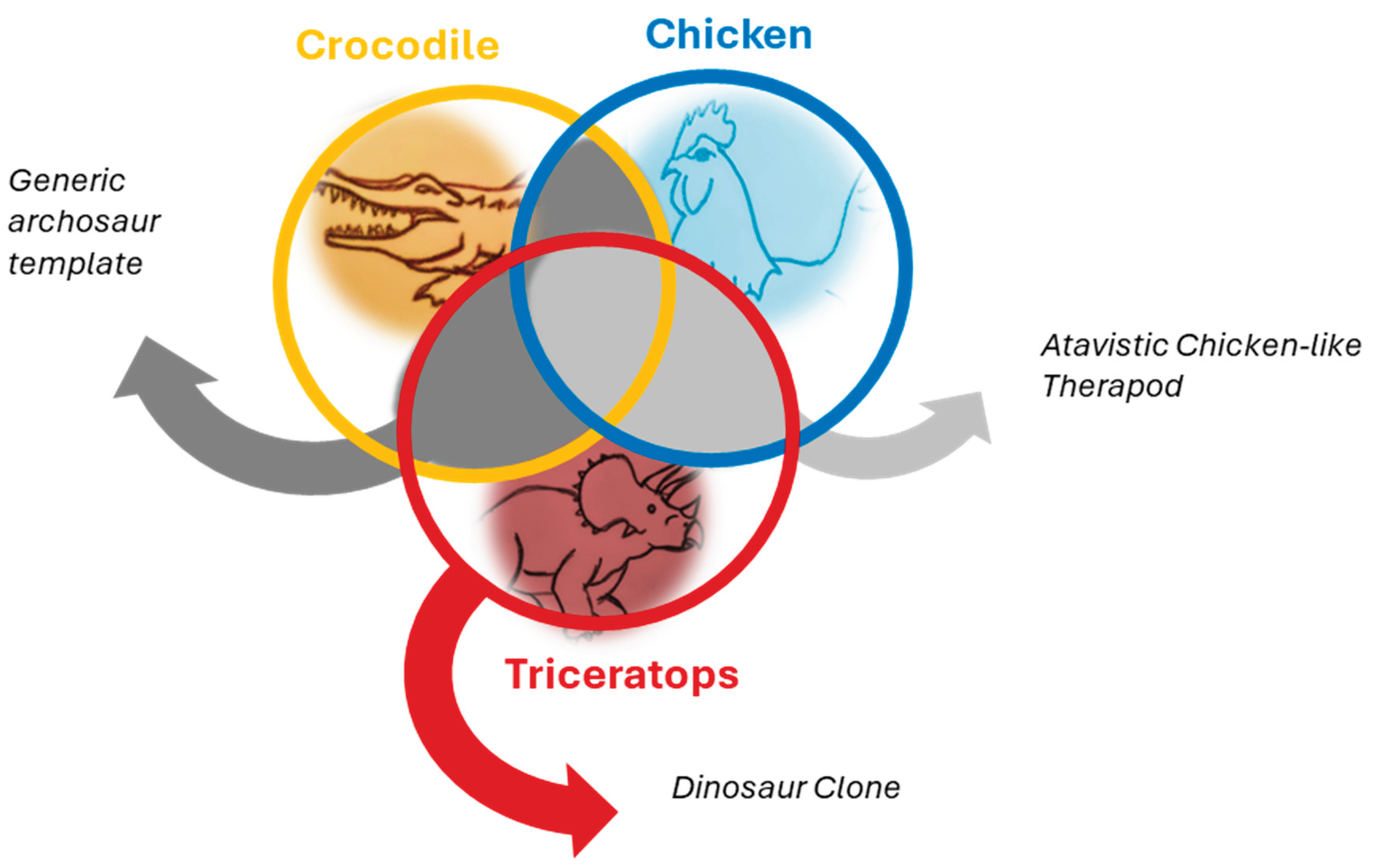
Figure 2.
The Genetic Shrapnel Model. Upward movement depicts genetic information transferred vertically (from parent to offspring) from prehistoric times to the modern day. Meanwhile, horizontal (red dashed lines) represents genes transferred hypothetically through HGT events via microorganisms.
Figure 2.
The Genetic Shrapnel Model. Upward movement depicts genetic information transferred vertically (from parent to offspring) from prehistoric times to the modern day. Meanwhile, horizontal (red dashed lines) represents genes transferred hypothetically through HGT events via microorganisms.
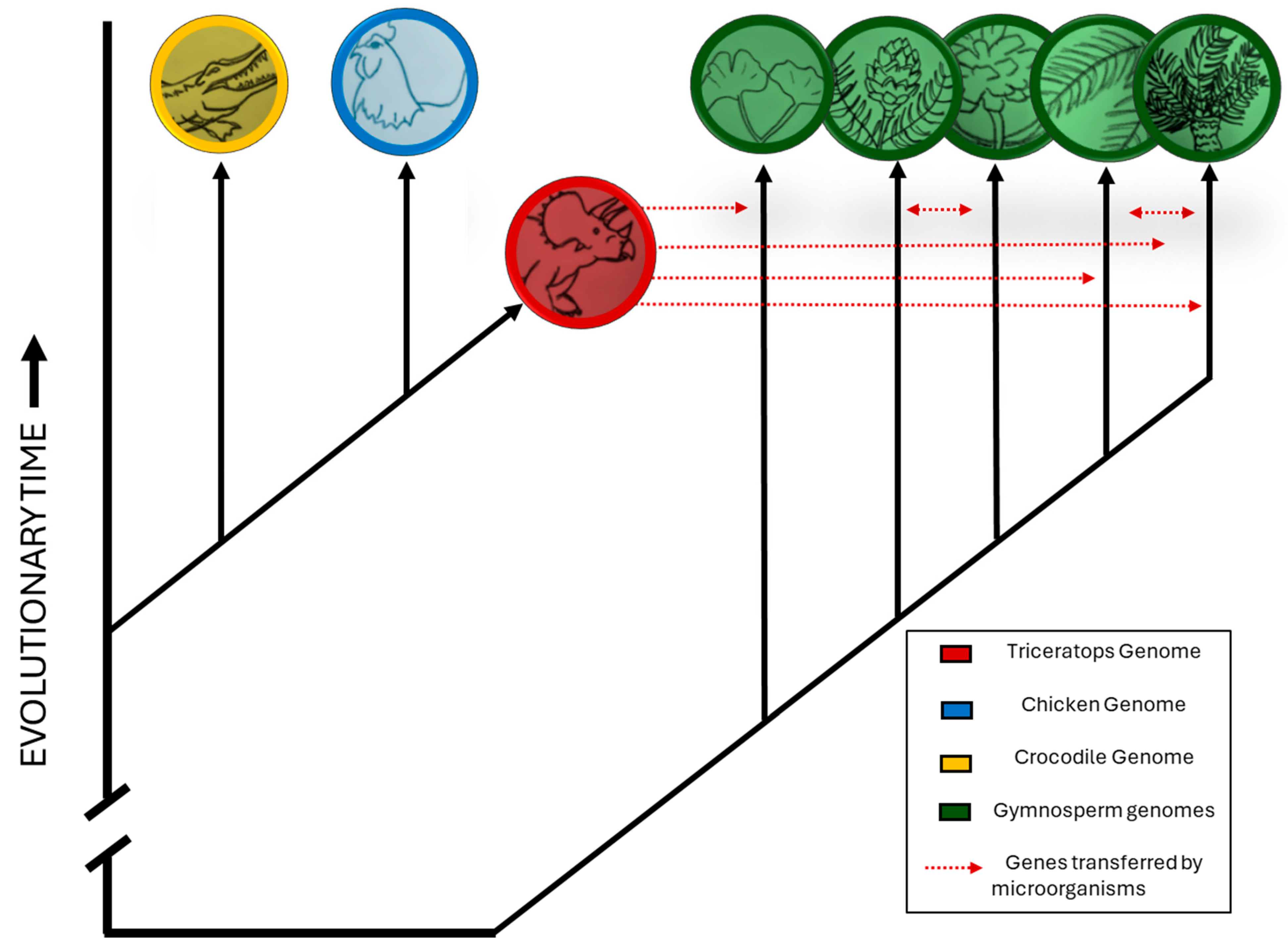
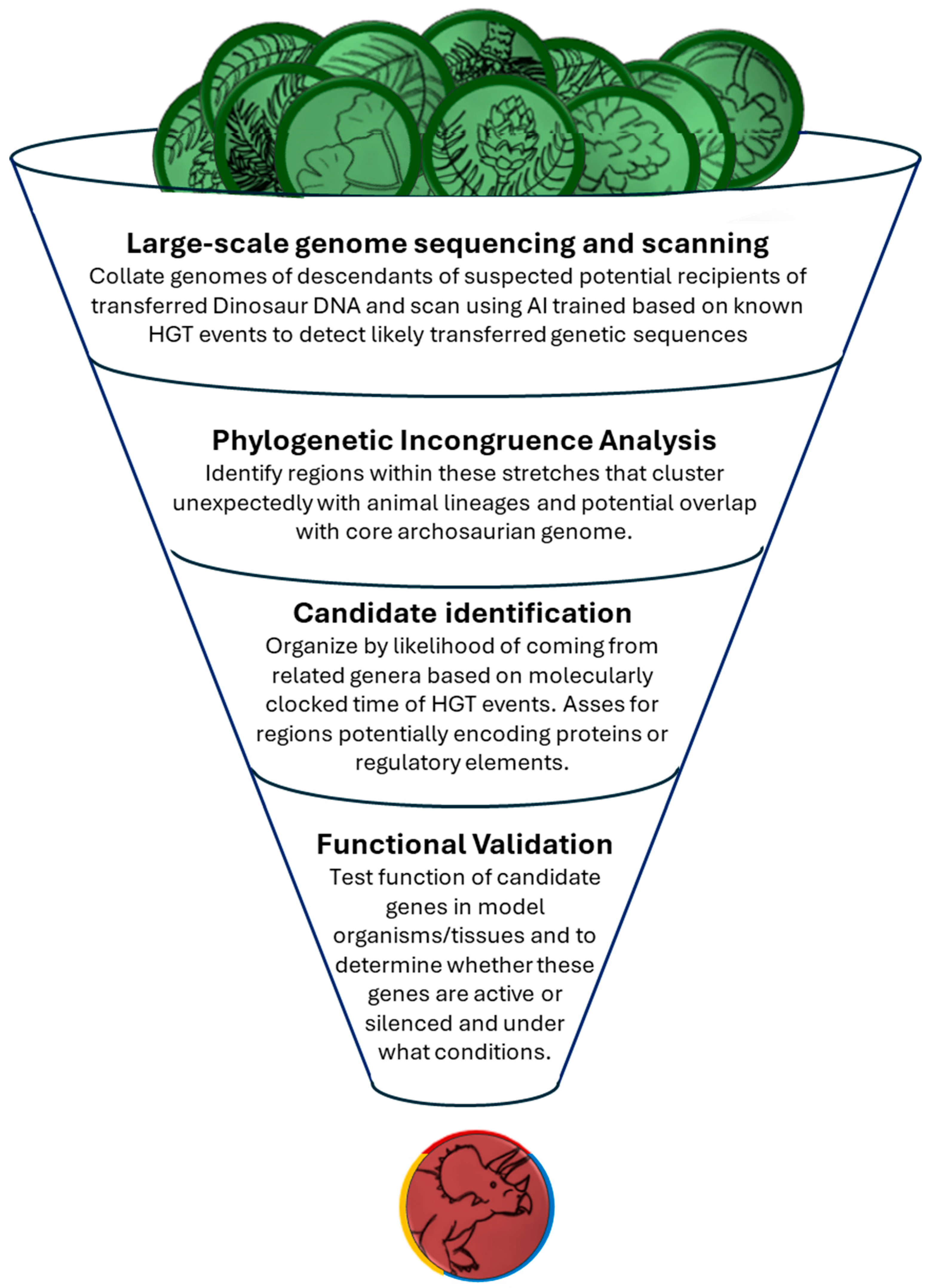
Figure 3.
Suggested filtering system for identifying functional genetic information originating in dinosaurs. Starting with mass sequencing of gymnosperm (and possible candidate organisms such as fungi and tics), various filters are applied until genetic material can be organized to be tested in biological tissues. If successful, components needed to recreate uniquely dinosaurian genomes may be accumulated, piece by piece, and combined with an archosaur template provided from modern birds and crocodilians.
Figure 3.
Suggested filtering system for identifying functional genetic information originating in dinosaurs. Starting with mass sequencing of gymnosperm (and possible candidate organisms such as fungi and tics), various filters are applied until genetic material can be organized to be tested in biological tissues. If successful, components needed to recreate uniquely dinosaurian genomes may be accumulated, piece by piece, and combined with an archosaur template provided from modern birds and crocodilians.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



