Submitted:
03 February 2026
Posted:
04 February 2026
You are already at the latest version
Abstract
Signal transducer and activator of transcription 3 (STAT3) is a central oncogenic hub in several tumors including the Triple-Negative Breast Cancer (TNBC) subtype, where its constitutive activity supports proliferation, metabolic flexibility, tumor progression, immune evasion, and therapeutic resistance. Therapeutic development has largely focused on canonical STAT3 activation driven by tyrosine 705 phosphorylation (p-Y705), which enables dimerization and transcriptional programs. However, accumulating evidence indicates that phosphorylation at serine 727 (p-S727) defines a functionally distinct STAT3 axis, underpinning non-canonical activities across extranuclear compartments that include mitochondria and endoplasmic reticulum/mitochondria-associated membranes. In TNBC, p-S727 STAT3 is frequently prevalent and may sustain oncogenic signaling when p-Y705 is low or pharmacologically suppressed, contributing to metabolic rewiring, redox control, apoptosis resistance, and metastatic fitness. Here, we review the mechanistic basis and clinical correlations of STAT3 p-S727 across cancers with emphasis on TNBC, and discuss how compartmentalized STAT3 pools may integrate kinase signaling, nutrient sensing, and stress responses. We also summarize emerging therapeutic strategies that modulate p-S727—often in conjunction with p-Y705—highlighting proof-of-concept for dual targeting or specific p-S727 to overcome limitations of Y705-centric approaches. Finally, we propose that integrating p-S727/p-Y705 distribution and activity into patient stratification could improve the efficacy–toxicity balance of STAT3-directed therapies in TNBC.
Keywords:
STAT3
; S727
; Y705
; cancer
; triple-negative breast cancer
; breast cancer
1. Introduction
Signal transducer and activator of transcription 3 (STAT3) is a pleiotropic transcription factor that integrates cytokine and growth factor signals to regulate gene expression programs controlling cell proliferation, survival, differentiation, and immune regulation, thus functioning as a central integrator of inflammatory and oncogenic cues [1,2]. Canonical STAT3 activation occurs downstream of cytokine receptors, notably those of the interleukin 6 (IL-6) family, through Janus kinases and non-receptor tyrosine kinases, leading to phosphorylation at tyrosine 705 (Y705, p-Y). This promotes dimerization through reciprocal SH2-pY domains interaction, nuclear accumulation, and binding to γ-activated sequence (GAS) motifs to regulate transcription of STAT3 target genes [1,2,3,4]. Under physiological conditions, this signaling is transient and tightly controlled by negative regulators, including suppressor of cytokine signaling (SOCS) proteins, protein inhibitor of activated STAT3 (PIAS3), and protein tyrosine phosphatases (PTPs), ensuring timely signal termination and preventing uncontrolled gene expression [5,6,7,8].
Despite sparse cases where STAT3 activating somatic mutations have been detected in cancer, in most instances constitutive activation occurs in the presence of the wild type protein [9]. Accordingly, persistent p-Y705 can be driven by chronic stimulation from tumor- and stroma-derived cytokines (e.g., IL-6, IL-11, OSM), aberrant activation of receptor tyrosine kinases such as Epidermal Growth Factor Receptor (EGFR) and Human Estrogen Receptor 2 (HER2), non-receptor tyrosine kinases including Src and Abl, and deregulated G protein–coupled receptor (GPCR) pathways [8,10]. Loss or functional impairment of inhibitory molecules such as SOCS1/3, PIAS3, SHP-1/2, or PTPN2 further stabilize STAT3 activation prolonging its nuclear residency [5,8]. These alterations collectively rewire STAT3 from a transient inflammatory responder to a chronically active oncogenic driver, a state documented across multiple solid tumors and hematologic malignancies [8,10]. Of note, STAT3 activity can contribute to malignant transformation in primary cells, supporting its role as a bona fide oncogenic driver [10,11,12].
However, multiple independent lines of evidence indicate that STAT3-dependent phenotypes cannot be fully accounted for by the canonical transcriptional model. Across diverse systems, genetic ablation of Y705 phosphorylation or disruption of STAT3 DNA binding fails to completely suppress STAT3-driven effects on cellular metabolism, stress tolerance, or survival [13,14,15]. These observations have contributed to the concept of non-canonical STAT3 signaling, encompassing STAT3 functions that are not strictly dependent on Y705-driven nuclear transcription [16,17,18]. Non-canonical STAT3 activities are closely related to its ability to engage distinct subcellular compartments. Beyond its p-Y driven nuclear localization and the diffused cytoplasmic localization of the non-phosphorylated form, specific and functional STAT3 localization was detected in the mitochondria and the endoplasmic reticulum (ER)–associated compartments, including mitochondria-associated ER membranes (MAMs) [17,18,19,20].
A key functional determinant of these non-nuclear STAT3 activities is phosphorylation at serine 727 (S727, p-S) [17,18,19,21]. Historically, p-S727 was viewed primarily as an accessory modification that enhances the transcriptional efficiency of Y705-phosphorylated STAT3, e.g. by facilitating recruitment of co-activators and supporting productive transcriptional elongation at a subset of promoters [14,22,23]. Accordingly, while germline STAT3 gene inactivation is embryonic lethal, mice carrying a S727A homozygous mutation are normal and viable [23]. More recent evidence, however, supports a broader model in which S727 phosphorylation modulates STAT3 protein–protein interactions and specific activities in diverse subcellular compartments. Indeed, S727 phosphorylation independently from DNA binding or Y705 phosphorylation was shown to be required both for mitochondrial STAT3 activities and for its participation in ER-associated signaling complexes [17,19,21,24]. In this framework, S727 is positioned downstream of pathways such as MAPK and mTOR and links growth factor signaling, nutrient sensing, and cellular stress to STAT3-dependent outputs in metabolism, survival, and cellular fitness [13,15,25,26,27].
The functional distinction between Y705 and S727-dependent STAT3 activities has been dissected using phosphorylation-site mutants (Figure 1). Substitution of Y705 with phenylalanine (Y705F) abolishes STAT3 dimerization and transcriptional activity while preserving specific extranuclear functions [15,19,24,28,29]. In contrast, mutation of S727 to alanine (S727A) selectively disrupts non-canonical STAT3 activities at both mitochondria and the ER compartment, without affecting nuclear transcription [15,19,24,28,29,30]. Phospho-mimetic S727 mutants (S727D or S727E), as well as combinatorial mutants (Y705F/S727A or Y705F/S727E) were designed to isolate serine-driven outputs from canonical transcription [24,28,29]. In addition to phosphorylation-site mutants, a constitutively active STAT3 variant, STAT3-C, where two cysteine substitutions in the SH2 domain promote stable dimerization independently of Y705 phosphorylation, was generated. This mutation results in persistent nuclear localization and transcriptional activity and is able to support tumor transformation of immortalized cells [3,12]. In vivo, this mutation enhances breast cancer cell migration and metastases in mouse mammary tumor virus (MMTV)-Neu transgenic mice [3,31]. A fusion protein with a mitochondrial localization sequence (MLS-STAT3), driving localization to the inner mitochondrial membrane, was generated in order to explore mitochondrial functions [17,24] (Figure 1).
Breast cancer (BC) represents a clinically and biologically relevant context to apply this framework. STAT3 expression and activation are frequently elevated in mammary tumors and particularly in triple-negative breast cancer (TNBC), where constitutive STAT3 signaling has been linked to aggressive behavior, metabolic plasticity, immune evasion, and therapeutic resistance [32,33,34]. Indeed, STAT3 mRNA levels are up-regulated in breast tumors compared with normal mammary tissue, with particularly high expression observed in the TNBC subtype [35]. Importantly, elevated STAT3 expression correlates with reduced relapse-free survival (RFS) in both BC in general and TNBC patients [36]. However, no clear correlation was observed with the levels of pY705-STAT3 [37]. Interestingly, p-S727 STAT3 is increased in breast infiltrating ductal carcinoma and correlates with ER-negative status and tumor aggressiveness [38], while pY705-STAT3 expression was shown to correlate with longer overall survival (OS) and RFS in ER-positive breast tumors [39]. Moreover, STAT3 S727 and Y705 phosphorylation have been associated with distinct tumor cell phenotypes in TNBC patients, suggesting distinct functional roles in breast tumor biology [40]. In this vein, STAT3 activity in TNBC was reported to be predominantly driven by S727 phosphorylation with limited involvement of Y705 [41]. This pattern appears to be subtype-specific, as STAT3 expression and activation are rarely detected in HER2-positive and luminal breast tumors, underscoring a TNBC-specific reliance on p-S727 STAT3 signaling [41].
In this review, we first summarize canonical Y705-mediated STAT3 signaling and its established roles in cancer (Section 2). We then discuss emerging non-canonical STAT3 functions, with particular emphasis on alternative subcellular localizations and the distinct biological activities associated with S727 phosphorylation (Section 3), focusing on cancer in general (Section 4), and on BC and TNBC (Section 5). Finally, we review experimental evidence underling the potential translation relevance of targeting p-S727 STAT3 signaling, particularly in mitochondria (Section 6) and TNBC (Section 7).
2. Canonical Y-705-Mediated STAT3 Signaling in Cancer
As mentioned above, in most instances constitutive STAT3 Y705 phosphorylation in cancer occurs downstream of aberrantly active upstream inducers, such as IL-6-type cytokines and growth factors. In this vein, particularly relevant is the original observation that STAT3 is required for tumor transformation downstream of the Src oncogene [42]. The oncogenic potential of canonical STAT3 signaling is largely attributable to its extensive transcriptional output, which mostly aligns with classical cancer hallmarks [8,10,43]. STAT3 promotes cell-cycle progression by inducing cyclins (e.g., cyclin D1/D2) and c-MYC, thereby facilitating the G1/S transition and sustaining proliferative signaling [1,44]. In parallel, STAT3 enhances survival and resistance to apoptosis by upregulating anti-apoptotic BCL-2 family members (BCL-2, BCL-XL, MCL-1) and inhibitors of apoptosis such as survivin (BIRC5), XIAP, and c-IAP2 [1,10]. Moreover, STAT3 participates in metabolic rewiring and interfaces with mitochondrial signaling pathways that shape apoptotic thresholds and transformation [10,11,45]. Both transcriptional and non-transcriptional outputs collectively blunt intrinsic and extrinsic apoptotic pathways, contributing to resistance to chemotherapy, radiotherapy, and targeted agents [8,10,11,12,43,46,47].
Canonical STAT3 signaling also plays a central role in tumor angiogenesis and metastatic dissemination. STAT3 directly induces Vascular Endothelial Growth Factor (VEGF) expression and, under hypoxic conditions, cooperates with Hypoxia-Inducible Factor 1α (HIF-1α) to promote neovascularization [8,10,44]. Of relevance, STAT3 constitutive activity was shown to induce a metabolic switch to aerobic glycolysis via the up-regulation of HIF-1α itself [45]. During invasion and metastasis, STAT3 interacts with epithelial-mesenchymal transition (EMT)-associated transcription factors and with TGF-β/SMAD signaling to repress epithelial markers such as E-cadherin and to induce mesenchymal markers, including twist, vimentin and N-cadherin. Simultaneously, STAT3 induces the expression of matrix metalloproteinases (MMP2, MMP9) [8,10,43,46], thus contributing to extra-cellular matrix remodeling, known to be crucial for tumor growth and metastases. These processes support local invasion, intravasation, and colonization at distant organs and are particularly relevant in aggressive breast cancer subtypes such as TNBC [43,46,48,49].
STAT3 signaling also regulates the maintenance of cancer stem cells (CSC), where its persistent activation sustains self-renewal, survival and tumor-initiating capacity [50]. Through the IL-6/JAK/STAT3 axis STAT3 induces the expression of stemness-associated genes, including OCT4, SOX2, NANOG, KLF4 and MYC thereby enhancing CSC self-renewal and plasticity, invasiveness and therapy resistance [50,51,52]. Finally, due to its significant correlation with the expression of CSC phenotype markers, STAT3 was proposed as a marker and regulator of CSCs, together with ΔNp63, particularly in the triple-negative subtype [52].
Beyond tumor cell–intrinsic functions, transcriptional STAT3 activities exert profound non-cell-autonomous effects within the tumor microenvironment. STAT3 activation in both immune (dendritic cells, macrophages, neutrophils, myeloid-derived suppressor cells) and mesenchymal components (fibroblasts, endothelial cells, mesenchymal stem cells) of the tumor stroma promotes an immunosuppressive and pro-tumorigenic milieu [1,8,43,44,48,53]. In cancer cells, STAT3 dampens antigen presentation, upregulates immune checkpoint molecules such as PD-L1, and drives secretion of IL-6, IL-10, and TGF-β, which in turn reinforce the expansion and activity of regulatory T cells and myeloid-derived suppressor cells [1,8,44,53]. This establishes a feed-forward loop in which tumor- and stroma-derived cytokines maintain chronic STAT3 activation, that, in turn, reciprocally stabilizes an immunosuppressive and inflammatory tumor-promoting microenvironment [1,8,44,53].
At the chromatin level, canonical Y705-dependent STAT3 dimers bind GAS motifs in promoters and distal regulatory elements. Genome-wide analyses show that STAT3-occupied regions frequently overlap with active enhancers enriched for p300/CBP, BRD4, and H3K27ac, indicating that STAT3 not only drives transcription initiation but also contributes to enhancer remodeling and higher-order chromatin organization [54]. Cooperative binding with NF-κB, AP-1, and nuclear hormone receptors further shapes STAT3-dependent transcriptional specificity, explaining how STAT3 can exert context-dependent oncogenic functions across diverse tumor types [10,54,55]. In breast cancer, including TNBC, these interactions integrate growth factor, hormone, and inflammatory cues into consolidated transcriptional programs that promote tumor growth, stemness, and therapeutic resistance [8,43,46].
Given its central role in tumor biology, STAT3 has long been pursued as a therapeutic target. Strategies include inhibiting upstream cytokines and their receptors (e.g., IL-6/IL-6R blockade), targeting JAK kinases, interfering with SH2-mediated STAT3 Y705 phosphorylation and dimerization, blocking DNA binding, or promoting STAT3 degradation [56,57,58]. Several small-molecule inhibitors have shown preclinical activity in TNBC and other malignancies, but clinical translation has been challenging due to issues of specificity, pharmacokinetics, and on-target toxicities [44,56,57,58]. Notably, most of these efforts have historically focused on Y705-mediated activities and canonical nuclear signaling, reflecting the generalized view that Y705 is the predominant driver of STAT3 oncogenic functions [4,10,56,57,58].
3. STAT3 Alternative Subcellular Localizations and Non-Canonical Functions
STAT3 phosphorylation on serine 727 (p-S727) has for decades been regarded as an accessory post-translational modification (PTM), primarily functioning to fine-tune signaling cascades initiated by tyrosine 705 (Y705) phosphorylation [42,59]. In this canonical framework, p-S727 was assigned a minor modulatory role in nuclear STAT3 signaling, where it enhances transcriptional efficiency by facilitating recruitment of transcriptional co-activators, stabilizing STAT3-containing transcriptional complexes, and promoting chromatin engagement at target gene loci [22]. Mechanistically, S727 phosphorylation has been linked to increased interaction with histone acetyltransferases (HAT) such as p300/CBP, improved RNA Pol II recruitment, and enhanced transcriptional elongation, thereby maximizing the output of Y705-dependent STAT3 dimers without altering DNA-binding specificity or nuclear localization [22,23]. Interestingly, STAT3 S727 phosphorylation was on the other hand shown to contrast subsequent Y705 phosphorylation, as in a sort of negative feedback. However, this initial observation was not further pursued [22,23,27].
This Y705-centric view was substantially broadened by evidence indicating that p-S727 can define qualitatively distinct STAT3 functions, including non-canonical, non-nuclear ones. A pivotal shift arose from studies reporting localization of STAT3 to mitochondria, where its p-S727 form regulates processes unrelated to transcription [17,21,60]. Seminal work demonstrated that mitochondrial STAT3 requires S727—but not Y705—phosphorylation to modulate electron transport chain (ETC) activity, particularly at complexes I and II, thereby enhancing oxidative phosphorylation efficiency while limiting excessive reactive oxygen species (ROS) production [17,21,60]. At the molecular level, p-S727 STAT3 was shown to associate with mitochondrial membranes and interact with ETC components, influencing NADH oxidation, membrane potential, and ATP production [17,21,60]. These activities promote metabolic flexibility, resistance to oxidative stress, and oncogenic transformation downstream of Ras oncogenes and MAP kinases, particularly under conditions in which mitochondrial function becomes rate-limiting for tumor cell survival and proliferation [17,21,24,25,27]. Importantly, these mitochondrial effects occur independently of STAT3 DNA binding or transcriptional activity, challenging the paradigm of STAT3 as an exclusively nuclear transcription factor [17,21,60].
Although early models proposed that S727 phosphorylation is required for mitochondrial recruitment of STAT3 [60], subsequent evidence has challenged this view. Studies in primary mouse tissues demonstrate that loss of S727 phosphorylation does not abolish STAT3 mitochondrial localization [61]. Similar observations were reported in proliferating embryonic zebrafish stem cell niches and mouse stem cells, where Y705—but not S727—appeared to influence sub-mitochondrial localization [62], but S727 alone was instead required for transcriptional activation of mitochondrial genes and proliferation. Contrasting results describing reduced mitochondrial STAT3 levels in S727A mutants suggest that perhaps the contribution of S727 phosphorylation may be context-dependent and influenced by metabolic state or chaperone availability, including GRIM-19 [60]. Marié and colleagues demonstrated that STAT3 mitochondrial targeting is governed by both the amino-terminal domain (aa 1–132), well known to engage in protein-protein interactions [16], and the carboxyl-terminal α-helical region (aa 536–583) of the linker domain [61]. The former is required for effective mitochondrial localization but not sufficient to confer targeting on its own. In contrast, the latter contains an internal mitochondrial targeting sequence that is sufficient for translocation. However, the authors did not test the relevance of Y705 phosphorylation in this context.
Beyond the modulation of mitochondrial respiration and the transcriptional activation of mitochondrial genes [21,63], p-S727 STAT3 has been implicated in broader metabolic rewiring. Serine phosphorylation integrates signals from MAPK, mTOR, CDK, and stress-activated kinases, positioning STAT3 as a downstream effector of growth factor signaling, nutrient availability, and cellular stress responses [21,22,27,60,64]. Through these pathways, p-S727 STAT3 contributes to the coordination of glycolysis, glutamine metabolism, and mitochondrial biogenesis, thereby aligning metabolic programs with proliferative and survival pathways, both in physiological and pathological contexts [17,21,60]. Recent transcriptomic and functional studies further support the notion that p-S727 drives distinct, Y705-independent gene-expression programs associated not only with tumor aggressiveness but also with metabolic adaptation [65,66].
It is worth mentioning that some studies have questioned the extent, regulation, and relevance of mitochondrial STAT3 localization, highlighting technical limitations of mitochondrial fractionation leading to contamination by cytosolic or ER-associated membranes, and variability in antibody specificity for p-S727 detection [20,67]. Indeed, demonstration of the functional mechanisms explaining intramitochondrial STAT3 functions remains experimentally challenging, particularly given the low abundance of STAT3 relative to core mitochondrial proteins [20,67]. One potential source of artifacts with early studies demonstrating STAT3 mitochondrial activities is that they mostly relied on the over-expression of wild type and mutant STAT3 proteins artificially tagged to the mitochondrion thanks to a fusion with a mitochondrial signaling peptide at the N-terminus [17,68,69,70], whose functional outputs might be misleading. Recently, via rigorous subcellular fractionation and super-resolution imaging analyses Su and colleagues have shown that endogenous STAT3 is lowly represented in purified mitochondrial fractions while abundantly localized to the MAMs [20]. These findings raise the possibility that at least part of the phenotypes attributed to mitochondrial STAT3 may originate from functions exerted at the ER-mitochondria contact sites, where the protein is much more abundant rather than within the mitochondrial matrix.
Consistently, our group has identified STAT3 as an ER- and MAM-localized protein regulating calcium fluxes between the ER and mitochondria through interaction with the IP3R3 calcium channel and its degradation [19], thereby controlling mitochondrial Ca²⁺ uptake, permeability transition pore opening, and apoptotic sensitivity [19,71,72]. IP3R3 degradation and apoptosis protection, but not ER/MAM localization or interaction with IP3R3, require S727 phosphorylation [19]. These data support an integrative model where STAT3 acts as a versatile signaling hub whose non-nuclear activities extend beyond mitochondria to include ER and MAM domains [18,19,20] (Figure 2).
Alongside its ER–MAM functions, STAT3 was shown to localize to distinct cytoplasmic locations, including the lysosomal system. Early evidence for extranuclear STAT3 showed that Y705-independent variants shuttle in the cytoplasm and modulate stress responses without entering the nucleus [73]. More recently, lysosome-focused studies revealed that STAT3 dynamically associates to the cytosolic surface of LAMP1/LAMP2-positive lysosomes, where it binds the V-ATPase complex via its coiled-coil domain [74]. This positioning enables STAT3 to regulate proton gradients by stimulating V-ATPase activity, thereby maintaining an alkaline cytosolic pH and an acidic lysosomal lumen and promoting tumor cell homeostasis under metabolic stress. Pharmacological perturbations of lysosomes further support this view: cationic amphiphilic drugs that dissipate lysosomal acidity trigger cytosolic acidification, dephosphorylation of STAT3, and its redistribution toward lysosomes, thereby functionally inactivating STAT3 oncogenic signaling [75]. Finally, cytoplasmic STAT3 has also been shown to repress autophagy by interacting with PKR and inhibiting its eIF2α phosphorylating activity [73]. Interestingly, Y705 phosphorylation was not required for this activity, while S727 phosphorylation was not assessed.
Together, these findings extend the spatial map of non-canonical STAT3 beyond mitochondria and ER/MAMs to include a lysosome-related regulatory axis through which STAT3 integrates pH homeostasis, autophagy, and stress signaling within the cytoplasm. It is worth noting that also canonical transcriptional activation of STAT3 was shown to take part in lysosome regulation. Indeed, STAT3 induces the expression of cathepsins, achieving either cell adaptation to protein overload [76] or lysosome-mediated programmed cell death in the context of post-lactational involution of the mammary gland [76,77,78].
Further complicating the picture, several studies suggest that nuclear, cytosolic, ER-associated, and mitochondrial pools of STAT3, whether phosphorylated on S727 or not, can coexist and are subject to dynamic and sometimes reciprocal regulation [69,75,79]. In this frame, p-S727 acts as a molecular integrator of environmental cues rather than a fixed determinant of subcellular localization and function. It is moreover worth noting that at present the actual localization of S727 phosphorylation events has not been investigated. Determining whether phosphorylation is also localized at the specific compartments where p-S727 is physically detected and functionally characterized, or whether there is a cytoplasmic pool of p-S727 STAT3 that is then sorted to the different cellular compartments in response to specific cues will be instrumental to understand how its different functions are regulated and whether they interact to each other.
Collectively, these findings underline an ongoing debate regarding the mechanistic scope of p-S727 STAT3 in cancer biology. Rather than representing a binary nuclear-versus-mitochondrial switch, also in cancer S727 phosphorylation is increasingly recognized as a dynamic regulatory layer that modulates STAT3 functions across multiple cellular compartments, metabolic requirements and signaling axes, often synergizing with but sometimes opposing the canonical functions of nuclear STAT3.
4. Roles of STAT3 S727 Phosphorylation in Cancer
In recent years, STAT3 serine 727 phosphorylation has emerged as a clinically relevant signaling event in cancer, with multiple studies associating it with tumor grade, patient outcome, and metastatic progression. The main clinical findings are discussed below and summarized in Table 1.
The first evidence that STAT3 S727 phosphorylation can actively promote tumorigenesis independently of its Y705 counterpart emerged from the work of Qin and colleagues demonstrating a pro-oncogenic role of a phosphomimetic S727E mutant [29]. In their work, this mutation in combination with a Y705F mutation that abolishes Y phosphorylation conferred significant in vitro and in vivo growth advantage to human prostate cancer (PCa) LNCaP cells, potentially mediated by downstream activation of c-MYC, MCL-1, and survivin. Moreover, the Y705F/S727E mutant—but not a Y705F/S727A mutant where also S727 phosphorylation is abolished—induced anchorage-independent growth in non-tumorigenic prostate epithelial RWPE-1 cells, further supporting a pro-tumorigenic role for STAT3 S727 phosphorylation [29]. Accordingly, a positive correlation between the levels of p-S727 STAT3 and Gleason score was observed in prostate cancer tissues [29]. The finding that S727, but not Y705, phosphorylation is required for Ras-mediated tumor transformation reported by Gough et al [21] is in line with these observations.
More recently, high levels of cytoplasmic p-S727 STAT3 in the PCa stroma were identified as an independent prognostic marker associated with reduced OS and 5-year metastasis-free survival (MFS) following androgen deprivation therapy (ADT) in a cohort of 111 patients with advanced PCa [80]. Consistently, two independent studies reported that elevated p-S727 STAT3 was associated with higher Gleason scores [81,82]. Moreover, Thaper et al demonstrated that STAT3 inhibition enhanced the sensitivity of PCa LNCaP cells to the androgen receptor (AR) inhibitor enzalutamide (ENZ) and that cell lines derived from ENZ-resistant tumors displayed up-regulation of p-S727, but not of pY705-STAT3, correlating with enhanced interactions with the AR and consequent increase of its activity [83]. All together, these findings suggest a role for p-S727 STAT3 in acquired therapeutic resistance and metastatic progression of PCa.
STAT3 S727 phosphorylation has also been linked to disease progression in gynecological malignancies. Indeed, in cervical intraepithelial neoplasia (CIN), p-S727 STAT3 levels correlated with lesion grade and expression of the proliferation marker Ki-67 in 56 untreated patients [84]. Similarly, p-S727 STAT3 was highly expressed in seven advanced-stage human endometrial tumor samples, whereas Y705 phosphorylation was undetectable [85]. Moreover, several human endometrial cancer (EC) cell lines exhibited strong cytosolic p-S727 STAT3 expression in the absence of Y705 phosphorylation, suggesting a distinctive role for S727 phosphorylation in EC development [85].
A comparable phosphorylation pattern has been reported in melanoma. STAT3 S727 was found to be constitutively phosphorylated in seven out of seven human melanoma cell lines analyzed, downstream of the bRaf–MEK–ERK1/2 pathway, while p-Y705 STAT3 levels were much more variable [86]. Moreover, p-S727—but not p-Y705—STAT3 was present in five out of nine in situ lesions of acral lentiginous melanoma (ALM) patients, whereas both phosphorylation events were detected in dermally invasive ALM lesions [86].
A Y705-independent prognostic value for STAT3 S727 phosphorylation was also demonstrated in clear cell renal cell carcinoma (ccRCC). In a cohort of 98 patients with 100-month follow-up, high p-S727 STAT3 levels correlated with higher Fuhrman grade and poorer OS [87]. Consistently, in an independent cohort of 82 ccRCC patients followed for 10 years after nephrectomy, elevated p-S727 STAT3 levels—both nuclear (p=0.002) and cytosolic (p=0.040)—were associated with reduced OS [88].
In lung cancer (LC), immunohistochemical (IHC) analysis of 40 primary tumor specimens revealed that high p-S727 STAT3 levels, together with elevated leukemia inhibitory factor receptor (LIFR) expression, significantly correlated with higher tumor grade and reduced OS [89]. In the same study, STAT3 S727 and Y705 phosphorylation were shown to differentially regulate EMT and mesenchymal-epithelial transition (MET) programs, with p-Y705 STAT3 enriched in primary tumors and p-S727 STAT3 predominant in metastatic regions [89]. In this context, the LIFR/p-ERK/p-S727-STAT3 signaling axis drove MET, whereas EMT was associated with IL6R/p-Y705 STAT3 signaling in bone marrow-derived mesenchymal stem cells (BM-MSCs). Importantly, these effects were validated in vivo using a bone homing assay with human lung adenocarcinoma A549 cells, and abolished upon STAT3 inactivation [89].
Finally, in glioblastoma (GBM), STAT3 S727 phosphorylation was detected in 100% and 70% of tumors from a cohort of 30 and 88 patients, respectively [90,91], being associated with poor OS in both univariate and multivariate analyses [91]. Patients exhibiting high levels of both p-S727 and p-Y705 STAT3 experienced the worst clinical outcomes, suggesting potential synergistic effects [91]. Interestingly, in temozolomide (TMZ)-resistant U373 and U251 glioma cells, p-S727 STAT3 levels were increased, whereas p-Y705 STAT3 levels were reduced. STAT3 silencing restored TMZ sensitivity, supporting p-S727 STAT3 as a promising therapeutic target in TMZ-resistant gliomas [92]. Consistently, STAT3 S727 phosphorylation correlated with intrinsic radioresistance across 15 GBM cell lines, and treatment with the multi-kinase inhibitor Gö6976 restored radiosensitivity at least in part through selective down-modulation of p-S727 STAT3 in Y705-negative or weakly positive GBM cells, highlighting S727 phosphorylation as a potential therapeutic target in this context [90].
In conclusion, while most of the clinical studies were correlative in nature, their convergence with functional genetic and pharmacological data supports a casual contribution of p-S727 STAT3 to aggressive tumor phenotypes.
5. p-S727 STAT3 Activities in BC and TNBC
Although as described above p-S727 is likely to exert pro-tumoral action in multiple malignancies, it appears to be particularly enriched and functionally relevant in TNBC, which we therefore dissect in better detail in this section.
The clinical relevance of STAT3 S727 phosphorylation in BC remained largely unexplored until Yeh and colleagues reported that high nuclear and cytoplasmic p-S727 STAT3 levels—detected in 62% of infiltrating ductal carcinoma samples compared with adjacent non-tumoral tissues—significantly correlated with estrogen receptor (ER)-negative status, larger tumor size, and advanced disease stage [38]. This inverse correlation was further validated in vitro using a panel of BC cell lines, and substantiated by the observation that knocking down ERα in ER-positive MCF7 cells increased p-S727 STAT3 levels [38].
In contrast, a positive correlation between nuclear p-S727 STAT3 and progesterone receptor (PR) expression was reported in a cohort of 39 infiltrating ductal breast carcinomas (p=0.027) [25]. Mechanistically, the synthetic progestin medroxyprogesterone acetate (MPA) induced STAT3 S727 phosphorylation by activating the p42/p44 MAPK signaling pathway and promoted its nuclear localization. This event was required for STAT3 binding to the cyclin D1 promoter in vivo, thereby stimulating BC cell proliferation [25]. Notably, MPA-induced growth was abrogated by expression of the STAT3 S727A mutant in both murine progestin-dependent C4HD cells and human luminal A T-47D BC cells, resulting in G1 cell-cycle arrest [25].
A recent study analyzing 173 TNBC patient samples demonstrated that STAT3 S727 and Y705 phosphorylation occur independently in 34% of cases, implying distinct functional roles in breast tumor biology [40]. The aggressive basal-like tumors were characterized by high p-S727 and low p-Y705 STAT3 levels, whereas dual ERβ and AR positivity correlated with low p-S727 and high p-Y705 STAT3 expression [40]. Interestingly, both STAT3 phosphorylation events were associated with lower tumor size and clinical stage but not with improved or reduced survival, suggesting that high STAT3 S727/Y705 phosphorylation can skew the good prognosis conferred by clinicopathological characteristics [40], possibly due to the ability of STAT3 to induce drug resistance [93].
STAT3 S727 phosphorylation has recently been linked to STAT3 activation and dimerization in a Y705 independent manner, as demonstrated using a bioluminescence resonance energy transfer (BRET) biosensor in luminal A MCF7 and TNBC MDA-MB-231 cells [41], consistent with previous observations in chronic lymphocytic leukemia (CLL) [94]. Accordingly, MCF7 cells expressing the STAT3 S727A mutant displayed significantly reduced proliferation and clonogenic capacity compared with cells expressing the Y705F mutant [41]. In line with these findings, a retrospective IHC analysis of 76 TNBC patient samples revealed that over 92% of STAT3-positive tumors exhibited constitutive S727 phosphorylation, whereas Y705 phosphorylation was detected in only 15% of cases [41]. In contrast, STAT3 expression—and phosphorylation—was absent or weakly cytoplasmic in HER2-positive and luminal BC subtypes, supporting a subtype-specific role of STAT3 and its S727 phosphorylated form in TNBC [41].
Mitochondria-localized STAT3 S727 phosphorylation was shown to promote TNBC growth and metastasis in vivo in a Y705-independent manner using the murine 4T1 model [24]. 4T1 cells expressing a mitochondrially tagged MLS-STAT3 S727A mutant formed significantly fewer colonies in soft agar, whereas expression of the STAT3 S727D phosphomimetic mutant markedly increased anchorage-independent growth. In vivo, tumors derived from cells expressing MLS-STAT3 S727A/Y705F were smaller and exhibited reduced liver and lung metastasis compared with those expressing MLS-STAT3 S727D/Y705F. Importantly, inactivation of STAT3 transcriptional functions through Y705F mutation, SH2 disruption, or deletion of the DNA-binding domain did not affect tumor growth, supporting a transcription-independent role for mitochondrial STAT3 in TNBC [24]. Accordingly, a construct lacking the nuclear localization sequence enhanced mitochondrial STAT3–driven invasion in Matrigel assays, confirming that nuclear activities are not involved and suggesting reciprocal regulation between the nuclear and mitochondrial forms. Mechanistically, p-S727 STAT3 improved mitochondrial complex I coupling, thereby reducing ROS production. These effects were observed under hypoxic but not normoxic conditions, supporting a model in which mitochondrial phosphorylated STAT3 S727 functions as a rheostat to fine-tune ROS levels to promote TNBC cell survival and metastatic fitness rather than apoptosis [24]. Despite not experimentally proven, the observation that mitochondrial p-S727 STAT3 is required for the proliferation of stem cells [62] might extend this feature to de-differentiated cancer cells.
STAT3 S727 phosphorylation has also emerged as a critical determinant for life/death decisions in BC cells upon silencing of the lactate dehydrogenase C (LDHC) gene [95], which in turn was previously associated with poor OS in basal-like and HER2-enriched breast tumors [96]. Indeed, LDHC knockdown markedly impaired the survival of basal-like MDA-MB-468 and BT-549 TNBC cells through p-S727 STAT3 downregulation, inducing DNA damage accumulation, microtubule destabilization, and cell-cycle dysregulation through p-S727 STAT3 downregulation. In contrast, LDHC silencing in HER2-enriched HCC-1954 cells increased p-S727 STAT3 levels and conferred resistance to cell death, which was reversed by STAT3 silencing [95], highlighting how regulatory networks are determined by biological conditions. p-S727 STAT3 was also shown to form a transcriptional complex with BCL9 at enhancer regions of target genes, driving invasive progression in HER2-enriched SUM225 and DCIS.COM BC cell lines [97].
Furthermore, STAT3 phosphorylation downstream of the AKT-mTOR signaling pathway was implicated in the activation of the CC motif chemokine receptor 1 (CCR1) promoter following epidermal EGF stimulation, thereby promoting invasion and metastasis in MDA-MB-231 TNBC cells [98]. In turn, CCR1 silencing reduced invasive sprouting in 3D spheroid cultures in vitro and diminished MDA-MB-231 lung metastatic colonization in vivo [98].
Finally, STAT3 S727 phosphorylation was found to be upregulated by approximately 1.8-fold in the side population of luminal A MCF7 cells—a rare subpopulation enriched in cancer stem-like cells with enhanced clonogenic capacity in vitro and increased tumorigenicity in vivo—further supporting a role for p-S727 STAT3 in breast cancer aggressiveness [26].
6. Targeting Mitochondria-Associated STAT3 S727 Signaling
Substantial efforts have been devoted to pharmacologically target mitochondrial STAT3, leading to the development of multiple small molecules and experimental strategies aimed at disrupting its non-canonical functions.
The first of such agents was phospho-valproic acid (P-V; MDC-1112), which inhibited the growth of human pancreatic cancer xenografts both by inhibiting STAT3 Y705 phosphorylation by JAK and Src kinases and by reducing total and S727-phosphorylated STAT3 levels in mitochondria, leading to increased mitochondrial ROS accumulation and induction of apoptosis. [99]. Reduced STAT3 in mitochondria could be due to P-V-mediated impairment of STAT3 interactions with HSP90 and TOM20, the latter involved in STAT3 mitochondrial translocation in addition to GRIM-19 [100]. Importantly, inhibition of mitochondrial STAT3 was required for P-V antitumor activity, as overexpression of mitochondrially targeted STAT3 rescued tumor growth in vivo. Although P-V reduced IL-6–induced STAT3 Y705 phosphorylation in BxPC-3 cells, it failed to suppress the growth of tumors overexpressing either wild-type or Y705F STAT3, indirectly suggesting that its antitumor efficacy might depend on targeting mitochondrial STAT3 S727 phosphorylation rather than canonical Y705-mediated signaling [99].
Another strategy to target mitochondria-localized STAT3 involved conjugating the mitochondria-targeting fluorophore N,N-diethyl-7-aminocoumarin to a benzo[b]thiophene 1,1-dioxide (BTP) moiety, generating compound 7a [101]. This compound co-localized with the mitochondrial marker MitoTracker in MDA-MB-231 TNBC and luminal A MCF7 cells, although partial lysosomal localization was also observed. Notably, compound 7a dose-dependently inhibited both STAT3 S727 and Y705 phosphorylation in MDA-MB-231 cells without affecting upstream STAT3 regulators such as p-JAK2, p-Src, or p-ERK1/2 [101]. 7a suppressed in vitro MDA-MB-231 cell viability, mediated by downregulation of STAT3 target genes such as BCL-2 and cyclin D1, increased ROS production and activated the mitochondrial apoptotic pathway. Antitumor activity and inhibition of p-Y705 STAT3 were further confirmed in syngeneic 4T1 tumors in vivo [101].
Mitochondrial STAT3 has also been implicated in the toxicity and antitumor activity of the STAT3 inhibitor OPB-51602 [102], which was previously shown to inhibit the growth of DU145 PCa cancer cells by inducing mitochondrial dysfunction and proteotoxic aggregates formation through SH2-domain binding and dual inhibition of STAT3 phosphorylation on both S727 and Y705 [103]. The related compound OPB-31121 displayed 100–1000-fold higher potency than other STAT3 inhibitors (i.e. Cryptotanshinone and S3I.201) in suppressing S727 and Y705 phosphorylation and proliferation of DU145 and LNCaP PCa cells, likely due to its ability to occupy a broader and distinct hydrophobic region of the STAT3 SH2 domain [104]. However, OPB-31121 failed in a phase 1 clinical trial in subjects with advanced solid tumors due to adverse effects and therapeutic resistance [58,105]. Brambilla and colleagues later demonstrated that OPB-51602 inhibits mitochondrial complex I, leading to ROS accumulation, mitophagy, cytoskeletal remodeling, and cell death in A549 non-small cell lung carcinoma (NSCLC) and in MDA-MB-468 and -231 TNBC cells [102]. However, partial residual sensitivity to the compound in STAT3 knockout cells and technical limitations in isolating pure mitochondrial fractions suggest that OPB-51602 effects may not be exclusively mediated by mitochondrial STAT3.
More recently, a potentially more selective strategy to target STAT3 S727 phosphorylation was proposed by Wan and colleagues using Sculponeatin A (sptA) [106]. STAT3 was identified among the top 10 predicted targets of this compound, which indeed selectively reduced S727—but not Y705—phosphorylation in whole-cell lysates and purified mitochondrial fractions of H460, A549 and PC9 NSCLC cells, also confirmed by immunofluorescence analysis [106]. SptA preferentially inhibited proliferation of NSCLC cells over normal lung epithelial cells and reduced tumor growth in zebrafish and mouse A549 xenograft models, accompanied by decreased p-S727 and total STAT3 levels [106]. Mechanistically, sptA promoted proteasomal degradation of mitochondrial STAT3 via WWP2-mediated ubiquitination, a process supported by molecular dynamics simulations showing enhanced STAT3–WWP2 binding. Overexpression of mitochondrially targeted STAT3 antagonized sptA-induced mitochondrial dysfunction and rescued cell viability, confirming mitochondrial STAT3 as a critical target of sptA. Although sptA showed no overt hepatotoxicity, cardiotoxic effects were observed at higher doses in zebrafish models [106]. Notably, sptA was also reported to induce ferroptosis in breast cancer cells, suggesting broader therapeutic potential beyond NSCLC [107].
Despite the paucity of specific inhibitors, these studies confirm an important role of mitochondrial pS727 STAT3 in driving cancer, supporting efforts to develop specific, effective inhibitors.
7. Targeting STAT3 S727 Phosphorylation in TNBC
Several STAT3 inhibitors have shown efficacy in TNBC models in vitro and in vivo [32,56,58]. In this section, we specifically focus on therapeutic strategies that modulate STAT3 S727 phosphorylation in TNBC, with the main results summarized in Table 2. Of note, although compound 7a and OPB-51602 were discussed above in the context of mitochondrial p-S727 STAT3 targeting, both agents were also evaluated in TNBC models and are therefore included in Table 2 as compounds interfering with STAT3 S727 signaling in TNBC [101,102]. While the present section focuses on the effects of S727 inhibition in TNBC, for a broader overview of STAT3 inhibitors—mainly directed against Y705 phosphorylation—we refer readers to the recent comprehensive review by Berkley and colleagues [58].
The rationale of targeting STAT3 S727 phosphorylation in TNBC was initially established by Dimri and colleagues [41], who demonstrated that the anti-helminth drug compound Niclosamide (NSA) significantly reduced primary tumor growth and distant metastasis in an orthotopic xenograft model of MDA-MB-231 TNBC cells. Mechanistically, NSA was shown to prevent STAT3 dimer formation in a S727-dependent way in live cells while displaying inhibitory activity on both STAT3 phosphorylation events. In contrast, the widely used STAT3 inhibitor Stattic, an SH2-binding compound that inhibits both Y705 phosphorylation and dimerization, preferentially inhibited Y705 phosphorylation and failed to achieve comparable effects [41]. However, niclosamide is known to target multiple signaling pathways—including Wnt/β-catenin, mTORC1, NF-κB, and Notch—which raises the possibility of off-target effects contributing to STAT3 dimer disruption and tumor suppression and disqualify it as a clinically suitable compound [108].
The benzothiazole-based compound B19 has been reported to simultaneously inhibit STAT3 S727 and Y705 phosphorylation, thereby suppressing STAT3-dependent transcription of c-MYC and MCL-1, inducing G2/M cell-cycle arrest and apoptosis in MDA-MB-468 TNBC and HEL erythroleukemia cells [109]. Molecular docking analyses, along with surface plasmon resonance (SPR) and fluorescence polarization (FP) assays, revealed that like Stattic B19 binds the STAT3 SH2 domain, at residues Leu706–Phe710. It is moreover poorly soluble, and its oral viability would require considerable optimization to improve pharmacokinetic properties and in vivo stability [109].
A synthetic derivative of the SH2-domain STAT3 inhibitor SLSI-1, named SLSI-1216, was shown to dose-dependently inhibit both STAT3 S727 and Y705 phosphorylation in MDA-MB-231 TNBC cells [36]. SLSI-1216 effectively suppressed the in vitro growth of MDA-MB-231 and HCC38 TNBC cells, while displaying an approximately 10-fold higher IC₅₀ in non-tumorigenic human mammary epithelial MCF-10A cells [36]. Moreover, SLSI-1216 exhibited antitumor efficacy comparable to paclitaxel in an MDA-MB-231 xenograft model, where IHC analysis confirmed p-Y705 STAT3 downregulation in treated tumors; STAT3 S727 phosphorylation was not assessed in this study [36].
SMY002, a naphthalene-based compound derived from selective estrogen receptor modulator (SERM) scaffolds, demonstrated potent inhibition of primary tumor growth and lung metastasis formation in a syngeneic 4T1 TNBC mouse model by suppressing STAT3 phosphorylation, dimerization, nuclear localization, and transcriptional activity [110]. Notably, SMY002 exhibited selective cytotoxicity toward mouse 4T1 and human MDA-MB-468 and -231 TNBC cells, while sparing normal human mammary epithelial MCF-10A cells [110]. However, it remains unclear whether SMY002-mediated inhibition of STAT3 phosphorylation was specifically assessed at Y705, as the antibody described in this paper targets p-S727 rather than p-Y705, raising some doubts about the author’s conclusions that antitumor activities are due to inhibition of Y705-dependent STAT3 activation.
A SMY002 analog with improved STAT3 SH2-domain affinity, RDp002, was subsequently developed and shown to inhibit TNBC cell proliferation, survival, invasion, and migration by inducing G0/G1 cell-cycle arrest and lysosome-dependent cell death through dual suppression of STAT3 S727 and Y705 phosphorylation [111]. RDp002 exhibited minimal cytotoxicity in normal mammary epithelial MCF-10A cells and human kidney epithelial HEK-293T cells, while also demonstrating activity in multiple myeloma (MM) cell lines, including U266 and MM.1S [111]. Importantly, RDp002 induced significant tumor regression in MDA-MB-468 xenograft models, with effective inhibition of both STAT3 phosphorylation events confirmed by IHC and Western blot analysis [111]. This compound also impaired lung dissemination of the TNBC 4T1 cells in a syngeneic mouse model.
More recently, Chen and colleagues developed YY002, a highly selective SH2-domain STAT3 inhibitor capable of simultaneously suppressing STAT3 S727 and Y705 phosphorylation at nanomolar concentrations without affecting the activity of known upstream STAT3 kinases, including ERK1, JNK1, SRC, p38δ, EGFR, JAK1, and JAK2 [112]. Mechanistically, YY002 inhibited both nuclear p-Y705 STAT3 signaling and mitochondrial p-S727 STAT3 functions, resulting in pronounced cell death in MDA-MB-231 and MDA-MB-468 TNBC cells. YY002 also exhibited superior antitumor efficacy compared with the orally available STAT3 inhibitor napabucasin (BBI608) in an MDA-MB-231 xenograft model. The latter was in turn previously shown to attenuate drug resistance of the ER+ MCF7 cells via down-regulating p-S727 and p-Y705 STAT3 levels [112,113]. Similar antitumor and anti-metastatic effects were observed in an orthotopic pancreatic cancer xenograft model, where YY002 reduced tumor growth and metastasis in a dose-dependent manner and significantly prolonged survival [112]. Moreover, two novel orally bioavailable dual STAT3 phosphorylation inhibitors directed against the SH2-domain were recently proven to be effective in preventing tumor growth of gastric and pancreatic cancer xenograft models through the inhibition of both STAT3 nuclear- and mitochondrial-related pro-tumorigenic functions [114,115].
Finally, Zheng and colleagues demonstrated that dual inhibition of STAT3 S727 and Y705 phosphorylation using the saponin pulchinenoside E2 (PSE2) in MDA-MB-231 and HS-578T TNBC cells significantly suppressed lung and liver metastases in a xenograft model [116]. PSE2 displayed greater anti-metastatic efficacy than Stattic, as evidenced by a reduced number of metastatic nodules and more potent inhibition of both p-S727 and p-Y705 STAT3 in metastatic lesions, as assessed by IHC [116]. Importantly, the anti-metastatic effects of PSE2 were restricted to the TNBC subtype, as no significant activity was observed in luminal A MCF-7 and T47D BC cells or in normal MCF-10A cells, whereas migration and invasion were markedly suppressed in multiple TNBC cell lines, including MDA-MB-231 and -468, HS-578T, and BT-549 [116].
Overall, the data reviewed identify STAT3 S727 phosphorylation as a functionally distinct and therapeutically relevant vulnerability in TNBC, whether linked to mitochondrial functions or not. Unlike Y705 phosphorylation, S727 is frequently enriched and constitutively active in the TNBC subtype, where it drives STAT3 dimerization, transcriptional activity, mitochondrial functions, and metastatic progression. Targeting STAT3 S727 phosphorylation—in combination with Y705 inhibition—consistently suppresses TNBC growth and dissemination with limited effects on other breast cancer subtypes, supporting S727 as a TNBC-selective therapeutic target.
8. Conclusions
STAT3 is a well-established oncogene whose pro-tumorigenic activity has classically been attributed to Y705-dependent transcriptional signaling [42]. However, accumulating experimental and clinical evidence now indicates that serine 727 phosphorylation represents a functionally independent regulatory axis that contributes to STAT3-driven tumor progression [65,66]. Elevated p-S727 STAT3 levels have been associated with aggressive disease features, metastatic dissemination, and poor clinical outcome in multiple cancer types, including PCa, ccRCC, and TNBC, often uncoupled from Y705 phosphorylation [29,41,87,88]. These findings highlight S727 phosphorylation as a critical determinant of non-canonical STAT3 functions that support tumor cell survival, metabolic adaptation, and invasiveness in TNBC [19,101,102]. Importantly, recent preclinical studies demonstrate that targeting p-S727 STAT3 can effectively suppress tumor growth and metastasis in NSCLC and TNBC models, positioning this form as a promising therapeutic vulnerability [36,41,101,106,111,112,116].
While Berkley and colleagues raised the possibility that inhibition of p-S727 STAT3 may contribute to treatment-related toxicity [58], the evidence cited derives from a dual STAT3 inhibitor (OPB-31121) and does not clearly isolate S727 inhibition as the causative factor. Indeed, toxicity observed in early-phase clinical studies is frequently influenced by multiple variables, including disease context, tumor burden, prior treatments, pharmacokinetics, and off-target effects. As such, currently available clinical data does not allow definitive attribution of toxicity specifically to STAT3 p-S727 inhibition. Indeed, as shown in Table 2, all compounds tested to date largely behave as dual inhibitors of both p-Y705 and p-S727 STAT3, and most are known to engage the SH2 domain, which is required for Y705 phosphorylation, dimerization, and canonical transcriptional activity [1,2,3,4,56,57,58]. Because the SH2 domain has not been directly implicated in mediating STAT3 p-S727–dependent extranuclear functions, reductions in p-S727 levels observed with SH2-directed agents are perhaps to be interpreted as indirect, potentially arising from upstream signaling rewiring, altered feedback loops, changes in phosphatase/kinase balance, or global effects on STAT3 stability and turnover. In addition, the compounds discussed here are heterogeneous in their dominant mode of action: some primarily reduce total STAT3 abundance (including mitochondrial pools), others chiefly impair mitochondrial function and/or subcellular distribution, and others mainly decrease the detectable p-S727 signal without clearly establishing direct engagement of the S727 regulatory axis. Therefore, to truly dissect the specific functions, therapeutic potential, and toxicity attributable to p-S727 STAT3, new compounds enabling selective and mechanistically validated modulation of p-S727 levels/activities are required.
Finally, while canonical and non-canonical STAT3 functions are mediated by different phosphorylated forms located at various sub-cellular compartments, in most instances both p-Y705 and p-S727 are known to contribute to tumor transformation and progression [3,12,15,21,24,25,29,59,65,89]. Because all alternative STAT3 species derive from a single pool of total STAT3, their balance is likely to represent a critical element of reciprocal cross-regulation (Figure 2), as also highlighted in selected publications [69,75,79]. Although still largely inferential, this concept of competition for a shared protein pool provides a useful framework to interpret context-dependent STAT3 outputs.
As a concluding remark, we strongly believe that integrating p-S727 and p-Y705 STAT3 distribution and activity into biomarker-driven patient stratification and coupling it with phosphorylation-specific inhibitors may enable more effective and better-tolerated STAT3-targeted therapeutic strategies.
Author Contributions
D.V., A.R.M. and N.S. wrote the original draft and prepared the figures. D.V. and V.P. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Italian Cancer Research Association (AIRC IG24851 to V.P.); the Italian Ministry of University and Research (MUR PRIN 2022BFF2CJ to V.P.), National Center for Gene Therapy and Drugs based on RNA Technology, Spoke2, PNRR M4C2-Investimento 1.4-CN00000041-VP, and the Truus and Gerrit van Riemsdijk Foundation, Liechtenstein, donation to V.P. D.V. was supported by an Italian Cancer Research Foundation (FIRC) post-doctoral fellowship (Project No 31186).
Data Availability Statement
No new data was created or analyzed thus study. Data sharing is not applicable.
Acknowledgments
Regarding technical assistance, figures were created with Biorender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADT ALM AR BC BM-MSC BRET BTP CCR1 CCRCC CIN CLL CSC EC EGFR EMT ENZ ER ER ETC FP GAS GBM GPCR HAT HER2 HIF-1α IHC IL-6 LC LDHC LIFR MAM MET MFS MLS MM MMP MMTV MPA NSA NSCLC OS PCa PIAS3 PR PSE2 PTM PTP PV RFS ROS S727 S727A SERM SPTA SOCS SPR STAT3 STAT3-C TKI TMZ TNBC VEGF |
Androgen Deprivation Therapy Acral Lentiginous Melanoma Androgen Receptor Breast Cancer Bone Marrow-derived Mesenchymal Stem Cell Bioluminescence Resonance Energy Transfer Benzothiophene CC motif Chemokine Receptor 1 Clear Cell Renal Cell Carcinoma Cervical Intraepithelial Neoplasia Chronic Lymphocytic Leukemia Cancer Stem Cells Endometrial Cancer Epidermal Growth Factor Receptor Epithelial Mesenchymal Transition Enzalutamide Estrogen Receptor Endoplasmic Reticulum Electron Transport Chain Fluorescence Polarization Γ-Activated Sequence Glioblastoma G Protein-Coupled Receptor Histone Acetyltransferase Human Estrogen Receptor 2 Hypoxia-inducible factor 1α Immunohistochemistry Interleukin 6 Lung Cancer Lactate Dehydrogenase C Leukemia Inhibitory Factor Receptor Mitochondria-Associated Membranes Mesenchymal Epithelial Transition Metastasis-Free Survival Mitochondria Localization Sequence Multiple Myeloma Matrix Metalloproteinases Mouse Mammary Tumor Virus Medroxy Progesterone Acetate Niclosamide Non-Small Cell Lung Carcinoma Overall Survival Prostate Cancer Protein Inhibitor of Activated STAT 3 Progesterone Receptor Pulchinenoside E2 Post Translational Modification Protein Tyrosine Phosphatases Phospho-Valproic acid Relapse-Free Survival Reactive Oxygen Species Serine 727 Serine 727 Alanine Selective Estrogen Receptor Modulator Sculponeatin A Suppressors Of Cytokines Signaling Surface Plasmon Resonance Signal Transducer and Activator of Transcription 3 Constitutively active Signal Transducer and Activator of Transcription 3 Tyrosine Kinase Inhibitor Temozolomide Triple-Negative Breast Cancer Vascular Endothelial Growth Factor |
| Y705 Y705F |
Tyrosine 705 Tyrosine 705 Phenylalanine |
References
- Yu, H; Pardoll, D; Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Darnell, JE, Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Bromberg, JF; Wrzeszczynska, MH; Devgan, G; Zhao, Y; Pestell, RG; Albanese, C; et al. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Schindler, C; Levy, DE; Decker, T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Krebs, DL; Hilton, DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Xu, D; Qu, CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Levy, DE; Darnell, JE, Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Avalle, L; Camporeale, A; Camperi, A; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M; Cascio, A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Levy, DE; Inghirami, G. STAT3: a multifaceted oncogene. Proc Natl Acad Sci U S A 2006, 103, 10151–10152. [Google Scholar] [CrossRef]
- Demaria, M; Camporeale, A; Poli, V. STAT3 and metabolism: how many ways to use a single molecule? Int J Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M; Misale, S; Giorgi, C; Miano, V; Camporeale, A; Campisi, J; et al. STAT3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ 2012, 19, 1390–1397. [Google Scholar] [CrossRef]
- Gough, DJ; Marie, IJ; Lobry, C; Aifantis, I; Levy, DE. STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood 2014, 124, 2252–2261. [Google Scholar] [CrossRef]
- Schuringa, JJ; Jonk, LJ; Dokter, WH; Vellenga, E; Kruijer, W. Interleukin-6-induced STAT3 transactivation and Ser727 phosphorylation involves Vav, Rac-1 and the kinase SEK-1/MKK-4 as signal transduction components. Biochem J 2000, 347 Pt 1, 89–96. [Google Scholar] [CrossRef]
- Alhayyani, S; McLeod, L; West, AC; Balic, JJ; Hodges, C; Yu, L; et al. Oncogenic dependency on STAT3 serine phosphorylation in KRAS mutant lung cancer. Oncogene 2022, 41, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y; Dong, Z; Liu, K. Unraveling the complexity of STAT3 in cancer: molecular understanding and drug discovery. J Exp Clin Cancer Res 2024, 43, 23. [Google Scholar] [CrossRef]
- Wegrzyn, J; Potla, R; Chwae, YJ; Sepuri, NB; Zhang, Q; Koeck, T; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Avalle, L; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Avalle, L; Camporeale, A; Morciano, G; Caroccia, N; Ghetti, E; Orecchia, V; et al. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ 2019, 26, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Su, Y; Huang, X; Huang, Z; Huang, T; Xu, Y; Yi, C. STAT3 Localizes in Mitochondria-Associated ER Membranes Instead of in Mitochondria. Front Cell Dev Biol 2020, 8, 274. [Google Scholar] [CrossRef]
- Gough, DJ; Corlett, A; Schlessinger, K; Wegrzyn, J; Larner, AC; Levy, DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Decker, T; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef]
- Shen, Y; Schlessinger, K; Zhu, X; Meffre, E; Quimby, F; Levy, DE; et al. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol 2004, 24, 407–419. [Google Scholar] [CrossRef]
- Zhang, Q; Raje, V; Yakovlev, VA; Yacoub, A; Szczepanek, K; Meier, J; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Tkach, M; Rosemblit, C; Rivas, MA; Proietti, CJ; Diaz Flaque, MC; Mercogliano, MF; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr Relat Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef]
- Zhou, J; Wulfkuhle, J; Zhang, H; Gu, P; Yang, Y; Deng, J; et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Chung, J; Uchida, E; Grammer, TC; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 1997, 17, 6508–6516. [Google Scholar] [CrossRef]
- Tesoriere, A; Dinarello, A; Argenton, F. The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Qin, HR; Kim, HJ; Kim, JY; Hurt, EM; Klarmann, GJ; Kawasaki, BT; et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res 2008, 68, 7736–7741. [Google Scholar] [CrossRef] [PubMed]
- Chen, L; Chen, D; Li, J; He, L; Chen, T; Song, D; et al. Ciclopirox drives growth arrest and autophagic cell death through STAT3 in gastric cancer cells. Cell Death Dis 2022, 13, 1007. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I; Pensa, S; Pannellini, T; Quaglino, E; Maritano, D; Demaria, M; et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res 2010, 70, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Qin, JJ; Yan, L; Zhang, J; Zhang, WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res 2019, 38, 195. [Google Scholar] [CrossRef] [PubMed]
- Bai, X; Ni, J; Beretov, J; Graham, P; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles' heel? Cancer Lett 2021, 497, 100–111. [Google Scholar] [CrossRef]
- Zagami, P; Carey, LA. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef]
- Zhong, Y; Deng, L; Shi, S; Huang, QY; Ou-Yang, SM; Mo, JS; et al. The novel STAT3 inhibitor WZ-2-033 causes regression of human triple-negative breast cancer and gastric cancer xenografts. Acta Pharmacol Sin 2022, 43, 1013–1023. [Google Scholar] [CrossRef]
- Park, SK; Byun, WS; Lee, S; Han, YT; Jeong, YS; Jang, K; et al. A novel small molecule STAT3 inhibitor SLSI-1216 suppresses proliferation and tumor growth of triple-negative breast cancer cells through apoptotic induction. Biochem Pharmacol 2020, 178, 114053. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y; Wang, Q; Tang, CH; Chen, HD; Hu, GN; Shao, JK; et al. p-STAT3 expression in breast cancer correlates negatively with tumor size and HER2 status. Medicine (Baltimore) 2021, 100, e25124. [Google Scholar] [CrossRef]
- Yeh, YT; Ou-Yang, F; Chen, IF; Yang, SF; Wang, YY; Chuang, HY; et al. STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. Int J Cancer 2006, 118, 2943–2947. [Google Scholar] [CrossRef]
- Radenkovic, S; Konjevic, G; Gavrilovic, D; Stojanovic-Rundic, S; Plesinac-Karapandzic, V; Stevanovic, P; et al. pSTAT3 expression associated with survival and mammographic density of breast cancer patients. Pathol Res Pract 2019, 215, 366–372. [Google Scholar] [CrossRef]
- Stenckova, M; Nenutil, R; Vojtesek, B; Coates, PJ. Stat3 Tyrosine 705 and Serine 727 Phosphorylation Associate With Clinicopathological Characteristics and Distinct Tumor Cell Phenotypes in Triple-Negative Breast Cancer. Pathol Oncol Res 2022, 28, 1610592. [Google Scholar] [CrossRef]
- Dimri, S; Malhotra, R; Shet, T; Mokal, S; Gupta, S; De, A. Noncanonical pS727 post translational modification dictates major STAT3 activation and downstream functions in breast cancer. Exp Cell Res 2020, 396, 112313. [Google Scholar] [CrossRef]
- Bromberg, JF; Horvath, CM; Besser, D; Lathem, WW; Darnell, JE, Jr. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol 1998, 18, 2553–2558. [Google Scholar] [CrossRef]
- Avalle, L; Pensa, S; Regis, G; Novelli, F; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. Jakstat 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Yu, H; Lee, H; Herrmann, A; Buettner, R; Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Demaria, M; Giorgi, C; Lebiedzinska, M; Esposito, G; D'Angeli, L; Bartoli, A; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010, 2, 823–842. [Google Scholar] [CrossRef]
- Hughes, K; Watson, CJ. The Multifaceted Role of STAT3 in Mammary Gland Involution and Breast Cancer. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Yang, PL; Liu, LX; Li, EM; Xu, LY. STAT3, the Challenge for Chemotherapeutic and Radiotherapeutic Efficacy. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L; Raggi, L; Monteleone, E; Savino, A; Viavattene, D; Statello, L; et al. STAT3 induces breast cancer growth via ANGPTL4, MMP13 and STC1 secretion by cancer associated fibroblasts. Oncogene 2022, 41, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Bullock, E; Rozyczko, A; Shabbir, S; Tsoupi, I; Young, AIJ; Travnickova, J; et al. Cancer-associated fibroblast driven paracrine IL-6/STAT3 signaling promotes migration and dissemination in invasive lobular carcinoma. Breast Cancer Res 2025, 27, 121. [Google Scholar] [CrossRef]
- Godugu, D; Chilamakuri, R; Agarwal, S. STAT3 axis in cancer and cancer stem cells: From oncogenesis to targeted therapies. Biochim Biophys Acta Rev Cancer 2025, 1880, 189461. [Google Scholar] [CrossRef]
- Wei, S; Li, J; Tang, M; Zhang, K; Gao, X; Fang, L; et al. STAT3 and p63 in the Regulation of Cancer Stemness. Front Genet 2022, 13, 909251. [Google Scholar] [CrossRef]
- Galoczova, M; Coates, P; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett 2018, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, T; Niu, G; Kortylewski, M; Burdelya, L; Shain, K; Zhang, S; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Vallania, F; Schiavone, D; Dewilde, S; Pupo, E; Garbay, S; Calogero, R; et al. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc Natl Acad Sci U S A 2009, 106, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T; Garcia, R; Turkson, J; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Dong, J; Cheng, XD; Zhang, WD; Qin, JJ. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J Med Chem 2021, 64, 8884–8915. [Google Scholar] [CrossRef]
- Yang, J; Wang, L; Guan, X; Qin, JJ. Inhibiting STAT3 signaling pathway by natural products for cancer prevention and therapy: In vitro and in vivo activity and mechanisms of action. Pharmacol Res 2022, 182, 106357. [Google Scholar] [CrossRef]
- Berkley, K; Zalejski, J; Sharma, A. Targeting STAT3 for Cancer Therapy: Focusing on Y705, S727, or Dual Inhibition? Cancers (Basel) 2025, 17. [Google Scholar] [CrossRef]
- Wen, Z; Zhong, Z; Darnell, JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Tammineni, P; Anugula, C; Mohammed, F; Anjaneyulu, M; Larner, AC; Sepuri, NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Marié, IJ; Lahiri, T; Önder, Ö; Elenitoba-Johnson, KSJ; Levy, DE. Structural determinants of mitochondrial STAT3 targeting and function. Mitochondrial Commun 2024, 2, 1–13. [Google Scholar] [CrossRef]
- Peron, M; Dinarello, A; Meneghetti, G; Martorano, L; Betto, RM; Facchinello, N; et al. Y705 and S727 are required for the mitochondrial import and transcriptional activities of STAT3, and for regulation of stem cell proliferation. Development 2021, 148. [Google Scholar] [CrossRef]
- Carbognin, E; Betto, RM; Soriano, ME; Smith, AG; Martello, G. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. Embo j 2016, 35, 618–634. [Google Scholar] [CrossRef]
- Yokogami, K; Wakisaka, S; Avruch, J; Reeves, SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 2000, 10, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J; Campoy, I; Duran, M; Nemours, S; Areny, A; Vall-Palomar, M; et al. STAT3 phosphorylation at serine 727 activates specific genetic programs and promotes clear cell renal cell carcinoma (ccRCC) aggressiveness. Sci Rep 2023, 13, 19552. [Google Scholar] [CrossRef]
- Jiang, RY; Zhu, JY; Zhang, HP; Yu, Y; Dong, ZX; Zhou, HH; et al. STAT3: Key targets of growth-promoting receptor positive breast cancer. Cancer Cell Int 2024, 24, 356. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D; Reilley, MJ; Aponte, AM; Wang, G; Boja, E; Gucek, M; et al. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem 2010, 285, 23532–23536. [Google Scholar] [CrossRef] [PubMed]
- Xu, YS; Liang, JJ; Wang, Y; Zhao, XJ; Xu, L; Xu, YY; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci Rep 2016, 6, 39517. [Google Scholar] [CrossRef]
- Meier, JA; Hyun, M; Cantwell, M; Raza, A; Mertens, C; Raje, V; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal 2017, 10. [Google Scholar] [CrossRef]
- Lahiri, T; Brambilla, L; Andrade, J; Askenazi, M; Ueberheide, B; Levy, DE. Mitochondrial STAT3 regulates antioxidant gene expression through complex I-derived NAD in triple negative breast cancer. Mol Oncol 2021, 15, 1432–1449. [Google Scholar] [CrossRef]
- Mendes, CC; Gomes, DA; Thompson, M; Souto, NC; Goes, TS; Goes, AM; et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005, 280, 40892–40900. [Google Scholar] [CrossRef]
- Dhaouadi, N; Vitto, VAM; Pinton, P; Galluzzi, L; Marchi, S. Ca(2+) signaling and cell death. Cell Calcium 2023, 113, 102759. [Google Scholar] [CrossRef]
- Shen, S; Niso-Santano, M; Adjemian, S; Takehara, T; Malik, SA; Minoux, H; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Liu, B; Palmfeldt, J; Lin, L; Colaço, A; Clemmensen, KKB; Huang, J; et al. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Research 2018, 28, 996–1012. [Google Scholar] [CrossRef]
- Liu, B; Chen, R; Zhang, Y; Huang, J; Luo, Y; Rosthøj, S; et al. Cationic amphiphilic antihistamines inhibit STAT3 via Ca(2+)-dependent lysosomal H(+) efflux. Cell Rep 2023, 42, 112137. [Google Scholar] [CrossRef]
- Martínez-Fábregas, J; Prescott, A; van Kasteren, S; Pedrioli, DL; McLean, I; Moles, A; et al. Lysosomal protease deficiency or substrate overload induces an oxidative-stress mediated STAT3-dependent pathway of lysosomal homeostasis. Nature Communications 2018, 9, 5343. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, PA; Staniszewska, AD; Li, W; Omidvar, N; Kedjouar, B; Turkson, J; et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 2011, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, TJ; Lloyd-Lewis, B; Resemann, HK; Ramos-Montoya, A; Skepper, J; Watson, CJ. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat Cell Biol 2014, 16, 1057–1068. [Google Scholar] [CrossRef]
- Shi, D; Tao, J; Man, S; Zhang, N; Ma, L; Guo, L; et al. Structure, function, signaling pathways and clinical therapeutics: The translational potential of STAT3 as a target for cancer therapy. Biochim Biophys Acta Rev Cancer 2024, 1879, 189207. [Google Scholar] [CrossRef]
- Bialas, P; Kobayashi, T; Hellsten, R; Krzyzanowska, A; Persson, M; Marginean, F; et al. pSTAT3 Expression is Increased in Advanced Prostate Cancer in Post-Initiation of Androgen Deprivation Therapy. Prostate 2025, 85, 252–264. [Google Scholar] [CrossRef]
- Cocchiola, R; Romaniello, D; Grillo, C; Altieri, F; Liberti, M; Magliocca, FM; et al. Analysis of STAT3 post-translational modifications (PTMs) in human prostate cancer with different Gleason Score. Oncotarget 2017, 8, 42560–42570. [Google Scholar] [CrossRef]
- Hsu, FN; Chen, MC; Lin, KC; Peng, YT; Li, PC; Lin, E; et al. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser⁷²⁷ on STAT3 in prostate cancer cells. Am J Physiol Endocrinol Metab 2013, 305, E975-986. [Google Scholar] [CrossRef]
- Thaper, D; Vahid, S; Kaur, R; Kumar, S; Nouruzi, S; Bishop, JL; et al. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Scientific Reports 2018, 8, 17307. [Google Scholar] [CrossRef]
- Yang, SF; Yuan, SS; Yeh, YT; Wu, MT; Su, JH; Hung, SC; et al. The role of p-STAT3 (ser727) revealed by its association with Ki-67 in cervical intraepithelial neoplasia. Gynecol Oncol 2005, 98, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Tierney, BJ; McCann, GA; Naidu, S; Rath, KS; Saini, U; Wanner, R; et al. Aberrantly activated pSTAT3-Ser727 in human endometrial cancer is suppressed by HO-3867, a novel STAT3 inhibitor. Gynecol Oncol 2014, 135, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M; Oka, M; Iwasaki, T; Fukami, Y; Nishigori, C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J Invest Dermatol 2012, 132, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Cuadros, T; Trilla, E; Sarro, E; Vila, MR; Vilardell, J; de Torres, I; et al. HAVCR/KIM-1 activates the IL-6/STAT-3 pathway in clear cell renal cell carcinoma and determines tumor progression and patient outcome. Cancer Res 2014, 74, 1416–1428. [Google Scholar] [CrossRef]
- Arevalo, J; Lorente, D; Trilla, E; Salcedo, MT; Morote, J; Meseguer, A. Nuclear and cytosolic pS727-STAT3 levels correlate with overall survival of patients affected by clear cell renal cell carcinoma (ccRCC). Sci Rep 2021, 11, 6957. [Google Scholar] [CrossRef]
- Lin, WH; Chang, YW; Hong, MX; Hsu, TC; Lee, KC; Lin, C; et al. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene 2021, 40, 791–805. [Google Scholar] [CrossRef]
- Ouédraogo, ZG; Müller-Barthélémy, M; Kemeny, JL; Dedieu, V; Biau, J; Khalil, T; et al. STAT3 Serine 727 Phosphorylation: A Relevant Target to Radiosensitize Human Glioblastoma. Brain Pathol 2016, 26, 18–30. [Google Scholar] [CrossRef]
- Lin, GS; Chen, YP; Lin, ZX; Wang, XF; Zheng, ZQ; Chen, L. STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int J Clin Exp Pathol 2014, 7, 3141–3149. [Google Scholar]
- Lee, ES; Ko, KK; Joe, YA; Kang, SG; Hong, YK. Inhibition of STAT3 reverses drug resistance acquired in temozolomide-resistant human glioma cells. Oncol Lett 2011, 2, 115–121. [Google Scholar] [CrossRef]
- Ma, JH; Qin, L; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun Signal 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Hazan-Halevy, I; Harris, D; Liu, Z; Liu, J; Li, P; Chen, X; et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Naik, A; Thomas, R; Sikhondze, M; Babiker, A; Lattab, B; Qasem, H; et al. The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Naik, A; Decock, J. Targeting of lactate dehydrogenase C dysregulates the cell cycle and sensitizes breast cancer cells to DNA damage response targeted therapy. Mol Oncol 2022, 16, 885–903. [Google Scholar] [CrossRef]
- Elsarraj, HS; Hong, Y; Limback, D; Zhao, R; Berger, J; Bishop, SC; et al. BCL9/STAT3 regulation of transcriptional enhancer networks promote DCIS progression. NPJ Breast Cancer 2020, 6, 12. [Google Scholar] [CrossRef]
- Shin, SY; Lee, DH; Lee, J; Choi, C; Kim, JY; Nam, JS; et al. C-C motif chemokine receptor 1 (CCR1) is a target of the EGF-AKT-mTOR-STAT3 signaling axis in breast cancer cells. Oncotarget 2017, 8, 94591–94605. [Google Scholar] [CrossRef]
- Mackenzie, GG; Huang, L; Alston, N; Ouyang, N; Vrankova, K; Mattheolabakis, G; et al. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS One 2013, 8, e61532. [Google Scholar] [CrossRef]
- Boengler, K; Hilfiker-Kleiner, D; Heusch, G; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 2010, 105, 771–785. [Google Scholar] [CrossRef]
- Cai, G; Yu, W; Song, D; Zhang, W; Guo, J; Zhu, J; et al. Discovery of fluorescent coumarin-benzo[b]thiophene 1, 1-dioxide conjugates as mitochondria-targeting antitumor STAT3 inhibitors. Eur J Med Chem 2019, 174, 236–251. [Google Scholar] [CrossRef]
- Brambilla, L; Lahiri, T; Cammer, M; Levy, DE. STAT3 Inhibitor OPB-51602 Is Cytotoxic to Tumor Cells Through Inhibition of Complex I and ROS Induction. iScience 2020, 23, 101822. [Google Scholar] [CrossRef] [PubMed]
- Genini, D; Brambilla, L; Laurini, E; Merulla, J; Civenni, G; Pandit, S; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc Natl Acad Sci U S A 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L; Genini, D; Laurini, E; Merulla, J; Perez, L; Fermeglia, M; et al. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol Oncol 2015, 9, 1194–1206. [Google Scholar] [CrossRef]
- Bendell, JC; Hong, DS; Burris, HA, 3rd; Naing, A; Jones, SF; Falchook, G; et al. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol 2014, 74, 125–130. [Google Scholar] [CrossRef]
- Wan, F; Qian, C; Liu, X; Zhong, Y; Peng, W; Zhang, L; et al. Sculponeatin A induces mitochondrial dysfunction in non-small cell lung cancer through WWP2-mediated degradation of mitochondrial STAT3. Br J Pharmacol 2025, 182, 2662–2681. [Google Scholar] [CrossRef]
- Peng, P; Ren, Y; Wan, F; Tan, M; Wu, H; Shen, J; et al. Sculponeatin A promotes the ETS1-SYVN1 interaction to induce SLC7A11/xCT-dependent ferroptosis in breast cancer. Phytomedicine 2023, 117, 154921. [Google Scholar] [CrossRef]
- Li, Y; Li, PK; Roberts, MJ; Arend, RC; Samant, RS; Buchsbaum, DJ. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett 2014, 349, 8–14. [Google Scholar] [CrossRef]
- Gao, D; Jin, N; Fu, Y; Zhu, Y; Wang, Y; Wang, T; et al. Rational drug design of benzothiazole-based derivatives as potent signal transducer and activator of transcription 3 (STAT3) signaling pathway inhibitors. Eur J Med Chem 2021, 216, 113333. [Google Scholar] [CrossRef]
- Yang, Z; Xu, H; Yang, Y; Duan, C; Zhang, P; Wang, Y; et al. Synthesis and evaluation of naphthalene derivatives as potent STAT3 inhibitors and agents against triple-negative breast cancer growth and metastasis. Breast Cancer Res Treat 2023, 197, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, MX; Liu, X; Zhang, HL; Xu, H; Ma, X; Yang, Y; et al. A novel synthesised STAT3 inhibitor exerts potent anti-tumour activity by inducing lysosome-dependent cell death. Br J Pharmacol 2025, 182, 4041–4057. [Google Scholar] [CrossRef]
- Chen, H; Bian, A; Zhou, W; Miao, Y; Ye, J; Li, J; et al. Discovery of the Highly Selective and Potent STAT3 Inhibitor for Pancreatic Cancer Treatment. ACS Cent Sci 2024, 10, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, X; Huang, J; Xie, Y; Zhou, Y; Wang, R; Lou, J. Napabucasin Attenuates Resistance of Breast Cancer Cells to Tamoxifen by Reducing Stem Cell-Like Properties. Med Sci Monit 2019, 25, 8905–8912. [Google Scholar] [CrossRef]
- He, P; Miao, Y; Sun, Y; Bian, A; Jin, W; Chen, H; et al. Discovery of a Novel Potent STAT3 Inhibitor HP590 with Dual p-Tyr(705)/Ser(727) Inhibitory Activity for Gastric Cancer Treatment. J Med Chem 2022, 65, 12650–12674. [Google Scholar] [CrossRef] [PubMed]
- He, P; Bian, A; Miao, Y; Jin, W; Chen, H; He, J; et al. Discovery of a Highly Potent and Orally Bioavailable STAT3 Dual Phosphorylation Inhibitor for Pancreatic Cancer Treatment. J Med Chem 2022, 65, 15487–15511. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D; Shu, J; Gao, B; Yu, H; Liu, K; Zheng, M; et al. Pulchinenoside E2: A dual-functional STAT3 and autophagy inhibitor with potent anti-metastatic activity in triple-negative breast cancer. Phytomedicine 2025, 148, 157485. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of STAT3 structural domains and mutant constructs, including mitochondrial-targeted STAT3, used to dissect the functional roles of S727 and Y705 phosphorylation, including STAT3-C (constitutively active STAT3), STAT3-YF (Y705F phosphorylation-defective mutant), STAT3-SA (S727A phosphorylation-defective mutant), STAT3-SE (S727E phosphomimetic mutant), and mitochondria-targeted STAT3 (MLS-STAT3, bearing a mitochondrial localization sequence). See text for details and references.
Figure 1.
Schematic overview of STAT3 structural domains and mutant constructs, including mitochondrial-targeted STAT3, used to dissect the functional roles of S727 and Y705 phosphorylation, including STAT3-C (constitutively active STAT3), STAT3-YF (Y705F phosphorylation-defective mutant), STAT3-SA (S727A phosphorylation-defective mutant), STAT3-SE (S727E phosphomimetic mutant), and mitochondria-targeted STAT3 (MLS-STAT3, bearing a mitochondrial localization sequence). See text for details and references.
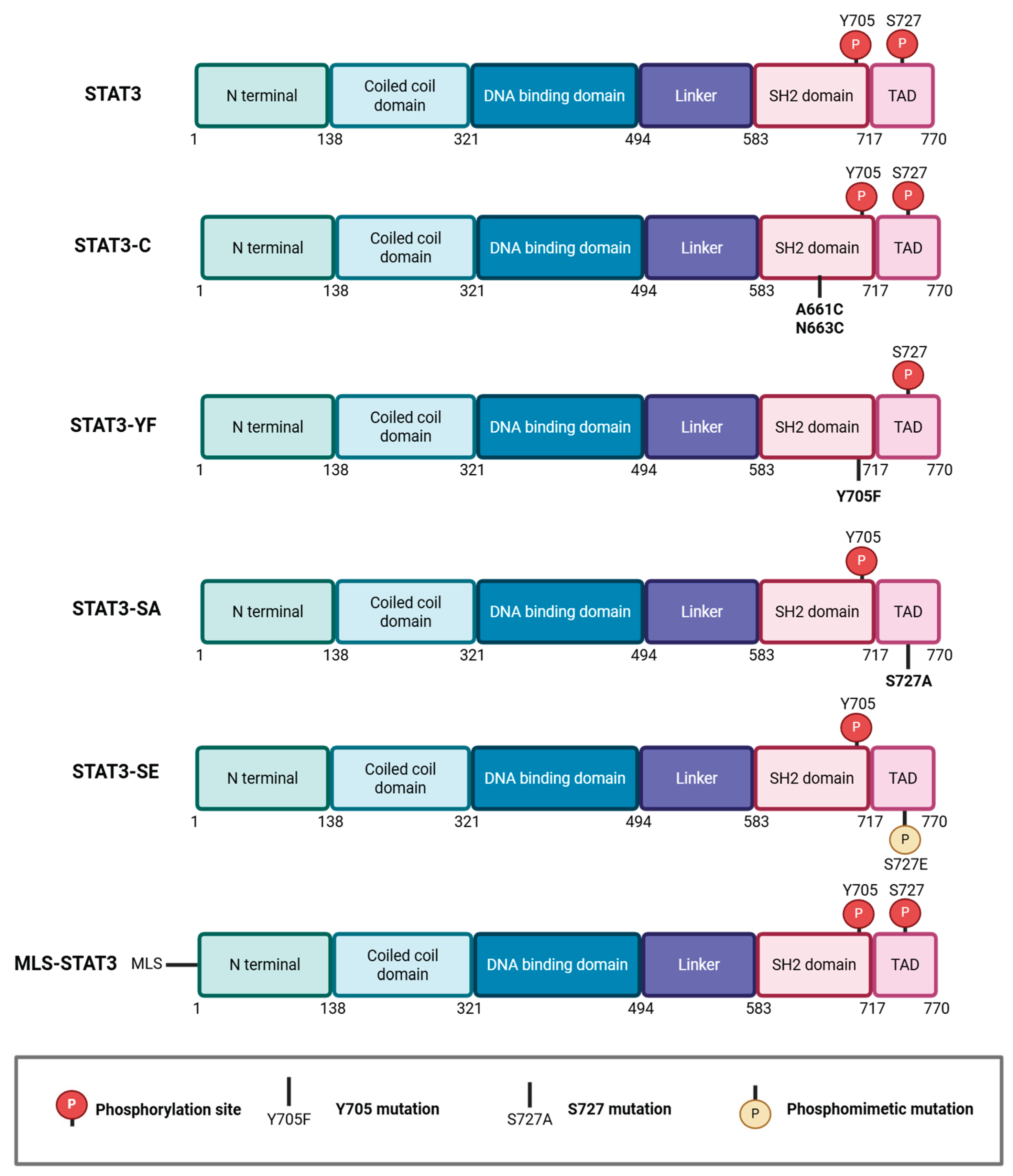
Figure 2.
Comprehensive model depicting the balance between canonical and non-canonical STAT3 functions. Left (Blue): Canonical activation by stimuli such as IL-6, growth factors, or c-Src leads to tyrosine phosphorylation (p-Y705) and nuclear translocation. Right (Orange): Non-canonical activation triggered by cellular stress and kinase signaling (MAPK, mTOR, RAS) directs STAT3 to various organelles. In the mitochondria, STAT3 regulates the electron transport chain (ETC), ROS production, mtDNA transcription, and SC proliferation; at the ER/MAMs, it modulates calcium flux and apoptosis resistance; and in lysosomes, it increases autophagy and maintains lysosomal pH.
Figure 2.
Comprehensive model depicting the balance between canonical and non-canonical STAT3 functions. Left (Blue): Canonical activation by stimuli such as IL-6, growth factors, or c-Src leads to tyrosine phosphorylation (p-Y705) and nuclear translocation. Right (Orange): Non-canonical activation triggered by cellular stress and kinase signaling (MAPK, mTOR, RAS) directs STAT3 to various organelles. In the mitochondria, STAT3 regulates the electron transport chain (ETC), ROS production, mtDNA transcription, and SC proliferation; at the ER/MAMs, it modulates calcium flux and apoptosis resistance; and in lysosomes, it increases autophagy and maintains lysosomal pH.
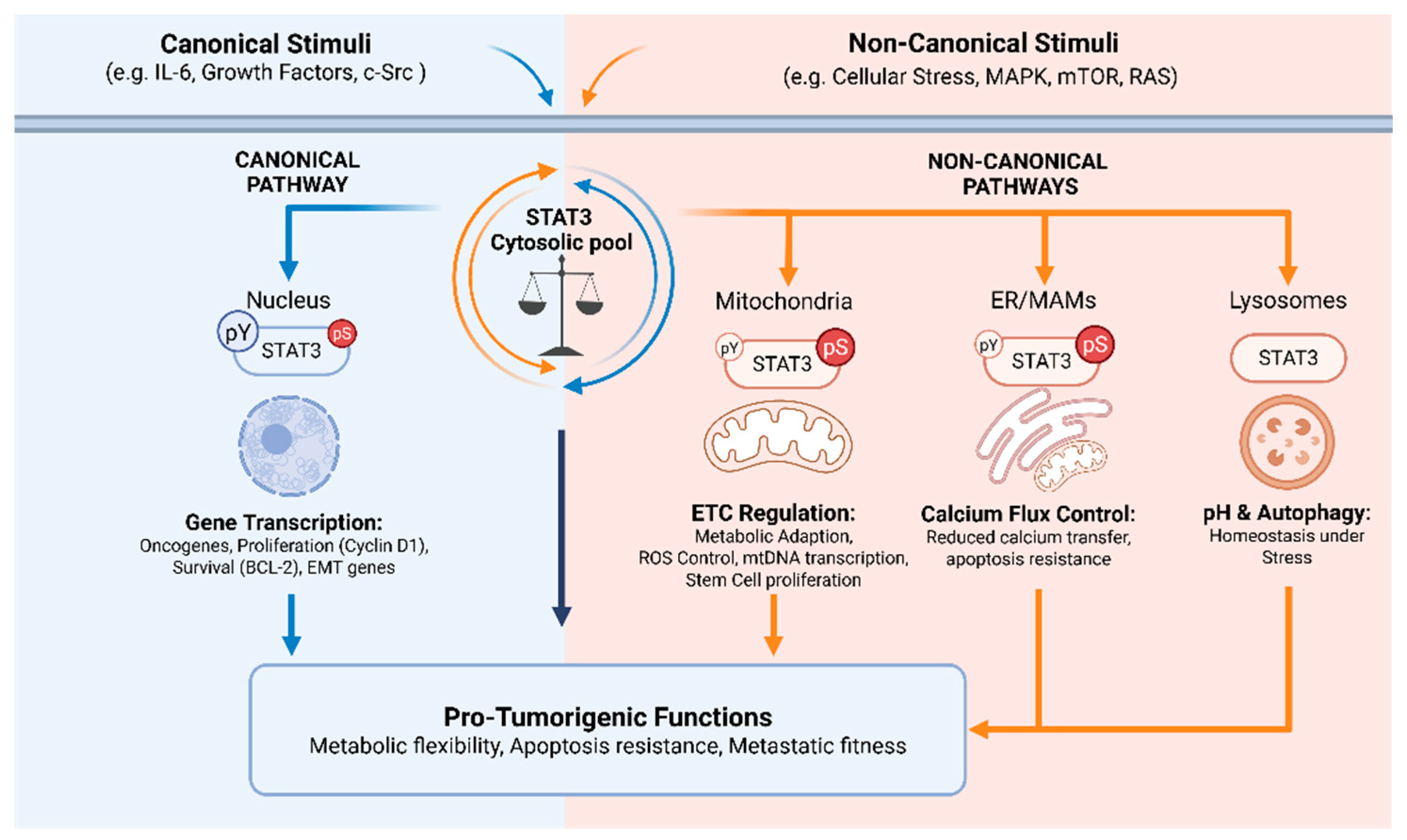

Table 1.
List of tumor types where p-S727 STAT3 levels were analyzed and correlated with tumor grade, overall survival (OS), or metastasis-free survival (MFS). PCa, prostate cancer; CIN, cervical intraepithelial neoplasia; ALM, acral lentiginous melanoma; ccRCC, clear cell renal cell carcinoma; LC, lung cancer, GBM, glioblastoma; BC, breast cancer; TNBC, triple negative breast cancer; POS, positive; NEG, negative; N/A, not available.
Table 1.
List of tumor types where p-S727 STAT3 levels were analyzed and correlated with tumor grade, overall survival (OS), or metastasis-free survival (MFS). PCa, prostate cancer; CIN, cervical intraepithelial neoplasia; ALM, acral lentiginous melanoma; ccRCC, clear cell renal cell carcinoma; LC, lung cancer, GBM, glioblastoma; BC, breast cancer; TNBC, triple negative breast cancer; POS, positive; NEG, negative; N/A, not available.
| Tumor type | Patients (n) | p-S727 STAT3 + | Correlation with grade | Correlation with OS | Correlation with MFS | Reference |
|---|---|---|---|---|---|---|
| PCa | 20 | 65% | Pos (p=0.05) | N/A | N/A | [29] |
| PCa | 111 | 49% | N/A | Neg (p=0.013) | Neg (p=0.034) | [80] |
| CIN | 56 | 100% | Pos (p<0.001) | N/A | N/A | [84] |
| ALM | 15 | 73% | N/A | N/A | N/A | [86] |
| ccRCC | 98 | N/A | Pos (p<0.01) | Neg (p<0.001) | N/A | [87] |
| ccRCC | 82 | N/A | N/A | Neg (p=0.002) | N/A | [88] |
| LC | 40 | N/A | Pos | Neg (p=0.017) | N/A | [89] |
| GBM | 30 | 100% | N/A | Not affected | N/A | [90] |
| GBM | 88 | 70% | N/A | Neg (p=0.002) | N/A | [91] |
| BC | 68 | 62% | Pos (p=0.024) | N/A | N/A | [38] |
| BC | 48 | 56% | N/A | N/A | N/A | [25] |
| TNBC | 173 | N/A | Neg (p=0.016) | Not affected | N/A | [40] |
| TNBC | 76 | 80% | N/A | N/A | N/A | [41] |
Table 2.
STAT3 inhibitors reported to interfere with S727 phosphorylation in TNBC, including the targeted domain, affected phosphorylated forms, and effects on both primary tumors and metastases. <, reduced; SUP, suppressed; LU, lung; BN, bone; LV, liver; N/A, not available.
Table 2.
STAT3 inhibitors reported to interfere with S727 phosphorylation in TNBC, including the targeted domain, affected phosphorylated forms, and effects on both primary tumors and metastases. <, reduced; SUP, suppressed; LU, lung; BN, bone; LV, liver; N/A, not available.
| Compound | Domain | p-S727 | p-Y705 | Exp. system | Primary tumors | Metastases | Reference |
|---|---|---|---|---|---|---|---|
| 7a | N/A | Yes | Yes | 4T1 | < | N/A | [101] |
| OPB-51602 | SH2 | Yes | Yes | N/A | N/A | [102] | |
| SLSI-1216 | SH2 | Yes | Yes | MDA-MB-231 | < | N/A | [36] |
| Niclosamide (NSA) | N/A | Yes | Yes | MDA-MB-231 | < | < (LU, BN) | [41] |
| B19 | SH2 | Yes | Yes | N/A | N/A | [109] | |
| RDp002 | SH2 | Yes | Yes | MDA-MB-468 4T1 |
< | < LU dissemination | [111] |
| Y002 | SH2 | Yes | Yes | MDA-MB-231 | < | N/A | [112] |
| PSE2 | SH2 | Yes | Yes | MDA-MB-231 | N/A | SUP (LU, LV) | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



