Submitted:
01 February 2026
Posted:
03 February 2026
You are already at the latest version
Abstract
Background: Chronic diabetic wounds are characterized by persistent inflammatory arrest driven by redox collapse, mitochondrial dysfunction, and extracellular matrix (ECM) instability. Sulfur deficiency has emerged as a central upstream defect linking these pathological domains. Impaired cystine uptake via SLC7A11/xCT limits intracellular cysteine, suppresses glutathione synthesis, and destabilizes redox buffering. Oxidative stress disrupts cytoskeletal signaling, suppresses angiogenic pathways, impairs mitochondrial bioenergetics, and perpetuates chronic inflammation. Concurrent extracellular sulfur depletion compromises disulfide bonding and crosslinking in collagen, elastin, and keratin, weakening tissue mechanics and impairing cell–matrix communication.Hypothesis: A dual-sulfur regenerative approach simultaneously addresses intracellular and extracellular sulfur deficits, restoring redox balance, mitochondrial function, and ECM integrity to convert the wound microenvironment from inflammatory arrest to regenerative competence.Therapeutic Strategy: A topical formulation combining N-acetylcysteine (NAC; ~2–3% w/v) and methylsulfonylmethane (MSM; ~5–8% w/v) targets complementary compartments. NAC replenishes cysteine, restores glutathione, stabilizes mitochondria, and reprograms inflammatory pathways. MSM enhances ECM crosslinking, restores biomechanical integrity, reduces proteolytic degradation, and reactivates mechanotransduction. Application involves gentle wound cleansing, thin layer topical delivery under semi-occlusive dressing once to twice daily for 2–4 weeks, with monitoring of granulation, epithelialization, exudate, and local inflammation.Implications: Intracellular redox normalization, mitochondrial recovery, macrophage phenotype transition, angiogenic restoration, and ECM stabilization converge to initiate coordinated tissue regeneration. Sulfur homeostasis thus serves as a unifying upstream regulator and therapeutic target.Conclusion: Targeting both intracellular and extracellular sulfur deficits provides a rational, mechanism-driven strategy to accelerate wound closure, restore tissue quality, and enhance vascular stability. This dual-sulfur approach redefines the biological potential of diabetic wound therapy.
Keywords:
Sulfur homeostasis
; SLC7A11–cysteine transport axis
; Glutathione redox collapse
; Mitochondrial iron–sulfur cluster dysfunction
; Chronic inflammatory persistence
; Diabetic wound regeneration
1. Introduction
Diabetic wounds represent one of the most severe and disabling chronic complications of diabetes mellitus and remain a leading cause of infection-related hospitalization and non-traumatic lower-limb amputation worldwide [1]. Despite major advances in vascular reconstruction, antimicrobial therapy, pressure off-loading, and bioengineered wound dressings, a significant proportion of diabetic ulcers fail to progress toward closure [2]. This persistent therapeutic failure indicates that prevailing clinical models, which predominantly attribute non-healing to ischemia, neuropathy, or infection, do not sufficiently explain the profound biological resistance to tissue repair observed in diabetic wounds [3].
Emerging molecular evidence supports a more fundamental interpretation in which diabetic wounds are sites of metabolic and proteostatic collapse characterized by redox disequilibrium, mitochondrial dysfunction, extracellular matrix (ECM) disintegration, and unresolved inflammation [4,5]. Chronic hyperglycemia drives excessive production of reactive oxygen species (ROS) through mitochondrial electron transport chain over-reduction, activation of NADPH oxidases, increased polyol pathway flux, and signaling through advanced glycation end-product (AGE) receptors [6,7,8]. These processes disrupt intracellular homeostasis at multiple levels, resulting in oxidative damage to macromolecules, defective protein folding, impaired cellular energetics, and loss of coordinated intercellular signaling required for regeneration [9,10].
A unifying but underrecognized contributor to these abnormalities is disruption of sulfur metabolism. Sulfur is indispensable for intracellular redox buffering, stabilization of protein structure through disulfide bond formation, enzymatic catalysis, and mitochondrial respiration [11,12]. In peripheral diabetic tissues, cysteine bioavailability is frequently limited due to impaired uptake of extracellular cystine via the cystine–glutamate antiporter system xCT, encoded by SLC7A11 [13,14]. This transporter constitutes the principal mechanism by which most non-hepatic cells acquire cysteine equivalents necessary for glutathione synthesis and structural disulfide bonding [15]. Functional suppression of SLC7A11 in the diabetic wound microenvironment leads to intracellular cysteine depletion, exhaustion of glutathione reserves, and uncontrolled accumulation of ROS [16]. These events propagate oxidative damage to proteins, lipids, and nucleic acids, destabilize ECM architecture, impair mitochondrial ATP generation, and perpetuate inflammatory signaling. From a pathophysiological perspective, diabetic wounds can therefore be conceptualized as localized sulfur-deficient, redox-collapsed tissues that are structurally, metabolically, and immunologically incapable of repair. This framework supports a shift away from exclusively symptomatic wound management toward targeted restoration of fundamental molecular deficits, reframing diabetic wounds as a manifestation of SLC7A11-mediated sulfur dysregulation and opening new avenues for regenerative strategies grounded in redox biology, mitochondrial medicine, and proteostasis restoration [12,16].
1. Sulfur Biology as a Foundation of Tissue Integrity
Sulfur occupies a central role in maintaining tissue integrity by governing antioxidant defense systems, protein conformation, mitochondrial bioenergetics, and immune modulation [17]. During wound repair, sulfur availability determines whether resident and infiltrating cells can withstand oxidative stress, synthesize structurally competent matrix proteins, generate sufficient ATP, and successfully transition through the inflammatory, proliferative, and remodeling phases of healing [18,19,20]. Disruption of sulfur metabolism therefore exerts system-wide consequences that compromise regenerative capacity at molecular, cellular, and tissue levels [21].
Cysteine is the rate-limiting substrate for the synthesis of glutathione, the dominant intracellular antioxidant responsible for maintaining thiol redox balance and detoxifying reactive oxygen species through the glutathione peroxidase and glutaredoxin systems [22]. In most somatic cells, cysteine is derived primarily from extracellular cystine imported via SLC7A11 and subsequently reduced in the cytosol [23].
In diabetic wounds, multiple hyperglycemia-driven mechanisms markedly increase ROS production, including mitochondrial electron leakage at complexes I and III, activation of NADPH oxidase isoforms, and AGE–RAGE signaling cascades that amplify inflammatory oxidant generation [24,25,26,27].
When cysteine availability declines, glutathione synthesis becomes insufficient, leading to oxidation of critical thiol groups in proteins, lipids, and transcriptional regulators [12,28,29]. A major downstream consequence is disruption of Nrf2 signaling, a master regulator of antioxidant gene expression. Under physiological conditions, redox-sensitive modification of KEAP1 permits Nrf2 stabilization and transcription of cytoprotective genes such as HMOX1, NQO1, GCLC, superoxide dismutase, and catalase [30,31,32]. In sulfur-depleted diabetic wounds, persistent oxidative stress and thiol imbalance impair proper Nrf2 activation while simultaneously enhancing NF-κB signaling, resulting in sustained transcription of pro-inflammatory mediators including TNF-α, IL-1β, and IL-6. This establishes a self-perpetuating redox-inflammatory cycle in which oxidative stress reinforces inflammation and inflammatory cells further amplify ROS production, effectively preventing the transition toward tissue repair [33,34,35,36].
Beyond its antioxidant role, sulfur is essential for structural protein integrity through the formation of disulfide bonds between cysteine residues, which stabilize the tertiary and quaternary structures of numerous secreted and membrane proteins [12]. Proper disulfide bond formation within the endoplasmic reticulum, catalyzed by protein disulfide isomerases, is critical for the maturation of collagen, elastin, fibronectin, laminin, and integrins that collectively form the extracellular matrix scaffold [12,37]. In sulfur-deficient conditions, impaired oxidative protein folding leads to accumulation of misfolded ECM precursors in the endoplasmic reticulum and activation of the unfolded protein response via PERK, IRE1α, and ATF6 pathways [38]. Although initially adaptive, chronic unfolded protein response signaling promotes translational attenuation, CHOP-mediated apoptosis, and reduced fibroblast viability and collagen synthesis. Histologically, this manifests as disorganized collagen fibrils, fragmented elastin networks, and diminished matrix tensile strength [39].
The altered matrix composition disrupts integrin engagement and focal adhesion kinase signaling, impairing mechanotransduction processes required for fibroblast migration, myofibroblast differentiation, and wound contraction. Sulfur deficiency therefore directly undermines the structural and signaling framework upon which tissue regeneration depends [40,41,42].
Sulfur is also indispensable for mitochondrial respiration through its incorporation into iron–sulfur clusters, which serve as essential cofactors for enzymes within complexes I, II, and III of the electron transport chain as well as tricarboxylic acid cycle enzymes such as aconitase [43,44].
Iron–sulfur cluster assembly depends on cysteine-derived sulfur mobilized by mitochondrial cysteine desulfurase systems [45]. In sulfur-restricted diabetic wound cells, defective iron–sulfur cluster biogenesis impairs electron transport efficiency, increases electron leakage and secondary ROS formation, disrupts mitochondrial membrane potential, and reduces ATP synthesis [46,47].
Energy failure has profound functional implications because keratinocyte migration requires ATP-dependent cytoskeletal remodeling, fibroblast proliferation and collagen secretion demand high biosynthetic energy, and endothelial sprouting during angiogenesis depends on mitochondrial signaling and ATP availability. Sulfur-linked mitochondrial dysfunction thus creates a metabolic bottleneck that prevents coordinated progression from inflammation to tissue formation [48,49].
Effective wound healing additionally requires resolution of inflammation and establishment of a pro-repair immune environment, processes that are strongly influenced by intracellular thiol status [50,51]. Adequate cysteine and glutathione levels promote redox conditions that restrain NF-κB activation and favor polarization of macrophages toward a reparative phenotype characterized by secretion of IL-10, TGF-β, and vascular endothelial growth factor [52,53]. In sulfur-deficient diabetic wounds, persistent oxidative stress maintains macrophages in a pro-inflammatory state marked by high production of TNF-α, IL-1β, and matrix metalloproteinases such as MMP-9, which degrade extracellular matrix components and inhibit granulation tissue formation [54,55,56]. Impaired clearance of neutrophils further prolongs protease and oxidant release, amplifying collateral tissue damage [57]. Failure of sulfur-dependent redox signaling to resolve inflammation therefore prevents the coordinated activation of fibroblasts, endothelial cells, and keratinocytes required for the proliferative phase, locking the wound in a chronic inflammatory state that is biologically incapable of regeneration [58,59].

Table 1.
Multisystem Consequences of Sulfur Deficiency in the Diabetic Wound Microenvironment.
| Biological System | Sulfur-Dependent Component | Molecular Disruption in Diabetes | Cellular Consequence | Tissue/Clinical Outcome |
|---|---|---|---|---|
| Redox homeostasis | Cysteine → Glutathione synthesis via SLC7A11 | Reduced cystine uptake, GSH depletion, ROS accumulation | Oxidative macromolecular damage, redox signaling failure | Persistent oxidative stress, delayed healing |
| Antioxidant defense regulation | Nrf2–KEAP1 thiol-sensitive pathway | Impaired Nrf2 activation and antioxidant gene transcription | Decreased HO-1, NQO1, SOD, catalase expression | Loss of cytoprotection |
| Inflammatory control | Redox modulation of NF-κB | ROS-driven NF-κB hyperactivation | Sustained TNF-α, IL-1β, IL-6 production | Chronic non-resolving inflammation |
| Proteostasis | Disulfide bond formation and ER folding | Misfolded ECM proteins, chronic UPR activation | Apoptosis, reduced fibroblast function | Poor granulation tissue formation |
| ECM structural integrity | Disulfide-stabilized collagen and matrix proteins | Defective cross-linking and structural weakness | Impaired adhesion, migration, mechanotransduction | Fragile matrix, poor wound contraction |
| Mitochondrial bioenergetics | Iron–sulfur cluster biogenesis | ETC dysfunction, ↓ membrane potential, ↑ ROS | Reduced ATP production | Impaired keratinocyte, fibroblast, endothelial function |
| Immune resolution | GSH-dependent macrophage polarization | Persistence of M1 phenotype, impaired M2 transition | Excess TNF-α, IL-1β, MMP-9; low IL-10, TGF-β | ECM degradation, poor angiogenesis |
| Protease balance | Redox regulation of MMP/TIMP systems | Oxidative activation of MMPs | Excess ECM breakdown | Chronic ulcer persistence |
2. Sulfur Dysregulation as a Core Pathophysiological Driver in Diabetic Wounds
Central to sulfur dysregulation in diabetic wounds is impaired function of the cystine–glutamate antiporter system xCT, encoded by SLC7A11, which governs cellular uptake of extracellular cystine and thereby regulates intracellular cysteine availability [13,14]. In the diabetic microenvironment, persistent hyperglycemia, accumulation of advanced glycation end products, and chronic exposure to inflammatory cytokines converge to suppress SLC7A11 expression and functional activity through transcriptional and post-translational mechanisms linked to oxidative stress and inflammatory signaling [60]. Reduced transporter function limits cystine import, leading to intracellular cysteine depletion and progressive exhaustion of glutathione reserves, which undermines the cell’s primary redox buffering system [13,14,60]. Loss of glutathione-mediated control over intracellular thiol redox balance amplifies oxidative stress, promoting irreversible oxidation of extracellular matrix proteins and cell-surface receptors [61]. Integrins and associated focal adhesion complexes become structurally and functionally compromised, disrupting focal adhesion kinase signaling and cytoskeletal organization required for directed cell migration [62]. Fibroblast proliferation declines due to redox-sensitive cell cycle arrest, while keratinocyte motility becomes impaired as actin dynamics and adhesion turnover lose coordination [63]. The resulting cellular immobility directly contributes to stalled re-epithelialization and poor granulation tissue formation [64].
Concurrently, oxidative stress interferes with stabilization of hypoxia-inducible factor-1α, a transcription factor essential for adaptive responses to tissue hypoxia [65].
Under sulfur-deficient redox conditions, prolyl hydroxylase activity remains aberrantly elevated due to altered redox cofactor availability, promoting premature HIF-1α degradation and blunting transcription of angiogenic mediators such as vascular endothelial growth factor [66]. Endothelial cells within the wound bed therefore fail to mount adequate angiogenic responses, leading to poor neovascularization and persistent tissue hypoxia [67].
From a pathophysiological standpoint, impaired SLC7A11-mediated cysteine transport represents a nodal molecular defect linking metabolic stress to structural failure and immune dysregulation [13,14]. Targeting this sulfur bottleneck offers a rational strategy for restoring the biochemical conditions necessary for tissue regeneration rather than merely addressing downstream consequences of wound chronicity.
Table 2.
Molecular Consequences of SLC7A11 Dysfunction in Diabetic Wound Cells.
| Cellular Process | Normal Role of SLC7A11 | Effect of Diabetic Suppression | Functional Outcome in Wound |
|---|---|---|---|
| Cystine uptake | Supplies cysteine for GSH synthesis | Intracellular cysteine depletion | Redox collapse, ROS accumulation |
| Antioxidant defense | Maintains glutathione-dependent detoxification | Impaired ROS neutralization | Oxidative macromolecular damage |
| Cell migration | Preserves integrin and cytoskeletal thiol integrity | Oxidative modification of adhesion proteins | Reduced keratinocyte and fibroblast motility |
| Angiogenic signaling | Supports redox balance for HIF-1α stabilization | HIF-1α degradation, ↓ VEGF expression | Poor neovascularization |
| Mitochondrial function | Provides sulfur for Fe–S cluster assembly | ETC dysfunction, ↓ ATP | Energy deficit in repair cells |
| Inflammatory regulation | Enables redox-mediated resolution signaling | NF-κB persistence, cytokine excess | Chronic inflammation |
3. N-Acetylcysteine as an Intracellular Sulfur Restorative in Diabetic Wounds
N-acetylcysteine (NAC) represents a mechanistically targeted intracellular sulfur donor whose therapeutic relevance in diabetic wound healing derives from its ability to correct cysteine-dependent molecular dysfunction rather than functioning solely as a nonspecific antioxidant [68]. In the diabetic wound microenvironment, impaired cystine uptake through SLC7A11 restricts intracellular cysteine availability and constrains glutathione synthesis, precipitating redox collapse [69]. NAC bypasses this transport limitation by entering cells through alternative uptake pathways and undergoing intracellular deacetylation to release cysteine, thereby directly replenishing intracellular thiol pools [70].
Restoration of cysteine availability permits reconstitution of glutathione synthesis and normalization of intracellular redox buffering capacity. As redox equilibrium is re-established, oxidative stress–mediated inhibition of cytoprotective pathways is relieved, allowing reactivation of Nrf2-dependent transcriptional programs and attenuation of NF-κB–driven inflammatory gene expression [71,72].
The resulting decline in oxidative and inflammatory pressure enables redox-sensitive signaling networks to recover, facilitating the transition from chronic inflammation to a reparative cellular phenotype [73]. At the mitochondrial level, cysteine restoration supports iron–sulfur cluster biogenesis and limits oxidative damage to components of the electron transport chain [74]. Recovery of mitochondrial membrane potential and improved oxidative phosphorylation efficiency increase ATP production, which is essential for actin remodeling during keratinocyte migration, biosynthetic activity in fibroblasts, and angiogenic function in endothelial cells. By linking thiol restoration to bioenergetic recovery, NAC directly bridges molecular redox repair with functional cellular regeneration [75].
Stabilization of intracellular redox status also protects structural and signaling proteins from oxidative modification, preserving receptor conformation and downstream responsiveness to growth factors such as EGF, PDGF, and TGF-β [76,77]. Collectively, these effects position NAC as a pathophysiologically rational intervention aimed at correcting intracellular sulfur deficiency as a primary driver of diabetic wound chronicity.
Table 3.
Mechanistic Effects of N-Acetylcysteine in Diabetic Wound Cells.
| Target Domain | Sulfur-Dependent Mechanism Restored by NAC | Downstream Biological Effect | Relevance to Healing |
|---|---|---|---|
| Redox balance | Replenishes cysteine for GSH synthesis | Decreased intracellular ROS | Protection from oxidative injury |
| Antioxidant signaling | Enables Nrf2 pathway recovery | Increased cytoprotective gene expression | Cellular stress resistance |
| Inflammatory control | Redox suppression of NF-κB | Reduced TNF-α, IL-1β, IL-6 | Transition toward reparative phase |
| Mitochondrial bioenergetics | Supports Fe–S cluster formation | Improved ATP production | Enhanced migration and proliferation |
| Growth factor signaling | Preserves receptor thiol integrity | Restored responsiveness to repair signals | Improved granulation and re-epithelialization |
4. Methylsulfonylmethane as a Structural Sulfur Donor for Extracellular Matrix Repair
Effective wound healing requires not only restoration of intracellular metabolic competence but also reconstruction of the extracellular matrix that provides structural and biochemical support for regenerating tissue [78].
Methylsulfonylmethane (MSM) serves as a bioavailable organic sulfur source with particular relevance to extracellular protein synthesis and matrix stability in sulfur-deficient diabetic wounds. Whereas intracellular sulfur donors primarily restore redox and metabolic functions, MSM contributes sulfur for structural integration within newly synthesized extracellular proteins [79].
Structural macromolecules critical to wound repair, including collagen, elastin, laminin, and keratin, depend on sulfur-mediated crosslinking and disulfide bond formation to achieve mechanical strength and resilience [80,81]. In diabetic wounds characterized by sulfur insufficiency, newly synthesized matrix proteins often exhibit defective structural maturation, rendering the extracellular matrix vulnerable to degradation and incapable of sustaining effective cell–matrix interactions [82]. Increased sulfur availability in the extracellular compartment supports proper folding, crosslinking, and stabilization of these proteins, improving tensile strength and resistance to proteolysis [12].
Restoration of matrix structural integrity enhances the biomechanical properties of the wound bed, reinforcing the scaffold required for fibroblast attachment, keratinocyte migration, and mechanotransduction. Improved matrix quality also supports integrin signaling and focal adhesion formation, facilitating coordinated cellular movement and wound contraction [83]. In addition to its structural role, MSM modulates inflammatory signaling within the wound milieu by reducing expression of matrix-degrading enzymes and limiting amplification of inflammatory cascades, thereby contributing to stabilization of the extracellular environment necessary for regeneration. From a pathophysiological perspective, MSM acts predominantly at the tissue and matrix level, addressing extracellular sulfur deficits that cannot be corrected by intracellular redox restoration alone. Its function is therefore complementary to intracellular thiol donors, forming part of a multi-compartment sulfur restoration strategy aimed at rebuilding both the metabolic and structural foundations of tissue repair [84].
Table 4.
Complementary Roles of Intracellular and Extracellular Sulfur Donors.
| Therapeutic Agent | Primary Compartment | Main Molecular Target | Principal Biological Effect | Role in Regenerative Strategy |
|---|---|---|---|---|
| N-Acetylcysteine | Intracellular | Cysteine → Glutathione → Redox systems | Restores antioxidant defense and mitochondrial function | Reverses metabolic and redox collapse |
| Methylsulfonylmethane | Extracellular / Matrix | Sulfur for structural protein stabilization | Enhances ECM strength and integrity | Rebuilds structural scaffold for repair |
5. The Dual-Sulfur Regenerative Hypothesis
Chronic non-healing diabetic wounds arise from the convergence of intracellular metabolic failure and extracellular structural instability, two pathologies that are mechanistically linked through sulfur dysregulation. Restoration of only one biological compartment is insufficient to overcome this integrated failure because cellular recovery cannot translate into tissue regeneration in the absence of a stable extracellular framework, and conversely, matrix repair cannot proceed when cellular metabolism remains redox-collapsed and energy-deprived. A regenerative strategy must therefore correct sulfur-dependent dysfunction across intracellular and extracellular domains in a coordinated manner.
Within this framework, N-acetylcysteine and methylsulfonylmethane fulfill distinct but complementary biological roles. NAC primarily restores intracellular sulfur availability by replenishing cysteine pools required for glutathione synthesis, redox buffering, and iron–sulfur cluster biogenesis. These corrections normalize intracellular redox signaling, revive mitochondrial bioenergetics, and suppress maladaptive inflammatory transcriptional programs, thereby reprogramming keratinocytes, fibroblasts, endothelial cells, and immune cells toward a pro-regenerative phenotype. However, these cellular improvements remain functionally constrained when the surrounding extracellular matrix is mechanically fragile and biochemically disordered.
In contrast, MSM exerts its dominant effects within the extracellular compartment by supplying bioavailable sulfur necessary for protein crosslinking and stabilization of disulfide bonds in structural macromolecules such as collagen, elastin, and keratin. Restoration of extracellular matrix integrity re-establishes tensile strength, elasticity, and spatial organization of the wound scaffold while simultaneously preserving biochemical signaling niches that regulate cell adhesion, migration, and mechanotransduction. Without this extracellular repair, improvements in cellular redox balance and mitochondrial function cannot be translated into coordinated tissue architecture. The dual-sulfur regenerative hypothesis therefore proposes that simultaneous intracellular and extracellular sulfur restoration is required to reverse the chronic non-healing state of diabetic wounds, directly targeting the upstream molecular bottlenecks that prevent progression from persistent inflammation to active tissue repair (Figure 1).
Table 5.
Conceptual Framework of the Dual-Sulfur Regenerative Hypothesis.
| Pathological Domain | Primary Sulfur Defect | Biological Consequence | Targeted Sulfur Donor | Expected Regenerative Effect |
|---|---|---|---|---|
| Intracellular redox collapse | Cysteine depletion, low glutathione | Oxidative stress, inflammatory persistence | N-acetylcysteine | Restored redox control and cytoprotection |
| Mitochondrial dysfunction | Impaired Fe–S cluster biogenesis | Reduced ATP, impaired cell function | N-acetylcysteine | Bioenergetic recovery |
| ECM structural failure | Defective disulfide bonding in matrix proteins | Weak scaffold, poor mechanotransduction | Methylsulfonylmethane | Matrix stabilization and strength |
| Impaired cell–matrix signaling | Oxidative damage to adhesion proteins | Reduced migration and organization | Combined NAC + MSM | Coordinated tissue regeneration |
6. Integrated Inflammatory Resolution, Mitochondrial Recovery, and Tissue Regeneration: Discussion and Therapeutic Implications
The persistent non-healing phenotype of diabetic wounds can be more accurately understood as the consequence of a unified sulfur-dependent pathophysiological collapse rather than as the additive result of ischemia, neuropathy, or infection. Within the diabetic wound microenvironment, chronic hyperglycemia drives excessive production of reactive oxygen species through mitochondrial electron leakage, activation of NADPH oxidases, and accumulation of advanced glycation end products [85].
This sustained oxidative burden progressively consumes intracellular cysteine reserves, suppresses glutathione synthesis, and disrupts thiol-dependent redox buffering systems that normally constrain inflammatory signaling and preserve protein structure [86]. As redox homeostasis collapses, inflammatory transcriptional programs remain pathologically active, mitochondrial function deteriorates, and extracellular matrix integrity becomes progressively destabilized, creating a self-reinforcing molecular state incompatible with regeneration [87].
From a mechanistic standpoint, sulfur deficiency represents the nodal defect that synchronizes these failures. Impaired cystine uptake and cysteine availability limit glutathione synthesis, allowing oxidative stress to persist and continuously activate redox-sensitive inflammatory pathways. This redox imbalance sustains nuclear factor-κB signaling while suppressing cytoprotective transcriptional programs, preventing immune cells from transitioning from tissue-destructive to reparative phenotypes [88]. In parallel, cysteine depletion compromises the biogenesis and stability of mitochondrial iron–sulfur clusters, impairing electron transport chain function and reducing ATP generation [89]. Energy deprivation directly constrains cytoskeletal remodeling, focal adhesion turnover, and directional migration of keratinocytes and fibroblasts, while endothelial cells lose angiogenic competence due to insufficient bioenergetic support and disrupted hypoxia-responsive signaling [90,91].
These intracellular defects are further amplified at the tissue level by sulfur-dependent destabilization of extracellular matrix proteins, where impaired disulfide bond formation and crosslinking weaken collagen and elastin architecture, disrupt mechanotransduction signaling, and prevent coordinated cellular assembly within the wound bed [12].
Within this framework, the dual-sulfur regenerative hypothesis proposes that effective reversal of diabetic wound chronicity requires simultaneous restoration of sulfur availability across both intracellular and extracellular compartments. Replenishment of intracellular cysteine through sulfur donation reconstitutes glutathione synthesis, restores redox buffering capacity, and attenuates oxidative stress-driven inflammatory signaling. As redox equilibrium is progressively re-established, immune cells regain the ability to resolve inflammation, macrophages shift toward reparative phenotypes, and oxidative injury to mitochondrial components is reduced [12]. Recovery of mitochondrial membrane potential and oxidative phosphorylation efficiency restores ATP availability, thereby enabling energy-dependent processes such as keratinocyte migration, fibroblast proliferation, collagen biosynthesis, and endothelial sprouting that are essential for wound closure and neovascularization [92].
Concurrently, restoration of extracellular sulfur availability stabilizes the structural framework required for regeneration. Enhanced sulfur incorporation into matrix proteins facilitates proper disulfide bonding and crosslinking within collagen, elastin, and keratin, improving tensile strength, elasticity, and resistance to proteolytic degradation. A mechanically competent extracellular matrix reactivates integrin-mediated signaling and mechanotransduction pathways, allowing intracellular bioenergetic recovery to be translated into organized tissue architecture rather than disordered cell accumulation. Stabilization of the matrix also preserves growth factor gradients and signaling niches, reinforcing coordinated spatial repair processes.
From a translational perspective, these mechanistic insights support a rational topical therapeutic strategy based on simultaneous intracellular and extracellular sulfur repletion. A dual-sulfur topical formulation combining N-acetylcysteine (NAC) and methylsulfonylmethane (MSM) is proposed to target complementary compartments of wound pathophysiology. Based on existing dermatologic and wound-healing literature, NAC may be incorporated at concentrations of approximately 2–3% (w/v), which have demonstrated efficacy in reducing oxidative stress, restoring glutathione synthesis, and improving cellular migration while maintaining favorable local tolerability [93,94].
MSM may be included at concentrations of approximately 5–8% (w/v), consistent with its documented topical safety profile and its capacity to support structural protein stabilization, reduce matrix degradation, and modulate inflammatory signaling [95].
The proposed method of application involves gentle wound cleansing with sterile saline, followed by application of a thin layer of the dual-sulfur formulation to the wound bed and margins, and coverage with a non-adherent, semi-occlusive dressing to preserve moisture balance while permitting gas exchange. Application may be performed once to twice daily depending on wound severity, exudate levels, and tissue tolerance. A treatment duration of at least two to four weeks is proposed, with periodic clinical assessment of granulation tissue formation, epithelialization, exudate reduction, and local inflammatory signs.
The convergence of intracellular redox normalization, mitochondrial recovery, and extracellular matrix stabilization shifts the diabetic wound microenvironment from a state dominated by inflammatory stress and metabolic insufficiency toward one permissive for coordinated regeneration. This sulfur-centered perspective reframes diabetic wounds as locally sulfur-depleted tissues trapped in a redox-collapsed and structurally misassembled state, rather than as lesions limited by perfusion or microbial burden alone. Therapeutic restoration of sulfur homeostasis therefore functions as an upstream molecular reconditioning strategy that enables downstream regenerative pathways to operate effectively. By correcting the fundamental biochemical and bioenergetic constraints that arrest healing, sulfur-targeted interventions hold the potential not only to accelerate wound closure but also to restore tissue quality, vascular stability, and resistance to recurrence, thereby redefining the biological expectations of diabetic wound therapy.
7. Conclusion
Diabetic wound chronicity arises from a convergence of intracellular oxidative failure and extracellular structural instability rooted in sulfur deficiency. Correcting only one compartment leaves the regenerative process incomplete. The proposed dual-sulfur strategy integrates intracellular redox restoration through NAC with extracellular matrix stabilization via MSM, thereby reconnecting metabolic recovery with structural repair. By acting upstream of inflammation, mitochondrial dysfunction, and ECM degradation, sulfur restoration reconditions the wound microenvironment to support coordinated healing rather than merely accelerating closure. This mechanism-driven framework reframes diabetic wound therapy from symptomatic management to metabolic and structural reprogramming, offering a biologically coherent foundation for next-generation regenerative interventions.
Author Contributions
Maher Monir Akl: Conceived and developed the central research hypothesis, designed the immunopathophysiological framework, performed data integration and analysis, and drafted the original manuscript. Ahmed Ali. El-Nagar: Contributed to literature review, mechanistic interpretation related to mucosal immunity and epithelial barrier biology, and participated in manuscript revision. Amr Ahmed: Provided clinical insight, supervised the translational and public health relevance of the work, and critically revised the manuscript for clinical accuracy. All authors contributed to the conception, design, data collection, analysis, and manuscript preparation equally.
Funding Information
No financial support was received for this study.
Ethical Approval Statement
This study did not involve human participants or animal experiments.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors declare that there are no conflicts of interest.
References
- Akkus, G.; Sert, M. Diabetic foot ulcers: A devastating complication of diabetes mellitus continues non-stop in spite of new medical treatment modalities. World journal of diabetes 2022, 13(12), 1106–1121. [Google Scholar] [CrossRef]
- Ramirez-Acuña, J. M.; Cardenas-Cadena, S. A.; Marquez-Salas, P. A.; Garza-Veloz, I.; Perez-Favila, A.; Cid-Baez, M. A.; Flores-Morales, V.; Martinez-Fierro, M. L. Diabetic Foot Ulcers: Current Advances in Antimicrobial Therapies and Emerging Treatments. Antibiotics (Basel, Switzerland) 2019, 8(4), 193. [Google Scholar] [CrossRef]
- Wang, R.; Gu, S.; Kim, Y. H.; Lee, A.; Lin, H.; Jiang, D. Diabetic Wound Repair: From Mechanism to Therapeutic Opportunities. MedComm 2025, 6(10), e70406. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, L.; Chen, Y.; Thomas, E. R.; Zhou, S.; Yang, Y.; Liu, K.; Wu, J.; Li, X. Mitochondrial dysfunction in diabetic ulcers: pathophysiological mechanisms and targeted therapeutic strategies. Frontiers in cell and developmental biology 2025, 13, 1625474. [Google Scholar] [CrossRef]
- Kunkemoeller, B.; Kyriakides, T. R. Redox Signaling in Diabetic Wound Healing Regulates Extracellular Matrix Deposition. Antioxidants & redox signaling 2017, 27(12), 823–838. [Google Scholar] [CrossRef]
- Yan, L. J. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. Journal of diabetes research 2014, 2014, 137919. [Google Scholar] [CrossRef]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10(5), 727. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A. Chronic hyperglycemia and cardiovascular dysfunction: an in-depth exploration of metabolic and cellular pathways in type 2 diabetes mellitus. Cardiovasc. Diabetol. – Endocrinol. Rep. 2025, 11, 39. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L. S.; Reverdatto, S.; Gugger, P. F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A. M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Frontiers in cardiovascular medicine 2020, 7, 37. [Google Scholar] [CrossRef]
- Wang, L; Jiang, Y; Zhao, C. The effects of advanced glycation end-products on skin and potential anti-glycation strategies. Exp Dermatol 2024, 33, e15065. [Google Scholar] [CrossRef]
- Hou, Y.; Lv, B.; Du, J.; et al. Sulfide regulation and catabolism in health and disease. Sig Transduct Target Ther 2025, 10, 174. [Google Scholar] [CrossRef]
- Akl, MM; Ahmed, A. Disruption of the transsulfuration pathway as a sulfur-driven etiology of insulin resistance: proinsulin misfolding, disulfide bond deformation, and PDI dysregulation. Explor Endocr Metab Dis. 2025, 2, 101444. [Google Scholar] [CrossRef]
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. The Journal of biological chemistry 2017, 292(48), 19721–19732. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S. J.; Huang, Y.; Lambros, M.; Gout, P. W.; Kalivas, P. W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; Smith, S. B.; Ganapathy, V.; Maher, P. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & redox signaling 2013, 18(5), 522–555. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Communications 2018, 38, 1–13 12. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, J.; Ni, Y.; Li, G.; Wang, Y.; Cao, Y.; Zhou, M.; Zhao, C. The role of SLC7A11 in diabetic wound healing: novel insights and new therapeutic strategies. Frontiers in immunology 2024, 15, 1467531. [Google Scholar] [CrossRef]
- Mukwevho, E.; Ferreira, Z.; Ayeleso, A. Potential role of sulfur-containing antioxidant systems in highly oxidative environments. Molecules (Basel, Switzerland) 2014, 19(12), 19376–19389. [Google Scholar] [CrossRef]
- Mukwevho, E.; Ferreira, Z.; Ayeleso, A. Potential role of sulfur-containing antioxidant systems in highly oxidative environments. Molecules (Basel, Switzerland) 2014, 19(12), 19376–19389. [Google Scholar] [CrossRef] [PubMed]
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N. T. Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. International wound journal 2017, 14(1), 89–96. [Google Scholar] [CrossRef]
- Lopes, F. B.; Sarandy, M. M.; Novaes, R. D.; Valacchi, G.; Gonçalves, R. V. OxInflammatory Responses in the Wound Healing Process: A Systematic Review. Antioxidants 2024, 13(7), 823. [Google Scholar] [CrossRef] [PubMed]
- Francioso, Antonio; Baseggio Conrado, Alessia; Mosca, Luciana; Fontana, Mario. Chemistry and Biochemistry of Sulfur Natural Compounds: Key Intermediates of Metabolism and Redox Biology. Oxidative Medicine and Cellular Longevity 2020, 2020(8294158), 27 pages. [Google Scholar] [CrossRef]
- Lu, S. C. Regulation of glutathione synthesis. Molecular aspects of medicine 2009, 30(1-2), 42–59. [Google Scholar] [CrossRef]
- Yan, Y.; Teng, H.; Hang, Q.; Kondiparthi, L.; Lei, G.; Horbath, A.; Liu, X.; Mao, C.; Wu, S.; Zhuang, L.; James You, M.; Poyurovsky, M. V.; Ma, L.; Olszewski, K.; Gan, B. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nature communications 2023, 14(1), 3673. [Google Scholar] [CrossRef]
- Elajaili, H.; Lyttle, B. D.; Lewis, C. V.; Bardill, J. R.; Dee, N.; Seal, S.; Nozik, E. S.; Liechty, K. W.; Zgheib, C. Increased ROS and Persistent Pro-Inflammatory Responses in a Diabetic Wound Healing Model (db/db): Implications for Delayed Wound Healing. International journal of molecular sciences 2025, 26(10), 4884. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, L.; Chen, Y.; Thomas, E. R.; Zhou, S.; Yang, Y.; Liu, K.; Wu, J.; Li, X. Mitochondrial dysfunction in diabetic ulcers: pathophysiological mechanisms and targeted therapeutic strategies. Frontiers in cell and developmental biology 2025, 13, 1625474. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Yang, Y.; Wang, X.; Chung, M.; Zhang, L.; Cai, S.; Pan, X.; Pan, Y. Targeting the AGEs-RAGE axis: pathogenic mechanisms and therapeutic interventions in diabetic wound healing. Frontiers in medicine 2025, 12, 1667620. [Google Scholar] [CrossRef]
- Guo, Y. L.; Niu, W. J.; Jiao, H. R.; Li, Y. P.; Xu, C.; Zhou, X.; Wang, J. Crosstalk between oxidative stress and inflammatory pathways: Natural therapeutic approaches for diabetic wound healing. World journal of diabetes 2025, 16(11), 111400. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Lemley, K. V.; Andrus, J. P.; De Rosa, S. C.; Holmgren, A.; Jones, D.; Jahoor, F.; Kopke, R.; Cotgreave, I.; Bottiglieri, T.; Kaplowitz, N.; Nakamura, H.; Staal, F.; Ela, S. W.; Atkuri, K. R.; Tirouvanziam, R.; Heydari, K.; Sahaf, B.; Zolopa, A.; Frye, R. E.; Herzenberg, L. A. Cysteine/Glutathione Deficiency: A Significant and Treatable Corollary of Disease. The Therapeutic Use of N-Acetylcysteine (NAC) in Medicine 2018, 349–386. [Google Scholar] [CrossRef]
- Fujii, J.; Osaki, T.; Soma, Y.; Matsuda, Y. Critical Roles of the Cysteine–Glutathione Axis in the Production of γ-Glutamyl Peptides in the Nervous System. International Journal of Molecular Sciences 2023, 24(9), 8044. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z. Y.; Kong, A. N. The complexity of the Nrf2 pathway: beyond the antioxidant response. The Journal of nutritional biochemistry 2015, 26(12), 1401–1413. [Google Scholar] [CrossRef]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Medicinal research reviews 2012, 32(4), 687–726. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T. W.; Motohashi, H. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiological reviews 2018, 98(3), 1169–1203. [Google Scholar] [CrossRef]
- Sun, R.; Xu, Y.; Ji, Z.; Li, X.; Tao, Z.; Luo, W.; Yao, Y.; Chen, L.; Ma, G. Update on the impact of lipid and glucose control on diabetic wound healing. Metabolism open 2025, 28, 100408. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, J.; Wang, Y.; Deng, F.; Deng, Z. Oxidative Stress: Signaling Pathways, Biological Functions, and Disease. MedComm 2025, 6(7), e70268. [Google Scholar] [CrossRef]
- Tan, S. M.; de Haan, J. B. Combating oxidative stress in diabetic complications with Nrf2 activators: how much is too much? Redox report: communications in free radical research 2014, 19(3), 107–117. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Tong, Z.; Han, B.; Zhang, Z.; Xian, Z.; Yuan, Y.; Duan, X.; Han, S.; Liu, P.; Wang, Z. Synergistic Wound Healing: Unraveling the Multi-Target Effects of Traditional Chinese Medicine and Its Biomaterials on Chronic Wound Pathways. International journal of nanomedicine 2025, 20, 12889–12912. [Google Scholar] [CrossRef]
- Winter, A. D.; McCormack, G.; Page, A. P. Protein disulfide isomerase activity is essential for viability and extracellular matrix formation in the nematode Caenorhabditis elegans. Developmental biology 2007, 308(2), 449–461. [Google Scholar] [CrossRef]
- Kupsco, A.; Schlenk, D. Oxidative stress, unfolded protein response, and apoptosis in developmental toxicity. International review of cell and molecular biology 2015, 317, 1–66. [Google Scholar] [CrossRef] [PubMed]
- Bachar-Wikstrom, E.; Manchanda, M.; Bansal, R.; Karlsson, M.; Kelly-Pettersson, P.; Sköldenberg, O.; Wikstrom, J. D. Endoplasmic reticulum stress in human chronic wound healing: Rescue by 4-phenylbutyrate. International wound journal 2021, 18(1), 49–61. [Google Scholar] [CrossRef]
- Urciuoli, E.; Peruzzi, B. Involvement of the FAK Network in Pathologies Related to Altered Mechanotransduction. International Journal of Molecular Sciences 2020, 21(24), 9426. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harbor perspectives in biology 2023, 15(1), a041231. [Google Scholar] [CrossRef] [PubMed]
- Rustad, K. C.; Wong, V. W.; Gurtner, G. C. The role of focal adhesion complexes in fibroblast mechanotransduction during scar formation. Differentiation; research in biological diversity 2013, 86(3), 87–91. [Google Scholar] [CrossRef]
- Read, A. D.; Bentley, R. E.; Archer, S. L.; Dunham-Snary, K. J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox biology 2021, 47, 102164. [Google Scholar] [CrossRef]
- Stiban, J.; So, M.; Kaguni, L. S. Iron-Sulfur Clusters in Mitochondrial Metabolism: Multifaceted Roles of a Simple Cofactor. Biochemistry. Biokhimiia 2016, 81(10), 1066–1080. [Google Scholar] [CrossRef]
- Read, A. D.; Bentley, R. E.; Archer, S. L.; Dunham-Snary, K. J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox biology 2021, 47, 102164. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, S.; Tian, J.; Yang, F.; Chen, H.; Bai, S.; Kang, J.; Pang, K.; Huang, J.; Dong, M.; Dong, S.; Tian, Z.; Fang, S.; Fan, H.; Lu, F.; Yu, B.; Li, S.; Zhang, W. Impaired Iron-Sulfur Cluster Synthesis Induces Mitochondrial PARthanatos in Diabetic Cardiomyopathy. Advanced science (Weinheim, Baden-Wurttemberg, Germany) 2025, 12(1), e2406695. [Google Scholar] [CrossRef]
- Wu, L.; Huang, F.; Sun, Z.; Zhang, J.; Xia, S.; Zhao, H.; Liu, Y.; Yang, L.; Ding, Y.; Bian, D.; Li, K.; Sun, Y. Downregulation of Iron–Sulfur Cluster Biogenesis May Contribute to Hyperglycemia-Mediated Diabetic Peripheral Neuropathy in Murine Models. Antioxidants 2024, 13(9), 1036. [Google Scholar] [CrossRef]
- Guo, S.; Dipietro, L. A. Factors affecting wound healing. Journal of dental research 2010, 89(3), 219–229. [Google Scholar] [CrossRef]
- Wang, S.; Liang, Y.; Dai, C. Metabolic Regulation of Fibroblast Activation and Proliferation during Organ Fibrosis. Kidney diseases (Basel, Switzerland) 2022, 8(2), 115–125. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D. H.; Lee, H.; Park, C.; Hong, S. H.; Hong, S. H.; Kim, G. Y.; Cha, H. J.; Kim, S.; Kim, H. S.; Hwang, H. J.; Choi, Y. H. Glutathione Induced Immune-Stimulatory Activity by Promoting M1-Like Macrophages Polarization via Potential ROS Scavenging Capacity. Antioxidants (Basel, Switzerland) 2019, 8(9), 413. [Google Scholar] [CrossRef]
- Lopes, F. B.; Sarandy, M. M.; Novaes, R. D.; Valacchi, G.; Gonçalves, R. V. OxInflammatory Responses in the Wound Healing Process: A Systematic Review. Antioxidants (Basel, Switzerland) 2024, 13(7), 823. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxidants & redox signaling 2013, 19(6), 595–637. [Google Scholar] [CrossRef]
- Kwon, D. H.; Lee, H.; Park, C.; Hong, S. H.; Hong, S. H.; Kim, G. Y.; Cha, H. J.; Kim, S.; Kim, H. S.; Hwang, H. J.; Choi, Y. H. Glutathione Induced Immune-Stimulatory Activity by Promoting M1-Like Macrophages Polarization via Potential ROS Scavenging Capacity. Antioxidants (Basel, Switzerland) 2019, 8(9), 413. [Google Scholar] [CrossRef]
- Sharma, S.; Kishen, A. Dysfunctional crosstalk between macrophages and fibroblasts under LPS-infected and hyperglycemic environment in diabetic wounds. Sci Rep 2025, 15, 17233. [Google Scholar] [CrossRef]
- Jindatanmanusan; Punyanuch; Luanraksa; Sivat; Boonsiri; Tanit; Nimmanon; Thirayost; Arnutti; Pasra. Wound Fluid Matrix Metalloproteinase-9 as a Potential Predictive Marker for the Poor Healing Outcome in Diabetic Foot Ulcers. Pathology Research International 2018, 2018(1631325), 5 pages. [Google Scholar] [CrossRef]
- Jones, J. I.; Nguyen, T. T.; Peng, Z.; Chang, M. Targeting MMP-9 in Diabetic Foot Ulcers. Pharmaceuticals (Basel, Switzerland) 2019, 12(2), 79. [Google Scholar] [CrossRef]
- Filep, J. G. Targeting Neutrophils for Promoting the Resolution of Inflammation. Frontiers in immunology 2022, 13, 866747. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, Y. Targeting persistently activated inflammatory microenvironment to promote chronic wound healing. Frontiers in immunology 2025, 16, 1708358. [Google Scholar] [CrossRef] [PubMed]
- Talbott, H. E.; Mascharak, S.; Griffin, M.; Wan, D. C.; Longaker, M. T. Wound healing, fibroblast heterogeneity, and fibrosis. Cell stem cell 2022, 29(8), 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Shan, X. M.; Chen, C. W.; Zou, D. W.; Gao, Y. B.; Ba, Y. Y.; He, J. X.; Zhu, Z. Y.; Liang, J. J. Suppression of ferroptosis through the SLC7A11/glutathione/glutathione peroxidase 4 axis contributes to the therapeutic action of the Tangshenning formula on diabetic renal tubular injury. Chinese medicine 2024, 19(1), 151. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules (Basel, Switzerland) 2015, 20(5), 8742–8758. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Raghavan, S.; Nikolova, M.; Polak, L.; Pasolli, H. A.; Beggs, H. E.; Reichardt, L. F.; Fuchs, E. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. The Journal of cell biology 2007, 176(5), 667–680. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Lofaro, F. D.; Bonacorsi, S.; Mazzilli, A.; Garcia-Fernandez, M.; Quaglino, D. The Role of Fibroblasts in Skin Homeostasis and Repair. Biomedicines 2024, 12(7), 1586. [Google Scholar] [CrossRef]
- Landén, N. X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: a critical step during wound healing. Cellular and molecular life sciences: CMLS 2016, 73(20), 3861–3885. [Google Scholar] [CrossRef]
- Li, H. S.; Zhou, Y. N.; Li, L.; Li, S. F.; Long, D.; Chen, X. L.; Zhang, J. B.; Feng, L.; Li, Y. P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox biology 2019, 25, 101109. [Google Scholar] [CrossRef]
- Pan, Y.; Mansfield, K. D.; Bertozzi, C. C.; Rudenko, V.; Chan, D. A.; Giaccia, A. J.; Simon, M. C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Molecular and cellular biology 2007, 27(3), 912–925. [Google Scholar] [CrossRef]
- Heun, Y.; Pogoda, K.; Anton, M.; Pircher, J.; Pfeifer, A.; Woernle, M.; Ribeiro, A.; Kameritsch, P.; Mykhaylyk, O.; Plank, C.; Kroetz, F.; Pohl, U.; Mannell, H. HIF-1α Dependent Wound Healing Angiogenesis In Vivo Can Be Controlled by Site-Specific Lentiviral Magnetic Targeting of SHP-2. Molecular therapy: the journal of the American Society of Gene Therapy 2017, 25(7), 1616–1627. [Google Scholar] [CrossRef]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T. P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell chemical biology 2018, 25(4), 447–459.e4. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, S.; Tanaka, T.; Mano, R.; Kondo, S.; Kodama, S. SLC7A11/xCT-mediated Cystine Uptake Regulates Intracellular Glutathione and Promotes Antioxidant Defense in Lymphatic Endothelial Cells. Anticancer research 2025, 45(1), 65–71. [Google Scholar] [CrossRef]
- Tenório, M. C. D. S.; Graciliano, N. G.; Moura, F. A.; Oliveira, A. C. M.; Goulart, M. O. F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants (Basel, Switzerland) 2021, 10(6), 967. [Google Scholar] [CrossRef]
- McGinley, C.; Adeyemi, O.; Oyolola, O.; Ford, B. D.; Ford, G. D. Redox State of Glutathione and Cysteine in Plasma Following Acute Stroke. Antioxidants 2026, 15(1), 117. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules (Basel, Switzerland) 2020, 25(22), 5474. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Coda, A. R. D.; Trecca, M. I.; Lo Buglio, A.; Serviddio, G.; Vendemiale, G. Redox Imbalance in Inflammation: The Interplay of Oxidative and Reductive Stress. Antioxidants (Basel, Switzerland) 2025, 14(6), 656. [Google Scholar] [CrossRef]
- Read, A. D.; Bentley, R. E.; Archer, S. L.; Dunham-Snary, K. J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox biology 2021, 47, 102164. [Google Scholar] [CrossRef]
- Amador-Martínez, I.; Aparicio-Trejo, O. E.; Aranda-Rivera, A. K.; Bernabe-Yepes, B.; Medina-Campos, O. N.; Tapia, E.; Cortés-González, C. C.; Silva-Palacios, A.; Roldán, F. J.; León-Contreras, J. C.; Hernández-Pando, R.; Saavedra, E.; Gonzaga-Sánchez, J. G.; Ceja-Galicia, Z. A.; Sánchez-Lozada, L. G.; Pedraza-Chaverri, J. Effect of N-Acetylcysteine in Mitochondrial Function, Redox Signaling, and Sirtuin 3 Levels in the Heart During Cardiorenal Syndrome Type 4 Development. Antioxidants (Basel, Switzerland) 2025, 14(3), 367. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, J.; Wang, Y.; Deng, F.; Deng, Z. Oxidative Stress: Signaling Pathways, Biological Functions, and Disease. MedComm 2025, 6, e70268. [Google Scholar] [CrossRef]
- Nigam, M.; Punia, B.; Dimri, D. B.; Mishra, A. P.; Radu, A.-F.; Bungau, G. Reactive Oxygen Species: A Double-Edged Sword in the Modulation of Cancer Signaling Pathway Dynamics. Cells 2025, 14(15), 1207. [Google Scholar] [CrossRef]
- Diller, R. B.; Tabor, A. J. The Role of the Extracellular Matrix (ECM) in Wound Healing: A Review. Biomimetics (Basel, Switzerland) 2022, 7(3), 87. [Google Scholar] [CrossRef]
- Butawan, M.; Benjamin, R. L.; Bloomer, R. J. Methylsulfonylmethane: Applications and Safety of a Novel Dietary Supplement. Nutrients 2017, 9(3), 290. [Google Scholar] [CrossRef]
- Karimi, M.; Ignasiak, M.; Chan, B.; et al. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Sci Rep 2016, 6, 38572. [Google Scholar] [CrossRef]
- Yamauchi, M.; Taga, Y.; Hattori, S.; Shiiba, M.; Terajima, M. Analysis of collagen and elastin cross-links. Methods in cell biology 2018, 143, 115–132. [Google Scholar] [CrossRef]
- Huang, Y.; Kyriakides, T. R. The role of extracellular matrix in the pathophysiology of diabetic wounds. Matrix biology plus 2020, 6–7, 100037. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L. E.; Minasian, R. A.; Caterson, E. J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Advances in wound care 2016, 5(3), 119–136. [Google Scholar] [CrossRef]
- Jeong, H.-L.; Kang, E.-B.; Yun, S.-G.; Park, D.-b.; Lim, J.-O.; Suh, J.-S. Effect of a Silk Sericin and Methylsulfonylmethane (MSM) Blends on Inflammatory Response and Wound Healing. Applied Sciences 2023, 13(1), 288. [Google Scholar] [CrossRef]
- Hong, L.; Li, M.; Fan, Y. Oxidative stress and programmed cell death in diabetic wounds: A comprehensive review. Science progress 2025, 108(3), 368504251370676. [Google Scholar] [CrossRef] [PubMed]
- Baba, S. P.; Bhatnagar, A. ROLE OF THIOLS IN OXIDATIVE STRESS. Current opinion in toxicology 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Galán, Y. I.; Guzmán-Silahua, S.; Trujillo-Rangel, W. Á.; Rodríguez-Lara, S. Q. Role of Ischemia/Reperfusion and Oxidative Stress in Shock State. Cells 2025, 14(11), 808. [Google Scholar] [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V.; et al. Redox regulation of the immune response. Cell Mol Immunol 2022, 19, 1079–1101. [Google Scholar] [CrossRef]
- Braymer, J. J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. The Journal of biological chemistry 2017, 292(31), 12754–12763. [Google Scholar] [CrossRef]
- DeWane, G.; Salvi, A. M.; DeMali, K. A. Fueling the cytoskeleton - links between cell metabolism and actin remodeling. Journal of cell science 2021, 134(3), jcs248385. [Google Scholar] [CrossRef]
- DeWane, G.; Salvi, A. M.; DeMali, K. A. Fueling the cytoskeleton - links between cell metabolism and actin remodeling. Journal of cell science 2021, 134(3), jcs248385. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Torres, M.; Bachar-Wikström, E.; Wikström, J. D. Multifaceted roles of mitochondria in wound healing and chronic wound pathogenesis. Frontiers in cell and developmental biology 2023, 11, 1252318. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M. L.; Huang, H. P.; Hsu, J. D.; Lai, Y. R.; Hsiao, Y. P.; Lu, F. J.; Chang, H. R. Topical N-acetylcysteine accelerates wound healing in vitro and in vivo via the PKC/Stat3 pathway. International journal of molecular sciences 2014, 15(5), 7563–7578. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M. L.; Huang, H. P.; Hsu, J. D.; Lai, Y. R.; Hsiao, Y. P.; Lu, F. J.; Chang, H. R. Topical N-acetylcysteine accelerates wound healing in vitro and in vivo via the PKC/Stat3 pathway. International journal of molecular sciences 2014, 15(5), 7563–7578. [Google Scholar] [CrossRef]
- Butawan, M.; Benjamin, R. L.; Bloomer, R. J. Methylsulfonylmethane: Applications and Safety of a Novel Dietary Supplement. Nutrients 2017, 9(3), 290. [Google Scholar] [CrossRef]
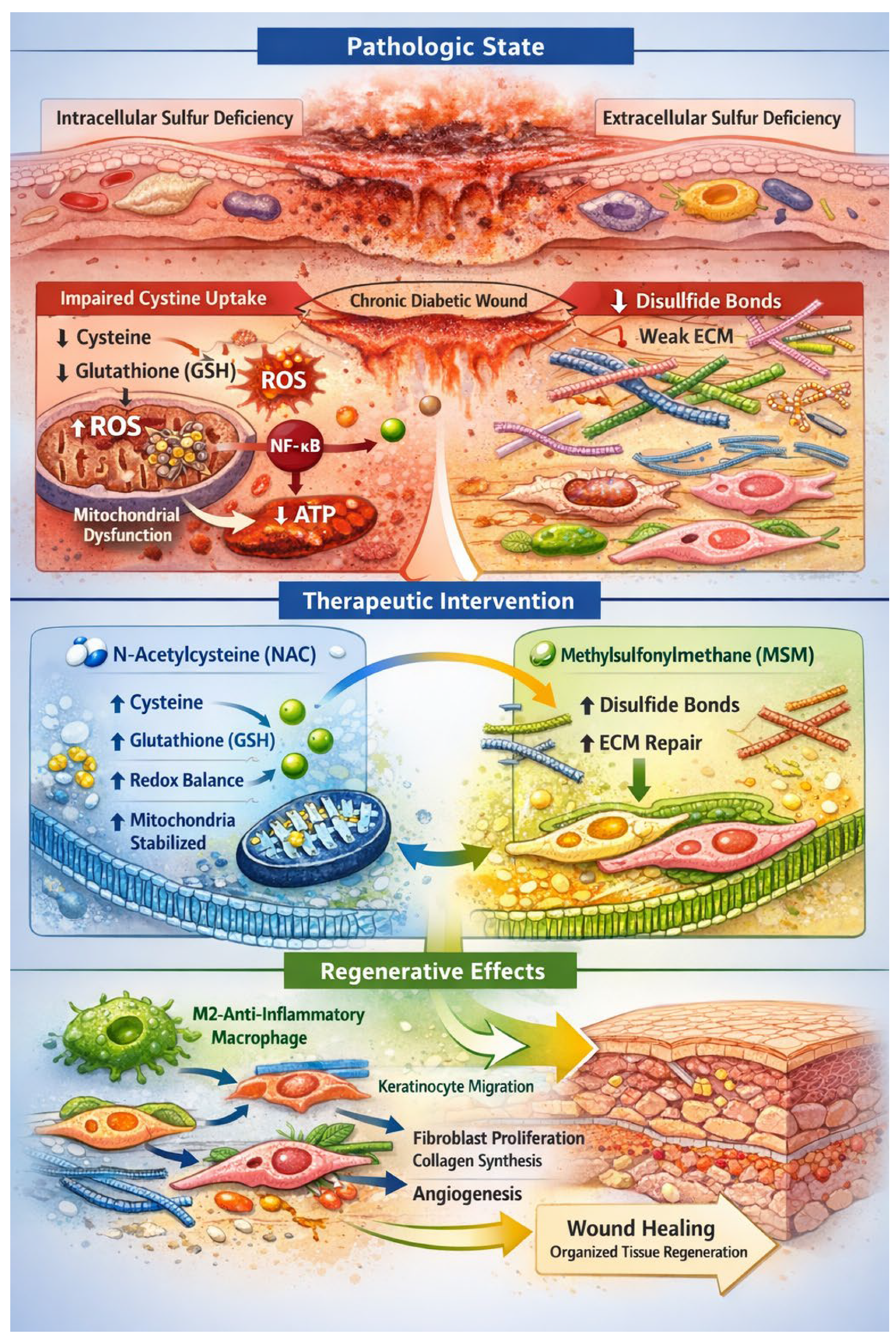
Figure 1.
Dual-Sulfur Pathophysiology and Regenerative Hypothesis in Chronic Diabetic Wounds. Schematic representation of the proposed pathophysiological mechanism underlying chronic diabetic wounds and the dual-sulfur regenerative strategy. The diagram illustrates the diabetic wound microenvironment, showing keratinocytes, fibroblasts, endothelial cells, and immune cells (macrophages and neutrophils) in a state of persistent inflammation and oxidative stress. Intracellular sulfur deficiency is depicted as impaired cystine uptake via the SLC7A11/xCT transporter, leading to cysteine depletion, reduced glutathione (GSH) synthesis, reactive oxygen species (ROS) accumulation, NF-κB activation, mitochondrial iron–sulfur cluster destabilization, and impaired ATP production. Extracellular sulfur deficiency is shown in the extracellular matrix (ECM), with collagen, elastin, and keratin exhibiting disrupted disulfide bonds, weak crosslinking, impaired mechanotransduction, and reduced cell adhesion and migration. Therapeutic intervention includes topical N-acetylcysteine (NAC) restoring intracellular cysteine, GSH synthesis, redox balance, and mitochondrial function, and methylsulfonylmethane (MSM) reinforcing ECM disulfide bonding and structural integrity. The regenerative cascade highlights macrophage polarization (M1 → M2), keratinocyte migration, fibroblast proliferation, collagen synthesis, endothelial sprouting, and coordinated wound closure. Arrows indicate the sequential flow of molecular and cellular events. Color-coded compartments distinguish intracellular and extracellular domains, emphasizing the integrated sulfur-dependent repair mechanism.
Figure 1.
Dual-Sulfur Pathophysiology and Regenerative Hypothesis in Chronic Diabetic Wounds. Schematic representation of the proposed pathophysiological mechanism underlying chronic diabetic wounds and the dual-sulfur regenerative strategy. The diagram illustrates the diabetic wound microenvironment, showing keratinocytes, fibroblasts, endothelial cells, and immune cells (macrophages and neutrophils) in a state of persistent inflammation and oxidative stress. Intracellular sulfur deficiency is depicted as impaired cystine uptake via the SLC7A11/xCT transporter, leading to cysteine depletion, reduced glutathione (GSH) synthesis, reactive oxygen species (ROS) accumulation, NF-κB activation, mitochondrial iron–sulfur cluster destabilization, and impaired ATP production. Extracellular sulfur deficiency is shown in the extracellular matrix (ECM), with collagen, elastin, and keratin exhibiting disrupted disulfide bonds, weak crosslinking, impaired mechanotransduction, and reduced cell adhesion and migration. Therapeutic intervention includes topical N-acetylcysteine (NAC) restoring intracellular cysteine, GSH synthesis, redox balance, and mitochondrial function, and methylsulfonylmethane (MSM) reinforcing ECM disulfide bonding and structural integrity. The regenerative cascade highlights macrophage polarization (M1 → M2), keratinocyte migration, fibroblast proliferation, collagen synthesis, endothelial sprouting, and coordinated wound closure. Arrows indicate the sequential flow of molecular and cellular events. Color-coded compartments distinguish intracellular and extracellular domains, emphasizing the integrated sulfur-dependent repair mechanism.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



