
Submitted:
01 February 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Primary sarcopenia is an age-associated degenerative disorder marked by progressive loss of skeletal muscle mass, strength, and function, representing a major driver of frailty and morbidity after midlife. Convergent evidence from human muscle biopsies, aged rodents, Drosophila, and myogenic cell models identifies mitochondrial dysfunction as the proximal cause of this decline, with impaired mitophagy emerging as the central mechanistic failure. Aging muscle exhibits reduced mitochondrial content, compromised oxidative phosphorylation, dysregulated dynamics favoring excessive fission, and accumulation of oxidized, depolarized mitochondria. These defects closely associate with a collapse in mitophagy flux, characterized by coordinated downregulation of PINK1–PARKIN signaling, receptor-mediated pathways (BNIP3, NIX, FUNDC1), autophagosome formation, and lysosomal clearance, resulting in defective mitochondrial turnover and bioenergetic insufficiency. Genetic or pharmacological restoration of mitophagy reverses these phenotypes, preserving muscle mass, respiratory capacity, and functional performance while extending lifespan in multiple model organisms. Notably, the natural compounds urolithin A and spermidine consistently activate mitophagy, improve mitochondrial quality control, and enhance muscle strength and endurance in aged animals and sedentary middle-aged humans. Collectively, these data position age-related mitophagy suppression as the pivotal driver of skeletal muscle aging and define mitochondrial quality control as a tractable, mechanistic therapeutic target to delay or reverse primary sarcopenia.
Keywords:
primary sarcopenia
; skeletal muscle aging
; mitochondrial dysfunction
; mitophagy
; mitochondrial quality control
; urolithin A
; spermidine
; natural mitophagy inducers
; Drosophila melanogaster
; C57BL/6J mice
; C2C12 myotubes
; human skeletal muscle biopsies
; therapeutics
I. Introduction
Mitochondria are key organelles for cellular function in aging. Mitochondria are double-membrane eukaryotic organelles that produce ATP, with a potential membrane around -140mV to -180mV, and that are indispensable for life and health in the majority of the eukaryotic organisms [1,2,3]. Decades of study on cellular destinies (apoptosis, necrosis, division, and differentiation), age-associated diseases, and aging have expanded vastly the comprehension of mitochondria as multifaceted organelles that show critical roles in metabolic homeostasis, innate immunity, calcium regulation, mitochondria-nucleus communication, the decision of cellular survival, its resistance to stress and death [4,5,6,7]. To fulfill these roles, it’s essential to maintain a healthy population of mitochondria, with their adequate functions and membrane potential.
The maintenance of the function of mitochondria depends on 2 mitochondrial systems that collaborate, these are the oxidative phosphorylation system (OXPHOS) and the mitochondrial life cycle, also named mitochondrial quality control system (MLC/MQC) [8]. Both systems complement each other to sustain mitochondrial function, energy production (ATP), detoxification of oxygen reactive species (ROS), appropriate metabolism, and retrograde signaling. The oxidative phosphorylation system, corresponding to the electron transport chain, which is composed of ubiquinone, cytochrome C, 4 protein complexes, and one ATP-synthase, is coded by two separate genetic materials: mitochondrial DNA (mtDNA) and nuclear DNA [9].
Mitochondria are extremely dynamic organelles and have to keep functioning through life, for that reason mitochondria submit to a quality control system that corresponds to their life cycle, this cycle includes damaged mitochondrial basal replacement for new, functional, and healthy mitochondria. Mitochondria life cycle is balanced by its biogenesis, regulated mainly by PGC1-α protein (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha), remodeling (consisting of fission, mainly by dynamin-related protein 1 (DRP1), and fission 1 (FIS1) proteins. While for fusion events, mainly by autosomal-dominant optic atrophy (OPA1) and mitofusin 1-2 (MFN1-2) proteins, also known as mitochondrial dynamics [10,11], and the selective mitochondria autophagy, known as mitophagy [10,12,13,14]. A flaw in the mitochondrial life cycle is associated with aging and diverse age-related diseases such as primary sarcopenia [4,10,11,15,16]. Hence, the cells can maintain a healthy mitochondrial network through their adequate functioning and the balance of their mitochondrial dynamics [14,17,18,19]. Mitochondria are subjected to these processes, degrading poorly-functional mitochondria on the go, and while specific proteins vary between model organisms, these processes are highly conserved [20].
Mitochondrial selective autophagy or mitophagy is essential for deficient mitochondrial recycling and protecting functional mitochondria, keeping cellular homeostasis and the other mitochondrial functions. Mitophagy is a type of macroautophagy that initiates due to different signals from the mitochondria or the cellular environment that leads to the formation of a double-membrane phagophore which elongates and closes to generate a mature double-membrane autophagosome that engulfs the mitochondria, then the autophagosomes fuse with the lysosomes to degrade their content [21]. Although the homeostasis pathways mediate cellular responses to mitochondrial damage, persisting damage triggers the elimination of the entire organelle via mitophagy [18]. Mitophagy allows the removal of dysfunctional, depolarized, and/or damaged mitochondria for their renovation, ROS management, mitochondrial programmed clearance, and cell development [22,23,24,25,26]. When mitophagy is induced mitochondrial biomass degradation is higher, resulting in less mitochondrial protein abundance, as complex IV cytochrome oxidase (COX) and mitochondrial outer membrane translocator 20 (TOM-20) proteins, and the opposite occur when mitophagy is inhibited [27]. The decrease of the mitophagy process leads to a deficient removal of dysfunctional, depolarized, and/or damaged mitochondria [28] being this process essential for tissue functioning. The dysfunction of mitophagy is implicated in developing diseases like primary sarcopenia in skeletal muscle, cancer, and aging [4,10,11,15,16].
The process of mitophagy requires two systems: autophagy’s main machinery, composed partly of Beclin-1, ATG-1/7/8 (Autophagy-related proteins 1,7 and 8), for the phagophore formation, Lamp-1/2 (Lysosome-associated membrane proteins 1 and 2) for the acidification of the lysosome, LC3-I (Microtubule-associated 1A/1B-light chain protein I), which is cytosolic, and LC3-II, when LC3-I conjugates with the phagophore phosphatidylinositol-3-phosphate lipids, that anchors the phagophore to the autophagy/mitophagy adaptors like p62 (Sequestosome 1), sending different cargo for degradation, common to all types of autophagy [21]. The second system are mitochondrial receptors, which consist in part by PTEN-induced kinase 1 (PINK1), PARKIN, FUN14 domain protein containing 1 (FUNDC-1), BCL2 interacting protein 3 (BNIP3) and BNIP3 like protein (NIX), a group of mitochondrial or cytosolic proteins, and adaptors like p62, and optineurin (OPTN), needed for the assembly of the mitochondria with the main autophagic machinery via their LC3 interacting region (LC3) [29].
Mitophagy is characterized by having non-redundant pathways [7,30]. Depending on the physiological context mitophagy is classified as basal, which corresponds to the ubiquitin-dependent pathway, mainly by PINK1/PARKIN proteins, for the continuous maintenance by the renewal of mitochondria which ensures the recycling of damaged and depolarized organelles [27,31]. The stress-induced response is composed partly of FUNDC-1 and BNIP3 proteins, and the programmed pathways, where NIX protein belongs. Corresponding these to the ubiquitin independent pathways through mitochondrial receptors. Also, NIX, BNIP3, and FUNDC1 possess BH3 domains, LC3 regions, respond to hypoxia, and are proapoptotic [7]. The stress-induced pathway corresponds to extracellular stress signals that affect the mitochondrial physiology and can induce an acute mitochondrial clearance via mitophagy, as is the loss of mitochondrial membrane potential, starvation due to lack of nitrogen, or hypoxia due to the lack of oxygen. And finally, the programmed mitophagy activates in different types of cells during development [22,32,33].
Without continuous renewal by mitophagy, mitochondria lose their function [34]. Mitophagy flux, efficiency, and proteins are reduced throughout the aging process, and conversely, inducing mitophagy through genetic or pharmacological means leads to a longer lifespan [35,36]. As cells and organisms age, the efficacy of the respiratory chain tends to diminish, thus increasing electron leakage, ROS, and reducing ATP generation [37]. The relationship between mitochondrial dysfunction and aging has been long suspected, and at present human aging is generally linked to progressive mitochondrial dysfunction [38]. As mitophagy is a complex process that has not been blocked entirely due to the participation of several actors and non-redundant pathways [30], new actors may emerge in this process.
II. Mitophagy Decline Drives Primary Sarcopenia
Skeletal Muscle tissue is a major and the largest body tissue characterized by its higher mitochondrial content and metabolism. Skeletal muscle corresponds to around 40% of body mass [39], and is a contractile machine that allows the locomotion and movement of the organism’s body. The prior is achieved by the muscle-skeletal system where muscles contract and relax to move bones to which they are inserted via tendons through the body’s articulations. Skeletal muscles are surrounded by an outer layer of connective tissue called epimysium, and beneath the epimysium are bundles of myofibers, also named fascicles [40]. Inside fascicles individual myofibers are found, surrounded by connective tissue, interstitial cells for regulation, and satellite cells for muscle regeneration. Each myofiber is surrounded by several myonuclei and the sarcolemma, the myofiber cell membrane reservoir of calcium that when released allows the fiber’s contraction, and possesses mitochondria localized between myofibrils. Several individual myofibrils are composed mainly of myosin heavy chains (MYHC) and actin protein filaments, responsible for muscle contraction-relaxation [41]. Skeletal muscles have different fiber types, classified among slow and fast-twitch. Myofiber types are defined by their composition of myosin heavy chain isoforms: MYH-7, type I, which is slow-twitch, MYH-2 and 1, type IIA and IIB respectively, which are fast-twitch, distinguishing each different myofiber type from the slowest, with a preponderant oxidative metabolism, to fastest, with a preponderant glycolytic metabolism: 1, 1/2A, 2A, 2A/2X and 2X in humans [42,43]. Skeletal muscle fiber types are commonly found in different proportions in a single muscle group [44,45].
Primary sarcopenia is the main aging disease of skeletal muscle and corresponds to the progressive, accelerated, variable, involuntary loss of skeletal muscle mass and strength/function that occurs due to aging alone in elderly people, starting at 50 years old [46,47]. It was observed and described decades earlier in 1931, and many years later it was noticed that with aging muscle loss occurs accompanied by the loss of type II myofibers, observed in studies of human skeletal muscle biopsies, starting in the third decade of human life [48,49]. Then, in 1989 the term “Sarcopenia” was formed, from the Greek “Sarco” (meaning flesh/muscle) and the Greek word “Penia” (meaning loss), and presented officially as a disease by the author [50]. Later, in 1995 sarcopenia became considered a clinical entity after it was coined in 1989, because of the highly significant increase in incidence and prevalence of sarcopenia due to the increase in the elderly population during the 20th and 21st centuries [51]. According to the main sarcopenia work groups (EWGSOP, AWGS, IWGS, FNIH), primary sarcopenia is diagnosed by measuring muscle strength/function together with muscle mass, although low muscle strength is the principal factor to determine the diagnosis of sarcopenia in humans [46,52,53,54].
Primary sarcopenia becomes more prevalent with age [55]. From 25 to 80 years old humans lose around 25% of muscle mass [56,57], the onset and progression of sarcopenia are highly variable among different subjects because of genetic, lifestyle, and environmental factors [58,59,60], whereas muscle strength and mass in adults are due to genetic factors up to more than 50% [61]. Primary sarcopenia is a major threat and risk factor for several age-associated diseases [62,63,64,65], its deterioration accelerates over time [66,67], is accompanied and produces other morbidities, like cardiovascular diseases, type 2 diabetes, high blood pressure, obesity, and impairments like frailty, reduced mobility and disability [62,63,64,65,68], leads to an increased risk and fall-related injuries, hospitalization, loss of independence, diseases and death [53,69,70,71,72,73]. Accordingly, primary sarcopenia has become a major geriatric disease due to the increase of life expectancy and the growth of the older population worldwide, where the fast aging of the human population increases the incidence and prevalence of sarcopenia [71,74], being clinically assessable from 50 years old with a prevalence of more than 5%, with an annual progression of 0.5 to 2% each year, and accelerating progressively after the 7th decade of life with a prevalence of more than 10% [74,75,76].
Vastly in the literature, primary sarcopenia has been described as a loss, atrophy, and grouping of muscle fibers [49,51,68,77,78,79,80], with myofiber types proportions impacting significatively in primary sarcopenia, where type II fibers loss, smaller diameter, and atrophy occur, with a transition from fast to slow-twitch type I myofibers [51,80,81]. These changes in fiber type correlate with the reduced expression of MYHC-1 (IIX type), and MYHC-2 (IIA type), while MYHC-7 (type I) expression remains normal [82,83]. The MHC composition of single myofibers revealed a 12% higher proportion of more mixed I/IIA and II/IIB types, and groupings of type I and II myofibers through the skeletal muscles in elderly individuals [51,82,84]. Fast type II, MHC-1 and 2, myofibers’ size and amount decreased significatively, including satellite cell content and myonuclear domain, decreasing their adaptation to exercise, and MHC-2 and 1 protein levels declined by 3% and 1-5% respectively per decade, which was inversely associated with age, shifting from fastest IIB to IIX and IIA to I slower myofiber, and none of these parameters were improved with heavy resistance training during advanced aging [51,79,80,83,85,86,87]. Whereas, slow type I myofibers increased by 6%, positively associated with age, together with a corresponding increase in MHC-7 mRNA [83].
The fundamental change that occurs in skeletal muscle myofiber cells with aging and primary sarcopenia, in humans, D. melanogaster, mice, C. elegans, and C2C12 sarcopenia cell line models, is mitochondrial dysfunction, which corresponds to the functional decline of mitochondrial quantity, quality, and activity due to the oxidation and destruction of its structure [88]. First, a significant reduction in mitochondrial biomass and content in primary sarcopenia and aging is observed in COX, TOM-20 proteins, and mtDNA levels, where mtDNA, relative to nuclear DNA, was 38% lower [89,90]. Second, glycolytic metabolism also declines, seen by the decline in mRNA of MHC-1 and 2, of 14 and 10%, of myofibers 2A and 2X respectively, which primarily use glycolytic metabolism, whereas the mRNA abundance of MHC-7 remained unaltered with age [83,91]. Third, mitochondrial oxidative phosphorylation, oxidative metabolism, resting and maximal oxygen consumption, and oxidative capacity are impaired, reduced, and decline in men and women, between 25 and 80 years old. Peak aerobic capacity declines 8% per decade with age, oxidative capacity is 50% lower, oxidative and phosphorylation activity is reduced by around 40%, tricarboxylic acid enzymes activity is reduced, and mitochondrial volume is 24% lower [88,92,93,94,95,96]. Fourth, a decrease in lipid metabolism occurs as older adults have fewer mitochondria in contact with lipid droplets, producing larger and more intramyocellular lipid droplets, due to the lack of mitochondrial metabolism and function [89]. Fifth, mitochondrial ATP synthesis, mtDNA, and mitochondrial mRNA abundance are decreased and declined among women and men between 18 and 89 years old. Furthermore, mtDNA levels were associated positively with mtATP production rate, being closely related to aerobic capacity [83]. Sixth, a decrease in mitochondrial biogenesis was observed due to less PGC1-α protein abundance. And, seventh, the dysregulation of mitochondrial dynamics with an excess of DRP1 fission relative to fusion OPA1 proteins [71,88].
Mitochondrial dysfunction seems to be the main cause of primary sarcopenia and is a well-established hallmark of aging [4,88,97], which can accelerate aging in mammals [98,99,100], particularly observed in studies with mice deficient in DNA polymerase γ, responsible for mtDNA replication [101,102]. The accumulation of defective, damaged, and/or dysfunctional mitochondria in skeletal muscle myofiber cells are associated with aging, primary sarcopenia, decreased mtDNA damage repair, increased ROS, cellular apoptosis, a decrease in skeletal muscle mass, strength, and function, prolonging and increasing damage like excessive structure oxidation due to ROS, impairment of contraction excitation of the muscle fiber, altered calcium homeostasis, energy deficiency, altered signaling to the nucleus, dysfunction of myofibers, death of myofibers, and lack of myofiber regeneration [71,103,104,105]. Therefore, the lack of mitochondrial homeostasis has been suggested to perform a causal role in age-associated skeletal muscle decline [94,106,107,108]. Accordingly, mitophagy deficiency has been associated with primary sarcopenia mitochondrial dysfunction causing defective bioenergetics [4,10,11,15,16,38]. The accumulation of dysfunctional mitochondria would be caused by an impairment in the mitophagy flux, seen as the decrease of mitophagy-related proteins, or the interruption of mitophagy, which alters the mitochondrial function progressively, accumulating deficient mitochondrial organelles, damaging cells and tissues [7,109]. Notably, biological aging affects negatively the mitophagy homeostasis in the skeletal muscle system [110,111,112], where impaired mitophagy performs a key role in the loss of mitochondrial functions [111]. The combination of increased mitochondria damage (like oxidized mitochondrial DNA, lipids, and proteins), reduced mitochondrial turnover (seen by mitophagy flux and efficiency with LC3-II/I ratio, ATG-1,7,8 and Lamp-1 protein levels, plus mitophagy receptor and adaptor protein levels as p62, BNIP3, PINK1, and PARKIN), lower mitochondrial biogenesis (seen as a decrease in PGC1-α protein and mtDNA levels), plus reduced mitochondrial clearance (seen as depolarized mitochondria accumulation), contribute to the aging process of skeletal muscle [106,113,114,115]. Hence, the stimulation of the mitophagy process could possibly reverse mitochondrial dysfunction in primary sarcopenia by degrading dysfunctional mitochondria, promoting mitochondrial biogenesis and protecting mitochondria from oxidative stress.
The main pathway for mitochondrial dysfunction to cause primary sarcopenia, which counts with the most evidence, also as the best potential treatment for primary sarcopenia, is the downregulation of mitophagy with aging (Figure 1). Mitophagy in skeletal muscle is reduced during normal aging in humans, D. melanogaster, mice, C2C12 and L6 rodents skeletal muscle cell lines, where impairment in mitophagy flux is due to a decrease in mitophagy-related proteins: PINK, PARKIN, AMPK, ULK1, BECLIN-1, BCL2, ATG-3, 5 and 12, LC3-I and II, LAMP-1 and 2 is suggested to accumulate dysfunctional mitochondria, accompanied with a decrease in PGC1-α protein levels for mitochondrial biogenesis reciprocal balance [71,109,116]. Accordingly, in old humans there is a downregulation of mitophagy-related genes BNIP3, PARKIN, BCL2L13, LC3-II/I ratio, Beclin-1, Atg-7, and E3 ubiquitin ligases, which causes insufficient skeletal muscle mass, poor muscle strength, severe decrease in mitochondrial respiration, uncoupling and susceptibility to opening permeability transition pores, showing a positive correlation between the decrease in physical function, leg lean mass and mitophagy genes [117,118,119,120,121]. Also in humans and rodents, the progressive age-related decline of the autophagy/lysosome system, which mitophagy depends on, presents lower protein levels of autophagy core components and mitophagy regulators LC3-II/I ratio, p62, Beclin-1, Atg-3 and 4, and Bcl2 [122,123].
III. Evidence of Mitophagy Impairment in Aged Skeletal Muscle
In mice aged skeletal muscle presents a decreased mitophagy flux (LC3-II and p62 ratio protein levels) and efficiency (accumulation of BNIP3, NIX, Beclin-1, Lamp-1/2, transcription factor for lysosomal biogenesis EB, TFEB, and Cathepsin D, a lysosomal protease) compared to young mice skeletal muscle even in response to chronic contractile activity [124]. Also, when measuring mitophagy in aged mice skeletal muscle in vivo, again a decrease in mitophagy flux was observed, where a mitochondrial pH-dependent fluorescent reporter transitions from the mitochondria into the lysosome [27]. Even more, in mice and rats’ certain alterations in the mitophagy regulators PARKIN, PARKIN/VIDAC ratio, and PGC1-α lead to mitophagy deficiency and the following accumulation of dysfunctional mitochondria in primary sarcopenia [117,125]. Notably, parkin gene deletion in mice causes insufficient muscle mass and poor muscle strength [117,118]. Consequently, the overexpression and upregulation of parkin gene expression in old mice ameliorated the loss of skeletal muscle mass and function due to aging, unexpectedly causing hypertrophy and an increase in fibers size via the increase in mitochondrial content and enzymatic activities [118,126].
IV. Autophagy–Mitophagy Axis in Muscle Degeneration
Autophagy is closely related to the deficiency in the mitophagy process that affects skeletal muscle in primary sarcopenia. Expressing the atg-7 gene in aged mice skeletal and cardiac muscle ameliorates skeletal muscle mass and cardiac function, while overexpressing atg-5 prevents the age-induced decrease in muscle function, preserving muscle strength and extending lifespan [127,128]. On the contrary, atg-7 gene deletion, necessary for mitophagy occurrence, leads to defective mitophagy, causing an increase in accumulation of enlarged, swollen mitochondria, inducing muscle atrophy, loss of force, myopathy, accelerated aging, and the accumulation of p62, LC3, and NIX/BNIP3L mitophagy related protein levels due to the blockage of the mitophagy process [127]. Even more, in mice, it has been described that activating autophagy facilitates muscle regeneration by reversing senescence in skeletal muscle satellite cells, where atg-7 gene deletion results in mitochondrial dysfunction [129]. Autophagy also allows the degradation of dysfunctional mitochondria, preventing myofiber atrophy, therefore stimulating basal autophagy protects from primary sarcopenia in humans and mice [130]. Remarkably, the stimulation of selective autophagy in sarcopenia preserves and even recovers muscle strength and function in aged organisms by promoting the degradation of faulty mitochondria and preventing muscle mass loss [131].
V. Natural Mitophagy Inducers: Urolithin A and Spermidine
Following the previous studies’ results, the utilization of mitophagy stimulants natural compounds Urolithin A and Spermidine, extracted from pomegranate fruit and wheat germs respectively, shown to act via the PINK1/PARKIN mitophagy pathway, had beneficial effects on D. melanogaster, mice and worms aged skeletal muscle, increasing mitophagy related proteins PINK1, PARKIN, BNIP3, p62 and Beclin-1 expression levels [36,132,133,134,135]. In aged mice muscle function, grip strength and running capacity increased, while in aged worms increased motility, lifespan, delayed aging, and inhibited neurodegeneration [36,135]. Even more, Urolithin A administration in aged mice heart skeletal muscle increased heart blood ejection, and mitochondrial and myofiber volume [36,134]. Spermidine increased mitophagy in aged D. melanogaster, promoted stress resistance, and reduced aging locomotor loss [132,133].
VI. Translational Evidence in Humans and Animal Models
In humans, Urolithin A and Spermidine could protect, prevent and/or ameliorate primary sarcopenia and age-associated diseases. In 40 to 64 years old, sedentary with low physical performance, overweight individuals Urolithin A increased: muscle strength, exercise performance, TCA enzymes protein levels, mitochondrial content, and biomarkers of mitochondrial health, were seen, with lowered blood pressure and systemic inflammation [136,137,138]. While, Urolithin A and Spermidine lowered cardiovascular disease, pondering them as potential therapeutic compounds [36,139,140]. Similar natural compounds: Resveratrol, Curcumin, Tomatidine, Ascorbic acid, Schizandrin A, Anthocyanidin, and Astragaloside IV have been found to stimulate mitophagy in mice and have neuroprotective effects against strokes, Alzheimer’s and Parkinson’s disease, which are age-associated diseases also presenting mitochondrial dysfunction [141].
VII. Therapeutic Perspective: Targeting Mitophagy to Reverse Sarcopenia
It is still necessary to understand the mechanisms that change skeletal muscle strength/function and mass to develop specific intervention strategies against primary sarcopenia [68], considering that at the moment conflicting results exist when studying primary sarcopenia due to methodological limitations [68]. The study of primary skeletal muscle cells from different human donors with a variety of biological conditions, such as natural aging, genomes, diseases, lifestyles, and health conditions, are the best in vitro models to study primary sarcopenia [71]. So, the use of these appropriate in vitro models is a good strategy to screen molecular targets to identify therapeutic compounds against primary sarcopenia [71]. Despite this, molecular and cellular mechanisms have yet to be characterized to design an effective treatment for primary sarcopenia, where most of our understanding of myofibers and the muscle scaffold come from work performed in animals [67]. Therefore, there is a need for clinical investigation in humans to integrate with state-of-the-art basic research in laboratory animals [68]. Due to the latter, pre-clinical research in aged laboratory animals is critical and still has to be performed to provide sufficient evidence to move forward to clinical research and achieve translational value [71]. Hence, the use of naturally aged animals and cell line models has the advantage of studying primary sarcopenia at the systemic effects level, molecular patterns at the organs level, and finally the cellular level for biochemical biomarkers [71]. Therefore, the use of aged D. melanogaster and mice C2C12 cell line is recommended to study sarcopenia effects at all levels [142,143,144], where the D. melanogaster model resembles the mammalian skeletal muscle fibers organization and metabolism [145,146,147], and the mammalian mice C2C12 cell line differentiated into skeletal muscle myotubes treated with hydrogen peroxide to induce sarcopenia has been extensively utilized as a sarcopenia model for studying sarcopenia [148,149,150,151].
VIII. Future Directions and Clinical Relevance
The previous findings indicate that mitophagy deficiency contributes to primary sarcopenia through a complex mitochondrial quality control network. As there are multiple pathways and roles of mitophagy proteins in mitochondrial quality control, it is to be expected that mitophagy-related proteins like, PARKIN, PINK, BNIP3, NIX, BCL2L13, p62, LC3-I and II, AMPK, ULK1, Beclin-1, Bcl-2, TFEB, Cathepsin D, Lamp-1 and 2, ATG-1,5, 7 and 8, together with the natural compounds Urolithin A and Spermidine, become new therapeutic targets and options for primary sarcopenia, preventing and ameliorating the loss of strength and mass of skeletal muscle during aging.
Corresponding to the different possible causes of sarcopenia in humans, its markers are: forkhead box O3a transcription factor (FoxO3a), indicative of protein degradation, mammalian target of rapamycin (mTOR), whose activation inhibits autophagy [152], tumor necrosis factor-alpha (TNF-α) and interleukin 6 (IL-6), inflammatory cytokines that inhibit the synthesis and degrade muscle proteins [153], which present positively correlated higher levels. Oppositely, autophagy-related kinase 1 (ULK1), a regulator of autophagy in aging skeletal muscle [154], adenosine monophosphate-activated protein kinase (AMPK), which mediate mitochondria quality control [152], insulin-like growth factor 1 (IGF-1), that regulates fiber’s growth via protein synthesis [155], and Myosin protein, present negatively correlated lower levels.
The design and production of a natural mitophagy compound in primary sarcopenia would be useful for most age-related diseases, especially primary sarcopenia, mtDNA mutation diseases, and the treatment of aged cells, tissues, and organs deterioration, where the main cause of aging and damage is the accumulation of dysfunctional mitochondria, as in: diabetes, cancers, cardiovascular diseases, neurodegenerative disorders, amyotrophic lateral sclerosis, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, autoimmune disorders, hypertrophic and dilated cardiomyopathies, cardiac conduction defects, increases in the size of myocardial infarction and Leber’s Hereditary Optic Neuropathy [156].
Also, the application of a Urolithin A/Spermidine mitophagy natural compound would reveal its relevance in primary sarcopenia where dysfunctional mitochondria are present and mitophagy is decreased, responsible for the aging phenotype [29,157]. The use of the natural mitophagy compound would improve the healthspan and lifespan of all aged people above 50 years old, preventing hospitalizations, and fall injuries, treating and decreasing other morbidities such as obesity, hypertension, diabetes, and mortality.
IX. Acknowledgments and Conflicts of Interest
Both authors are partners in “Elyxia Vita”, a biotechnology startup developing personalized aging-assessment and lifestyle optimization services. The authors received no financial compensation for the preparation of this review, which was completed entirely on personal time. No external funding influenced the content, analysis, or conclusions presented.
Soffia JP contributed with conceptualization, investigation, resources, writing—original draft preparation, writing—review and editing, visualization, supervision, project administration. Dawson AP contributed with editing and visualization. An artificial intelligence large language model (LLM) was used to assist with language editing and improvement of clarity. All scientific content, data interpretation, and conclusions were developed and verified solely by the authors. The figure was created in https://BioRender.com.
References
- Lane, N.; Martin, W., “The energetics of genome complexity,” Nature, vol. 467, no. 7318, pp. 929–934, Oct. 2010. [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A., “Protecting the mitochondrial powerhouse,” Trends Cell Biol., vol. 25, no. 3, pp. 158–170, Mar. 2015. [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A., “Mitochondrial membrane potential probes and the proton gradient: a practical usage guide,” Biotechniques, vol. 50, no. 2, pp. 98–115, Feb. 2011. [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G., “The hallmarks of aging,” Cell, vol. 153, no. 6, p. 1194, Jun. 2013. [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A., “Nuclear DNA damage signalling to mitochondria in ageing,” Nat. Rev. Mol. Cell Biol., vol. 17, no. 5, pp. 308–321, Apr. 2016. [CrossRef]
- Mattson, M.P.; Gleichmann, M.; Cheng, A., “Mitochondria in Neuroplasticity and Neurological Disorders,” Neuron, vol. 60, no. 5, pp. 748–766, Dec. 2008. [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N., “Mechanisms of mitophagy in cellular homeostasis, physiology and pathology,” Nat. Cell Biol., vol. 20, no. 9, pp. 1013–1022, Sep. 2018. [CrossRef]
- Elorza, A.A.; Soffia, J.P., “mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology,” Front. Cell Dev. Biol., vol. 9, Feb. 2021. [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B., “Mitochondrial electron transport chain, ROS generation and uncoupling (Review),” Int. J. Mol. Med., vol. 44, no. 1, pp. 3–15, 2019. [CrossRef]
- Liesa, M.; Shirihai, O.S., “Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure,” Cell Metab., vol. 17, no. 4, pp. 491–506, Apr. 2013. [CrossRef]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A., “Mitochondria and endothelial function,” Circ. Res., vol. 112, no. 8, pp. 1171–1188, Apr. 2013. [CrossRef]
- Westermann, B., “Mitochondrial fusion and fission in cell life and death,” Nat. Rev. Mol. Cell Biol., vol. 11, no. 12, pp. 872–884, Dec. 2010. [CrossRef]
- Novak, I., “Mitophagy: a complex mechanism of mitochondrial removal,” Antioxid. Redox Signal., vol. 17, no. 5, pp. 794–802, Sep. 2012. [CrossRef]
- Youle, R.J.; Narendra, D.P., “Mechanisms of mitophagy,” Nat. Rev. Mol. Cell Biol., vol. 12, no. 1, pp. 9–14, Jan. 2011. [CrossRef]
- Santos et al, R.X., “Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes,” Mol. Cell. Biochem., vol. 394, no. 1–2, pp. 13–22, Sep. 2014. [CrossRef]
- Kornmann, B., “Quality control in mitochondria: use it, break it, fix it, trash it,” F1000Prime Rep., vol. 6, Mar. 2014. [CrossRef]
- Held, N.M.; Houtkooper, R.H., “Mitochondrial quality control pathways as determinants of metabolic health,” Bioessays, vol. 37, no. 8, pp. 867–876, Aug. 2015. [CrossRef]
- Ashrafi, G.; Schwarz, T.L., “The pathways of mitophagy for quality control and clearance of mitochondria,” Cell Death Differ., vol. 20, no. 1, pp. 31–42, Jan. 2013. [CrossRef]
- Ruiz et al, L.M., “Non-cytotoxic copper overload boosts mitochondrial energy metabolism to modulate cell proliferation and differentiation in the human erythroleukemic cell line K562,” Mitochondrion, vol. 29, pp. 18–30, Jul. 2016. [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H., “Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease,” Mech. Ageing Dev., vol. 186, Mar. 2020. [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E., “LC3 and Autophagy,” Methods Mol. Biol., vol. 445, pp. 77–88, 2008. [CrossRef]
- Schweers et al, R.L., “NIX is required for programmed mitochondrial clearance during reticulocyte maturation,” Proc. Natl. Acad. Sci. U. S. A., vol. 104, no. 49, pp. 19500–19505, Dec. 2007. [CrossRef]
- Bassnett, S., “Lens organelle degradation,” Exp. Eye Res., vol. 74, no. 1, pp. 1–6, 2002. [CrossRef]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.-W., “Autophagy is essential for mitochondrial clearance in mature T lymphocytes,” J. Immunol., vol. 182, no. 7, pp. 4046–4055, Apr. 2009. [CrossRef]
- Sharpley et al, M.S., “Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition,” Cell, vol. 151, no. 2, pp. 333–343, Oct. 2012. [CrossRef]
- Rojansky, R.; Cha, M.Y.; Chan, D.C., “Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1,” Elife, vol. 5, no. NOVEMBER2016, Nov. 2016. [CrossRef]
- Sun et al, N., “Measuring In Vivo Mitophagy,” Mol. Cell, vol. 60, no. 4, pp. 685–696, Nov. 2015. [CrossRef]
- Mortensen et al, M., “Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo,” Proc. Natl. Acad. Sci. U. S. A., vol. 107, no. 2, pp. 832–837, 2010. [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N., “Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover,” Pharmacol. Ther., vol. 178, pp. 157–174, Oct. 2017. [CrossRef]
- Zhang et al, Y., “Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing,” Nat. Immunol., vol. 20, no. 4, pp. 433–446, Apr. 2019. [CrossRef]
- McWilliams et al, T.G., “mito-QC illuminates mitophagy and mitochondrial architecture in vivo,” J. Cell Biol., vol. 214, no. 3, pp. 333–345, Aug. 2016. [CrossRef]
- Esteban-Martínez et al, L., “Programmed mitophagy is essential for the glycolytic switch during cell differentiation,” EMBO J., vol. 36, no. 12, pp. 1688–1706, Jun. 2017. [CrossRef]
- Sandoval et al, H., “Essential role for Nix in autophagic maturation of erythroid cells,” Nature, vol. 454, no. 7201, pp. 232–235, Jul. 2008. [CrossRef]
- López-Otín, C.; Kroemer, G., “Hallmarks of Health,” Cell, vol. 184, no. 1, pp. 33–63, Jan. 2021. [CrossRef]
- Wu et al, W., “ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy,” EMBO Rep., vol. 15, no. 5, pp. 566–575, 2014. [CrossRef]
- Ryu et al, D., “Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents,” Nat. Med., vol. 22, no. 8, pp. 879–888, Aug. 2016. [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G., “Mitochondria and the autophagy-inflammation-cell death axis in organismal aging,” Science, vol. 333, no. 6046, pp. 1109–1112, Aug. 2011. [CrossRef]
- Wang, K.; Klionsky, D.J., “Mitochondria removal by autophagy,” Autophagy, vol. 7, no. 3, pp. 297–300, 2011. [CrossRef]
- McLeod, M.; Breen, L.; Hamilton, D.L.; Philp, A., “Live strong and prosper: the importance of skeletal muscle strength for healthy ageing,” Biogerontology, vol. 17, no. 3, pp. 497–510, Jun. 2016. [CrossRef]
- Frontera, W.R.; Ochala, J., “Skeletal muscle: a brief review of structure and function,” Calcif. Tissue Int., vol. 96, no. 3, pp. 183–195, Mar. 2015. [CrossRef]
- Jorgenson, K.W.; Phillips, S.M.; Hornberger, T.A., “Identifying the Structural Adaptations that Drive the Mechanical Load-Induced Growth of Skeletal Muscle: A Scoping Review,” Cells, vol. 9, no. 7, Jul. 2020. [CrossRef]
- Schiaffino, S.; Reggiani, C., “Fiber types in mammalian skeletal muscles,” Physiol. Rev., vol. 91, no. 4, pp. 1447–1531, Oct. 2011. [CrossRef]
- Bassel-Duby, R.; Olson, E.N., “Signaling pathways in skeletal muscle remodeling,” Annu. Rev. Biochem., vol. 75, pp. 19–37, 2006. [CrossRef]
- Tajsharghi, H., “Thick and thin filament gene mutations in striated muscle diseases,” Int. J. Mol. Sci., vol. 9, no. 7, pp. 1259–1275, Jul. 2008. [CrossRef]
- Johnson, M.A.; Polgar, J.; Weightman, D.; Appleton, D., “Data on the distribution of fibre types in thirty-six human muscles. An autopsy study,” J. Neurol. Sci., vol. 18, no. 1, pp. 111–129, 1973. [CrossRef]
- Fielding et al, R.A., “Sarcopenia: An Undiagnosed Condition in Older Adults. Current Consensus Definition: Prevalence, Etiology, and Consequences. International Working Group on Sarcopenia,” J. Am. Med. Dir. Assoc., vol. 12, no. 4, pp. 249–256, 2011. [CrossRef]
- Roubenoff, R.; Heymsfield, S.B.; Kehayias, J.J.; Cannon, J.G.; Rosenberg, I.H., “Standardization of nomenclature of body composition in weight loss.,” Am. J. Clin. Nutr., vol. 66, no. 1, pp. 192–196, 1997. [CrossRef]
- Critchley, M., “THE NEUROLOGY OF OLD AGE.,” The Lancet, vol. 217, no. 5623, pp. 1221–1231, Jun. 1931. [CrossRef]
- Lexell, J.; Henriksson-Larsén, K.; Winblad, B.; Sjöström, M., “Distribution of different fiber types in human skeletal muscles: effects of aging studied in whole muscle cross sections,” Muscle Nerve, vol. 6, no. 8, pp. 588–595, 1983. [CrossRef]
- Rosenberg, I.H., “Summary comments,” Am. J. Clin. Nutr., vol. 50, no. 5, pp. 1231–1233, Nov. 1989. [CrossRef]
- Lexell, J., “Human aging, muscle mass, and fiber type composition,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 50 Spec No, no. SPEC. ISSUE, pp. 11–16, 1995. [CrossRef]
- Cruz-Jentoft et al, A.J., “Sarcopenia: revised European consensus on definition and diagnosis,” Age Ageing, vol. 48, no. 4, p. 601, Jul. 2019. [CrossRef]
- Chen, H.; Ma, J.; Liu, A.; Cui, Y.; Ma, X., “The association between sarcopenia and fracture in middle-aged and elderly people: A systematic review and meta-analysis of cohort studies,” Injury, vol. 51, no. 4, pp. 804–811, Apr. 2020. [CrossRef]
- Chen et al, L.K., “Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment,” J. Am. Med. Dir. Assoc., vol. 21, no. 3, pp. 300-307.e2, Mar. 2020. [CrossRef]
- Doherty, T.J., “Invited review: Aging and sarcopenia,” J. Appl. Physiol. (1985)., vol. 95, no. 4, pp. 1717–1727, Oct. 2003. [CrossRef]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M., “Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review,” Front. Physiol., vol. 3, 2012. [CrossRef]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L., “Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases,” Lancet Diabetes Endocrinol., vol. 2, no. 10, pp. 819–829, 2014. [CrossRef]
- Degens, H.; Korhonen, M.T., “Factors contributing to the variability in muscle ageing,” Maturitas, vol. 73, no. 3, pp. 197–201, Nov. 2012. [CrossRef]
- Bann et al, D., “Light Intensity physical activity and sedentary behavior in relation to body mass index and grip strength in older adults: cross-sectional findings from the Lifestyle Interventions and Independence for Elders (LIFE) study,” PLoS One, vol. 10, no. 2, Feb. 2015. [CrossRef]
- Khanal et al, P., “The Association of Multiple Gene Variants with Ageing Skeletal Muscle Phenotypes in Elderly Women,” Genes (Basel)., vol. 11, no. 12, pp. 1–18, Dec. 2020. [CrossRef]
- Zempo, H.; Miyamoto-Mikami, E.; Kikuchi, N.; Fuku, N.; Miyachi, M.; Murakami, H., “Heritability estimates of muscle strength-related phenotypes: A systematic review and meta-analysis,” Scand. J. Med. Sci. Sports, vol. 27, no. 12, pp. 1537–1546, Dec. 2017. [CrossRef]
- Roubenoff, R., “Sarcopenia: a major modifiable cause of frailty in the elderly,” J. Nutr. Health Aging, vol. 4, no. 3, pp. 140–142, 2000, Accessed: Nov. 14, 2025. [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/10936900/.
- Organization, W.H., “The implications for training of embracing : a life course approach to health,” Dec. 31, 2000, World Health Organization. Accessed: Nov. 14, 2025. [Online]. Available: https://iris.who.int/handle/10665/69400.
- Organization, W.H., “Decade of Healthy Ageing: Plan of Action 2021-2030,” World Health Organisation, pp. 1–26, 2020, Accessed: Nov. 14, 2025. [Online]. Available: https://cdn.who.int/media/docs/default-source/decade-of-healthy-ageing/final-decade-proposal/decade-proposal-final-apr2020-en.pdf?sfvrsn=b4b75ebc_25&download=true.
- Harrison, J.E.; Weber, S.; Jakob, R.; Chute, C.G., “ICD-11: an international classification of diseases for the twenty-first century,” BMC Med. Inform. Decis. Mak., vol. 21, Nov. 2021. [CrossRef]
- Rolland, Y.; Van Kan, G.A.; Gillette-Guyonnet, S.; Vellas, B., “Cachexia versus sarcopenia,” Curr. Opin. Clin. Nutr. Metab. Care, vol. 14, no. 1, pp. 15–21, Jan. 2011. [CrossRef]
- Pascual-Fernández, J.; Fernández-Montero, A.; Córdova-Martínez, A.; Pastor, D.; Martínez-Rodríguez, A.; Roche, E., “Sarcopenia: Molecular Pathways and Potential Targets for Intervention,” Int. J. Mol. Sci., vol. 21, no. 22, pp. 1–16, Nov. 2020. [CrossRef]
- Larsson et al, L., “Sarcopenia: Aging-Related Loss of Muscle Mass and Function,” Physiol. Rev., vol. 99, no. 1, pp. 427–511, Jan. 2019. [CrossRef]
- Beaudart, C.; Zaaria, M.; Pasleau, F.; Reginster, J.Y.; Bruyère, O., “Health Outcomes of Sarcopenia: A Systematic Review and Meta-Analysis,” PLoS One, vol. 12, no. 1, Jan. 2017. [CrossRef]
- Shou, J.; Chen, P.J.; Xiao, W.H., “Mechanism of increased risk of insulin resistance in aging skeletal muscle,” Diabetol. Metab. Syndr., vol. 12, no. 1, Feb. 2020. [CrossRef]
- Mankhong, S.; Kim, S.; Moon, S.; Kwak, H.B.; Park, D.H.; Kang, J.H., “Experimental Models of Sarcopenia: Bridging Molecular Mechanism and Therapeutic Strategy,” Cells, vol. 9, no. 6, Jun. 2020. [CrossRef]
- Hemenway, D.; Solnick, S.J.; Koeck, C.; Kytir, J., “The incidence of stairway injuries in Austria,” Accid. Anal. Prev., vol. 26, no. 5, pp. 675–679, 1994. [CrossRef]
- “[A study of falls experienced by institutionalized elderly] - PubMed.” Accessed: Nov. 19, 2025. [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/1292738/.
- Gustafsson, T.; Ulfhake, B., “Sarcopenia: What Is the Origin of This Aging-Induced Disorder?,” Front. Genet., vol. 12, Jul. 2021. [CrossRef]
- Phillip, S.M., “Physiologic and molecular bases of muscle hypertrophy and atrophy: impact of resistance exercise on human skeletal muscle (protein and exercise dose effects),” Appl. Physiol. Nutr. Metab., vol. 34, no. 3, pp. 403–410, Jun. 2009. [CrossRef]
- Hughes, V.A.; Frontera, W.R.; Roubenoff, R.; Evans, W.J.; Singh, M.A.F., “Longitudinal changes in body composition in older men and women: Role of body weight change and physical activity,” American Journal of Clinical Nutrition, vol. 76, no. 2, pp. 473–481, 2002. [CrossRef]
- Bowen, T.S.; Schuler, G.; Adams, V., “Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training,” J. Cachexia Sarcopenia Muscle, vol. 6, no. 3, pp. 197–207, Sep. 2015. [CrossRef]
- LARSSON, L.; BIRAL, D.; CAMPIONE, M.; SCHIAFFINO, S., “An age-related type IIB to IIX myosin heavy chain switching in rat skeletal muscle,” Acta Physiol. Scand., vol. 147, no. 2, pp. 227–234, 1993. [CrossRef]
- Larsson, L., “Motor units: remodeling in aged animals,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 50 Spec No, no. SPEC. ISSUE, pp. 91–95, 1995. [CrossRef]
- Nilwik et al, R., “The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size,” Exp. Gerontol., vol. 48, no. 5, pp. 492–498, May 2013. [CrossRef]
- Talbot, J.; Maves, L., “Skeletal muscle fiber type: using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease,” Wiley Interdiscip. Rev. Dev. Biol., vol. 5, no. 4, pp. 518–534, Jul. 2016. [CrossRef]
- Klitgaard, H.; Zhou, M.; Schiaffino, S.; Betto, R.; Salviati, G.; Saltin, B., “Ageing alters the myosin heavy chain composition of single fibres from human skeletal muscle,” Acta Physiol. Scand., vol. 140, no. 1, pp. 55–62, 1990. [CrossRef]
- Short et al, K.R., “Changes in myosin heavy chain mRNA and protein expression in human skeletal muscle with age and endurance exercise training,” J. Appl. Physiol. (1985)., vol. 99, no. 1, pp. 95–102, Jul. 2005. [CrossRef]
- Lexell, J.; Downham, D.Y., “The occurrence of fibre-type grouping in healthy human muscle: a quantitative study of cross-sections of whole vastus lateralis from men between 15 and 83 years,” Acta Neuropathol., vol. 81, no. 4, pp. 377–381, Mar. 1991. [CrossRef]
- Karlsen et al, A., “Lack of muscle fibre hypertrophy, myonuclear addition, and satellite cell pool expansion with resistance training in 83-94-year-old men and women,” Acta Physiol. (Oxf)., vol. 227, no. 1, 2019. [CrossRef]
- Fry et al, C.S., “Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis,” Skelet. Muscle, vol. 1, no. 1, Mar. 2011. [CrossRef]
- Holloszy, J.O.; Chen, M.; Cartee, G.D.; Young, J.C., “Skeletal muscle atrophy in old rats: Differential changes in the three fiber types,” Mech. Ageing Dev., vol. 60, no. 2, pp. 199–213, 1991. [CrossRef]
- Chan, D.C., “Mitochondria: Dynamic Organelles in Disease, Aging, and Development,” Cell, vol. 125, no. 7, pp. 1241–1252, Jun. 2006. [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A., “The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 65, no. 2, pp. 119–128, 2010. [CrossRef]
- Welle, S.; Bhatt, K.; Shah, B.; Needler, N.; Delehanty, J.M.; Thornton, C.A., “Reduced amount of mitochondrial DNA in aged human muscle,” J. Appl. Physiol. (1985)., vol. 94, no. 4, pp. 1479–1484, Apr. 2003. [CrossRef]
- Betts et al, J.G., “Anatomy and physiology,” p. 1300, 2022.
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Nair, K.S., “Age and aerobic exercise training effects on whole body and muscle protein metabolism,” Am. J. Physiol. Endocrinol. Metab., vol. 286, no. 1, pp. 92–101, 2004. [CrossRef]
- Coen et al, P.M., “Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 68, no. 4, pp. 447–455, Apr. 2013. [CrossRef]
- Joseph et al, A.M., “The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals,” Aging Cell, vol. 11, no. 5, pp. 801–809, Oct. 2012. [CrossRef]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C., “Oxidative capacity and ageing in human muscle,” J. Physiol., vol. 526 Pt 1, no. Pt 1, pp. 203–210, Jul. 2000. [CrossRef]
- Petersen et al, K.F., “Mitochondrial dysfunction in the elderly: possible role in insulin resistance,” Science, vol. 300, no. 5622, pp. 1140–1142, May 2003. [CrossRef]
- Wallace, D.C., “A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine,” Annu. Rev. Genet., vol. 39, pp. 359–407, 2005. [CrossRef]
- Kujoth et al, C.C., “Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging,” Science, vol. 309, no. 5733, pp. 481–484, Jul. 2005. [CrossRef]
- Trifunovic et al, A., “Premature ageing in mice expressing defective mitochondrial DNA polymerase,” Nature, vol. 429, no. 6990, pp. 417–423, May 2004. [CrossRef]
- Vermulst et al, M., “DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice,” Nat. Genet., vol. 40, no. 4, pp. 392–394, Apr. 2008. [CrossRef]
- Edgar et al, D., “Random Point Mutations with Major Effects on Protein-Coding Genes Are the Driving Force behind Premature Aging in mtDNA Mutator Mice,” Cell Metab., vol. 10, no. 2, pp. 131–138, Aug. 2009. [CrossRef]
- Hiona et al, A., “Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice,” PLoS One, vol. 5, no. 7, 2010. [CrossRef]
- Masiero, E.; Sandri, M., “Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles,” Autophagy, vol. 6, no. 2, pp. 307–309, Feb. 2010. [CrossRef]
- Marzetti et al, E., “Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials,” International Journal of Biochemistry and Cell Biology, vol. 45, no. 10, pp. 2288–2301, 2013. [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B., “Role of Age-Related Mitochondrial Dysfunction in Sarcopenia,” Int. J. Mol. Sci., vol. 21, no. 15, pp. 1–12, Aug. 2020. [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A., “Mitochondrial function and apoptotic susceptibility in aging skeletal muscle,” Aging Cell, vol. 7, no. 1, pp. 2–12, Feb. 2008. [CrossRef]
- Kruse et al, S.E., “Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner,” Aging Cell, vol. 15, no. 1, pp. 89–99, Feb. 2016. [CrossRef]
- Marcinek, D.J.; Schenkman, K.A.; Ciesielski, W.A.; Lee, D.; Conley, K.E., “Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle,” J. Physiol., vol. 569, no. Pt 2, pp. 467–473, Dec. 2005. [CrossRef]
- Drake, J.C.; Yan, Z., “Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing,” J. Physiol., vol. 595, no. 20, pp. 6391–6399, Oct. 2017. [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Aghanejad, A.; Zhang, Y.; Ren, J., “Mitophagy Receptors and Mediators: Therapeutic Targets in the Management of Cardiovascular Ageing,” Ageing Res. Rev., vol. 62, Sep. 2020. [CrossRef]
- Gustafsson, Å.B.; Dorn, G.W., “Evolving and Expanding the Roles of Mitophagy as a Homeostatic and Pathogenic Process,” Physiol. Rev., vol. 99, no. 1, pp. 853–892, Jan. 2019. [CrossRef]
- Tian, H.; Chen, P.; Ren, J., “Physical exercise, autophagy and cardiometabolic stress in aging,” Aging, vol. 11, no. 15, pp. 5287–5288, Aug. 2019. [CrossRef]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M., “The ageing neuromuscular system and sarcopenia: a mitochondrial perspective,” J. Physiol., vol. 594, no. 16, p. 4499, Aug. 2016. [CrossRef]
- Lundt, S.; Zhang, N.; Wang, X.; Polo-Parada, L.; Ding, S., “The effect of NAMPT deletion in projection neurons on the function and structure of neuromuscular junction (NMJ) in mice,” Sci. Rep., vol. 10, no. 1, Dec. 2020. [CrossRef]
- Lin, M.T.; Beal, M.F., “Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases,” Nature, vol. 443, no. 7113, pp. 787–795, Oct. 2006. [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T., “The Mitochondrial Basis of Aging,” Mol. Cell, vol. 61, no. 5, pp. 654–666, Mar. 2016. [CrossRef]
- Gouspillou et al, G., “Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans,” FASEB J., vol. 28, no. 4, pp. 1621–1633, 2014. [CrossRef]
- Gouspillou et al, G., “Protective role of Parkin in skeletal muscle contractile and mitochondrial function,” J. Physiol., vol. 596, no. 13, pp. 2565–2579, Jul. 2018. [CrossRef]
- Arribat et al, Y., “Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training,” Acta Physiol. (Oxf)., vol. 225, no. 2, Feb. 2019. [CrossRef]
- Mejías-Peña et al, Y., “Effects of aerobic training on markers of autophagy in the elderly,” Age (Dordr)., vol. 38, no. 2, Apr. 2016. [CrossRef]
- Drummond et al, M.J., “Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 69, no. 8, pp. 1040–1048, 2014. [CrossRef]
- Russ, D.W.; Boyd, I.M.; McCoy, K.M.; McCorkle, K.W., “Muscle-specificity of age-related changes in markers of autophagy and sphingolipid metabolism,” Biogerontology, vol. 16, no. 6, pp. 747–759, Dec. 2015. [CrossRef]
- Sebastián et al, D., “Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway,” EMBO J., vol. 35, no. 15, pp. 1677–1693, Aug. 2016. [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-khat, D.; Hood, D.A., “Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity,” J. Physiol., vol. 596, no. 16, pp. 3567–3584, Aug. 2018. [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A., “Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle,” Am. J. Physiol. Cell Physiol., vol. 304, no. 5, pp. 422–430, 2013. [CrossRef]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G., “Parkin overexpression protects from ageing-related loss of muscle mass and strength,” J. Physiol., vol. 597, no. 7, pp. 1975–1991, Apr. 2019. [CrossRef]
- Masiero et al, E., “Autophagy Is Required to Maintain Muscle Mass,” Cell Metab., vol. 10, no. 6, pp. 507–515, Dec. 2009. [CrossRef]
- Pyo et al, J.O., “Overexpression of Atg5 in mice activates autophagy and extends lifespan,” Nature Communications 2013 4:1, vol. 4, no. 1, pp. 2300-, Aug. 2013. [CrossRef]
- García-Prat et al, L., “Autophagy maintains stemness by preventing senescence,” Nature, vol. 529, no. 7584, pp. 37–42, Jan. 2016. [CrossRef]
- Jiao, J.; Demontis, F., “Skeletal muscle autophagy and its role in sarcopenia and organismal aging,” Curr. Opin. Pharmacol., vol. 34, pp. 1–6, Jun. 2017. [CrossRef]
- Noda, N.N.; Inagaki, F., “Selective Autophagy: Role of Interaction between the Atg8 Family Interacting Motif and Atg8 Family Proteins,” Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging, pp. 39–48, Jan. 2014. [CrossRef]
- Minois et al, N., “Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways,” Cell Death & Disease 2012 3:10, vol. 3, no. 10, pp. e401–e401, Oct. 2012. [CrossRef]
- Minois, N.; Rockenfeller, P.; Smith, T.K.; Carmona-Gutierrez, D., “Spermidine feeding decreases age-related locomotor activity loss and induces changes in lipid composition,” PLoS One, vol. 9, no. 7, Jul. 2014. [CrossRef]
- Tong, D.; Hill, J.A., “Spermidine Promotes Cardioprotective Autophagy,” Circ. Res., vol. 120, no. 8, pp. 1229–1231, Apr. 2017. [CrossRef]
- Yang et al, X., “Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans,” Aging, vol. 12, no. 17, pp. 16852–16866, Sep. 2020. [CrossRef]
- Singh et al, A., “Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults,” Cell Rep. Med., vol. 3, no. 5, May 2022. [CrossRef]
- Liu et al, S., “Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial,” JAMA Netw. Open, vol. 5, no. 1, Jan. 2022. [CrossRef]
- Andreux et al, P.A., “The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans,” Nat. Metab., vol. 1, no. 6, pp. 595–603, Jun. 2019. [CrossRef]
- Eisenberg et al, T., “Induction of autophagy by spermidine promotes longevity,” Nat. Cell Biol., vol. 11, no. 11, pp. 1305–1314, 2009. [CrossRef]
- Eisenberg et al, T., “Cardioprotection and lifespan extension by the natural polyamine spermidine,” Nat. Med., vol. 22, no. 12, pp. 1428–1438, Dec. 2016. [CrossRef]
- Ahsan et al, A., “Natural compounds modulate the autophagy with potential implication of stroke,” Acta Pharm. Sin. B, vol. 11, no. 7, pp. 1708–1720, Jul. 2021. [CrossRef]
- Christian, C.J.; Benian, G.M., “Animal models of sarcopenia,” Aging Cell, vol. 19, no. 10, Oct. 2020. [CrossRef]
- Rana et al, A., “Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster,” Nat. Commun., vol. 8, no. 1, Dec. 2017. [CrossRef]
- Piccirillo, R.; Demontis, F.; Perrimon, N.; Goldberg, A.L., “Mechanisms of muscle growth and atrophy in mammals and Drosophila,” Dev. Dyn., vol. 243, no. 2, pp. 201–215, Feb. 2014. [CrossRef]
- Demontis, F.; Piccirillo, R.; Goldberg, A.L.; Perrimon, N., “Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models,” Dis. Model. Mech., vol. 6, no. 6, pp. 1339–1352, Nov. 2013. [CrossRef]
- Taylor, M.V., “Comparison of Muscle Development in Drosophila and Vertebrates,” 2013, Accessed: Nov. 14, 2025. [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK6226/.
- Nikonova, E.; Kao, S.Y.; Spletter, M.L., “Contributions of alternative splicing to muscle type development and function,” Semin. Cell Dev. Biol., vol. 104, pp. 65–80, Aug. 2020. [CrossRef]
- Park, C.; Jeong, J.-W.; Choi, Y.H., “Induction of Muscle Atrophy by Dexamethasone and Hydrogen Peroxide in Differentiated C2C12 Myotubes,” J. Life Sci., vol. 27, no. 12, pp. 1479–1485, 2017. [CrossRef]
- Siu, P.M.; Wang, Y.; Alway, S.E., “Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes,” Life Sci., vol. 84, no. 13–14, pp. 468–481, Mar. 2009. [CrossRef]
- Pierre, N.; Barbé, C.; Gilson, H.; Deldicque, L.; Raymackers, J.M.; Francaux, M., “Activation of ER stress by hydrogen peroxide in C2C12 myotubes,” Biochem. Biophys. Res. Commun., vol. 450, no. 1, pp. 459–463, Jul. 2014. [CrossRef]
- Fan, X.; Hussien, R.; Brooks, G.A., “H2O2-induced mitochondrial fragmentation in C2C12 myocytes,” Free Radic. Biol. Med., vol. 49, no. 11, pp. 1646–1654, Dec. 2010. [CrossRef]
- Zeng, Z.; Liang, J.; Wu, L.; Zhang, H.; Jun, L.V.; Chen, N., “Exercise-Induced Autophagy Suppresses Sarcopenia Through Akt/mTOR and Akt/FoxO3a Signal Pathways and AMPK-Mediated Mitochondrial Quality Control,” Front. Physiol., vol. 11, 2020. [CrossRef]
- Bian, A.L.; Hu, H.Y.; Rong, Y.D.; Wang, J.; Wang, J.X.; Zhou, X.Z., “A study on relationship between elderly sarcopenia and inflammatory factors IL-6 and TNF-α,” Eur. J. Med. Res., vol. 22, no. 1, Jul. 2017. [CrossRef]
- Nichenko et al, A.S., “Lifelong Ulk1-Mediated Autophagy Deficiency in Muscle Induces Mitochondrial Dysfunction and Contractile Weakness,” Int. J. Mol. Sci., vol. 22, no. 4, pp. 1–20, Feb. 2021. [CrossRef]
- Bian et al, A., “Association between sarcopenia and levels of growth hormone and insulin-like growth factor-1 in the elderly,” BMC Musculoskelet. Disord., vol. 21, no. 1, Apr. 2020. [CrossRef]
- Haas, R.H., “Mitochondrial Dysfunction in Aging and Diseases of Aging,” Biology (Basel)., vol. 8, no. 2, Jun. 2019. [CrossRef]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M., “The pharmacological regulation of cellular mitophagy,” Nat. Chem. Biol., vol. 13, no. 2, pp. 136–146, Jan. 2017. [CrossRef]
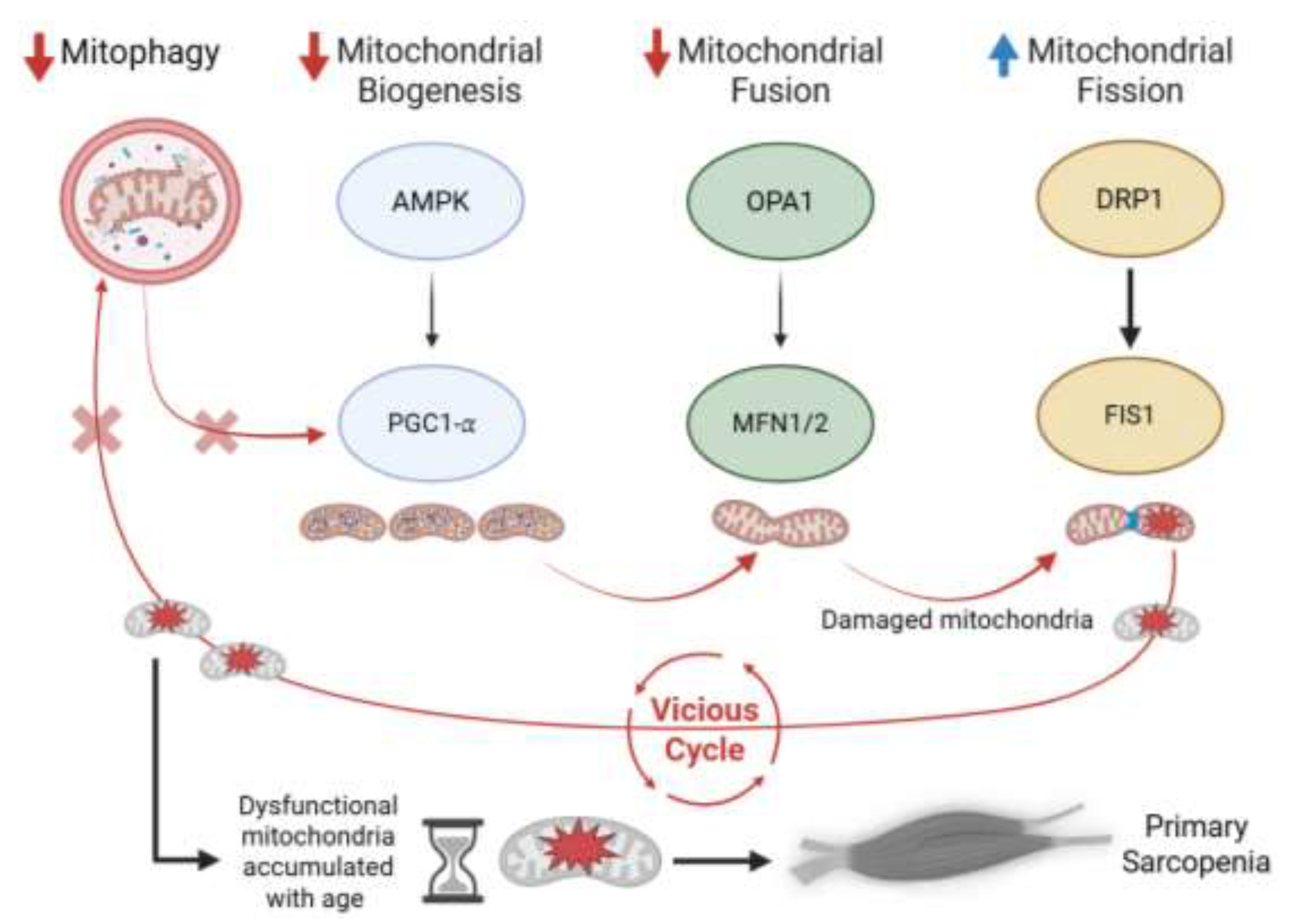
Figure 1.
Pathway of mitochondrial dysfunction driving primary sarcopenia. Aging reduces AMPK–SIRT1 signaling, downregulating PGC-1α and mitochondrial biogenesis. Impaired fusion follows via decreased OPA1 and MFN1/2, while DRP1 and FIS1 are upregulated, promoting excessive fission. This imbalance in mitochondrial dynamics leads to fragmented, damaged mitochondria. However, mitophagy is concurrently suppressed, preventing their clearance. The failed turnover marks the critical blockade in mitochondrial quality control, driving mitochondrial dysfunction and culminating in muscle atrophy and weakness—primary sarcopenia.
Figure 1.
Pathway of mitochondrial dysfunction driving primary sarcopenia. Aging reduces AMPK–SIRT1 signaling, downregulating PGC-1α and mitochondrial biogenesis. Impaired fusion follows via decreased OPA1 and MFN1/2, while DRP1 and FIS1 are upregulated, promoting excessive fission. This imbalance in mitochondrial dynamics leads to fragmented, damaged mitochondria. However, mitophagy is concurrently suppressed, preventing their clearance. The failed turnover marks the critical blockade in mitochondrial quality control, driving mitochondrial dysfunction and culminating in muscle atrophy and weakness—primary sarcopenia.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



