
Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
Calcium imaging with miniscopes allows researchers to record the activity of many neurons over long periods in freely moving animals. While data collection has become easier, analysis have not. Typical calcium imaging analysis requires many processing steps, uses multiple software tools, and depends heavily on parameter choices. In practice, these details are often poorly recorded, rely on proprietary software, or are lost when large intermediate files are deleted to save disk space. As a result, analyses are hard to reproduce, compare, or rerun reliably. This paper describes an open, Python-based framework designed to make calcium imaging analysis clearer, more reproducible, and easier to manage. The framework treats each analysis step as an explicit, recorded operation with defined inputs, outputs, parameters, and software providers. All steps are logged in lightweight trace files saved to disk, allowing analyses to be resumed, audited, or exactly reproduced later, even if large intermediate data have been removed. Algorithm-specific code is isolated behind standardized wrappers, so users can switch between proprietary and open-source tools without changing the overall workflow.The framework also supports branching to compare different methods, batch processing across multiple animals or sessions, controlled cleanup to reduce disk usage, and a modular design. The result is a practical system that makes calcium imaging analyses easier to follow, repeat, and reuse.
Keywords:
neuroscience open-source calcium imaging
1. Introduction
Genetically encoded calcium indicators such as GCaMP have revolutionized the visualization of neural activity by translating activity-linked calcium dynamics into fluores- cence signals that can be recorded over time [1]. While early in vivo calcium imaging was constrained by hardware and experimental setup, miniaturized, head-mounted mi- croscopes paired with implantable gradient index (GRIN) microendoscopes have enabled recordings from freely behaving animals and from deeper brain structures that are inaccessible with standard surface imaging [1,2]. These miniscope systems support longitudinal monitoring of large neural populations over weeks to months, which is particularly valuable for studying behavior and disease related circuit changes under naturalistic conditions [1]. Miniscope datasets have been collected across multiple brain regions and behavioral domains, and the field has increasingly relied on automated algorithms to transform raw movies into cell footprints and activity traces at scale [1].
As recording capacity has grown, producing increasingly rich and voluminous datasets, the primary bottleneck has shifted from data acquisition to computation. Extracting reliable cellular signals typically requires multi-step analy- sis pipelines, including motion correction, segmentation, event detection, deconvolution, and trace extraction1. Dif- ferent algorithmic choices at each stage can materially affect the final outputs, including the number of detected cells and the quality of extracted traces, particularly for one-photon microendoscopic data where out-of-focus background fluorescence is substantial1. This sensitivity means that calcium imaging results can become diffi- cult to compare across laboratories unless the full work- flow, parameterization, and execution order are explicit, recorded and shareable [3].
These challenges are part of a broader reproducibility problem in science. A landmark survey in Nature reported that more than 70% of researchers struggle to reproduce others’ findings, and over half report difficulty reproduc- ing their own previous results4. In neuroscience, where analyses often involve long chains of processing steps, multiple interacting parameters, and ad hoc custom scripts, reproducibility failures commonly arise from miss- ing parameter records, undocumented intermediate files, or software environments that cannot be reconstructed [3,5]. As a result, two research groups can begin with identical recordings yet arrive at divergent sets of detected cells and neural activity traces because their pipelines differ in small but compounding ways [1].
Open and transparent computational practice is one response to this problem. Recommendations for compu- tational reproducibility emphasize sharing code, tracking versions, recording execution environments, and mak- ing the full sequence of processing steps inspectable [3,6]. Jupyter notebooks align naturally with these principles by combining narrative explanation, executable code, and visible intermediate outputs in a single document, resulting in “computational essays” that are easier to un- derstand, audit, and rerun [7]. Furthermore, these notebooks support exporting outputs to publication-ready formats.
Version-controlled code sharing platforms such as GitHub further support open science by providing a public, collab- orative environment for sharing, reviewing, and auditing analyses. This infrastructure makes it possible for others to recreate complete analysis workflows rather than infer- ring missing details from written descriptions alone. Fi- nally, using Python lowers barriers to access and verifica- tion compared with proprietary, closed-source platforms that require paid licenses (e.g., MATLAB) [8]. Python also provides a mature ecosystem of scientific libraries for nu- merical computing and data handling, including NumPy, SciPy, and Pandas, which support flexible and extensible neuroscience workflow [9].
Despite the expanding ecosystem of calcium imaging tools, such as Minian, an open-source, notebook-based pipeline for miniscope analysis [10], many real-world work- flows continue to fall short in practice. Disk storage is a persistent limitation: most pipelines generate large intermediate video files (e.g., motion-corrected) that can quickly consume hundreds of gigabytes per experiment.
Customizability presents another challenge. Although standard analysis steps are often available within individ- ual tools, combining or modifying them in non-standard ways frequently requires switching between multiple software frameworks. This fragmentation disrupts repro- ducibility, complicates automation, and makes it difficult to trace complete workflows end to end.
Finally, Batch processing is also poorly supported. Many existing tools are optimized for single-session analyses and provide limited infrastructure for managing large-scale experiments spanning multiple animals or recording days.
Taken together, these limitations reflect a structural weak- ness in current calcium imaging workflows. While final outputs may remain interpretable to the original analyst, critical analytical context, such as parameter choices, in- termediate results, execution order, and software environ- ment, is often insufficiently captured for reuse or system- atic comparison. These shortcomings become increasingly problematic as datasets are shared across laboratories or revisited for secondary analyses.
This framing aligns with the FAIR Guiding Principles, which state that scientific outputs should be findable, ac- cessible, interoperable, and reusable by both humans and machines [11]. In practice, however, many miniscope work- flows remain fragmented: laboratories commonly stitch together separate tools for motion correction, ROI extrac- tion, and downstream analysis, with handoffs that are difficult to track, standardize, or audit1. Without explicit records of how intermediate outputs are generated and consumed, it becomes unclear which processing branch or parameter set produced a given result, limiting repro- ducibility and hindering systematic method comparison5.
To understand what motivated us to create this new frame- work along with its design we summarize the technical challenges commonly encountered when running and maintaining calcium imaging analysis.
- Multiple providers. Equivalent processing stages may be implemented by different providers with distinct interfaces and default behaviors, making the notion of “the pipeline” ambiguous unless provider choice is explicitly recorded.
- Multiple inputs of the same type. Real analyses fre- quently involve many recordings, where individual steps may produce zero, one, or multiple outputs per input, requiring an explicit mechanism for maintain- ing these associations.
- Multiple data types. Pipelines operate on heteroge- neous data types (e.g., videos, cell sets, summary tables), each with different semantics. Steps must therefore know both the content and the type of each input to behave correctly.
- Multiple formats per data type. The same conceptual data type may be stored in different file formats de- pending on the algorithm or provider, requiring these formats to be distinguishable and explicitly tracked.
- High disk usage. Large imaging datasets and their in- termediate products consume substantial disk space. Since many steps generate outputs comparable in size to their inputs, accumulated intermediates can quickly exceed available storage.
- Input selection. When multiple outputs of the same type are available, selection rules must be explicit to prevent accidental cherry-picking or inconsistent reuse.
- Algorithm parameterization. Reproducibility depends on recording parameter values. Default parameters must be easy to define, and the mapping between steps and parameters must remain unambiguous.
- Experiment reproducibility. Retaining only final out- puts is insufficient without a systematic mechanism that records which algorithms ran, with which para- meters, on which inputs.
- Testability. From a developer standpoint, the system must remain easy to modify without breaking existing behavior. From a development perspective, the system must remain easy to modify without breaking existing behavior. Rapid and automated testing is essential to support safe, iterative development.
- Repeated analyses across subjects. Users often need to run identical analyses on data from different animals or sessions. Simple pipeline abstractions frequently assume a single subject, requiring more flexible exe- cution models.
Motivated by these constraints, this project aims to build a modular, reproducible, and resource-efficient library for calcium imaging pipelines, in which every processing step and parameter is explicitly exposed and tracked within a notebook-based workflow. The goal is to enable users to run a pipeline, branch and compare alternative methods at any stage, and maintain a reliable execution record so that results can be reproduced exactly at a later time, even when large intermediate files have been removed. Figure 1 summarizes the core design objectives.
2. Methods
To address these challenges, we developed an open, Python-based, modular library for building standardized calcium imaging pipelines with explicit tracking of all processing steps. The design goal is to make workflows au- ditable and repeatable by recording which algorithm was run, on which inputs, with which parameters, and what outputs were produced at each stage. The library structure follows reproducible research principles that emphasize clear documentation, automation, and the ability to rerun analyses without manual reconstruction [5].
A run starts by creating a pipeline object initialized with a set of input files. Each algorithm is treated as a pipeline “step” that is executed by calling a method on the pipeline object. When a step runs, it consumes one or more inputs and produces outputs that can be used by later steps. Each step returns the same pipeline object, allowing steps to be chained. This chaining pattern keeps analyses readable while ensuring that each operation remains explicit and inspectable. In this way, the pipeline is initialized with a defined set of inputs, and downstream steps explicitly consume outputs produced by earlier steps.
Since calcium imagine pipelines are file heavy and many algorithms read from and write directly to disk, all inputs and outputs are represented as file paths. Every step writes its outputs into its own dedicated directory. When an external output directory is specified, the pipeline mirrors the input directory structure under the output root and appends step-specific suffixes to each generated file. This organization allows users to understand the full process- ing history by inspecting the output directory alone.
2.1. Traces
A trace is a single, persistent record of a pipeline run that is updated after each successfully completed step and can be used to reconstruct or resume execution at any point. Each pipeline run maintains a global trace, stored on disk as a JSON file. Figure 2 illustrates an example of a pipeline trace is created with multiple branches. Storing the trace on disk rather than only in memory ensures that it remains recoverable after crashes or unexpected interruptions.
The trace is initialized when the pipeline is created and updated after every successfully completed step. It records:
- • Step number and branch
- • Algorithm or provider implementation executed
- • Parameter values used
- • Input and output file paths
- • Identifiers linking outputs back to original inputs
Although this metadata may appear redundant, it serves two practical purposes. First, it confirms that a step com- pleted successfully and produced the expected outputs. Second, it enables output reuse by detecting whether results already exist for the same combination of input files, algorithm, and parameters, allowing the step to be skipped. This behavior is particularly useful in batch runs that are interrupted, for example due to exhausted disk space.
2.2. Algorithm Libraries and Providers
Provider libraries are often difficult to install across vari- ous environments, and most laboratories rely on a mixture of tools rather than a single software ecosystem. To avoid tight coupling, algorithm providers such as Inscopix and CaImAn, as well as future implementations, are injected externally rather than treated as mandatory dependencies. This follows standard software design practice in which provider-specific code is isolated so that changes in one tool’s behavior or output formats do not propagate through the entire pipeline.
As a result, the core library remains usable even when only a single provider is available, and users are not forced to install every supported backend. Provider integrations are implemented as separate modules, with one module per provider, rather than through a single combined interface.
This makes provider choice explicit at each step, localizes maintenance, and ensures that adding a new provider does not require modifying unrelated parts of the code.
2.3. Wrappers Around Provider Algorithms
Provider algorithms are always invoked through a wrapper layer that standardizes required inputs, parameter passing and recording, output locations, and saved metadata. This wrapper layer is also where format conversions are imple- mented, such as converting between movie containers when a downstream algorithm requires a specific format.
The motivation for using wrappers is that, although provider tools can be powerful, they become difficult to adopt when parameter handling and data transformations are implicit, hidden, or inconsistent. Wrappers make these operations explicit, inspectable, and reproducible.
2.4. Parameter Handling
Parameters can be specified at three levels, with the fol- lowing priority order:
- Parameters passed directly to a pipeline step
- Pipeline-level default parameters
- Provider-level defaults
This hierarchy ensures that explicit user choices always override implicit defaults, while still allowing reasonable behavior when parameters are not manually specified.
2.5. Keys and Identifiers
Each pipeline step explicitly declares the data it consumes and the data it produces. Data are passed between steps using keys, which serve as explicit labels for both data type and functional role within the workflow. A single step may consume multiple inputs and produce one or more outputs of different data types.
When a step produces an output associated with a given key, that output becomes the active value for that key within the pipeline state. If a later step produces the same key, the new output replaces the previous one. If a step re- quests a key that has not been defined by an earlier step or provided as an initial input, execution fails with an explicit error, rather than silently substituting an incorrect file. This behavior is intended to prevent implicit or ambiguous data flow.
Different file formats representing the same conceptual data type are distinguished using format-specific keys (e.g., videos_isxd and videos_tiff). Conversions between formats are implemented as explicit pipeline steps, ensur- ing that all format transformations are captured in the trace.
Initial inputs are assigned unique identifiers that propa- gate through the pipeline. This preserves correct associa- tions in multi-input steps and batch workflows, such as linking extracted traces back to their source recordings across multiple sessions.
2.6. Storage Efficiency, Cleanup and Recovery
In calcium imaging intermediate files can quickly con- sume the available storage. To deal with this, our pipeline supports optional automatic cleanup, where older outputs for a key can be deleted once a newer output replaces them in order to save disk space. If needed manual control is also an option.
In addition, if an execution of a step stops due to an error, system restart or depleted disk space, the pipeline can resume from the last successfully recorded step using the trace, without recomputing earlier steps.
2.7. Branching and Batch Execution
Pipelines can be branched to explore alternative algo- rithms or parameter settings starting from a common analysis point. Branches create independent pipeline ob- jects that share upstream results but diverge downstream.
For large experiments, a multi-pipeline abstraction en- ables batch execution of the same analysis across multiple animals or sessions while keeping files, traces, and meta- data isolated per dataset.
2.8. Processing Tools and Interoperability
The current implementation interfaces with Inscopix out- put formats and algorithms, reflecting their widespread use in experimental workflows. However, the pipeline is designed to support additional tools, such as CaImAn [12], through the same provider and wrapper mechanisms.
2.9. Quality Control
To validate the pipeline in a controlled setting, we used a synthetic data simulator that generates calcium imag- ing videos with configurable ground-truth cell masks and activity traces. The simulator can introduce degra- dations such as noise, motion artifacts, and corrupted frames. Running a full pipeline on these datasets enables automated, quantitative validation of motion correction, corrupted-frame handling, segmentation, and calcium trace extraction, and provides a consistent framework for regression testing.
2.10. Tests Driven Development
Each feature developed for this framework was imple- mented following test driven development, a process where tests are written before the code and development progresses through short, iterative cycles [13]. This approach allowed us to write the minimum code required while
keeping track of the capabilities of each feature within the codebase itself.
These tests not only allow us to test in a controlled envi- ronment how the pipeline should work, but also enable us to understand how the pipeline behaves in its base state and when integrated with external dependencies, such as ISX and CaImAn. By mocking these external dependencies, along with the file system, we significantly accelerated development, since there was no need to run real ISX or CalmAn algorithms, which would have been time consuming. Instead, we leveraged the described test architecture to validate everything faster.
Through this we have not only consolidated a considerable test suite, but we have also created a set of runnable exam- ples. These examples provided through Jupyter notebook allow new users to quickly understand the pipeline and the purpose of specific features.
3. Results
The primary contribution of this work is a fully imple- mented, inspectable, and reusable analysis pipeline. All components required to run, evaluate, and extend the pipeline are publicly available.
3.1. Source Repository
The complete CIPipeline implementation is maintained as an open-source repository on GitHub, which serves as the reference for the code, development history, and issue tracking: https://github.com/CGK-Laboratory/ci_pipe
3.2. Documentation
User and developer documentation is hosted separately to give a clear, stable description of how the pipeline is structured and run. It lists the inputs and outputs at each step, configuration options, and where the pipeline can be extended: https://cipipe.gitbook.io/cipipe-docs
3.3. Package Distribution
For installation and explicit dependency management, the pipeline is distributed as a Python package via PyPI: https://py p i.org/project/cipipeline
4. Discussion
This work was motivated by a practical gap between how calcium imaging analyses are described and how they are actually run. Modern miniscope datasets are processed through long chains of steps, each with algorithm choices, parameters, and intermediate outputs that substantively affect the results. In practice, much of this information is either scattered across scripts or lost once large interme- diate files are deleted. What we add here is not a new algorithm, but an execution infrastructure that makes the pipeline explicit, reproducible, and comparable across branches and batches, while keeping the workflow prac- tical for day-to-day use.
Each operation is treated as a recorded step with defined inputs, outputs, and parameters, and this information is documented in lightweight traces, making it possible to reconstruct exactly how a result was produced without relying on fragile manual records or opaque project files. At the same time, the framework deliberately separates workflow logic from specific algorithm providers. Many labs rely on vendor software because it is often the most practical way to work with real datasets but closed-source tools make it difficult to audit behavior, compare methods, or debug unexpected results. By isolating provider-specific code behind wrappers and injecting these dependencies only when needed, the pipeline avoids locking analyses to a single ecosystem, supports direct comparison between proprietary and open tools, and allows gradual transitions without rewriting the surrounding workflow or invalidat- ing previous results.
Finally, the emphasis on traceability and storage efficiency reflects constraints that are common but rarely addressed explicitly. Large intermediate videos quickly exhaust disk space, and restarting partial analyses after failures is often a source of errors. Here, per-step metadata, resumable ex- ecution, and optional cleanup allow users to trade storage for reproducibility in a controlled way. The result is not a new analysis method, but an execution structure that better matches how calcium imaging experiments are iter- ated, compared, and revisited over time. In this sense, the framework implements FAIR-style principles: results can be located, rerun, compared, and reused directly, without the need to reconstruct analysis decisions or reverse-engi- neer workflows long after the original run [11].
References
- Stamatakis, A. M.; et al. Miniature microscopes for manipulating and recording in vivo brain activity. Microscopy 2021, 70, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; et al. Long-term dynamics of CA1 hippocampal place codes. Nature Neuroscience 2013, 16, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Grüning, B.; et al. Practical Computational Reproducibility in the Life Sciences. Cell Systems 2018, 6, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 2016. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Roy, B. Challenges of Provenance in Scientific Workflow Management Systems. 2022 IEEE/ACM Workshop on Workflows in Support of Large-Scale Science (WORKS), 2022; pp. 10–18. [Google Scholar] [CrossRef]
- Ince, D. C.; Hatton, L.; Graham-Cumming, J. The case for open computer programs. Nature 2012, 482, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Bayarri, G.; Andrio, P.; Gelpí, J. L.; Hospital, A.; Orozco, M. Using in- teractive Jupyter Notebooks and BioConda for FAIR and reproducible biomolecular simulation workflows. PLOS Computational Biology 2024, 20, e1012173. [Google Scholar] [CrossRef] [PubMed]
- Jurica, P.; van Leeuwen, C. OMPC: an Open-Source MATLAB-to- Python Compiler. Frontiers in Neuroinformatics 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Viejo, G.; et al. Pynapple, a toolbox for data analysis in neuroscience. eLife 2023, 12, RP85786. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; et al. Minian, an open-source miniscope analysis pipeline. eLife 2022, 11, e70661. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M. D.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, A.; Friedrich, J.; Gunn, P.; Kalfon, J.; Et., A. CaImAn an open source tool for scalable calcium imaging data analysis. eLife 2019. [Google Scholar] [CrossRef] [PubMed]
- Beck, K. Test-Driven Development: By Example; Addison-Wesley Pro- fessional, 2003. [Google Scholar]
Figure 1.
Main objectives and storage strategy of the CIPipeline framework. (A) Core features of CIPipeline. The framework is designed to be FAIR- compliant by enforcing structured metadata and explicit configuration files. Cross-lab sharing is enabled through standardized inputs, outputs, and parameter definitions. Full reproducibility is achieved by ensuring that every processing step is deterministic and fully parameterized. Storage efficiency is obtained by allowing intermediate files to be discarded and regenerated on demand. Batch processing support enables scalable analysis across large datasets and multiple experiments. (B) Conceptual overview of the implemented solution. In a typical pipeline from raw video data to ΔF/F transformation, multiple processing steps are applied, each producing a video output. When only the final ΔF/F video is required for downstream analyses, intermediate videos can be removed as long as the arguments and configuration files of each algorithm are preserved. Because each transformation is deterministic and explicitly defined, any intermediate video can be recomputed when needed, enabling reproducibility and facilitating alternative processing strategies.
Figure 1.
Main objectives and storage strategy of the CIPipeline framework. (A) Core features of CIPipeline. The framework is designed to be FAIR- compliant by enforcing structured metadata and explicit configuration files. Cross-lab sharing is enabled through standardized inputs, outputs, and parameter definitions. Full reproducibility is achieved by ensuring that every processing step is deterministic and fully parameterized. Storage efficiency is obtained by allowing intermediate files to be discarded and regenerated on demand. Batch processing support enables scalable analysis across large datasets and multiple experiments. (B) Conceptual overview of the implemented solution. In a typical pipeline from raw video data to ΔF/F transformation, multiple processing steps are applied, each producing a video output. When only the final ΔF/F video is required for downstream analyses, intermediate videos can be removed as long as the arguments and configuration files of each algorithm are preserved. Because each transformation is deterministic and explicitly defined, any intermediate video can be recomputed when needed, enabling reproducibility and facilitating alternative processing strategies.
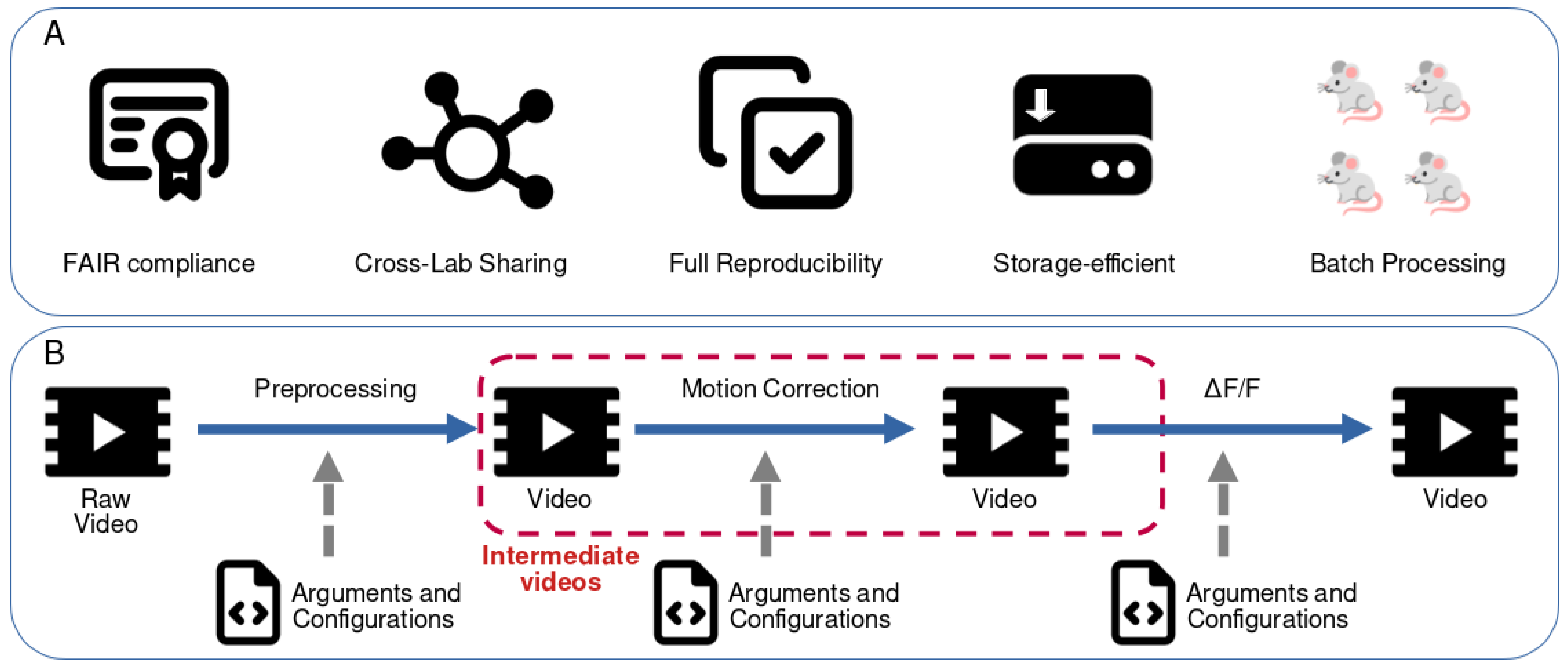
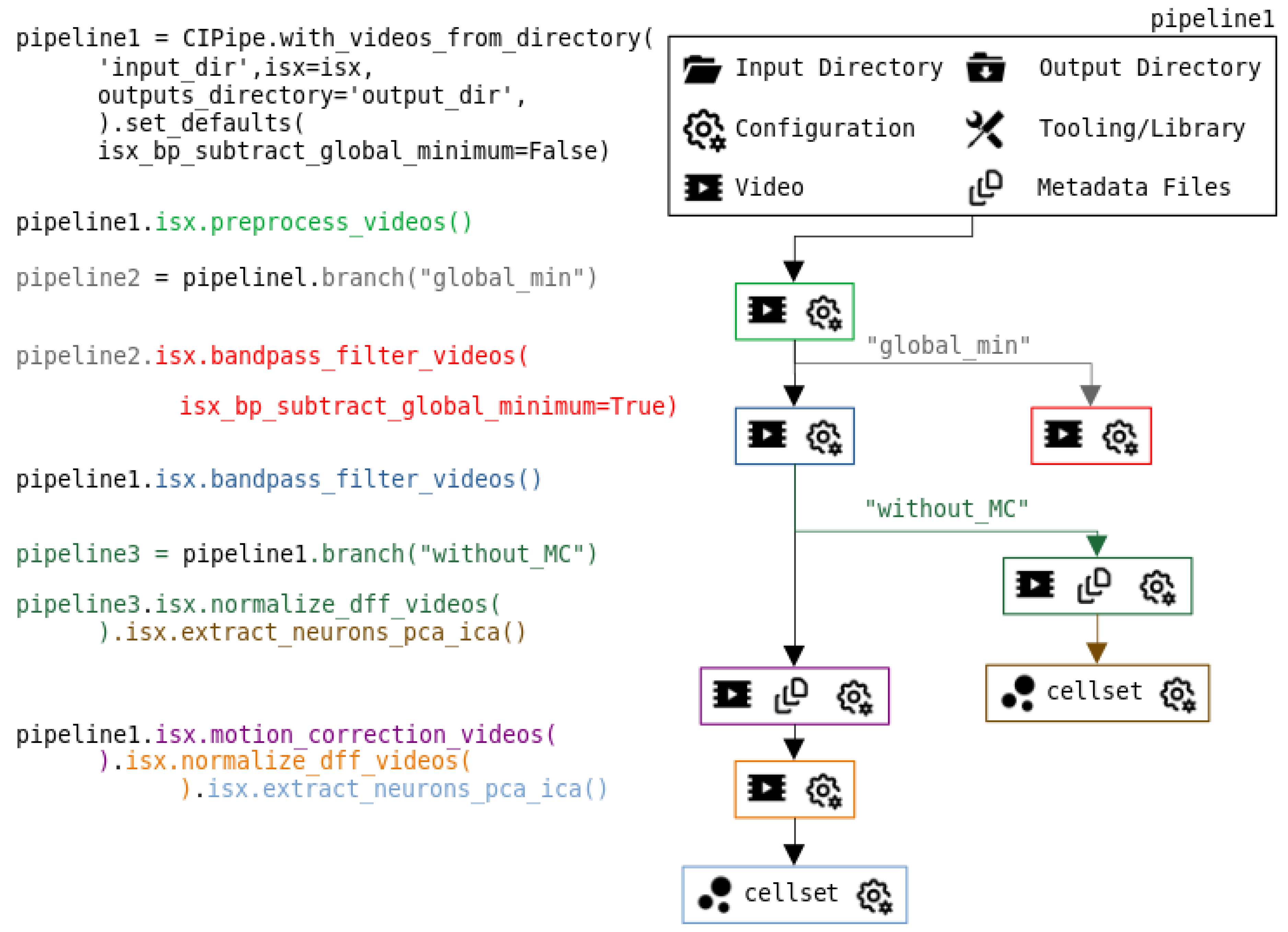
Figure 2.
Example of the execution of a pipeline with multiple branches. The pipeline is constructed by loading videos from an input folder, selecting an output directory, specifying external tools such as the private ISX library, and defining default parameters. In this framework, every operation produces its own output together with its configuration. The first processing step performs preprocessing, after which the user creates a new branch to test different parameters. In pipeline 2, the global minimum is subtracted during the bandpass filtering step, while the original pipeline 1 keeps the default parameter settings. The execution continues adding a new branch (pipeline3) in which cells are extracted immediately after the ΔF/F normalization, without motion correction. Finally, pipeline 1 is completed with the motion correction step, followed by the ΔF/F and PCA-ICA algorithms, enabling an easy comparison between both approaches.
Figure 2.
Example of the execution of a pipeline with multiple branches. The pipeline is constructed by loading videos from an input folder, selecting an output directory, specifying external tools such as the private ISX library, and defining default parameters. In this framework, every operation produces its own output together with its configuration. The first processing step performs preprocessing, after which the user creates a new branch to test different parameters. In pipeline 2, the global minimum is subtracted during the bandpass filtering step, while the original pipeline 1 keeps the default parameter settings. The execution continues adding a new branch (pipeline3) in which cells are extracted immediately after the ΔF/F normalization, without motion correction. Finally, pipeline 1 is completed with the motion correction step, followed by the ΔF/F and PCA-ICA algorithms, enabling an easy comparison between both approaches.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



