Submitted:
30 January 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Hemophagocytic lymphohistiocytosis (HLH) remains one of the greatest diagnostic challenges in pediatric critical care because of its high mortality and the complexity of its clinical presentations. We report the case of an 18-month-old boy who presented with persistent fever, a respiratory tract infection, pancytopenia, hepatosplenomegaly, and progressive neurological deterioration, fulfilling the diagnostic criteria of the HLH-2004 protocol [2]. His clinical course was complicated by concurrent viral infections (adenovirus and parainfluenza virus type III) and antibody-mediated neurological autoimmunity (anti-NMDA receptor encephalitis), an association rarely reported in the current literature [4,5]. A broad differential diagnosis was evaluated with immunologic, genetic, and neuroimaging studies. The combination of autoimmunity, recurrent infections, and positive family history suggested secondary HLH on the background of an underlying primary immunodeficiency [9]. The patient's outcome and the death of a sibling with a similar clinical picture indicate that this episode was not merely secondary HLH, but the first manifestation of a severe primary immune dysregulation, possibly related to mutations in genes such as CTLA4, LRBA, or STAT3—genes recognized within the spectrum of autoimmune lymphoproliferative immunodeficiency disorders (ALPID) [9,10]. This case illustrates the most aggressive facet of pediatric HLH and underscores the need to suspect hidden primary immunodeficiencies in patients with recurrent infections and early autoimmunity. Early recognition of this phenotype can make the difference between a fatal outcome and the possibility of offering targeted immunomodulatory or potentially curative therapies [12,13]. In critical pediatrics, HLH should no longer be considered a final diagnosis but rather a warning sign of an underlying genetic immune dysregulation.
Keywords:
Hemophagocytic Lymphohistiocytosis (HLH)
; isovaleric acidemia
; Anti-N-Methyl-D-Aspartate Receptor Encephalitis
; inborn errors of metabolism
; immune dysregulation
1. Introduction and Objectives
To identify diagnostic criteria and analyze the clinical course of hemophagocytic syndrome/hemophagocytic lymphohistiocytosis (HPS/HLH) in a pediatric patient with concomitant Adenovirus and Parainfluenza virus III infection [2,3]. To examine the causal relationship between respiratory viral infections and the development of secondary HPS/HLH, considering the possibility of an underlying congenital immunodeficiency as a predisposing factor [7,8].
To describe the coexistence of HPS/HLH and post-infectious anti-NMDAR autoimmune encephalitis, assessing its impact on immune dysregulation and neurological manifestations [4,5,6].
2. Clinical Case Presentation
An 18-month-old boy presented to a private clinic in June 2025 with a five-day history of persistent fever. Initially managed as unspecified tonsillitis, he showed no response to parenteral antibiotics and was subsequently transferred to a local hospital, where pneumonia of undetermined etiology and encephalopathy—possibly related to the respiratory infection—were diagnosed.
Four days later, he was referred to the pediatric trauma and shock unit due to progressive respiratory and neurological deterioration accompanied by significant gastrointestinal symptoms. On admission, he scored 9/15 on the Glasgow Coma Scale, exhibited generalized hypotonia, and showed no meningeal signs. Vital signs remained relatively stable with supplemental oxygen requirement.
Given worsening respiratory compromise, invasive mechanical ventilation and broad-spectrum empirical antimicrobial coverage were initiated. PCR-based microbiological studies of sputum samples were positive for Adenovirus and Parainfluenza virus III, while CSF analysis was negative [3,7,8]. Significant elevations in procalcitonin and interleukin-6 were documented, indicative of systemic inflammatory response [3].
Magnetic resonance neuroimaging revealed abnormalities in cortico-subcortical differentiation and lesions consistent with encephalitis. Subsequently, complete blood count showed persistent pancytopenia and elevated ferritin levels, associated with hepatosplenomegaly. Bone marrow aspiration confirmed hemophagocytosis, establishing the diagnosis of HLH [2].
A history of a sibling who died from a similar clinical presentation prompted investigation for an underlying primary immunodeficiency. The patient received comprehensive intensive care management for 11 days, with favorable outcomes after intensive support and immunomodulation, followed by ongoing multidisciplinary follow-up.
3. Methodology and Results
Serological, immunological, microbiological, cytological, histopathological, and imaging studies were performed. Findings included pancytopenia, hypogammaglobulinemia, elevated ferritin, high IL-6 levels, and bone marrow confirmation of hemophagocytosis. Flow cytometry ruled out neoplastic infiltration. Detection of anti-NMDAR antibodies confirmed the diagnosis of autoimmune encephalitis [4,5,6].
4. Discussion
In general terms, the presence of Hemophagocytic Syndrome – HPS (regardless of its origin) is typically associated with a poor prognosis. Secondary forms of HPS (commonly linked to primary/congenital immunodeficiencies (PID), infections (usually viral), neoplasms, or autoimmune processes) are more frequent than primary forms [1].
Initially, our patient meets more than five diagnostic criteria (in this case, 10) of the Hemophagocytic Lymphohistiocytosis Protocol or Hemophagocytic Syndrome (HPS/HLH-2004) (see Table 1) [2]. In our case of HPS/HLH, and based on the clinical history (specifically, a family history of death due to a similar clinical presentation), the aggressive progression of the condition, and the detection of viral agents such as Adenovirus and Parainfluenza, we are led to consider the development of secondary HPS/HLH due to viral infection, as well as the possibility of an underlying primary immunodeficiency.
In this case, the development of HPS/HLH represents a critical juncture in the clinical course and suggests possible dysregulation of immune activation in response to a viral trigger. Although HPS/HLH can arise secondary to infectious processes, this patient is notable for the intensity of the inflammatory picture (in response to viral agents not usually regarded as lethal) and for the striking appearance of numerous pathognomonic features of the syndrome (see Table 1). Histologic confirmation of hemophagocytosis in bone marrow supports the diagnosis. However, the fact that the patient had a sibling who died from a similar clinical picture, together with progressive neurological deterioration and the need for prolonged immunomodulation (see Table 4 and Table 6), obliges us to consider that this process is not merely secondary. This perspective is especially relevant because certain immunodeficiencies can debut with HPS/HLH as their first clinical manifestation.
Referencing the above, in the cohort study by Astudillo et al. [3], one case tested positive for Adenovirus and Parainfluenza; indeed, the index case had a coinfection with both viruses. That study enrolled 23 hospitalized pediatric patients diagnosed with HPS/HLH and used broad microbiological panels like those in our patient. Our infant likewise tested positive for both agents. Recognition of these agents is important in the context of HPS/HLH because they can trigger the uncontrolled inflammatory process (see Table 1). This underscores the importance of timely introduction of specific antiviral therapy, which, combined with immunomodulatory treatment, may improve prognosis, and reduce associated complications (see Table 4).
Furthermore, highlighting the presence of anti-NMDA antibodies in our case (see Table 1), the presence of imaging findings consistent with paraneoplastic or postinfectious autoimmune encephalitis (see Table 1 and Table 6) should be correlated with the diagnosis of SHF/HLH. This entity originates etiopathogenically from antibodies against the GluN1 subunit of the NMDAR receptor, causing profound synaptic dysfunction and glutamatergic system hypofunction [4]. In the systematic review by Yam et al. [5], which included 283 pediatric patients, the most common presentations were seizures (63.3%) and extrapyramidal symptoms (36.7%), with both EEG and MRI showing abnormalities in a significant proportion; these findings align with those observed in our case (see Table 6).

Table 1.
Analytical findings and data interpretation.
| Parameters | Findings | Interpretation |
| Complete blood count | Lymphocytes: 0.2 x 10³/mm³ Leukocytes: 0.8 x 10³/mm³ Neutrophils: 58.5% Monocytes: 0.14 x 10³/mm³ Hemoglobin: 8.5 g/dl Hematocrit: 25.5% Platelets: 10,000 Red cell distribution width: 14.8% |
Pancytopenia (anemia + thrombocytopenia + leukopenia) - suggests bone marrow involvement Neutropenia Severe lymphopenia Monocytopenia Anisocytosis |
| Procalcitonin (pct) | 4.57 ng/ml | Strong suspicion of severe bacterial infection or sepsis High probability of bacteremia |
| Primary immunodeficiency panel | ***** | ****despite the negative result obtained in the primary immunodeficiency screening panel And in contrast to the clinical and laboratory findings (distinct from the aforementioned panel), immunological abnormalities consistent with primary immunodeficiency are suspected |
| Pcr (genexpert – ultra) in sputum sample | Not detected | Negative for tuberculosis |
| Infectious diseases | Cytomegalovirus–igg: 500.00 u/ml Toxoplasma- igg: 204.00 iu/ml |
Positive igg for toxoplasma and cmv: indicates Prior infection Acquired immunity from prior exposure |
| Pcr panel (lower respiratory tract) | Parainfluenza virus type 3 Adenovirus |
Active infection due to: Parainfluenza virus type 3 Adenovirus |
| IL-6 | 97.40 pg/ml | Marked elevation suggests: Systemic bacterial infection Acute inflammatory response |
| Hepato-biliary enzyme profile | Serum amylase: 870.00 u/l Ast: 157.00 u/l Alt: 135.00 u/l Alkaline phosphatase: 137.00 iu/l |
All of the above show significant serum elevation |
| Blood chemistry | Serum creatinine: 0.46 mg/dl | Serum creatinine + physical findings: - indicates sarcopenia |
| Serum electrolytes | Total calcium: 6.74 mg/dl Serum magnesium: 1.19 mg/dl Potassium: 2.36 mg/dl |
Hypocalcemia Hypomagnesemia Hypokalemia |
| Immunology | Immunoglobulin igg: 484.00 mg/dl Immunoglobulin iga: 22.40 mg/dl |
Hypogammaglobulinemia |
| Anti-nmda (n-methyl-d-aspartate) antibodies | 3.1 iu/ml | Positive for anti-nmda receptor autoimmune encephalitis |
| Ferritin | 755 μg/l | Elevated ferritin - indicative of acute inflammatory process present |
| Fibrinogen | 155.9 mg/dl | Normal fibrinogen (approaching borderline values) |
Table 2.
Flow cytometry immunophenotyping.
| Test | Findings | Interpretation |
| Flow cytometry | Erythroid series: Neutrophils: 66.6% Monocytes: 9.9% Eosinophils: 0.47% lymphoid series: T lymphocytes: 16.6% Nk cells: 0.32% B lymphocytes: 2.1% |
Adequate bone marrow sample The described populations – do not exhibit phenotypic alterations for the neoplastic markers studied Conclusion: No evidence of pathological cell infiltration in bone marrow (with the markers studied) |
| Sample type: bone marrow – 3ml | ||
Table 3.
Cytological findings of bone marrow aspirate.
| Exam | Finding | Interpretation |
| Bone marrow aspirate |
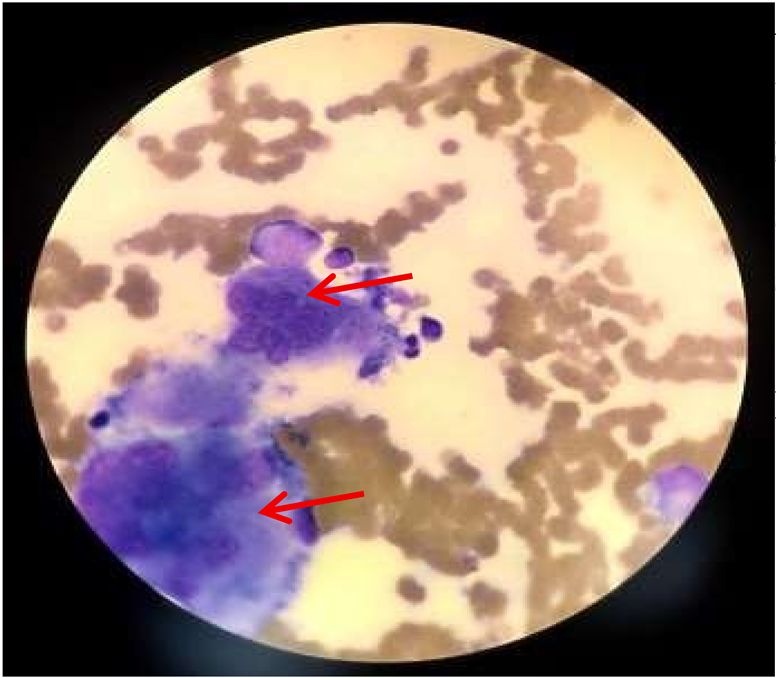 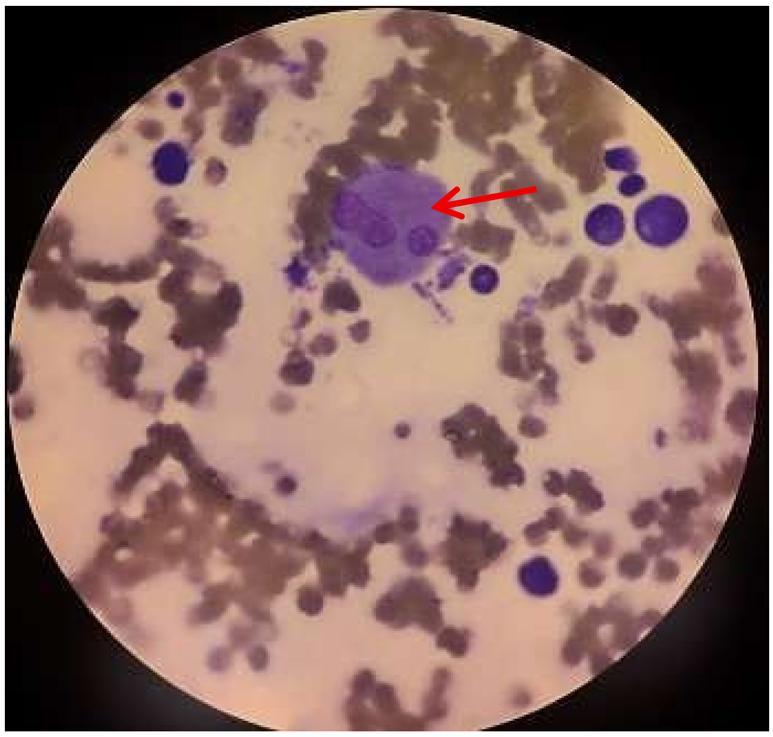 
|
Hemophagocytosis in bone marrow Suggestive of possible secondary hemophagocytic syndrome Findings include:
|
Table 4.
Supportive and pharmacological interventions applied.
| Drug/intervention | Dosage and administration | Duration | Rationale | Changes and explanation |
| Ceftriaxone | IV - unspecified dose | 4 days (june 19–23) |
Empirical initiation for confirmed sepsis Gram-negative coverage |
Discontinued on improved infectious profile Add extended coverage |
| Vancomycin | IV - unspecified dose | 11 days (june 19–30) |
Coverage for resistant gram-positive bacteria Suspected meningitis |
Maintained during prolonged febrile course |
| Meropenem | IV - unspecified dose | 7 days June 23–30 |
Broad coverage for suspected nosocomial or resistant infection | Added due to: Elevated pct Pancytopenia |
| TMP-SMX | IV - unspecified dose | 7 days June 23–30 |
Prophylaxis for suspected immunodeficiency under investigation | Indicated after: Pancytopenia Hlh |
| Acyclovir | IV - unspecified dose | 2 days (june 23–25) |
Suspected viral meningoencephalitis | Discontinued after negative viral study results |
| Oseltamivir | Oral - unspecified dose | 2 days (june 23–25) |
Empirical initiation for viral respiratory symptoms | Discontinued: influenza ruled out |
| Fluconazole | IV - unspecified dose | 5 days (june 25–30) |
Fungal prophylaxis due to immunosuppression | Maintained by: Pancytopenia Persistent fever |
| IMV with ett (invasive mechanical ventilation with endotracheal tube) | Ett: 3.5mm fio2: 30 – 65% | 7 days (june 19 – 26) |
Ventilatory support for acute respiratory failure | Gradual weaning based on clinical improvement |
| Niv (non-invasive ventilation) | Mask or Cannula - Unspecified parameters |
4 days (june 26 – 30) |
Transition after mechanical ventilation withdrawal | Withdrawal based on respiratory tolerance |
| Midazolam | IV - continuous infusion | During mechanical ventilation | Deep sedation | Gradual withdrawal as glasgow coma scale score improves |
| Fentanyl | IV - continuous infusion | During mechanical ventilation | Sedation analgesia | Gradual withdrawal in phase of recovery |
| Dexmedetomidine | IV 0.3–1 Mcg/kg/h |
During post-extubation mechanical ventilation | Light sedation for neuroprotection | Adjusted based on level of consciousness |
Table 5.
Supportive interventions applied.
| Intervention | Details | Duration | Effectiveness |
| Mechanical ventilation | Protective Parameters (PEEP – 8 / FiO2 – 60%) | Day 1 – 7 | Progressive improvement of PaO2/FiO2 Successful Extubation on Day 7 |
| Norepinephrine | 0.1 – 0.3 µg/kg/min IV | Day 1 – 5 | Maintenance of MAP > 65 mmHg Successful progressive Weaning |
| Early rehabilitation | Respiratory and Motor Physiotherapy (Passive and Active) | Day 5 – 23 | Strength/motor recovery Glasgow Coma Scale Score 9 → 13-15 |
| Enteral nutrition | Orogastric Tube Calories and Unspecified Formula | Day 3 – 28 | Good Tolerance Continued nutritional support without complications |
Table 6.
Brain imaging findings in magnetic resonance sequences.
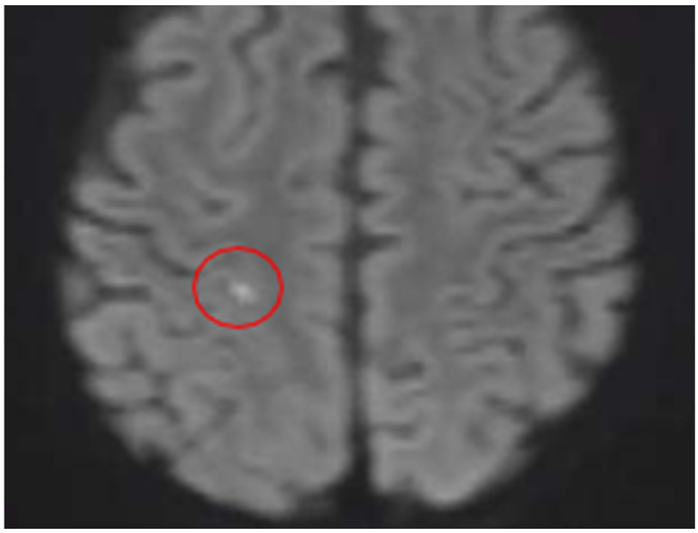
Quadrant A: DWI sequence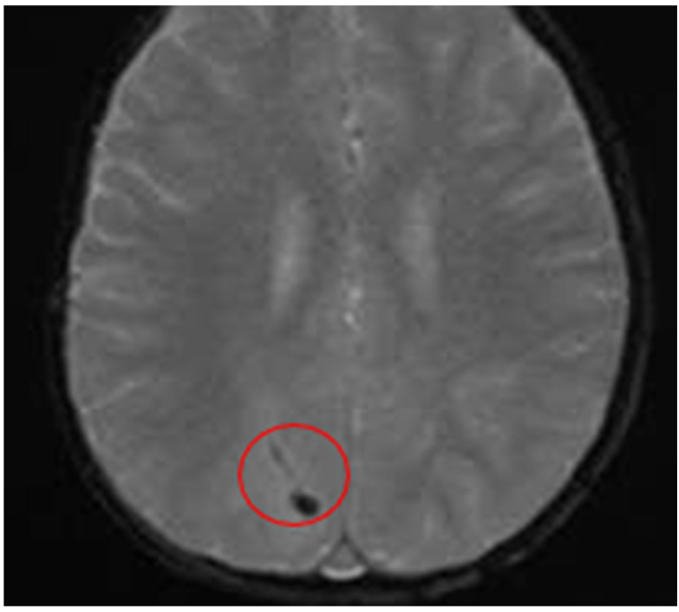
|
Quadrant B: T2 – FLAIR sequence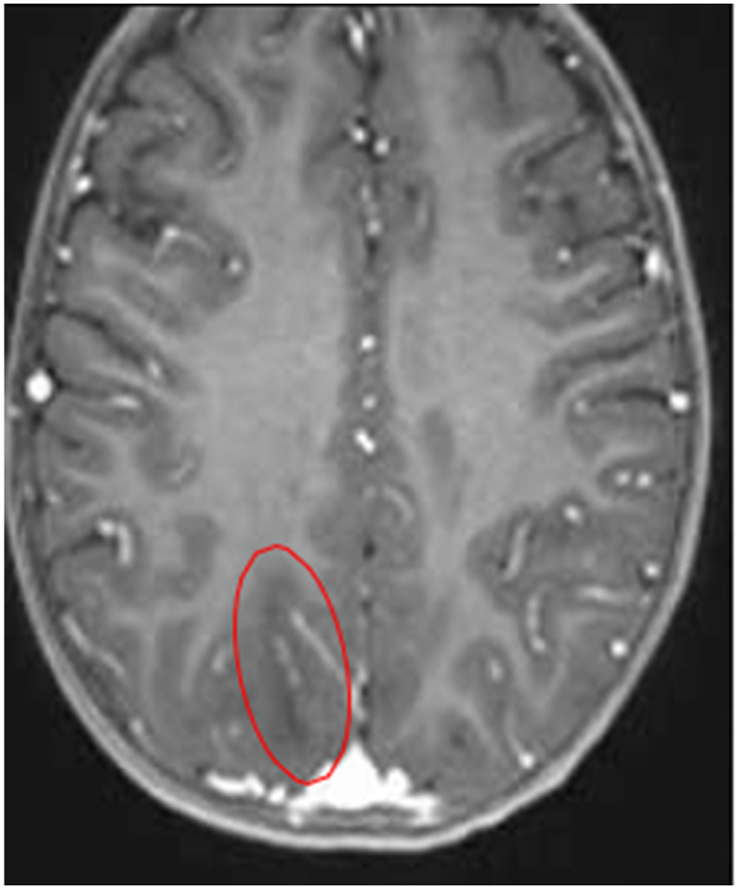
|
| A Minimal Right Subcortical Postcentral Parietal Hyperintensity Is Observed This shows restricted diffusion and signal drop on the ADC map Consistent with a Hyperacute Ischemic Focus
|
Hypointense Spots Are Evident in the Right Occipital Region ∙ Suggestive Findings of Hemorrhagic Collection |
Quadrant C: T1/ADC sequence – contrast-enhanced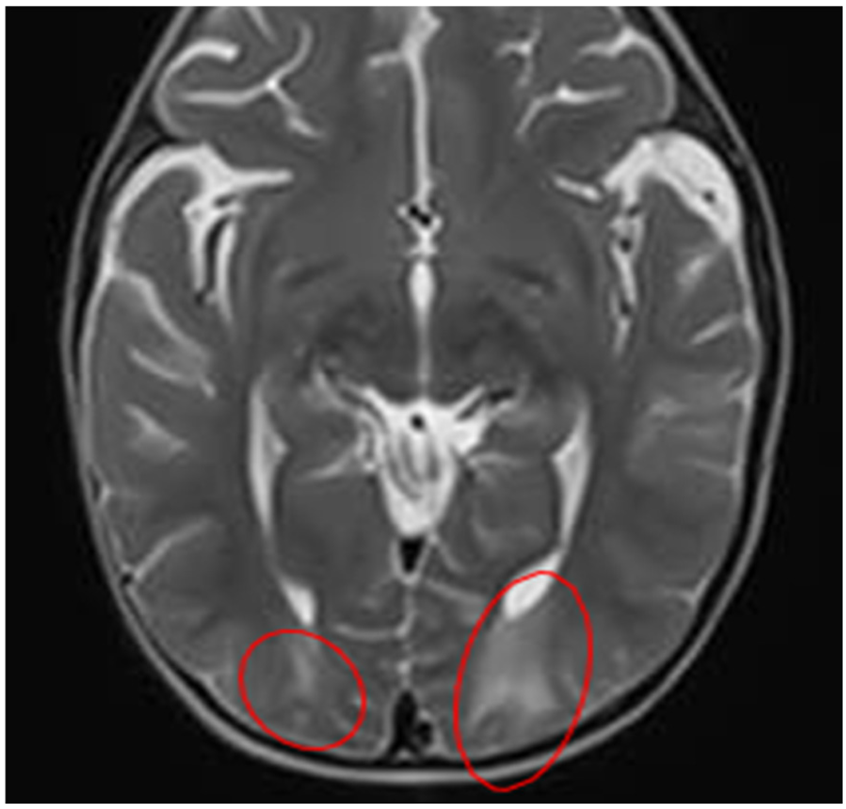
|
Quadrant D: T2 – FLAIR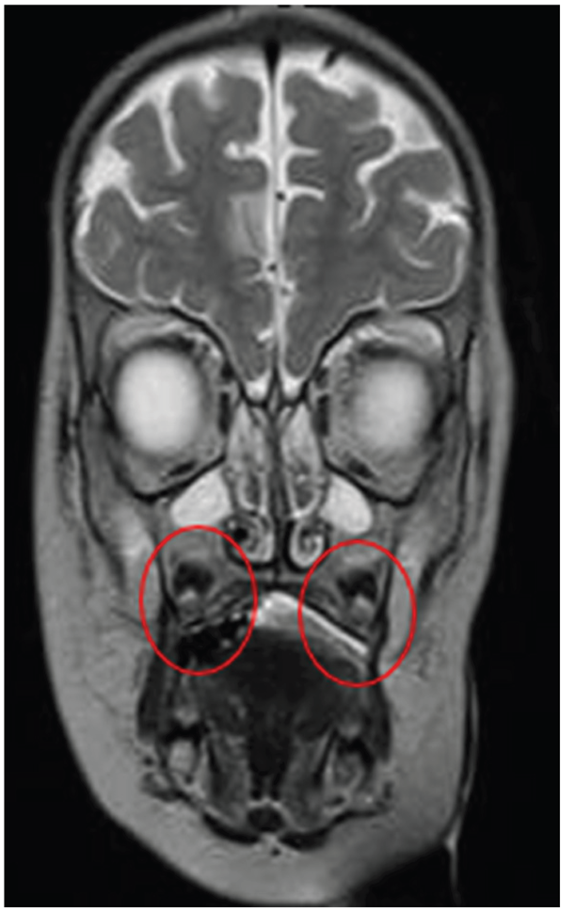 |
| A hypointense focus is observed in the right subcortical postcentral parietal region Probable areas of demyelination are suspected - Suggesting a possible diagnosis of encephalitis |
Abnormalities in gray-white matter differentiation are observed Hyperintense images are observed in the subcortical white matter – in the following regions: Bilateral occipital Bilateral parieto-occipital |
Quadrant E: T2 – FLAIR Hyperintense Tissue is Evident in Bilateral Maxillary Sinuses Hyperintense Tissue is Evident in Bilateral Maxillary SinusesConsistent with Sinusitis |
In our case, a complex phenotype of autoimmune encephalitis developing against a background of primary immune dysregulation should be considered. A study from the Spanish Hospital of Mexico emphasizes that autoimmune encephalitis should be suspected in cases of acute symptoms with infectious prodromes, including fever, malaise, headache, respiratory or gastrointestinal discomfort, neurological disturbances, and even autonomic dysfunction [6]. These findings correlate with the infant’s presentation. Additionally, the positive sputum PCR for Adenovirus and Parainfluenza supports a respiratory viral trigger. In cases of human Parainfluenza virus, pediatric patients commonly present with fever, cough, respiratory distress, and oxygen therapy requirements [7]. Similarly, Adenoviruses are notable for their multisystemic clinical involvement (respiratory, gastrointestinal, and neurological), consistent with our case [8].
After reviewing the relationship between SHF/HLH and autoimmune encephalitis, we will highlight the correlation of both manifestations with their occurrence within the framework of a congenital/primary immunodeficiency. According to Tangye et al. [9], primary immunodeficiencies with immune dysregulation (PID-ID) constitute a growing group of monogenic syndromes that predispose not only to infections but also to autoimmunity, inflammation, lymphoproliferation, and cancer predisposition. Phenotypic heterogeneity, with intrafamilial variability, incomplete penetrance, and epigenetic/environmental modifiers, may explain similar clinical events in both siblings [9]. Despite such variability, hallmarks such as hypogammaglobulinemia, T/B lymphocytopenia, and persistent cytopenias are often observed, findings that correlate with our case. In this context, genes such as STAT3, CTLA4, or LRBA could be considered [9]. Toskov and Ehl also describe the ALPID spectrum as part of PID-ID, associated with multisystem autoimmunity, susceptibility to infections, and cytopenias [10]. Although our case meets several criteria, lymphoproliferation was not evident, so further investigation is required.
Nevertheless, even in the absence of classic lymphoproliferation, the inability to control viral replication is notable, allowing sustained replication and dissemination, perpetuating immune activation, and favoring progression toward SHF/HLH [11].
General and primary treatment measures include progressive antimicrobial coverage tailored to the degree of immunosuppression, neuroprotective sedation, early ventilatory support, and prompt enteral nutrition. Another strategy employed is replacement therapy with immunoglobulins, which was used in our patient with clinical improvement. However, future therapeutic choices must also be based on the specific genotype and the altered immunological pathway [12]. All of the above, combined with a multidisciplinary approach, constitutes a fundamental pillar in the management of these conditions [13].
This case highlights the clinical complexity of HLH/MAS in pediatric patients, where neurological, infectious, and vascular complications closely coexist. High clinical suspicion, early diagnosis, and multidisciplinary management are crucial to improving prognosis and reducing the incidence of complications such as Posterior Reversible Encephalopathy Syndrome (PRES). In our patient, neurological hypoactivity correlated with cerebral edema on MRI justified a diagnosis of PRES; this was supported by the presence of hematologic disease and the use of immunosuppressive drugs as risk factors [14]. Early recognition of PRES is key, as its course can be reversed if the underlying cause is addressed.
This case illustrates the complex interplay between inborn errors of metabolism, particularly isovaleric acidemia, and pediatric hyperinflammatory syndromes, specifically hemophagocytic lymphohistiocytosis (HLH). Whole-exome sequencing identified a pathogenic homozygous variant in the IVD gene (c.1174C>T; p. Arg392Cys), confirming the molecular diagnosis of isovaleric acidemia caused by isovaleryl-CoA dehydrogenase deficiency [15,16]. This finding explains the clinical phenotype, characterized by recurrent infections, thrombocytopenia, and a family history of a deceased sibling with a similar presentation [15].
From an epidemiological standpoint, isovaleric acidemia is a rare disease (≈1/100,000 live births in expanded newborn screening programs), with geographic variability [16,19]. However, when organic acidemias are considered as a group, their incidence increases; population-based neonatal screening studies have reported rates close to 1/10,000 newborns, underscoring their clinical relevance in pediatrics [17,18].
On the other hand, hemophagocytic lymphohistiocytosis is a rare but potentially fatal hyperinflammatory disorder. National epidemiological studies describe low rates, with possible underdiagnosis depending on the population and criteria used [20]. In pediatric populations, HLH can present as primary or secondary, the latter often triggered by severe infections, malignancies, or autoimmune diseases [21,22].
The relevance of this case lies in the clinical and pathophysiological overlap between the metabolic crises of isovaleric acidemia and secondary HLH. During metabolic decompensations, patients with isovaleric acidemia experience intense catabolic stress, systemic inflammation, mitochondrial dysfunction, and accumulation of toxic metabolites, which may manifest as cytopenias, hepatic impairment, multiorgan failure, and a sepsis-like presentation [16,19]. This scenario can mimic an HLH-like phenotype or, in extreme cases, act as a trigger for secondary HLH, particularly with concurrent infections.
Recent literature acknowledges that inborn errors of metabolism, including organic acidemias, may be associated with phenotypes consistent with HLH or severe hyperinflammatory syndromes [21,22,23]. Although the association with isovaleric acidemia is less documented than with other acidemias, the proposed mechanism is shared: dysregulated immune activation due to persistent inflammation, cellular damage, and excessive cytokine release [22,23]. In this context, the described thrombocytopenia constitutes a key finding and a point of convergence between severe metabolic crises and HLH criteria [2,15].
From a clinical perspective, comparative epidemiology proves useful. Following genetic confirmation by whole-exome sequencing, the most parsimonious explanation for episodes of infection and cytopenia corresponds to recurrent metabolic crises [15]. However, secondary HLH should be considered when the inflammatory response is disproportionate, persistent, or refractory, especially if accompanied by marked hyperferritinemia, hypofibrinogenemia, or progressive multiorgan dysfunction [21,22].
Collectively, this case reinforces the need for a hierarchical and integrative diagnostic approach in pediatrics: recognizing the underlying metabolic disease while maintaining a high index of suspicion for secondary hyperinflammatory syndromes when the clinical course exceeds what is expected for an isolated metabolic crisis. Early identification of this overlap has direct implications for treatment, prognosis, and mortality reduction.
Finally, the patient’s clinical improvement following intensive support highlights the efficacy of early intervention. However, the risk of recurrence or neurological progression remains high in the absence of definitive treatment; thus, close neurological follow-up is required, and immunomodulatory or curative therapies should be considered based on the underlying etiology, particularly in the context of an underlying primary immunodeficiency.
5. Conclusions
This case demonstrates that hemophagocytic syndrome in pediatrics should not be interpreted solely as an isolated event or secondary to common infections. The combination of a fulminant inflammatory presentation, neurological autoimmunity, and suggestive family history necessitates consideration of an underlying primary immune dysregulation [9,10,11].
Early recognition of this phenotype transforms the diagnostic and therapeutic approach, enabling consideration of potentially curative strategies such as targeted immunomodulatory therapies or hematopoietic stem cell transplantation [12,13].
In resource-limited settings, such as in developing countries, these cases present an additional challenge due to restricted access to advanced genetic studies, which negatively impacts prognosis. Raising awareness of these cases is crucial to strengthening health policies and improving access to diagnostic technologies.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gómez, VG; Martínez, AP. Síndrome hemofagocítico. Pediatr Integral 2021, 25(6), 326.e1–326.e9. [Google Scholar]
- Canna, SW; Marsh, RA. Pediatric hemophagocytic lymphohistiocytosis. Blood 2020, 135(16), 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, P; Parejas, T; Wietstruck, MA; Morales, P; Abarca, K. Hemophagocytic syndrome: clinical characterization and follow-up of a Chilean pediatric cohort. Rev Chil Infectol. 2021, 38(3), 423–431. [Google Scholar]
- Ortiz Saldaña, S; Jaramillo, G. Análisis de exoma completo por NGS: informe preliminar; Hospital de Especialidades Eugenio Espejo: Quito, 12 Sep 2025. [Google Scholar]
- Yam, G; Parrill, A; Damodar, S; Fogel, J; Ayed, N; Syed, M; et al. Anti-N-methyl-D-aspartate receptor encephalitis in children and adolescents: a systematic review and quantitative analysis. J Can Acad Child Adolesc Psychiatry 2021, 30(4), 236–248. [Google Scholar]
- Carreto-Espinosa, C; Enciso-Peláez, S; Iglesias-Leboreiro, J; Bernárdez-Zapata, I; Silva-Ramírez, H; Escobedo-Berumen, L; et al. Autoimmune encephalitis: experience in the pediatric service of Hospital Español de México. Rev Hosp Juarez Mex. 2023, 90(1), 1–7. [Google Scholar]
- Abu-Helalah, M; Abu Lubad, M; Al-Hanaktah, M; Al Tibi, A; Alhousani, M; Drysdale, SB. Epidemiology and clinical burden of human parainfluenza virus infections among hospitalized children under 5 years of age in Jordan. Viruses 2025, 17(2), 170. [Google Scholar] [CrossRef] [PubMed]
- Soler Wenglein, J; Scarsella, L; Kotlewski, C; Heim, A; Aydin, M. Current trends of human adenovirus types among hospitalized children: a systematic review. Viruses 2025, 17(7), 914. [Google Scholar] [CrossRef] [PubMed]
- Tangye, SG; Al-Herz, W; Bousfiha, A; Cunningham-Rundles, C; Franco, JL; Holland, SM; et al. Human inborn errors of immunity: 2022 update. J Clin Immunol. 2022, 42(1), 147–164. [Google Scholar] [CrossRef] [PubMed]
- Toskov, V; Ehl, S. Autoimmune lymphoproliferative immunodeficiencies in childhood: breakdown of immune homeostasis and immune dysregulation. Mol Cell Pediatr. 2023, 10(1), 11. [Google Scholar] [CrossRef] [PubMed]
- Benevenuta, C; Mussinatto, I; Orsi, C; Timeus, FS. Secondary hemophagocytic lymphohistiocytosis in children. Exp Ther Med. 2023, 26(3), 423. [Google Scholar] [CrossRef] [PubMed]
- Paris, K; Wall, LA. The treatment of primary immune deficiencies: lessons learned and future opportunities. Clin Rev Allergy Immunol. 2023, 65, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Raymond, LS; Leiding, J; Forbes-Satter, LR. Diagnostic modalities in primary immunodeficiency. Clin Rev Allergy Immunol. 2022, 63, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Vélez, R; Benítez, A. Historical series of posterior reversible encephalopathy syndrome at a national pediatric referral hospital. Gac Med Junta Benefic Guayaquil 2023, 1(1), 27–36. [Google Scholar] [CrossRef]
- Ortiz Saldaña, S; Jaramillo, G. Whole-exome sequencing analysis by next-generation sequencing (NGS): preliminary report; Hospital de Especialidades Eugenio Espejo: Quito, Ecuador, 12 Sep 2025. [Google Scholar]
- Mütze, U; Reischl-Hajiabadi, A; Kölker, S. Classic isovaleric acidemia. In GeneReviews® [Internet]; Adam, MP, Feldman, J, Mirzaa, GM, et al., Eds.; University of Washington: Seattle (WA), 2024. [Google Scholar]
- Lin, Y; Zhou, Y; Wang, J; et al. Large-scale newborn screening for organic acidemias: incidence and genetic profiles from 2014 to 2023. Sci Rep. 2025, 15, 15625. [Google Scholar]
- Couce, ML; Castiñeiras, DE; Bóveda, MD; et al. Newborn screening programmes for inborn errors of metabolism: prevalence and outcomes. Orphanet J Rare Dis. 2024, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Thimm, E; Nuoffer, JM; Baumgartner, MR. Isovaleric acidemia: epidemiology, diagnosis and outcome in the era of newborn screening. J Rare Dis. 2025, 11(4), 92. [Google Scholar]
- Kuron, D; Höglund, P; Henter, JI; et al. Epidemiology of haemophagocytic lymphohistiocytosis at a national level. Br J Haematol. 2023, 201(3), 475–483. [Google Scholar] [CrossRef] [PubMed]
- Benevenuta, C; Colafati, GS; D’Argenio, P; et al. Secondary hemophagocytic lymphohistiocytosis in children: a literature review. Children (Basel) 2023, 10(8), 1341. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y; Chen, Y; Wang, J; et al. Hemophagocytic lymphohistiocytosis: current treatment advances and emerging targeted therapies. Front Immunol. 2024, 15, 1387452. [Google Scholar]
- Gioia, C; Roncarolo, MG; Gattorno, M. Pathogenesis of hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Int J Mol Sci. 2024, 25(11), 592. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



