
Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
Transthyretin amyloidosis (ATTR) is a progressive disease characterized by systemic deposition of transthyretin-derived amyloid. Although the recent advent of disease-modifying therapies has expanded treatment options, substantial unmet needs remain, such as understanding disease heterogeneity, predicting treatment response, and prognostic stratification. In this review, we highlight the current and emerging roles of omics technologies in both clinical and research settings of ATTR, including genomics and its integration with other modalities. Currently, omics technologies are applied in clinical settings for accurate disease typing. Clinical samples are utilized to identify risk factors beyond specific transthyretin mutations via genomics and to discover promising biomarkers via proteomics. Accumulating findings from omics analyses using cell and animal models are also facilitating the elucidation of the complex pathology of ATTR. Nevertheless, the application of omics analysis in ATTR research is still developing. Moving forward, it is expected to play a central role in accumulating datasets, leveraging cutting-edge technologies, utilizing integrated multi-omics, and bridging basic and clinical research. These advancements are expected to further accelerate the implementation of next-generation therapeutic strategies and precision medicine.
Keywords:
transthyretin amyloidosis
; transthyretin
; omics
; genomics
; proteomics
; biomarker
; cardiomyopathy
; neuropathy
1. Overview of Tansthyretin Amyloidosis
Amyloidosis is a collective term for a group of diseases in which fibrous proteins rich in β-sheet structures, known as amyloid, deposit in various organs throughout the body, causing organ dysfunction. It is known that over 30 types of precursor proteins can undergo protein misfolding to form amyloid. Among these, transthyretin (TTR) amyloidosis (ATTR) is a clinically significant disease alongside light-chain amyloidosis (AL) caused by immunoglobulin light chains. In ATTR, TTR deposits in various organs such as the heart, peripheral nerves, gastrointestinal tract, and eyes, causing progressive impairment [1,2,3]. TTR is primarily produced in the liver and circulates in the blood as a tetramer, functioning to transport thyroxine and retinol-binding protein [4]. It is hypothesized that when the stability of this tetrameric structure is compromised, it dissociates into monomers, undergoes misfolding, and ultimately forms insoluble TTR amyloid fibrils that deposit in various tissues [5,6].
ATTR is broadly classified based on its etiology into wild-type ATTR (ATTRwt) and hereditary (variant) ATTR (ATTRv). ATTRwt is caused by wild-type TTR without genetic mutation, likely triggered by unknown factors including the breakdown of homeostatic mechanisms associated with aging, and it predominantly affects elderly males [7,8]. In contrast, ATTRv follows an autosomal dominant inheritance pattern caused by TTR gene mutations; to date, over 130 TTR gene mutations have been reported. The age of onset, clinical symptoms, severity, and prognosis of the disease vary widely [9,10].
In ATTRwt, TTR deposition occurs systemically across various organs but mainly affects joints and ligaments—causing carpal tunnel syndrome and spinal stenosis—and the heart, leading to cardiac hypertrophy, arrhythmia, and heart failure [3,10,11,12]. Bilateral carpal tunnel syndrome is one of the symptoms appearing in the early stages of the disease and generally precedes cardiac lesions [13]. Cardiac involvement contributes significantly to mortality and morbidity, resulting in cardiac dysfunction, heart failure, repeated hospitalizations, and ultimately death [7]. In clinical practice, it is an important differential diagnosis for heart failure with preserved ejection fraction (HFpEF) and left ventricular hypertrophy, and it has been revealed that comorbidity with aortic stenosis is frequent [14,15,16].
The clinical presentation of ATTRv is diverse, with polyneuropathy and cardiomyopathy being the typical phenotypes; depending on the mutation, one may predominate, or both may coexist. Symptoms of polyneuropathy include sensory blunting, numbness, and pain, as well as orthostatic hypotension and gastrointestinal symptoms due to autonomic dysfunction [17]. Definitive diagnosis requires the demonstration of amyloid deposition via tissue biopsy and the identification of the precursor protein through immunohistochemistry or mass spectrometry. However, bone scintigraphy (using Tc-99m PYP, DPD, or HMDP) has been established as a tool capable of diagnosing ATTR non-invasively with high sensitivity and specificity, contributing significantly to clinical and early diagnosis [18].
The therapeutic strategy for ATTR has undergone a dramatic transformation over the last decade, shifting from an era centered on symptomatic treatment to the establishment of disease-modifying therapies that intervene in the fundamental process of amyloid formation [7,12,19,20]. Currently, clinical application is centered on two main approaches. The first involves TTR stabilizers that inhibit the dissociation of TTR tetramers. In addition to Tafamidis, which has established efficacy in improving long-term prognosis, the next-generation drug Acoramidis, which provides more potent structural stabilization, has achieved favorable results in a Phase III trial (ATTRibute-CM) [21]. The second approach involves TTR production inhibitors (Silencers) that inhibit TTR synthesis in the liver using RNA interference (RNAi) or antisense oligonucleotides (ASO). In this field, Vutrisiran and Eplontersen, which feature improved administration convenience, have emerged. Notably, Vutrisiran suggested efficacy in suppressing all-cause mortality and cardiovascular events in cardiomyopathy in the recent HELIOS-B trial, indicating an expansion of its indication from neuropathy to cardiac involvement[22]. Furthermore, a paradigm shift is underway, moving beyond merely suppressing disease progression toward more curative approaches. In vivo gene editing therapy using CRISPR-Cas9 technology (NTLA-2001) is being developed with the goal of "One-and-done" treatment via permanent TTR knockdown through a single dose, raising hopes for liberation from the burden of lifelong treatment [23,24]. Additionally, development is proceeding on anti-amyloid antibodies (such as NI006) aimed at removing amyloid already deposited in tissues and restoring organ function [25]. Going forward, along with the clinical application of these novel modalities, the establishment of combination therapies using drugs with different mechanisms of action and optimal treatment sequences according to disease stage are becoming critical challenges.
As described above, while the management of ATTR has achieved dramatic progress with the advent of disease-modifying therapies, new challenges have surfaced, such as the thorough implementation of early diagnosis and the prediction of treatment responsiveness in individual cases [2,7]. To address these clinical issues and elucidate the complex pathological mechanisms at the molecular level—mechanisms that extend beyond mere "amyloid deposition in organs"—it is essential to utilize omics analysis to enable comprehensive data acquisition, rather than relying solely on conventional fragmentary analyses. In Alzheimer's disease, another representative amyloid-related disorder, the application of omics is advancing, leading to the identification of risk genes other than APOE and processes related to disease onset [26,27,28]. Conversely, the use of omics in ATTR is still developing, and the number of reports remains limited. In this review, we outline current findings regarding the use of omics in the diagnosis, elucidation of pathology, and biomarker exploration of ATTR amyloidosis, and discuss future perspectives for ATTR research utilizing these technologies.
2. Application of Proteomics for Amyloid Typing in Clinical Practice
In actual clinical practice, proteomics is utilized for determining the amyloid type (amyloid typing) and is incorporated into the diagnostic workflow [3,29]. Although bone scintigraphy is a useful non-invasive tool for ATTR, tissue biopsy is required when definitive diagnosis cannot be obtained. While amyloid deposition is confirmed by biopsy and amyloid typing is attempted via immunohistochemistry, diagnosis is often difficult in a significant number of cases due to limitations in antibody sensitivity and specificity. Implementing proteomics on collected samples at this stage significantly improves sensitivity and specificity, enabling the unbiased and comprehensive identification of amyloid precursor proteins [29,30,31,32] (Figure 1). The method of performing proteomics after harvesting amyloid deposition areas using laser microdissection has become standard practice [30,33].
The following case demonstrates the clinical utility of proteomics combined with laser microdissection in overcoming diagnostic ambiguity in amyloid typing. In a patient suspected of having both ATTR-CM and AL-CM based on clinical symptoms and screening tests, an endomyocardial biopsy was performed for definitive diagnosis. Although amyloid deposition was confirmed in the biopsy sample, immunohistochemistry showed positive results for both TTR and immunoglobulin light chain in different areas of the same sample, making pathological typing difficult. Therefore, the respective areas were cut out using laser microdissection, and separate proteomics analyses detected TTR and immunoglobulin light chain proteins, respectively. Consequently, the patient was diagnosed with concurrent ATTR and AL [34].
Proteomics of biopsy samples also enables the detection of rare amyloid types that were difficult to detect with immunohistochemistry. A comprehensive review of 16,175 specimens derived from various organs analyzed by shotgun proteomics at the Mayo Clinic identified numerous rare amyloid types and presented a "type-by-organ map" demonstrating the organ tropism of 18 rare types [29]. Furthermore, the same study demonstrated that combining this with bioinformatics strategies makes it possible to identify protein mutations in cases of hereditary amyloidosis.
3. Research Applications of Omics in Patient Sample Analysis
3.1. Serum Biomarker Discovery
The discovery of serum biomarkers in ATTR is a critical theme addressing multifaceted clinical and scientific needs, including non-invasive diagnosis, early diagnosis and screening, prediction of disease onset and progression, monitoring of therapeutic efficacy, promotion of precision medicine, and the identification of novel therapeutic targets. This section introduces research examples employing omics technologies for serum biomarker discovery.
In early studies performing proteomics using serum from 8 patients with ATTR-CM, 8 patients with ATTR-polyneuropathy, and 10 controls, proteins specific to ATTR-CM (including low levels of TTR) were identified [35]. Similarly, a study comparing the serum profiles of 8 ATTRv patients, 10 ATTRwt patients, and 10 healthy controls revealed that serum protein profiles differ between the two disease types despite both being ATTR. In particular, the ATTRv group showed specific changes in 14 proteins, including retinol-binding protein 4 (RBP4), while the ATTRwt group exhibited specific variations in 27 proteins, including apolipoprotein E and complement component C7 [36].
A prime example of biomarker discovery where omics analysis proved useful and led to practical application is the identification of neurofilament light chain (NfL) as a marker for neuronal damage in ATTRv. In a study utilizing serum samples collected during the Phase 3 APOLLO study (a randomized, double-blind, placebo-controlled trial of patisiran in patients with ATTRv with polyneuropathy; NCT01960348) [37], proteomics identified NfL as a valuable biomarker for ATTRv [38]. NfL is a protein that maintains the structure and shape of nerve cells and functions as a marker for neuronal axonal injury or degeneration. In this study, serum levels of over 1,000 proteins were measured in 136 ATTRv patients treated with patisiran (samples at baseline, 9, and 18 months), placebo-treated ATTRv patients, and 57 healthy controls. Among the 66 proteins that showed significant differences in the patisiran group compared to the placebo group, NfL exhibited the most significant change. The proteome at 18 months in the patisiran treatment group tended to approach that of the healthy control group. Furthermore, longitudinally, NfL levels increased in the placebo group while demonstrating a decrease in the patisiran group. These findings suggested that NfL functions as a biomarker for nerve damage and polyneuropathy in ATTRv, potentially enabling early diagnosis of ATTR amyloidosis patients and facilitating the monitoring of disease progression. The utility of NfL as a robust indicator of disease progression and treatment response has been confirmed in subsequent analyses of real-world clinical samples [39].
In a small-scale study performing blood proteome analysis targeting ATTRv patients with the V30M mutation, which predominantly presents as polyneuropathy, a systemic inflammatory signature characteristic of ATTR was revealed. Inflammation and acute-phase proteins such as C-reactive protein (CRP), serum amyloid A (SAA), and complement factors were shown to be significantly elevated in untreated patients. In patients undergoing tafamidis treatment, these abnormalities were partially normalized, and the proteome pattern approached that of the healthy control group, suggesting that the plasma proteome may serve as a useful biomarker for reflecting disease pathology and therapeutic response [40].
Data regarding serum metabolomics in ATTRv patients have also been reported. A study utilizing GC-MS and LC-MS metabolomics on serum samples from 27 ATTRv patients (V30M mutation), asymptomatic TTR V30M carriers, and 26 controls demonstrated the existence of significantly different metabolic profiles in ATTRv patients, revealing decreased levels of multiple amino acids such as tryptophan and phenylalanine [41]. However, this study could not identify differences in metabolic profiles between controls and asymptomatic carriers.
The number of patients with ATTR-CM has increased rapidly in recent years, making the appropriate identification of ATTR-CM among patients presenting with left ventricular hypertrophy (LVH) a clinically important issue. Serum proteome analysis was performed on pre-treatment ATTR-CM patients, ATTR-CM patients after the induction of tafamidis treatment, and patients with myocardial hypertrophy in whom ATTR was excluded, in an attempt to identify potential biomarkers for ATTR-CM diagnosis and the evaluation of tafamidis treatment efficacy [42]. Characteristic protein changes were observed in each group; specifically, prior to the initiation of specific therapy, ATTR-CM patients showed elevated levels of multiple proteins, including ceruloplasmin, apolipoprotein E, SERPINA1, and cDNA FLJ54111 (which is highly similar to serum transferrin). Notably, levels of SERPINA1 and cDNA FLJ54111 were found to decrease in the tafamidis treatment group.
Furthermore, Akita et al. conducted large-scale proteomics on serum samples from a total of 1,415 patients with heart diseases presenting with LVH, including ATTR-CM, to analyze the characteristics of each disease. Although the paper primarily focuses on hypertrophic cardiomyopathy (HCM), it identified a group of molecules showing significant changes in ATTR-CM compared to HCM [43].
3.2. Tissue Sample Analysis
In recent years, omics analysis utilizing clinical samples obtained via biopsy has been actively pursued, significantly contributing to the elucidation of disease pathology.
Amyloid deposition areas are characterized by the accumulation of not only precursor proteins but also specific proteins known as the "amyloid signature" [44,45]. Kourelis et al. conducted a comprehensive proteomic analysis of amyloid plaques within cardiac tissue samples from 292 ATTR-CM patients and 139 AL-CM patients to elucidate the specific proteome composition of cardiac amyloid plaques and its clinical significance [44]. The results revealed characteristic differences: ATTR plaques were rich in complement and contraction-related proteins, whereas AL plaques were rich in keratin. Furthermore, in a subset of ATTR patients, elevated levels of PIK3C3, an autophagy-related protein, were identified to be associated with reduced survival, suggesting the importance of autophagy pathways in the mechanism of cardiac tissue injury.
Netzel et al. performed proteomic analysis on whole endomyocardial biopsy samples from 76 ATTR patients and 27 AL patients [46]. They identified pathways common to both ATTR-CM and AL-CM, as well as pathways specific to ATTR-CM. In both ATTR-CM and AL-CM, extracellular matrix remodeling, fibrosis, epithelial-mesenchymal transition, and complement system activation were observed; these features were revealed to be associated with severe disease and poor prognosis. In ATTR-CM, particularly robust activation of the complement system was observed, suggesting the importance of immune-mediated reactions and antibody-related mechanisms. Based on these findings, the group reported that they have embarked on proof-of-concept research regarding complement inhibition for patients with advanced cardiac amyloidosis.
3.3. Genomic Analysis of Patients with Wild-Type Transthyretin Amyloidosis
As part of large-scale genomic analyses of ATTR, studies exploring TTR gene variants in general population cohorts have been reported [47,48]. Meanwhile, ATTRwt is defined as ATTR lacking TTR gene mutations. Efforts have begun to elucidate the complex genetic background and regulatory mechanisms involved in its pathogenesis. Recently, studies utilizing genomics to identify risk factors other than the TTR gene have started to emerge.
In a report conducting genetic testing (covering over 50 genes using multiple panels) on familial cases from two ATTRwt families, no clear genetic variants were identified within the scope verified [49]. On the other hand, Moreno-Gázquez et al. performed targeted sequencing of 84 coding genes suspected of disease involvement using samples from 27 ATTRwt patients. Their analysis classified 17 variants across 14 genes—including those related to proteolysis (F2, PLAT, PSEN2), extracellular chaperones (FGA, FGB), and genes associated with other amyloid diseases (ABCA1, APP, GC)—as Variants of Uncertain Significance (VUS). This finding highlighted the possibility that genomic variants other than those in the TTR gene contribute to the onset and progression of ATTRwt [50].
Future research utilizing omics approaches is expected to advance more comprehensive genomic analyses in ATTRwt, such as genome-wide association studies (GWAS) and whole-exome sequencing, as well as the analysis of TTR non-coding regions (e.g. promoter/enhancer regions) and the investigation of potential somatic mutations.
4. Application of Omics in Preclinical Models
Although the in vitro reconstruction of TTR-derived amyloid fibrils that accurately replicate patient-derived structures remains challenging, research efforts are underway to elucidate the pathophysiology of ATTR by examining cellular structure, function, and responses following exposure to precursor proteins or amyloid fibrils.
To investigate the impact of amyloid fibrils on cardiac fibroblasts, Dittloff et al. cultured primary fibroblasts on glass substrates coated with TTR fibrils for 48 hours and performed transcriptome analysis [51]. The results demonstrated a strong upregulation of inflammation-related genes such as ACOD1 and IL1b, confirming the enrichment of terms and pathways associated with innate immune responses. Cytokine proteomics of the culture supernatant revealed a greater than 5-fold increase in the secretion of multiple cytokines, including CXCL1 and CXCL2, which are known inducers of inflammation in neutrophils. These findings suggest that fibroblasts may potentiate the inflammatory cascade in the presence of amyloid fibrils, thereby contributing to the pathology of cardiomyopathy and amyloidosis.
Qin et al. evaluated in vitro cellular responses to amyloid fibrils in the three major cardiac cell types: cardiomyocytes, endothelial cells, and fibroblasts [52]. They generated human recombinant TTR proteins (WT, V122I variant, and V30M variant) and their respective amyloid fibrils. The study demonstrated that exposure to TTR tetramers did not exhibit cytotoxicity. In contrast, upon exposure to TTR fibrils, cardiomyocytes exhibited cell death and the disruption of contractile structures, while endothelial cells showed gene expression changes associated with vascular remodeling alongside a significant decrease in survival rates. Conversely, fibroblasts demonstrated robust resistance while promoting collagen formation and the overproduction of extracellular matrix. These findings suggest that the cumulative effect of these distinct cell-type-specific responses contributes to the formation of the unique heart failure pathology observed in ATTR-CM.
Ghosh et al. investigated cellular responses to variant TTR proteins by exposing neuronal cells (SH-SY5Y) and cardiomyocytes (AC16) to wild-type TTR and variant TTR (V122I and L55P) and subsequently performing RNA sequencing and ATAC sequencing [53]. The study revealed that cellular responses vary depending on the specific TTR variant, suggesting a potential link to the diverse clinical profiles observed among different mutations. Furthermore, treatment with tafamidis in the presence of variant TTR was found to suppress chromatin structural alterations and stress-response gene expression. Distinct gene expression patterns were observed between neuronal and cardiac cells; for instance, neuronal cells exhibited alterations in genes related to fatty acid metabolism and protein secretion, whereas cardiomyocytes showed an upregulation of gene sets associated with cardiac hypertrophy. Although previous reports indicate that TTR monomers and oligomers are cytotoxic [54,55], this study did not include steps to directly isolate and verify the denaturation or folding states of the protein. Consequently, the authors refrained from drawing definitive conclusions regarding which specific structural form—monomer, oligomer, or fibril—was responsible for the observed cellular responses.
Research utilizing human induced pluripotent stem cells to elucidate the role of the liver—the primary TTR-producing organ—in disease pathology has also been reported [56]. Single-cell RNA sequencing performed on hepatocyte-like cells expressing amyloidogenic TTR revealed that the production of variant TTR triggers the restructuring of intracellular proteostasis (protein homeostasis). Furthermore, the activation of the transcription factor ATF6 was found to induce chaperones (such as BiP and PDIA4), resulting in the retention of variant TTR within the endoplasmic reticulum and a selective reduction in its secretion. These findings suggest the potential for a novel therapeutic strategy that prevents the release of toxic TTR by enhancing the protein quality control mechanisms of the liver.
In contrast to such cell-based analyses, adequately reproducing ATTR in animal models has historically been considered difficult [57,58], and research reports utilizing omics in in vivo models remain scarce. However, with the recent development of humanized models [59,60], it is expected that future omics analyses using in vivo models will yield new insights.
5. Future Directions
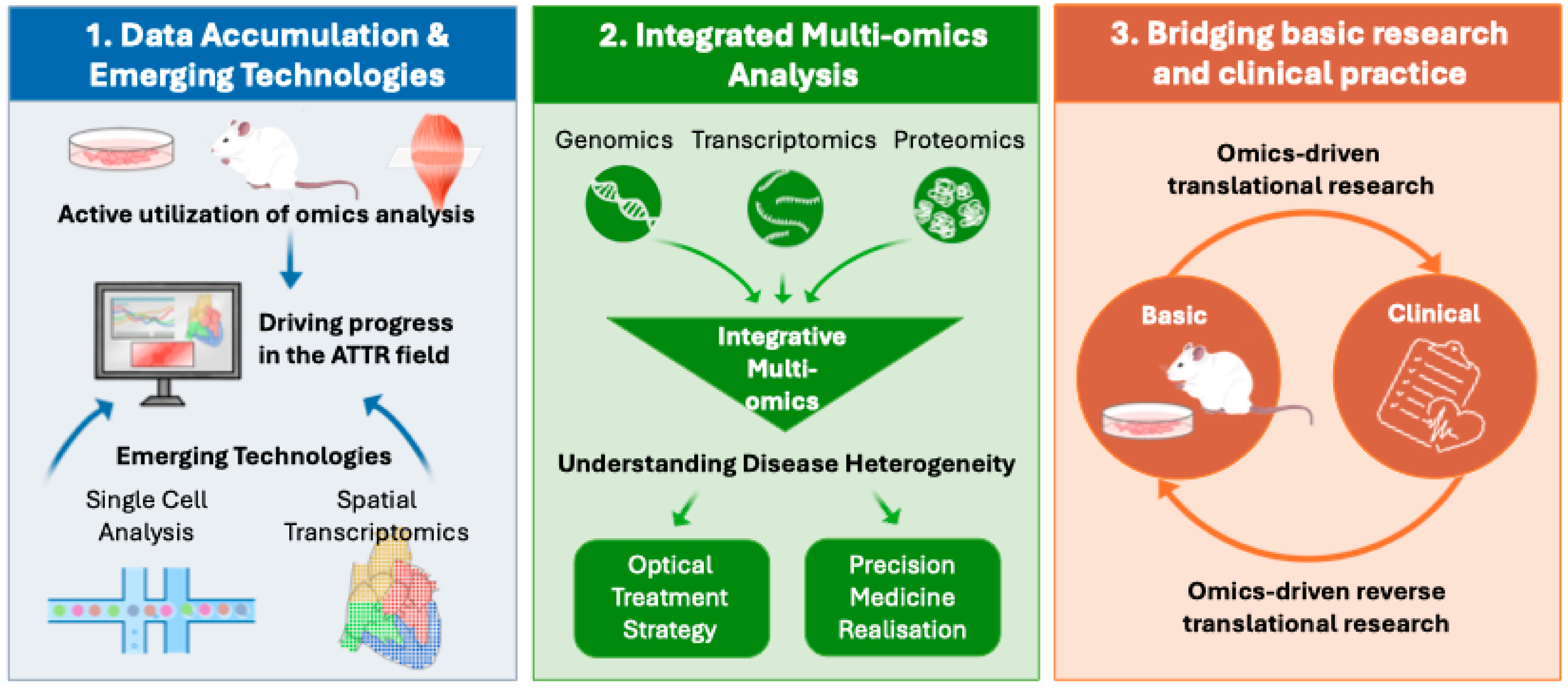
In the field of ATTR, omics-based approaches have already achieved tangible clinical impact, including their incorporation into diagnostic workflows for amyloid typing and their contribution to the identification of clinically relevant biomarkers such as NfL. At the same time, growing evidence indicates that ATTR-CM requires disease-specific therapeutic strategies distinct from conventional heart failure management [61,62], and that ATTR pathophysiology is considerably more complex than previously appreciated. These advances underscore the need for deeper molecular characterization of the disease. In this context, three key challenges emerge as important future directions for omics-based research in ATTR (Figure 2).
First, the systematic accumulation of omics datasets and the broader adoption of advanced analytical technologies are essential. To date, the number of omics studies and available datasets in ATTR remains limited. Expanded application of omics analyses across cellular models, animal models, and clinical samples is therefore critical to advancing the field as a whole. One particularly noteworthy initiative is the TTR GWAS, an international collaborative project that includes several thousand patients with ATTRv and ATTRwt and is currently establishing a large cohort for GWAS [63]. This project is expected to become a valuable data resource for future research. Moreover, emerging technologies such as single-cell and spatial transcriptomics—now widely applied in other disease areas—have scarcely been explored in ATTR. Applying these approaches to human biopsy specimens has the potential to elucidate microenvironmental changes surrounding amyloid deposits at high resolution, thereby uncovering previously unrecognized disease mechanisms and novel therapeutic targets.
Second, integrated multi-omics analyses are needed to define disease subtypes at the molecular level. A major future challenge lies in the integration of independently generated datasets, including transcriptomic, proteomic, and metabolomic data, to better capture disease heterogeneity [64,65]. At the genomic level, not only the presence or absence of mutations in the TTR gene but also data derived from ATTRwt patient samples will be informative. Such integrative multi-omics approaches may enable molecular stratification of patients with differing rates of disease progression, organ tropism, and therapeutic responsiveness. This framework would provide a foundation for precision medicine in ATTR, facilitating the selection of optimal, individualized treatment strategies.
Third, omics analyses are expected to play a central role in bridging basic research and clinical practice. Indispensable to this goal are not only translational efforts—driving therapeutic development from cellular and animal models—but also reverse translational strategies that validate patient-derived hypotheses in experimental systems. In this reverse translational context, the rapid expansion of therapeutic options for ATTR offers a unique opportunity. Longitudinal omics profiling before and after treatment serves as a powerful approach to delineate how disease-modifying therapies reshape molecular networks at both tissue and systemic levels. Such analyses may ultimately lead to the identification of novel pharmacodynamic biomarkers and predictors of treatment response.
In summary, continued advances in omics-based research promise to propel our understanding of ATTR into a new era, accelerating the development of next-generation therapeutic strategies and the implementation of precision medicine for this complex disease.
Author Contributions
Conceptualization, H.M. and T.Y.; writing—original draft preparation, H.M. and F.A.S.; writing—review and editing, H.M. F.A.S and T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. T.Y. is supported by the Friends of Garrett Cumming Research Fund, Muscular Dystrophy Canada, the Canadian Institute of Health Research (CIHR), the Women and Children’s Health Research Institute (WCHRI), the HM Toupin Neurological Science Research Fund, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, the U.S. Department of Defense, and the Heart and Stroke Foundation Canada. H.M. is supported by scholarships from the University of Alberta Faculty of Graduate and Postdoctoral Studies and postdoctoral fellowship from WCHRI.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the University of Alberta Faculty of Graduate and Postdoctoral Studies, the Friends of Garrett Cumming Research Fund, Muscular Dystrophy Canada, the Canadian Institute of Health Research (CIHR), the Women and Children’s Health Research Institute (WCHRI), the HM Toupin Neurological Science Research Fund, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, and the Heart and Stroke Foundation Canada for their support.
Conflicts of Interest
T.Y. is a cofounder and shareholder of OligomicsTx Inc. which aims to commercialize antisense technology. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Fontana, M.; Berk, J.L.; Drachman, B.; Garcia-Pavia, P.; Hanna, M.; Lairez, O.; Witteles, R. Changing Treatment Landscape in Transthyretin Cardiac Amyloidosis. Circ Heart Fail 2025, 18, e012112. [Google Scholar] [CrossRef]
- Kitaoka, H.; Izumi, C.; Izumiya, Y.; Inomata, T.; Ueda, M.; Kubo, T.; Koyama, J.; Sano, M.; Sekijima, Y.; Tahara, N.; Tsukada, N.; Tsujita, K.; Tsutsui, H.; Tomita, T.; Amano, M.; Endo, J.; Okada, A.; Oda, S.; Takashio, S.; Baba, Y.; Misumi, Y.; Yazaki, M.; Anzai, T.; Ando, Y.; Isobe, M.; Kimura, T.; Fukuda, K. JCS 2020 Guideline on Diagnosis and Treatment of Cardiac Amyloidosis. Circ J 2020, 84, 1610–1671. [Google Scholar] [CrossRef]
- Foss, T.R.; Wiseman, R.L.; Kelly, J.W. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 2005, 44, 15525–15533. [Google Scholar] [CrossRef]
- Lai, Z.; Colón, W.; Kelly, J.W. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 1996, 35, 6470–6482. [Google Scholar] [CrossRef] [PubMed]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.; Brito, R.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez, E.; Maurer, M.S.; Garcia-Pavia, P. Transthyretin amyloid cardiomyopathy: a paradigm for advancing precision medicine. Eur Heart J 2025, 46, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Dispenzieri, A.; Sher, T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol 2015, 12, 91–102. [Google Scholar] [CrossRef]
- Siemer, J.M.; Grote-Levi, L.; Hänselmann, A.; Sassmann, M.L.; Nay, S.; Ratuszny, D.; Körner, S.; Seeliger, T.; Hümmert, M.W.; Dohrn, M.F.; Huss, A.; Tumani, H.; Gödecke, V.; Heuser, M.; Bauersachs, J.; Bavendiek, U.; Skripuletz, T.; Gingele, S. Polyneuropathy in hereditary and wildtype transthyretin amyloidosis, comparison of key clinical features and red flags. Sci Rep 2025, 15, 35028. [Google Scholar] [CrossRef]
- Muchtar, E.; Dispenzieri, A.; Magen, H.; Grogan, M.; Mauermann, M.; McPhail, E.D.; Kurtin, P.J.; Leung, N.; Buadi, F.K.; Dingli, D.; Kumar, S.K.; Gertz, M.A. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med 2021, 289, 268–292. [Google Scholar] [CrossRef]
- Sueyoshi, T.; Ueda, M.; Jono, H.; Irie, H.; Sei, A.; Ide, J.; Ando, Y.; Mizuta, H. Wild-type transthyretin-derived amyloidosis in various ligaments and tendons. Hum Pathol 2011, 42, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Maurer, M.S. Cardiac Amyloidosis Due to Transthyretin Protein: A Review. Jama 2024, 331, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sekijima, Y.; Yazaki, M.; Tojo, K.; Yoshinaga, T.; Doden, T.; Koyama, J.; Yanagisawa, S.; Ikeda, S. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; Garcia-Pavia, P. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; George, I.; Kodali, S.; Leon, M.B.; Hahn, R.; Bokhari, S.; Maurer, M.S. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017, 38, 2879–2887. [Google Scholar] [CrossRef]
- AbouEzzeddine, O.F.; Davies, D.R.; Scott, C.G.; Fayyaz, A.U.; Askew, J.W.; McKie, P.M.; Noseworthy, P.A.; Johnson, G.B.; Dunlay, S.M.; Borlaug, B.A.; Chareonthaitawee, P.; Roger, V.L.; Dispenzieri, A.; Grogan, M.; Redfield, M.M. Prevalence of Transthyretin Amyloid Cardiomyopathy in Heart Failure With Preserved Ejection Fraction. JAMA Cardiol 2021, 6, 1267–1274. [Google Scholar] [CrossRef]
- Poli, L.; Labella, B.; Cotti Piccinelli, S.; Caria, F.; Risi, B.; Damioli, S.; Padovani, A.; Filosto, M. Hereditary transthyretin amyloidosis: a comprehensive review with a focus on peripheral neuropathy. Front Neurol 2023, 14, 1242815. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; Lachmann, H.J.; Bokhari, S.; Castano, A.; Dorbala, S.; Johnson, G.B.; Glaudemans, A.W.; Rezk, T.; Fontana, M.; Palladini, G.; Milani, P.; Guidalotti, P.L.; Flatman, K.; Lane, T.; Vonberg, F.W.; Whelan, C.J.; Moon, J.C.; Ruberg, F.L.; Miller, E.J.; Hutt, D.F.; Hazenberg, B.P.; Rapezzi, C.; Hawkins, P.N. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Vergaro, G.; Ferrari Chen, Y.F.; Ioannou, A.; Panichella, G.; Castiglione, V.; Aimo, A.; Emdin, M.; Fontana, M. Current and emerging treatment options for transthyretin amyloid cardiomyopathy. Heart 2026, 112, 129–138. [Google Scholar] [CrossRef]
- Nguyen, O.; Kamna, D.; Masri, A. New therapies to treat cardiac amyloidosis. Curr Opin Cardiol 2025, 40, 98–106. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Judge, D.P.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Gibbs, S.; Grogan, M.; Hanna, M.; Hoffman, J.; Masri, A.; Maurer, M.S.; Nativi-Nicolau, J.; Obici, L.; Poulsen, S.H.; Rockhold, F.; Shah, K.B.; Soman, P.; Garg, J.; Chiswell, K.; Xu, H.; Cao, X.; Lystig, T.; Sinha, U.; Fox, J.C. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2024, 390, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Berk, J.L.; Gillmore, J.D.; Witteles, R.M.; Grogan, M.; Drachman, B.; Damy, T.; Garcia-Pavia, P.; Taubel, J.; Solomon, S.D.; Sheikh, F.H.; Tahara, N.; González-Costello, J.; Tsujita, K.; Morbach, C.; Pozsonyi, Z.; Petrie, M.C.; Delgado, D.; Van der Meer, P.; Jabbour, A.; Bondue, A.; Kim, D.; Azevedo, O.; Hvitfeldt Poulsen, S.; Yilmaz, A.; Jankowska, E.A.; Algalarrondo, V.; Slugg, A.; Garg, P.P.; Boyle, K.L.; Yureneva, E.; Silliman, N.; Yang, L.; Chen, J.; Eraly, S.A.; Vest, J.; Maurer, M.S. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N Engl J Med 2025, 392, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O'Connell, D.; Walsh, K.R.; Wood, K.; Phillips, J.; Xu, Y.; Amaral, A.; Boyd, A.P.; Cehelsky, J.E.; McKee, M.D.; Schiermeier, A.; Harari, O.; Murphy, A.; Kyratsous, C.A.; Zambrowicz, B.; Soltys, R.; Gutstein, D.E.; Leonard, J.; Sepp-Lorenzino, L.; Lebwohl, D. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Solomon, S.D.; Kachadourian, J.; Walsh, L.; Rocha, R.; Lebwohl, D.; Smith, D.; Täubel, J.; Gane, E.J.; Pilebro, B.; Adams, D.; Razvi, Y.; Olbertz, J.; Haagensen, A.; Zhu, P.; Xu, Y.; Leung, A.; Sonderfan, A.; Gutstein, D.E.; Gillmore, J.D. CRISPR-Cas9 Gene Editing with Nexiguran Ziclumeran for ATTR Cardiomyopathy. N Engl J Med 2024, 391, 2231–2241. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Aus dem Siepen, F.; Donal, E.; Lairez, O.; van der Meer, P.; Kristen, A.V.; Mercuri, M.F.; Michalon, A.; Frost, R.J.A.; Grimm, J.; Nitsch, R.M.; Hock, C.; Kahr, P.C.; Damy, T. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid. N Engl J Med 2023, 389, 239–250. [Google Scholar] [CrossRef]
- Campanelli, L.; Sendoya, J.M.; Brody, S.; Galeano, P.; Do Carmo, S.; Cuello, A.C.; Castaño, E.M.; Gonzalés-Jimenez, A.; Verheul, J.; Medina-Vera, D.; de Fonseca, F.R.; Wernersson, R.; Morelli, L. New insights into the molecular biology of Alzheimer's-like cerebral amyloidosis achieved through multi-omics approaches. PLoS One 2025, 20, e0330859. [Google Scholar]
- Sancesario, G.M.; Bernardini, S. Alzheimer's disease in the omics era. Clin Biochem 2018, 59, 9–16. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; Holmans, P.A.; Boland, A.; Damotte, V.; van der Lee, S.J.; Costa, M.R.; Kuulasmaa, T.; Yang, Q.; de Rojas, I.; Bis, J.C.; Yaqub, A.; Prokic, I.; Chapuis, J.; Ahmad, S.; Giedraitis, V.; Aarsland, D.; Garcia-Gonzalez, P.; Abdelnour, C.; Alarcón-Martín, E.; Alcolea, D.; Alegret, M.; Alvarez, I.; Álvarez, V.; Armstrong, N.J.; Tsolaki, A.; Antúnez, C.; Appollonio, I.; Arcaro, M.; Archetti, S.; Pastor, A.A.; Arosio, B.; Athanasiu, L.; Bailly, H.; Banaj, N.; Baquero, M.; Barral, S.; Beiser, A.; Pastor, A.B.; Below, J.E.; Benchek, P.; Benussi, L.; Berr, C.; Besse, C.; Bessi, V.; Binetti, G.; Bizarro, A.; Blesa, R.; Boada, M.; Boerwinkle, E.; Borroni, B.; Boschi, S.; Bossù, P.; Bråthen, G.; Bressler, J.; Bresner, C.; Brodaty, H.; Brookes, K.J.; Brusco, L.I.; Buiza-Rueda, D.; Bûrger, K.; Burholt, V.; Bush, W.S.; Calero, M.; Cantwell, L.B.; Chene, G.; Chung, J. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet 2022, 54, 412–436. [Google Scholar]
- Dasari, S.; Theis, J.D.; Vrana, J.A.; Rech, K.L.; Dao, L.N.; Howard, M.T.; Dispenzieri, A.; Gertz, M.A.; Hasadsri, L.; Highsmith, W.E.; Kurtin, P.J.; McPhail, E.D. Amyloid Typing by Mass Spectrometry in Clinical Practice: a Comprehensive Review of 16,175 Samples. Mayo Clin Proc 2020, 95, 1852–1864. [Google Scholar] [CrossRef]
- Vrana, J.A.; Gamez, J.D.; Madden, B.J.; Theis, J.D.; Bergen, H.R., 3rd; Dogan, A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 2009, 114, 4957–4959. [Google Scholar] [CrossRef]
- Vrana, J.A.; Theis, J.D.; Dasari, S.; Mereuta, O.M.; Dispenzieri, A.; Zeldenrust, S.R.; Gertz, M.A.; Kurtin, P.J.; Grogg, K.L.; Dogan, A. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica 2014, 99, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Vrana, J.A.; Theis, J.D.; Leung, N.; Sethi, A.; Nasr, S.H.; Fervenza, F.C.; Cornell, L.D.; Fidler, M.E.; Dogan, A. Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int 2012, 82, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Rezk, T.; Gilbertson, J.A.; Mangione, P.P.; Rowczenio, D.; Rendell, N.B.; Canetti, D.; Lachmann, H.J.; Wechalekar, A.D.; Bass, P.; Hawkins, P.N.; Bellotti, V.; Taylor, G.W.; Gillmore, J.D. The complementary role of histology and proteomics for diagnosis and typing of systemic amyloidosis. J Pathol Clin Res 2019, 5, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, H.; Kitakata, H.; Endo, J.; Ikura, H.; Sano, M.; Tasaki, M.; Sakai, S.; Ueda, M.; Fukuda, K. Step-by-step typing for the accurate diagnosis of concurrent light chain and transthyretin cardiac amyloidosis. ESC Heart Fail 2022, 9, 1474–1477. [Google Scholar] [CrossRef]
- Chan, G.G.; Koch, C.M.; Connors, L.H. Serum Proteomic Variability Associated with Clinical Phenotype in Familial Transthyretin Amyloidosis (ATTRm). J Proteome Res 2017, 16, 4104–4112. [Google Scholar] [CrossRef]
- Chan, G.G.; Koch, C.M.; Connors, L.H. Blood Proteomic Profiling in Inherited (ATTRm) and Acquired (ATTRwt) Forms of Transthyretin-Associated Cardiac Amyloidosis. J Proteome Res 2017, 16, 1659–1668. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O'Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; Lin, K.P.; Vita, G.; Attarian, S.; Planté-Bordeneuve, V.; Mezei, M.M.; Campistol, J.M.; Buades, J.; Brannagan, T.H., 3rd; Kim, B.J.; Oh, J.; Parman, Y.; Sekijima, Y.; Hawkins, P.N.; Solomon, S.D.; Polydefkis, M.; Dyck, P.J.; Gandhi, P.J.; Goyal, S.; Chen, J.; Strahs, A.L.; Nochur, S.V.; Sweetser, M.T.; Garg, P.P.; Vaishnaw, A.K.; Gollob, J.A.; Suhr, O.B. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Ticau, S.; Sridharan, G.V.; Tsour, S.; Cantley, W.L.; Chan, A.; Gilbert, J.A.; Erbe, D.; Aldinc, E.; Reilly, M.M.; Adams, D.; Polydefkis, M.; Fitzgerald, K.; Vaishnaw, A.; Nioi, P. Neurofilament Light Chain as a Biomarker of Hereditary Transthyretin-Mediated Amyloidosis. Neurology 2021, 96, e412–e422. [Google Scholar] [CrossRef]
- Carroll, A.S.; Razvi, Y.; O'Donnell, L.; Veleva, E.; Heslegrave, A.; Zetterberg, H.; Vucic, S.; Kiernan, M.C.; Rossor, A.M.; Gillmore, J.D.; Reilly, M.M. Serum neurofilament light chain in hereditary transthyretin amyloidosis: validation in real-life practice. Amyloid 2024, 31, 95–104. [Google Scholar] [CrossRef]
- Nugroho, K.; Lin, C.Y.; Monteiro, C.; Coelho, T.; Moresco, J.J.; Pinto, A.F.M.; Powers, E.T.; Yates, J.R., 3rd; Diedrich, J.K.; Kelly, J.W. Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy. Int J Mol Sci 2025, 26((12)). [Google Scholar] [CrossRef]
- Olsson, M.; Hellman, U.; Wixner, J.; Anan, I. Metabolomics analysis for diagnosis and biomarker discovery of transthyretin amyloidosis. Amyloid 2021, 28, 234–242. [Google Scholar] [CrossRef]
- Waś, J.; Gawor-Prokopczyk, M.; Sioma, A.; Szewczyk, R.; Pel, A.; Krzysztoń-Russjan, J.; Niedolistek, M.; Sokołowska, D.; Grzybowski, J.; Mazurkiewicz, Ł. Proteomic Analysis of Serum in Cardiac Transthyretin Amyloidosis: Diagnostic and Prognostic Implications for Biomarker Discovery. Biomedicines 2025, 13((7)). [Google Scholar] [CrossRef] [PubMed]
- Akita, K.; Maurer, M.S.; Tower-Rader, A.; Fifer, M.A.; Shimada, Y.J. Comprehensive Proteomics Profiling Identifies Circulating Biomarkers to Distinguish Hypertrophic Cardiomyopathy From Other Cardiomyopathies With Left Ventricular Hypertrophy. Circ Heart Fail 2025, 18, e012434. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Dasari, S.S.; Dispenzieri, A.; Maleszewski, J.J.; Redfield, M.M.; Fayyaz, A.U.; Grogan, M.; Ramirez-Alvarado, M.; Abou Ezzeddine, O.F.; McPhail, E.D. A Proteomic Atlas of Cardiac Amyloid Plaques. JACC CardioOncol 2020, 2, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Theis, J.D.; Vrana, J.A.; Fervenza, F.C.; Sethi, A.; Qian, Q.; Quint, P.; Leung, N.; Dogan, A.; Nasr, S.H. Laser microdissection and proteomic analysis of amyloidosis, cryoglobulinemic GN, fibrillary GN, and immunotactoid glomerulopathy. Clin J Am Soc Nephrol 2013, 8, 915–921. [Google Scholar] [CrossRef]
- Netzel, B.C.; Charlesworth, M.C.; Johnson, K.L.; French, A.J.; Dispenzieri, A.; Maleszewski, J.J.; McPhail, E.D.; Grogan, M.; Redfield, M.M.; Weivoda, M.; Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Misra, P.; Vrana, J.; Theis, J.; Hayman, S.R.; Ramirez-Alvarado, M.; Dasari, S.; Kourelis, T. Whole tissue proteomic analyses of cardiac ATTR and AL unveil mechanisms of tissue damage. Amyloid 2025, 32, 72–80. [Google Scholar] [CrossRef]
- Carry, B.J.; Young, K.; Fielden, S.; Kelly, M.A.; Sturm, A.C.; Avila, J.D.; Martin, C.L.; Kirchner, H.L.; Fornwalt, B.K.; Haggerty, C.M. Genomic Screening for Pathogenic Transthyretin Variants Finds Evidence of Underdiagnosed Amyloid Cardiomyopathy From Health Records. JACC CardioOncol 2021, 3, 550–561. [Google Scholar] [CrossRef]
- Aung, N.; Nicholls, H.L.; Chahal, C.A.A.; Khanji, M.Y.; Rauseo, E.; Chadalavada, S.; Petersen, S.E.; Munroe, P.B.; Elliott, P.M.; Lopes, L.R. Prevalence, Cardiac Phenotype, and Outcomes of Transthyretin Variants in the UK Biobank Population. JAMA Cardiol 2024, 9, 964–972. [Google Scholar] [CrossRef]
- Westin, O.M.; Clemmensen, T.S.; Hansen, A.T.; Gustafsson, F.; Poulsen, S.H. Familial occurrences of cardiac wild-type transthyretin amyloidosis: a case series. Eur Heart J Case Rep 2024, 8, ytae199. [Google Scholar] [CrossRef]
- Moreno-Gázquez, I.; Pérez-Palacios, R.; Abengochea-Quílez, L.; Lahuerta Pueyo, C.; Roteta Unceta Barrenechea, A.; Andrés Gracia, A.; Aibar Arregui, M.A.; Menao Guillén, S. Targeted sequencing of selected functional genes in patients with wild-type transthyretin amyloidosis. BMC Res Notes 2023, 16, 249. [Google Scholar] [CrossRef]
- Dittloff, K.T.; Iezzi, A.; Zhong, J.X.; Mohindra, P.; Desai, T.A.; Russell, B. Transthyretin amyloid fibrils alter primary fibroblast structure, function, and inflammatory gene expression. Am J Physiol Heart Circ Physiol 2021, 321, H149–H160. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Yang, Q.; Ullate-Agote, A.; Sampaio-Pinto, V.; Florit, L.; Dokter, I.; Mathioudaki, C.; Middelberg, L.; Montero-Calle, P.; Aguirre-Ruiz, P.; de Las Heras Rojo, J.; Lei, Z.; Qiu, Z.; Wei, J.; van der Harst, P.; Prosper, F.; Mazo, M.M.; Iglesias-García, O.; Minnema, M.C.; Sluijter, J.P.G.; Oerlemans, M.; van Mil, A. Uncovering cell type-specific phenotypes using a novel human in vitro model of transthyretin amyloid cardiomyopathy. Stem Cell Res Ther 2025, 16, 352. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Villacorta-Martin, C.; Lindstrom-Vautrin, J.; Kenney, D.; Golden, C.S.; Edwards, C.V.; Sanchorawala, V.; Connors, L.H.; Giadone, R.M.; Murphy, G.J. Mapping cellular response to destabilized transthyretin reveals cell- and amyloidogenic protein-specific signatures. Amyloid 2023, 30, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A 2004, 101, 2817–2822. [Google Scholar] [CrossRef]
- Leung, A.; Nah, S.K.; Reid, W.; Ebata, A.; Koch, C.M.; Monti, S.; Genereux, J.C.; Wiseman, R.L.; Wolozin, B.; Connors, L.H.; Berk, J.L.; Seldin, D.C.; Mostoslavsky, G.; Kotton, D.N.; Murphy, G.J. Induced pluripotent stem cell modeling of multisystemic, hereditary transthyretin amyloidosis. Stem Cell Reports 2013, 1, 451–463. [Google Scholar] [CrossRef]
- Giadone, R.M.; Liberti, D.C.; Matte, T.M.; Rosarda, J.D.; Torres-Arancivia, C.; Ghosh, S.; Diedrich, J.K.; Pankow, S.; Skvir, N.; Jean, J.C.; Yates, J.R., 3rd; Wilson, A.A.; Connors, L.H.; Kotton, D.N.; Wiseman, R.L.; Murphy, G.J. Expression of Amyloidogenic Transthyretin Drives Hepatic Proteostasis Remodeling in an Induced Pluripotent Stem Cell Model of Systemic Amyloid Disease. Stem Cell Reports 2020, 15, 515–528. [Google Scholar] [CrossRef]
- Ibrahim, R.B.; Liu, Y.T.; Yeh, S.Y.; Tsai, J.W. Contributions of Animal Models to the Mechanisms and Therapies of Transthyretin Amyloidosis. Front Physiol 2019, 10, 338. [Google Scholar] [CrossRef]
- Qin, J.; Qiu, Z.; Fan, Y.; Xiong, Q.; Lei, Z.; Wei, J.; van der Harst, P.; Minnema, M.C.; Sluijter, J.P.G.; van Mil, A.; Oerlemans, M. In vitro and in vivo disease models of cardiac amyloidosis: progress, pitfalls, and potential. Cardiovasc Res 2025(121), 1997–2013. [CrossRef]
- Li, Z.; Kanazashi, H.; Tokashiki, Y.; Fujikawa, R.; Okagaki, A.; Katoh, S.; Kojima, K.; Haruna, K.; Matsushita, N.; Ishikawa, T.O.; Chen, H.; Yamamura, K. TTR exon-humanized mouse optimal for verifying new therapies for FAP. Biochem Biophys Res Commun 2022, 599, 69–74. [Google Scholar] [CrossRef]
- Ishida, S.; Sato, Y.; Chosa, K.; Ezawa, E.; Yamauchi, Y.; Oyama, M.; Kozuka-Hata, H.; Ito, R.; Sato, R.; Maeki, M.; Ishikawa, T.O.; Yamamura, K.; Takeshita, K.; Yamaguchi, K.; Kochi, Y.; Hashiya, F.; Liu, Y.; Abe, N.; Abe, H.; Sekijima, Y.; Yoshimi, K.; Mashimo, T. CRISPR-Cas3-based editing for targeted deletions in a mouse model of transthyretin amyloidosis. Nat Biotechnol 2026. [Google Scholar] [CrossRef]
- Ioannou, A.; Massa, P.; Patel, R.K.; Razvi, Y.; Porcari, A.; Rauf, M.U.; Jiang, A.; Cabras, G.; Filisetti, S.; Bolhuis, R.E.; Bandera, F.; Venneri, L.; Martinez-Naharro, A.; Law, S.; Kotecha, T.; Virsinskaite, R.; Knight, D.S.; Emdin, M.; Petrie, A.; Lachmann, H.; Wechelakar, A.; Petrie, M.; Hughes, A.; Freemantle, N.; Hawkins, P.N.; Whelan, C.; McMurray, J.J.V.; Gillmore, J.D.; Fontana, M. Conventional heart failure therapy in cardiac ATTR amyloidosis. Eur Heart J 2023, 44, 2893–2907. [Google Scholar] [CrossRef]
- Cheng, R.K.; Vasbinder, A.; Levy, W.C.; Goyal, P.; Griffin, J.M.; Leedy, D.J.; Maurer, M.S. Lack of Association Between Neurohormonal Blockade and Survival in Transthyretin Cardiac Amyloidosis. J Am Heart Assoc 2021, 10, e022859. [Google Scholar] [CrossRef]
- TTR GWAS. Available online: https://ttrgwas.wordpress.com/?utm_source=chatgpt.com (accessed on 26 January 2026).
- Clark, C.; Rabl, M.; Dayon, L.; Popp, J. The promise of multi-omics approaches to discover biological alterations with clinical relevance in Alzheimer's disease. Front Aging Neurosci 2022, 14, 1065904. [Google Scholar] [CrossRef]
- Hu, Y.; Zou, Y.; Qiao, L.; Lin, L. Integrative proteomic and metabolomic elucidation of cardiomyopathy with in vivo and in vitro models and clinical samples. Mol Ther 2024, 32, 3288–3312. [Google Scholar] [CrossRef]
Figure 1.
Role of proteomics in the diagnosis of amyloidosis. When amyloid deposition is confirmed in biopsy specimens, but definitive amyloid typing cannot be achieved by immunohistochemistry, proteomic analysis is useful for identifying the amyloid precursor protein. LMD, laser microdissection; LC-MS/MS, liquid chromatography–tandem mass spectrometry.
Figure 1.
Role of proteomics in the diagnosis of amyloidosis. When amyloid deposition is confirmed in biopsy specimens, but definitive amyloid typing cannot be achieved by immunohistochemistry, proteomic analysis is useful for identifying the amyloid precursor protein. LMD, laser microdissection; LC-MS/MS, liquid chromatography–tandem mass spectrometry.
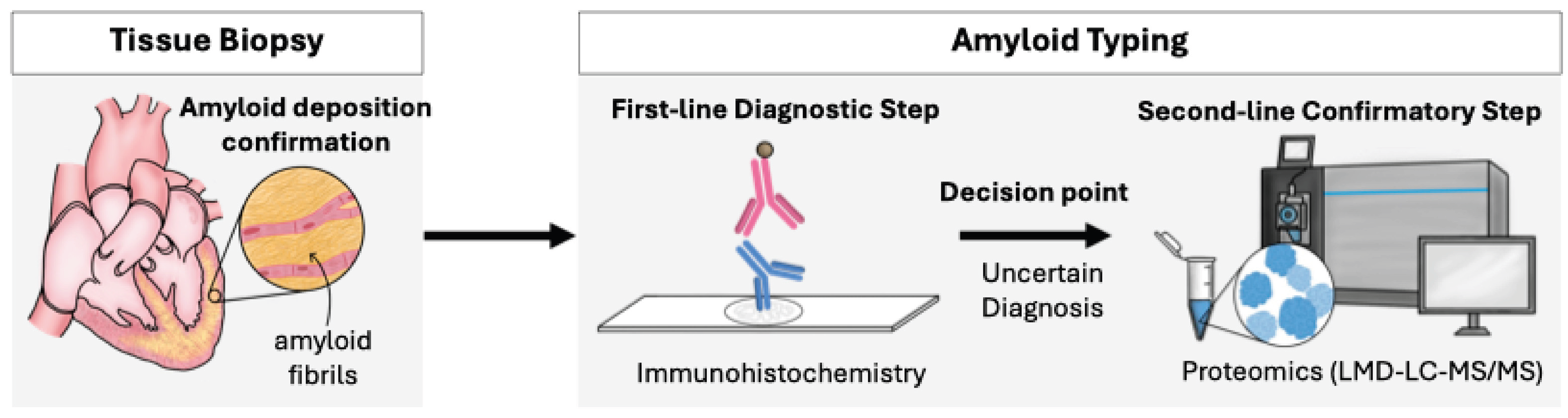
Figure 2.
Future directions for omics-based research in transthyretin amyloidosis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



