
Submitted:
20 December 2024
Posted:
23 December 2024
You are already at the latest version
Abstract
Background/Objectives: Gaucher disease (GD) is characterized by significant phenotypic heterogeneity, even among patients with identical GBA1 genotypes, suggesting the role of genetic and/or epigenetic modifiers. The enzymatic defect and pathological accumulation of glucosylceramide (GlcCer) lead to chronic metabolic inflammation, potentially interacting with other biological pathways to influence disease expression. Methods: This study leveraged one of the world’s most deeply phenotyped cohorts of GD patients, drawn from a major tertiary referral center, with prolonged longitudinal follow-up. Whole exome sequencing (WES) was conducted on 275 extensively characterized patients, focusing on those exhibiting complex phenotypes. Results: Eighteen patients (6.5%) presented with atypical manifestations not fully explained by GD. WES revealed additional genetic diagnoses, including hereditary hemochromatosis (n=5), Familial Mediterranean Fever (n=4), homozygous MSH6 mutation-associated hereditary cancer predisposition (n=2), and others. These concurrent genetic diseases can modify GD presentation and complicate clinical management. Conclusions: This work underscores the importance of recognizing complex phenotypes in GD, identifying modifier genes, and informing precision medicine strategies for improved patient outcomes.
Keywords:
Gaucher disease
; modifier genes
; complex phenotypes
; longitudinal deep phenotyping
; whole exome sequencing
1. Introduction
Gaucher disease (GD) is a prototype lysosomal storage disorder caused by biallelic mutations in the GBA1 gene. These mutations lead to a deficiency of the lysosomal enzyme acid β-glucosidase [1]. This enzymatic defect results in the accumulation of bioactive lipids, glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph), resulting in a generalized dysfunction of the lysosomal system and driving chronic metabolic inflammation. The hallmark of GD pathology is the multisystemic buildup of lipid-laden macrophages, known as Gaucher cells. GD is broadly classified into three phenotypes: type 1 (non-neuronopathic lacking childhood-onset neurodegenerative disease), and the childhood-onset neuronopathic forms, type 2 (acute, infantile onset) and type 3 (chronic) [2]. While some genotype-phenotype correlations exist—such as the association of the p.Leu483Pro homozygous mutation with neuronopathic disease (nGD) and the p.Asn409Ser mutation with type 1 GD (GD1)—phenotypic variability remains striking [3]. Even among individuals with identical GBA1 genotypes or among affected siblings, there can be profound differences in disease severity and presentation, challenging the traditional genotype-phenotype model [4].
This variability, despite GD being a monogenic disorder, suggests that additional factors, including genetic and/or epigenetic modifiers, may play significant roles in shaping the clinical manifestations of the disease. By modulating penetrance, expressivity, and pleiotropy, such modifiers can intricately adjust cellular and organismic responses to GBA1 mutations, thereby sculpting the clinical landscape of GD beyond the confines of traditional genotype-phenotype correlations. These modifiers may influence critical pathways, such as immune regulation, inflammation, and lysosomal function, further complicating disease expression. However, genotype-phenotype studies in GD have been limited by its rarity and extreme heterogeneity, making it difficult to draw robust conclusions from small cohorts. Some researchers have approached GD as a phenotypic continuum, ranging from asymptomatic GD1 to severe GD2 [5]. Genome-wide association studies (GWAS) and candidate gene studies have offered insights, but much remains unexplained [6,7,8].
A promising avenue for further exploration is the identification of modifier genes that modulate penetrance, expressivity, and pleiotropy in GD. These genetic modifiers can interact with GBA1 mutations and/or the accumulating bioactive lipids and/or the lysosomal system to adjust cellular responses, potentially shaping the clinical trajectory of the disease. Recognizing such modifiers could address the diagnostic and therapeutic challenges posed by complex phenotypes that deviate from classical GD presentations, offering more tailored approaches for effective management. Uncovering such modifiers could provide critical insights into the molecular mechanisms underlying GD and guide the development of precision medicine approaches that offer individualized monitoring and therapy.
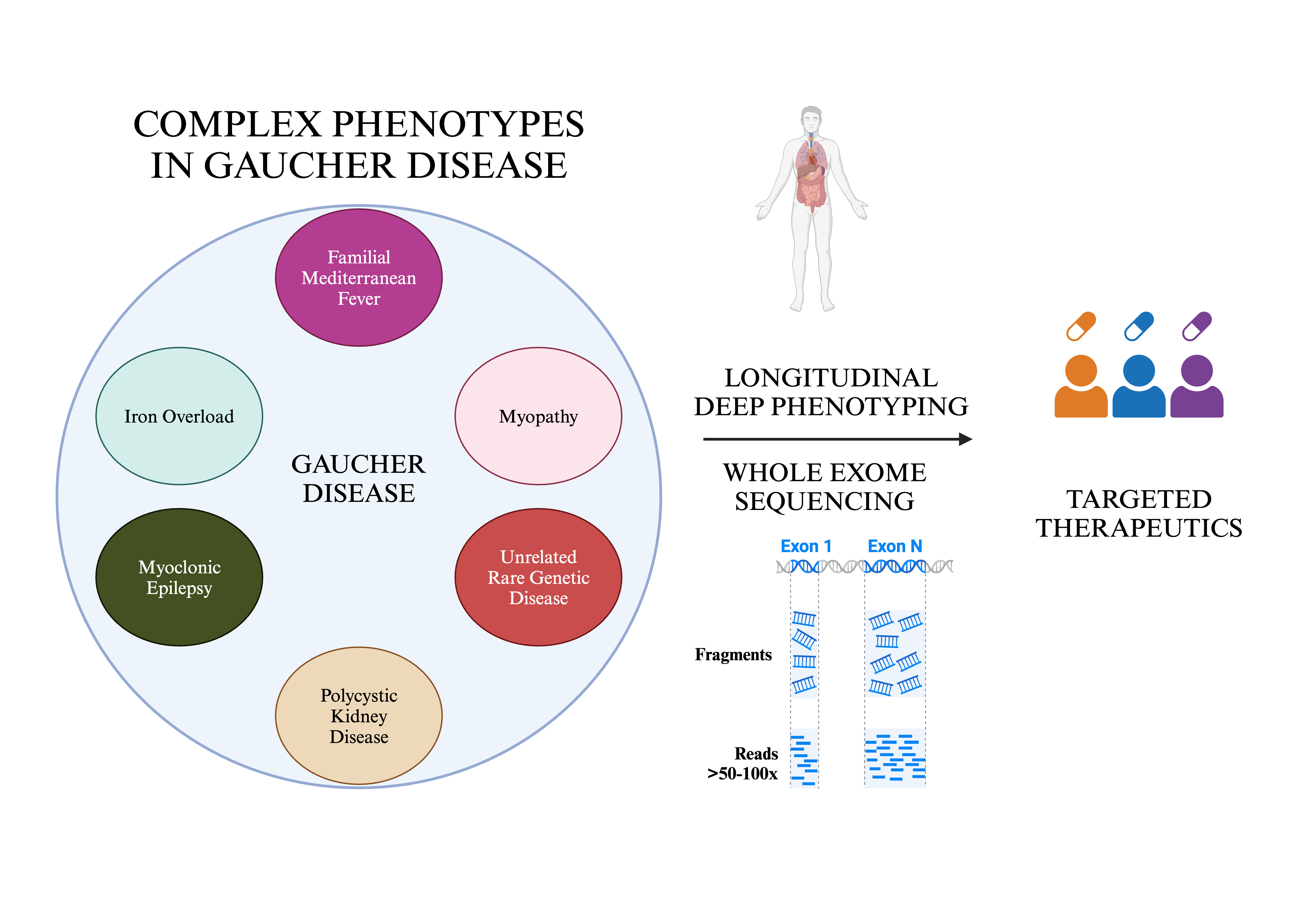
A phenotype-first approach is particularly valuable in conditions like Gaucher disease, where patients with identical genetic mutations can display a wide spectrum of clinical outcomes. Leveraging this approach, we systematically investigated the genomic basis of complex phenotypes in GD to uncover potential genetic modifiers and their role in shaping disease variability. Drawing on a cohort of 275 deeply phenotyped patients from a major tertiary referral center, we identified 18 patients (6.5%) with clinical features beyond those explained by the classical glycosphingolipid metabolic defect. These ’expanded phenotypes’ were linked to additional variants in genes that may interact with GD pathways, suggesting that GD can sometimes manifest as a multi-locus disorder. This study highlights the importance of recognizing complex phenotypes in GD to improve clinical management and deepen our understanding of the disease’s genetic landscape.
2. Materials and Methods
2.1. Study Design and Cohort
We conducted a study combining longitudinal deep phenotyping and whole exome sequencing (WES) in a large cohort of patients (n=275) from a major tertiary referral center. All patients were confirmed to have GD based on low acid β-glucosidase activity in peripheral blood leucocytes and biallelic GBA1 mutations. For precise GBA1 genotyping, we employed a combination of PacBio long-read sequencing, WES, and Sanger sequencing to ensure accurate variant detection [9].
2.2. Longitudinal Deep Phenotyping
The Yale Gaucher Disease Center is a destination center for GD patients who undergo comprehensive evaluations 1-2 times per year. This cohort (n=275) has been followed for up to 26 years (range: seven months to 26 years), with systematic collection of clinical, genetic, and laboratory data. Deep phenotyping included assessments of organ volumes, bone disease indicators, and extensive laboratory testing, including GD biomarkers. This longitudinal approach provided a robust dataset capturing the natural history of GD across various phenotypes.
2.3. Whole Exome Sequencing (WES) and Analysis
WES was performed as described previously, with additional modifications outlined in Supplement 1 [10]. We integrated clinicopathological correlations with genetic data to identify candidate cellular pathways implicated in GD pathogenesis. We evaluated gene variants encoding proteins involved in cellular pathways relevant to disease mechanisms. This integrative approach allowed us to draw connections between specific genetic variations and the complex phenotypic presentations observed in individual patients, thereby shedding light on potential disease-modifying mechanisms.
2.4. Ethical Considerations
This study adhered to all relevant ethical guidelines and was approved by the institutional review board at Yale University. All participants provided informed consent, and confidentiality and patient privacy were rigorously upheld throughout the research.
3. Results
We focused on eighteen patients who exhibited expanded phenotypes to assess for potential concurrent genetic disorders (Table 1 and Table 2). These patients broadly fell into six categories:
- I.
- Unusual inflammatory symptoms due to concurrent Familial Mediterranean Fever (FMF) in GD1 (n=4).
- II.
- Myopathy, osteoporosis and chronic fatigue in GD1 (n=2).
- III.
- Occurrence of complex phenotype due to another rare genetic disease (n=5, GD1).
- IV.
- Autosomal dominant polycystic kidney disease (ADPKD) (n=2, GD1).
- V.
- Myoclonic epilepsy (n=1, GD3).
- VI.
- Hyperferritinemia and iron overload (n=5, GD1).
WES was analyzed for candidate modifier genes. The genes and variants thus selected were annotated for ClinVar, SIFT, MAF, PolyPhen and MAF in several populations including the Ashkenazi Jewish population (about 90% of our cohort is of Ashkenazi Jewish ancestry). Additionally, we show the frequency of the identified variants in our entire GD cohort (Table 3).
This table depicts the genetic variants detected in 18 patients with GD in our cohort. For each variant, we report the associated disease, affected gene, mutation designation (SNP/coding change), mutation type, zygosity status, amino acid change, SIFT prediction score, MAF in general population, ASJ-specific MAF, PolyPhen predicted functional impact and Yale GD ratio. SIFT scores range from 0-1, with scores ≤0.05 considered deleterious. PolyPhen scores also range from 0-1, with higher scores indicating greater functional impact. The Yale GD ratio represents the relative frequency of variants in these 18 patients compared to our entire cohort of GD patients at Yale.
- I.
- Unusual inflammatory symptoms due to concurrent FMF
Patient 1: A 42 year-old male presented with mild splenomegaly, thrombocytopenia, and osteopenia and was diagnosed with GD1 due to homozygosity for p.Asn409Ser GBA1 mutation. He experienced recurrent severe serous pericarditis, necessitating repeated hospitalizations and steroid treatment. Despite imiglucerase enzyme replacement therapy (ERT) for GD, recurrent pericarditis persisted, warranting a pericardiectomy. However, episodes of recurrent pericarditis continued. Finally, he transitioned to eliglustat substrate reduction therapy (SRT), with a striking, sustained remission of pericarditis. WES, focusing on genes underlying inflammatory diseases, revealed a heterozygous p.Asp424Glu mutation in the MEFV gene [11].
Patient 2: A female presented at age 25 due to fatigue and bone pain. Genetic testing revealed homozygosity for p.Asn409Ser GBA1 mutation. Symptoms of fatigue, joint pain and bone pain were debilitating. WES revealed a heterozygous mutation of p.Val726Ala, in the MEFV gene previously reported to be associated with FMF [12]. Initiation of eliglustat SRT resulted in significant amelioration of symptoms.
Patient 3: An adult female with a history of thrombocytopenia, easy bruising, and recurrent mucosal bleeding was diagnosed with GD1 at age 39, due to homozygosity for p.Asn409Ser GBA1 mutation. She developed hepatosplenomegaly, cytopenia, and osteopenia, along with debilitating ankle and abdominal pain. WES revealed a heterozygous p.Val726Ala mutation in the MEFV gene. Her symptoms improved with eliglustat SRT.
Patient 4: A 25-year-old female presented with an unusual constellation of symptoms. The patient complained of abdominal distension, pain, nausea, malaise, polyarthralgia and splenomegaly. MRI abdomen revealed splenomegaly while lab testing showed normal blood counts. She was subsequently diagnosed with GD based on low leucocyte acid β-glucosidase activity. Based on the severity of her symptoms, treatment was initiated with eliglustat SRT with reduction in disease biomarker level to normal (<1 ng/mL) but the patient continued to complain of frequent episodes of fever, night sweats, persistent abdominal pain and mouth sores. WES analysis revealed p.Asp424Glu mutation in the MEFV gene. The patient reported significant improvement in symptoms on a combination of eliglustat and colchicine.
- II.
- Myopathy and chronic fatigue
Patient 5: An adult female presented with splenomegaly, thrombocytopenia, and elevated ferritin at age 20. The initial diagnosis was hemochromatosis, and in addition to hepatocyte siderosis, she had Gaucher cells on liver biopsy. She was diagnosed with GD1 due to homozygous p.Asn409Ser mutation in GBA1. With ERT, her symptoms initially improved. However, subsequently she developed myalgia and exertional dyspnea. Cardiopulmonary exercise testing suggested myopathy. A subsequent muscle biopsy showed cytochrome c oxidase-deficient myofibers with normal mitochondrial activity. WES revealed a heterozygous mutation of p.Asp5398Asn in the NEB gene that encodes the protein nebulin, involved in skeletal muscle function, and when mutated, results in Nemaline myopathy [13,14].
Patient 6: A 2-year-old male child presented with frequent fractures, low bone mass, and proximal muscle weakness. He was homozygous for p.Asn409Ser mutation in GBA1 gene. A right quadriceps biopsy revealed type II myofiber atrophy and cytoarchitectural changes consistent with myopathy. Some mobility was restored four years after beginning ERT; a reduction of GD biomarkers accompanied a decrease in the frequency of fractures and less bone pain. However, by 12-years-of age, his myopathic symptoms started to progress. He was wheelchair-bound and had dyspnea due to diaphragmatic weakness. WES revealed a heterozygous synonymous variant (p.Asp8345=) in the NEB gene, previously reported as VUS in Nemaline myopathy genes [15]. There were no variants in other myopathy genes to explain this phenotype.
- III.
- Occurrence of complex phenotype due to a second, apparently unrelated genetic disease
Patient 7: Metachromatic leukodystrophy (MLD)
A male child presented with increased tone and developmental regression in his first year of life. By 16 months, he had developed spasticity and ataxia with reduced mobility and loss of speech. Brain magnetic resonance imaging (MRI) showed diffuse white matter abnormality. MLD was diagnosed based on low leucocyte arylsulfatase A activity and abnormal urinary sulfatides. At age 4, he was noted to have thrombocytopenia and elevated liver enzymes. WES revealed compound heterozygous GBA1 mutations, p.Asn409Ser and 84GinsG. Additionally, he had homozygous p.Arg60Trp mutations in the ARSA gene that encodes arylsulfatase, known to cause late infantile MLD [16]. He received ERT and supportive care. Unfortunately, he was not a candidate for a bone marrow transplant, as his disease was too advanced to offer meaningful benefits.
Patients 8 and 9: Fibromuscular dysplasia (FMD) and Brugada Syndrome
Two patients in our cohort, homozygous for GBA1 p.Asn409Ser mutation, developed cardiovascular disease. Cardiovascular complications are recognized in homozygous p.Asp448His type 3C GD. However, it is rare for GD1 patients to develop coronary artery disease, presumably due to the profoundly low LDL cholesterol typically found in GD [17].
Patient 8: A 60-year-old female presented at age 21 with massive splenomegaly and thrombocytopenia and was found to be compound heterozygous for GBA1 mutations p.Asn409Ser and p.Val433Leu. ERT with alglucerase reversed her organomegaly and cytopenia. Subsequently, she transitioned to eliglustat SRT and maintained reasonable control of her GD. At 56, she had a spontaneous coronary artery dissection resulting in a non-ST elevation myocardial infarction (NSTEMI) with systolic cardiomyopathy. Cardiac catheterization showed increased tortuosity in distal coronary arteries without occlusion. Positron emission tomography (PET) and computed tomography angiography (CTA) imaging of the abdomen and pelvis revealed a beaded appearance in the right renal and external iliac arteries, suggesting FMD. WES revealed a c.724C>T (p.Gln242*) nonsense mutation in the YY1AP1 gene, previously implicated in Grange syndrome and FMD-like syndrome which involves vascular abnormalities [18]. While recognizing the possibility of this being a rare adverse effect of eliglustat, it is important to note that structural cardiovascular abnormalities have not been reported as a consequence of eliglustat SRT and moreover, patient was dosed pharmacologically with attention to drug-drug interactions, proven to ensure safety [19].
Patient 9: An adult male, who initially presented at age 14 with splenomegaly and thrombocytopenia, was diagnosed with GD1 due to homozygosity for p.Asn409Ser GBA1 mutation, and commenced ERT. A routine EKG during screening for eliglustat trial revealed a type I Brugada pattern in leads V1-V2 (Figure 1). WES, focusing on genes associated with Brugada syndrome, revealed he was heterozygous for g.15206G>T splice acceptor mutation in CACNB2b gene [20]. There were no mutations in SCN5A which is classically associated with Brugada syndrome, however CACNB2b gene defects also result in Brugada syndrome [20].
Patients 10 and 11: Constitutional Mismatch Repair Deficiency (CMMRD) Syndrome
Two siblings with GD1, developed T-cell acute lymphoblastic lymphoma (T-ALL), previously reported by us [10]. Both children presented with splenomegaly and mediastinal mass. Bone marrow aspirate revealed presence of T-ALL as well as Gaucher cells. Exome analysis in these siblings revealed homozygosity for a novel GD mutation (p.Asp137Asn) in the GBA1 gene and a homozygous c.3822dupA/c.3822dupA mutation in the MSH6 mismatch repair (MMR) gene [10].
- IV. ADPKD
Patient 12: An adult female with a known family history of polycystic kidney disease (PKD) was found to have hepatosplenomegaly and cytopenia at age 33, and she started ERT. She was homozygous for p.Asn409Ser mutation in GBA1 gene. WES, focusing on genes associated with PKD, revealed heterozygosity for p.Pro694Leu mutation in the PKD1 gene. Her GD has been well-controlled on ERT and there has been no progression of her PKD indicated by preserved renal function and no increase of renal cysts.
Patient 13: An adolescent male who was diagnosed with GD1 at the age of 2.5 years through family screening because his sibling was known to be affected. There was also a positive family history of PKD, and he was found to be affected by ultrasound. Exome analysis was confirmatory for diagnosis of ADPKD, revealing heterozygosity for the mutation p.R4150C in the PKD1 gene. During regular follow-up, he had up-trending indicators of GD activity and an increasing number of renal cysts, although his renal function was normal. At age 16, he was started on eliglustat SRT.
- V. Myoclonic epilepsy
Patient 14: A 17-year-old female was diagnosed with GD3 at the age of 20 months when she presented with pancytopenia, hepatosplenomegaly, developmental delay, and horizontal gaze palsy. Bone marrow biopsy showed Gaucher cells, and later investigations revealed the compound heterozygous mutations, p.Leu363Pro and Gly416Ser, in the GBA1 gene. Treatment was initiated with a combination of imiglucerase and miglustat. At age 16, she experienced subtle myoclonic jerks in her fingers, that evolved into episodes of generalized tonic-clonic seizures and myoclonic epilepsy requiring multiple anti-epileptic drugs. WES for epilepsy genes revealed heterozygous EFHC1 mutation, p.Arg294His, previously implicated in myoclonic epilepsy [21,22].
- VI.
- Hyperferritinemia and iron overload
Patient 5: This is the same patient who was described earlier with GD and myopathy. She presented with splenomegaly, thrombocytopenia, and elevated ferritin at age 20. Initially diagnosed with chronic liver disease, her liver biopsy revealed Gaucher cells and hepatocyte siderosis. She was found to have low leucocyte acid β-glucosidase activity, and she was homozygous for the p.Asn409Ser GBA1 mutation. Her symptoms improved on ERT. A routine MRI was performed to assess GD, which revealed iron overload in the liver and the bone marrow [23]. She underwent phlebotomy to reverse iron overload. WES analysis revealed homozygous p.His63Asp mutation in HFE gene [24]. Additionally, the patient exhibited homozygosity for both the c.44-24G>C and synonymous p.Val221= (alters the splice site) single nucleotide polymorphisms (SNPs) in the SLC40A1 gene, which have been linked to iron overload syndromes [25].
Patient 15: A male child presented with massive splenomegaly and avascular necrosis of the hips, and he was diagnosed with GD1. He underwent splenectomy at age 6 years and started ERT at age 23 years. He was homozygous for the p.Asn409Ser GBA1 mutation. Later, he developed pain and swelling in his proximal interphalangeal joints and knee chondrocalcinosis. He was found to have hyperferritinemia and a high iron saturation. MRI of the liver showed hepatic iron overload. WES revealed heterozygous p.Cys282Tyr mutation in the HFE gene. Additionally, the patient was homozygous for the c.44-24G>C and p.Val221= SNPs in the SLC40A1 gene [25]. He is undergoing regular phlebotomies to manage the iron overload while he continues ERT.
Patient 16: An adult female of Italian ancestry presented at age 48 with thrombocytopenia and was diagnosed with GD1 due to homozygous p.Asn409Ser GBA1 mutation. Despite responding well to ERT, she had persistently elevated ferritin and splenic iron overload on MRI (Figure 2). WES revealed heterozygous p.His63Asp mutation in the HFE gene [26]. Additionally, she was homozygous for the c.44-24G>C and synonymous p.Val221= SNPs in the ferroportin gene, SLC40A1.
This image represents a T1 weighted image with a uniformly hypointense spleen, which was associated with a signal dropout on in phase gradient echo sequence with longer time to echo (TE), suggestive of diffuse iron deposition.
Patient 17: A routine physical exam in an adult male at age 22 revealed elevated ferritin, thrombocytopenia, elevated liver function tests, and splenomegaly. Liver biopsy revealed hepatocyte siderosis as well as Gaucher cells. Leukocyte acid β-glucosidase activity was low and he was found to be compound heterozygous for p.Asn409Ser and c.115+1G>A GBA1 mutations. Despite a satisfactory response to ERT, he had persistent hyperferritinemia. Abdominal MRI revealed hepatic and splenic iron overload. WES revealed a heterozygous p.His63Asp HFE mutation. He was also found to have the SNPs c.44-24G>C, UTR c.C98G, and UTR c.G8C, in the SLC40A1 gene. The latter two SNPs have been reported in Brazilian patients with primary iron overload [27].
Patient 18: Patient 18 was a male, presenting at age 48 with thrombocytopenia and hyperferritinemia, and was diagnosed with GD1 due to a homozygous p.Asn409Ser GBA1 mutation. He was started on ERT and later switched to eliglustat SRT. Despite a good response to therapy, he had persistent hyperferritinemia and marked splenic iron overload on MRI. WES revealed homozygosity for the c.44-24G>C and p.Val221= variants in the SLC40A1 gene
4. Discussion
Analysis of several thousand exomes from clinical diagnostic laboratories suggests that up to 5% of patients carry more than one genetic disorder, which leads to blended phenotypes associated with multi-locus inheritance [28]. In our study, we analyzed the exomes of GD patients with expanded phenotypes and found that approximately 6% harbored variants in other genes that modified their overall clinical presentation. The extraordinary diversity of phenotypes observed within GBA1 genotype groups in GD cannot be solely explained by the severity of enzyme deficiency. The accumulation of GlcCer and GlcSph in GD activates a range of pathways, including those involving inflammasomes, iron metabolism, necroptosis, ferroptosis, lysosomal function, autophagy, and reactive oxygen species (ROS) [29,30]. This underscores the importance of considering genetic and/or epigenetic modifiers and the need for comprehensive biochemical and genetic evaluations, especially in atypical cases [31]. We show this approach enhances clinical management and improves outcomes.
Interplay of GD and FMF Disease Pathways
Recurrent pericarditis has been previously described in GD [32]. The co-occurrence of Gaucher disease (GD) and familial Mediterranean fever (FMF) highlights a significant intersection between lysosomal dysfunction and inflammatory pathways [33,34]. One patient in our cohort presented with recurrent serous pericarditis and abdominal pain, which raised suspicion of FMF. FMF, caused by mutations in the MEFV gene, is characterized by recurrent fevers and serositis [35]. In GD, impaired autophagy has been linked to inflammasome activation, resulting in elevated levels of interleukin (IL)-1β and IL-6, which may overlap with FMF-associated inflammatory mechanisms [36].
While heterozygous MEFV mutations can produce FMF-like symptoms, particularly in carriers of the p.Val726Ala mutation, the role of the p.Asp424Lys variant in FMF remains less well defined [37,38]. The high carrier frequency of MEFV mutations in the Ashkenazi Jewish population, along with the prevalence of GD in this group, increases the likelihood of co-occurrence, warranting further investigation [39,40]. Interestingly, genetic analysis of the medieval Ashkenazi Jewish founder population from the 14th century revealed the presence of the GBA1 mutation N370S – using the old hg19 reference genome version (which is p.Asn409Ser in the current hg38 reference genome), as well as the p.Val726Ala mutation in the MEFV gene, suggesting a historical genetic predisposition to both conditions in this population [41].
The therapeutic implication is that FMF-associated inflammation may amplify glucosylceramide (GlcCer) synthesis, in GD inflammatory cascade potentially exacerbating GD manifestations [42]. This is supported by the significant clinical improvement observed in our patients treated with glucosylceramide synthase (GCS) inhibitors, such as eliglustat, compared to enzyme replacement therapy (ERT), which appeared less effective in addressing the inflammatory component of the disease.
Muscle Weakness and Myopathy in GD
Approximately 25% of GD patients experience muscle weakness, suggesting an interaction between lysosomal dysfunction and neuromuscular pathology [43]. The etiology of myopathy in GD is debated, with one hypothesis implicating GlcCer-induced calcium release as a cause of myofibril damage [44]. However, this theory is challenged by the lack of creatine kinase elevation and normal muscle biopsy findings. Alternatively, chronic inflammation in GD may contribute to sarcopenia and myopathy-like symptoms [45]. In our cohort, NEB mutations, known to cause nemaline myopathy, were identified as a significant exacerbating factor of muscle weakness [46]. Chronic inflammation, as observed in sporadic late-onset nemaline myopathy, may play a role in this interaction [47]. Additionally, the coexistence of myopathy and osteopenia, particularly in patient 6, suggests that sarcopenia can contribute to bone fragility, as seen in other lysosomal storage disorders like Pompe disease [48]. The carrier frequency of NEB-related nemaline myopathy among Ashkenazi Jews is approximately 1 in 108, leading to an estimated disease incidence of about 1 in 47,000; while that for GD is 1 in 15 carrier rate with estimated frequency of 1 in 850. The Ashkenazi Jewish population’s founder mutations and carrier rate for NEB mutations, coupled with the prevalence of GD, suggest that dual diagnoses like these are rare but should be considered in patients manifesting atypical symptoms [49,50].
Cancer Risk and Genetic Modifiers
The systemic macrophage dysfunction and immune dysregulation in GD create a pro-cancerous environment [51]. Patients in our cohort with homozygous MSH6 mutations, responsible for DNA mismatch repair (MMR), exhibited an increased cancer risk due to the accumulation of random mutations [10]. This finding supports the concept that genetic modifiers may influence the heightened malignancy risk in GD [52]. Additionally, impaired T-cell maturation, potentially caused by GlcCer accumulation, further predisposes GD patients to lymphoid malignancies, as seen in our cohort [51,53].
Co-occurrence of Rare Disorders
Our study demonstrates the unexpected co-occurrence of GD with other rare disorders, including MLD, FMD, ADPKD, and Brugada syndrome. For instance, the neurological symptoms in patients with both GD and MLD may mimic neuronopathic GD, underscoring the importance of contextualized broader genetic diagnoses in selected populations [16]. Although patient 7 had GD1, white matter changes have been reported in GD1, and GBA1 deficiency may exacerbate MLD [54]. Patient 7 was homozygous for ARSA mutation, p.Arg60Trp; our recent study demonstrated a frequency of 1 in 1554 or 0.06% in the Ashkenazi Jewish population [16]. In our cohort, two patients had ADPKD and GD, and interestingly PKD has been linked to aberrant GlcCer metabolism [55,56]. Moreover, clinical trials have targeted GCS in PKD [56]. In the setting of GD, renal cysts could be exposed to increased GlcSph, the downstream bioactive lipid derived from GlcCer, which would likely affect interstitial inflammation and cyst formation [57]. These considerations have obvious therapeutic implications.
Myoclonic Epilepsy in GD3
A small subset of patients with GD3 develop myoclonic epilepsy [58,59]. Myoclonic epilepsy is characterized by marked elevations in somatosensory evoked potentials (SEPs), also known as giant potentials, consistent with a cortical origin [60,61]. Interestingly, even GD3 patients without myoclonic epilepsy have shown increased SEP amplitudes, likely due to elevated intracellular GlcCer levels, leading to an increased sensitivity of hippocampal neurons to glutamate [62]. Therefore, overt myoclonic epilepsy may represent an extreme manifestation of such neuronal hyperexcitability in GD3, invoking a possible role of modifiers this pathway [21,22]. Patient 14, for instance, harbored heterozygous p.Arg294His mutation in the EFHC1 gene, previously associated with myoclonic epilepsy [63]. EFHC1 is a non-ion channel protein that regulates neuronal transmission through mechano-sensation and modulation of calcium channels, especially in the dopaminergic system [64]. Notably, dopaminergic signaling has been reported to be dysregulated in myoclonic epilepsy [65]. Hence, EFHC1 mutations may synergistically affect the neuronal hyperexcitability in GD3, contributing to the relatively rare phenotype of myoclonic epilepsy in GD3. Notably, six other patients with GD3 in our cohort (age 11-52 years) had no history of myoclonic epilepsy, and they did not harbor EFHC1 mutations.
Iron Overload and GD
The co-occurrence of GD and hemochromatosis in five patients in our series highlights the potential crosstalk between iron metabolism and lysosomal dysfunction [66]. Indeed, the accumulation of GlcCer within macrophages in GD induces a pro-inflammatory state characterized by elevated cytokines, including IL-6, which upregulates hepcidin synthesis via the JAK-STAT signaling pathway, contributing to hyperferritinemia [67,68]. Previous studies have reported elevated IL-6 in GD but not hepcidin [69]. Lefebvre et al. proposed that elevated IL-6 induces local upregulation of hepcidin, leading to ferroportin downregulation on the macrophage membrane [70].
The pattern of hemochromatosis gene variants (HFE and SLC40A1) seen in our cohort, combined with radiological appearance of striking iron overload in the spleen point to ferroportin as a compelling player in iron dysregulation seen in GD. Elevation in IL-6 levels in GD has the potential to accentuate defects in ferroportin and HFE, contributing to iron dysregulation. Of note, the variants in SLC40A1 were in non-coding regions, but the same variants have been implicated in other hemochromatosis disorders [25,27].
Study Limitations
Our study is limited by its single-center nature and the rarity of GD, which may limit generalizability. The heterogeneity of GD and varying depths of phenotyping across centers make replication difficult. Additionally, the identification of heterozygous or non-coding variants in some patients necessitates further functional studies to establish their clinical significance. Emerging evidence suggests that synonymous variants may still impact gene expression, mRNA stability, and translation efficiency [71]. WES excludes most non-coding regions, making it challenging to detect complex alleles. Additionally, WES is less reliable for identifying copy number variants (CNVs) or mosaicism due to its inherent limitations in read depth, coverage, and difficulty capturing structural variants. Overall, our data underscore that rather than seeking a single genetic modifier that acts on the metabolic defect, there is a complex interplay of multiple genetic modifiers and significant occurrence of additional rare disorders in GD patients. Identifying these genetic predilections could help refine patient monitoring and enhance precision medicine in GD.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplement 1.
Author Contributions
Conceptualization, AS, PKM; methodology, AS, JR, PKM; software, JR, SM; validation, PKM; formal analysis, AS, JR, PKM; investigation, AS, PKM; resources, PKM; data curation, JR.; writing—original draft preparation, AS; writing—review and editing, AS, GI, JR, NB, NU Ain, PKM, SM, SN; visualization, AS, PKM; supervision, PKM; project administration, PKM; funding acquisition, PKM. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of Yale University.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
We extend our deepest gratitude to our patients, whose trust and willingness to participate made this study possible. We also thank our clinical and research teams for their dedicated support throughout this work. AS and NU Ain were recipients of the Rare Lysosomal Fellowship from Sanofi, and we acknowledge their valuable contribution to this research. Yale HPC cluster was used for preliminary sequencing data analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grabowski GA, Antommaria AHM, Kolodny EH, Mistry PK. Gaucher disease: Basic and translational science needs for more complete therapy and management. Mol Genet Metab. 2021;132(2):59-75. [CrossRef]
- Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease—diagnosis and disease management algorithms. Am J Hematol. 2011;86(1):110-115. [CrossRef]
- Grabowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher Disease: Phenotypic and Genetic Variation. (Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell G, eds.). The Online Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Education; 2019. Accessed December 9, 2024. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225546386.
- Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov Med. 2012;14(77):273-281.
- Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1-2):6-15. [CrossRef]
- Zhang CK, Stein PB, Liu J, et al. Genome-wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation. Am J Hematol. 2012;87(4):377-383. [CrossRef]
- Velayati A, DePaolo J, Gupta N, et al. A mutation in SCARB2 is a modifier in gaucher disease. Hum Mutat. 2011;32(11):1232-1238. [CrossRef]
- Lo SM, Liu J, Chen F, et al. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy. J Inherit Metab Dis. 2011;34(3):643-650. [CrossRef]
- Drelichman GI, Fernández Escobar N, Soberon BC, et al. Long-read single molecule real-time (SMRT) sequencing of GBA1 locus in Gaucher disease national cohort from Argentina reveals high frequency of complex allele underlying severe skeletal phenotypes: Collaborative study from the Argentine Group for Diagnosis and Treatment of Gaucher Disease. Mol Genet Metab Rep. 2021;29:100820. [CrossRef]
- Lo SM, Choi M, Liu J, et al. Phenotype diversity in type 1 Gaucher disease: discovering the genetic basis of Gaucher disease/hematologic malignancy phenotype by individual genome analysis. Blood. 2012;119(20):4731-4740. [CrossRef]
- The International FMF Consortium. Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever. Cell. 1997;90(4):797-807. [CrossRef]
- Aksentijevich I, Torosyan Y, Samuels J, et al. Mutation and Haplotype Studies of Familial Mediterranean Fever Reveal New Ancestral Relationships and Evidence for a High Carrier Frequency with Reduced Penetrance in the Ashkenazi Jewish Population. The American Journal of Human Genetics. 1999;64(4):949-962. [CrossRef]
- Lehtokari VL, Pelin K, Herczegfalvi A, et al. Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscular Disorders. 2011;21(8):556-562. [CrossRef]
- Moreno CAM, Artilheiro MC, Fonseca ATQSM, et al. Clinical Manifestation of Nebulin-Associated Nemaline Myopathy. Neurol Genet. 2023;9(1). [CrossRef]
- https://www.ncbi.nlm.nih.gov/clinvar/variation/95123/.
- Rabin R, Hirsch Y, Booth KTA, et al. ARSA Variant Associated With Late Infantile Metachromatic Leukodystrophy and Carrier Rate in Individuals of Ashkenazi Jewish Ancestry. Am J Med Genet A. Published online October 30, 2024. [CrossRef]
- Stein P, Yang R, Liu J, Pastores GM, Mistry PK. Evaluation of high density lipoprotein as a circulating biomarker of Gaucher disease activity. J Inherit Metab Dis. 2011;34(2):429-437. [CrossRef]
- Guo D chuan, Duan XY, Regalado ES, et al. Loss-of-Function Mutations in YY1AP1 Lead to Grange Syndrome and a Fibromuscular Dysplasia-Like Vascular Disease. The American Journal of Human Genetics. 2017;100(1):21-30. [CrossRef]
- Peterschmitt MJ, Freisens S, Underhill LH, Foster MC, Lewis G, Gaemers SJM. Long-term adverse event profile from four completed trials of oral eliglustat in adults with Gaucher disease type 1. Orphanet J Rare Dis. 2019;14(1):128. [CrossRef]
- Garcia-Elias A, Benito B. Ion Channel Disorders and Sudden Cardiac Death. Int J Mol Sci. 2018;19(3):692. [CrossRef]
- Suzuki T, Delgado-Escueta A V, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36(8):842-849. [CrossRef]
- de Nijs L, Wolkoff N, Coumans B, Delgado-Escueta A V., Grisar T, Lakaye B. Mutations of EFHC1, linked to juvenile myoclonic epilepsy, disrupt radial and tangential migrations during brain development. Hum Mol Genet. 2012;21(23):5106-5117. [CrossRef]
- Stein P, Yu H, Jain D, Mistry PK. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am J Hematol. 2010;85(7):472-476. [CrossRef]
- Yassin MA, Soliman AT, Desanctis V, Abusamaan S, Elsotouhy A, Aldewik N. Hereditary Hemochromatosis in an Adult Due to H63D Mutation: The Value of Estimating Iron Deposition By MRI T2* and Dissociation Between Serum Ferritin Concentration and Hepatic Iron Overload. Blood. 2014;124(21):4891-4891. [CrossRef]
- Duca L, Granata F, Di Pierro E, Brancaleoni V, Graziadei G, Nava I. Associated Effect of SLC40A1 and TMPRSS6 Polymorphisms on Iron Overload. Metabolites. 2022;12(10):919. [CrossRef]
- Hanson EH. HFE Gene and Hereditary Hemochromatosis: A HuGE Review. Am J Epidemiol. 2001;154(3):193-206. [CrossRef]
- Santos PCJL, Cançado RD, Pereira AC, et al. Hereditary hemochromatosis: Mutations in genes involved in iron homeostasis in Brazilian patients. Blood Cells Mol Dis. 2011;46(4):302-307. [CrossRef]
- Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. New England Journal of Medicine. 2017;376(1):21-31. [CrossRef]
- Boddupalli CS, Nair S, Belinsky G, et al. Neuroinflammation in neuronopathic Gaucher disease: Role of microglia and NK cells, biomarkers, and response to substrate reduction therapy. Elife. 2022;11. [CrossRef]
- Vardi A, Zigdon H, Meshcheriakova A, et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J Pathol. 2016;239(4):496-509. [CrossRef]
- Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77(1-2):91-98. [CrossRef]
- ROBERTS WC, FREDRICKSON DS. Gaucher’s Disease of the Lung Causing Severe Pulmonary Hypertension with Associated Acute Recurrent Pericarditis. Circulation. 1967;35(4):783-789. [CrossRef]
- Nair S, Boddupalli CS, Verma R, et al. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood. 2015;125(8):1256-1271. [CrossRef]
- Pandey MK, Grabowski GA. Immunological Cells and Functions in Gaucher Disease. Crit Rev Oncog. 2013;18(3):197-220. [CrossRef]
- Lancieri M, Bustaffa M, Palmeri S, et al. An Update on Familial Mediterranean Fever. Int J Mol Sci. 2023;24(11):9584. [CrossRef]
- Aflaki E, Moaven N, Borger DK, et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell. 2016;15(1):77-88. [CrossRef]
- Booty MG, Chae JJ, Masters SL, et al. Familial mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum. 2009;60(6):1851-1861. [CrossRef]
- Berkun Y, Eisenstein E, Ben-Chetrit E. FMF - clinical features, new treatments and the role of genetic modifiers: a critical digest of the 2010-2012 literature. Clin Exp Rheumatol. 2012;30(3 Suppl 72):S90-5.
- Stirnemann J, Belmatoug N, Camou F, et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int J Mol Sci. 2017;18(2):441. [CrossRef]
- LIDAR M, KEDEM R, BERKUN Y, LANGEVITZ P, LIVNEH A. Familial Mediterranean Fever in Ashkenazi Jews: The Mild End of the Clinical Spectrum. J Rheumatol. 2010;37(2):422-425. [CrossRef]
- Waldman S, Backenroth D, Harney É, et al. Genome-wide data from medieval German Jews show that the Ashkenazi founder event pre-dated the 14th century. Cell. 2022;185(25):4703-4716.e16. [CrossRef]
- Pandey MK, Burrow TA, Rani R, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. 2017;543(7643):108-112. [CrossRef]
- Pastores GM, Barnett NL, Bathan P, Kolodny EH. A neurological symptom survey of patients with type I Gaucher disease. J Inherit Metab Dis. 2003;26(7):641-645. [CrossRef]
- Tsai L, Chien Y, Yang C, Hwu W. Myopathy in Gaucher disease. J Inherit Metab Dis. 2008;31(S3):489-491. [CrossRef]
- Dalle S, Rossmeislova L, Koppo K. The Role of Inflammation in Age-Related Sarcopenia. Front Physiol. 2017;8. [CrossRef]
- Sewry CA, Laitila JM, Wallgren-Pettersson C. Nemaline myopathies: a current view. J Muscle Res Cell Motil. 2019;40(2):111-126. [CrossRef]
- González-Jamett A, Vásquez W, Cifuentes-Riveros G, Martínez-Pando R, Sáez JC, Cárdenas AM. Oxidative Stress, Inflammation and Connexin Hemichannels in Muscular Dystrophies. Biomedicines. 2022;10(2):507. [CrossRef]
- Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6. [CrossRef]
- Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat. 2010;31(11):1240-1250. [CrossRef]
- Emery A. Rare genetic disorders in certain populations. Neuromuscular Disorders. 2009;19(5):307. [CrossRef]
- Mistry PK, Liu J, Yang M, et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proceedings of the National Academy of Sciences. 2010;107(45):19473-19478. [CrossRef]
- Rosenbloom BE, Cappellini MD, Weinreb NJ, et al. Cancer risk and gammopathies in 2123 adults with Gaucher disease type 1 in the International Gaucher Group Gaucher Registry. Am J Hematol. 2022;97(10):1337-1347. [CrossRef]
- Liu J, Halene S, Yang M, et al. Gaucher disease gene GBA functions in immune regulation. Proceedings of the National Academy of Sciences. 2012;109(25):10018-10023. [CrossRef]
- Davies EH, Seunarine KK, Banks T, Clark CA, Vellodi A. Brain white matter abnormalities in paediatric Gaucher Type I and Type III using diffusion tensor imaging. J Inherit Metab Dis. 2011;34(2):549-553. [CrossRef]
- Natoli TA, Smith LA, Rogers KA, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med. 2010;16(7):788-792. [CrossRef]
- Gansevoort RT, Hariri A, Minini P, et al. Venglustat, a Novel Glucosylceramide Synthase Inhibitor, in Patients at Risk of Rapidly Progressing ADPKD: Primary Results of a Double-Blind, Placebo-Controlled, Phase 2/3 Randomized Clinical Trial. American Journal of Kidney Diseases. 2023;81(5):517-527.e1. [CrossRef]
- Nobakht N, Hanna RM, Al-Baghdadi M, et al. Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review. Kidney Med. 2020;2(2):196-208. [CrossRef]
- King JO. Progressive myoclonic epilepsy due to Gaucher’s disease in an adult. J Neurol Neurosurg Psychiatry. 1975;38(9):849-854. [CrossRef]
- Park JK, Orvisky E, Tayebi N, et al. Myoclonic Epilepsy in Gaucher Disease: Genotype-Phenotype Insights from a Rare Patient Subgroup. Pediatr Res. 2003;53(3):387-395. [CrossRef]
- Rothwell JC, Obeso JA, Marsden CD. On the significance of giant somatosensory evoked potentials in cortical myoclonus. J Neurol Neurosurg Psychiatry. 1984;47(1):33-42. [CrossRef]
- Manganotti P, Tamburin S, Zanette G, Fiaschi A. Hyperexcitable cortical responses in progressive myoclonic epilepsy. Neurology. 2001;57(10):1793-1799. [CrossRef]
- Garvey MA, Toro C, Goldstein S, et al. Somatosensory evoked potentials as a marker of disease burden in type 3 Gaucher disease. Neurology. 2001;56(3):391-394. [CrossRef]
- Thounaojam R, Langbang L, Itisham K, et al. EFHC1 mutation in Indian juvenile myoclonic epilepsy patient. Epilepsia Open. 2017;2(1):84-89. [CrossRef]
- Loucks CM, Park K, Walker DS, et al. EFHC1, implicated in juvenile myoclonic epilepsy, functions at the cilium and synapse to modulate dopamine signaling. Elife. 2019;8. [CrossRef]
- Ciumas C, Wahlin TBR, Jucaite A, Lindstrom P, Halldin C, Savic I. Reduced dopamine transporter binding in patients with juvenile myoclonic epilepsy. Neurology. 2008;71(11):788-794. [CrossRef]
- Regenboog M, van Kuilenburg ABP, Verheij J, Swinkels DW, Hollak CEM. Hyperferritinemia and iron metabolism in Gaucher disease: Potential pathophysiological implications. Blood Rev. 2016;30(6):431-437. [CrossRef]
- Allen MJ, Myer BJ, Khokher AM, Rushton N, Cox TM. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. QJM. 1997;90(1):19-25. [CrossRef]
- Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. Journal of Clinical Investigation. 2004;113(9):1271-1276. [CrossRef]
- Lorenz F, Pawłowicz E, Klimkowska M, et al. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol Dis. 2018;68:35-42. [CrossRef]
- Lefebvre T, Reihani N, Daher R, et al. Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease. Haematologica. 2018;103(4):587-596. [CrossRef]
- Kesner JS, Chen Z, Shi P, et al. Noncoding translation mitigation. Nature. 2023;617(7960):395-402. [CrossRef]
Figure 1.
Electrocardiogram of a patient with Gaucher disease and Brugada syndrome. The EKG shows a characteristic type I Brugada pattern, evident in leads V1 and V2. This pattern is defined by a coved-type ST segment elevation greater than 2 mm, followed by a descending negative T wave.
Figure 1.
Electrocardiogram of a patient with Gaucher disease and Brugada syndrome. The EKG shows a characteristic type I Brugada pattern, evident in leads V1 and V2. This pattern is defined by a coved-type ST segment elevation greater than 2 mm, followed by a descending negative T wave.
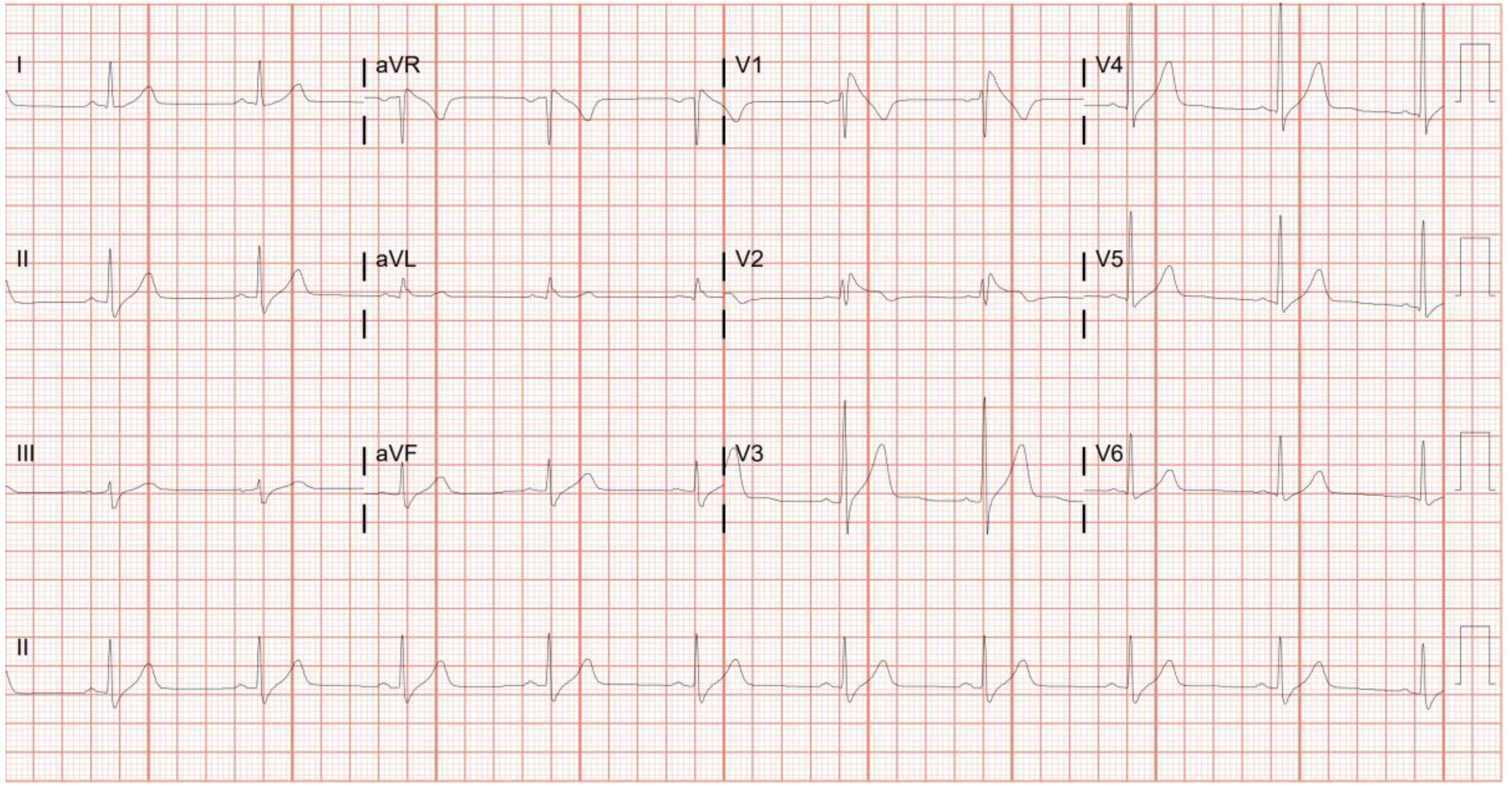
Figure 2.
Non-contrast MRI abdomen of a patient with Gaucher disease/hemochromatosis.
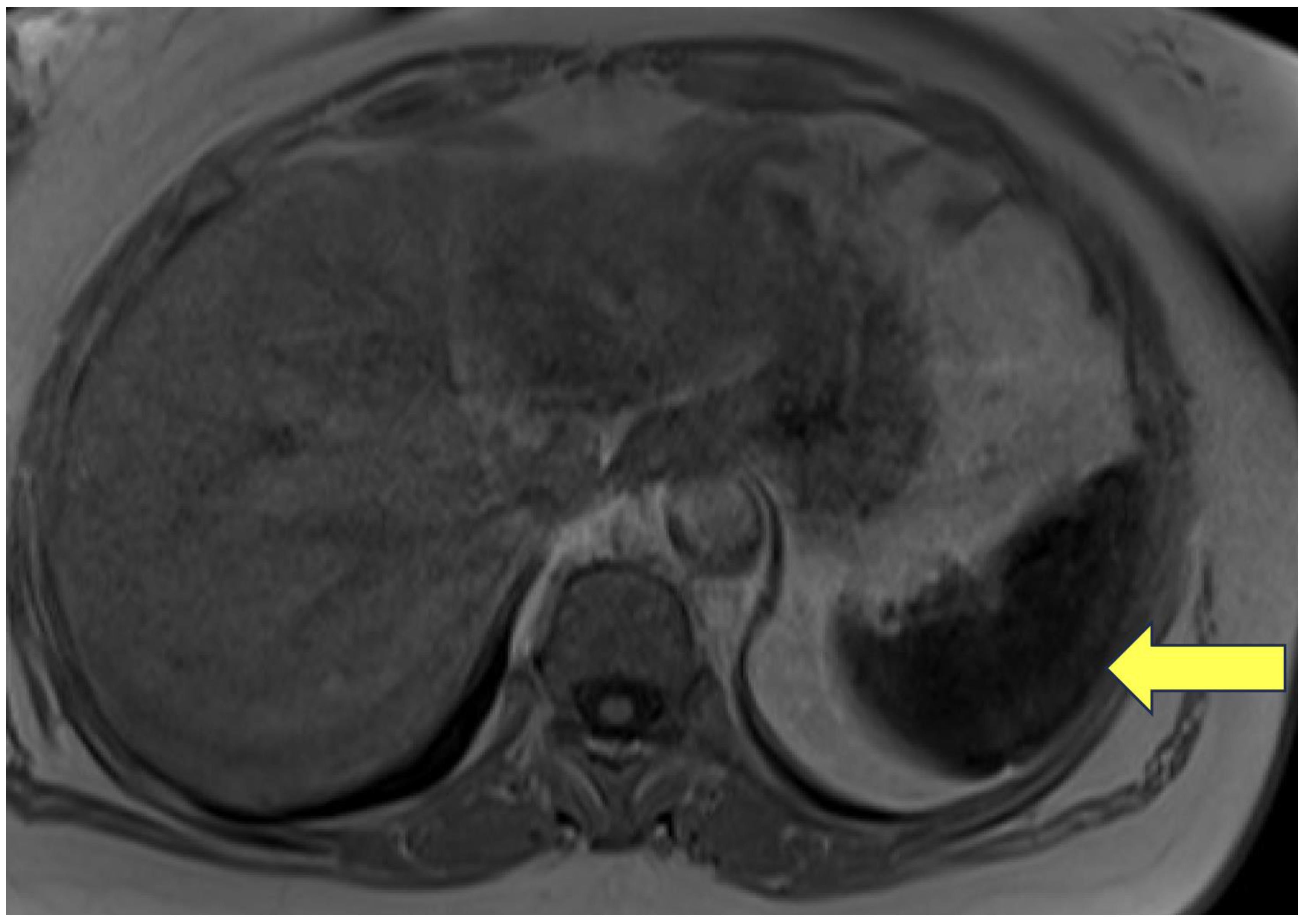

Table 1.
Our cohort of 18 patients harboring another rare disorder in addition to Gaucher disease.
| Concomitant Genetic Disorder | Number of Cases |
| Familial Mediterranean Fever | 4 |
| Nemaline Myopathy | 2 |
| Metachromatic Leukodystrophy | 1 |
| Fibromuscular Dysplasia | 1 |
| Constitutional Mismatch Repair Deficiency Syndrome | 2 |
| Myoclonic Epilepsy | 1 |
| Brugada syndrome | 1 |
| Autosomal Dominant Polycystic Kidney Disease | 2 |
| Hereditary Hemochromatosis | 5 |
Table 2.
Demographic details of our cohort of patients with complex phenotype GD. This table highlights the demographic and clinical characteristics of GD patients at our center, comparing those with complex phenotypes (N=18) to our total cohort (N=275).
Table 2.
Demographic details of our cohort of patients with complex phenotype GD. This table highlights the demographic and clinical characteristics of GD patients at our center, comparing those with complex phenotypes (N=18) to our total cohort (N=275).
| Demographic | Complex phenotype GD (N=18) | Total cohort (N=275) |
| Age (years) [Mean (Range)] | 42.4 (11-73) | 46.4 (1-94) |
| Gender (Female) N (%) | 18 (55.5) | 142 (51.6) |
| GBA1 genotype (p.Asn409Ser/p.Asn409Ser) N (%) | 11 (61.1) | 151 (54.9) |
| Splenectomy N (%) | 0 (0) | 15 (5.4) |
| ERT N (%) | 9 (50) | 133 (48.4) |
| SRT N (%) | 9 (50) | 107 (38.9) |
| Untreated N (%) | 0 (0) | 39 (14.2) |
GD – Gaucher disease; ERT -Enzyme Replacement Therapy; SRT – Substrate Reduction Therapy.
Table 3.
Genetic variants detected in our cohort of 18 patients.
| Disease | Patient | Gene | Mutation | Amino Acid Change | Type | Zygosity | SIFT | MAF | ASJ-MAF | PolyPhen | Yale GD Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Familial Mediterranean Fever | Patient 1 | MEFV | rs1231123 | p.Asp424Lys | Coding | Heterozygous | tolerated (0.34) | 0.3744 | 0.4184 | possibly damaging(0.631) | 0.63 |
| Patient 2 | MEFV | rs28940579 | p.Val726Ala | Coding | Heterozygous | tolerated(1) | 0.002167 | 0.0421 | benign(0.06) | 0.07 | |
| Patient 3 | MEFV | rs28940579 | p.Val726Ala | Coding | Heterozygous | tolerated(1) | 0.002167 | 0.0421 | benign(0.06) | 0.07 | |
| Patient 4 | MEFV | rs1231123 | p.Asp424Lys | Coding | Heterozygous | tolerated (0.34) | 0.3744 | 0.4184 | possibly damaging(0.631) | 0.63 | |
| Nemaline Myopathy | Patient 5 | NEB | rs750810441 | p.Asp5398Asn | Coding | Heterozygous | NA | 0.002193 | 0.0115 | unknown(0) | 0.01 |
| Patient 6 | NEB | rs201825451 | p.Asp8345= | Coding | Heterozygous | NA | 0.00157 | 0.017 | NA | 0.03 | |
| Metachromatic Leukodystrophy | Patient 7 | ARSA | rs867538940 | p.Arg60Trp | Coding | Homozygous | deleterious(0.03) | 5.82E-06 | 0 | probably damaging(0.989) | 0.01 |
| Fibromuscular Dysplasia | Patient 8 | YY1AP1 | rs41264945 | p.Gln242 | Coding | Heterozygous | deleterious(0) | 0.03638 | 0.0308 | probably damaging(0.999) | 0.04 |
| Brugada Syndrome | Patient 9 | CACNB2 | rs875989812 | NA | Splice acceptor | Heterozygous | NA | 0.0006187 | 0.0026 | NA | 0.007 |
| Constitutional Mismatch Repair Deficiency | Patient 10 | MSH6 | c.2822dupA | NA | Frame-shift | Homozygous | deleterious(0) | 0 | 0 | probably damaging(0.999) | 0.008 |
| Patient 11 | MSH6 | c.2822dupA | NA | Frame-shift | Homozygous | deleterious(0) | 0 | 0 | probably damaging(0.999) | 0.008 | |
| ADPKD | Patient 12 | PKD1 | rs138575342 | p.Pro694Leu | Coding | Heterozygous | deleterious(0) | 0.02435 | 0 | probably damaging(0.993) | 0.037 |
| Patient 13 | PKD1 | rs1282668884 | p.Arg4150Cys | Coding | Heterozygous | deleterious(0) | NA | 0 | probably damaging(1) | 0 | |
| Myoclonic Epilepsy | Patient 14 | EFHC1 | rs1570624 | p.Arg294His | Coding | Heterozygous | deleterious(0) | 0.01005 | 0.0043 | probably damaging(0.999) | 0.02 |
| Hemochromatosis | Patient 5 | HFE | rs1799945 | p.His63Asp | Coding | Homozygous | tolerated(0.09) | 0.1092 | 0.1208 | possibly damaging(0.704) | 0.22 |
| SLC40A1 | rs2304704 | p.Val221= | Splice site | Homozygous | NA | 0.6286 | 0.7137 | NA | 0.89 | ||
| SLC40A1 | rs1439816 | NA | Non-coding | Homozygous | NA | 0.7967 | 0.8192 | NA | 0.96 | ||
| Patient 15 | HFE | rs1800562 | p.Cys282Tyr | Coding | Heterozygous | deleterious(0) | 0.03321 | 0.0138 | possibly damaging(0.509) | 0.06 | |
| SLC40A1 | rs2304704 | p.Val221= | Splice site | Homozygous | NA | 0.6286 | 0.7137 | NA | 0.89 | ||
| SLC40A1 | rs1439816 | NA | Non-coding | Homozygous | NA | 0.7967 | 0.8192 | NA | 0.96 | ||
| Patient 16 | HFE | rs1799945 | p.His63Asp | Coding | Heterozygous | tolerated(0.09) | 0.1092 | 0.1208 | possibly damaging(0.704) | 0.22 | |
| SLC40A1 | rs1439816 | NA | Non-coding | Heterozygous | NA | 0.7967 | 0.8192 | NA | 0.96 | ||
| SLC40A1 | rs11568351 | NA | Non-coding | Heterozygous | NA | 0.168 | 0.1275 | NA | 0.28 | ||
| SLC40A1 | rs13008848 | NA | Non-coding | Heterozygous | NA | 0.1655 | 0.1252 | NA | 0.27 | ||
| Patient 17 | HFE | rs1799945 | p.His63Asp | Coding | Heterozygous | tolerated(0.09) | 0.1092 | 0.1208 | possibly damaging(0.704) | 0.22 | |
| SLC40A1 | rs2304704 | p.Val221= | Splice site | Homozygous | NA | 0.6286 | 0.7137 | NA | 0.89 | ||
| SLC40A1 | rs1439816 | NA | Non-coding | Homozygous | NA | 0.7967 | 0.8192 | NA | 0.96 | ||
| Patient 18 | SLC40A1 | rs2304704 | p.Val221= | Splice site | Homozygous | NA | 0.6286 | 0.7137 | NA | 0.89 | |
| SLC40A1 | rs1439816 | NA | Non-coding | Homozygous | NA | 0.7967 | 0.8192 | NA | 0.96 |
SIFT – Sorting Intolerant From Tolerant; MAF – Minor Allele Frequency; ASJ-MAF – Minor Allele Frequency in the Ashkenazi Jewish Population; GD – Gaucher disease; ADPKD – Autosomal Dominant Polycystic Kidney Disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



