
Submitted:
21 July 2025
Posted:
22 July 2025
You are already at the latest version
Abstract
Background: Transthyretin amyloidosis (ATTR) is a multisystem, progressive, and potentially fatal condition. It is significantly under-recognized, leading to delayed diagnosis and treatment and poor outcomes. The CARABELA-ATTR initiative aims to address the challenges of ATTR management in the Spanish healthcare system. Methods: This initiative promotes collaboration between scientific societies, clinicians, and the pharmaceutical industry to optimize ATTR care using lean methodology. It included four phases: (1) characterization of ATTR clinical pathways; (2) validation and prioritization of areas for improvement and selection of quality indicators (QIs); (3) co-creation of solutions; and (4) dissemination of results. Results: A generic ATTR patient journey was defined, identifying four healthcare models with varying coordination and resources. Multidisciplinary discussions identified 13 areas for improvement, including deficient multidisciplinary coordination, lack of standardized diagnostic protocols, and uneven resource distribution. A set of QIs covering all aspects of care was defined. Validated solutions included establishing multidisciplinary teams, standardizing diagnostic and treatment protocols, and ensuring equitable access to advanced diagnostics and treatments. Conclusions: The CARABELA-ATTR initiative provides a framework that emphasizes multidisciplinary coordination, diagnostic and treatment protocols, continuous education for healthcare professionals and patients, and equitable access to resources, aiming to improve patient outcomes and quality of life.
Keywords:
CARABELA
; transthyretin amyloidosis
; ATTR
; healthcare models
; indicators
; quality of care
; management
1. Introduction
Transthyretin amyloidosis (ATTR) is a progressive, debilitating, and multisystem disease characterized by amyloid fibril deposition from misfolded TTR protein in various organs and tissues [1]. This potentially fatal condition is often under-recognized, leading to significant delays in diagnosis and treatment, which can greatly impact patient outcomes [2,3,4,5,6].
Hereditary or variant (ATTRv) and wild-type (ATTRwt) ATTR are two forms of this pathology with distinct epidemiological and clinical features. ATTRv, caused by TTR gene mutations, is rare, typically manifests earlier, and affects multiple organs, most commonly the heart and nerves, although it may also impact the kidneys and eyes. In Spain, the V30M mutation in TTR is endemic in certain regions [7,8,9], resulting in an earlier onset and familial patterns. Notably, in a study conducted in the USA, the V30M variant showed a 83% cumulative multisystem involvement, particularly with regard to cardiac association [10]. It was also reported that 43% of patients with the V30M mutation had cardiac involvement at diagnosis, 95% had neurological involvement, and 60% had autonomic involvement [11]. These findings highlight the significant multisystem impact of the V30M variant. In addition to V30M, there are other variants present in Spain, such as Val142Ile and Glu89Lys [8,12]. In contrast to the hereditary form of the condition, ATTRwt may be quite common [13] and typically affects older adults. It presents predominantly as cardiomyopathy and is often underdiagnosed due to late onset and symptom overlap with other age-related conditions [14,15]. Up to 13% of elderly patients with heart failure (HF) and preserved ejection fraction (HFpEF) may have ATTRwt, which highlights its significant public health impact in Spain [16].
ATTR can present predominantly as cardiomyopathy (ATTR-CM) or polyneuropathy (ATTR-PN), but mixed phenotypes are more common than previously thought [2], even among patients initially presenting with predominantly neurological or cardiac phenotypes who, over time, may transition to a mixed phenotype [17]. The THAOS study found that approximately one-third of patients were classified as having a mixed phenotype either at the start of the study or during follow-up. Among the 344 cases reclassified as mixed during follow-up, 69.2% initially had a predominantly neurological phenotype, while 30.8% had a predominantly cardiac phenotype [17,18]. Indeed, depending on the specific TTR mutation, up to 80% of patients with ATTRv exhibit a mixed presentation with various clinical presentations [4,19,20].
ATTR-PN often begins with sensory symptoms such as pain, paraesthesia, or numbness in the feet, and autonomic dysfunction can significantly impact quality of life (QoL) and lead to severe morbidity. Symptoms of autonomic dysfunction include orthostatic hypotension, diarrhoea, constipation, and erectile dysfunction [21,22] and can result in malnutrition and weight loss, associated with poor prognosis. Cardiac involvement in ATTR is characterized by left ventricular wall thickening, diastolic dysfunction, and conduction system abnormalities, including atrioventricular blocks [2,23]. Patients with ATTR-CM have a notably poorer prognosis, with an estimated median survival ranging from 2 to 6 years post-diagnosis, while individuals with ATTR-PN have an estimated survival ranging from 5 to 15 years [5,24,25,26].
The multifaceted nature of ATTR reinforces the importance of its consideration as a multisystem disease, requiring a high index of suspicion for a timely and accurate diagnosis [2,27,28,29,30]. Several “red flags” have been established, including the presence of bilateral carpal tunnel syndrome, unexplained left ventricular hypertrophy in the absence of hypertension, HF, aortic stenosis, and a history of familial amyloidosis [2,31].
Although ATTR recognition and awareness have increased due to diagnostic advances [2,32,33,34], several barriers still need to be overcome regarding its care process. Delayed diagnosis highlights the need to enhance awareness and education among healthcare professionals (HCPs) not only of the condition (especially considering that the signs and symptoms associated with the disease are highly prevalent pathologies such as HF), but also of the rapid expansion of specific treatments that could prevent progression [32,33,34,35].
Given these challenges, the CARABELA-ATTR initiative aims to understand and transform ATTR management in the Spanish healthcare system, representing a significant step forward in optimizing care pathways, addressing areas for improvement, and proposing actionable solutions. By promoting the adoption of a multidisciplinary approach from the onset of disease signs or symptoms that integrates all specialists involved in the care of these patients, the CARABELA-ATTR initiative seeks to enhance the diagnosis, management, follow-up, and quality of care, thereby improving patient clinical outcomes.
2. Materials and Methods
Design and Participants
The general methodology for the CARABELA approach, based on lean methodology applied to healthcare ecosystems [36], has been described elsewhere [37]. The Spanish scientific societies SEC (Spanish Society of Cardiology), SEN (Spanish Society of Neurology), SEMI (Spanish Society of Internal Medicine), SECA (Spanish Society for Healthcare Quality), and SEDISA (Spanish Society of Health Managers), in collaboration with AstraZeneca and clinicians with expertise in ATTR participated in this initiative led by a Scientific Committee (SC) composed of the authors of this manuscript. See Supplementary Figure S1 and Supplementary Material for additional details.
3. Results
3.1. Characterization of ATTR Care Models
Initially, the SC discussed and established a general model for ATTR management. This model, based on a multidisciplinary approach, differentiates the patient pathway according to specific factors. During the initial phase, a thorough analysis conducted in archetypal centres identified eight key variables in ATTR care: 1) coordination and communication among medical specialties; 2) strategies for early patient identification and diagnosis; 3) existence of care protocols, guidelines, and referral pathways for standardized and improved patient care; 4) availability of diagnostic and follow-up tools; 5) presence of specialized and trained nursing support personnel; 6) resources for training HCPs; 7) resources and strategies to improve patient knowledge and disease management; and 8) tools for recording and analysing patient experiences and health outcomes. Based on these variables, four care models for ATTR patients were identified, which differed primarily in the level of coordination and communication between services, resource availability, and implementation of diagnostic and treatment protocols. An overview of the four care models is shown in Figure 1 and Supplemental Figures S2–S16.
3.2. Indicators for the Evaluation of ATTR Care Models
Establishing QIs is essential for continuously evaluating and enhancing patient care. Care model evaluation should use three categories of indicators: structural, quality of care, and transformation indicators, as outlined by Escalada et al. (2024) [37]. The CARABELA-ATTR SC initially proposed potential indicators and, after prioritization, 31 were selected for the Delphi voting process at the national workshop. The selection criteria were implementation, feasibility, and potential impact on patient care or healthcare outcomes. Ultimately, 25 indicators were agreed upon and confirmed by the SC (Supplementary Table S1).
The Future of ATTR Care: Improvements and Solutions
During the initial evaluation of ATTR care in the five archetypal centres, 13 areas for improvement were identified, which fell into the following categories: diagnosis, communication and coordination, pathways and protocols, availability of human resources and diagnostic tests, training of HCPs, patient education, information systems, and health outcomes registries (Figure 2). To address these needs, 23 solutions were finally validated (Figure 2). The identified areas for improvement were prioritized based on their impact and actionability (Figure 3A), while proposed solutions were prioritized according to their impact and speed of implementation (Figure 3B).
4. Discussion
Advancing our understanding of ATTR requires addressing unresolved questions. In this context, the CARABELA-ATTR initiative is a comprehensive effort to transform the understanding and management of this disease in Spain and to raise awareness among clinicians and HCPs about best practices. The initiative emphasizes the benefits of a multidisciplinary approach to enhance early diagnosis, referral, treatment, and comprehensive care for patients with ATTR. Although lacking a quantitative methodology, CARABELA-ATTR established a systematic, nationwide set of indicators covering all aspects of the ATTR care process, which were instrumental in evaluating care models.
Our findings reinforce the importance of understanding ATTR as a multisystem disease with potential manifestations beyond cardiac involvement. The substantial frequency of neurological and mixed phenotypes underscores the need for a multidisciplinary and comprehensive evaluation of all potential manifestations to ensure an accurate diagnosis and optimal approach.
Clinical guidelines assume the availability of diagnostic methods, specialist expertise, and adherence to quality standards [29,38,39,40,41,42], but daily clinical practice often differs. Our evaluation of currently operating ATTR models of care identified coordination and communication among specialties as significant impact factors for care process quality. We found high variability in ATTR management across healthcare areas, particularly in multidisciplinary coordination, diagnostic and treatment protocols, and resource allocation.
Several key issues arise from the CARABELA-ATTR initiative that need to be addressed. These include the lack of standardized diagnostic protocols for early and accurate diagnosis of ATTR and the need for 1) improved multidisciplinary coordination; 2) essential training for HCPs and patients; and 3) equitable access to resources such as advanced diagnostic tools, specialized HCPs (including expert nursing staff), and effective treatment options.
A major barrier to establishing clear diagnostic pathways is the lack of coordinated care between subspecialties. This results in fragmented care and compromised patient management. The establishment of multidisciplinary collaboration and optimal care pathways reduces diagnostic and treatment delays and improves patient prognosis [43]. One of the main findings of the CARABELA-ATTR initiative was the frequent lack of established multidisciplinary teams or regular meeting schedules. Importantly, the most advanced centres evaluated in the initial phases were characterized by robust multidisciplinary coordination, which facilitated comprehensive patient care and improved diagnosis and treatment outcomes. These models also benefited from specialized diagnostic tools and trained nursing staff, essential for early detection and continuous disease management. All these findings emphasize the importance of creating structured and coordinated multidisciplinary teams [2,44,45]. Coordination of cardiologists, neurologists, internal medicine specialists, and other relevant specialists, such as haematologists, nephrologists, nurses, pharmacists, and geneticists, is crucial for effective management of ATTR that improves health outcomes [46,47]. Specialized nurses play a key role in communication, patient care coordination, early detection of complications, and education of patients and families, thereby enhancing adherence to treatment and overall QoL. This multidisciplinary, integral approach addresses the complex needs of ATTR patients [45,46,47,48,49]. It also facilitates timely diagnosis, quick access to medications, and enrolment in ongoing clinical trials, promoting cutting-edge research on disease mechanisms. The CARABELA-ATTR initiative advocates optimizing and implementing referral protocols that facilitate patient diagnosis and treatment in specialized units or reference centres to benefit from their specialized multidisciplinary expertise and high-quality imaging, while promoting the continuous monitoring of patients by PC or hospital HCPs.
Accurate diagnosis is essential for personalized medicine and is particularly challenging in the context of ATTR, as it is often delayed due to nonspecific symptoms and overlap with other diseases. Early detection of ATTR enables prompt and tailored treatment, significantly improving clinical outcomes and QoL [50] and lessening the economic burden of the condition on the healthcare system [51,52]. The CARABELA-ATTR initiative revealed significant variability in access to diagnostic tools and available resources across centres, which affected the quality of care. Several reports seek expanded access to advanced diagnostic methods for ATTR, such as mass spectrometry, bone scintigraphy with technetium-labelled bisphosphonates (99mTc-DPD or 99mTc-PYP) for detection of cardiac amyloid deposition [53,54], genetic testing, and advanced imaging techniques like cardiac MRI [27,55]. MR neurography and peripheral nerve ultrasound are also valuable tools for detecting polyneuropathy, as is whole-body 18F-florbetapir PET for detecting systemic amyloid deposition [4,56]. However, these advanced techniques are not widely available in routine clinical practice. The CARABELA-ATTR initiative proposes the standardization of diagnostic protocols across all healthcare areas and the implementation of early screening strategies, aiming to ensure the highest standard of care for all patients.
Significant gaps were identified in the training of HCPs and the education of patients and caregivers. Limited or outdated knowledge about the disease or the latest advances in diagnosis and treatment contributes highly to misdiagnosis and delayed treatment. Regular training sessions and workshops are essential to update professionals on the latest evidence on ATTR, reducing underdiagnosis or misdiagnosis and preventing poor patient prognosis [57,58]. Reinforcing patient and caregiver education will also empower and engage them in participating in active self-care, improving adherence to treatment plans and overall outcomes [45]. Additionally, educating patients increases their likelihood of seeking medical care and participating in research studies. However, comprehensive educational resources on ATTR outside medical websites are limited [59]. The CARABELA-ATTR initiative aligns with the demands of patients and associations [60], emphasizing the implementation of regular training programs for HCPs to recognize ATTR signs and symptoms early in the disease course.
Quality assurance measures in ATTR have been limited due to its perceived rarity and lack of comprehensive care guidelines. A major challenge in current care models is the lack of a systematic and standardized collection of clinical information. The CARABELA-ATTR initiative recommends creating and standardizing patient registries as a QI. Patient registries can significantly improve diagnostic rates, reduce gaps in understanding disease heterogeneity, encourage research and collaboration, support clinical pathways, and facilitate practice quality and safety. In Europe, several national registries of rare diseases, including ATTR-CA, have been established [54,55]. In addition to clinical data, the collection of PROM and PREM allows the evaluation of patient QoL and treatment effectiveness. However, QoL assessment in ATTR patients has received limited focus, and specific evaluation tools are lacking [61,62,63]. The CARABELA-ATTR initiative brings the added value of systematic PROM/PREM collection in comprehensive patient care.
Lastly, healthcare systems often operate with separate administrative structures and electronic health records. In today's era of advanced technology, it is crucial to evaluate the current state of information systems and technological options in healthcare centres. The CARABELA-ATTR initiative calls for the implementation of unified clinical records, standardized communication tools, and cutting-edge technological resources within a shared healthcare framework. This approach will maximize benefits in the referral and monitoring processes of ATTR patients.
5. Conclusions
The CARABELA-ATTR initiative has identified critical areas for improvement in ATTR management and proposed implementable solutions to enhance multidisciplinary coordination, the standardization of protocols and care pathways, the promotion of continuous education of HCPs and patients, and equitable access to resources and advanced diagnostics, aiming to lead a meaningful transformation of the Spanish model of ATTR care. The framework provided aims to expedite diagnoses, improve treatment outcomes, and enhance QoL for all patients battling this debilitating disease. Future efforts should focus on implementing these recommendations and continuously monitoring their impact to ensure sustained improvement in ATTR care.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary materials are available in a PDF file.
Author Contributions
Conceptualization, Lucia Galán-Dávila, Mónica Angélica López, Inmaculada Mediavilla and José Francisco Soto; Investigation, Lucia Galán-Dávila, Mónica Angélica López, Tomás Ripoll-Vera, Inmaculada Mediavilla and José Francisco Soto; Validation, Lucia Galán-Dávila, Mónica Angélica López, Tomás Ripoll-Vera, Inmaculada Mediavilla and José Francisco Soto; Writing – review & editing, Lucia Galán-Dávila, Mónica Angélica López, Tomás Ripoll-Vera, Inmaculada Mediavilla and José Francisco Soto. All members of the CARABELA-ATTR Scientific Committee attended the meetings, reviewed the manuscript, and validated the results and content of the publication.
Acknowledgments
The authors acknowledge the participation of all the professionals involved in the pilot phase of the CARABELA-ATTR project from Hospital Universitario de Cabueñes (Gijón, Spain), Hospital General Universitario de Ciudad Real (Ciudad Real, Spain), Hospital Universitario Juan Ramón Jiménez (Huelva, Spain), Hospital Universitario Vall d´Hebron (Barcelona, Spain), Hospital Clínico Universitario Virgen de la Arrixaca (Murcia, Spain), as well as all the professionals attending the national and regional workshops (see Supplementary Information). The authors would also like to acknowledge the medical writing support provided by Susana Cañón-Sánchez, PhD, and Blanca Piedrafita, PhD, from Medical Statistics Consulting (MSC), Valencia, Spain, under the Good Publication Practice guidelines (https://www.ismpp.org/gpp3), which were funded by AstraZeneca.
Appendix A. CARABELA ATTR Scientific Committee
The CARABELA-ATTR SC consists of the following members: Pablo García-Pavía and Tomás Ripoll-Vera (Spanish Society of Cardiology, SEC), María del Prado Salamanca Bautista and Mónica Angélica López Rodríguez (Spanish Society of Internal Medicine, SEMI), Lucía Galán Dávila and María Teresa Sevilla Mantecón (Spanish Society of Neurology, SEN), Inmaculada Mediavilla Herrera (Spanish Society for Healthcare Quality, SECA), José Francisco Soto Bonel (Spanish Society of Health Managers, SEDISA), Eunice Fitas (AstraZeneca Farmacéutica Spain), Manuel Leal (AstraZeneca Farmacéutica Spain), Fiama Caimi (AstraZeneca Farmacéutica Spain), Alicia Eisman (AstraZeneca Farmacéutica Spain) and Lucía Regadera (AstraZeneca Farmacéutica Spain).
References
- Yun, S.; González-Costello, J.; Formiga, F. Amiloidosis por transtiretina: lo que ahora vemos es solo la punta del iceberg. Revista Española de Geriatría y Gerontología 2020, 55, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Nativi-Nicolau, J.N.; Karam, C.; Khella, S.; Maurer, M.S. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev 2022, 27, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, V.; Ferreira, A.; Lalu, T.; Zaros, C.; Lacroix, C.; Adams, D.; Said, G. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology 2007, 69, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol 2021, 268, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.N.; Ando, Y.; Dispenzeri, A.; Gonzalez-Duarte, A.; Adams, D.; Suhr, O.B. Evolving landscape in the management of transthyretin amyloidosis. Ann Med 2015, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Rozenbaum, M.H.; Large, S.; Bhambri, R.; Stewart, M.; Whelan, J.; van Doornewaard, A.; Dasgupta, N.; Masri, A.; Nativi-Nicolau, J. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol Ther 2021, 10, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Reinés, J.B.; Vera, T.R.; Martín, M.U.; Serra, H.A.; Campins, M.M.C.; Millán, J.M.D.; Lezaun, C.G.; Cruz, M.R. Epidemiology of transthyretin-associated familial amyloid polyneuropathy in the Majorcan area: Son Llàtzer Hospital descriptive study. Orphanet Journal of Rare Diseases 2014, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Álvarez Rubio, J.; Manovel Sánchez, A.J.; González-Costello, J.; García-Pavía, P.; Limeres Freire, J.; García-Pinilla, J.M.; Zorio Grima, E.; García-Álvarez, A.; Valverde Gómez, M.; Espinosa Castro, M.Á.; et al. Characterization of hereditary transthyretin cardiac amyloidosis in Spain. Revista Española de Cardiología (English Edition) 2022, 75, 488–495. [Google Scholar] [CrossRef] [PubMed]
- González-Moreno, J.; Losada-López, I.; Cisneros-Barroso, E.; Garcia-Pavia, P.; González-Costello, J.; Muñoz-Beamud, F.; Campistol, J.M.; Fernandez-Torron, R.; Chapman, D.; Amass, L. A Descriptive Analysis of ATTR Amyloidosis in Spain from the Transthyretin Amyloidosis Outcomes Survey. Neurol Ther 2021, 10, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Zampino, S.; Sheikh, F.H.; Vaishnav, J.; Judge, D.; Pan, B.; Daniel, A.; Brown, E.; Ebenezer, G.; Polydefkis, M. Phenotypes Associated With the Val122Ile, Leu58His, and Late-Onset Val30Met Variants in Patients With Hereditary Transthyretin Amyloidosis. Neurology 2023, 100, e2036–e2044. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Plante-Bordeneuve, V.; Schmidt, H.H.J.; Merlini, G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. Journal of the American College of Cardiology 2015, 66, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- de Frutos, F.; Ochoa, J.P.; Gómez-González, C.; Reyes-Leiva, D.; Aróstegui, J.I.; Casasnovas, C.; Barriales-Villa, R.; Sevilla, T.; Gonzalez-Lopez, E.; Ramil, E.; et al. Phenotype and clinical outcomes of Glu89Lys hereditary transthyretin amyloidosis: a new endemic variant in Spain. Amyloid 2023, 30, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail 2020, 8, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart Failure Resulting From Age-Related Cardiac Amyloid Disease Associated With Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. European Heart Journal 2017, 38, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. European Heart Journal 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- González-Moreno, J.; Dispenzieri, A.; Grogan, M.; Coelho, T.; Tournev, I.; Waddington-Cruz, M.; Wixner, J.; Diemberger, I.; Garcia-Pavia, P.; Chapman, D.; et al. Clinical and Genotype Characteristics and Symptom Migration in Patients With Mixed Phenotype Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey. Cardiol Ther 2024, 13, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.; Coelho, T.; Dispenzieri, A.; Conceição, I.; Waddington-Cruz, M.; Kristen, A.; Wixner, J.; Diemberger, I.; Gonzalez-Moreno, J.; Cariou, E.; et al. A 15-year consolidated overview of data in over 6000 patients from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Orphanet J Rare Dis 2023, 18, 350. [Google Scholar] [CrossRef] [PubMed]
- Swiecicki, P.L.; Zhen, D.B.; Mauermann, M.L.; Kyle, R.A.; Zeldenrust, S.R.; Grogan, M.; Dispenzieri, A.; Gertz, M.A. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid 2015, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Waddington-Cruz, M.; Wixner, J.; Amass, L.; Kiszko, J.; Chapman, D.; Ando, Y. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther 2021, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Wixner, J.; Mundayat, R.; Karayal, O.N.; Anan, I.; Karling, P.; Suhr, O.B. THAOS: gastrointestinal manifestations of transthyretin amyloidosis - common complications of a rare disease. Orphanet J Rare Dis 2014, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Lovley, A.; Raymond, K.; Guthrie, S.D.; Pollock, M.; Sanchorawala, V.; White, M.K. Patient-reported burden of hereditary transthyretin amyloidosis on functioning and well-being. J Patient Rep Outcomes 2021, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Rosenthal, J.L.; Grodin, J.L.; Maurer, M.S.; Grogan, M.; Cheng, R.K. ATTR Amyloidosis: Current and Emerging Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol 2021, 3, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Witteles, R.M.; Bokhari, S.; Damy, T.; Elliott, P.M.; Falk, R.H.; Fine, N.M.; Gospodinova, M.; Obici, L.; Rapezzi, C.; Garcia-Pavia, P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail 2019, 7, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Kristen, A.V.; Suhr, O.B.; Maurer, M.S.; Planté-Bordeneuve, V.; Yu, C.R.; Ong, M.L.; Coelho, T.; Rapezzi, C. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J 2022, 43, 391–400. [Google Scholar] [CrossRef] [PubMed]
- González-Duarte, A.; Conceição, I.; Amass, L.; Botteman, M.F.; Carter, J.A.; Stewart, M. Impact of Non-Cardiac Clinicopathologic Characteristics on Survival in Transthyretin Amyloid Polyneuropathy. Neurol Ther 2020, 9, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Adams, D.; Ando, Y.; Beirão, J.M.; Bokhari, S.; Coelho, T.; Comenzo, R.L.; Damy, T.; Dorbala, S.; Drachman, B.M.; et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract 2020, 21, 198. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Dasgupta, N.R.; Rao, R. Diagnosis and Screening of Patients with Hereditary Transthyretin Amyloidosis (hATTR): Current Strategies and Guidelines. Therapeutics and clinical risk management 2020, 16, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021, 42, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Conceição, I.; Coelho, T.; Rapezzi, C.; Parman, Y.; Obici, L.; Galán, L.; Rousseau, A. Assessment of patients with hereditary transthyretin amyloidosis – understanding the impact of management and disease progression. Amyloid 2019, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Silva-Hernández, L.; Horga Hernández, A.; Valls Carbó, A.; Guerrero Sola, A.; Montalvo-Moraleda, M.T.; Galán Dávila, L. Red flags in patients with hereditary transthyretin amyloidosis at diagnosis in a non-endemic area of Spain. Neurologia (Engl Ed) 2023, 38, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Elsanhoury, A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. Journal of Clinical Medicine 2022, 11, 2148. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O'Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Graban, M.; Toussaint, J. Lean Hospitals: Improving Quality, Patient Safety, and Employee Engagement; 2018.
- Escalada, J.; Carretero Gomez, J.; Anguita, M.; de Sequera, P.; García-Río, F.; Dávila, I.; Soto Bonel, J.F.; García, J.P.; Rodríguez Ledo, P.; Barrena, E.; et al. Enhancing the management of chronic diseases in clinical practice: The CARABELA methodology. J Healthc Qual Res 2024. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). European Heart Journal 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol 2019, 26, 2065–2123. [Google Scholar] [CrossRef] [PubMed]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020, 142. [Google Scholar] [CrossRef] [PubMed]
- Moody, W.E.; Turvey-Haigh, L.; Knight, D.; Coats, C.J.; Cooper, R.M.; Schofield, R.; Robinson, S.; Harkness, A.; Oxborough, D.L.; Gillmore, J.D.; et al. British Society of Echocardiography guideline for the transthoracic echocardiographic assessment of cardiac amyloidosis. Echo Res Pract 2023, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Casado, J.; Pérez-Silvestre, J.; Salamanca, P.; Llàcer, P.; Quirós, R.; Ruiz-Hueso, R.; Méndez, M.; Manzano, L.; Formiga, F. Sospecha clínica, diagnóstico y seguimiento de la amiloidosis cardíaca: documento de actualización y resumen ejecutivo. Revista Clínica Española 2024, 224, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.; Moody, W.E. The importance of pathways to facilitate early diagnosis and treatment of patients with cardiac amyloidosis. Ther Adv Cardiovasc Dis 2023, 17, 17539447231216318. [Google Scholar] [CrossRef] [PubMed]
- Brailovsky, Y.; Rajapreyar, I.; Alvarez, R. TTR Amyloidosis: Current State of Affairs and Promise for the Future. JACC Case Rep 2023, 10, 101759. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Callaghan, R.; Ablett, J.; Bibiloni, C.; Bueser, T.; Conceição, I.; Dongiglio, F.; Farrugia, A.; Knebel, F.; Lane, T.; et al. Consensus recommendations on holistic care in hereditary ATTR amyloidosis: an international Delphi survey of patient advocates and multidisciplinary healthcare professionals. BMJ Open 2023, 13, e073130. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Okumura, T.; Murohara, T.; Katsuno, M. Multidisciplinary Approaches for Transthyretin Amyloidosis. Cardiol Ther 2021, 10, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Losada, I.; González-Moreno, J.; Rodriguez, A.; Uson, M.; Ripoll-Vera, T.; Ferrer-Nadal, A.; Rigo, E.; Andreu, H.; Figuerola, A.; Montalà, J.C.; et al. Multidisciplinary approach in the management of hATTR. Eur J Clin Invest 2020, 50, e13296. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Kittleson, M.M.; Wechalekar, A.D.; Alvarez-Cardona, J.; Mitchell, J.D.; Scarlatelli Macedo, A.V.; Dutra, J.P.P.; Campbell, C.M.; Liu, J.E.; Landau, H.J.; et al. Moving towards establishing centres of excellence in cardiac amyloidosis: an International Cardio-Oncology Society statement. Heart 2024. [Google Scholar] [CrossRef] [PubMed]
- Bumma, N.; Kahwash, R.; Parikh, S.V.; Isfort, M.; Freimer, M.; Vallakati, A.; Redder, E.; Campbell, C.M.; Sharma, N.; Efebera, Y.; et al. Multidisciplinary amyloidosis care in the era of personalized medicine. Front Neurol 2022, 13, 935936. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Reilly, M.M.; Coats, C.J.; Cooper, R.; Cox, H.; Coyne, M.R.E.; Green, A.J.; McGowan, R.; Moody, W.E.; Hawkins, P.N. Clinical and Genetic Evaluation of People with or at Risk of Hereditary ATTR Amyloidosis: An Expert Opinion and Consensus on Best Practice in Ireland and the UK. Adv Ther 2022, 39, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Peral, C.; Formiga, F.; García, P.; Martín-Sánchez, J.; Navarro-Ruiz, A.; Tarilonte, P.; López, A.; Rubio-Rodriguez, D.; Rubio-Terrés, C. PCV24 Health and Economic IMPACT of the Correct Diagnosis of Transthyretin Cardiac Amyloidosis in Spain. Value in Health 2020, 23, S490–S491. [Google Scholar] [CrossRef]
- Stewart, M.; Shaffer, S.; Murphy, B.; Loftus, J.; Alvir, J.; Cicchetti, M.; Lenderking, W.R. Characterizing the High Disease Burden of Transthyretin Amyloidosis for Patients and Caregivers. Neurol Ther 2018, 7, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Giblin, G.T.; Cuddy, S.A.M. Multimodality Imaging in Cardiac Amyloidosis. Curr Cardiol Rep 2021, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Naharro, A.; Baksi, A.J.; Hawkins, P.N.; Fontana, M. Diagnostic imaging of cardiac amyloidosis. Nat Rev Cardiol 2020, 17, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Perez, R.; Vázquez-García, R.; Barge-Caballero, G.; Bouzas-Mosquera, A.; Soler-Fernandez, R.; Larrañaga-Moreira, J.M.; Crespo-Leiro, M.G.; Vazquez-Rodriguez, J.M. Diagnostic and prognostic value of cardiac imaging in amyloidosis. World J Cardiol 2020, 12, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Shouman, K.; Broski, S.M.; Muchtar, E.; Pendleton, C.A.; Johnson, G.B.; Tracy, J.; Engelstad, J.K.; Spinner, R.J.; Dyck, P.J.B. Novel imaging techniques using (18) F-florbetapir PET/MRI can guide fascicular nerve biopsy in amyloid multiple mononeuropathy. Muscle Nerve 2021, 63, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Elliott, P.; Comenzo, R.; Semigran, M.; Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation 2017, 135, 1357–1377. [Google Scholar] [CrossRef] [PubMed]
- Rintell, D.; Heath, D.; Braga Mendendez, F.; Cross, E.; Cross, T.; Knobel, V.; Gagnon, B.; Turtle, C.; Cohen, A.; Kalmykov, E.; et al. Patient and family experience with transthyretin amyloid cardiomyopathy (ATTR-CM) and polyneuropathy (ATTR-PN) amyloidosis: results of two focus groups. Orphanet J Rare Dis 2021, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Pack, A.P.; Zuleta, A.; Daugerdas, E.; Huang, W.; Batio, S.; Svoboda, S.; Zeitler, E.P.; Kumar, N.; Watt, S.; Fernandez-Arias, M.I.; et al. Developing, optimizing, and evaluating patient infographics for diagnosing cardiac amyloidosis. PEC Innovation 2023, 3, 100212. [Google Scholar] [CrossRef] [PubMed]
- FEDER. AMILO presenta en Palma su decálogo para mejorar la calidad de vida de los pacientes con amiloidosis. https://www.enfermedades-raras.org/movimiento-asociativo/actualidad-asociativa/amilo-presenta-en-palma-su-decalogo-para-mejorar-la-calidad-de-vida-de-los-pacientes-con-amiloidosis. 2023.
- Siu, A.; Sierra, I.; Zhang, M.; Dranow, L.; Hong, J.; Waldron, J.; Caballero, K.; Greene, T.; Kovacsovics, T.; Stehlik, J.; et al. Patient Reported Outcomes In Amyloidosis Cardiomyopathy. Journal of Cardiac Failure 2020, 26, S116–S117. [Google Scholar] [CrossRef]
- Aimo, A.; Rapezzi, C.; Perfetto, F.; Cappelli, F.; Palladini, G.; Obici, L.; Merlini, G.; Di Bella, G.; Serenelli, M.; Zampieri, M.; et al. Quality of life assessment in amyloid transthyretin (ATTR) amyloidosis. Eur J Clin Invest 2021, 51, e13598. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Teresi, L.; Castiglione, V.; Picerni, A.L.; Niccolai, M.; Severino, S.; Agazio, A.; Carnevale Baraglia, A.; Obici, L.; Palladini, G.; et al. Patient-reported outcome measures for transthyretin cardiac amyloidosis: the ITALY study. Amyloid 2024, 31, 52–61. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Models of ATTR care identified by the CARABELA-ATTR initiative. ATTR, transthyretin amyloidosis; C, cardiology; HCPs, healthcare professionals; IM, internal medicine; MD, multidisciplinary; N, neurology; PAG, patient advocacy groups; PC, primary care; PREM, patient-reported experience measures; PROM, patient-reported outcome measures.
Figure 1.
Models of ATTR care identified by the CARABELA-ATTR initiative. ATTR, transthyretin amyloidosis; C, cardiology; HCPs, healthcare professionals; IM, internal medicine; MD, multidisciplinary; N, neurology; PAG, patient advocacy groups; PC, primary care; PREM, patient-reported experience measures; PROM, patient-reported outcome measures.
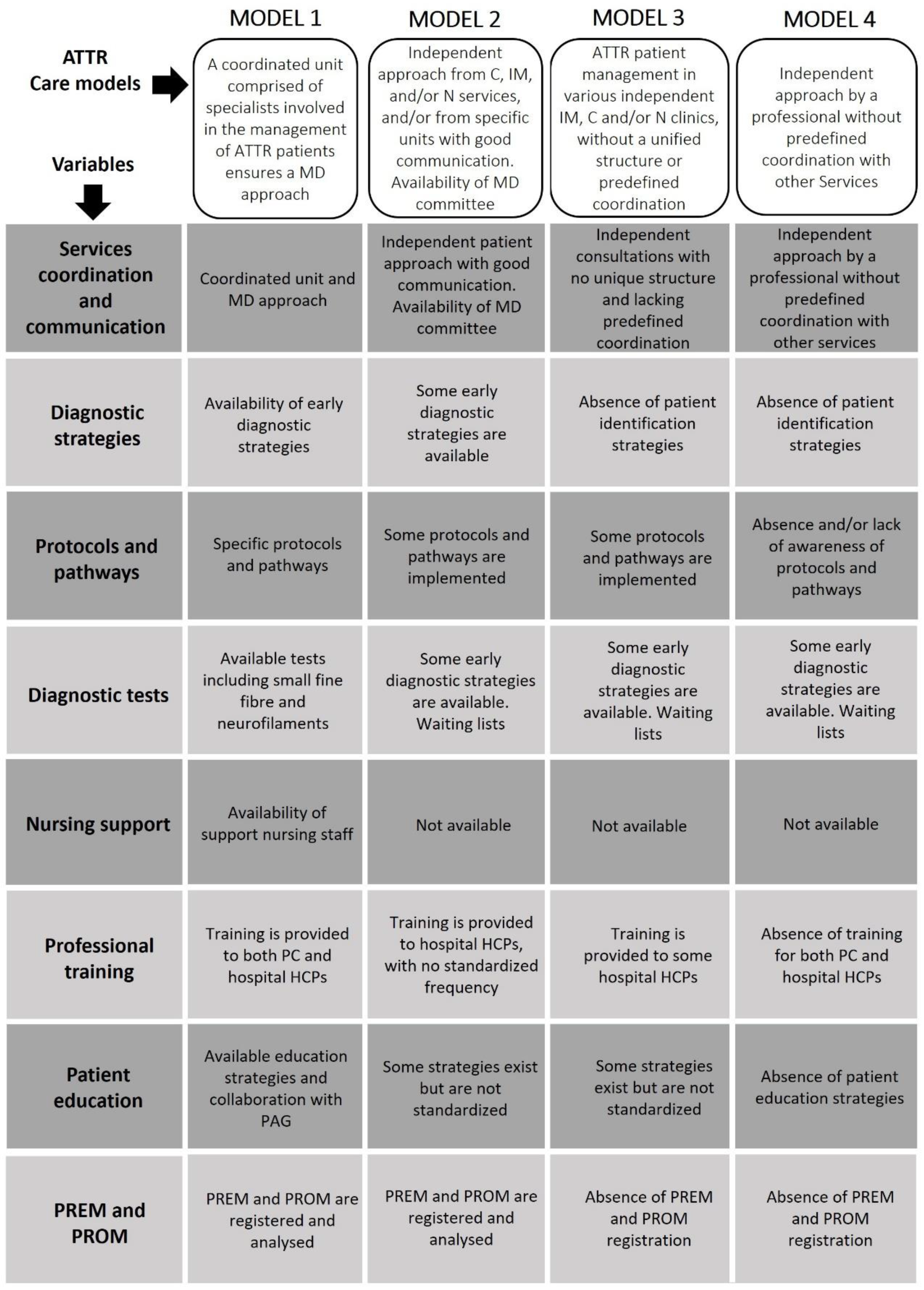
Figure 2.
Proposed solutions for each of the areas for improvement identified during CARABELA-ATTR. ATTR, amyloid transthyretin; IT, information technology; KPI, key performance indicators; PAG, patient advocacy groups; PC, primary care; PREM, patient-reported experience measures; PROM, patient-reported outcome measures.
Figure 2.
Proposed solutions for each of the areas for improvement identified during CARABELA-ATTR. ATTR, amyloid transthyretin; IT, information technology; KPI, key performance indicators; PAG, patient advocacy groups; PC, primary care; PREM, patient-reported experience measures; PROM, patient-reported outcome measures.
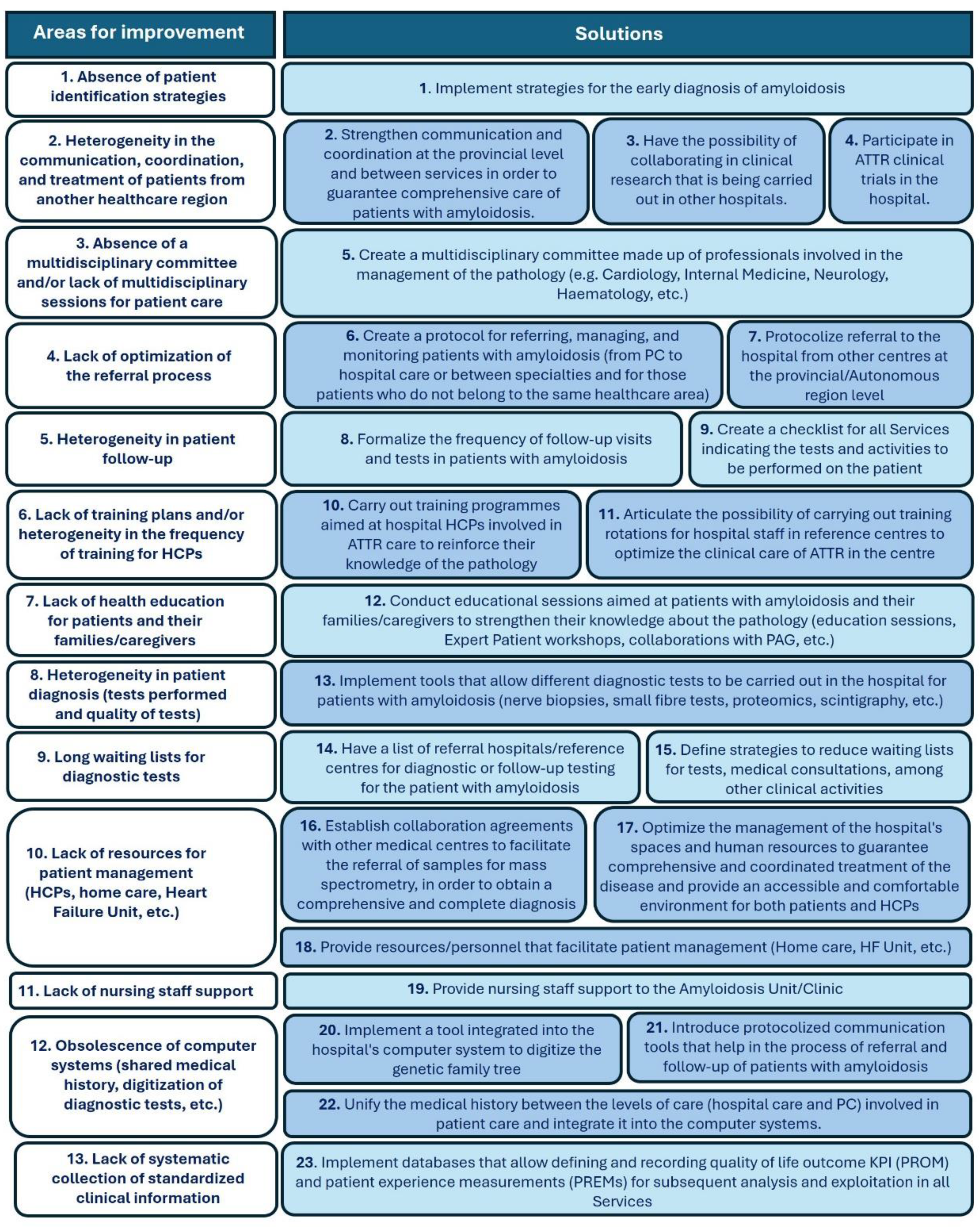
Figure 3.
Identified areas for improvement and validated solutions. (A) The top ten areas for improvement are prioritized based on their impact or actionability. The top 5 are highlighted in blue. (B) Top ten recommended solutions for transforming Spanish ATTR care, prioritized according to their impact and speed of implementation. The top 5 are highlighted in red colour. ATTR, transthyretin amyloidosis; HCPs, healthcare professionals; PC, primary care.
Figure 3.
Identified areas for improvement and validated solutions. (A) The top ten areas for improvement are prioritized based on their impact or actionability. The top 5 are highlighted in blue. (B) Top ten recommended solutions for transforming Spanish ATTR care, prioritized according to their impact and speed of implementation. The top 5 are highlighted in red colour. ATTR, transthyretin amyloidosis; HCPs, healthcare professionals; PC, primary care.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



