
Submitted:
28 January 2026
Posted:
29 January 2026
You are already at the latest version
Abstract
Eight H-bonded complexes of arsenic acid with nitrogen bases (diethylamine, 4-methoxypyridine, pyridine-2,6-diamine, 2,4,6-trimethylpyridine, N1,N1,N2,N2-tetraethylethane-1,2-diamine, 1,3,5-triazine-2,4,6-triamine, pyridin-4-ol and 4-methoxyaniline) were studied in the solid state by single crystal X-ray diffraction technique and DFT calculations. In all cases quite short (≤ 2.65 Å) OHO bonds were found in the self-assembled supramolecular infinite chains or ribbons of dihydrogen arsenates, constituting a repertoire of five different H-bonding patterns (motifs). The electron localization function maps revealed the spots of the nucleophilic sites on oxy-gen atoms that determine the preferable directions for H-bonding of H2AsO4– anions observed in the crystal packing. Analysis of the electrostatic potential maps for isolated species has demonstrated that upon H-bonding between H2AsO4– anions and proto-nated nitrogen bases, NH+···–OAsO(OH)2, the redistribution of electron density within the anion provides otherwise virtually non-existent electrophilic sites on hydrogen atoms, which balances the Coulomb repulsion and allows for the anion···anion pairing within the crystal. The topological analysis of calculated electron density after relaxation of the hydrogen atoms’ positions was used to classify the OHO bonds as moderately strong ones (with an interaction energy up to 65 kJ/mol) and revealed a high degree of ionicity of molecular moieties within zwitterions (with an absolute charge up to 0.87 e). For the strongest OHO and NHO bonds the noticeable covalent character was shown by using the crystal orbital Hamiltonian population analysis.
Keywords:
proton conduction
; anion self-assembly
1. Introduction
In recent years, inorganic acidic anions and organic–inorganic hybrids have attracted increasing attention for their role in functional materials, particularly for applications related to proton conductivity and, in the presence of non-centrosymmetric crystal structures, nonlinear optical properties [1]. In this context, it is now well recognized that efficient proton conduction requires well-organized hydrogen-bond networks capable of facilitating proton transfer via delocalization and hopping mechanisms along supramolecular chains [2,3,4,5,6,7]. A particularly interesting aspect of these systems is the hydrogen-bond-mediated anion···anion self-assembly, an apparently counterintuitive phenomenon as it involves overcoming the Coulombic repulsion between negatively charged species. This behavior has been rationalized in terms of anti-electrostatic hydrogen bonds (AEHB), which enable the formation of dimers, infinite chains, or extended anionic networks. Depending on the dimensionality of these supramolecular architectures, hydrogen-bond networks can be classified as 0D (dimers or trimers), 1D, or 2D, according to the extent of the anionic assembly [8,9]. Hydrogen-bond-driven anion···anion self-assembly is well documented for systems containing anions such as HSO₄⁻, H₂PO₄⁻, and HCO₃⁻ [10,11,12]. More recent studies have also shown that alternative interactions, such as σ-hole interactions, can contribute to the formation of anion···anion supramolecular adducts, occasionally giving rise to hybrid motifs in which multiple types of interactions cooperate in stabilizing the crystal structure [13]. Structural data reported in the Cambridge Structural Database indicate that such phenomena are not rare and can lead to infinite supramolecular chains or anionic dimers stabilized by hydrogen-bond networks [14,15]. In this context, dihydrogen arsenate anions (H₂AsO₄⁻) represent a system of great interest but remain relatively less explored compared to their sulfate and selenate analogues. Several literature examples show that H₂AsO₄⁻ anions can self-assemble into extended supramolecular structures through AsO–H···OAs hydrogen bonds in the presence of nitrogen-containing organic cations, both aromatic and aliphatic. These architectures, often featuring non-centrosymmetric infinite chains, appear particularly promising as candidates for high proton-conductivity materials [16,17,18,19,20,21]. Moreover, extended hydrogen-bond networks based on arsenate anion···anion assembly have also been linked to proton delocalization phenomena relevant for photonic applications [22,23]. Proton conductivity in these materials is strongly influenced by several factors, including hydrogen-bond strength, cooperative coupling between adjacent bonds, and the degree of proton delocalization within O–H···O bonds [24]. A thorough understanding of these aspects is essential for the rational design and functional optimization of materials based on H₂AsO₄⁻···H₂AsO₄⁻ supramolecular synthons. In this work, we present a systematic study of hydrogen-bond-mediated anion···anion motifs in the supramolecular assemblies of dihydrogen arsenates. Eight salts obtained from arsenic acid with various nitrogen bases (Scheme 1) were investigated in the solid state using single-crystal X-ray diffraction and density functional theory (DFT) calculations. Structural analysis revealed the presence of particularly short O–H···O bonds, organized into infinite chains or supramolecular ribbons, defining distinct hydrogen-bond motifs. The integration of crystallographic data with theoretical analyses of electron density, electrostatic potential, and bonding interactions allows elucidation of the electronic and structural factors underlying anion···anion self-assembly in dihydrogen arsenates, as well as the key role of proton delocalization in stabilizing these architectures.
2. Materials and Methods
2.1. Materials
All compounds were purchased from commercial suppliers (Sigma-Aldrich, TCI America, and Merck) and used without further purification. The arsenic acid (H₃AsO₄) solution was prepared by dissolving 1.150 g of arsenic pentoxide (As₂O₅) in 100 mL of distilled water, followed by reflux at 100 °C for 5 weeks [25]. The concentration of the resulting acid solution was determined by titration with NaOH using a pH meter and found to be 0.1 M, consistent with the quantitative hydrolysis of As₂O₅ to H₃AsO₄.
Crystal sample preparation: All salts were prepared according to the following procedure. Separate aqueous solutions (0.1 mmol) containing equimolar amounts of arsenic acid and the corresponding monoprotic base were mixed under constant stirring. The solutions were then left to slowly evaporate at room temperature in clear 4 mL borosilicate vials. Single crystals suitable for X-ray diffraction were obtained within two weeks.
2.2. Methods
2.2.1. Characterization
X-ray diffraction analysis: Data collections were performed at 300K or 100 K using an XtaLAB Synergy diffractometer equipped with a HyPix detector. Unit cell refinement and data reduction were performed using CrysAlisPro (version 1.171.41.98a) software [26]. Structures were solved by direct methods using SHELXT [27] program and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for the non-hydrogen atoms using the SHELXL [28] program incorporated in the Olex2 (version 1.5) software [29]; hydrogen atoms were placed in idealized positions and firstly refined using a riding model, and subsequently additionally refined as described in the next subsection. Absorption correction was performed based on the multiscan procedure. X-ray structures were visualized using capped sticks representation built in the Mercury (version 2022.3.0) software [30]. CCDC deposition numbers: 2521265, 2521276, 2521277, 2521278, 2521279, 2521280, 2521281, 2521284 contain the validated [31] supplementary crystallographic data for this paper (see ESI). These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Solid-state calculations. Electronic calculations of 1a-8a were performed using the CRYSTAL17 (version 1.0.2)[32] software. Constrained geometry optimizations were done at the B3LYP-D*/POB-TZVP [33,34,35,36,37,38] level of theory. The chosen combination of the DFT functional and the atomic basis set has been shown to reproduce the H-bond geometric features of the supramolecular chains of hydrogen selenates (HSeO4–) with sufficient accuracy, see our recent work [14]. Accuracy of calculation of the Coulomb and Hartree–Fock exchange series was controlled by a set of overlap tolerances, which were taken to be (10–9, 10–9, 10–9, 10–9, 10–18).[39] The maximum order of multipole expansion [41,4][] in the long-range zone for the electron-electron Coulomb interaction was set to 6. Calculation of the DFT exchange-correlation contribution over the unit cell volume was done with the XLGRID DFT grid specification; [39,40,41,42] tolerances for the DFT density and the DFT grid weight were taken to be 10–10 and 10–18, respectively. Integration in the Brillouin zone was performed using the Monkhorst–Pack scheme [43] for an anisotropic 8×4×4 (for 6a), 6×8×3 (for 4a), 4×8×2 (for 3a), 8×6×6 (for 2a and 8a), 4×6×6 (for 5a) and 8×5×8 (for 7a) k-point grids, and for an isotropic 6×6×6 k-point grid for 1a. DIIS [44] tolerance on energy controlling the SCF convergence for geometry optimizations and single-point calculations was set to 10–8 and 10–9 Hartree, respectively.
Single-crystal structure parameters of the above crystals from the X-ray diffraction data at 300 and 100 K were used as the starting points for the DFT calculations. While keeping fixed the unit cell parameters and heavy atoms’ positions, all hydrogen atoms’ positions were optimized with tight convergence criteria (maximum/RMS forces and displacements smaller than 0.000075/0.000050 a.u. and 0.000225/0.000150 a.u., respectively) and symmetry constraint imposed by the corresponding space group: triclinic P-1 for 2a, 6a, and 8a; monoclinic P21/n for 4a and 7a, P21/m for 1a, and P21/c for 3a and 5a. For 1a featuring dynamically disordered hydrogen atoms of methyl groups of the protonated 2,4,6-trimethylpyridine molecules, the structures of eight different ordered isomorphs were subjected to the above constrained geometry optimization; afterwards, only one of the optimized structures (see Figures S27 and S28) was selected for further calculations which are described onwards. Analytical energy gradients [45] within a quasi-Newton approach combined with the BFGS [46] algorithm for Hessian updating were used. Consistency between the optimized geometries and the aforementioned overlap tolerances for integrals’ evaluation was ensured by the FINALRUN option with the value of 4 [39].
Topological analysis [47,48] of the periodic electron density (ED) was performed using quantum theory of atoms in molecules (QTAIM) by means of the TOPOND14 module [49] implemented in the CRYSTAL17 code [50]. AIM atomic charges were evaluated by integration of the three-dimensional density distribution over the atomic basins, whose enclosing zero-flux surfaces were determined manually to the nearest preliminary retrieved bond critical point for a given nucleus using an eigenvector following approach with the tolerance value of 0.002 a.u.[51,52,53].
Crystal orbital Hamiltonian population projected onto local atomic orbitals pertaining to two selected groups of H and A (A = O or N) atoms, involved in the H···A hydrogen-bonding, was calculated over the energy range spanned from the first valence band, occupied by the non-core electrons, up to the highest occupied valence band, i.e. up to the Fermi energy level. The Hamiltonian eigenvectors were preliminary recalculated using a fine isotropic 12×12×12 k-point grid.
Molecular calculations. Electronic calculations of the molecular adducts analyzed in this work were performed using the Gaussian16 (version A.01) software [54]. Single-point calculations with standard tolerance on energy controlling the SCF convergence were performed at the PW6B95-D3(BJ)/def2-TZVPD level of theory [55,56,57,58]. The basis set superposition error for the calculation of complexation (or interaction) energies was corrected using the counterpoise method [59]. The ED, molecular ESP [60], and electron localization function (ELF [61]) surfaces were calculated using the MultiWFN (version 3.8) software [62,63]. The ELF isosurfaces were mapped at 0.87 isovalue.
3. Results and Discussion
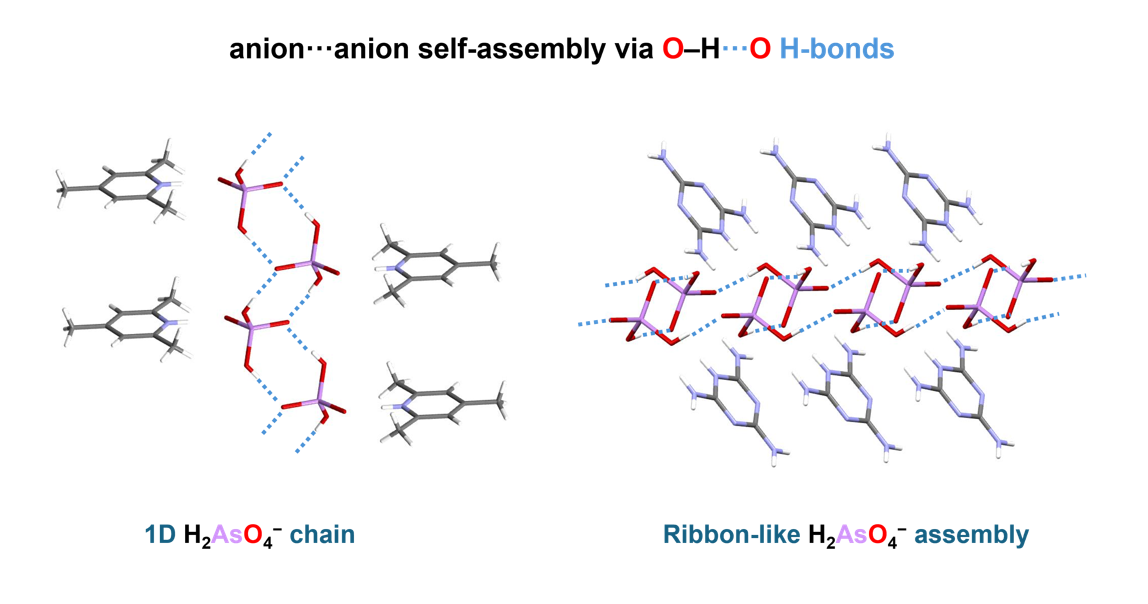
In this work, eight crystal structures are reported, all containing a single unique zwitterion consisting of the H₂AsO₄⁻ anion hydrogen-bonded to the corresponding protonated nitrogen base within the primitive unit cell. The crystals belong to triclinic (2a, 6a, 8a) or monoclinic (1a, 3a, 4a, 5a, 7a) lattices. The presence of only one independent zwitterion in the unit cell accounts for the observation of only two—or, in some cases, only one—distinct O–H···O hydrogen bonds between the anions, depending on the relative positions of the cations. From a structural viewpoint, compounds 1a, 2a, 4a, 7a, and 8a adopt anion···anion assemblies in the form of infinite linear chains, whereas 3a and 6a exhibit ribbon-like anion assemblies. Compound 5a represents a unique case, displaying an anion···anion self-assembly organized into a two-dimensional (2D) network.
2,4,6-Trimethylpyridinium dihydrogen arsenate (1a), crystallizes in the P2₁/m space group. In this compound, the average O···O distance within the AsO–H···O hydrogen bond is 2.622 Å, a remarkably short value indicative of a strong hydrogen bond, which enables the H₂AsO₄⁻ anions to self-assemble while overcoming electrostatic anion–anion repulsion. Shorter O···O distances are observed in compounds 2a (already known in another polymorphic form [67]), 4a, 7a, and 8a, as summarized in Table 1. This consistent trend toward short O···O separations suggests the presence of a robust network of H₂AsO₄⁻ anions stabilized by so-called “anti-electrostatic” hydrogen bonding.
The crystal structures are stabilized by electrostatic attraction between oppositely charged cations and anions, as well as by a variety of non-covalent interactions spanning a wide range of lengths and strengths. These include: (i) major O–H···O and N–H···O hydrogen bonds (ii) π-stacking interactions between N-heterocycles, observed in compounds 1a-6a; and (iii) numerous additional minor C–H···O contacts. Among the studied crystals, 1a, 2a, and 4a clearly show that aromatic cations engage in π–π stacking interactions, leading to the formation of spatially segregated cationic and anionic regions. This arrangement facilitates anion···anion pairing through hydrogen bonding, as directly observed in their crystal structures. In contrast, crystals containing 4-hydroxypyridinium (3a) melaminium (6a) and 4-methoxy anilinium (5a) cations display ribbon-like anion···anion assemblies. In these cases, H₂AsO₄⁻ dimers propagate along the crystallographic b axis, with O···O distances ranging from 2.554 to 2.602 Å. Close contacts between anions and cations, characterized by N···O distances of 2.61–2.78 Å, contribute to the redistribution of negative charge within the anion, thereby enabling the formation of short inter-anionic O–H···O hydrogen bonds (2.56–2.64 Å). All major hydrogen bonds involving H₂AsO₄⁻ anions are fairly linear, with angles in the ranges 169° < ∠(OHO) < 179° and 131° < ∠(NHO) < 179° in computationally optimized structures. The crystal packings of the studied compounds are shown in Figures S1–S8 in the Electronic Supporting Information (ESI). The association motifs of the amphoteric dihydrogen arsenate anions, which can simultaneously act as hydrogen-bond donors and acceptors, are more readily discussed in conjunction with the results of density functional theory (DFT) calculations performed on the same crystals. Acknowledging the intrinsic difficulty of X-ray diffraction techniques in accurately locating hydrogen atoms, the hydrogen positions were selectively relaxed while keeping the remaining unit-cell parameters fixed (see Table S1 and the resulting structures in Figure 1, Figure 2, Figure 3 and Figure 4). Analysis of these optimized structures allows five distinct motifs of “anti-electrostatic” [68] hydrogen bonding to be identified: tightly linked chains of cyclic dimers (motifs 1 and 2 in Figure 1 and Figure 2), loosely linked chains and slabs of cyclic dimers (motifs 3 and 5 in Figure 3 and Figure 4), and a tightly linked chain of cyclic trimers (motif 4 in Figure 3). The non-equivalent O–H···O hydrogen bonds between anions are highlighted in different colors in Figure 1, Figure 2, Figure 3 and Figure 4. The spatial arrangement of the hydrogen-bond networks in the studied crystals is further constrained by the anisotropy of the outer electronic shell of the proton-accepting oxygen atoms, which dictates the preferred directions for the formation of O–H···O and N–H···O bonds involving the H₂AsO₄⁻ anions. This anisotropy originates from lone-pair domains, which give rise to toroidal and bean-shaped isosurfaces of the electron localization function (ELF) in the As–Oδ– and As–O–H groups, respectively (see Figures S9–S16). The ELF isosurfaces are very similar for the isolated anion and for the corresponding zwitterion considered in the X-ray-derived geometry.
The 3D ESP maps represented via ED isosurfaces color-coded with the ESP values are shown in Figure 5 for isolated H2AsO4– anions and their complexes with counter cations in crystalline geometry. Note that the color-coded ranges of ESP values are not symmetric and different in each case, being selected for maximum contrast between various regions. The key difference between isolated anions and zwitterions is in the electrophilicity of H2AsO4– fragment. The entire surface is negative for the isolated anions, but polarization effects in the zwitterions creates electrophilic sites on the hydrogen atoms of the As–O–H groups: the negative extremum of ESP around H atoms changes to the positive one (see the values given in Figure 5). Thus, the ESP maps demonstrate that redistribution of the negative charge of H2AsO4– anion by adjacent cation is crucial to drive the self-assembly of the anions in all studied crystals. Indeed, the complexation between isolated H2AsO4– anions in crystalline geometry is energetically unfavorable: significant electrostatic repulsion hampers self-association, as evidenced by additional calculations results of which are shown in Figure S17.
Within the QTAIM methodology, the nature of an H-bond can be described by the topological properties of the electron density (ED), ρ, and various functions derived from it at the bond critical point (BCP) between the bridging proton and the proton-accepting atom.[69,70,71,72,73,74,75,76,77] For example, the signs of ▽2ρbcp (i.e. the Laplacian of the ED) and Hbcp = Gbcp + Vbcp (i.e. the total electronic energy density as a sum of the kinetic and potential energy densities Gbcp and Vbcp at the BCP) are used to differentiate between three types of interactions: non-covalent (region I, ▽2ρbcp > 0, Hbcp > 0), covalent (region II, ▽2ρbcp < 0, Hbcp < 0), and partially covalent (region III, ▽2ρbcp > 0, Hbcp < 0). Since V < 0 and G > 0, the sign of Hbcp reveals whether Vbcp or Gbcp dominate in the bonding region: a dominance of Vbcp (Hbcp < 0) indicates that accumulation of electronic charge in the internuclear region is stabilizing; conversely, if Gbcp > Vbcp, (Hbcp > 0), the internuclear charge accumulation is destabilizing, which is typical for a non-covalent interaction. The boundaries between regions I, II, and III are defined by Hbcp = 0 (i.e. |Vbcp|/Gbcp = 1) and by ▽2ρbcp = 0 (i.e. |Vbcp|/Gbcp = 2 from the virial equation). Within region I, the higher is the bond degree index (BDI) Hbcp/ρbcp, the weaker the interaction. Similarly, in regions II and III, the higher is the BDI, the higher the covalent character of the interaction.
For all studied structures, the calculated values of the above outlined parameters at the intermolecular BCPs of the OHO bonds between H2AsO4– anions are listed in Table 2. These bonds, being similar to each other, exhibit quite large ρbcp values of approximately 0.06 a.u. but positive ▽2ρbcp values indicating a locally charge depleted domain, which places these bonds in region III at the border with region I, characterizing them as non-covalent interactions with a small degree of covalency.[78] This finding is in agreement with the relative values of other H-bond descriptors, meeting the inequalities 1 < |Vbcp|/Gbcp < 2, Gbcp/ρbcp < 1 (the kinetic energy per electron, in a.u.), and Hbcp/ρbcp < 0. The hydrogen bond energies, EOHO, estimated using the equation EOHO = 0.429 Gbcp,[79,80] fall in the range 51 < EOHO < 65 kJ/mol with an average value of 56 kJ/mol. Noteworthy, the full set of QTAIM parameters, the estimated H-bond energies, and the O...O interatomic distances for the interacting H2AsO4– anions are similar to those for the recently studied interacting HSeO4– anions [24], for which we have predicted the thermally accessible vibrational motion of the bridging protons from one heavy atom to another in double-well potentials. For completeness, in ESI we provide QTAIM parameters at the intermolecular BCPs of the NHO bonds between cations and anions (as well as analogous OHO bond in case of 3a), and also NHN bonds between cations in 6a, see Figures S18–S25 and Table S2. In a nutshell, the ρ, ▽2ρ, V, G, |V|/G, H and H/ρ values indicate that appertaining H-bonds in studied crystals can be classified as follows: (i) OHO, moderately strong bond (72 kJ/mol); (ii) NHO, from weak to moderately strong bonds (9 < ENHO < 66 kJ/mol); (iii) NHN, weak bonds (21 < ENHN < 24 kJ/mol). Despite the short N...O distances between cations and anions, the strong proton displacements towards nitrogen atoms (almost complete proton transfers) lead to a large degree of charge separation: the absolute values of AIM charges of the interacting molecular moieties are 0.81–0.87 e (see Table S3). Furthermore, an AIM charge of the bridging hydrogen atom might be considered as an additional descriptor of the H-bond strength that performs well over a sufficiently wide sampling, which is shown for NHO bonds in Figure S26a.
It is well-known that the electronic density of states (DOS) analysis provides valuable insight into the contribution of particular local atomic orbitals (AOs) to the overall electronic structure of solids over a specified energy range [81,82,83,84,85]. Projected crystal orbital Hamiltonian population (COHP) analysis is an extension of the DOS analysis aimed to characterize pairwise interactions between two atoms (two discrete groups of AOs) in solids [86,87,88,89]. By weighing DOS by the Hamiltonian matrix, COHP partitions the band energies into bonding (stabilizing) and antibonding (destabilizing) regions, thus providing information regarding the partial covalent character of hydrogen···acceptor interactions in molecular solids [90,91]. In this context, COHP analysis has proven itself to be complementary to the QTAIM analysis. Both approaches, being fundamentally different, share the same concept of gauging the nature and strength of H-bonds as a tangible quantity in a solid directly from its underlying electronic structure. An estimate of the covalent contribution to H-bonding interaction can be made by integrating stabilizing (negative) and destabilizing (positive) regions of the projected COHP values up to the highest occupied band, i.e. the Fermi energy level (εF), giving so-called IpCOHP values. In Figure 6, –IpCOHP values are plotted against the corresponding H-bond lengths R(AH···O) and ρbcp densities for pairs of atoms involved in the AH···O interactions (A = O or N) for all studied structures. One obtains a variety of stabilizing interaction energies in the range 0–260 kJ/mol. On the one hand, the larger values in this set correlate reasonably well with R(AH···O) and ρbcp values, similarly to what was previously observed for short OHO bonds, (see Figure 2 and Figure 3 in Ref. 90). It is also appreciable that weak NHN bonds in 6a, although there are only two unique ones of them, follow the same trend (green symbols in Figure 6). On the other hand, there is a group of −IpCOHP values that tightly cluster around zero (circled data points in Figure 6) regardless the OH···O (NH···O) interatomic distance or the electronic charge concentration at the BCP. For OHO bonds (red symbols in Figure 6) this means that stabilizing and destabilizing pCOHP regions effectively cancel each other over the entire valence region, which indicates that the H-bonding anion–anion interactions are essentially closed-shell electrostatic in nature. An exception is the following three OHO bonds with significant covalent contribution: the strongest among motifs 1–5 OHO bonds linking H2AsO4– dimers in 6a (highlighted in green in Figure 3), the strongest and unique among studied structures OHO bonds between protonated pyridinium-4-ol cations and H2AsO4– anions (see Figure S20) in 3a, and moderately strong OHO bonds in the tightly linked chains of H2AsO4– cyclic trimers in the same structure (highlighted in pale orange in Figure 3). In other words, it seems that the covalent character of an anion···anion interaction becomes detectable by the –IpCOHP values if either the bond is very strong (short) or the negative charge of the anion is strongly shifted towards the tightly bound cation. In case of 3a such charge redistribution is facilitated by the fact that both OHO bonds in question (anion···anion and anion···cation ones) share the same acceptor atom. For NHO bonds (blue symbols in Figure 6), the −IpCOHP values are close to zero for three data points (circled in Figure 6), which correspond to H-bonds that involve one H atom of the NH2+ group in 7a and two H atoms of the NH3+ group in 5a (see Figures S24 and S22). Now it is not clear which feature of these interactions leads to such small −IpCOHP values. Comparison of the OH···O, NH···O, and NH···N interaction energies obtained by QTAIM and COHP analyses reveals that an increase of the covalent contribution to the interaction energy upon the bond strengthening is not always fulfilled.
4. Conclusions
Eight crystalline salts of arsenic acid with nitrogen-containing bases were investigated to analyze hydrogen-bond-mediated anion···anion self-assembly in dihydrogen arsenates. Single-crystal X-ray diffraction shows that, in all structures, H₂AsO₄⁻ anions are involved in short and nearly linear O–H···O hydrogen bonds (O···O ≤ 2.65 Å), leading to the formation of extended supramolecular architectures. Depending on the crystal packing, these interactions generate infinite chains, ribbon-like assemblies, or a two-dimensional anionic network. On this basis, five recurrent hydrogen-bonding motifs were identified. Density functional theory calculations were used to rationalize the experimental observations. Electron localization function analysis indicates that the anisotropic distribution of lone-pair electron density on oxygen atoms governs the preferred directions of hydrogen bonding. Electrostatic potential maps show that interaction with protonated nitrogen bases induces a redistribution of electron density within the H₂AsO₄⁻ anion, creating electrophilic regions on the As–O–H hydrogen atoms that enable anion···anion association despite Coulombic repulsion. Topological analysis of the electron density classifies the inter-anionic O–H···O hydrogen bonds as moderately strong interactions, with estimated energies in the range of 50–65 kJ/mol and a limited covalent contribution. The zwitterionic building blocks display a high degree of ionicity, with AIM charges approaching ±0.87 e. Complementary COHP analysis indicates that most anion···anion hydrogen bonds are predominantly electrostatic, while a measurable covalent component is observed only for the shortest O–H···O contacts or in cases of pronounced cation-induced charge redistribution. Overall, this study provides a systematic structural and electronic description of hydrogen-bond-driven anion···anion self-assembly in dihydrogen arsenates. The identified motifs and bonding characteristics contribute to the understanding of anti-electrostatic hydrogen bonding in inorganic acidic anions and may assist in the analysis of related hydrogen-bonded arsenate systems.
Data Availability Statement
The data supporting this article have been included as part of the Supplementary Information. Crystallographic data of compounds described in this paper have been deposited at the CCDC under Deposition Numbers 2521278 (for 1a), 2521277 (for 2a), 2521281 (for 3a), 2521276 (for 4a), 2521284 (for 5a), 2521280 (for 6a), 2521265 (for 7a) and 2521279 (for 8a) can be obtained free of charge from https://www.ccdc.cam.ac.uk/
Conflicts of Interest
There is no conflict of interest.
Appendix B
All appendix sections must be cited in the main text. In the appendices, Figures, Tables, etc. should be labeled starting with “A”—e.g., Figure A1, Figure A2, etc.
References
- Stejskal, J.; Havlíček, D.; Císařová, I.; Matulková, I. Vibrational Spectroscopic and X-Ray Single Crystal Diffraction Investigation of Tetra-n-Alkylammonium Hydrogen Selenates. J. Chem. Crystallogr. 2017, 47, 59–68. [CrossRef]
- Belushkin, A. V.; Carlile C. J.; Shuvalov, L. A. A quasielastic neutron scattering study of protonic transport in hydrogen-bonded alkali metal hydrogen sulphates and selenates. Ferroelectrics 1995, 167, 21–31. [CrossRef]
- Ikeda, S.; Yamada, Y. Phase transition in hydrogen bonded ferroelectric compounds—Quantum fluctuations versus thermal fluctuations. Phys. B Condens. Matter 1995, 213–214, 652–657. [CrossRef]
- Pavlenko, N. I. Protonic conductivity at the superionic phase transitions in the M3H(XO4)2 crystal group. J. Phys.: Condens. Matter 1999, 11, 5099–5110. [CrossRef]
- Pavlenko, N.; Pietraszko, A.; Pawlowski, A.; Polomska, M.; Stasyuk I. V.; Hilczer, B. Hydrogen transport in superionic system Rb3H(SeO4)2: a revised cooperative migration mechanism. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 1–10. [CrossRef]
- Matsui, H.; Shimatani, K.; Ikemoto, Y.; Sasaki T.; Matsuo, Y. Phonon-assisted proton tunneling in the hydrogen-bonded dimeric selenates of Cs3H(SeO4)2. J. Chem. Phys. 2020, 152, 154502. [CrossRef]
- Matsui, H.; Fukuda, K.; Takano, S.; Ikemoto, Y.; Sasaki T.; Matsuo, Y. Mechanisms of the antiferro-electric ordering in superprotonic conductors Cs3H(SeO4)2 and Cs3D(SeO4)2. J. Chem. Phys. 2022, 156, 204504. [CrossRef]
- Rajbanshi, A.; Wan, S.; Custelcean, R. Dihydrogen Phosphate Clusters: Trapping H2PO4 − Tetramers and Hexamers in Urea-Functionalized Molecular Crystals. Cryst Growth Des 2013, 13, 2233–2237. doi:10.1021/cg400336q.
- Zhao, W.; Flood, A.H.; White, N.G. Recognition and Applications of Anion–Anion Dimers Based on Anti-Electrostatic Hydrogen Bonds (AEHBs). Chem Soc Rev 2020, 49, 7893–7906. doi:10.1039/D0CS00486C.
- Pizzi, A.; Dhaka, A.; Beccaria, R.; Resnati, G. Anion⋅⋅⋅anion self-assembly under the control of σ- and π-hole bonds. Chem. Soc. Rev. 2024, 53, 6654. [CrossRef]
- Beccaria, R.; Dhaka, A.; Calabrese, M.; Pizzi, A.; Frontera, A.; Resnati, G. Chalcogen and Hydrogen Bond Team up in Driving Anion⋅⋅⋅Anion Self-Assembly. Chem. Eur. J. 2024, 30, e202303641. [CrossRef]
- Drozd M.; Baran, J. Polarized IR-microscope spectra of guanidinium hydrogenselenate single crystal. Spectrochim. Acta Part A 2005, 61, 2953–2965. [CrossRef]
- Lorenc, J.; Bryndal, I.; Marchewka, M.; Kucharska, E.; Lis. T.; Hanuza, J. Crystal and molecular structure of 2-amino-5-chloropyridinium hydrogen selenate—its IR and Raman spectra, DFT calculations and physicochemical properties. J. Raman Spectrosc. 2008, 39, 863–872. [CrossRef]
- Akriche S.; Rzaigui, M. 2-Amino-3-nitropyridinium hydrogen selenate. Acta Crystallogr., Sect. E. 2009, 65, o1648. [CrossRef]
- Maalej, W.; Ben Rached, A.; Mhiri, T.; Daoud, A.; Zouari, N.; Elaoud, Z. Vibrational study, phase transitions and electrical properties of 4-benzylpyridinium monohydrogenselenate. J. Phys. Chem. Solids 2016, 96–97, 92–99. [CrossRef]
- Janczak, J.; Perpétuo, G. J. Hydrogen-Bonded Networks in Crystals of 1-(Diaminomethylene)Thiouron-1-Ium Perchlorate, Hydrogen Sulfate, Dihydrogen Phosphate and Dihydrogen Arsenate. Acta Crystallogr., Sect. C 2008, 64, o330–o334. doi:10.1107/S0108270108013504.
- Chtioui, A.; Benhamada, L.; Jouini, A. Crystal Structure, Thermal Analysis and IR Spectroscopic Investigation of (C6H9N2)H2XO4 (X=As, P). Mater. Res. Bull. 2005, 40, 2243–2255. doi:10.1016/j.materresbull.2005.06.006.
- Oueslati, A.; Rayes, A.; Ben Nasr, C.; Lefebvre, F. Synthesis and Characterization of 2-Amino-3-Methylpyridinium Dihy-drogenomonoarsenate. Mater. Res. Bull. 2005, 40, 1680–1689. doi:10.1016/j.materresbull.2005.05.029.
- Wilkinson, H.S.; Harrison, W.T.A. Creatininium Dihydrogenarsenate. Acta Crystallogr., Sect. E 2005, 61, 1228-1230. doi:10.1107/S1600536805016144.
- Wilkinson, H.S.; Harrison, W.T.A. Guanidinium Dihydrogenarsenate. Acta Crystallogr., Sect. E 2005, 61, 2023–2025. doi:10.1107/S1600536805028825.
- Kanagathara, N.; MaryAnjalin, F.; Ragavendran, V.; Dhanasekaran, D.; Usha, R.; Rao, R.G.S.; Marchewka, M.K. Experi-mental and Theoretical (DFT) Investigation of Crystallographic, Spectroscopic and Hirshfeld Surface Analysis of Anilin-ium Arsenate. J. Mol. Struct. 2021, 1223, 128965. doi:10.1016/j.molstruc.2020.128965.
- Anbalagan, G.; Marchewka, M.K.; Pawlus, K.; Kanagathara, N. Crystal Structure and Vibrational Spectra of Melaminium Arsenate. J. Mol. Struct. 2015, 1079, 407–413. doi:10.1016/j.molstruc.2014.09.006.
- Roshini, S.R.A.; Kanagathara, N.; Marchewka, M.K.; Janczak, J.; Jayalakshmi, D. Growth, Structural, Optical, Morpholog-ical, Thermal, Laser Damage Threshold and Hardness Properties of Organic-Inorganic Crystal: L-Argininium (Bis)Dihydrogenarsenate. J. Mol. Struct. 2026, 1352, 144499. doi:10.1016/j.molstruc.2025.144499.
- Beccaria, R.; Pizzi, A.; Chakalov, E.; Resnati, G.; Tolstoy, P. Proton Delocalization in Short Hydrogen Bonds Assembling HSeO4− Anions into Supramolecular Adducts. Phys. Chem. Chem. Phys. 2025, 27, 13601–13617. doi:10.1039/D5CP01211B.
- Shikina, N. D.; Zotov, A. B.; Tagirov, B. R. Influence of Pressure in the 0.1–100 MPa Interval on the First Dissociation Constant of Arsenous Acid in Water Solutions at 298.15 K. Russ. J. Phys. Chem. A 2010, 84, 1076–1078. [CrossRef]
- CrysAlisPro 1.171.43.136a. Rigaku Oxford Diffraction 2024.
- Sheldrick, G. M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [CrossRef]
- Sheldrick, G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [CrossRef]
- Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [CrossRef]
- MacRae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [CrossRef]
- Spek, A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C. M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; Kirtman, B. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, 1–36. [CrossRef]
- Becke, A. D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [CrossRef]
- Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [CrossRef]
- Vilela Oliveira, D.; Laun, J.; Peintinger, M. F.; Bredow, T. BSSE-Correction Scheme for Consistent Gaussian Basis Sets of Double- and Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2019, 40, 2364–2376. [CrossRef]
- CRYSTAL - Basis Sets Library. https://www.crystal.unito.it/basis_sets.html.
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [CrossRef]
- Civalleri, B.; Zicovich-Wilson, C. M.; Valenzano, L.; Ugliengo, P. B3LYP Augmented with an Empirical Dispersion Term (B3LYP-D*) as Applied to Molecular Crystals. CrystEngComm 2008, 10, 405–410. [CrossRef]
- Dovesi, R.; Saunders, V. R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C. M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N. M.; Bush, I. J.; D’Arco, P.; Llunell, M.; Causà, M.; Noël, Y.; Maschio, L.; Erba, A.; Rerat, M.; Casassa, S. CRYSTAL17 User’s Manual, University of Torino, Torino, 2017; 2018.
- Ewald, P. P. Die Berechnung Optischer Und Elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [CrossRef]
- Dovesi, R.; Pisani, C.; Roetti, C. Treatment of Coulomb Interactions in Hartree-Fock Calculations of Periodic Systems. Phys. Rev. B Condens. Matter Mater. Phys. 1983, 28, 5781–5792. [CrossRef]
- Towler, M. D.; Zupan, A.; Causà, M. Density Functional Theory in Periodic Systems Using Local Gaussian Basis Sets. Comput. Phys. Commun. 1996, 98, 181–205. [CrossRef]
- Monkhorst, H. J.; Pack, J. D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [CrossRef]
- Maschio, L. Direct Inversion of the Iterative Subspace (DIIS) Convergence Accelerator for Crystalline Solids Employing Gaussian Basis Sets. Theor. Chem. Acc. 2018, 137, 1–5. [CrossRef]
- Doll, K.; Saunders, V. R.; Harrison, N. M. Analytical Hartree–Fock Gradients for Periodic Systems. Int. J. Quantum Chem. 2001, 82, 1–13. [CrossRef]
- Schlegel, H. B. Geometry Optimization. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 790–809. [CrossRef]
- Bader, R. F. W. Atoms in Molecules - A Quantum Theory, Vol. 22 of International Series of Monographs in Chemistry, Oxford, UK; Oxford University Press, 1990.
- Bertini, L.; Cargnoni, F.; Gatti, C. Chemical Insight into Electron Density and Wave Functions: Software Developments and Applications to Crystals, Molecular Complexes and Materials Science. Theor. Chem. Acc. 2007, 117, 847–884. [CrossRef]
- Gatti, C.; Casassa, S. TOPOND14 User’s Manual (CNR-ISTM Milano, Milano, 2014); 2017.
- Casassa, S.; Erba, A.; Baima, J.; Orlando, R. Electron Density Analysis of Large (Molecular and Periodic) Systems: A Parallel Implementation. J. Comput. Chem. 2015, 36, 1940–1946. [CrossRef]
- Banerjee, A.; Adams, N.; Simons, J. Search for Stationary Points on Surfaces. J. Phys. Chem. 1985, 89, 52–57. [CrossRef]
- Keith, T. A. PhD Thesis, McMaster University, Hamilton, Ontario, Canada, 1993.
- Popelier, P. L. A. A Robust Algorithm to Locate Automatically All Types of Critical Points in the Charge Density and Its Laplacian. Chem. Phys. Lett. 1994, 228, 160–164. [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr. Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision A.03.
- Zhao, Y.; Truhlar, D. G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [CrossRef]
- Rappoport, D.; Furche, F. Property-Optimized Gaussian Basis Sets for Molecular Response Calculations. J. Chem. Phys. 2010, 133, 134105. [CrossRef]
- Pritchard, B. P.; Altarawy, D.; Didier, B.; Gibson, T. D.; Windus, T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [CrossRef]
- Boys, S. F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [CrossRef]
- Murray, J. S.; Politzer, P. The Electrostatic Potential: An Overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [CrossRef]
- Becke, A. D.; Edgecombe, K. E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [CrossRef]
- Zhang, J.; Lu, T. Efficient Evaluation of Electrostatic Potential with Computerized Optimized Code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [CrossRef]
- Materials Studio 2017 (17.1.0.48). BIOVIA, Dassault Systèmes. San Diego, CA, USA.
- Dennington II, R. D.; Keith, T. A.; Millam, J. M. GaussView 6.0.16. Semichem Inc., Shawnee Mission, KS 2016.
- Origin 2018. OriginLab. Northampton, MA, USA.
- Bouaziz, E; Hassen, C. B.; Chniba-Boudjada, N.; Daouda, A.; Mhiri T.; Boujelbene M. Crystal structure, Hirshfeld surface analysis, vibrational, thermal behavior and UV spectroscopy of (2,6-diaminopyridinium) dihydrogen arsenate. J. Mol. Struct. 2017, 1145, 121e131. [CrossRef]
- White, N. G. Antielectrostatically Hydrogen Bonded Anion Dimers: Counter-Intuitive, Common and Consistent. CrystEngComm 2019, 21, 4855–4858. [CrossRef]
- Bader, R. F. W.; Essén, H. The Characterization of Atomic Interactions. J. Chem. Phys. 1983, 80, 1943–1960. [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281.
- Koch, U.; Popelier, P. L. A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [CrossRef]
- Jenkins, S.; Morrison, I. The Chemical Character of the Intermolecular Bonds of Seven Phases of Ice as Revealed by Ab Initio Calculation of Electron Densities. Chem. Phys. Lett. 2000, 317, 97–102. [CrossRef]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CH··O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(Dimethylamino)-3-Cyclobutene-1,2-Dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [CrossRef]
- Gatti, C. Chemical Bonding in Crystals: New Directions. Z. Krist. 2005, 220, 399–457. [CrossRef]
- Grabowski, S. J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X-H…F-Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [CrossRef]
- Vener, M. V.; Manaev, A. V.; Egorova, A. N.; Tsirelson, V. G. QTAIM Study of Strong H-Bonds with the O−H···A Fragment (A = O, N) in Three-Dimensional Periodical Crystals. J. Phys. Chem. A 2007, 111, 1155–1162. [CrossRef]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [CrossRef]
- Vener, M. V.; Egorova, A. N.; Churakov, A. V.; Tsirelson, V. G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [CrossRef]
- Bandura, A. V.; Kubicki, J. D.; Sofo, J. O. Comparisons of Multilayer H2O Adsorption onto the (110) Surfaces of α-TiO2 and SnO2 as Calculated with Density Functional Theory. J. Phys. Chem. B 2008, 112, 11616–11624. [CrossRef]
- Evarestov, R. A.; Bandura, A. V.; Losev, M. V.; Piskunov, S.; Zhukovskii, Y. F. Titania Nanotubes Modeled from 3- and 6-Layered (1 0 1) Anatase Sheets: Line Group Symmetry and Comparative Ab Initio LCAO Calculations. Physica E Low Dimens. Syst. Nanostruct. 2010, 43, 266–278. [CrossRef]
- Laskowski, R.; Blaha, P. Understanding of 33S NMR Shielding in Inorganic Sulfides and Sulfates. J. Phys. Chem. C 2015, 119, 731–740. [CrossRef]
- Treviño, P.; Garcia-Castro, A. C.; López-Moreno, S.; Bautista-Hernández, A.; Bobocioiu, E.; Reynard, B.; Caracas, R.; Romero, A. H. Anharmonic Contribution to the Stabilization of Mg(OH)2 from First Principles. Phys. Chem. Chem. Phys. 2018, 20, 17799–17808. [CrossRef]
- Evarestov, R. A.; Kuzmin, A. Topological Analysis of Chemical Bonding in the Layered FePSe3 upon Pressure-Induced Phase Transitions. J. Comput. Chem. 2020, 41, 2610–2623. [CrossRef]
- Dronskowski, R.; Peter, E. B. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [CrossRef]
- Deringer, V. L.; Tchougréeff, A. L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [CrossRef]
- Maintz, S.; Deringer, V. L.; Tchougréeff, A. L.; Dronskowski, R. Analytic Projection from Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. [CrossRef]
- Ruggiero, M. T.; Erba, A.; Korter, T. M. Origins of Contrasting Copper Coordination Geometries in Crystalline Copper Sulfate Pentahydrate. Phys. Chem. Chem. Phys. 2015, 17, 31023–31029. [CrossRef]
- Deringer, V. L.; Englert, U.; Dronskowski, R. Covalency of Hydrogen Bonds in Solids Revisited. Chem. Commun. 2014, 50, 11547–11549. [CrossRef]
- Deringer, V. L.; George, J.; Dronskowski, R.; Englert, U. Plane-Wave Density Functional Theory Meets Molecular Crystals: Thermal Ellipsoids and Intermolecular Interactions. Acc. Chem. Res. 2017, 50, 1231–1239. [CrossRef]
Scheme 1.
Scheme 1. Structural formulas of the components in the crystal structures described in this work.
Scheme 1.
Scheme 1. Structural formulas of the components in the crystal structures described in this work.
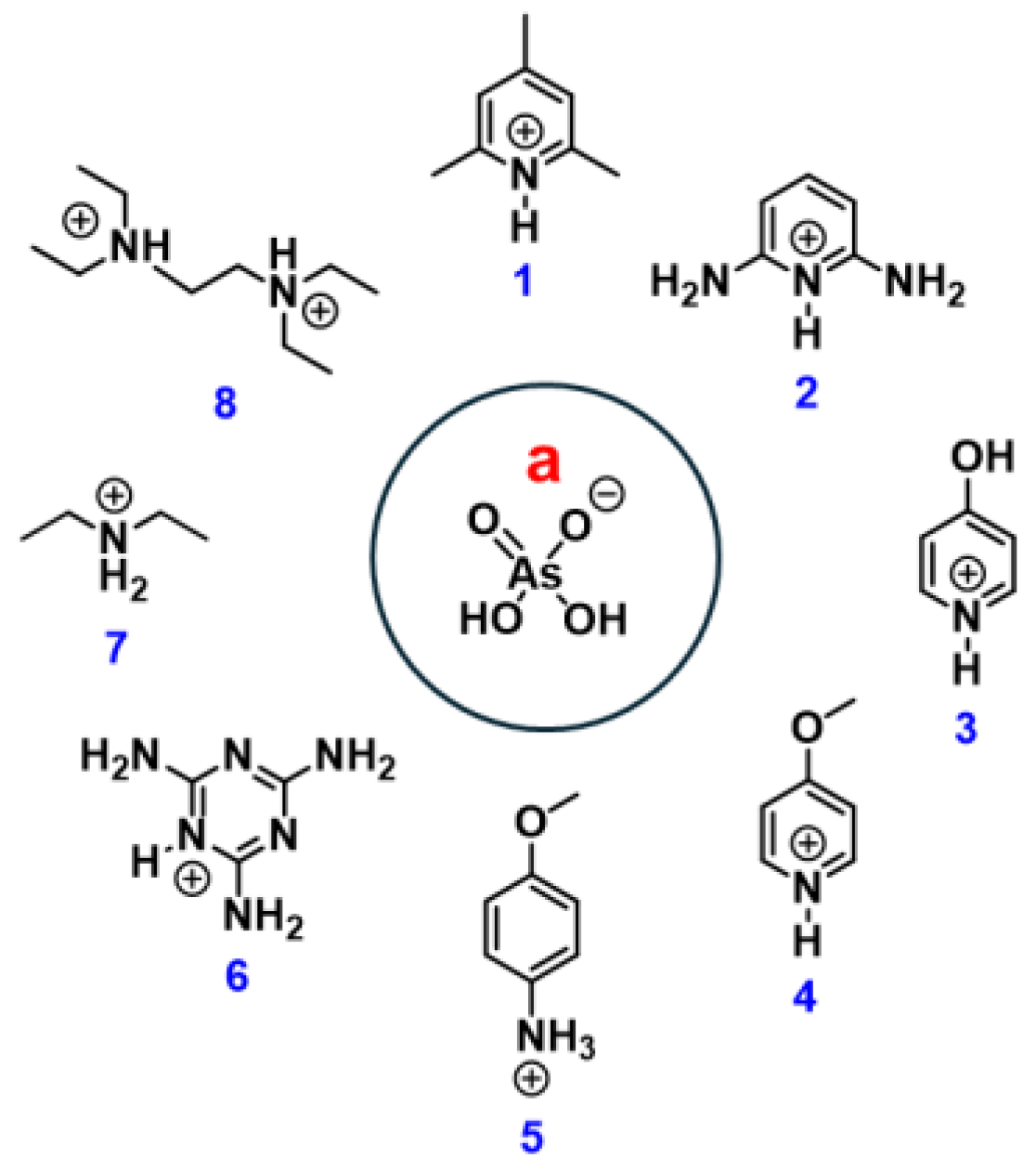
Figure 1.
Partial view of the H-bonded chains in 7a with optimized positions of the hydrogen atoms (motif 1 of H2AsO4– anions’ assembling). Interatomic distances are given in Å. Bond energies, EOHO, are estimated from the electronic kinetic energy density values at BCP, Gbcp (see Table 2).
Figure 1.
Partial view of the H-bonded chains in 7a with optimized positions of the hydrogen atoms (motif 1 of H2AsO4– anions’ assembling). Interatomic distances are given in Å. Bond energies, EOHO, are estimated from the electronic kinetic energy density values at BCP, Gbcp (see Table 2).
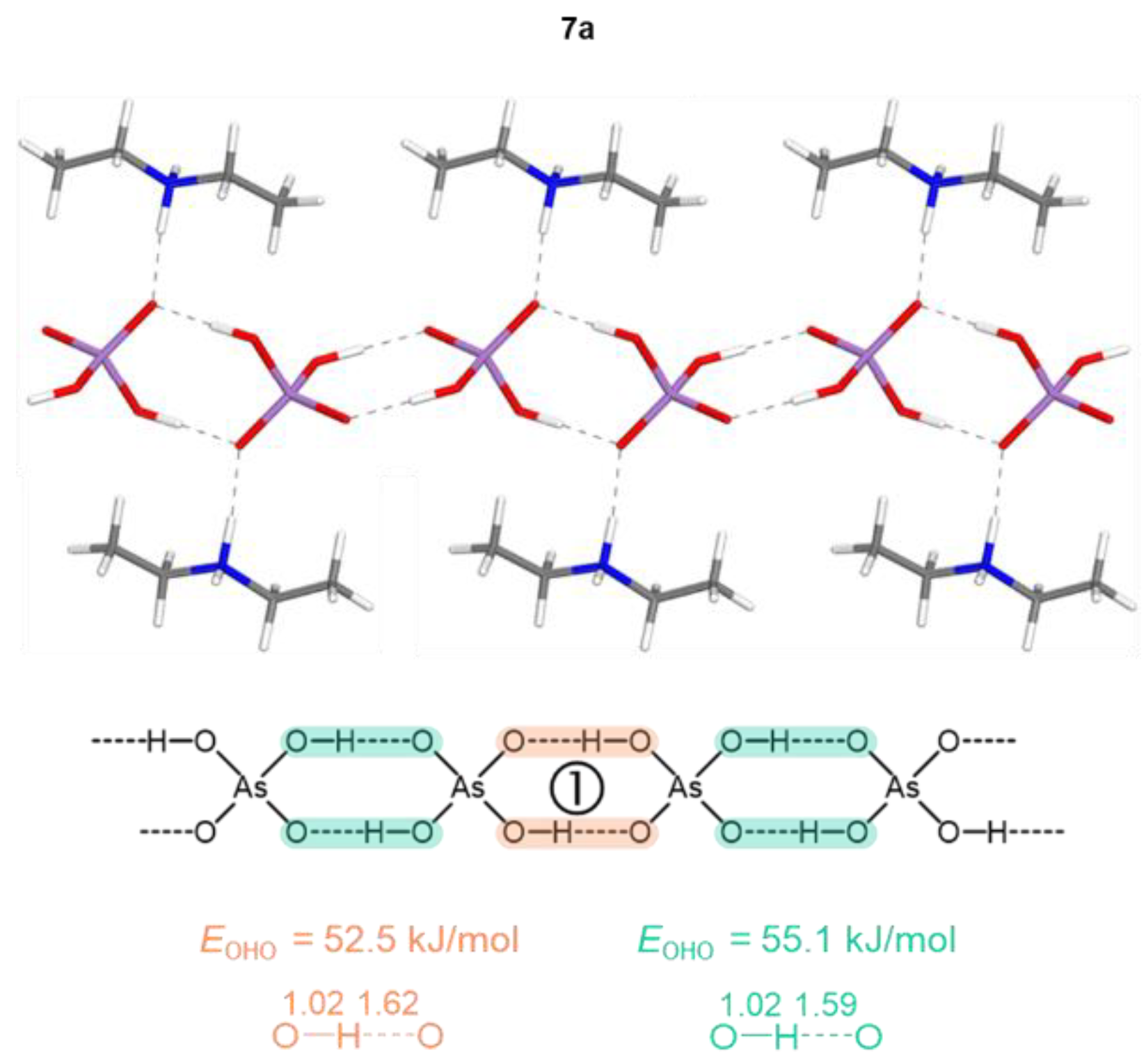
Figure 2.
Partial view of the H-bonded chains in 1a, 2a, 4a and 8a with optimized positions of the hydrogen atoms (motif 2 of H2AsO4– anions’ assembling). Interatomic distances are given in Å. EOHO values are estimated from the Gbcp values (see Table 2).
Figure 2.
Partial view of the H-bonded chains in 1a, 2a, 4a and 8a with optimized positions of the hydrogen atoms (motif 2 of H2AsO4– anions’ assembling). Interatomic distances are given in Å. EOHO values are estimated from the Gbcp values (see Table 2).
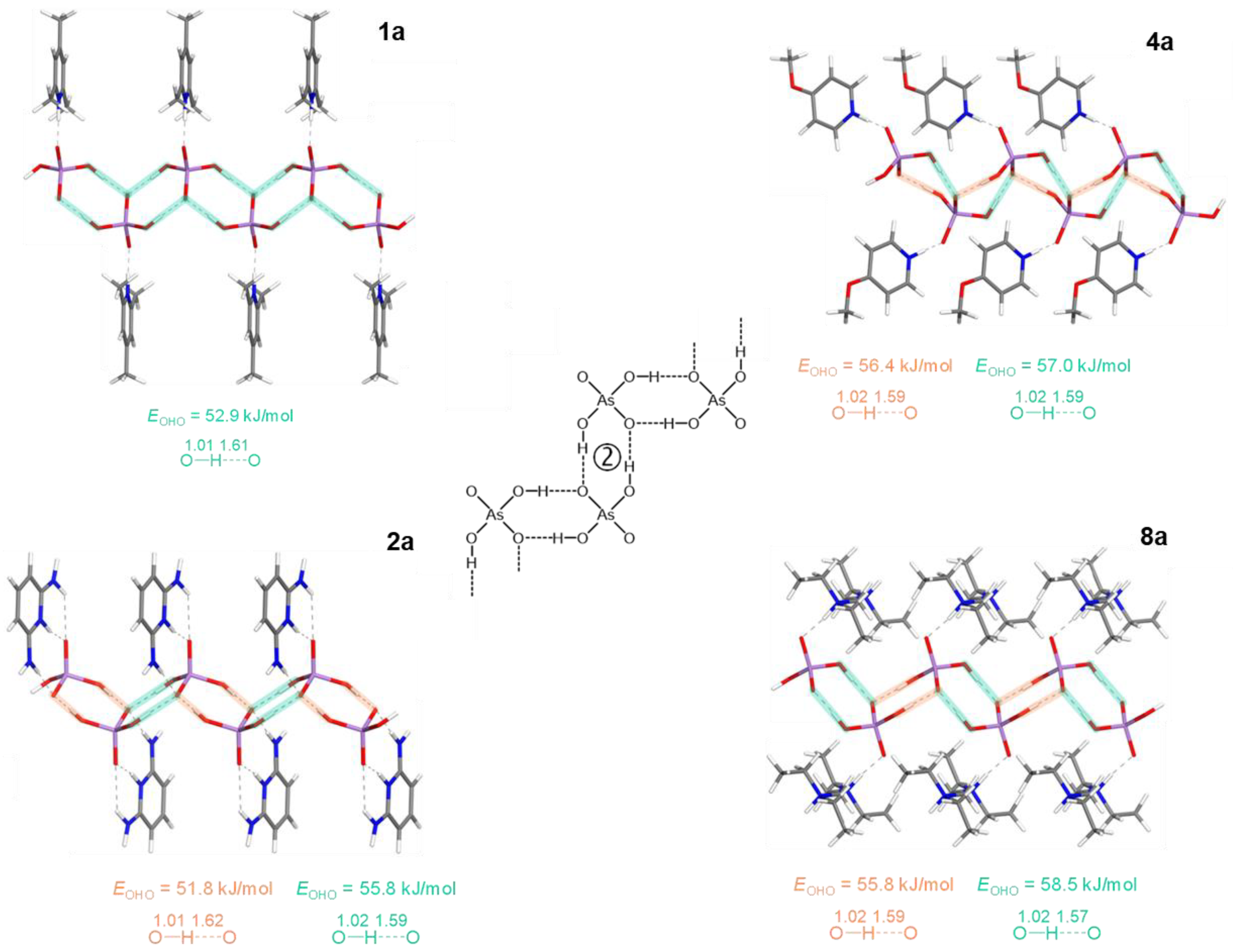
Figure 3.
Partial view of the H-bonded chains in 6a and 3a with optimized positions of the hydrogen atoms (motifs 3 and 4 of H2AsO4– anions’ assembling, respectively). Interatomic distances are given in Å. EOHO values are estimated from the Gbcp values (see Table 2).
Figure 3.
Partial view of the H-bonded chains in 6a and 3a with optimized positions of the hydrogen atoms (motifs 3 and 4 of H2AsO4– anions’ assembling, respectively). Interatomic distances are given in Å. EOHO values are estimated from the Gbcp values (see Table 2).
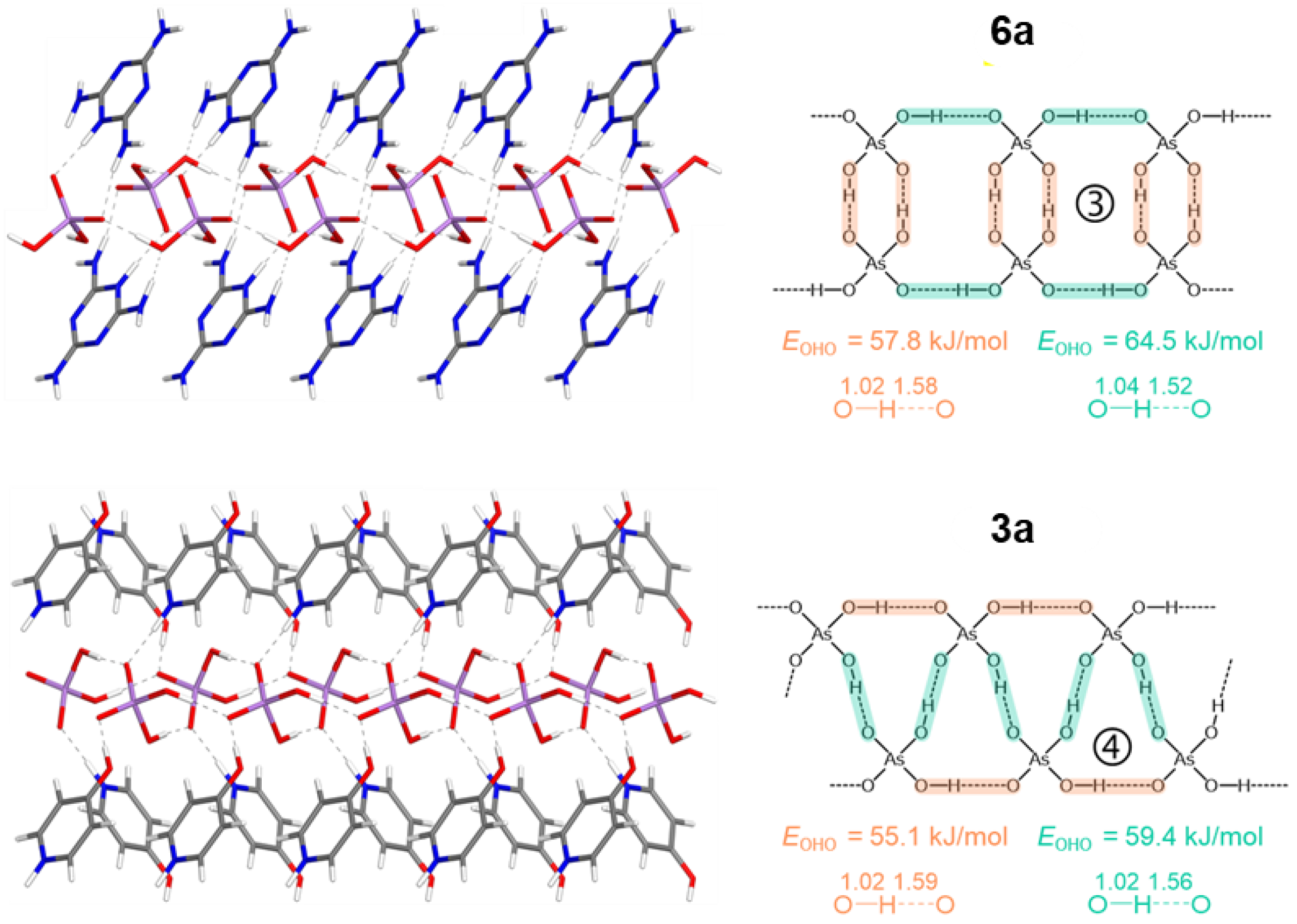
Figure 4.
Partial view of the chainmail-like H-bonded slabs in 5a with optimized positions of the hydrogen atoms (motif 5 of H2AsO4– anions’ assembling). (a): side view, (b): top view. Interatomic distances are given in Å. EOHO values (c) are estimated from the Gbcp values (see Table 2).
Figure 4.
Partial view of the chainmail-like H-bonded slabs in 5a with optimized positions of the hydrogen atoms (motif 5 of H2AsO4– anions’ assembling). (a): side view, (b): top view. Interatomic distances are given in Å. EOHO values (c) are estimated from the Gbcp values (see Table 2).
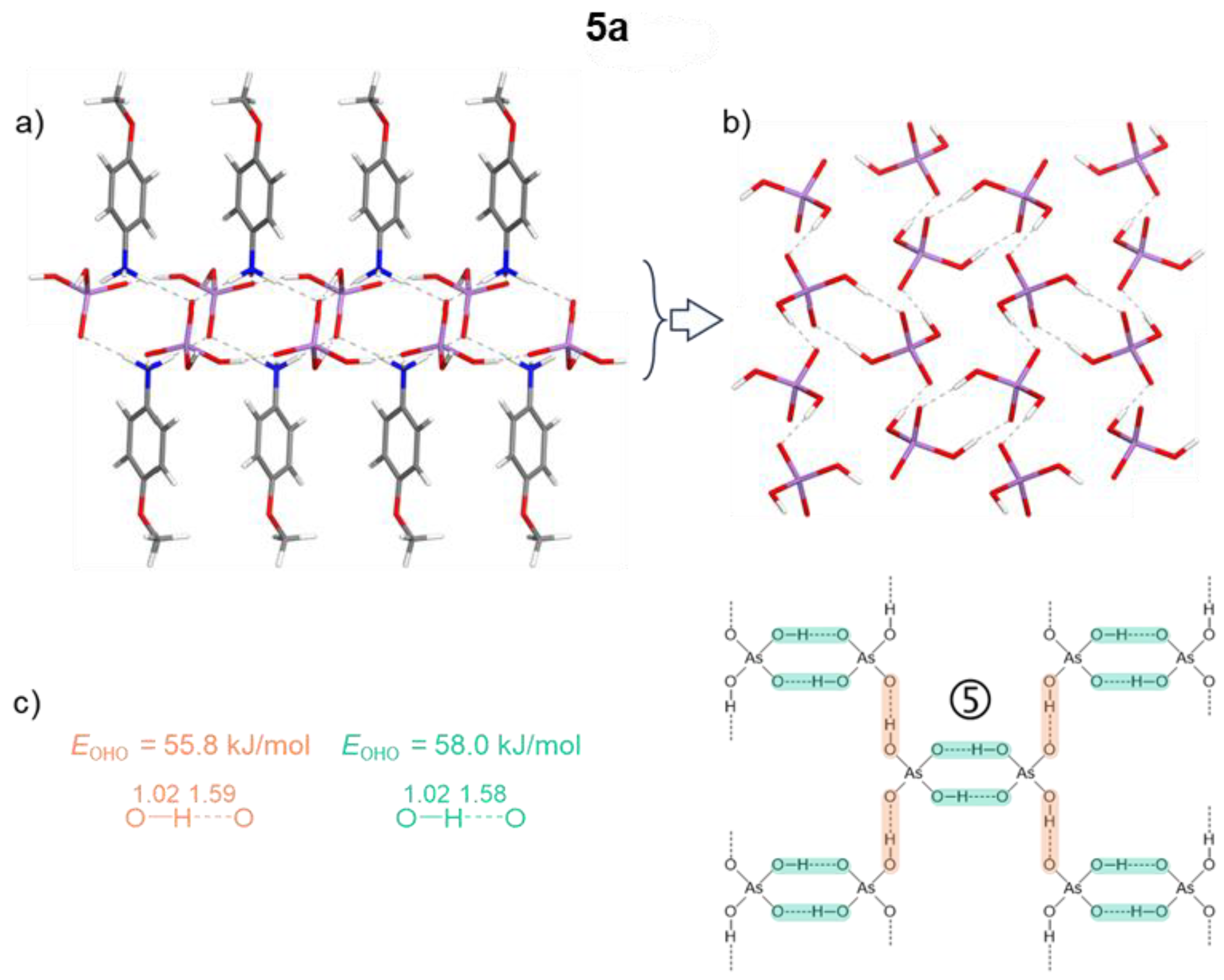
Figure 5.
ED 0.001 a.u. isosurfaces mapped by the molecular electrostatic potential (MESP) for H2AsO4– anion and the corresponding zwitterion in the X-ray geometries of the studied crystals (calculated at PW6B95-D3(BJ)/def2-TZVPD level of theory) but with preoptimized positions of the hydrogen atoms (at B3LYP-D*/POB-TZVP level of theory). Different color scales are used for the MESP (in a.u.).
Figure 5.
ED 0.001 a.u. isosurfaces mapped by the molecular electrostatic potential (MESP) for H2AsO4– anion and the corresponding zwitterion in the X-ray geometries of the studied crystals (calculated at PW6B95-D3(BJ)/def2-TZVPD level of theory) but with preoptimized positions of the hydrogen atoms (at B3LYP-D*/POB-TZVP level of theory). Different color scales are used for the MESP (in a.u.).
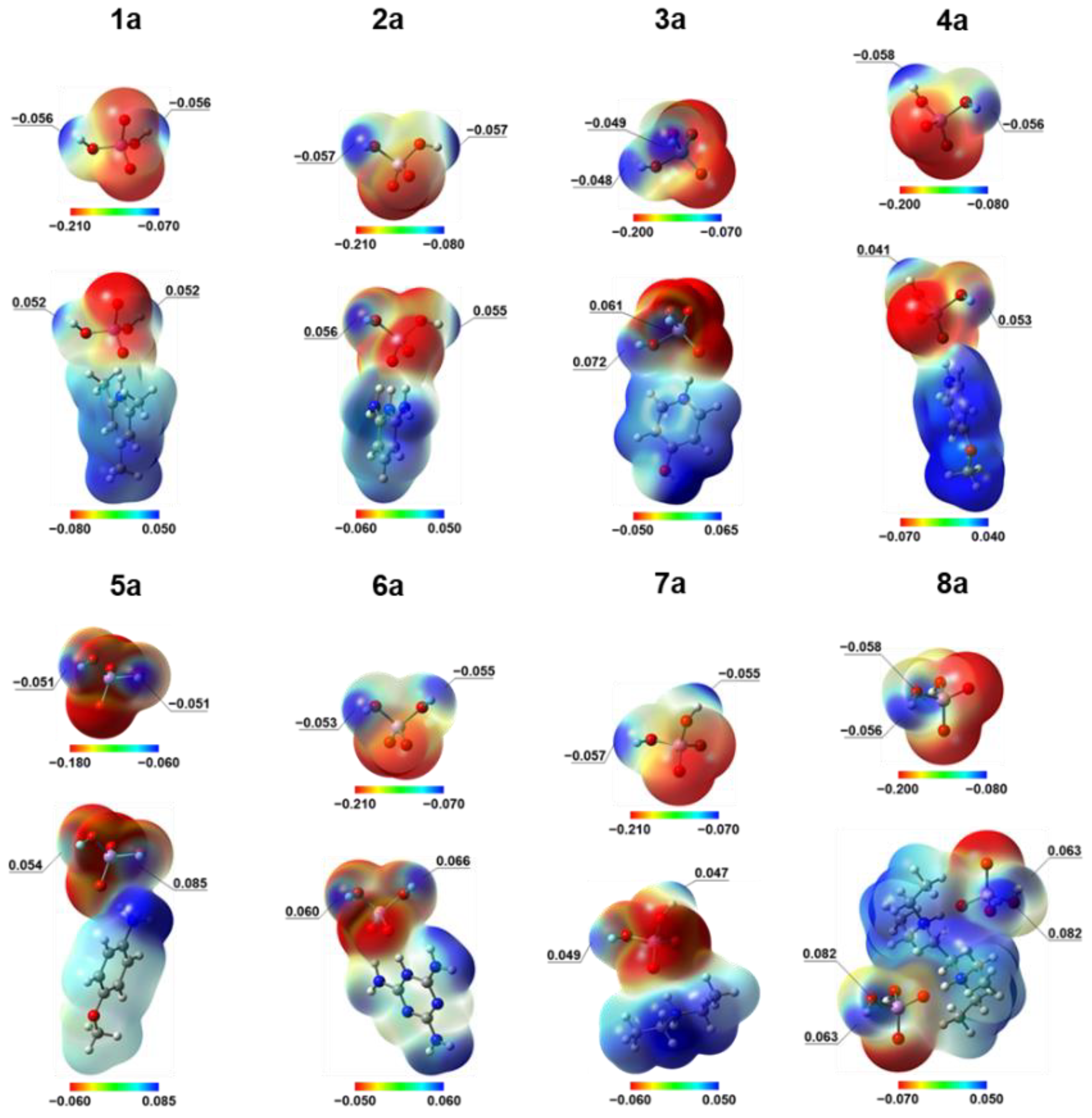
Figure 6.
Dependencies of the negative integrated to the εF level projected COHP values (−IpCOHP) for the OH···O, NH···O, and NH···N interactions in all studied crystals on the corresponding interatomic R(H···A) distances (A = O, N) (a) and ρbcp densities (b).
Figure 6.
Dependencies of the negative integrated to the εF level projected COHP values (−IpCOHP) for the OH···O, NH···O, and NH···N interactions in all studied crystals on the corresponding interatomic R(H···A) distances (A = O, N) (a) and ρbcp densities (b).
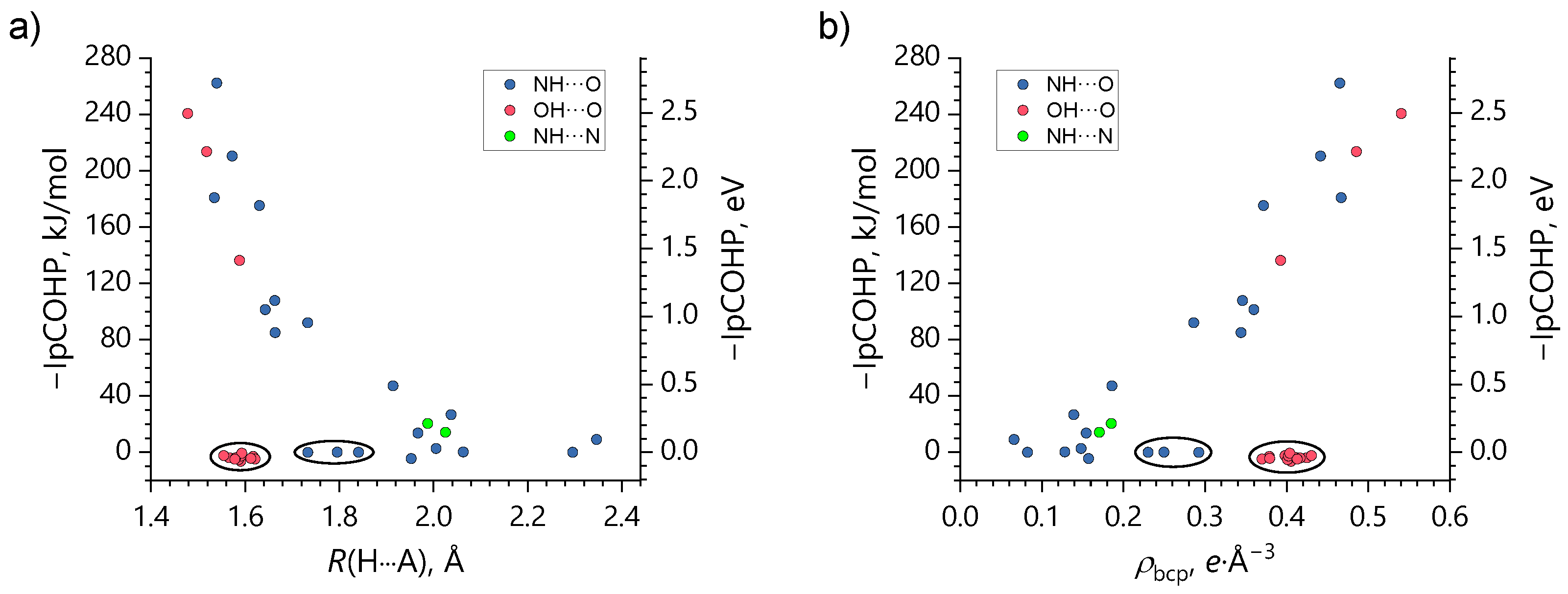

Table 1.
Space group and the list of the O⋅⋅⋅O distances.
| Salt | Space group | O⋅⋅⋅O distances |
|---|---|---|
| 1a | P 21/m | 2.622 Å |
| 2a | P ̅1 | 2.632 Å 2.600 Å |
| 3a | P 21/c | 2.575 Å 2.602 Å |
| 4a | P 21/n | 2.603 Å 2.608 Å |
| 5a | P 21/c | 2.587 Å 2.605 Å |
| 6a | P ̅1 | 2.593 Å 2.554 Å |
| 7a | P ̅1 | 2.585 Å 2.609 Å |
| 8a | P 21/n | 2.604 Å 2.634 Å |
Table 2.
QTAIM parameters (in a.u.) at the intermolecular BCPs of the OHO bonds between H2AsO4– anions, and estimated corresponding bond energies (in kJ/mol), EOHO = 0.429 Gbcp [79,80] for 1a-8a.
| 1a | 2a | 3a | 4a | 5a | 6a | 7a | 8a | |
|---|---|---|---|---|---|---|---|---|
| ρbcp | 0.0562 | 0.0548 0.0594 |
0.0582 0.0638 |
0.0601 0.0607 |
0.0598 0.0612 |
0.0617 0.0719 |
0.0561 0.0590 |
0.0596 0.0630 |
| ▽2ρbcp | 0.1311 | 0.1307 0.1344 |
0.1358 0.1376 |
0.1347 0.1355 |
0.1337 0.1388 |
0.1367 0.1334 |
0.1299 0.1330 |
0.1340 0.1357 |
| Gbcp | 0.0469 | 0.0460 0.0496 |
0.0489 0.0528 |
0.0500 0.0506 |
0.0495 0.0515 |
0.0513 0.0572 |
0.0466 0.0489 |
0.0496 0.0520 |
| Gbcp/ρbcp | 0.8345 | 0.8394 0.8350 |
0.8402 0.8276 |
0.8319 0.8336 |
0.8278 0.8415 |
0.8314 0.7955 |
0.8307 0.8288 |
0.8322 0.8254 |
| Vbcp | -0.0611 | -0.0594 -0.0655 |
-0.0638 -0.0711 |
-0.0664 -0.0673 |
-0.0656 -0.0683 |
-0.0684 -0.0812 |
-0.0607 -0.0646 |
-0.0656 -0.0700 |
| |Vbcp|/ Gbcp | 1.3016 | 1.2901 1.3219 |
1.3057 1.3481 |
1.3270 1.3304 |
1.3250 1.3263 |
1.3334 1.4176 |
1.3028 1.3204 |
1.3241 1.3473 |
| Hbcp | -0.0142 | -0.0134 -0.0160 |
-0.0149 -0.0184 |
-0.0164 -0.0167 |
-0.0161 -0.0168 |
-0.0171 -0.0239 |
-0.0141 -0.0157 |
-0.0161 -0.0181 |
| Hbcp /ρbcp | -0.2520 | -0.2437 -0.2686 |
-0.2569 -0.2880 |
-0.2725 -0.2752 |
-0.2688 -0.2747 |
-0.2770 -0.3324 |
-0.2514 -0.2659 |
-0.2695 -0.2867 |
| EOHO | 52.87 | 51.84 55.83 |
55.07 59.43 |
56.37 56.99 |
55.76 58.03 |
57.75 64.48 |
52.48 55.13 |
55.83 58.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



