Submitted:
27 January 2026
Posted:
28 January 2026
Read the latest preprint version here
Abstract
Neurodegenerative diseases, including Alzheimer's Disease (AD), Parkinson's Disease (PD), Lewy Body Disease (LBD), and related dementias, represents a global health challenge, particularly in aging populations. The simultaneous occurrence of neurodegenerative diseases in an aging population suggests a potential link between causative proteins. Such neurodegenerative proteins, including amyloid-β (Aβ), τ-protein (tau), αsynuclein, TAR DNA-binding protein 43 (TDP-43), and Fused in Sarcoma (FUS), share key characteristics of intrinsically disordered proteins (IDPs), which can explain promiscuous physical interactions, cross-seeding, co-occurrence, pathological synergy, and shared upstream and downstream mechanisms. This review synthesizes current evidence on (1) shared biophysical features of neurodegeneration-associated proteins, (2) mechanisms driving mixed neuropathology, (3) experimental and therapeutic implications of disorder-driven interactions, and (4) key unresolved questions shaping future research. By framing neurodegeneration as a network of interacting, disorder-driven proteinopathies rather than isolated entities, this perspective highlights the need for integrative, systems-level approaches to better understand disease heterogeneity and to identify novel targets for intervention.
Keywords:
protein intrinsic disorder
; neurodegeneration
; proteinopathies
; liquid-liquid phase separation
; amyloid-β
; tau
; α-synuclein
; TDP-43
; FUS
1. Introduction
1.1. Clinical and Pathological Motivation
Neurodegenerative diseases are a complex group of disorders that vary by pathology, genetics, and clinical symptoms. Currently, they lack effective disease-modifying treatments. Pathologically, they are classified as proteinopathies, characterized by the accumulation of specific, abnormal proteins in neurons or glial cells. Many of these diseases share similar pathologies, where the same misfolded protein deposits in different brain regions, causing distinct cognitive and motor impairments. This is illustrated by synucleinopathies e.g., Parkinson’s Disease (PD), Dementia with Lewy Bodies (DLB), and Multiple System Atrophy (MSA) [1] and tauopathies e.g., Alzheimer’s disease (AD), frontotemporal dementia (FTD), Pick’s disease, chronic traumatic encephalopathy (CTE), and corticobasal degeneration [2], where different pathologies are associated with the accumulation of aggregated forms of α-synuclein and τ-protein (tau) protein in different brain regions. However, it is increasingly recognized that combinations of one or more proteinopathies often occur in individuals with neurodegenerative diseases, rather than happening in isolation [3]. In fact, around 75% of autopsies reveal multiple neuropathologies in older adults, highlighting a pressing global health concern in a growing aging population [4]. This mixed (neuro-)pathology seems to be the rule rather than the exception [5] and makes diagnosis and treatment difficult, e.g., for AD, PD, LBD, Vascular Brain injury (VBI), or dementia. Since such mixed pathology is most seen in dementia, it is frequently referred to as mixed dementia. It describes the simultaneous occurrence of several distinct brain diseases or abnormalities in one patient. In these cases, hallmark markers of AD (amyloid plaques and tau tangles) often overlap with other issues, such as vascular damage (blood vessel disease) or the accumulation of additional proteins, α-synuclein (Lewy bodies), TAR DNA-binding protein 43 (TDP-43), and Fused in Sarcoma (FUS) [3]. The simultaneous occurrence of neuropathologies suggests that these proteinopathies are not independent, but rather interconnected, with mechanistic interactions between aggregates driving, and not merely accompanying, disease progression [6,7]. The synergistic effect of multiple pathologies commonly results in more rapid, severe dementia, underlining why many cases involve complex, multifactorial, or “mixed” dementia rather than pure AD. Since the prevalence of mixed pathologies increases in the context of an aging population, and since the related pathologies are associated with misbehavior of specific proteins, these findings need to be better understood, particularly considering the phenomenon of protein intrinsic disorder (PID) which can shed additional light on neuropathologies.
PID refers to the property of many proteins or protein regions not to adopt a single, stable three-dimensional (3D) structure under physiological conditions, but are fully functional, with intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) being the categories within this framework [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. IDPs are proteins that entirely lack a stable fold, while IDRs are segments within otherwise structured proteins that exhibit disorder. Key characteristics of IDPs are (1) structural features, e.g., high conformational flexibility, interconverting conformations, lack of a stable folded 3D structure, and sensitivity to environmental conditions, such as pH, ions, and crowding [9,11,13,14,26,27]; (2) sequence peculiarities, e.g., high content of charged and polar amino acids ((Glutamic acid (E), Aspartic acid (D), Lysine (K), Arginine (R), Glutamine (Q), Serine (S)), therefore low hydrophobic content and depletion in bulky hydrophobic residues (Isoleucine (I), Leucine (L), Valine (V), Phenylalainine (F), Tryptophan (W)), low sequence complexity, and presence of repetitive motifs [10,28,29,30,31]. Therefore, the absence of regular structure in these proteins has been explained by these specific features of their amino acid sequences including the presence of numerous uncompensated charged groups (often negative); i.e., a high net charge at neutral pH, arising from the extreme pI values in such proteins [32,33,34], and a low content of hydrophobic amino acid residues [32,33].
IDPs/IDRs are known as promiscuous binders capable of interaction with a variety of binding partners, including other proteins, membranes, nucleic acids, and various small molecules, and as a result, they have multiple biological functions [27]. Many IDPs/IDRs are capable of undergoing at least partial disorder-to-order transition upon binding [27], whereas others retain a considerable level of disorder even in their bound states forming “fuzzy” complexes [35,36,37,38,39,40,41,42,43,44]. IDPs/IDRs are commonly engaged in formation of complex protein-protein interaction (PPI) networks, often acting as hub proteins with many connections [45,46,47,48,49]. Among the important biological functions of IDPs/IDRs, which are complementary to the activities of ordered proteins and domains [8,10,12,16,50], are regulation of cell division, transcription and translation, signal transduction, storage of small molecules, and chaperone activity [51,52,53,54,55,56,57,58]. These proteins regulate the function of binding partners and promote the assembly of various complexes [59,60,61], ranging from BAF complex [62], mediator complex [63], and mitochondrial enzymatic machines [64], to spliceosomes [65], nucleosomes [66], and ribosomes [24]. Biological functions of IDPs/IDRs are fine-tuned by various post-translational modifications (PTMs) [67,68,69].
Recent studies indicated that IDPs/IDRs serve as fundamental drivers of liquid-liquid phase separation (LLPS), a biophysical process that yields diverse physiological results. Specifically, it is associated with the biogenesis of numerous membrane-less organelles (MLOs, which are also known as biomolecular condensates (BCs), granules, intracellular microdomains, speckles, bodies, puncta, coacervates, and naked cellular organelles among others), which are ubiquitous throughout the cytoplasm and nucleus [70,71,72,73,74,75,76,77,78,79,80]. Often, LLPS is activated when cells experience stress, and thereby it represents a protective mechanism [80,81]. The droplet-like structures formed as a result of LLPS limit the interaction volume of the molecules and increase the probability of interaction [80]. Many MLOs are expected to be present in a certain location at a certain time [80]. Beyond merely operating within favorable conditions, MLOs are characterized by a distinct timeframe and specific requirements for secure existence, alongside the “comfort zone” of the conditions favorable to LLPS [80]. When MLOs persist beyond their intended functional lifespan, they can undergo a “pathological aging” process. This transformation may trigger neurodegenerative diseases by turning these organelles into hubs for the accumulation of toxic amyloidogenic proteins [80]. Other triggers for pathological LLPS and abnormal MLOs include increased levels of proteins undergoing LLPS, irregular PTMs, specific disease-linked mutations, or chromosomal translocations [80].
1.2. Purpose and Scope of the Review
This review provides a comprehensive evaluation of the five major neurodegeneration-related, aggregating proteins involved in mixed pathology such as Amyloid beta (Aβ), tau, α-synuclein, TDP-43, and FUS, within the context of PID. Figure 1 illustrates an accepted model linking misbehavior and aggregation of the neurodegeneration-related proteins with transition from a healthy to a diseased brain state. Misbehavior of these proteins that can occur individually or in combination, can be better understood by applying the PID concept, which aids in explaining the underlying regulatory failures. This raises questions about physical interactions, cross-seeding, co-occurrence, pathological synergy, and shared upstream and downstream mechanisms with a unique view on PID and its neuropathological impact. Here, we will discuss those proteins in the light of functional advantages and disadvantages such as (1) binding promiscuity: IDPs can interact with many different partners and one region can bind multiple targets using different conformations; (2) molecular recognition flexibility where binding often occurs via disorder-to-order transitions and enables context-dependent interactions; (3) regulatory versatility as ideal substrates for various PTMs, e.g., phosphorylation, acetylation, ubiquitination, and SUMOylation; and (4) multifunctionality, in which the same protein participates in transcription, RNA metabolism, signaling, and stress responses forming stress granules. All these interactions need to be considered when referring to neuropathological proteins. In the past, the population prevalence of the co-occurrence of the hallmarks of different proteinopathies was reported in mixed pathologies [3,87,88], what seems not to be sufficient.
2. Overview of the Five Neurodegeneration-Associated Protein and Their Intrinsic Disorder Status
2.1. Amyloid-β (Aβ)
Amyloid-β (Aβ) is a peptide abundantly produced in the brain. It is derived from the type I transmemebrane amyloid precursor protein (APP) sequential cleaved by β-secretase (BACE1) and γ-secretases. Low concentrations of soluble forms of amyloid-β contribute to normal neuronal activity [89]. Various forms, such as Aβ40 (40-residue-long peptide that corresponds to the residues 672-711 of the APP), Aβ42 (42-residue-long peptide corresponding to the APP residues 671-712), N-truncated Aβ4-42, and amyloid-α (also known as Aβ17-40/42 or p3) and different oligomeric and aggregated states, such as non-fibrillar or soluble forms, amyloid fibrils and amorphous aggregates are known. Among these, Aβ42 aggregates more readily, and oligomeric Aβ species are particularly associated with neurotoxicity and the development of AD and related neuropathologies [90]. It is an intrinsically disordered peptide as a monomer, whose amino acid sequence is characterized by a charged, flexible N-terminus and a hydrophobic C-terminal region containing aggregation-prone motifs [91]. This sequence organization underlies its conformational plasticity, binding promiscuity, and strong tendency toward β-sheet–rich self-assembly [92]. The presence of additional hydrophobic residues in Aβ42 further enhances aggregation propensity, providing a molecular explanation for its increased pathogenicity [93]. Furthermore, PTMs, such as phosphorylation and acetylation, can significantly influence the aggregation of Aβ. It has been shown, that PTMs could modulate the polymerization rates of Aβ and thus impact its neurotoxicity. Thus, the peculiarities of the amino acid sequence of Aβ directly link PID with aggregation and neurotoxicity [94]. Aβ aggregation emerges from the plasticity of a disordered peptide, not from destabilization of a folded state [95,96,97]. It was also indicated that clinical variability of AD, at least in part, can be associated with the possibility of misfolded Aβ to acquire different conformations (referred to as “Aβ strains”) [98]. High levels of aggregated Aβ are associated with cognitive decline, dementia, and AD, and misfolded Aβ species can promote the misfolding of other aggregation-prone proteins, particularly tau, thereby accelerating disease progression through prion-like mechanisms [99,100,101,102].
AD, affecting approximately 35-40 million individuals worldwide, is neuropathologically characterized by the accumulation of extracellular Aβ plaques and intracellular tau-containing neurofibrillary tangles (NFTs), both of which contribute to impaired neuronal communication and synaptic dysfunction [103]. Under physiological conditions, Aβ is continuously produced through Blood-Brain Barrier Transport, microglial phagocytosis, or chorid plexus function [104,105,106] and efficiently cleared, indicating a role in normal brain homeostasis. An imbalance between Aβ production and clearance leads to the accumulation of extracellular Aβ, while abnormal tau phosphorylation results in intracellular neurofibrillary tangle formation; either pathology alone, but also their interaction, drives AD progression [107,108].
Importantly, AD pathology rarely occurs in isolation, particularly in aging populations. Neuropathological and biomarker-based studies indicate that Aβ plaques and tau NFTs frequently coexist with additional proteinopathies, including α-synuclein–positive Lewy pathology and TDP-43 inclusions. Moreover, increasing evidence implicates RNA-binding proteins, such as FUS, in overlapping neurodegenerative processes, linking classical amyloid and tau pathology to dysregulated RNA metabolism and stress-granule dynamics. Aggregation-prone biomolecules including Aβ, tau, α-synuclein, TDP-43, and FUS can interact through prion-like seeding, cross-aggregation, and shared proteostatic pathways, thereby amplifying cellular dysfunction [109]. These convergent mechanisms support the evidence that AD are central, but function within a broader network of interacting IDPs rather than a disorder driven by a single misfolded protein [110]. Those connections will be elucidated further below.
Figure 2 illustrates the intrinsic disorder status of human amyloid precursor protein (APP). This protein operates as a cell surface receptor on neurons, facilitating critical physiological processes such as neurite outgrowth, neuronal adhesion, and axonogenesis [111]. Furthermore, trans-cellular interaction between APP molecules on adjacent cells can promote synaptogenesis [111]. Position of the Aβ peptide, which is located within the C-terminal part of the protein is shown by black box. Figure 2 shows that APP is predicted to have a high level of intrinsic disorder. This is evidenced by the presence of long regions with low and very low confidence scores (pLDDT (Predicted Local Distance Difference Test) below 70) (see Figure 2A) and by the multiple IDRs confidently predicted (see Figure 2B). IDRs are very prominent especially in the central part of APP, which is also predicted to contain 12 disorder-based PPI sites (molecular recognition features (MoRFs), that are disordered regions undergoing folding at binding to specific partners), and multiple different PTMs, indicating that intrinsic disorder is utilized by APP for its binding functions, which are regulated by PTMs. Likely, because of these features, APP is capable of interaction with a very broad spectrum of proteins (e.g., according to BioGRID [112,113], it has more than 2400 protein partners). Although Aβ peptides are lipophilic, they are also predicted to contain some disorder, which might be utilized in coordination of Cu2+ and Zn2+ ions by these metal chelators with metal-reducing activity [114]. As per the FuizDrop analysis [115], APP is expected to spontaneously undergo LLPS (i.e., being characterized by a probability of spontaneous liquid-liquid phase separation (LLPS), pLLPS, of 0.7463, it can act as a droplet driver) and contain five droplet-promoting regions (DPRs, residues 188-216, 230-285, 353-373, 437-451, and 624-657), indicating that LLPS is included in its functional repertoire. Curiously, although most disease-causal mutations of APP occur within the Aβ-coding region or in its immediate proximity, it was recently shown that mutations in the N-terminus of APP protein might have pathological consequences as well, as they can promote AD-like tau pathology and notably alter the LLPS of intracellular tau [116].
2.2. τ-Protein (Tau)
Tau, a microtubule-associated protein, is crucial for the stabilization and maintenance of structure and function of neurons and a prominent example of an IDP that plays a critical role in neuronal function and stability. Understanding the structural and sequence features of tau, particularly in the context of its intrinsic disorder, can provide significant insights into its biological functions and pathological implications. Tau is characterized by high conformational flexibility combined with the presence charged and polar amino acids, such as glutamic acid (E), aspartic acid (D), lysine (K), arginine (R), and serine (S), while being low in hydrophobic amino acids. It also contains low-complexity regions and is characterized by the ability to interconvert between multiple structural states [119,120,121]. These amino acid sequence peculiarities are critical in preventing the formation of stable secondary and tertiary structures typical of ordered proteins. The low hydrophobic content and depletion of bulky hydrophobic amino acids like isoleucine (I), leucine (L), valine (v), tyrosine (Y), tryptophan (W), and phenylalanine (F) further underscore tau’s propensity for intrinsic disorder, allowing it to maintain a dynamic flexibility essential for its cellular functions [120,122]. Unlike globular proteins that possess stable 3D structures, tau does not fold into a well-defined conformation and instead samples a wide range of shapes under physiological conditions, a hallmark of IDPs [119,120]. This structural plasticity allows tau to engage in diverse interactions with microtubules and other proteins, vital for maintaining neuronal architecture [67]. Furthermore, tau’s dynamic structure is highly sensitive to various environmental factors, such as pH, ionic strength, and macromolecular crowding, further complicating its behavior in cellular environments [123].
Tau undergoes LLPS, which can lead to the formation of liquid-like droplets that subsequently transition to more condensed amyloid structures under certain conditions [124,125,126,127,128,129,130,131,132,133,134,135,136]. This is significantly influenced by tau’s charged, polar residues, which are crucial for its interactions with other molecules [137]. For instance, the positively charged microtubule-binding domains of tau contribute to its LLPS properties, essential for its role in cell signaling and pathology [80].
In neurodegenerative disease, e.g., AD and tauopathies, the protein undergoes hyperphosphorylation, which leads to aggregation and formation of NFTs [138]. After chronic traumatic encephalopathy (CTE) NFTs accumulate abnormally and disrupt normal cellular function. Interestingly, disease models suggesting that Aβ initiate a pathophysiological change leading to tau misfolding. Human studies showed that NFTs in the neocortex of the brain are more related to cognitive decline than amyloid plaques [139]. For example, one study demonstrated that Aβ plaques enhance tau pathologies by facilitating tau aggregation in the presence of misfolded tau seeds, indicating a significant interaction promoting neurodegeneration [140].
In conclusion, tau’s low sequence complexity, characterized by repetitive motifs, contributes to its flexibility and functional diversity. Such properties enable tau to participate in various cellular processes, including signal transduction and cytoskeletal organization [141,142]. In the context of neurodegenerative diseases like AD, the disordered regions of tau are particularly important, as they can aggregate into neurotoxic forms, highlighting the dual nature of PIDs [16].
Figure 3 provides an outlook on the prevalence of functional disorder in human tau proteins and clearly shows that this protein is mostly disordered and contains multiple PTMs. Furthermore, according to Figure 3B, tau’s almost entire sequence is expected to be involved in disorder-based interactions and therefore can serve as a disordered scaffold. In line with these predictions, BioGRID [112,113] indicates that tau has more than 1100 protein partners. According to FuzDrop, human tau protein is clearly defined as a strong droplet driver, since it is predicted to have a very high pLLPS of 0.9985 and contain four DPRs (residues 1-30, 309-589, 608-622, and 719-739) that cover almost entire sequence of this protein.
2.3. α-Synuclein
α-synuclein (also known as non-Aβ component (NAC) precursor (NACP) of AD) is a 140-amino acid transport protein primarily found in the brain, particularly in presynaptic terminals. It has three functional regions, such as an amphipathic N-terminal region (residues 1–60) containing 11-residue repeats including the KTKEGV motif, allowing the protein to bind to acidic lipid membranes and form α-helices, a central domain (residues 61–95) known as NAC, which is a highly hydrophobic, aggregation-prone region, and highly acidic and proline-rich C-terminal region (residues 96-140) involved in regulating solubility, interacting with metal ions, and binding to protein partners [143]. α-synuclein is predominantly found in neuronal cell bodies, where it is involved in synaptic transmission and vascular regulation. This “chameleon” protein [144] is highly conformational flexible and is characterized by the propensity to exist in various structural forms, a characteristic feature of IDPs [145]. Under physiological conditions, α-synuclein is largely unstructured, lacking a stable 3D conformation [34,145,146,147]. This intrinsic disorder allows the protein to adopt different conformations depending on environmental factors, such as the presence of membranes, which favor an α-helical conformation [148]. The protein is also prone to undergo LLPS [149,150,151,152,153], and is capable of shifting from a soluble disordered state to aggregated forms, such as oligomers and fibrils, which are implicated in neurodegeneration [154,155]. This protein has strong aggregation potential and its ability to form amyloid fibrils is enhanced by a variety of factors, such as interaction with pesticides and herbicides [156,157,158]. The primary sequence of α-synuclein demonstrates several features typical of IDPs. The protein is enriched in polar and charged amino acids, particularly in its N-terminus, which includes a high density of serine (S), lysine (K), and glutamic acid (E) residues [159]. This contributes to its low hydrophobicity and helps maintain a flexible structure without the stable α-helices or β-sheets observed in globular proteins [159].
Furthermore, α-synuclein features repetitive motifs, specifically the NAC region, which has been associated with aggregation [160,161]. This region’s sequence composition aids in its ability to interact with lipid membranes and other cellular components, while simultaneously modulating its propensity to aggregate into pathogenic forms. The low sequence complexity of α-synuclein reflects the properties commonly associated with IDPs, enabling it to engage in multiple PPI crucial for synaptic function [162]. The protein’s tendency to aggregate into filamentous structures is a result of its intrinsic flexibility, which allows it to adopt multiple conformations that can stabilize intermolecular interactions [154,155]. Furthermore, PTMs, such as N-terminal acetylation can affect the protein’s binding properties and overall structure, thereby influencing its aggregation dynamics [163]. Additionally, studies indicate that various factors, such as PTM modifications and interactions with chaperones, can alter the aggregation propensity of α-synuclein, thereby influencing its structure and stability in cellular environments [123,146].
Research has shown that α-synuclein can form oligomeric intermediates during its aggregation process, which precedes the formation of amyloid fibrils seen in Lewy bodies and Lewy neurites [154]. These pre-fibrillar oligomers are often more toxic than mature fibrils, emphasizing the pathological implications of transient conformational states [155].
Mutations in the SNCA gene are responsible for synucleinopathies and PD. In PD, α-synuclein can induce cellular toxicity through mechanisms such as mitochondrial dysfunction and impaired protein degradation pathways. It’s transmission and spread resembles prion-like behavior. Misfolded α-synuclein induces misfolding of proteins in neighboring cells [164]. α-synuclein interacts with Aβ and tau, which impacts neurodegenerative processes [165]. It was also shown that heterotypic droplets composed of tau and α-synuclein are formed at physiologically relevant molar ratios that mimic neurons’ soma and terminal buttons of neurons, indicating that heterotypic LLPS of tau and α-synuclein can be implicated in overlapping neuropathologies and contribute to mixed pathology [166]. Some strains of α-synuclein promote tau aggregations in neurons and coexists with AD pathology [167], thereby α-synuclein is a biomarker in cerebrospinal fluid (CSF) for cognitive decline. Combinations of Aβ, tau, and α-synuclein biomarkers improves diagnosis of PD, dementia, or LBD [168].
Understanding α-synuclein’s intrinsic disorder through its structural and sequence features reveals crucial insights into its biological functions and pathological roles in diseases like PD. The protein’s conformational flexibility allows it to participate in vital synaptic processes while simultaneously predisposing it to aggregation, leading to neurodegeneration. A deeper comprehension of these properties provides a framework for targeted therapeutic strategies aimed at mitigating α-synuclein-associated neurodegenerative diseases. Different roles of intrinsic disorder in multifunctionality and polypathogenicity of were discussed in a comprehensive review, where it was emphasized that the remarkable structural, functional, and dysfunctional multifaceted nature of this protein can be understood using the intrinsic disorder-based proteoform concept [143]. Figure 4 illustrates these points by showing a conformational ensemble generated for human α-synuclein by AFflecto (Figure 4A) and functional disorder profile generated by D2P2 (Figure 4B). High binding promiscuity of α-synuclein is illustrated by the fact that according to BioGRID, it is involved in interaction with more than 1500 protein partners. With the pLLPS of 0.6249 and a long IDR (residues 101-140), human α-synuclein is expected to serve as a droplet driver capable of spontaneous LLPS.
2.4. TAR DNA-Binding Protein 43 (TDP-43)
TAR DNA-binding protein 43 (TDP-43) is a predominantly nuclear RNA-binding protein that regulates alternative splicing, mRNA stability, transport, and translation, thereby playing a central role in neuronal RNA homeostasis and stress responses under normal conditions. It is predominantly composed of IDRs that allow significant conformational flexibility. This flexibility enables TDP-43 to dynamically interact with various RNAs and protein partners [83]. Under physiological conditions, TDP-43 does not adopt a stable 3D structure; instead, it exists in a range of conformations, facilitating its involvement in the regulation of gene expression and RNA processing [83]. The sequence of TDP-43 reflects its intrinsic disorder, featuring a high content of charged and polar residues, contributing to its low hydrophobicity. Such a composition is typical for IDPs and facilitates the protein’s flexible and multifunctional nature, promoting interactions critical for its roles in cellular signaling and regulation [145]. TDP-43 contains two RNA recognition motifs (RRMs) that facilitate its binding to RNA, showcasing how specific domains can play a vital role in modulating its interactions while retaining overall disordered characteristics [83,145]. TDP-43 exemplifies the role of intrinsic disorder in disease pathology and cellular function [72,171,172]. Its structural flexibility and unique sequence features are fundamental to the normal biological functions of this protein, while also predisposing it to aggregation in neurodegenerative contexts. A deeper understanding of TDP-43’s intrinsic disorder can foster new therapeutic approaches to address the challenges posed by TDP-43 proteinopathies.
Importantly, TDP-43 can undergo LLPS, a process that leads to the formation of membrane-less organelles, aging of which results in the accumulation of TDP-43 in cytoplasmic aggregates [72,73,81,173,174,175,176,177,178,179,180,181,182,183,184,185,186]. This abnormal aggregation is a hallmark of several neurodegenerative disorders. The aggregation propensity of TDP-43 is closely linked to its charge distributions and IDRs, which can interact with various cellular components to modulate its function and stability [83].
Notably, TDP-43’s sequence integrity is crucial for its function; mutations within its coding region have been implicated in familial forms of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD), leading to disruptions in its normal cellular roles and promoting pathological aggregation [187]. The presence of low-complexity regions in TDP-43 further underlines its propensity to form aggregates under pathological conditions, pointing toward a link between intrinsic disorder and neurodegeneration [187].
Under pathophysiological conditions, TDP-43 is hyperphosphorylated and ubiquitinated in the cytoplasm of neurons and glial cells and plays a role in AD. TDP-43 misfolding is connected to translocation of the protein from nucleus to cytoplasm due to stress like it has been seen for tau [188]. Evidence suggests, that TDP-43 occur alongside Aβ, tau, and α-synuclein in older individuals and has been shown to contribute to mixed pathology within the aging brain [189]. TDP-43 pathology is a defining feature of ALS and FTLD, but it is also highly prevalent in aging-related neuropathologies. Autopsy studies indicate that TDP-43 inclusions are present in approximately 40–50% of Alzheimer’s disease cases and in 20–30% of cognitively normal individuals over 80 years [190]. This age-associated presentation, termed limbic-predominant age-related TDP-43 encephalopathy (LATE) [191], frequently coexists with Aβ and tau pathology and is associated with disproportionate hippocampal atrophy and accelerated cognitive decline. These epidemiological findings identify TDP-43 as one of the most common contributors to mixed neuropathologies and proteinopathies in late-life dementia [192,193].
Figure 5 illustrates the highly disordered nature of human TDP-43 and shows that its IDRs serve as targets for various PTMs and are also used for disorder-based interaction with partners (as per BioGRID, there are at least 572 protein partners of TDP-43). Furthermore, as per FuzDrop analysis, human TDP-43 has a high probability of spontaneous LLPS (pLLPS = 0.8981) and contains a long, C-terminally located DPR (residues 251-414), confirming that the amino acid sequence features of this protein are consistent with its ability to undergo spontaneous LLPS.
2.5. Fused in Sarcoma (FUS)
Fused in Sarcoma (FUS, also known as translocated in liposarcoma, TLS) is a nuclear DNA/RNA-binding protein essential for RNA metabolism and stress responses that plays important roles in various cellular processes ranging from transcription regulation, RNA splicing, and RNA transport to DNA repair and damage response [194]. Mutations in FUS can lead to its cytoplasmatic mislocalization, accumulation and aggregation which contribute to neurodegeneration such as ALS, FTLD and display prion-like properties in experimental systems [110,195]. Abnormal aggregation contribute to dysfunctional protein homeostasis to neurodegeneration, with FUS anomalies exacerbating these conditions [6]. FUS is characterized by an intrinsically disordered structure, which allows it to exist in a dynamic ensemble of conformations rather than adopting a stable 3D structure [72,171,196,197]. This facilitates its interaction with multiple partners, essential for its roles in RNA-binding and regulation of gene expression [123]. Moreover, FUS undergoes phase separation, forming membrane-less organelles that play critical roles in stress response and RNA processing [198,199,200,201,202,203,204,205,206,207,208,209,210]. The ability of FUS to condense into these structures underscores the significance of its dynamic properties in cellular contexts. FUS contains a high proportion of charged and polar amino acid residues, typical of IDPs, which promotes its interaction with RNA and other proteins [211]. The protein’s sequence includes two RNA recognition motifs (RRMs), facilitating its specific binding to RNA molecules while remaining largely disordered overall. The presence of low-complexity domains allows FUS to engage in various interactions while retaining flexibility, which is essential for its multifunctional role within the cell [212].
While not a defining pathological hallmark of AD, FUS dysregulation may indirectly exacerbate mixed neuropathologies by impairing proteostasis and RNA homeostasis [213]. Approximately 5% of mutations in FUS/TLS account for familial ALS cases [214]. Around 10% of FTLD cases show significant involvement of FUS, linking it to the broader spectrum of proteinopathies characteristic of neurodegenerative diseases [215].
Additionally, mutations in the FUS gene can lead to forms of ALS and FTD, linking specific sequence variations to pathogenic behavior and further suggesting that the functional capacity of FUS is heavily dictated by its disordered nature [216]. Understanding these sequence characteristics provides insights into how FUS can maintain functional flexibility while being prone to misfolding and aggregation under pathological conditions. Furthermore, FUS aggregates can form cytoplasmic inclusions associated with neurodegeneration. The transitions between soluble and aggregated states are influenced by its IDRs, highlighting the pathogenic potential of FUS misfolding [83]. This behavior exemplifies the connection between intrinsic disorder and neurodegeneration, as the aggregated forms of FUS are implicated in cellular toxicity.
FUS exemplifies how intrinsic disorder can be pivotal for protein functionality, as structural and sequence features of this protein enable it to play diverse roles in cellular processes. However, the same characteristics that facilitate its normal functions can also predispose it to aggregation, contributing to neurodegenerative disease pathology. A detailed understanding of FUS’s intrinsic disorder can lead to potential therapeutic approaches for diseases associated with its aggregation.
Figure 6 illustrates the prevalence of intrinsic disorder in human FUS and suggests that structural pliability is important for function of this protein, which is heavily decorated by multiple different PTMs and contains 15 MoRFs that covers a very significant part of its sequence. Intrinsic disorder can contribute to multifunctionality of this protein and its ability to interact with multiple partners. According to BioGRID, FUS can interact with almost 850 proteins. FuzDrop analysis revealed that human FUS has an extremely high pLLPS of 0.9999 and contains three DPRs (residues 1-294, 360-437, and 443-526) that cover almost 90% of its sequence, providing strong support to the capability of this protein to act as a powerful droplet driver capable of undergoing spontaneous LLPS.
2.6. Commonality of Individualities
Table 1 provides a systematic overview of some of the characteristic features of human Aβ, tau, α-synuclein, TDP-43 and FUS and shows that these proteins exhibit substantial IDRs that support physiological flexibility but also predispose to aggregation and cross-interaction under pathological conditions.
2.7. Mightly Alliance: Beyond the Individual Armies
Facts considered so far illustrated individual importance of Aβ/APP, tau, α-synuclein, TDP-43, and FUS in both physiological and pathological processes. However, these proteins are not acting alone but do interact with each other forming a network with seven edges and an average local clustering coefficient of 0.767. Since the expected number of edges for the random network of this size is 0, this intra-set PPI network has significantly more interactions than expected (PPI enrichment p-value 3×10-11) (see Figure 7A).
Crucially, these five proteins not only possess individual “armies” of numerous interactors, but they also exhibit collective interactivity, featuring multiple shared binding partners. This idea is illustrated by Figure 7B showing that these proteins function not just as individual “hubs”, but as a unified group with high interconnectivity. In fact, although this network was generated using the highly restringing settings (the highest confidence level of 0.9 for a minimal interaction score), it includes 190 proteins involved in 807 interactions, which significantly exceeds the 289 interactions expected to occur in a random set of proteins of the same size and degree distribution drawn from the genome (PPI enrichment p-value < 10-16). The average local clustering coefficient of this network is 0.654, and its average node degree is 8.5. Among the 190 proteins in this network, 83 have at least 8 interacting partners, with 26 proteins interacting with more than 15 partners each. The most significant number of interactors is ascribed to the “army commanders”, APP, MAPT/TAU, SNCA/synuclein, TARDBP/TDP-43, and FUS, which interact with 63, 49, 45, 43, and 34 partners, respectively. Next in the interactivity ranks within this united network are HNRNPA1, HNRNPC, HNRNPM, HSP90AA1, HNRNPA2B1, HNRNPH1, HNRNPK, and GSK3B interacting with 30, 24, 24, 23, 22, 22, 22, and 21 partners. There are also 26 proteins that interact exclusively with one of the “army commanders”. There are no proteins that would serve as joint interactors for five and four connect human proteins linked to mixed pathologies, and only one “outside” protein, APOE, is connecting three “army commanders” (APP, SNCA, and MAPT). However, TARD and FUS have 15 joint interactors DROSHA, EWSR1, HNRNPA1, HNRNPA2B1, HNRNPA3, HNRNPC, HNRNPH1, HNRNPK, HNRNPM, MATR3-2, OPTN, RBMX, SFPQ, SOD1, and UBQLN2); IAPP, KLC1, KLC2, and PRNP are common binding partners of APP and SNCA; MAPT and APP share CASP3, GSK3A, and PSEN1; LRRK2, PRKN and VDAC1 are shared by MAPT and SNCA; whereas HSPA4 and HTT serve as joint interactors for TARDBP and SNCA. Obviously, if less restrictive settings are used, the resulting PPI network will include more shared partners. However, conducting such analysis is outside the scopes of this review.
The most enriched biological processes, molecular functions, and cellular components (as per Gene Ontology annotations) of the members of joined network are shown in Figure 8, along with the most enriched KEGG pathways and the most enriched subcellular localizations and disease-gene associations. Figure 8A shows that among the 1055 significantly enriched biological process GO terms associated with the members of this network, the most enriched are related to modulation of chemical synaptic transmission, amyloid precursor protein metabolic process, regulation of calcium-mediated signaling, cellular response to Aβ, regulation of calcium ion import across plasma membrane, negative regulation of mRNA metabolic process, and regulation of neurotransmitter levels. Figure 8B shows that among the 150 significantly enriched molecular function GO terms, and the most enriched are Aβ binding, tau protein binding, peptide binding, structural constituent of cytoskeleton, and single-stranded RNA binding. As per Figure 8C, of the 188 cellular component GO terms, the most enriched are distal axon, growth cone, inclusion body, cell body, somatodendritic compartment, dendrite, and microtubule. Analysis of the most significantly enriched KEGG pathways revealed that the members of the studied network are related to PD, AD, ALS, HD, prion disease, gap junction, amphetamine addiction, dopaminergic synapse, and long-term potentiation (Figure 8D). Figure 8E shows that among the 186 significantly enriched subcellular compartments are inclusion body, Aβ complex, extracellular membrane-bounded organelle, growth cone, distal axon, calcineurin complex, and cell body. The multifunctionality of the analyzed five proteins and their interactors indicates that misbehavior and deregulation of these proteins can be associated with various pathological processes. In agreement with this notion, Figure 8F shows that among the 80 significantly enriched diseases linked to the members of this network are dementia, cognitive disorder, AD, ALS, FTD, PD, neurodegenerative disease, central nervous system disease, motor neuron disease, and synucleinopathy.
Importantly, not only APP, tau, α-synuclein, TDP-43, and FUS are characterized by high intrinsic disorder content; many of their interactors are disordered as well. This is illustrated by Figure 9A representing the PONDR® VSL2 score (average disorder score, ADS) vs. PONDR® VSL2 (%) (percent of predicted intrinsically disordered residues, PPIDR) plot. Both PPIDR and ADS values are used to rank proteins. Based on their PPIDR scores, proteins are classified as highly ordered, moderately disordered, or highly disordered, if their PPIDR values are below 10%, between 10% and 30%, and above 30%, respectively [223,224]. Alternative classification of proteins as highly ordered, moderately disordered/flexible, or highly disordered is derived from their ADS values, with ADS < 0.15, 0.15 ≤ ADS < 0.5, and ADS ≥ 0.5, respectively.
Based on these criteria, almost all interactors are clearly classified as moderately or highly disordered by PONDR® VSL2, with just one protein being predicted as highly ordered by PPIDR and not by ADS (i.e., is located within cyan area). For comparison, the analogous analysis of the entire human proteome (20,317 proteins) revealed that 0.4%, 5.1%, 33.7%, 21.0%, and 39.8% proteins were located within dark blue, cyan, dark pink, light pink, and red areas, respectively [225]. Therefore, with their 0.0% – 0.53% – 37.04% – 20.10% – 42.33% distribution, interactors are generally more disordered than the whole proteome. Note that APP, tau, α-synuclein, TDP-43, and FUS are all predicted as highly disordered (see Figure 9). Figure 9B represents the results of global disorder analysis of human proteins interacting with APP, tau, α-synuclein, TDP-43, and FUS in the form of the ΔCH-ΔCDF plot that can be used for further classification of proteins as mostly ordered, molten globule-like or hybrid, or highly disordered based on their positions within the resulting CH-CDF phase space [226,227,228,229]. This analysis provided further support to the idea that the human proteins interacting with APP, tau, α-synuclein, TDP-43, and FUS include noticeable levels of disorder, being a bit more disordered than human proteome in general, which contains 59.1%, 25.5%, 12.3%, and 3.1% proteins in quadrants Q1, Q2, Q3, and Q4, respectively [225]. Figure 9C shows correlation between interactability of the analyzed proteins (in terms of their node degree) with their intrinsic disorder status and demonstrates that there is a weak positive correlation between these two parameters. This is an expected behavior, as disordered proteins are typically prone to be more promiscuous binders. Finally, Figure 9D shows correlation between the propensity of all the query proteins to undergo LLPS and their intrinsic disorder status in a form of the pLLPS vs. PPIDR plot. This analysis revealed that many studied proteins, which are predicted as highly disordered, are capable of spontaneous LLPS. In fact, in addition to APP, tau, α-synuclein, TDP-43, and FUS 60 of their interactors are expected to serve as droplet drivers, and 74 interactors can operate as droplet clients. In other words, only about one-third of these proteins are not related to LLPS.
3. And Intrinsic Disorder to Rule Them All
Despite their diverse primary sequences and physiological roles, Aβ, tau, α-synuclein, TDP-43, and FUS share a pronounced degree of intrinsic disorder, low sequence complexity, and enrichment in charged and polar residues. These common biophysical features promote multivalent and dynamic protein–protein interactions, extensive PTMs, and participation in highly connected interaction networks. Importantly, the same disorder-driven properties that enable functional flexibility under physiological conditions also predispose these proteins to undergo LLPS and aberrant phase transitions. Consequently, intrinsic disorder provides a mechanistic bridge linking protein-specific biology with the formation of biomolecular condensates and, under conditions of stress or aging, their conversion into pathological aggregates.
Mixed proteinopathies and neuropathologies occur frequently rather than being rare exceptions due to the structural and functional similarities of IDPs. These proteins often share IDRs and can interact through multiple pathways, facilitating their co-aggregation [230]. As such, neurodegenerative diseases are more likely to present symptoms stemming from multiple protein aggregations [231].The pathological interplay among tau and α-synuclein exemplifies this phenomenon, reinforcing the need for a holistic understanding of neurodegeneration. Understanding the roles of intrinsic disorder in mixed proteinopathies and neuropathologies enhances the comprehension of neurodegeneration. Sections below provide more focused considerations of the involvement of intrinsic disorder in various functional and dysfunctional aspects of these proteins.
3.1. Intrinsic Disorder and LLPS
LLPS is a cellular phenomenon that plays a fundamental role in forming biomolecular condensates, which are crucial for various biological processes. LLPS occurs when biomolecules spontaneously demix into coexisting liquid phases, forming dynamic, membrane-less compartments that concentrate specific proteins and RNAs. This process is driven by multivalent, weak interactions among biomolecules that become energetically favored, leading to the formation of a dense phase and a dilute phase to minimize free energy [232].
For example, APP [116], tau [80,124,125,126,127,128,129,130,131,132,133,134,135,136,137], α-synuclein [149,150,151,152,153], TDP-43 [72,73,81,173,174,175,176,177,178,179,180,181,182,183,184,185,186,233], and FUS [198,199,200,201,202,203,204,205,206,207,208,209,210,234] undergo LLPS because of their IDRs, leading to the formation of membrane-less organelles that are essential for cellular function. Weak and multivalent interactions among IDRs enable reversible assembly and dynamic compartmentalization without membranes and play an important role in neurodegenerative proteins [235]. Aberrant phase separation can also lead to the pathological aggregation of these proteins, contributing to neurodegeneration [236]. In neurodegeneration, many disease-associated proteins undergo LLPS separation as part of normal cellular organization, but chronic stress, aging, or impaired proteostasis can drive these normally reversible condensates toward pathology [237].
3.2. Intrinsic Disorder and Aggregation
Intrinsic disorder confers a dual role in neurodegeneration-associated proteins, supporting physiological flexibility and regulation while simultaneously increasing vulnerability to aberrant phase transitions and aggregation. Many proteins, implicated in neurodegenerative disease, including Aβ [238], tau [125], α-synuclein [239], TDP-43, and FUS [201] are highly intrinsically disordered, a property that enables dynamic interactions, extensive PTM, and participation in LLPS required for neuronal plasticity [240]. Structural plasticity and exposed interaction motifs further facilitate aggregation and cross-seeding among IDPs, providing a mechanistic explanation for the frequent emergence of mixed proteinopathies in neurodegeneration [241].
3.3. Intrinsic Disorder as a Driver of Mixed Proteinopathies and Neuropathologies
The role of intrinsic disorder among neurodegeneration-associated proteins highlights its significance in understanding mixed proteinopathies and neuropathologies. This exposition explores shared features of disorder across various proteins, the implications of disorder-facilitated cross-seeding, prion-like behaviors, and convergence of aggregation pathways.
3.4. Shared Disorder Features Across Neurodegeneration-Associated Proteins
Potentially neurodegenerative proteins typically exhibit high conformational flexibility, a lack of stable secondary and 3D structure, and a propensity for aggregation, characteristics often linked to their neurodegenerative properties [165,242] The shared disorder features suggest that these proteins may utilize similar molecular mechanisms that facilitate LLPS and aggregation and interaction with other cellular components, underscoring connectivity in pathological processes across different diseases.
3.5. Disorder-Facilitated Cross-Seeding and Co-Aggregation
IDRs in proteins can promote cross-seeding and co-aggregation—phenomena where one protein facilitates the aggregation of another. Evidence shows that α-synuclein can enhance tau inclusions in neurons, indicating that the aggregation of these proteins is not isolated but rather interconnected [165]. This interplay further supports the concept of mixed proteinopathies and neuropathologies, wherein multiple proteins can aggregate together, contributing to complex disease manifestations [109]. Studies with Aβ and tau suggest similar cross-seeding mechanisms [243,244].
3.6. Prion-Like Behaviors in Disordered Protein Systems
Proteins like α-synuclein and tau demonstrate prion-like behaviors, meaning they can template their misfolded conformations onto native forms, thus propagating their aggregated states [245]. This prion-like propagation can occur in IDP systems, with shared structural features aiding this process. For instance, studies have shown the ability of α-synuclein aggregates to induce the fibrillization of tau, framing both proteins as part of a shared pathological landscape where aberrant conformations are transmitted [109,243].
3.7. Convergence of Aggregation Pathways in Mixed Pathology
There is considerable overlap in the aggregation pathways of different neurodegenerative proteins. This convergence may allow for shared oligopeptide structures to act as nucleation sites for aggregates, accelerating the aggregation process across proteins [246]. For instance, α-synuclein and tau have been shown to influence each other’s aggregation behaviors, leading to a mutual exacerbation of proteinopathies that can be detected in mixed pathology conditions, emphasizing the interconnectedness of these pathways [243].
3.8. Intrinsic Disorder, Aging, and Proteostasis Failure
The intersection of intrinsic disorder, aging, and proteostasis failure provides significant insights into the mechanisms underlying proteinopathies and neurodegeneration. This section discusses the decline in proteostasis with age, the vulnerability of IDPs to age-related stress, the stabilization of aberrant condensates, and the systems-level failures in long-lived neurons [4].
Proteostasis, a balance between protein synthesis, folding, and degradation, declines significantly by age. This failure leads to an accumulation of misfolded proteins, increasing the risk of neurodegenerative diseases like AD and PD [247,248].The decline in proteostasis capacity is marked by decreased efficiency of molecular chaperones and degradation pathways, making it harder for cells to manage protein integrity effectively [247,249].
IDPs are particularly susceptible to aging-related stress, as their structural flexibility makes them more prone to misfolding and aggregation under stress [238,250]. The progressive loss of proteostasis efficiency leads to the aggregation of IDPs, which play pivotal roles in cellular functions but can lead to toxicity when misfolded [247]. For instance, neuronal proteins, including tau and α-synuclein, are susceptible to the decline of proteostasis, further linking age-related impairments with neurodegenerative diseases [250,251].
3.9. Aberrant Stabilization of Condensates and Irreversible Aggregation
Proteins that undergo LLPS can, under pathological conditions, transition into solid-like states and form irreversible aggregates that disrupt normal cellular function [249,252]. During aging, the stabilization of aberrant condensates can lead to irreversible protein aggregation. This transition is particularly troubling in neurodegenerative diseases where proteins like tau and FUS are implicated, as the formation of stable aggregates can lead to neurotoxicity and cellular dysfunction [238,253]. The stress-induced formation of these pathological aggregates highlights the importance of understanding phase transitions in the context of aging-related neurodegeneration and therefore long-lived neurons [249,254].
3.10. Systems-Level Failure to Regulate Disorder in Long-Lived Neurons
Long-lived neurons face compounded challenges in regulating protein homeostasis due to their post-mitotic nature, which limits their ability to eliminate damaged proteins. This leads to an accumulation of disordered proteins and aggregates that interfere with neuronal function [247,255]. The inability of the proteostasis network to adaptively respond to the accumulation of misfolded proteins in neurons leads to the risk for neurodegenerative diseases, indicating a failure in managing intrinsic disorder during aging [249,256].
3.11. Implications for Disease Classification and Mechanistic Understanding: Rethinking Neurodegenerative Diseases as Disorder-Driven Network Failures
Neurodegenerative disorders can be reframed as failures in disorder-driven networks. The intrinsic disorder of various neurodegenerative proteins suggests that their aggregated forms often propagate through similar pathways, leading to interconnected disease manifestations [250,256]. This perspective emphasizes the role of protein disorder in the development and progression of neurodegeneration, necessitating a holistic understanding of these diseases and its therapeutical and experimental implications.
4. Therapeutic and Experimental Implications
4.1. Challenges of Targeting IDPs
4.2. Modulating Phase Behavior and Proteostasis as Therapeutic Strategies
4.3. Disorder-Aware Drug Discovery and Intervention Approaches
Drug discovery efforts should be informed by the properties of intrinsic disorder, enabling the identification of compounds that specifically target disordered and aggregation-prone structures [258,259]. These strategies may help mitigate the detrimental effects of protein aggregates in neurodegeneration.
4.4. Experimental Tools for Studying Intrinsic Disorder in Neurodegeneration
Neuropathological PPI among Aβ, tau, α-synuclein, TDP-43, and FUS are measured experimentally using a diverse array of methodologies that include in vitro, in vivo, and in situ approaches. Each methodology addresses a distinct level of biological inference. In vitro biochemical and biophysical assays—including co-immunoprecipitation (Co-IP), pull-down assays, surface plasmon resonance, and isothermal titration calorimetry—are utilized to measure direct physical binding and binding kinetics. Aggregation and cross-seeding are quantified using Thioflavin-T kinetics, seeded fibrillization assays, real-time quaking-induced conversion (RT-QuIC), and protein misfolding cyclic amplification [260,261].
Cell-based systems enhance physiological relevance through confocal immunofluorescence, proximity ligation assays, Förster resonance energy transfer (FRET), and split-reporter systems to detect intracellular proximity and interactions. These methodologies are supplemented by functional cell-based assays like cytotoxicity assays, calcium imaging, and proteostasis reporters that assess downstream consequences of interactions [261].
In vivo studies utilize transgenic and viral-vector animal models, stereotaxic injection of preformed fibrils, and time-resolved pathology mapping to evaluate synergistic toxicity, directionality, and prion-like propagation of protein aggregates, with behavioral testing and electrophysiology providing systems-level correlates [262,263]. Finally, human neuropathological studies, including post-mortem immunohistochemistry and multiplex immunofluorescence, provide the gold standard. Techniques such as spatial proteomics and cryo-electron microscopy (cryo-EM) are critical for discerning aggregate strains and PTM [264,265].
Importantly, no single method can establish causality independently; robust inference of pathogenic interactions requires convergent evidence across biochemical, cellular, animal, and human tissue studies, along with perturbation or rescue experiments that extend beyond mere correlation [263,266]. These studies lack real-time qualitative and quantitative causality in humans, while animal models only provide a hint about the direction of causality.
4.5. Intrinsic Disorder Versus Protein-Specific Disease Models
Current models often focus on specific proteins implicated in neurodegeneration, which may overlook the contributions of intrinsic disorder and protein interconnectedness. Recognizing the shared features of disordered proteins across different pathologies encourages a shift toward disorder-informed classifications that better represent their complexity [247,267].
4.6. Integrating Intrinsic Disorder into Models of Disease Progression
Incorporating intrinsic disorder into models of disease progression can enhance our understanding of neurodegeneration. By emphasizing the roles of misfolded and aggregated proteins, this integration allows for the identification of novel therapeutic targets and strategies to mitigate the progression of associated disorders [258,268].
4.7. Conceptual Implications for Biomarker Interpretation
The consideration of intrinsic disorder in neurodegeneration has significant implications for biomarker interpretation. Biomarkers might require re-evaluation based on the understanding that aggregate formation and protein misfolding may not be strictly linear processes but rather complex interactions influenced by protein disorder [268,269].
5. Open Questions and Future Directions
Despite strong associations and correlations between PID and neurodegeneration, major conceptual gaps remain. For tau, α-synuclein, TDP-43, and FUS, it is unresolved which disordered conformational sub-ensembles are toxic and how physiological phase separation transitions into irreversible aggregation. It is also unclear whether intrinsic disorder itself enables cross-seeding and mixed pathology or merely amplifies vulnerability under conditions of aging and proteostasis decline. A further unresolved issue is why broadly expressed disordered proteins produce highly selective neuronal degeneration. Aβ, while less intrinsically disordered, may act as an initiator that destabilizes cellular environments rich in disordered proteins. Overall, intrinsic disorder appears to function as a risk amplifier rather than a singular cause, with pathogenic outcomes emerging from context-dependent failures of regulation, buffering, and cellular resilience.
5.1. Quantitative Thresholds Between Functional Disorder and Pathology
A central unresolved question is whether a quantitative or qualitative threshold of intrinsic disorder exists that permits prediction of neuropathological severity or clinical disease. Importantly, the presence of neuropathological proteins in the brain does not uniformly translate into disease, and conversely, clinical neurodegenerative syndromes do not always correspond to a single, well-defined pathology. Much of the current prevalence data relies on post-mortem studies with limited sample sizes and substantial uncertainty arising from retrospective assessment, comorbidities, and confounders. These limitations complicate efforts to draw causal or predictive conclusions and highlight the need for longitudinal and ethically conducted human studies incorporating a risk-benefit assessment with risky elements.
Another major open issue concerns the determinants of reversibility versus irreversibility in protein condensates. Experimental and pathological studies suggest that IDPs can form dynamic, reversible condensates under physiological conditions, but may transition into irreversible aggregates in disease. Factors implicated in this transition include:(1) biophysical properties such as composition, charge distribution, pH, and ionic environment; (2) cross-seeding and direct interactions between different disordered proteins that co-localize within the same cellular compartments; (3) pathological synergy arising from the simultaneous presence of multiple proteinopathies; (4) shared upstream stressors, including inflammation, oxidative stress, and proteostasis failure; and (5) shared downstream consequences such as synaptic dysfunction and neuronal loss. The relative contribution of these factors, and their interaction over time, remains incompletely understood and this knowledge could be an important advantage for systems neuroscience.
6. Conclusions
6.1. Intrinsic Disorder as a Unifying Biophysical Principle
This review highlights PID as a unifying biophysical framework for the understanding of neurodegenerative disease. IDPs play essential physiological roles in the brain, where synaptic plasticity, rapid signaling, and dynamic PPI require structural flexibility. However, these same properties render such proteins vulnerable to dysregulation, misfolding, and pathological interaction. The evidence reviewed here suggests that PID does not represent a pathological feature per se, but rather a context-dependent risk factor whose consequences depend on cellular environment, aging, and network-level interactions.
6.2. From Isolated Proteinopathies to Interacting Disorder-Driven Networks
The isolated look on proteinopathies without considering interacting neuropathological proteins and their PID leads to misunderstanding of the whole picture and must lead to a holistic understanding of neuropathology and neurodegeneration, especially in an aging population.
6.3. Outlook for Disorder-Centric Neurodegeneration Research
The understanding as interacting disorder-driven networks can lead to research which could consider determinants of molecular biological, biophysical, and interconnectedness to move basic research forward and convert it into treatment options in interdisciplinary manner.
Author Contributions
Conceptualization, A.S.S. and V.N.U.; methodology, A.S.S. and V.N.U.; software, V.N.U.; validation, A.S.S. and V.N.U.; investigation, A.S.S. and V.N.U.; data curation, A.S.S. and V.N.U.; writing—original draft preparation, A.S.S. and V.N.U.; writing—review and editing, A.S.S. and V.N.U.; visualization, A.S.S. and V.N.U.; supervision, V.N.U.; project administration, A.S.S. and V.N.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Dataset available on request from the corresponding author.
Acknowledgments
Citation contexts were assessed using the Scite database (Scite, Brooklyn, NY, USA). During the preparation of this work the author(s) used ChatGPT (OpenAI, San Francisco) to improve readability and language of the work. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication. A.S.S. gratefully acknowledges Bennet Lutz for his unwavering support, patience, and encouragement throughout this work in the Christmas break.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Aβ | Amyloid-β |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| CAA | Cerebral amyloid angiopathy |
| CBD | Corticobasal degeneration |
| CSF | Cerebrospinal fluid |
| CTE | Chronic traumatic encephalopathy |
| FTLD | Frontotemporal lobar degeneration |
| FTLD-FUS | Frontotemporal lobar degeneration with FUS pathology |
| FTLD-TDP | Frontotemporal lobar degeneration with TDP-43 pathology |
| FUS | Fused in sarcoma |
| IDP | Intrinsically disordered protein |
| IDR | Intrinsically disordered region |
| LATE | Limbic-predominant age-related TDP-43 encephalopathy |
| LLPS | Liquid–liquid phase separation |
| MSA | Multiple system atrophy |
| NFT | Neurofibrillary tangle |
| NAC | Non-amyloid component |
| NACP | Non-amyloid component precursor |
| PD | Parkinson’s Disease |
| PID | Protein intrinsic disorder |
| PiD | Pick’s disease |
| PSP | Progressive supranuclear palsy |
| PTM | Post-translational modification |
| RNA | Ribonucleic acid |
| SNCA | Synuclein alpha gene |
| TDP-43 | TAR DNA-binding protein 43 |
References
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J Parkinsons Dis 2017, 7, S51–S69. [Google Scholar] [CrossRef]
- Hattiholi, A.; Hegde, H.; Shetty, S.K. Tauopathies: Emerging discoveries on tau protein, with a special focus on Alzheimer’s disease. Neuropeptides 2025, 112, 102536. [Google Scholar] [CrossRef]
- Forrest, S.L.; Kovacs, G.G. Current Concepts of Mixed Pathologies in Neurodegenerative Diseases. Can J Neurol Sci 2023, 50, 329–345. [Google Scholar] [CrossRef]
- Brenowitz, W.D.; Hubbard, R.A.; Keene, C.D.; Hawes, S.E.; Longstreth, W.T., Jr.; Woltjer, R.L.; Kukull, W.A. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement 2017, 13, 654–662. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlapping spectrum of Alzheimer disease, Lewy body disease and vascular pathology. Lancet Neurology 2014. [Google Scholar]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathologica 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J Mol Graph Model 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat Biotechnol 2001, 19, 805–806. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem Sci 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Uversky, V.N. What does it mean to be natively unfolded? Eur J Biochem 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding Protein Non-Folding. Biochimica Et Biophysica Acta (Bba) - Proteins and Proteomics 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics 2008, 9 Suppl 2, S1. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr Opin Struct Biol 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform 2000, 11, 161–171. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. Journal of biomolecular structure & dynamics 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Peng, Z.; Oldfield, C.J.; Xue, B.; Mizianty, M.J.; Dunker, A.K.; Kurgan, L.; Uversky, V.N. A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome. Cellular and molecular life sciences: CMLS 2014, 71, 1477–1504. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cellular and molecular life sciences: CMLS 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cellular and molecular life sciences: CMLS 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 2001, 89–100. [Google Scholar]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys J 2007, 92, 1439–1456. [Google Scholar] [CrossRef]
- 10.1529/biophysj.106.094045.
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics 2007, 8, 211. [Google Scholar] [CrossRef]
- 10.1186/1471-2105-8-211.
- Hemmings, H.C., Jr.; Nairn, A.C.; Aswad, D.W.; Greengard, P. DARPP-32, a dopamine- and adenosine 3’:5’-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. II. Purification and characterization of the phosphoprotein from bovine caudate nucleus. J Neurosci 1984, 4, 99–110. [Google Scholar] [CrossRef]
- Gast, K.; Damaschun, H.; Eckert, K.; Schulze-Forster, K.; Maurer, H.R.; Muller-Frohne, M.; Zirwer, D.; Czarnecki, J.; Damaschun, G. Prothymosin alpha: a biologically active protein with random coil conformation. Biochemistry 1995, 34, 13211–13218. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Freiberger, M.I.; Wolynes, P.G.; Ferreiro, D.U.; Fuxreiter, M. Frustration in Fuzzy Protein Complexes Leads to Interaction Versatility. J Phys Chem B 2021, 125, 2513–2520. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzziness: linking regulation to protein dynamics. Mol Biosyst 2012, 8, 168–177. [Google Scholar] [CrossRef]
- Fuxreiter, M. Towards a Stochastic Paradigm: From Fuzzy Ensembles to Cellular Functions. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Fuxreiter, M. Fuzzy protein theory for disordered proteins. Biochem Soc Trans 2020, 48, 2557–2564. [Google Scholar] [CrossRef]
- Fuxreiter, M. Context-dependent, fuzzy protein interactions: Towards sequence-based insights. Curr Opin Struct Biol 2024, 87, 102834. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol 2012, 725, 1–14. [Google Scholar] [CrossRef]
- Miskei, M.; Gregus, A.; Sharma, R.; Duro, N.; Zsolyomi, F.; Fuxreiter, M. Fuzziness enables context dependence of protein interactions. FEBS Lett 2017, 591, 2682–2695. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett 2015, 589, 2533–2542. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Welch, G.R. “Fuzziness” in the celular interactome: a historical perspective. Adv Exp Med Biol 2012, 725, 184–190. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res 2006, 5, 2985–2995. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol 2006, 2, e100. [Google Scholar] [CrossRef]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9 Suppl 1, S1. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol 2011, 43, 1090–1103. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv Protein Chem 2002, 62, 25–49. [Google Scholar]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. Faseb J 2004, 18, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. On the roles of intrinsically disordered proteins and regions in cell communication and signaling. Cell Commun Signal 2021, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun Signal 2022, 20, 20. [Google Scholar] [CrossRef]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem Rev 2022, 122, 6614–6633. [Google Scholar] [CrossRef]
- Kulkarni, P.; Leite, V.B.P.; Roy, S.; Bhattacharyya, S.; Mohanty, A.; Achuthan, S.; Singh, D.; Appadurai, R.; Rangarajan, G.; Weninger, K.; et al. Intrinsically disordered proteins: Ensembles at the limits of Anfinsen’s dogma. Biophys Rev (Melville) 2022, 3, 011306. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol 2008, 4, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Toth-Petroczy, A.; Kraut, D.A.; Matouschek, A.; Lim, R.Y.; Xue, B.; Kurgan, L.; Uversky, V.N. Disordered proteinaceous machines. Chem Rev 2014, 114, 6806–6843. [Google Scholar] [CrossRef]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett 2015, 589, 2498–2506. [Google Scholar] [CrossRef]
- El Hadidy, N.; Uversky, V.N. Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Toth-Petroczy, A.; Oldfield, C.J.; Simon, I.; Takagi, Y.; Dunker, A.K.; Uversky, V.N.; Fuxreiter, M. Malleable machines in transcription regulation: the mediator complex. PLoS Comput Biol 2008, 4, e1000243. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Plokhikh, K.S.; Manuylov, V.D.; Chesnokov, Y.M.; Vasilov, R.G.; Kuznetsova, I.M.; Turoverov, K.K.; Gordeliy, V.I.; Fonin, A.V.; et al. Order wrapped in chaos: On the roles of intrinsically disordered proteins and RNAs in the arrangement of the mitochondrial enzymatic machines. Int J Biol Macromol 2024, 267, 131455. [Google Scholar] [CrossRef]
- Coelho Ribeiro Mde, L.; Espinosa, J.; Islam, S.; Martinez, O.; Thanki, J.J.; Mazariegos, S.; Nguyen, T.; Larina, M.; Xue, B.; Uversky, V.N. Malleable ribonucleoprotein machine: protein intrinsic disorder in the Saccharomyces cerevisiae spliceosome. PeerJ 2013, 1, e2. [Google Scholar] [CrossRef]
- Peng, Z.; Mizianty, M.J.; Xue, B.; Kurgan, L.; Uversky, V.N. More than just tails: intrinsic disorder in histone proteins. Mol Biosyst 2012, 8, 1886–1901. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci 2014, 23, 1077–1093. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front Genet 2018, 9, 158. [Google Scholar] [CrossRef]
- Meng, F.; Na, I.; Kurgan, L.; Uversky, V.N. Compartmentalization and Functionality of Nuclear Disorder: Intrinsic Disorder and Protein-Protein Interactions in Intra-Nuclear Compartments. Int J Mol Sci 2015, 17. [Google Scholar] [CrossRef]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193 (1700112 pages. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The roles of intrinsic disorder-based liquid-liquid phase transitions in the “Dr. Jekyll-Mr. Hyde” behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 2017, 13, 2115–2162. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv Colloid Interface Sci 2017, 239, 97–114. [Google Scholar] [CrossRef]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: overview of the evolution of the cell compartmentalization concept. Cellular and molecular life sciences: CMLS 2022, 79, 251. [Google Scholar] [CrossRef]
- Fonin, A.V.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B.Y.; Kulkarni, P.; Uversky, V.N. Biological soft matter: intrinsically disordered proteins in liquid-liquid phase separation and biomolecular condensates. Essays Biochem 2022, 66, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Intrinsically disordered proteins in crowded milieu: when chaos prevails within the cellular gumbo. Cellular and molecular life sciences: CMLS 2018, 75, 3907–3929. [Google Scholar] [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem Sci 2019, 44, 716–728. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett 2015, 589, 15–22. [Google Scholar] [CrossRef]
- Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Intrinsic Disorder-Based Emergence in Cellular Biology: Physiological and Pathological Liquid-Liquid Phase Transitions in Cells. Polymers 2019, 11, 990. [Google Scholar] [CrossRef]
- Darling, A.L.; Shorter, J. Combating deleterious phase transitions in neurodegenerative disease. Biochim Biophys Acta Mol Cell Res 2021, 1868, 118984. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2Concept. Annual Review of Biophysics 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Gadhave, K.; Gehi, B.R.; Kumar, P.; Xue, B.; Uversky, V.N.; Giri, R. The dark side of Alzheimer’s disease: unstructured biology of proteins from the amyloid cascade signaling pathway. Cellular and molecular life sciences: CMLS 2020, 77, 4163–4208. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Front Mol Biosci 2014, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Rebellion of the deregulated regulators: What is the clinical relevance of studying intrinsically disordered proteins? Expert Rev Proteomics 2022, 19, 279–282. [Google Scholar] [CrossRef]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014, 6, 82. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Milenkovic, I.; Wohrer, A.; Hoftberger, R.; Gelpi, E.; Haberler, C.; Honigschnabl, S.; Reiner-Concin, A.; Heinzl, H.; Jungwirth, S.; et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013, 126, 365–384. [Google Scholar] [CrossRef]
- Chen, G.-f.; Xu, T.-H.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacologica Sinica 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental Constraints on Quaternary Structure in Alzheimer’s Β-Amyloid Fibrils. Biochemistry 2005, 45, 498–512. [Google Scholar] [CrossRef]
- Selkoe, D.J. Translating Cell Biology Into Therapeutic Advances in Alzheimer’s Disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid Β-Protein (Aβ) Assembly: Aβ40 and Aβ42 Oligomerize Through Distinct Pathways. Proceedings of the National Academy of Sciences 2002, 100, 330–335. [Google Scholar] [CrossRef]
- Hamley, I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chemical Reviews 2012, 112, 5147–5192. [Google Scholar] [CrossRef]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem Biophys Res Commun 2011, 411, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Shao, H.; Zhang, Y.; Li, H.; Menon, N.K.; Neuhaus, E.B.; Brewer, J.M.; Byeon, I.J.; Ray, D.G.; Vitek, M.P.; et al. Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 2004, 126, 1992–2005. [Google Scholar] [CrossRef] [PubMed]
- Lazo, N.D.; Grant, M.A.; Condron, M.C.; Rigby, A.C.; Teplow, D.B. On the nucleation of amyloid beta-protein monomer folding. Protein Sci 2005, 14, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Saieva, S.; Morales, R. Misfolded amyloid-beta conformational variants (strains) as drivers of Alzheimer’s disease neuropathology. Neural Regen Res 2025, 20, 3219–3220. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014, 20, 130–138. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Morales, R.; Green, K.M.; Soto, C. Cross currents in protein misfolding disorders: interactions and therapy. CNS Neurol Disord Drug Targets 2009, 8, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate beta-Amyloid Aggregation in Alzheimer’s Disease. Front Aging Neurosci 2018, 10, 359. [Google Scholar] [CrossRef]
- Collaborators, G.D.F. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Sagare, A.P.; Deane, R.; Zloković, B.V. Low-Density Lipoprotein Receptor-Related Protein 1: A Physiological Aβ Homeostatic Mechanism With Multiple Therapeutic Opportunities. Pharmacology & Therapeutics 2012, 136, 94–105. [Google Scholar] [CrossRef]
- Xin, S.-H.; Tan, L.; Cao, X.P.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotoxicity Research 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Alvira, X.; Carro, E. Clearance of Amyloid-Β Peptide Across the Choroid Plexus in Alzheimers Disease. Current Aging Science 2010, 3, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Verma, A.; Bhaskar, U.; Sarkar, B.; Kallianpur, M.; Vishvakarma, V.; Das, A.K.; Garai, K.; Mukherjee, O.; Ishii, K.; et al. A Toxicogenic Interaction Between Intracellular Amyloid-Β and Apolipoprotein-E. Acs Chemical Neuroscience 2024, 15, 1265–1275. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Padilla-Godínez, F.J.; Vázquez-García, E.R.; Trujillo-Villagrán, M.I.; Soto-Rojas, L.O.; Palomero-Rivero, M.; Hernández-González, O.; Pérez-Eugenio, F.; Collazo-Navarrete, O.; Arias-Carrión, O.; Guerra-Crespo, M. A-Synuclein and Tau: Interactions, Cross-Seeding, and the Redefinition of Synucleinopathies as Complex Proteinopathies. Frontiers in Neuroscience 2025, 19. [Google Scholar] [CrossRef]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-Derived Frontotemporal Lobar Degeneration Brain Extracts Induce Formation and Spreading of TDP-43 Pathology in Vivo. Nature Communications 2018, 9. [Google Scholar] [CrossRef]
- Baumkotter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J Neurosci 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, B.J.; Stark, C.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Livstone, M.; Oughtred, R.; Lackner, D.H.; Bahler, J.; Wood, V.; et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res 2008, 36, D637–640. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 2001, 276, 20466–20473. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A 2020, 117, 33254–33262. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.; Zhang, F.; Chen, J.; Cai, C.; Guo, Y.; Lei, Z.; Zeng, L.H.; Zi, D.; Shen, Y.; et al. The pathogenic APP N-terminal Val225Ala mutation alters tau protein liquid-liquid phase separation and exacerbates synaptic damage. Mol Psychiatry 2025, 30, 2316–2334. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 2013, 41, D508–516. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem Rev 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N. A Decade and a Half of Protein Intrinsic Disorder: Biology Still Waits for Physics. Protein Science 2013, 22, 693–724. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Frontiers in Physics 2019, 7. [Google Scholar] [CrossRef]
- Bianchi, G.; Mangiagalli, M.; Ami, D.; Ahmed, J.; Lombardi, S.; Longhi, S.; Natalello, A.; Tompa, P.; Brocca, S. Condensation of the N-Terminal Domain of Human Topoisomerase 1 Is Driven by Electrostatic Interactions and Tuned by Its Charge Distribution. International Journal of Biological Macromolecules 2024, 254, 127754. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their Environment: Effects of Strong Denaturants, Temperature, pH, Counter Ions, Membranes, Binding Partners, Osmolytes, and Macromolecular Crowding. The Protein Journal 2009, 28, 305–325. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. RNA stores tau reversibly in complex coacervates. PLoS Biol 2017, 15, e2002183. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 2018, 37. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.H.; Surewicz, K.; Surewicz, W.K. Liquid-liquid phase separation of tau protein: The crucial role of electrostatic interactions. J Biol Chem 2019, 294, 11054–11059. [Google Scholar] [CrossRef]
- Margittai, M. Driving tau into phase-separated liquid droplets. J Biol Chem 2019, 294, 11060–11061. [Google Scholar] [CrossRef]
- Wegmann, S. Liquid-Liquid Phase Separation of Tau Protein in Neurobiology and Pathology. Adv Exp Med Biol 2019, 1184, 341–357. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, K.; Surewicz, W.K. Regulatory mechanisms of tau protein fibrillation under the conditions of liquid-liquid phase separation. Proc Natl Acad Sci U S A 2020, 117, 31882–31890. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun 2020, 11, 2809. [Google Scholar] [CrossRef] [PubMed]
- Ukmar-Godec, T.; Wegmann, S.; Zweckstetter, M. Biomolecular condensation of the microtubule-associated protein tau. Semin Cell Dev Biol 2020, 99, 202–214. [Google Scholar] [CrossRef]
- Zhang, X.; Vigers, M.; McCarty, J.; Rauch, J.N.; Fredrickson, G.H.; Wilson, M.Z.; Shea, J.E.; Han, S.; Kosik, K.S. The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J Cell Biol 2020, 219. [Google Scholar] [CrossRef]
- Dong, X.; Bera, S.; Qiao, Q.; Tang, Y.; Lao, Z.; Luo, Y.; Gazit, E.; Wei, G. Liquid-Liquid Phase Separation of Tau Protein Is Encoded at the Monomeric Level. J Phys Chem Lett 2021, 12, 2576–2586. [Google Scholar] [CrossRef]
- Lin, Y.; Fichou, Y.; Longhini, A.P.; Llanes, L.C.; Yin, P.; Bazan, G.C.; Kosik, K.S.; Han, S. Liquid-Liquid Phase Separation of Tau Driven by Hydrophobic Interaction Facilitates Fibrillization of Tau. J Mol Biol 2021, 433, 166731. [Google Scholar] [CrossRef]
- Chakraborty, P.; Zweckstetter, M. Phase separation of the microtubule-associated protein tau. Essays Biochem 2022, 66, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Prince, P.R.; Hochmair, J.; Brognaro, H.; Gevorgyan, S.; Franck, M.; Schubert, R.; Lorenzen, K.; Yazici, S.; Mandelkow, E.; Wegmann, S.; et al. Initiation and modulation of Tau protein phase separation by the drug suramin. Sci Rep 2023, 13, 3963. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Velazquez, C.L.; Vellosillo, T.; Guadalupe, K.; Schmidt, H.B.; Yu, F.; Moses, D.; Brophy, J.A.N.; Cosio-Acosta, D.; Das, A.; Wang, L.; et al. Intrinsically Disordered Protein Biosensor Tracks the Physical-Chemical Effects of Osmotic Stress on Cells. Nature Communications 2021, 12. [Google Scholar] [CrossRef]
- Ayers, J.I.; Giasson, B.I.; Borchelt, D. Prion-Like Spreading in Tauopathies. Biological Psychiatry 2018, 83, 337–346. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology 1992, 42, 631–631. [Google Scholar] [CrossRef]
- Eisele, Y.S. From Soluble Aβ to Progressive Aβ Aggregation: Could Prion-Like Templated Misfolding Play a Role? Brain Pathology 2013, 23, 333–341. [Google Scholar] [CrossRef]
- Lin, Y.H.; Chan, H.S. Phase Separation and Single-Chain Compactness of Charged Disordered Proteins Are Strongly Correlated. Biophysical Journal 2017, 112, 2043–2046. [Google Scholar] [CrossRef]
- McFadden, W.M.; Yanowitz, J.L. Idpr: A Package for Profiling and Analyzing Intrinsically Disordered Proteins in R. Plos One 2022, 17, e0266929. [Google Scholar] [CrossRef]
- Uversky, V.N. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000Res 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. Journal of biomolecular structure & dynamics 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.; Stuiver, M.; Verzini, S.; Lorenz, D.; Rossum, M.v.; Goldfarb, D.; et al. Structural Disorder of Monomeric A-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of A-Synuclein by Chaperones in Mammalian Cells. Nature 2019, 577, 127–132. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Trexler, A.; Rhoades, E. A-Synuclein Binds Large Unilamellar Vesicles as an Extended Helix. Biochemistry 2009, 48, 2304–2306. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Piroska, L.; Fenyi, A.; Thomas, S.; Plamont, M.A.; Redeker, V.; Melki, R.; Gueroui, Z. alpha-Synuclein liquid condensates fuel fibrillar alpha-synuclein growth. Sci Adv 2023, 9, eadg5663. [Google Scholar] [CrossRef]
- Rodriguez, L.C.; Foressi, N.N.; Celej, M.S. Liquid-liquid phase separation of tau and alpha-synuclein: A new pathway of overlapping neuropathologies. Biochem Biophys Res Commun 2024, 741, 151053. [Google Scholar] [CrossRef]
- Ruiz-Ortega, E.D.; Wilkaniec, A.; Adamczyk, A. Liquid-liquid phase separation and conformational strains of alpha-Synuclein: implications for Parkinson’s disease pathogenesis. Front Mol Neurosci 2024, 17, 1494218. [Google Scholar] [CrossRef] [PubMed]
- Sternke-Hoffmann, R.; Sun, X.; Menzel, A.; Pinto, M.D.S.; Venclovaite, U.; Wordehoff, M.; Hoyer, W.; Zheng, W.; Luo, J. Phase Separation and Aggregation of alpha-Synuclein Diverge at Different Salt Conditions. Adv Sci (Weinh) 2024, 11, e2308279. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That A-Synuclein Oligomers Are Toxic. Proceedings of the National Academy of Sciences 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.; Uversky, V.N. A-Synuclein Misfolding and Parkinson’s Disease. Biochimica Et Biophysica Acta (Bba) - Molecular Basis of Disease 2012, 1822, 261–285. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett 2001, 500, 105–108. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 2002, 277, 1641–1644. [Google Scholar] [CrossRef]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Elzen, R.V.; Johannessen, C.; Maudsley, S.; Lambeir, A.M.; Sobott, F. Metal Ions Shape A-Synuclein. Scientific Reports 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.; Oueslati, A.; Masliah, E. The Many Faces of A-Synuclein: From Structure and Toxicity to Therapeutic Target. Nature Reviews Neuroscience 2012, 14, 38–48. [Google Scholar] [CrossRef]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinsons Disease: Structure and Aggregation of &Amp;#945;- Synuclein. Current Protein and Peptide Science 2009, 10, 483–499. [Google Scholar] [CrossRef]
- Stephens, A.D.; Zacharopoulou, M.; Moons, R.; Fusco, G.; Seetaloo, N.; Chiki, A.; Woodhams, P.J.; Mela, I.; Lashuel, H.A.; Phillips, J.J.; et al. Extent of N-Terminus Exposure of Monomeric Alpha-Synuclein Determines Its Aggregation Propensity. Nature Communications 2020, 11. [Google Scholar] [CrossRef]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-Terminal Acetylation of A-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nature Cell Biology 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Guo, J.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct A-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Foressi, N.N.; Rodriguez, L.C.; Celej, M.S. Heterotypic liquid-liquid phase separation of tau and alpha-synuclein: Implications for overlapping neuropathologies. Biochim Biophys Acta Proteins Proteom 2023, 1871, 140950. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. A-Synuclein in the Pathophysiology of Alzheimer’s Disease. Molecular Neurodegeneration 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Bellomo, G.; Giannandrea, D.; Carlo, C.D.; Qureshi, M.M.; Ardah, M.T.; Varghese, S.; Bonanni, L.; Borroni, B.; et al. Cerebrospinal Fluid Tau/A-synuclein Ratio in Parkinson’s Disease and Degenerative Dementias. Movement Disorders 2011, 26, 1428–1435. [Google Scholar] [CrossRef]
- Pajkos, M.; Clerc, I.; Zanon, C.; Bernado, P.; Cortes, J. AFflecto: A web server to generate conformational ensembles of flexible proteins from AlphaFold models. J Mol Biol 2025, 437, 169003. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J Mol Graph 1996, 14, 33-38, 27-38. [Google Scholar] [CrossRef]
- Santamaria, N.; Alhothali, M.; Alfonso, M.H.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cellular and molecular life sciences: CMLS 2017, 74, 1297–1318. [Google Scholar] [CrossRef]
- Loughlin, F.E.; Wilce, J.A. TDP-43 and FUS-structural insights into RNA recognition and self-association. Curr Opin Struct Biol 2019, 59, 134–142. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.A.; Unger, T.L.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated Gene TDP-43 Leads to Neuronal Death and Degeneration in Mice. Journal of Clinical Investigation 2011, 121, 726–738. [Google Scholar] [CrossRef]
- Lang, R.; Hodgson, R.E.; Shelkovnikova, T.A. TDP-43 in nuclear condensates: where, how, and why. Biochem Soc Trans 2024, 52, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc Natl Acad Sci U S A 2017, 114, E2466–E2475. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Chiang, W.C.; Chou, P.C.; Wang, W.J.; Huang, J.R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J Biol Chem 2018, 293, 6090–6098. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J Biol Chem 2019, 294, 6306–6317. [Google Scholar] [CrossRef]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338 e328. [Google Scholar] [CrossRef]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S.; et al. Stress Induces Dynamic, Cytotoxicity-Antagonizing TDP-43 Nuclear Bodies via Paraspeckle LncRNA NEAT1-Mediated Liquid-Liquid Phase Separation. Mol Cell 2020, 79, 443–458 e447. [Google Scholar] [CrossRef]
- Webber, C.J.; Lei, S.E.; Wolozin, B. The pathophysiology of neurodegenerative disease: Disturbing the balance between phase separation and irreversible aggregation. Prog Mol Biol Transl Sci 2020, 174, 187–223. [Google Scholar] [CrossRef]
- Pakravan, D.; Michiels, E.; Bratek-Skicki, A.; De Decker, M.; Van Lindt, J.; Alsteens, D.; Derclaye, S.; Van Damme, P.; Schymkowitz, J.; Rousseau, F.; et al. Liquid-Liquid Phase Separation Enhances TDP-43 LCD Aggregation but Delays Seeded Aggregation. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- Bhopatkar, A.A.; Dhakal, S.; Abernathy, H.G.; Morgan, S.E.; Rangachari, V. Charge and redox states modulate granulin-TDP-43 coacervation toward phase separation or aggregation. Biophys J 2022, 121, 2107–2126. [Google Scholar] [CrossRef] [PubMed]
- Staderini, T.; Bigi, A.; Mongiello, D.; Cecchi, C.; Chiti, F. Biophysical characterization of full-length TAR DNA-binding protein (TDP-43) phase separation. Protein Sci 2022, 31, e4509. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Molecular Mechanisms of Phase Separation and Amyloidosis of ALS/FTD-linked FUS and TDP-43. Aging Dis 2024, 15, 2084–2112. [Google Scholar] [CrossRef]
- Sergeeva, O.S.; Neklesova, M.V.; Reushev, V.A.; Artemov, A.V.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N.; Fonin, A.V. On the potential roles of TDP-43 in the formation of membraneless organelles and their transformation into toxic aggregates. Biochem Biophys Res Commun 2025, 788, 152808. [Google Scholar] [CrossRef]
- Benajiba, L.; Ber, I.L.; Camuzat, A.; Lacoste, M.; Thomas-Antérion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Décousus, M.; et al. Annals of Neurology 2009, 65, 470–473. [CrossRef] [PubMed]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White, C.L.; Schneider, J.A.; Kretzschmar, H.A.; et al. TDP-43 in Familial and Sporadic Frontotemporal Lobar Degeneration With Ubiquitin Inclusions. American Journal of Pathology 2007, 171, 227–240. [Google Scholar] [CrossRef]
- Robinson, J.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Deerlin, V.M.V.; Yan, N.; Yousef, A.H.; et al. Neurodegenerative Disease Concomitant Proteinopathies Are Prevalent, Age-Related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Tosakulwong, N. TDP-43 pathology in aging and Alzheimer’s disease. Lancet Neurology 2016, 15, 129–138. [Google Scholar] [CrossRef]
- Nelson, P.T.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; Coyle-Gilchrist, I.T.S.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE). Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and aging. Brain Pathology 2017, 27, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Mackenzie, I.R.A. Review: Neuropathology of TDP-43 proteinopathies. Neuropathology and Applied Neurobiology 2019, 45, 521–537. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takanashi, K. FUS interacts with nuclear matrix-associated protein SAFB1 as well as Matrin3 to regulate splicing and ligand-mediated transcription. Sci Rep 2016, 6, 35195. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS/TDP-43 paradigm. Cold Spring Harbor Perspectives in Biology 2009, 1, a003689. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front Aging Neurosci 2015, 7, 18. [Google Scholar] [CrossRef]
- Bonucci, A.; Murrali, M.G.; Banci, L.; Pierattelli, R. A combined NMR and EPR investigation on the effect of the disordered RGG regions in the structure and the activity of the RRM domain of FUS. Sci Rep 2020, 10, 20956. [Google Scholar] [CrossRef]
- Soranno, A.; Koenig, I.; Borgia, M.B.; Hofmann, H.; Zosel, F.; Nettels, D.; Schuler, B. Single-Molecule Spectroscopy Reveals Polymer Effects of Disordered Proteins in Crowded Environments. Proceedings of the National Academy of Sciences 2014, 111, 4874–4879. [Google Scholar] [CrossRef]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol Cell 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Strohl, F.; et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 2018, 173, 720–734 e715. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat Struct Mol Biol 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Boczek, E.E.; Fursch, J.; Niedermeier, M.L.; Jawerth, L.; Jahnel, M.; Ruer-Gruss, M.; Kammer, K.M.; Heid, P.; Mediani, L.; Wang, J.; et al. HspB8 prevents aberrant phase transitions of FUS by chaperoning its folded RNA-binding domain. Elife 2021, 10. [Google Scholar] [CrossRef]
- Davis, R.B.; Kaur, T.; Moosa, M.M.; Banerjee, P.R. FUS oncofusion protein condensates recruit mSWI/SNF chromatin remodeler via heterotypic interactions between prion-like domains. Protein Sci 2021, 30, 1454–1466. [Google Scholar] [CrossRef]
- Joshi, A.; Walimbe, A.; Avni, A.; Rai, S.K.; Arora, L.; Sarkar, S.; Mukhopadhyay, S. Single-molecule FRET unmasks structural subpopulations and crucial molecular events during FUS low-complexity domain phase separation. Nat Commun 2023, 14, 7331. [Google Scholar] [CrossRef]
- Esteban-Hofer, L.; Emmanouilidis, L.; Yulikov, M.; Allain, F.H.; Jeschke, G. Ensemble structure of the N-terminal domain (1-267) of FUS in a biomolecular condensate. Biophys J 2024, 123, 538–554. [Google Scholar] [CrossRef]
- Thirumalai, D.; Kumar, A.; Chakraborty, D.; Straub, J.E.; Mugnai, M.L. Conformational fluctuations and phases in fused in sarcoma (FUS) low-complexity domain. Biopolymers 2024, 115, e23558. [Google Scholar] [CrossRef]
- Miller, A.; Toprakcioglu, Z.; Qamar, S.; St George-Hyslop, P.; Ruggeri, F.S.; Knowles, T.P.J.; Vendruscolo, M. Nanoscale profiling of evolving intermolecular interactions in ageing FUS condensates. Commun Chem 2025, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.L.; Mohanty, P.; Mittal, J. Elucidation of the Molecular Interaction Network Underlying Full-Length FUS Conformational Transitions and Its Phase Separation Using Atomistic Simulations. J Phys Chem B 2025, 129, 8843–8857. [Google Scholar] [CrossRef]
- Zheng, T.; Sojitra, K.A.; Cummings, S.; Chen, Q.; Mohanty, P.; Mittal, J.; Fawzi, N.L. RNA modulates FUS condensate assembly, dynamics, and aggregation through diverse molecular contacts. bioRxiv 2025. [Google Scholar] [CrossRef]
- Oliveira, N.R.; Siqueira, A.S.; Bueno, P.S.A.; Gonçalves, E.C.; Zanette, J. Metal-Coordination Specificity and Structural Dynamics of C. Elegans Metallothionein I: Insights From 3D Modeling and MD Simulations. Proteins Structure Function and Bioinformatics 2025. [Google Scholar] [CrossRef]
- Chen, H.; Meisburger, S.P.; Pabit, S.A.; Sutton, J.L.; Webb, W.W.; Pollack, L. Ionic Strength-Dependent Persistence Lengths of Single-Stranded RNA and DNA. Proceedings of the National Academy of Sciences 2011, 109, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia. Acta Neuropathologica 2016, 132, 891–918. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.M.; Kasarskis, E.J.; Munsat, T.L.; et al. Mutations in The. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R.A. A New Subtype of Frontotemporal Lobar Degeneration With FUS Pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef]
- Mehdiabadi, M. Modeling Intrinsically Disordered Regions From AlphaFold2 to AlphaFold3. Protein Science 2025, 35. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Journal 2016, 35, 595–608. [Google Scholar] [CrossRef]
- Lashuel, H.A. Structural biology of alpha-synuclein and its aggregation. Nature Reviews Molecular Cell Biology 2020. [Google Scholar]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Nastou, K.; Koutrouli, M.; Kirsch, R.; Mehryary, F.; Hachilif, R.; Hu, D.; Peluso, M.E.; Huang, Q.; Fang, T.; et al. The STRING database in 2025: protein networks with directionality of regulation. Nucleic Acids Res 2025, 53, D730–D737. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Uversky, V.N. Analyzing IDPs in interactomes. In Intrinsically Disordered Proteins;Volume Methods in Molecular Biology; Kragelund, B.B., Skriver, K., Eds.; Humana New York, NY, 2020; pp. 895–945. [Google Scholar]
- Mohammed, A.S.; Uversky, V.N. Intrinsic Disorder as a Natural Preservative: High Levels of Intrinsic Disorder in Proteins Found in the 2600-Year-Old Human Brain. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol Biol 2011, 77, 205–223. [Google Scholar] [CrossRef]
- Xue, B.; Oldfield, C.J.; Van, Y.Y.; Dunker, A.K.; Uversky, V.N. Protein intrinsic disorder and induced pluripotent stem cells. Mol Biosyst 2012, 8, 134–150. [Google Scholar] [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 2012, 128–139. [Google Scholar]
- Calculli, G.; Lee, H.J.; Shen, K.; Pham, U.; Herholz, M.; Trifunović, A.; Dillin, A.; Vı́lchez, D. Systemic Regulation of Mitochondria by Germline Proteostasis Prevents Protein Aggregation in the Soma Of C. Science Advances 2021, 7. [Google Scholar] [CrossRef]
- Sade, D.; Shaham-Niv, S.; Arnon, Z.A.; Tavassoly, O.; Gazit, E. Seeding of Proteins Into Amyloid Structures by Metabolite Assemblies May Clarify Certain Unexplained Epidemiological Associations. Open Biology 2018, 8, 170229. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid–liquid Phase Separation in Human Health and Diseases. Signal Transduction and Targeted Therapy 2021, 6. [Google Scholar] [CrossRef]
- Carey, J.L.; Guo, L. Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases. Frontiers in Molecular Biosciences 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-Like RNA Binding Proteins. Cell 2018, 174, 688–699.e616. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated With Neurodegeneration. International Journal of Molecular Sciences 2023, 24, 3380. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Dormann, D. Liquid–Liquid Phase Separation in Disease. Annual Review of Genetics 2019, 53, 171–194. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. International Journal of Molecular Sciences 2020, 21, 3028. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Смирнoва, Е.В.; Rakitina, T.V.; Ziganshin, R.; Arapidi, G.; Saratov, G.A.; Kudriaeva, A.A.; Belogurov, A.A. Comprehensive Atlas of the Myelin Basic Protein Interaction Landscape. Biomolecules 2021, 11, 1628. [Google Scholar] [CrossRef]
- Sandeep, K.; Savastano, A.; Singh, P.; Mukhopadhyay, S.; Zweckstetter, M. Liquid–liquid Phase Separation of Tau: From Molecular Biophysics to Physiology and Disease. Protein Science 2021, 30, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding Effects of Amyloid Β-protein and A-synuclein. Journal of Neurochemistry 2012, 122, 883–890. [Google Scholar] [CrossRef]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-Level Secondary Structure, Polymorphism, and Dynamics of Full-Length A-Synuclein Fibrils Studied by Solid-State NMR. Proceedings of the National Academy of Sciences 2005, 102, 15871–15876. [Google Scholar] [CrossRef]
- Vasconcelos, B.; Stancu, I.C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Kolen, K.V.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.N.; et al. Heterotypic Seeding of Tau Fibrillization by Pre-Aggregated Abeta Provides Potent Seeds for Prion-Like Seeding and Propagation of Tau-Pathology in Vivo. Acta Neuropathologica 2016, 131, 549–569. [Google Scholar] [CrossRef]
- Moreno-González, I.; Edwards, G.A.; Salvadores, N.; Shahnawaz, M.; Díaz-Espinoza, R.; Soto, C. Molecular Interaction Between Type 2 Diabetes and Alzheimer’s Disease Through Cross-Seeding of Protein Misfolding. Molecular Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef]
- Katorcha, E.; Makarava, N.; Lee, Y.J.; Lindberg, I.; Monteiro, M.J.; Kovács, G.G.; Baskakov, I.V. Cross-Seeding of Prions by Aggregated A-Synuclein Leads to Transmissible Spongiform Encephalopathy. Plos Pathogens 2017, 13, e1006563. [Google Scholar] [CrossRef] [PubMed]
- Cukalevski, R.; Yang, X.; Meisl, G.; Weininger, U.; Bernfur, K.; Frohm, B.; Knowles, T.P.J.; Linse, S. The Aβ40 and Aβ42 Peptides Self-Assemble Into Separate Homomolecular Fibrils in Binary Mixtures but Cross-React During Primary Nucleation. Chemical Science 2015, 6, 4215–4233. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and aggregation diseases. Nature Reviews Molecular Cell Biology 2014. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 Diabetes as a Protein Misfolding Disease. Trends in Molecular Medicine 2015, 21, 439–449. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The Proteostasis Network and Its Decline in Ageing. Nature Reviews Molecular Cell Biology 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular Strategies for Controlling Protein Aggregation. Nature Reviews Molecular Cell Biology 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Kikis, E.A. The Struggle by Caenorhabditis Elegans to Maintain Proteostasis During Aging and Disease. Biology Direct 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Berry, J.; Pannucci, N.L.; Haataja, M.; Toettcher, J.E.; Brangwynne, C.P. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 2017, 168, 159–171.e114. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annual Review of Biochemistry 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Zhou, Z.; Selvaratnam, T.; Lee, J.C.T.; Chao, Y.; Tan, E.K. Molecular Targets for Modulating the Protein Translation Vital to Proteostasis and Neuron Degeneration in Parkinson’s Disease. Translational Neurodegeneration 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. The Mammalian Disaggregase Machinery: Hsp110 Synergizes With Hsp70 and Hsp40 to Catalyze Protein Disaggregation and Reactivation in a Cell-Free System. Plos One 2011, 6, e26319. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Lumpkin, C.J.; Patel, H.S.; Potts, G.K.; Chaurasia, S.; Gibilisco, L.; Srivastava, G.; Lee, J.Y.; Brown, N.J.; Amarante, P.; Williams, J.D.; et al. Broad Proteomics Analysis of Seeding-Induced Aggregation of A-Synuclein in M83 Neurons Reveals Remodeling of Proteostasis Mechanisms That Might Contribute to Parkinson’s Disease Pathogenesis. Molecular Brain 2024, 17. [Google Scholar] [CrossRef]
- Lim, J.; Yue, Z. Neuronal Aggregates: Formation, Clearance, and Spreading. Developmental Cell 2015, 32, 491–501. [Google Scholar] [CrossRef]
- Karr, J.W.; Szalai, V.A. Cu(II) Binding to Monomeric, Oligomeric, and Fibrillar Forms of the Alzheimer’s Disease Amyloid-Β Peptide. Biochemistry 2008, 47, 5006–5016. [Google Scholar] [CrossRef]
- Limorenko, G.; Lashuel, H.A. Revisiting the Grammar of Tau Aggregation and Pathology Formation: How New Insights From Brain Pathology Are Shaping How We Study and Target Tauopathies. Chemical Society Reviews 2022, 51, 513–565. [Google Scholar] [CrossRef]
- Parekh, P.; Badachhape, A.; Tanifum, E.A.; Annapragada, A.; Ghaghada, K.B. Advances in Nanoprobes for Molecular MRI of Alzheimer’s Disease. Wiley Interdisciplinary Reviews Nanomedicine and Nanobiotechnology 2024, 16. [Google Scholar] [CrossRef]
- Mammadova, N.; Summers, C.M.; Kokemuller, R.; He, Q.; Ding, S.; Baron, T.; Yu, C.; Valentine, R.J.; Sakaguchi, D.S.; Kanthasamy, A.G.; et al. Accelerated Accumulation of Retinal A-Synuclein (pSer129) and Tau, Neuroinflammation, and Autophagic Dysregulation in a Seeded Mouse Model of Parkinson’s Disease. Neurobiology of Disease 2019, 121, 1–16. [Google Scholar] [CrossRef]
- Shahpasand-Kroner, H.; Siddique, I.; Malik, R.K.; Linares, G.; Ivanova, M.I.; Ichida, J.K.; Weil, T.; Münch, J.; Sánchez-García, E.; Klärner, F.G.; et al. Molecular Tweezers: Supramolecular Hosts With Broad-Spectrum Biological Applications. Pharmacological Reviews 2023, 75, 263–308. [Google Scholar] [CrossRef]
- Illes-Toth, E.; Rempel, D.L.; Gross, M.L. Exploration of Resveratrol as a Potent Modulator of A-Synuclein Fibril Formation. Acs Chemical Neuroscience 2024, 15, 503–516. [Google Scholar] [CrossRef]
- Lisi, I.; Mazzone, E.; Kobeissy, F.; Schneider, A.L.; Arrastia, R.D.; Menon, D.; Smith, D.H.; Zanier, E.R.; Abbasloo, E.; Agrawal, A.; et al. Exploiting Blood-Based Biomarkers to Align Preclinical Models With Human Traumatic Brain Injury. Brain 2024, 148, 1062–1080. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the Tightrope: Proteostasis and Neurodegenerative Disease. Journal of Neurochemistry 2016, 137, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein Homeostasis, Aging and Alzheimer’s Disease. Molecular Neurobiology 2012, 46, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Brandvold, K.; Morimoto, R.I. The Chemical Biology of Molecular Chaperones—Implications for Modulation of Proteostasis. Journal of Molecular Biology 2015, 427, 2931–2947. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Native protein monomers in healthy cells can reversibly assemble into oligomers or undergo LLPS to form dynamic, liquid-like condensates. Under certain conditions, these assemblies may mature into more rigid gel-like or glass-like condensates, which can serve as intermediates for the formation of protofibrils and ultimately insoluble amyloid fibrils. Accumulation of amyloid fibrils is associated with cellular dysfunction and neurodegeneration, as illustrated by the transition from a healthy to a diseased brain state. Arrows indicate reversible and irreversible transitions between aggregation states. Created with BioRender.com.
Figure 1.
Native protein monomers in healthy cells can reversibly assemble into oligomers or undergo LLPS to form dynamic, liquid-like condensates. Under certain conditions, these assemblies may mature into more rigid gel-like or glass-like condensates, which can serve as intermediates for the formation of protofibrils and ultimately insoluble amyloid fibrils. Accumulation of amyloid fibrils is associated with cellular dysfunction and neurodegeneration, as illustrated by the transition from a healthy to a diseased brain state. Arrows indicate reversible and irreversible transitions between aggregation states. Created with BioRender.com.
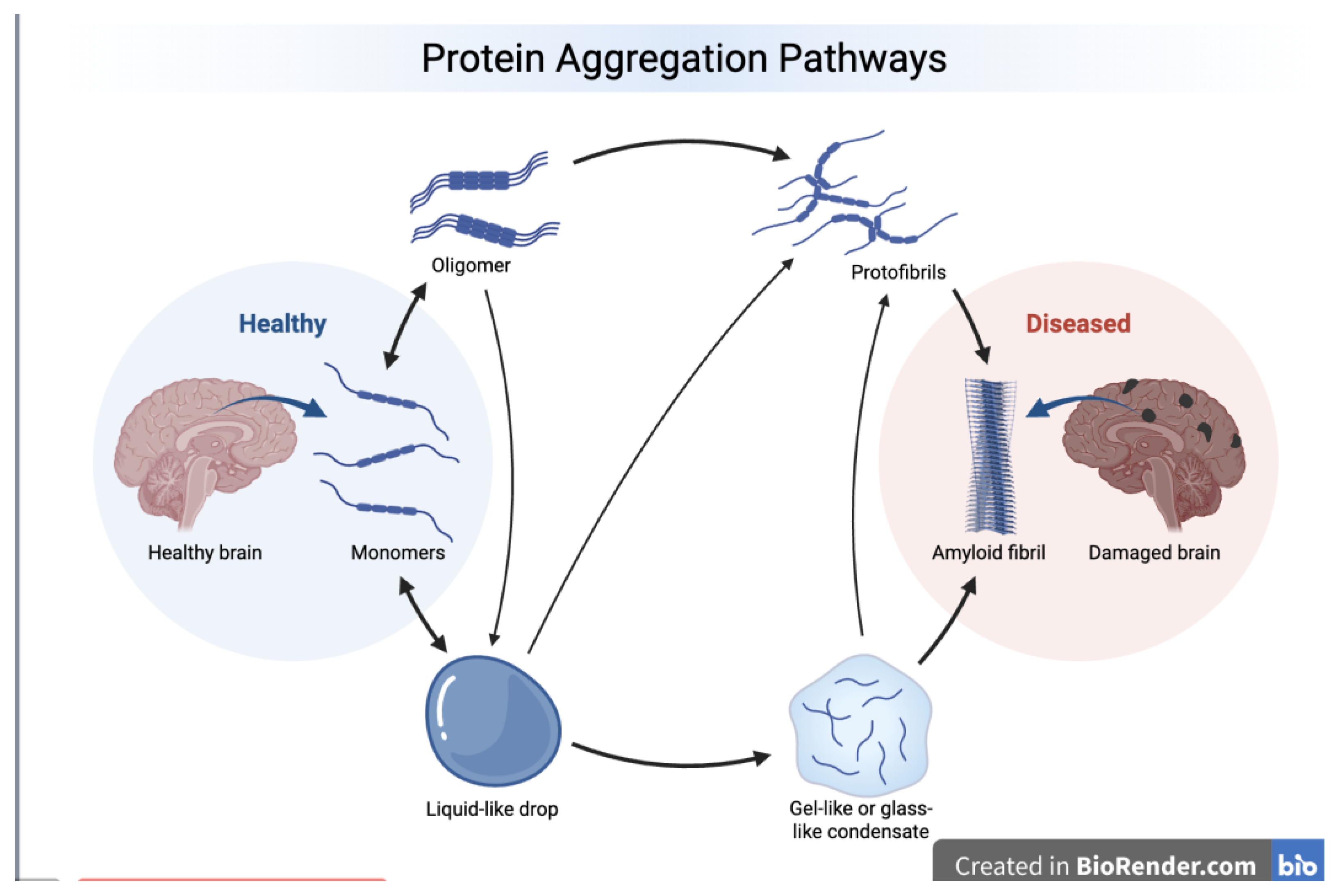
Figure 2.
Intrinsic disorder status of human amyloid precursor protein (APP) (UniProt ID: P05067). A. 3D structural model generated for human APP by AlphaFold [117]. The structure is colored based on the per-residue model confidence (pLDDT), which ranges between 0 and 100. Regions with very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low model confidence (pLDDT < 50) are shown by blue, cyan, yellow, and orange colors, respectively. Regions with low pLDDT may be unstructured in isolation. B. Functional disorder profile generated by D2P2 [118] shows the outputs of several disorder predictors such as PONDR® VLXT, PONDR® VSL2b, PrDOS, IU-Pred and Espritz. Consensus disorder predictions are shown by blue-green-white bar, where blue indicates regions are where the disorder predictions intersect the SCOP domain prediction and green indicates regions represent disorder that is not found within a predicted SCOP domain. The colored bar highlighted by blue and green shade represents the consensus disorder prediction. Above this consensus bar, line with numbered, colored bars show the predicted locations of SCOP (Structural Classifications of Proteins) domains. Yellow zigzagged bars show positions of MoRFs, whereas colored circles in the bottom of the plot show the positions of predicted PTMs. Position of Aβ, which is located within the C-terminal part of the protein is shown within the black box.
Figure 2.
Intrinsic disorder status of human amyloid precursor protein (APP) (UniProt ID: P05067). A. 3D structural model generated for human APP by AlphaFold [117]. The structure is colored based on the per-residue model confidence (pLDDT), which ranges between 0 and 100. Regions with very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low model confidence (pLDDT < 50) are shown by blue, cyan, yellow, and orange colors, respectively. Regions with low pLDDT may be unstructured in isolation. B. Functional disorder profile generated by D2P2 [118] shows the outputs of several disorder predictors such as PONDR® VLXT, PONDR® VSL2b, PrDOS, IU-Pred and Espritz. Consensus disorder predictions are shown by blue-green-white bar, where blue indicates regions are where the disorder predictions intersect the SCOP domain prediction and green indicates regions represent disorder that is not found within a predicted SCOP domain. The colored bar highlighted by blue and green shade represents the consensus disorder prediction. Above this consensus bar, line with numbered, colored bars show the predicted locations of SCOP (Structural Classifications of Proteins) domains. Yellow zigzagged bars show positions of MoRFs, whereas colored circles in the bottom of the plot show the positions of predicted PTMs. Position of Aβ, which is located within the C-terminal part of the protein is shown within the black box.
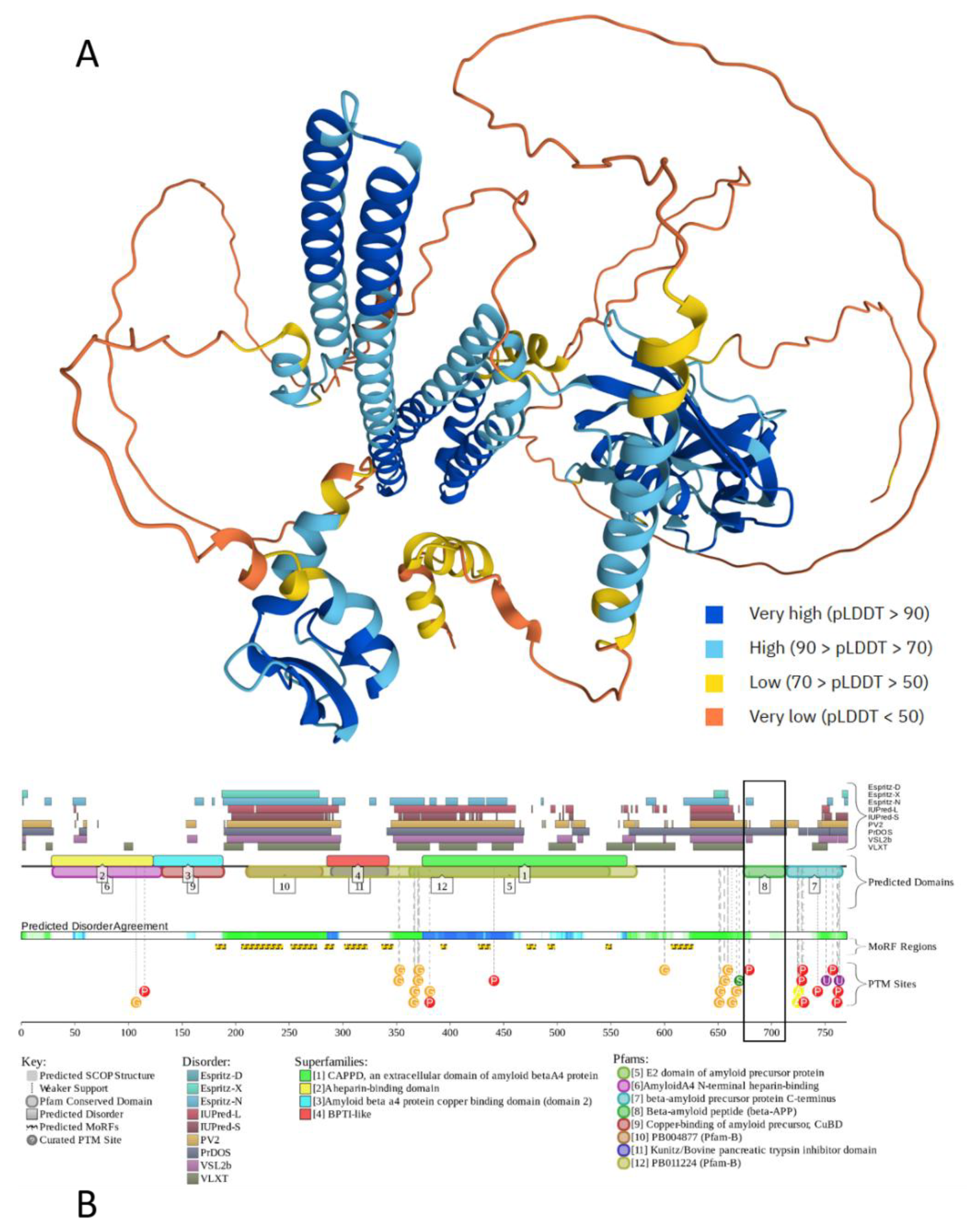
Figure 3.
Evaluation of intrinsic disorder in human microtubule-associated protein tau (UniProt ID: P10636). A. 3D structural model generated for human tau by AlphaFold. B. Functional disorder profile generated by D2P2.
Figure 3.
Evaluation of intrinsic disorder in human microtubule-associated protein tau (UniProt ID: P10636). A. 3D structural model generated for human tau by AlphaFold. B. Functional disorder profile generated by D2P2.
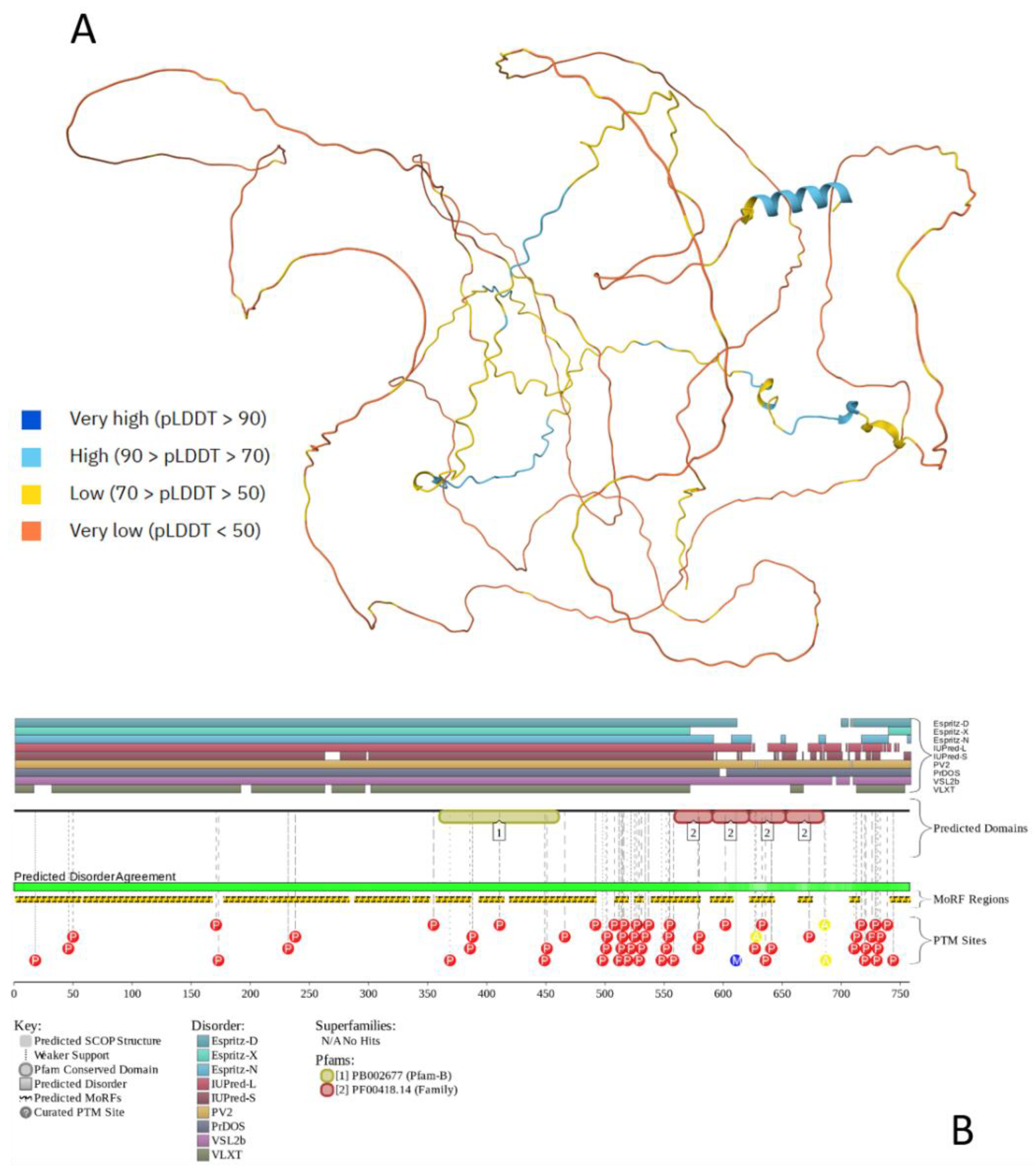
Figure 4.
Evaluation of the intrinsic disorder predisposition of human α-synuclein (UniProt ID: P37840). A. 3D conformational ensemble generated by AFflecto (https://moma.laas.fr/applications/AFflecto/) [169]. By analyzing the structural properties of the AlphaFold model, AFflecto identifies IDRs based on the pLDDT score (i.e., based on the analysis of structural context) and classifies them as tails, linkers, or loops. To explore the conformational diversity of these flexible regions, AFflecto employs computationally efficient stochastic sampling algorithms [169]. It also incorporates a method to identify conditionally folded IDRs that AF may incorrectly predict as natively folded elements [169]. Therefore, AFflecto generates protein ensembles that provide realistic representation of protein structural heterogeneity [169]. Ensemble includes 28 models. Plot was generated using Visual Molecular Dynamics (VMD) software for molecular visualization [170]. B. Functional disorder profile generated by D2P2.
Figure 4.
Evaluation of the intrinsic disorder predisposition of human α-synuclein (UniProt ID: P37840). A. 3D conformational ensemble generated by AFflecto (https://moma.laas.fr/applications/AFflecto/) [169]. By analyzing the structural properties of the AlphaFold model, AFflecto identifies IDRs based on the pLDDT score (i.e., based on the analysis of structural context) and classifies them as tails, linkers, or loops. To explore the conformational diversity of these flexible regions, AFflecto employs computationally efficient stochastic sampling algorithms [169]. It also incorporates a method to identify conditionally folded IDRs that AF may incorrectly predict as natively folded elements [169]. Therefore, AFflecto generates protein ensembles that provide realistic representation of protein structural heterogeneity [169]. Ensemble includes 28 models. Plot was generated using Visual Molecular Dynamics (VMD) software for molecular visualization [170]. B. Functional disorder profile generated by D2P2.
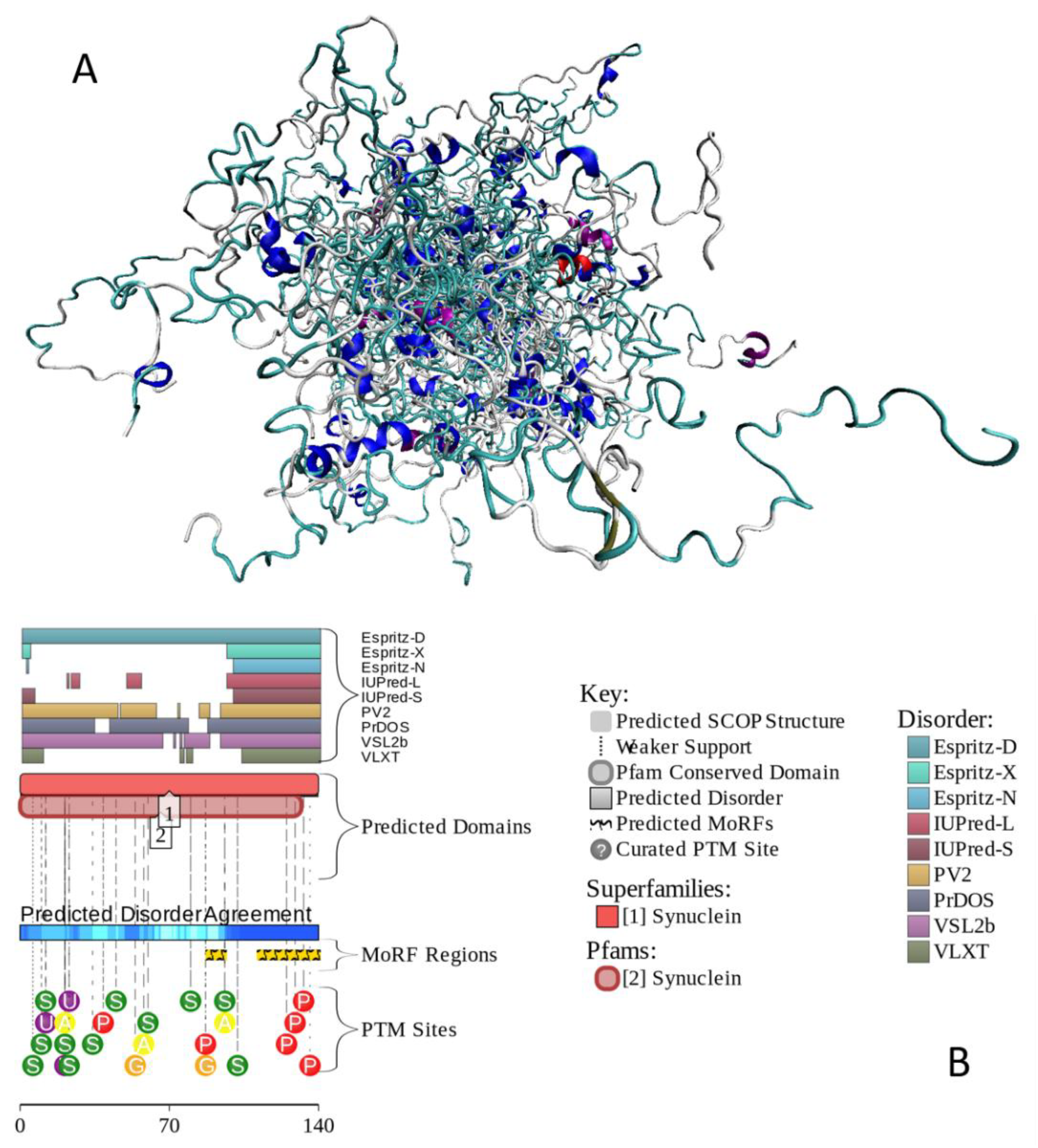
Figure 5.
Evaluation of intrinsic disorder in human TAR DNA-binding protein 43 (UniProt ID: Q13148). A. 3D structural model generated for human TDP-43 by AlphaFold. B. Functional disorder profile generated by D2P2.
Figure 5.
Evaluation of intrinsic disorder in human TAR DNA-binding protein 43 (UniProt ID: Q13148). A. 3D structural model generated for human TDP-43 by AlphaFold. B. Functional disorder profile generated by D2P2.
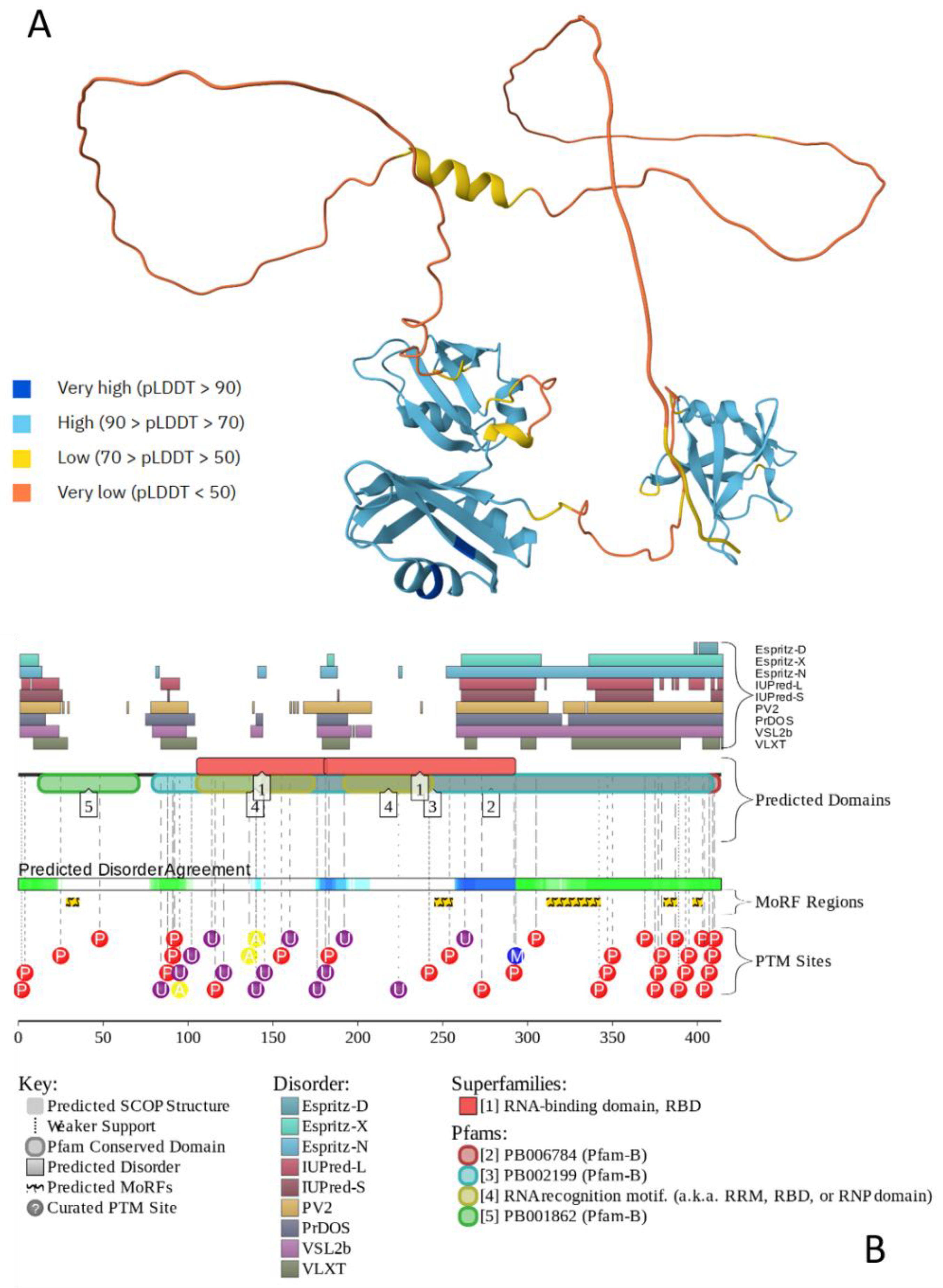
Figure 6.
Evaluation of intrinsic disorder in human RNA-binding protein FUS (UniProt ID: P35637). A. 3D structural model generated for human FUS by AlphaFold. B. Functional disorder profile generated by D2P2.
Figure 6.
Evaluation of intrinsic disorder in human RNA-binding protein FUS (UniProt ID: P35637). A. 3D structural model generated for human FUS by AlphaFold. B. Functional disorder profile generated by D2P2.
Figure 7.
STRING-generated protein-protein interaction (PPI) networks of human proteins linked to mixed pathologies. A. Intraset PPI network connecting APP, tau, α-synuclein, TDP-43, and FUS. B. Joint PPI network centered at five major mixed pathology-related proteins, highlighting shared interactors. Positions of APP, tau, α-synuclein, TDP-43, and FUS within this network are highlighted by yellow circles. This PPI network was generated by STRING database (https://string-db.org; [220,221,222]) using the maximum number of interactors in the first shell of 500 and a highest confidence level of 0.9 based on a minimum required interaction score. Here, individual proteins act as network nodes, whereas differently colored edges show protein-protein associations based on different types of evidence. The blue line represents information extracted from the curated databases, the black line represents co-expression, and the green line represents gene neighborhoods. Based on the STRING annotations, edges are expected to be specific and meaningful, indicating that linked proteins jointly contribute to a shared function. However, this does not necessarily mean that they are physically binding to each other. The interactive version of the joint PPI network can be viewed at the following permalink: https://version-12-0.string-db.org/cgi/network?networkId=bhz5YRrlPsno.
Figure 7.
STRING-generated protein-protein interaction (PPI) networks of human proteins linked to mixed pathologies. A. Intraset PPI network connecting APP, tau, α-synuclein, TDP-43, and FUS. B. Joint PPI network centered at five major mixed pathology-related proteins, highlighting shared interactors. Positions of APP, tau, α-synuclein, TDP-43, and FUS within this network are highlighted by yellow circles. This PPI network was generated by STRING database (https://string-db.org; [220,221,222]) using the maximum number of interactors in the first shell of 500 and a highest confidence level of 0.9 based on a minimum required interaction score. Here, individual proteins act as network nodes, whereas differently colored edges show protein-protein associations based on different types of evidence. The blue line represents information extracted from the curated databases, the black line represents co-expression, and the green line represents gene neighborhoods. Based on the STRING annotations, edges are expected to be specific and meaningful, indicating that linked proteins jointly contribute to a shared function. However, this does not necessarily mean that they are physically binding to each other. The interactive version of the joint PPI network can be viewed at the following permalink: https://version-12-0.string-db.org/cgi/network?networkId=bhz5YRrlPsno.
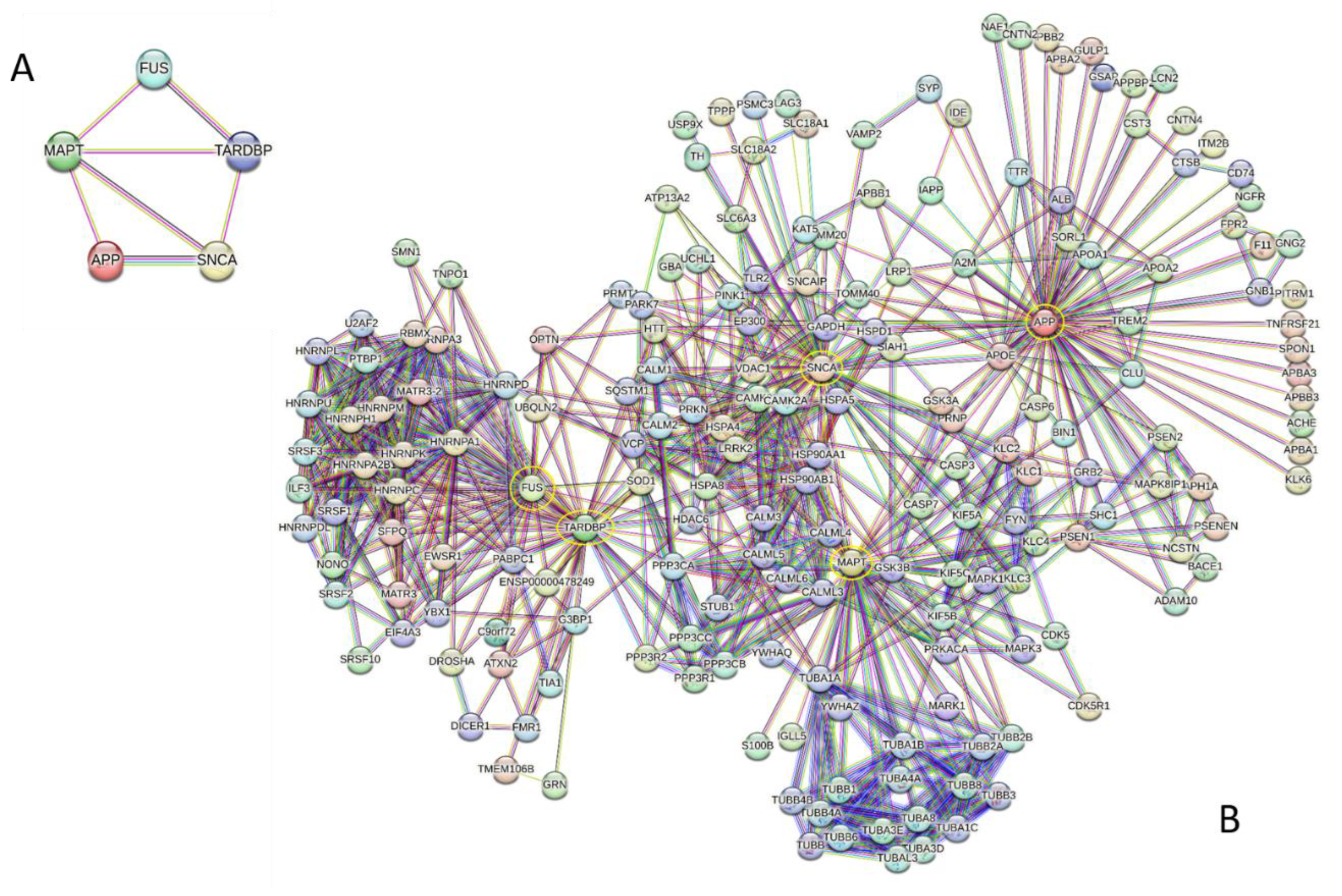
Figure 8.
Functional enrichment analysis of the joint interactome of APP, tau, α-synuclein, TDP-43, and FUS. A. 20 most enriched Biological Process (BP) Gene Ontology (GO) terms (of the 1055 significantly enriched BP terms). B. 20 most enriched Molecular Function (MF) GO terms (of 150 significantly enriched MF terms). C. 20 most enriched Cellular Component (CC) GO terms (of 188 significantly enriched CC terms). D. Enrichment analysis of the members of the interactome in KEGG Pathways (20 most enriched of 114 significantly enriched KEGG pathways). E. Enrichment analysis in the Subcellular Localization (COMPARTMENTS; 20 most enriched of 185 significantly enriched localizations). F. Enrichment analysis of interactome in the Disease Gene Association (DGA) terms (20 most enriched of 80 significantly enriched DGA terms).
Figure 8.
Functional enrichment analysis of the joint interactome of APP, tau, α-synuclein, TDP-43, and FUS. A. 20 most enriched Biological Process (BP) Gene Ontology (GO) terms (of the 1055 significantly enriched BP terms). B. 20 most enriched Molecular Function (MF) GO terms (of 150 significantly enriched MF terms). C. 20 most enriched Cellular Component (CC) GO terms (of 188 significantly enriched CC terms). D. Enrichment analysis of the members of the interactome in KEGG Pathways (20 most enriched of 114 significantly enriched KEGG pathways). E. Enrichment analysis in the Subcellular Localization (COMPARTMENTS; 20 most enriched of 185 significantly enriched localizations). F. Enrichment analysis of interactome in the Disease Gene Association (DGA) terms (20 most enriched of 80 significantly enriched DGA terms).
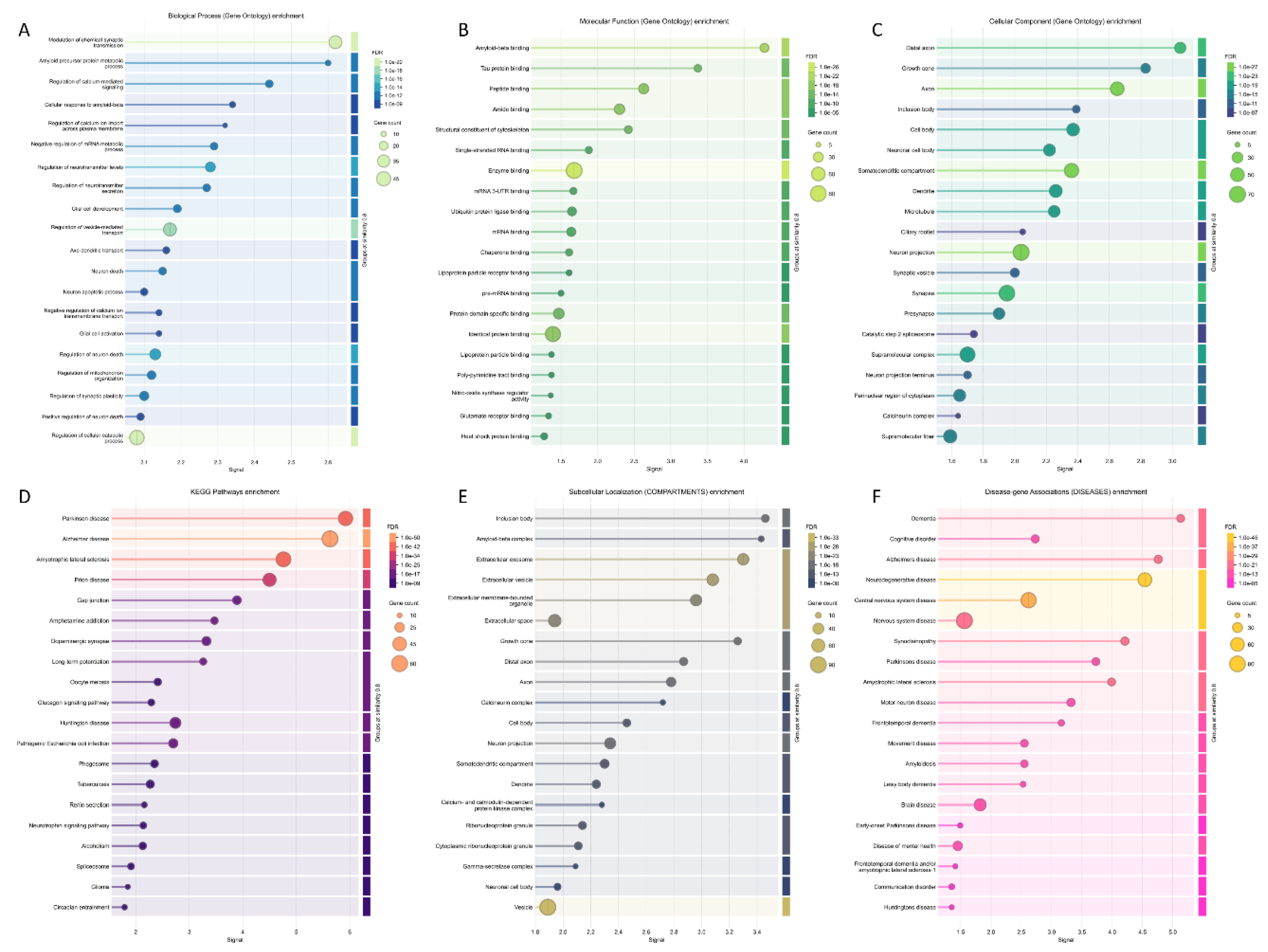
Figure 9.
Evaluation of global intrinsic disorder predisposition of human proteins associated with mixed pathologies and their interactors. A. The PONDR® VSL2 score (average disorder score, ADS) vs. PONDR® VSL2 (%) (percent of predicted intrinsically disordered residues, PPIDR) plot, where each point corresponding to a query protein. In this plot, the coordinates are derived from the PONDR® VSL2 data as the corresponding ADS and PPIDR values. The areas containing highly disordered, moderately disordered, and ordered proteins (based on the accepted classification, see text) are shown by red, pink/light pink, and blue/light blue colors, respectively. The regions where the ADS are PPIDR agree are indicated by dark (blue or pink) colors, whereas light blue or pink colors correspond to the areas, where only one of these criteria applies. B. Classification of query proteins based on the outputs of the charge-hydropathy (CH, which correlates a protein’s net charge with its hydrophobicity) and cumulative distribution function analyses (CDF, which correlates the cumulative frequency of disorder scores with the disorder scores). The resulting ΔCH-ΔCDF plot integrates the corresponding outputs as a two-dimensional graph, where the deviation of the disorder frequency of a query protein from the CDF boundary (ΔCDF) is shown at the X-axis, and the distance of a protein from the CH boundary (ΔCH) is shown at the Y-axis. Here, proteins, which are expected to be structured, molten globular/hybrid, highly disordered, or mixed are located within the Quadrant 1 (blue, bottom right), Quadrant 2 (pink, bottom left), Quadrant 3 (red, top left), and Quadrant 4 (violet, top right). C. Correlation between the interactivity (measured as the node degree in the STRING-generated PPI network shown in Figure 7B) and intrinsic disorder (measured as PONDR® VSL2-based PPIDR) among the members of the analyzed interactome. D. Comparison of the LLPS predisposition of the analyzed proteins with their intrinsic disorder propensity. Vertical lines show 10% and 30% PPIDR thresholds, whereas the horizontal line corresponds to the pLLPS threshold of 0.6. Proteins not related to LLPS (showing pLLPS below 0.6 and not containing DPRs) are shown by dotted circles. In these plots, the positions of individual major proteins associated with mixed pathologies are shown by differently shaped green symbols, whereas the gray circles indicate the corresponding data for their interactome.
Figure 9.
Evaluation of global intrinsic disorder predisposition of human proteins associated with mixed pathologies and their interactors. A. The PONDR® VSL2 score (average disorder score, ADS) vs. PONDR® VSL2 (%) (percent of predicted intrinsically disordered residues, PPIDR) plot, where each point corresponding to a query protein. In this plot, the coordinates are derived from the PONDR® VSL2 data as the corresponding ADS and PPIDR values. The areas containing highly disordered, moderately disordered, and ordered proteins (based on the accepted classification, see text) are shown by red, pink/light pink, and blue/light blue colors, respectively. The regions where the ADS are PPIDR agree are indicated by dark (blue or pink) colors, whereas light blue or pink colors correspond to the areas, where only one of these criteria applies. B. Classification of query proteins based on the outputs of the charge-hydropathy (CH, which correlates a protein’s net charge with its hydrophobicity) and cumulative distribution function analyses (CDF, which correlates the cumulative frequency of disorder scores with the disorder scores). The resulting ΔCH-ΔCDF plot integrates the corresponding outputs as a two-dimensional graph, where the deviation of the disorder frequency of a query protein from the CDF boundary (ΔCDF) is shown at the X-axis, and the distance of a protein from the CH boundary (ΔCH) is shown at the Y-axis. Here, proteins, which are expected to be structured, molten globular/hybrid, highly disordered, or mixed are located within the Quadrant 1 (blue, bottom right), Quadrant 2 (pink, bottom left), Quadrant 3 (red, top left), and Quadrant 4 (violet, top right). C. Correlation between the interactivity (measured as the node degree in the STRING-generated PPI network shown in Figure 7B) and intrinsic disorder (measured as PONDR® VSL2-based PPIDR) among the members of the analyzed interactome. D. Comparison of the LLPS predisposition of the analyzed proteins with their intrinsic disorder propensity. Vertical lines show 10% and 30% PPIDR thresholds, whereas the horizontal line corresponds to the pLLPS threshold of 0.6. Proteins not related to LLPS (showing pLLPS below 0.6 and not containing DPRs) are shown by dotted circles. In these plots, the positions of individual major proteins associated with mixed pathologies are shown by differently shaped green symbols, whereas the gray circles indicate the corresponding data for their interactome.
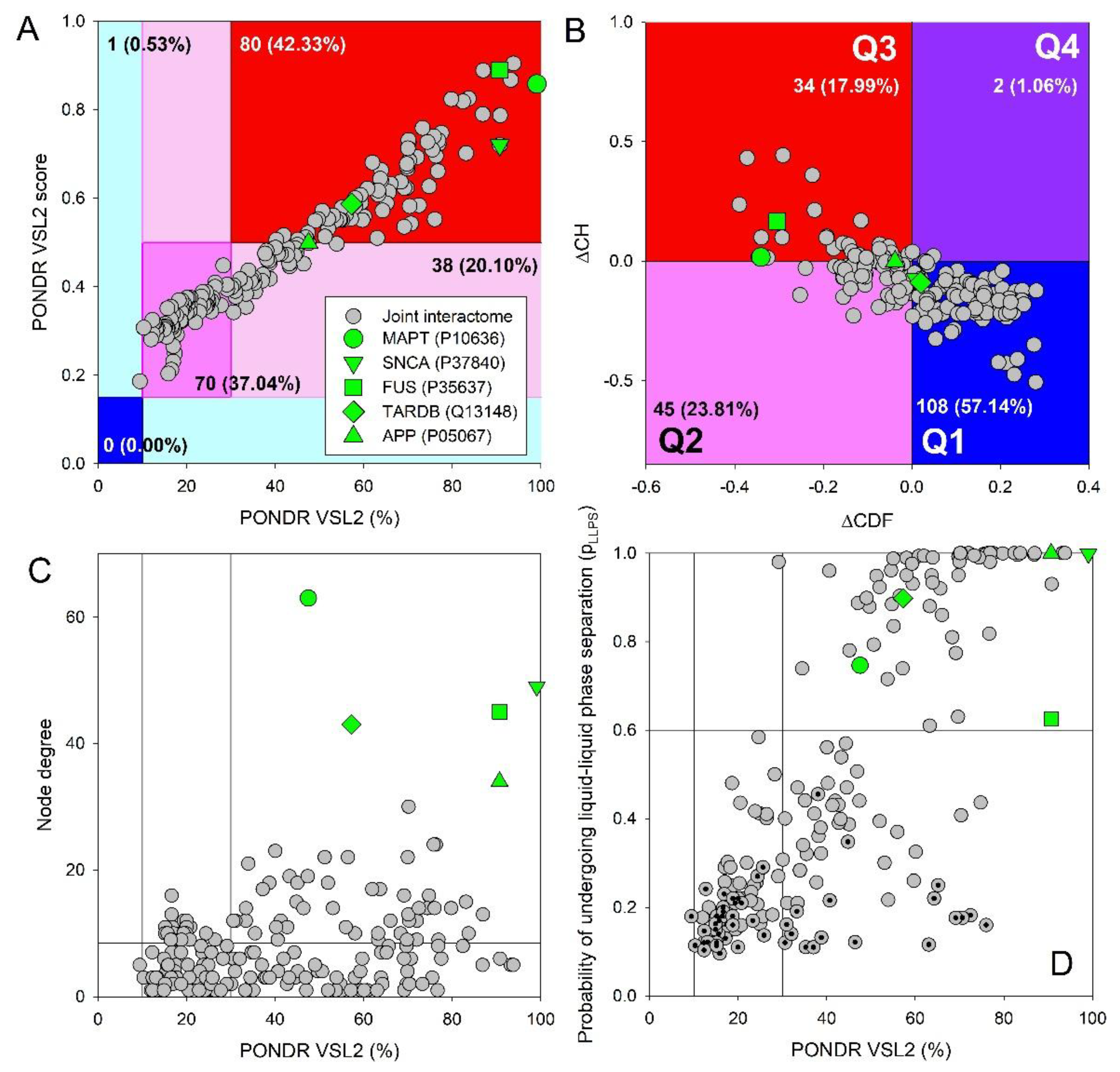

Table 1.
Comparative overview of major neurodegeneration-associated proteins with respect to intrinsic disorder, aggregation behavior, phase separation, and contribution to mixed neuropathology.
Table 1.
Comparative overview of major neurodegeneration-associated proteins with respect to intrinsic disorder, aggregation behavior, phase separation, and contribution to mixed neuropathology.
| Feature | Amyloid-β (Aβ) [90,92,94,217] | τ-protein (tau) [120,139] | α-synuclein [161,165,218] | TDP-43 [189,191,219] | FUS [123,193,195] |
|---|---|---|---|---|---|
| Physiological role | APP-derived peptide; modulates synaptic activity at low concentrations | Microtubule-associated protein stabilizing neuronal cytoskeleton | Presynaptic protein regulating vesicle trafficking | RNA-binding protein regulating splicing and RNA metabolism | RNA/DNA-binding protein involved in RNA metabolism and stress responses |
| Pathological aggregate | Extracellular plaques; soluble toxic oligomers | Intracellular neurofibrillary tangles (NFTs) | Lewy bodies and Lewy neurites; oligomers | Cytoplasmic inclusions with nuclear depletion | Cytoplasmic inclusions due to nuclear clearance defects |
| Intrinsic disorder features | Intrinsically disordered as a monomer | Highly intrinsically disordered; conformationally flexible | Almost entirely intrinsically disordered | Large intrinsically disordered regions | Highly intrinsically disordered; prion-like domains |
| Sequence determinants of disorder | Charged N-terminus; hydrophobic C-terminal motifs (Aβ42) | Low sequence complexity; charged and polar residues | NAC region; charged N-terminal repeats | Low-complexity C-terminal domain; charge asymmetry | Low-complexity, arginine-rich prion-like domains |
| Post-translational modifications (PTMs) | Phosphorylation and acetylation modulate aggregation kinetics | Hyperphosphorylation and acetylation | N-terminal acetylation and phosphorylation | Hyperphosphorylation and ubiquitination | PTMs modulate phase behavior and aggregation |
| Phase separation (LLPS) | No classical LLPS; aggregation emerges from disordered peptide plasticity | Undergoes LLPS; droplets can mature into fibrils | Forms stress-induced condensates | Stress-granule–associated LLPS precedes aggregation | Prominent LLPS; condensate hardening linked to disease |
| Prevalence in aged brain | Very high | Very high | Moderate to high | Common, especially in elderly | Rare in community autopsy series |
| Common co-pathologies | Tau, α-synuclein, TDP-43, vascular pathology | Aβ, α-synuclein, TDP-43 | Aβ, tau | Aβ, tau, α-synuclein | TDP-43; ALS/FTLD spectrum |
| Cross-seeding/ synergy | Seeds tau misfolding; interacts with α-synuclein | Synergizes with Aβ; interacts with α-synuclein | Cross-seeds tau and Aβ; strain-dependent | Often superimposed on AD pathology | Stress-granule and RNA-metabolism interactions |
| Impact on mixed-pathology dementia | Early driver and amplifier of downstream proteinopathies | Strong determinant of cognitive decline and neuronal loss | Exacerbates cognitive and neuropsychiatric symptoms | Accelerates dementia severity and memory impairment | Associated with earlier onset and aggressive disease |
| Associated diseases | AD, cerebral amyloid angiopathy (CAA), Down syndrome, Aβ–related angiitis (ABRA), CAA-related inflammation, cerebral amyloidoma, dementia with Lewy bodies (DLB), retinal disorders, traumatic brain injury (TBI) | AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD), frontotemporal dementia with parkinsonism-17 (FTDP-17), argyrophilic grain disease (AGD), chronic traumatic encephalopathy (CTE), down syndrome, Guam parkinsonism-dementia complex, postencephalitic parkinsonism, DLB, PD | PD, DLB, multiple system atrophy (MSA), pure autonomic failure (PAF), REM sleep behavior disorder (RBD), AD, Gaucher’s disease, neuroaxonal dystrophy, neurodegeneration with brain iron accumulation (NBIA) | ALS, FTLD-TDP, LATE, AD, CTE, LBD, Huntington’s disease (HD), multisystem proteinopathy (MSP), Perry syndrome, Alexander disease | ALS-FUS, FTLD-FUS, neuronal intermediate filament inclusion disease (NIFID), basophilic inclusion body disease (BIBD), essential tremor (ET), polyglutamine (PolyQ) diseases (HD and spinocerebellar ataxias SCA1 and SCA3), cancers (myxoid liposarcoma, ewing sarcoma, acute myeloid leukemia (AML) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



