Submitted:
23 January 2026
Posted:
26 January 2026
You are already at the latest version
Abstract
The integration of heterogeneous molecular data across multiple omics layers is essential for understanding complex disease biology, yet conventional analytical approaches struggle with the high dimensionality, non-linearity, missingness, and technical variability of multi-omics datasets. This review aims to critically evaluate how deep learning (DL) methodologies address these challenges and to assess their translational relevance within biomedical informatics. We systematically review contemporary DL-based approaches for multi-omics data integration and categorize them into two principal methodological strategies: (i) unsupervised latent-space learning models, such as variational autoencoders, which enable probabilistic feature fusion, denoising, and data imputation; and (ii) network-based integration frameworks, including graph neural networks, which incorporate biological prior knowledge through molecular interaction graphs. Supervised extensions, including attention-based models for clinical prediction tasks, are also examined with emphasis on architectural design, data fusion mechanisms, and validation practices. Deep learning-based multi-omics integration methods have demonstrated improved performance over conventional approaches in disease subtyping, biomarker discovery, and prognostic modeling by capturing complex, non-linear interactions across molecular layers. Latent-space models provide robust representations in the presence of incomplete data, while network-based approaches facilitate the identification of biologically coherent molecular subnetworks; however, most reported performance gains rely on internal validation, with limited evidence of external generalizability, interpretability robustness, and readiness for clinical deployment. Deep learning has emerged as a powerful paradigm for multi-omics integration in biomedical informatics, enabling clinically relevant molecular stratification and prediction. Nevertheless, effective translation into routine clinical practice requires a shift beyond predictive accuracy toward explainable modeling, standardized external validation on independent cohorts, and integration with real-world clinical data, which are essential for establishing trustworthy and actionable decision-support systems for precision medicine.
Keywords:
multi-omics integration
; deep learning
; variational autoencoders
; graph neural networks
; biomarker discovery
; precision medicine
1. Introduction
The contemporary medical paradigm is increasingly oriented toward precision medicine, which seeks to tailor prevention and treatment strategies to individual patients based on molecular and clinical characteristics [1]. Central to this shift is the integration of multi-omics data, reflecting the recognition that complex human diseases, including cancer, neurological disorders, and cardiovascular conditions, arise from coordinated perturbations across multiple molecular layers rather than isolated genetic alterations [2]. Advances in high-throughput profiling technologies have enabled the parallel measurement of genomics, epigenomics, transcriptomics, proteomics, and related modalities, providing complementary views of disease biology that are not attainable through single-omics analyses alone [2]. For example, genomic alterations alone often fail to capture adaptive resistance mechanisms, which may instead be driven by epigenetic reprogramming, transcriptional plasticity, or metabolic remodeling within disease-specific microenvironments. Consequently, integrating multi-layered molecular data is essential for improving biomarker robustness and enhancing clinical relevance.
Despite this promise, multi-omics integration poses substantial computational challenges that limit the effectiveness of traditional statistical and machine learning approaches. These challenges include pronounced heterogeneity across data types and measurement platforms, extreme high dimensionality in relation to sample size (p ≫ n), pervasive missingness, and complex non-linear and hierarchical dependencies among molecular features. Such characteristics violate the assumptions of many classical models and hinder their ability to extract clinically meaningful patterns. In response, deep learning (DL) architectures have emerged as powerful analytical tools capable of learning hierarchical representations directly from high-dimensional, heterogeneous data through end-to-end optimization and automated feature extraction [3,4]. By constructing unified latent representations, DL models can capture non-linear interactions across molecular layers that are critical for modeling disease mechanisms and clinical outcomes such as therapeutic response and patient survival [4]. Comparative studies have shown that advanced DL approaches, including graph neural networks, often outperform conventional machine learning methods, such as random forests, in complex biomedical prediction tasks [3].
Against this backdrop, this review examines the convergence of deep learning and multi-omics integration from a biomedical informatics perspective, with an emphasis on methodological design and translational applicability [5]. We first categorize foundational DL architectures for multi-omics integration into unsupervised latent-space learning approaches, such as variational autoencoders for feature fusion and data imputation, and network-based integration strategies, such as graph neural networks that incorporate biological prior knowledge. We then assess the translational applications of these models in disease subtyping and biomarker discovery, before critically discussing key barriers to clinical adoption, including the interpretability–performance trade-off and the lack of standardized external validation across diverse populations. Addressing these challenges is essential for enabling the reliable deployment of multi-omics deep learning models as clinical decision-support tools within precision medicine frameworks [5].
Figure 1.
Conceptual overview of key challenges and trade-offs governing the clinical translation of multi-omics deep learning.
Figure 1.
Conceptual overview of key challenges and trade-offs governing the clinical translation of multi-omics deep learning.
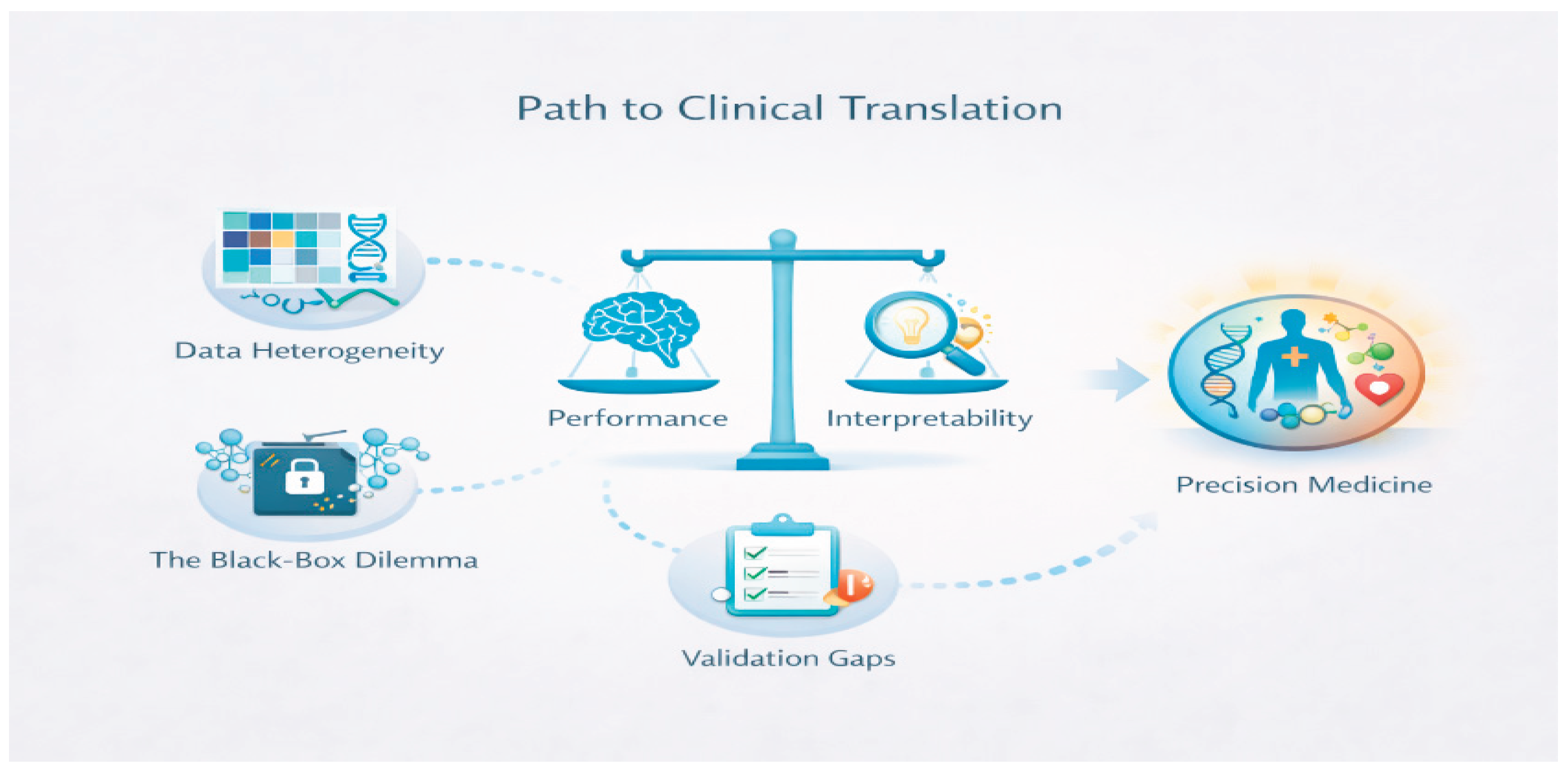
2. Characteristics of Multi-Omics Data and Core Challenges
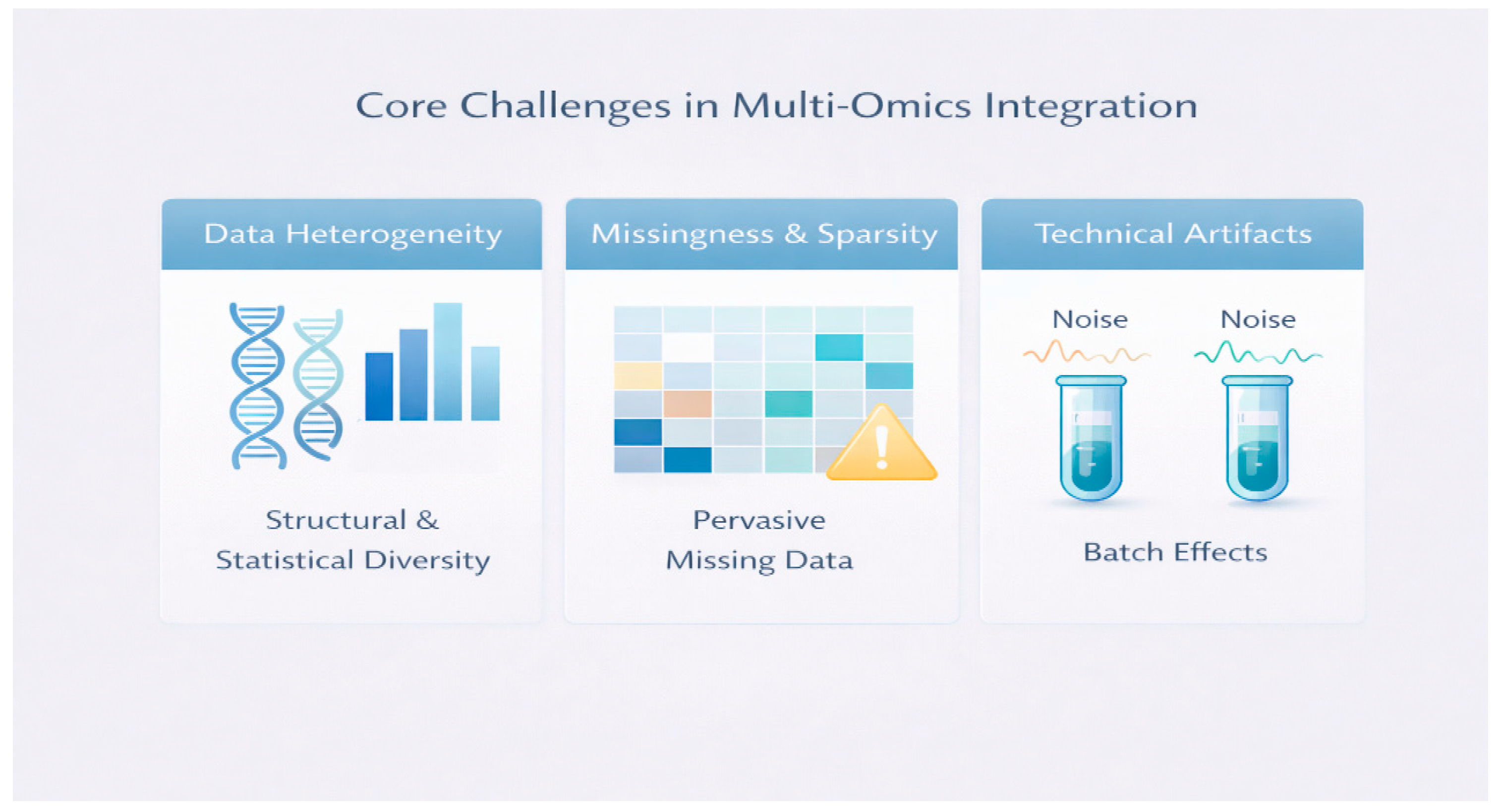
The promise of multi-omics data is intrinsically coupled with the substantial computational complexity inherent to its analysis. Effective multi-omics integration depends on the ability of analytical methods to accommodate pronounced heterogeneity, pervasive missingness, and technical variability across multiple molecular layers. These challenges can be broadly categorized into three interrelated domains: heterogeneity in data characteristics and statistical distributions, missing data and sparsity, and technical artifacts such as batch effects.
Data Heterogeneity and Statistical Distributions
Multi-omics data are generated using fundamentally distinct experimental technologies, resulting in marked structural and statistical diversity that complicates direct comparison and integration [2]. Genomic measurements, such as single-nucleotide variants and copy number variations, are typically discrete or categorical, whereas transcriptomic data derived from RNA sequencing are count-based and often require normalization and log transformation. In contrast, proteomic and metabolomic measurements are generally continuous, span wide dynamic ranges, and are subject to different sources of technical variability [6]. These disparities are further compounded by assay-specific noise profiles and detection limits. For example, a gene may be readily detected at the transcript level but remain undetectable at the protein level due to post-transcriptional regulation, protein degradation, or limited instrument sensitivity. Consequently, each omics layer requires modality-specific preprocessing pipelines, such as DESeq2-based normalization for RNA-seq data or z-score scaling for metabolomics. The absence of standardized preprocessing protocols across studies introduces additional variability and undermines cross-cohort comparability [2,6].
Missing Data and Sparsity
Missing data represent one of the most pervasive obstacles in multi-omics studies, arising from high assay costs, technical failures, and heterogeneous study designs. In most multi-omics cohorts, it is uncommon for all molecular layers to be measured for every sample, resulting in partially observed data where one or more modalities are absent. This issue is particularly pronounced in single-cell multi-omics datasets, such as scRNA-seq and scATAC-seq, which are characterized by extreme sparsity. In these settings, a large proportion of zero-valued measurements may reflect technical dropouts rather than true biological absence, obscuring underlying molecular signals [7]. Many conventional statistical and machine learning methods implicitly assume complete data and therefore require restricting analyses to fully paired samples. Such practices discard substantial amounts of patient data, introduce selection bias, and reduce statistical power, ultimately limiting the reliability of downstream biomarker discovery and clinical inference [8].
Technical Artifacts and Batch Effects
Even in the absence of missing data, technical artifacts can substantially confound multi-omics analyses. Batch effects arise from systematic, non-biological variations introduced during sample processing, reagent lot changes, instrument calibration differences, or temporal effects associated with experimental workflows. In multi-omics settings, these effects are amplified because each molecular layer is subject to distinct sources of technical noise, and their integration compounds the resulting variability. A particularly challenging scenario occurs when batch effects are confounded with biological variables of interest, such as disease status or treatment group. In such cases, standard batch correction approaches may inadvertently remove genuine biological signals along with technical noise, leading to spurious findings or loss of clinically relevant patterns.
Collectively, these challenges underscore the need for analytical frameworks capable of disentangling true biological variation from technical artifacts, accommodating incomplete and sparse data, and fusing heterogeneous molecular features in a principled manner. These requirements form the primary motivation for the adoption of deep learning approaches in multi-omics integration, as such models are explicitly designed to learn robust representations from complex, high-dimensional, and imperfect biomedical data.
Figure 2.
Core challenges underlying the complexity of multi-omics data integration.
3. Deep Learning Architectures for Multi-Omics Integration
The intrinsic complexity of multi-omics data has motivated the adoption of advanced deep learning (DL) methodologies capable of learning hierarchical representations, accommodating data sparsity, and integrating heterogeneous molecular features. In contrast to traditional machine learning approaches, DL models can perform end-to-end optimization and automatically extract non-linear feature representations that are well suited to the high dimensionality and heterogeneity of multi-omics datasets. The current landscape of DL-based multi-omics integrators can be broadly categorized into two principal strategies: unsupervised latent-space learning and network-based integration.
Unsupervised Latent-Space Learning
Unsupervised latent-space learning approaches aim to project high-dimensional, heterogeneous omics data into a shared, low-dimensional latent representation that captures the intrinsic biological structure of the data while suppressing technical noise [9]. These generative models are particularly well suited for multi-omics integration, as they can jointly model multiple data modalities and provide a principled framework for dimensionality reduction, denoising, and data imputation.
Variational Autoencoders (VAEs)
Variational autoencoders and their extensions represent the most widely adopted DL architecture for latent-space multi-omics integration. VAEs encode each omics modality, either independently or jointly, into a shared latent vector constrained by a probabilistic prior, typically a multivariate Gaussian distribution [9]. Model training optimizes a variational objective that balances reconstruction fidelity with latent-space regularization, resulting in a continuous, probabilistic representation of the underlying biological state [9,10]. These latent representations have been shown to support downstream tasks such as molecular subtyping and disease classification, often outperforming linear dimensionality reduction methods in complex oncological datasets [3,20].
Beyond representation learning, VAEs offer a natural solution to the pervasive problem of missing data in multi-omics studies. By leveraging statistical regularities encoded in the latent space, VAE-based models can impute missing features and, in some cases, entire missing modalities through cross-omics imputation. This capability is particularly relevant for sparse single-cell multi-omics data and large clinical cohorts with partially observed molecular profiles [8,10]. Frameworks such as OmiVAE and MoVAE have demonstrated task-oriented latent representations that effectively handle missingness and high dimensionality in pan-cancer classification settings.
Recent methodological advances have further extended VAEs to explicitly address confounding technical effects. Deconfounding VAEs incorporate additional latent variables or constraints to disentangle biological variation from technical noise, such as batch effects or unrelated sources of variability, thereby improving the stability and biological interpretability of learned clustering structures [11].
Generative Models and Domain Adaptation
Beyond VAEs, generative adversarial networks (GANs) have been increasingly explored for multi-omics integration, particularly for batch effect correction, data harmonization, and synthetic data augmentation [12]. Adversarial learning frameworks can be applied to domain adaptation problems, reducing platform- or laboratory-specific biases across omics datasets generated under heterogeneous experimental conditions. By aligning feature distributions across batches or institutions, these models aim to improve the robustness and portability of downstream predictive models in multi-center studies.
Network-Based Integration
Network-based integration strategies incorporate prior biological knowledge directly into the learning process by representing molecular systems as graphs. In this paradigm, deep learning models are constrained by molecular interaction networks, enabling predictions that are more biologically informed and potentially more interpretable than purely data-driven approaches.
Graph Neural Networks (GNNs)
Graph neural networks operate on graph-structured data in which nodes correspond to biological entities, such as genes or proteins, and edges represent known molecular interactions derived from protein–protein interaction networks, gene regulatory networks, or pathway databases [13]. Multi-omics measurements, including gene expression or epigenetic signals, are encoded as node features. Through iterative message passing and aggregation, GNNs learn context-aware representations that capture both molecular feature values and their relational dependencies within the biological network [13].
Several GNN variants have been applied to multi-omics integration, including graph convolutional networks (GCNs) [13], graph attention networks (GATs) [15], and graph transformer networks (GTNs) [14]. Among these, GATs have demonstrated particular utility by dynamically assigning attention weights to neighboring nodes, thereby prioritizing molecular interactions most relevant to the predictive task [15]. Frameworks such as MPK-GNN further extend this concept by integrating multiple biological knowledge graphs simultaneously, improving the robustness of cancer molecular subtype classification [16].
While many GNN-based models rely on fixed biological priors, recent approaches have explored data-driven graph construction. Models such as MoRE-GNN infer relational structures directly from the data, increasing adaptability to diverse single-cell multi-omics datasets and reducing dependence on predefined interaction networks that may be incomplete or context agnostic.
Advanced Supervised Strategies
For supervised tasks such as classification and survival prediction, deep learning architectures are often applied after or alongside feature fusion. Multi-modal attention mechanisms represent a prominent development in this context. By incorporating multi-head attention layers, these models dynamically weight the contribution of each omics modality or individual molecular feature, focusing representational capacity on inputs most predictive of the clinical outcome [15]. In addition to improving predictive performance, attention-based models provide an initial layer of interpretability, as learned attention weights can highlight candidate molecular features or pathways associated with disease phenotypes [18].
Figure 3.
Schematic illustration of a multi-modal attention mechanism for weighting omics modalities in biomarker discovery.
Figure 3.
Schematic illustration of a multi-modal attention mechanism for weighting omics modalities in biomarker discovery.
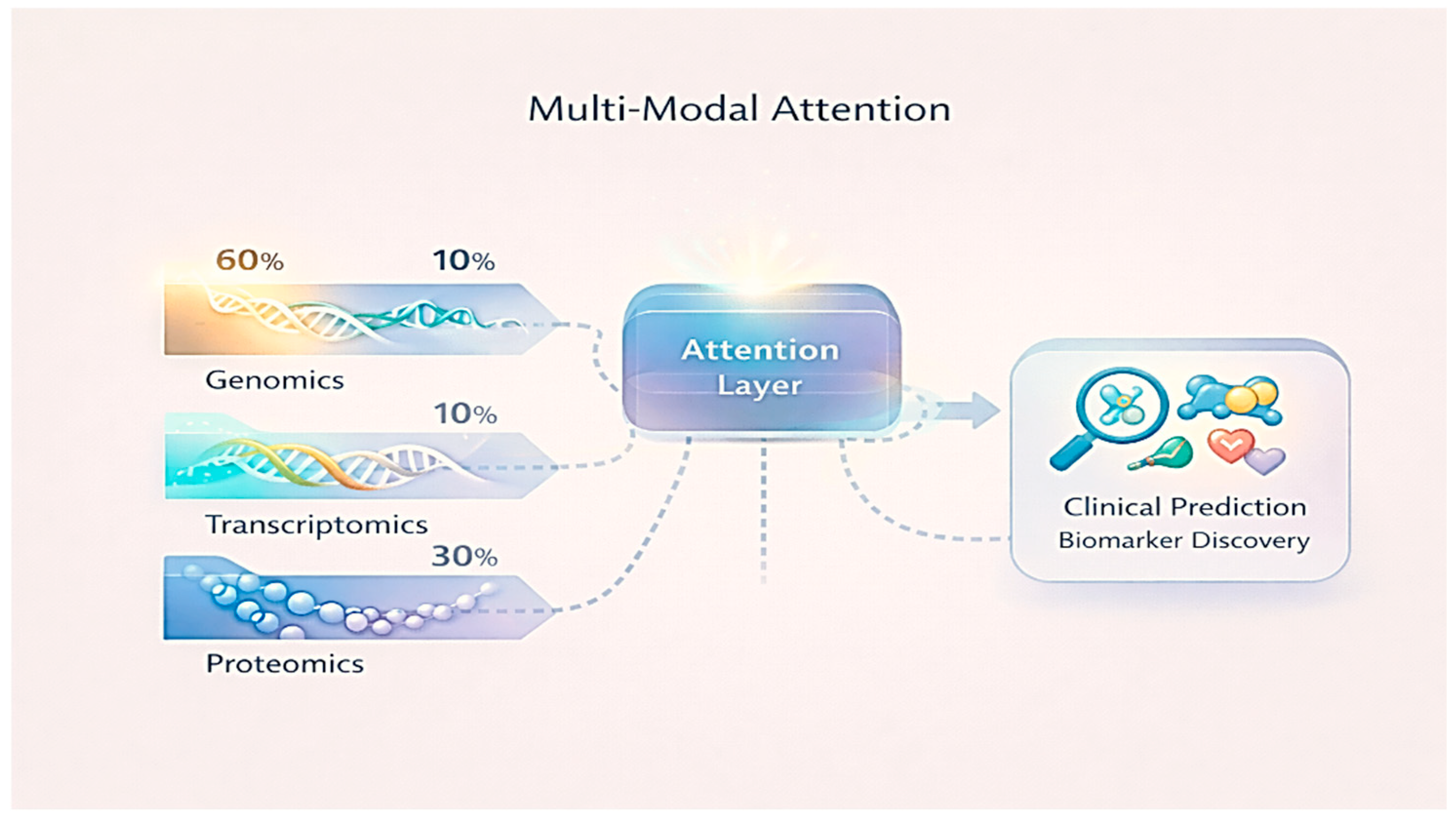

Table 1.
Taxonomy of Deep Learning Strategies for Multi-Omics Integration.
| Category | Latent-Space Learning | Deconfounding Generative Models | Adversarial Generative Models | Network-Based Integration | Data-Driven Graph Models | Attention-Based Models | Ensemble Prognostic Models | Decentralized Learning Frameworks |
| Fusion Level | Representation-level (latent) | Representation-level (latent) | Feature / representation-level | Network-level | Network-level | Decision-level | Decision-level | Training-level |
| Core Architecture | VAEs, Denoising Autoencoders | Disentangled VAEs, Conditional VAEs | GANs | GNNs (GCN, GAT, GTN) | Learned relational graphs (e.g., MoRE-GNN) | Multi-head attention, Transformers | Autoencoders + classical ML (e.g., DeepProg) | Federated learning, Transfer learning |
| Handling of Missing Data | Explicit (latent-space imputation) | Explicit (signal–confounder separation) | Indirect (distribution alignment) | Limited | Partial | Partial | Indirect | Indirect |
| Use of Biological Priors | Limited / implicit | Partial (constraints) | None | Explicit (PPI, pathways) | Minimal / none | None | None | None |
| Primary Translational Applications | Disease subtyping, classification | Batch-corrected clustering, scMulti-omics | Batch correction, harmonization | Biomarker discovery, subtype analysis | Single-cell multi-omics integration | Prognosis, drug response prediction | Survival stratification, pan-cancer prognosis | Multi-center survival prediction |
| Key Strengths | Robust to noise and sparsity; scalable | Separates biological and technical variation | Improves cross-cohort robustness | Biologically interpretable modules | Adaptive, prior-free modeling | Dynamic modality weighting | Improved robustness via model diversity | Privacy-preserving, improved generalizability |
| Key Limitations | Limited interpretability of latent features | Increased complexity; confounder specification | Training instability; low interpretability | Sensitive to prior knowledge quality | Reduced biological interpretability | Attention ≠ causality | Complex deployment; limited transparency | Communication overhead; data heterogeneity |
4. Translational Applications of Multi-Omics Deep Learning
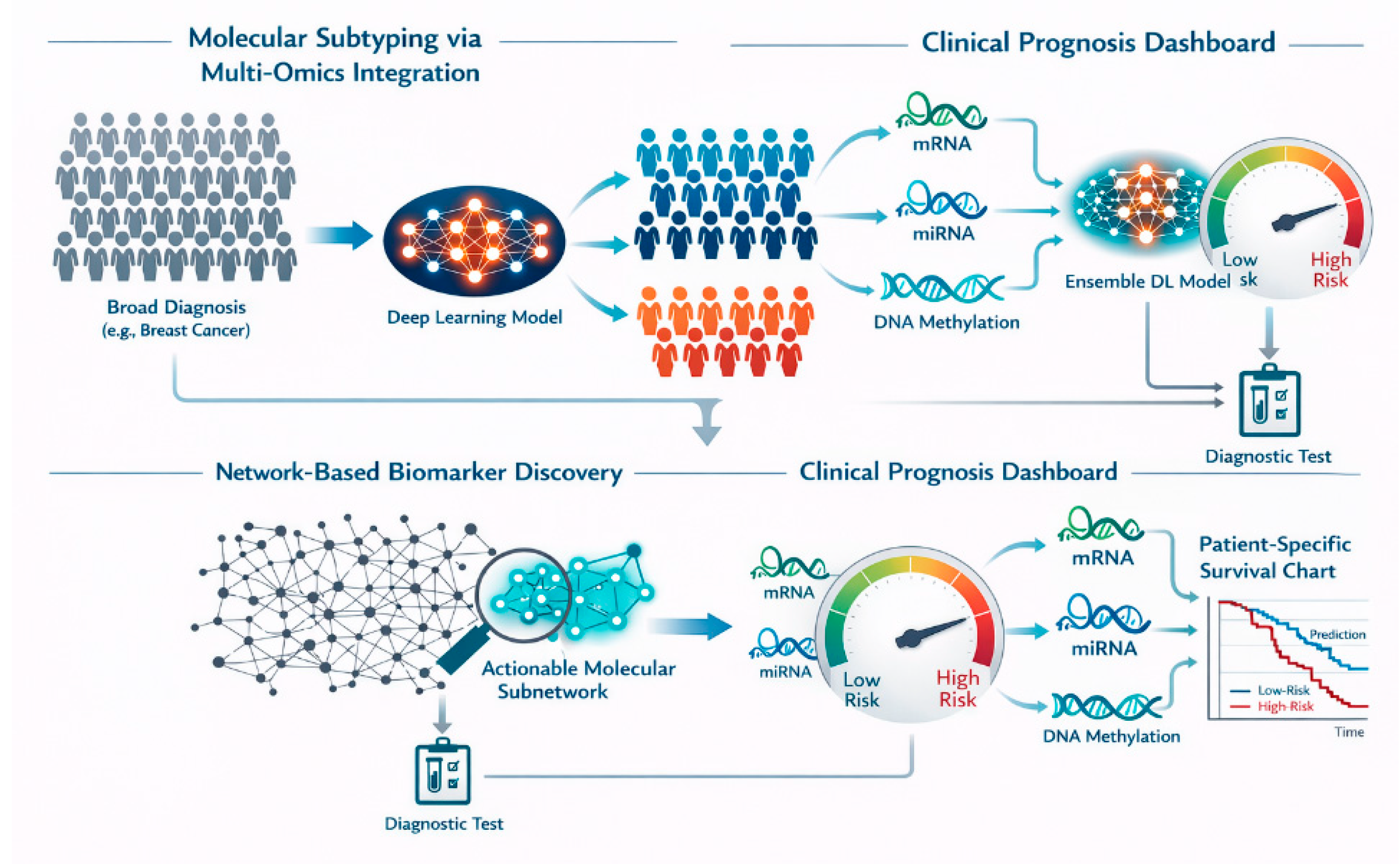
The overarching objective of deep learning (DL)–based multi-omics integration is to translate molecular complexity into clinically actionable insights that can inform diagnosis, prognosis, and therapeutic decision-making. In practice, translational applications of these models have focused primarily on refining disease stratification beyond conventional classifications and on developing predictive frameworks for biomarker discovery and patient outcome assessment.
Molecular Subtyping and Disease Classification
One of the most immediate translational impacts of multi-omics DL approaches is the refinement of disease subtyping. Traditional classification schemes, often based on histopathology or single molecular markers, frequently fail to capture the extensive molecular heterogeneity observed within clinically defined disease entities, resulting in suboptimal treatment stratification [20,32]. By integrating complementary molecular layers, DL models enable a more granular characterization of disease states.
Unsupervised architectures, particularly denoising autoencoders and variational autoencoders, are commonly used to cluster patients according to integrated molecular profiles [18,20]. These approaches have revealed molecularly coherent subtypes that are associated with distinct clinical outcomes, including significant differences in overall survival. For example, DAE-based multi-kernel learning frameworks have identified prognostically meaningful subgroups in cancers such as low-grade glioma and kidney renal clear cell carcinoma [19]. While multi-omics DL models often demonstrate improved classification performance relative to single-omics approaches, the primary translational value of these models lies in their ability to uncover biologically and clinically relevant disease substructures rather than in absolute predictive accuracy alone [15].
Robust Biomarker Discovery
A central translational goal of multi-omics integration is the identification of biomarkers that are sufficiently robust and reproducible for clinical use. Single biomarkers are often sensitive to technical noise and platform-specific effects, whereas multi-omics signatures can provide greater stability by integrating complementary molecular evidence across data modalities [23].
Network-based DL methods, particularly graph neural networks, are well suited for this task because they prioritize interconnected molecular features rather than isolated genes. GNN-based frameworks, such as the Expression Graph Network Framework, identify gene modules or molecular subnetworks whose collective activity is predictive of disease phenotypes, thereby improving biological interpretability and robustness [14]. In parallel, attention-based architectures and feature attribution methods have been applied to extract predictive molecular features by interrogating internal model representations. Although such approaches can highlight candidate biomarkers and pathways associated with clinical outcomes, their interpretability should be viewed as suggestive rather than causal.
Multi-omics DL models have also shown promise in predicting therapeutic response. For instance, composite scores derived from integrated DNA and RNA data, such as the Multi-Omics Tumor Immunogenicity Score, have been reported to outperform single markers like PD-L1 expression in predicting response to immune checkpoint inhibitors, underscoring the potential of integrated biomarkers in precision oncology.
Prognosis and Survival Prediction
Accurate prognosis is critical for clinical management, treatment planning, and patient counseling. Deep learning frameworks are increasingly applied to survival analysis by integrating multi-omics features with established statistical models capable of handling censored time-to-event data. In this context, DL-derived representations are often combined with Cox proportional hazards models to estimate patient-specific risk scores and stratify individuals into prognostic subgroups [17].
Ensemble approaches further enhance prognostic robustness by integrating multiple learning paradigms. Frameworks such as DeepProg combine autoencoder-based feature extraction with ensembles of machine learning models to predict survival subtypes across diverse cancer types, demonstrating improved performance relative to single-model or single-omics approaches [20,31]. Notably, some studies have reported the transferability of prognostic models trained on one cancer type to others, suggesting the potential for pan-cancer survival modeling. However, such generalization remains insufficiently validated across independent cohorts, highlighting the need for rigorous external validation before clinical deployment.
Figure 4.
Integrated multi-omics framework for precision oncology.
5. Open Challenges and Future Directions
Despite the substantial predictive advances achieved by deep learning (DL)–based multi-omics integration, the translation of these models from computational research into clinically deployed diagnostic and prognostic tools remains limited by several unresolved methodological and practical challenges. Addressing these limitations is essential for advancing clinically trustworthy biomedical informatics systems and defines a clear roadmap for future research.
Interpretability–Performance Trade-off
The most accurate multi-omics DL models are often the most complex, creating an inherent trade-off between predictive performance and interpretability that undermines clinical trust and adoption [21,22]. In high-stakes clinical settings, clinicians require transparency regarding the molecular basis of a model’s predictions to support informed decision-making, ensure accountability, and guide downstream therapeutic interventions [24]. However, the black-box nature of deep neural networks, which may contain millions of parameters, limits the ability to directly relate predictions to biologically meaningful mechanisms.
Explainable artificial intelligence (XAI) approaches have therefore emerged as a critical research priority for multi-omics DL [20,21]. Post hoc feature attribution techniques, such as SHAP and DeepLIFT, are increasingly used to assign importance scores to genes, pathways, or omics features contributing to model predictions [23]. While these methods improve transparency, their explanations are not inherently causal and may be unstable across datasets or model retrainings, necessitating careful validation and biological corroboration. Complementary strategies focus on intrinsically interpretable model design, including biologically constrained architectures such as graph neural networks and biologically informed variational autoencoders, which incorporate prior knowledge to restrict learning to plausible molecular interactions [24]. Disentangled representation learning in VAEs further enables the separation of biological signal from technical noise, particularly in single-cell multi-omics data [11].
Data Scarcity and Generalizability through Collaborative Learning
A persistent barrier to robust multi-omics DL modeling is the scarcity of large, well-annotated, paired multi-omics datasets. The high cost and logistical complexity of generating such data result in small, institution-specific cohorts, exacerbating the “big p, small n” problem and increasing susceptibility to overfitting [18,29]. Models trained under these conditions often exhibit sharp performance degradation when applied to independent cohorts from different populations or technical settings.
To address these limitations, decentralized and transfer-based learning paradigms are increasingly being explored. Federated learning enables collaborative model training across institutions without the exchange of raw patient data, thereby preserving privacy while leveraging distributed datasets [25,26,27]. This approach is particularly relevant under stringent regulatory frameworks such as the General Data Protection Regulation (GDPR) [28]. In parallel, transfer learning allows models pre-trained on large public repositories, such as The Cancer Genome Atlas, to be fine-tuned on smaller, task-specific clinical cohorts, facilitating knowledge transfer across domains [29]. Emerging foundation models for biological data extend this paradigm by learning generalizable molecular representations across diverse datasets [30]. The integration of federated and transfer learning strategies represents a promising pathway toward building robust and generalizable multi-omics predictive models [25,27].
Standardized Validation and Clinical Adoption
Perhaps the most significant gap between academic success and clinical impact lies in inadequate validation practices. The majority of multi-omics DL studies report performance based on internal cross-validation or limited train–test splits, which can substantially overestimate real-world performance [18]. There is a pressing need for standardized validation frameworks that mandate external evaluation on geographically, ethnically, and technically diverse cohorts to establish generalizability and clinical reliability.
Figure 5.
Addressing key barriers to real-world deployment of multi-omics artificial intelligence.
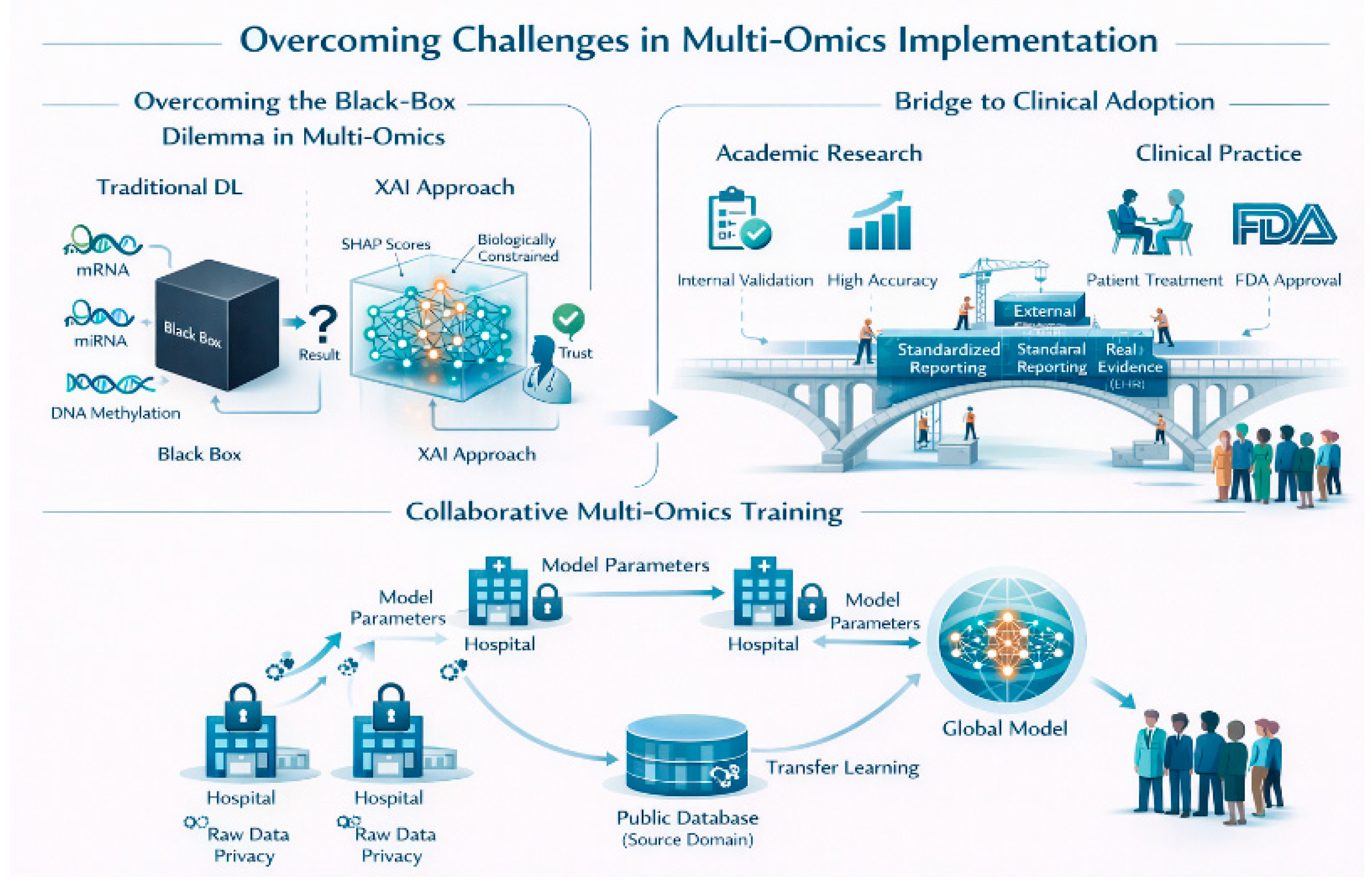
In addition, improved reporting standards are required to enhance reproducibility and transparency. Future studies should clearly document model architectures, training procedures, validation protocols, and evaluation metrics using reproducible code and standardized descriptions, analogous to reporting guidelines developed for medical imaging and clinical prediction models [32]. Finally, integrating real-world evidence, such as longitudinal electronic health record data, alongside multi-omics profiles can provide contextual validation and demonstrate that identified molecular biomarkers correlate with meaningful long-term clinical outcomes [31]. Such integration is essential for bridging the gap between algorithmic performance and real-world clinical utility.
6. Conclusions
The integration of multi-omics data is essential for advancing precision medicine by enabling a more comprehensive characterization of disease biology beyond single-layer molecular analyses [1]. As synthesized in this review, deep learning methodologies, particularly variational autoencoders for latent-space fusion and data imputation [9] and graph neural networks that incorporate biological prior knowledge into model architectures [13], have provided the analytical capacity required to address the complexity, heterogeneity, and high dimensionality inherent to multi-omics datasets. These approaches have demonstrated substantial promise in disease subtyping, biomarker discovery, and prognostic modeling, yielding molecular stratifications with greater clinical relevance than those obtained using conventional analytical methods [17,20].
Despite these advances, the field has reached a critical inflection point at which barriers to clinical translation are driven less by predictive performance than by challenges related to trust, interpretability, and validation. The widespread reliance on complex, opaque models limits clinical confidence and hampers adoption in high-stakes decision-making contexts. Addressing this gap requires a concerted shift toward explainable artificial intelligence frameworks that ensure model predictions are not only accurate but also biologically interpretable and clinically defensible [21,24]. In parallel, decentralized learning paradigms, such as federated learning, offer viable solutions to data scarcity and privacy constraints by enabling collaborative model development across institutions without the exchange of raw patient data [25,27]. Equally critical is the establishment of rigorous and standardized validation practices, including external evaluation on diverse and independent cohorts, to ensure generalizability and translational reliability [18,32].
By systematically addressing these challenges—interpretability, generalizability, and robust validation—computational biology and biomedical informatics are well positioned to translate multi-omics deep learning models from experimental research into clinically actionable decision-support tools. Achieving this transition will mark a substantive convergence of artificial intelligence and systems biology, with direct implications for individualized diagnosis, prognosis, and therapeutic decision-making in precision medicine.
References
- Wang, R.C.; Wang, Z. Precision medicine: Disease subtyping and tailored treatment. Cancers 2023, 15(15), 3837. [Google Scholar] [CrossRef]
- Athieniti, E.; Spyrou, G.M. A guide to multi-omics data collection and integration for translational medicine. Comput. Struct. Biotechnol. J. 2023, 21, 134–149. [Google Scholar] [CrossRef]
- Ballard, J.L.; Wang, Z.; Li, S.; Han, K.; Long, Q. Deep learning-based approaches for multi-omics data integration and analysis. BioData Min. 2024, 17(1), 22. [Google Scholar] [CrossRef] [PubMed]
- A. Yetgin, Revolutionizing multi-omics analysis with artificial intelligence and data processing. Quant. Biol. 2025, 13(3), 232–245.
- Ishida, J.P.; Huang, S.C.; Trostle, J.; Wang, K. A review of multi-omics data integration through deep learning approaches for disease diagnosis, prognosis, and treatment. Front. Genet. 2023, 14, 1199087. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Pan, D.; Lv, Y.; Ding, Y.; Sun, S.; Fan, D.; Liu, W.; Wei, R. Applications of multi-omics analysis in human diseases. MedComm 2023, 4(4), e315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Sun, T.; Jin, D. Review on the computational burden of single-cell multi-omics data: Sparsity, noise, and scalability. Quant. Biol. 2023, 11(4), 301–315. [Google Scholar]
- Nayak, S.; Khilar, P.M.; Roul, R.K. Deep generative models for multi-omics data imputation: A review, J. Supercomput 2025.
- Rana, M.; Seneviratne, S.; O’Connell, M.J. A technical review of multi-omics data integration methods: From classical statistical to deep generative approaches, Brief. Bioinform. 2025, 26(2), bbaf355. [Google Scholar]
- Kalafut, N.C.; Huang, X.; Wang, D. Joint variational autoencoders for multimodal imputation and embedding. Nat. Mach. Intell. 2023, 5(6), 631–642. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Li, G. Disentangled representation learning in VAEs for separating biological and technical variance in scRNA-seq. Nat. Methods 2025, 22(1), 11–19. [Google Scholar]
- Huang, Y.; Li, Q.; Wang, J. Deep generative models for synthetic multi-omics data augmentation. Cell Syst. 2024, 15(3), 221–235. [Google Scholar]
- Li, J.; Liu, B.; Wang, Y.; Zhu, Y.; Liu, Q. A multi-omics integration model based on graph convolutional network to analyze and classify cancer subtypes. Front. Genet. 2022, 13, 994087. [Google Scholar]
- Cheng, Y.; Zhao, Y.; Li, M. A hierarchical graph neural network integrating pathways for multi-omics analysis. Bioinform. Adv. 2024, 4(1), vbad150. [Google Scholar]
- Kim, K.; Lee, M.; Park, H. Graph attention networks for robust multi-omics drug sensitivity prediction. Nat. Comput. Sci. 2024, 4(5), 450–461. [Google Scholar]
- Ren, Y.; Gao, Y.; Du, W.; Qiao, W.; Li, W.; Yang, Q.; Liang, Y.; Li, G. Classifying breast cancer using multi-view graph neural network based on multi-omics data. Front. Genet. 2024, 15, 1363896. [Google Scholar] [CrossRef]
- Tan, S.; Yang, C.; Goudie, M.; Zhu, H.; Shi, Q. Enhancing non-small cell lung cancer survival prediction through multi-omics integration using graph attention network. Medicina 2024, 60(1), 2178. [Google Scholar]
- Wang, Y.; Li, Q.; Zhang, H. Benchmarking deep learning models for multi-omics integration on pan-cancer datasets. Genome Med. 2024, 16(1), 78. [Google Scholar]
- Park, T.; Kim, S.; Lee, J. Multi-omics model for early prediction of Alzheimer’s disease using deep learning. Alzheimers Dement. 2024, 20(4), 1–12. [Google Scholar]
- Wen, Y.; Zheng, L.; Leng, D.; Dai, C.; Lu, J.; Zhang, Z.; He, S.; Bo, X. Deep learning-based multiomics data integration methods for biomedical application. Adv. Intell. Syst. 2023, 5(5), 2200247. [Google Scholar] [CrossRef]
- Hussein, A.; Prasad, M.; Braytee, A. Explainable AI methods for multi-omics analysis: A survey. ACM Trans. Comput. Surv. 2024, 57(1), 1–39. [Google Scholar]
- Zhu, X.; Zhang, Y.; Wang, J. Explainable AI methods for multi-omics analysis: A survey. J. Biomed. Inform. 2024, 15(1), 384973895. [Google Scholar]
- Xie, X.; Liu, B.; Zhang, S. SHAP-based feature importance for graph neural networks in multi-omics biomarker discovery. Bioinformatics 2025, 41(2), btad899. [Google Scholar]
- Han, K.; Chen, Y.; Lee, S. Visible neural networks for multi-omics integration: A critical review. Front. Artif. Intell. 2025, 8, 1595291. [Google Scholar] [CrossRef]
- Andreux, M.; Wang, Y.; Yu, X.; N’guessan, A.; El-Haddad, R.; Sénécal, F.; Alimonti, J. Federated transfer learning with differential privacy for multi-omics survival analysis, Brief. Bioinform 2025, 26(2), bbaf555. [Google Scholar]
- Chen, S.; Li, M.; Zhao, H. AFEI: Adaptive optimized vertical federated learning for heterogeneous multi-omics data integration, Brief. Bioinform 2023, 24(10), bbac341. [Google Scholar]
- Zhang, Y.; Liu, Y.; Wang, B. Federated transfer learning with differential privacy for multi-omics survival analysis, Brief. Bioinform. 2025, 26(2), 390793804. [Google Scholar]
- Li, Q.; Zhang, Y.; Liu, B. Ethical and privacy challenges in federated learning for genomic data. Sci. Adv. 2023, 9(38), eadh4775. [Google Scholar]
- Cui, R.; Han, K.; Long, Q. A systematic review of transfer learning methods for multi-omics data. Bioinformatics 2023, 39(12), btad732. [Google Scholar]
- Qiao, W.; Wang, Y.; Zhang, X.; Zhou, Y. Foundation models in biology: A review on multi-omics data. Cell Rep. Phys. Sci. 2023, 4(10), 101569. [Google Scholar]
- Song, Y.; Zhang, W.; Sun, Y. Integrating electronic health records as a clinical ‘omic’ layer using deep learning. Nat. Biomed. Eng. 2024, 8(2), 150–162. [Google Scholar]
- Zhao, Y.; Shao, J.; Asmann, Y.W. Assessment and optimization of explainable machine learning models applied to transcriptomic data. Genomics Proteomics Bioinformatics 2022, 20(4), 899–911. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



