
Submitted:
22 January 2026
Posted:
23 January 2026
You are already at the latest version
Abstract
Ultraviolet (UV) radiation induces DNA damage and oxidative stress in melanocytes, shaping pigmentation phenotypes and elevating photocarcinogenesis risk. Human models that capture donor-specific genetic determinants of UV sensitivity remain limited. Here, we establish a genotype-driven UV response model using induced pluripotent stem cell (iPSC)-derived melanocytes from donors carrying defined MC1R variants. Differentiated cells recapitulated melanocytic morphology, marker expression, and pigmentation consistent with donor sun-sensitivity traits. Following narrowband UVB exposure, UV-sensitive lines exhibited reduced survival, prolonged checkpoint activation, and delayed cyclobutane pyrimidine dimer (CPD) repair. Mechanistic analysis revealed that the interferon-regulated GTPase MX2 amplifies UV-induced p53 and p38 activation while promoting apoptosis independently of AKT. These findings identify MX2 as a physiological enhancer of DNA damage signaling in normal melanocytes, distinct from its interferon-mediated role in melanoma. Our study provides a human-relevant platform linking pigmentation genotype to UV resilience and supports iPSC-derived systems as New Approach Methodologies (NAMs) for mechanistic and translational phototoxicology.
Keywords:
induced pluripotent stem cells
; iPSC-derived melanocytes
; ultraviolet radiation
; MC1R variants
; DNA damage response
; MX2
; p53 signaling
; new approach methodologies
1. Introduction
Ultraviolet (UV) radiation is a pervasive environmental stressor that inflicts direct and indirect damage on skin cells through cyclobutane pyrimidine dimers (CPDs), 6-4 photoproducts, and reactive oxygen species (ROS) [1,2]. ROS exacerbate cellular injury by oxidizing lipids and nucleic acids, activating the Nrf2-Keap1 antioxidant pathway, and modulating apoptosis via MAPK cascades [3,4,5]. The efficiency of these repair and detoxification mechanisms varies among individuals and is strongly influenced by pigmentation genotype.
The melanocortin-1 receptor (MC1R) governs melanogenesis, the eumelanin/pheomelanin ratio, and cAMP/PKA-CREB signaling after UV exposure [6,7]. Functional MC1R variants favor eumelanin synthesis and activate p53-mediated DNA repair, whereas loss-of-function alleles, common in red-hair and fair-skin phenotypes, impair nucleotide excision repair (NER) and increase melanoma susceptibility [8,9]. Beyond melanin photoprotection, MC1R signaling intersects with ATR/CHK1 checkpoint control and antioxidant responses, shaping cellular resilience to UV-induced DNA lesions [10,11].
Traditional studies rely on primary or transformed melanocytes, which are limited by availability and donor variability. Induced pluripotent stem cell (iPSC) technology enables reprogramming of fibroblasts from genetically defined donors into melanocytes, preserving pigmentation and repair traits [12,13]. Single-cell and transcriptomic analyses validate iPSC-derived melanocytes as faithful models of epidermal pigmentation and photobiology [14,15]. These systems also permit exploration of paracrine interactions with keratinocytes via α-melanocyte-stimulating hormone (α-MSH), endothelins, and stem cell factor (SCF), which modulate melanocyte UV responses [16,17,18].
At the molecular level, the interferon-inducible GTPase MX2 regulates antiviral defense and cell-cycle control [19,20,21,22]. In melanoma, MX2 influences therapeutic response and immune signaling through STAT1, XAF1, and p53 networks [21,22,23,24]. However, its physiological role in normal melanocytes and in the DNA damage response (DDR) to UV stress remains unclear. Preliminary evidence suggests MX2 integrates interferon, DDR, and apoptotic pathways via p38 and p53 crosstalk [24]. Elucidating this axis is critical for linking pigmentation genotype to stress signaling and disease risk.
Here, we establish an iPSC-based human melanocyte model to investigate how pigmentation genotype shapes UV-induced DNA damage responses. Using donor-specific lines carrying distinct MC1R variants, this study examines the relationship between genetic background, checkpoint activation, and repair fidelity. We also explore the role of the interferon-inducible factor MX2 in modulating UV stress responses. Together, these analyses provide a mechanistic framework for genotype-encoded UV sensitivity and support the relevance of iPSC-based NAMs for photobiology research [25,26].
2. Results
2.1. Generation, Validation, and Selection of Patient-Derived iPSC Lines
To establish a genetically diverse iPSC resource for modeling pigmentation biology and UV responses, dermal fibroblasts from melanoma patients and healthy donors were reprogrammed using Sendai virus vectors expressing OCT4, SOX2, KLF4, and c-MYC as originally described by Yamanaka and colleagues [27] and later adapted into non-integrating Sendai virus systems for footprint-free reprogramming [28]. Across all donors, we generated a panel of 28 human iPSC lines, each expanded as 4-5 clonal colonies and subjected to standardized pluripotency quality-control (QC) assessments (Supplementary Table S1). Lines passing RT-qPCR, TRA-1-60/SSEA-4 flow cytometry, and morphological criteria (Supplementary Figure S1) were further evaluated using the TaqMan hPSC Scorecard (see QC summary in Supplementary Table S1). Lines exhibiting unstable morphology or suboptimal pluripotency signatures were excluded from downstream work.
From this larger cohort, we prioritized a subset of iPSC lines with fully documented MC1R genotype, normal karyotype, and high-quality pluripotency profiles for melanocyte differentiation. These included donors SF003, SF004, SF006, SF007, SF008, and SF009, which consistently performed robustly across QC assays. Additional successfully reprogrammed lines (e.g., GM09943, GM00671-6, Wi38, FF160, FH202, FH217, FH310) were not advanced due to incomplete metadata, limited fibroblast availability, or inconsistent melanocyte yield.
Together, these data define the full reprogramming pipeline supporting this project and the criteria used to select the subset of iPSC lines for melanocyte differentiation (Supplementary Table S1).
2.2. Generation and Characterization of iPSC-Derived Melanocytes with Patient-Specific Pigmentation Genotypes
Fibroblasts from donors carrying distinct MC1R pigmentation genotypes were reprogrammed into iPSCs and differentiated into melanocytes following a standardized protocol (Figure 1A). The resulting cells exhibited characteristic dendritic morphology and expressed melanocytic markers MelanA and HMB45, confirmed by immunofluorescence (Figure 1B). Additional melanocytic markers, including S100, Mel-5 (TYRP1), and dual-marker combinations, were validated in representative lines (Supplementary Figure S2). Quantitative RT-PCR demonstrated robust expression of the lineage-specific genes MITF, PMEL, and TYR, comparable to that in normal human melanocytes (NHM160) (Figure 1C), consistent with previous reports [12,13,14]. Measurement of melanin content revealed variable pigmentation levels across donor lines, correlating with their MC1R genotypes and reported sun-sensitivity scores (Figure 1D–E; Supplementary Table S2). This genotype–phenotype relationship is consistent with previous work showing that pigmentation pathway activity, including MITF-dependent melanogenic programs, modulates UV resilience in human melanocytes [29].
A detailed summary of melanocyte differentiation outcomes for each selected SF line, including morphology, pigmentation level, marker expression, and culture timeline, is provided in Supplementary Table S2. These data document line-to-line variability among the SF iPSCs and justify their selection for subsequent mechanistic analyses.
Prior to differentiation, pluripotency was verified by flow cytometry and immunostaining for TRA-1-60 and SSEA-4, alongside high mRNA levels of Nanog, Oct4, and Sox2 (Supplementary Figure S1), confirming successful reprogramming. Collectively, these findings establish a reproducible platform for generating melanocytes that preserve donor-specific pigmentation traits, enabling mechanistic studies of UV response in a genetically defined context.
2.3. Donor-Encoded UV Sensitivity and Cell-Cycle Checkpoint Activation
To determine whether donor genotype influences UV stress resilience, iPSC-derived melanocytes were exposed to increasing doses of narrowband UVB (0–1.5 J/cm²). All lines exhibited a dose-dependent decrease in viability; however, survival differed markedly by MC1R genotype (Figure 2A,B). UV-sensitive lines (SF3, H1m) showed early activation of apoptosis, evidenced by cleaved caspase-3 and PARP within 2–6 h post-irradiation (Figure 2F), whereas resistant lines (SF4, NHM160) displayed delayed apoptotic signaling, suggesting more efficient damage recovery. Cell-cycle analysis revealed phosphorylation of histone H3 (Ser10) and CDK1 (Tyr15), indicating G2/M checkpoint engagement following UV exposure (Figure 2D,E). Flow cytometry confirmed accumulation of resistant lines in G2/M at 24 h, while sensitive lines progressed to apoptotic sub-G1 fractions.
Additionally, melanocytes incorporated into 3D skin reconstructs displayed donor-specific apoptotic responses after UV exposure, consistent with the monolayer assays (Figure 2G). These results demonstrate that UV sensitivity is an intrinsic, donor-encoded trait in melanocytes, integrating checkpoint activation and apoptotic dynamics consistent with pigmentation genotype.
2.4. UV-Induced DNA Damage Signaling and Repair Efficiency
To investigate genotype-dependent DNA damage response (DDR), melanocytes were irradiated with 1.5 J/cm² nbUVB and collected at defined intervals (2–24 h) (Figure 3A–D). Immunoblotting revealed rapid phosphorylation of ATM, CHK1, and CHK2 within 2 h (Figure 3B), confirming activation of canonical DDR pathways [4,15]. However, UV-sensitive lines exhibited sustained γH2AX and GADD45α accumulation up to 24 h, indicating incomplete lesion repair. This pattern parallels recent UVB studies in human skin-derived cell models showing that persistent γH2AX retention and delayed DDR resolution strongly predict apoptotic commitment [30].
Phosphorylation of AKT (S473) persisted longer in sensitive melanocytes compared to resistant lines (Figure 3B), consistent with checkpoint adaptation and survival signaling described in UV-exposed skin cells. CPD immunoassays showed peak damage immediately after irradiation, followed by gradual decline in all lines; however, residual CPDs at 24 h remained significantly higher in UV-sensitive melanocytes (Figure 3C, D). These findings indicate that inefficient CPD repair and persistent DDR signaling underlie increased UV susceptibility in cells derived from donors with loss-of-function MC1R variants, aligning with prior observations [9,15].
2.5. MX2 Expression Enhances UV-Induced Stress Signaling in Melanocytes
Given previous reports implicating MX2 in interferon responses and melanoma progression [21,22,23], we examined its role in UV-induced stress signaling in non-transformed melanocytes (Figure 4A–D). Basal MX2 expression varied across donor lines and positively correlated with UV sensitivity and p53 activation (Figure 4A, B). UV exposure induced further MX2 upregulation in sensitive lines, coinciding with strong phosphorylation of p38 and p53 (Ser15) (Figure 4B,D).
Functional perturbation studies in melanoma cells revealed that ectopic MX2 overexpression augmented UV-induced apoptosis and reduced cell viability (Supplementary Figure S3), whereas GFP controls exhibited higher survival. Importantly, MX2 modulation did not significantly alter AKT phosphorylation, suggesting a p53/p38-dependent but AKT-independent mechanism of stress amplification. These results identify MX2 as a physiological enhancer of UV-induced apoptosis and checkpoint signaling in human melanocytes, distinct from its interferon-mediated role in melanoma [21,22,31], highlighting its potential as a modulator of phototoxic stress and pigmentation-related DNA repair capacity.
3. Discussion
This study establishes a genotype-driven model of human UV sensitivity using iPSC-derived melanocytes from donors with defined MC1R pigmentation variants. The results demonstrate that UV responses are encoded at the genetic level and faithfully recapitulated in patient-derived melanocytes, linking pigmentation genotype to cellular resilience, checkpoint activation, and DNA repair efficiency. These findings advance our understanding of how inter-individual variation shapes phototoxicity risk and contribute to the definition of human-relevant, mechanistic frameworks for UV-induced disease research.
The ability to generate melanocytes from iPSCs enables reproducible and scalable modeling of human pigmentation biology [12,13,14]. Our data confirm that differentiation yields mature melanocytes expressing canonical lineage markers (MITF, TYR, PMEL, MelanA, HMB45), with pigmentation intensity reflecting the underlying MC1R genotype. The observed range of UV survival among donor-derived lines directly correlated with their sun-sensitivity variable, validating this system as a quantitative surrogate for in vivo responses. These consistencies further support the translational relevance of our model and its capacity to recapitulate patient-specific UV responses observed in vivo. These findings are consistent with previous reports showing that MC1R signaling regulates melanocyte survival and DNA-repair capacity through cAMP-dependent and p53-mediated pathways [9,32]. Importantly, the differential UV survival patterns observed among iPSC-derived melanocytes parallel those previously reported in primary melanocytes and clinical cohorts stratified by MC1R genotype and pigmentation phenotype [7,9,28,33].
UV-induced DNA damage is primarily manifested as cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts [1,2]. Efficient repair of these lesions through nucleotide excision repair (NER) prevents mutagenesis and photocarcinogenesis. In our model, UV-sensitive melanocytes exhibited delayed CPD clearance and sustained DDR signaling, suggesting that impaired checkpoint resolution is a determinant of sensitivity. Persistent activation of γH2AX, pATM, and pCHK1 in these lines mirrors the phenotype of MC1R-deficient cells, which show diminished ATR/p53 signaling and inefficient GADD45-mediated repair [33,34]. This phenotype parallels recent findings in human skin-derived models, where prolonged γH2AX retention and checkpoint persistence strongly correlate with apoptotic commitment [30]. Together, these results underscore the interplay between pigmentation genotype and DDR fidelity as a basis for differential UV outcomes.
Beyond direct DNA lesions, UV exposure also generates reactive oxygen species (ROS) and lipid peroxidation products that amplify stress signaling through Nrf2 and MAPK pathways [21,22,35,36]. This oxidative component of UV injury modulates melanocyte fate by tipping the balance between adaptive and apoptotic outcomes. Our findings indicate that loss-of-function MC1R alleles, which impair cAMP/PKA signaling, result in prolonged ROS accumulation and inefficient NER activation. This aligns with previous studies linking MC1R variants to redox imbalance, mitochondrial stress, and defective antioxidant responses in human skin [15,33,37]. Thus, pigmentation genotype integrates both pigment chemistry and redox regulation to define the cellular threshold for UV tolerance.
A central finding of this study is the identification of MX2 as a physiological enhancer of UV-induced apoptosis and stress signaling in normal melanocytes. While MX2 was previously characterized in melanoma as an interferon-inducible factor linked to cell-cycle arrest and immune signaling [21,22,23], our data reveal that its expression also amplifies p53/p38 activation under UV stress independently of AKT. This suggests that MX2 integrates stress and apoptotic signaling pathways beyond its canonical antiviral or IFN-regulated functions [21,38,39]. Elevated MX2 in UV-sensitive iPSC-derived melanocytes was accompanied by increased caspase activation and reduced viability, highlighting its potential role in reinforcing damage-induced checkpoint responses. Nevertheless, while our results establish a clear correlation between MX2 upregulation and enhanced apoptosis, the causal role of MX2 in regulating UV-induced cell death remains to be functionally validated. Future studies employing targeted MX2 knockdown or CRISPR-mediated gene editing in normal human melanocytes will be essential to confirm its direct mechanistic contribution. Such approaches will help delineate whether MX2 acts as a driver or amplifier of p53/p38-dependent apoptotic signaling.
Mechanistically, MX2 may influence apoptotic signaling by regulating pro-apoptotic factors such as XAF1 and modulating STAT1 phosphorylation, as demonstrated in melanoma cells under interferon stimulation [21]. Although direct links to p53 stabilization or mitochondrial apoptotic pathways remain to be elucidated, its correlation with UV sensitivity supports the notion that innate immune effectors can cross-regulate DDR and pigmentation pathways. This expands the physiological scope of MX2 from an interferon effector in melanoma to a UV-response modulator in normal melanocytes, integrating genotype, stress signaling, and DNA repair capacity. In this context, sustained MX2 activity may contribute to apoptotic clearance of damaged melanocytes through regulation of pro-apoptotic factors such as XAF1 and modulation of STAT1 phosphorylation [21]. Although similar checkpoint persistence mechanisms have been reported in other cell types, including fibroblasts and keratinocytes [40], their relevance to MX2 function in melanocytes remains to be confirmed.
Our findings also underscore how MC1R-driven pigment chemistry impacts ROS buffering and DDR activation. Eumelanin-rich cells scavenge radicals and efficiently couple cAMP-PKA signaling to p53-mediated repair, while pheomelanin-rich melanocytes generate additional ROS under UV exposure, enhancing mutagenic load [33,37]. This concept aligns with recent findings demonstrating that MC1R/MITF-driven melanogenic capacity modulates UV-induced stress signaling thresholds in human melanocytes [29]. The convergence of these redox and signaling differences with MX2 activity defines a spectrum of UV resilience across individuals. Notably, this framework integrates pigmentation genotype, DDR dynamics, and apoptosis thresholds as co-determinants of photoprotection.
From a broader mechanistic perspective, UV stress response in melanocytes exemplifies the integration of immune signaling, pigment biochemistry, and checkpoint control. The identification of MX2 as a regulator of XAF1 and STAT1 phosphorylation provides a potential link between interferon-regulated networks and stress-induced apoptotic signaling in melanoma cells [21].
Similar cross-talk between interferon-stimulated genes and DNA damage response pathways has been described in pancreatic islet cells under autoimmune stress, suggesting broader roles for interferons in cellular stress adaptation [41].
Our data extend this concept to melanocytes, positioning MX2 at the intersection of antiviral defense and photoprotection biology. Finally, this iPSC-based system provides a flexible experimental framework to investigate patient-specific UV responses under standardized conditions. Unlike conventional melanoma or keratinocyte models, iPSC-derived melanocytes maintain the donor’s genomic integrity and pigmentation genotype, enabling controlled studies of genetic and pharmacologic modifiers of UV sensitivity [12,14]. As regulatory agencies emphasize New Approach Methodologies (NAMs) for mechanistic toxicology, this model supports translational research in photoprotection, pigmentation disorders, and UV-induced disease susceptibility [42]. Recent advances in NAMs have demonstrated the utility of stem cell-derived skin models for phototoxicity testing, including pigmentation-related responses [43]. Integration of multi-omics profiling, high-content imaging, and predictive modeling could further refine these systems for NAM-based hazard assessment and personalized photoprotection strategies [44].
From a regulatory standpoint, our iPSC-based approach aligns with the principles of New Approach Methodologies (NAMs) endorsed by the OECD for non-animal safety testing and mechanistic toxicology (OECD Test Guidelines TG 432, TG 497). Incorporating genetically defined human cell systems into NAM frameworks may improve the predictivity of phototoxicity assays and strengthen translational applications in personalized photoprotection.
Collectively, our results reveal that MX2 enhances UV-induced p53/p38 signaling in a genotype-dependent manner, positioning it as a potential molecular link between pigmentation, stress responses, and DNA repair efficiency. This work establishes iPSC-derived melanocytes as a human-relevant tool for studying the mechanistic basis of UV sensitivity and opens new directions for developing personalized photoprotective and NAM-based translational approaches.
4. Materials and Methods
Cells and cell culture. Normal human melanocytes (NHM) were isolated as previously described [45] and cultured in 254CF media (Invitrogen) supplemented with Human Melanocyte Growth Supplement-2 (Gibco), Calcium chloride (Gibco), 5 mL Pen/Strep (cat. No. DE17-603E, Lonza) and 10 ng/mL PMA. Twenty-four hours before narrowband UVB (nbUVB) exposure, GFP/MX2 expression in transduced NHM134 was induced with 500 ng/mL doxycycline treatment. Melanoma cells were cultured in RPMI 1640 medium (Bio Whittaker, Verviers, Belgium) supplemented with 5% fetal bovine serum (cat.no F7524, Sigma) and 2 mM L-glutamine (cat. No. 17-605E, Lonza). Human dermal fibroblasts were derived and established from primary melanoma patients (Leeds, UK) by skin punch biopsy as previously described [46]. Fibroblasts were cultured in DMEM containing 10% FBS. Human embryonal stem cells H1, purchased from WiCell (cat. No. WA01), and derived iPSC lines were cultured with MEF feeders (cat. No. 6001G, GlobalStem) on tissue culture plates coated with 0.1% gelatin solution (cat. No. ES-006-B, Millipore) in stem cell culture media consisting of 400 mL DMEM/F-12 (cat. No. 31330-038, Invitrogen), 100 mL KnockOut Serum Replacement (KOSR) (cat. No. 10828-028, Invitrogen), 5 mL Pen/Strep (cat. No. DE17-603E; Lonza), 2 mM L-glutamine (cat. No. 17-605E, Lonza), 5 mL NEAA (cat. No. 11140035, Invitrogen), 100 mM 2-mercaptoethanol (cat. No. 31350010, Invitrogen) and 10 ng/mL bFGF (cat. No. PHG0023, Invitrogen). All cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Generation of Human Induced Pluripotent Stem Cells from human dermal fibroblasts.
Fibroblasts were reprogrammed to iPSCs using the CytoTune™-iPS Reprogramming kit (cat. No. A13780, Life Technologies) and following the manufacturer’s protocol. This method is based on the Sendai reprogramming platform, which follows the non-integrating OSKM Sendai system [28]. Selected iPSC colonies were characterized for pluripotency and trilineage differentiation potential using TaqMan® hPSC Scorecard™ (cat No. A15876, ThermoFisher Scientific). iPSC clones with optimal Scorcard results were also validated by flow cytometry for pluripotency markers and selected for melanocyte differentiation.
Embryoid Body (EB) formation. For embryoid body formation, selected iPSC clones and H1 cells were transferred to low attachment plates and cultured in EB medium, containing DMEM/F-12 (ref. 31330-038, Invitrogen) supplemented with 20% KnockOut Serum Replacement (cat. No. 10828-028, Gibco), 50 U/mL Pen/Strep (cat. No. DE17-603E, Lonza), 2 mM L-Glutamine (cat. No. 17-605E, Lonza), 1% MEM NEAA (cat. No. 11140035, Invitrogen) and 0.2% 2-mercaptoethanol (cat. No. 31350010, Invitrogen). EBs were formed and maintained in the EB medium for up to 6 days after which melanocyte differentiation was initiated.
Melanocyte differentiation. IPSC/H1 differentiation into melanocytes was performed as described previously [47]. Briefly, embryoid bodies were resuspended in melanocyte induction media (Supplementary Table 1) and plated on human fibronectin-coated 6-well plates. The media was changed every 2 days for up to 5 weeks. When bipolar and/or dendritic pigmented cells were observed in the wells, they were mechanically dissociated and maintained in melanocyte-supporting medium. Isolated cells were tested for pigmentation and melanocyte differentiation markers.
Flow Cytometry. To assess iPSC for the presence of pluripotency markers, after fixation cells were incubated with SSEA4 (cat. No. 09-0006, Stemgent) and TRA-1-60 (cat. No. MAB-4360, Merck Milipore) antibodies, followed by incubation with secondary anti-mouse Alexa Fluor 647 (cat. No. A32728, Invitrogen) and anti-rabbit PE (cat. No. 12-4739-81, Invitrogen).
For cell cycle analysis, 3×105 melanocytes per well were seeded into 6-well plates 24 hours before nbUVB irradiation. Twenty-four hours after UV treatment, cells were harvested by trypsinization, washed twice in ice-cold PBS, and fixed by resuspending cell pellets in 1 mL 100% ice-cold methanol. Fixated cells were stained with a ready-to-use DNA Labelling Solution (Cytognos, cat.no. CYT-PIR-25). Stained cells were analyzed on a BD FACSCaliburTM Flow cytometer (BD Biosciences). Data were analyzed with FlowJo v.7.6.1 software (Treestar Inc., Ashland, OR, USA).
Immunohistochemistry. Sections from formalin-fixed paraffin-embedded 3D reconstructs were immunostained using the Dako EnVision™+ system (K8012, Dako Cooperation, Carpinteria, CA) and Dako Autostainer. Deparaffinization, rehydration, and target retrieval were performed in a Dako PT link and EnVision™ FLEX Target Retrieval Solution, low pH (citrate buffer, pH 6.1). Endogenous peroxidase was blocked using Dako blocking reagent for 5 min, followed by incubation with rabbit monoclonal cleaved caspase 3 antibody (#9664; Cell Signaling (Danvers, MA, USA)) diluted 1:500 for 30 min. Thereafter, the sections were incubated with Dako EnVision™ FLEX+ rabbit linker for 15 min followed by incubation with Dako EnVision™ FLEX/HRP for an additional 30 min. For visualization of staining, the sections were treated with 3,3′-diaminobenzidine tetrahydrochloride (DAB), counterstained with hematoxylin, and mounted in Richard-Allan Scientific™ Cytoseal™ XYL (Thermo Scientific, Waltham, MA). Negative controls included substitution of the polyclonal primary antibody with normal rabbit IgG protein at the same concentration.
Immunofluorescence. Melanocytes were seeded onto 6-well plates with glass bottom at density 3×105 cells per well. For fixation, melanocytes were overlaid with 100% ice-cold methanol and allowed to fix for 15 minutes at -20⁰C. Methanol was aspirated, and cells were rinsed three times with PBS. To avoid non-specific antibody binding, cells were incubated with blocking solution (5% goat serum (cat. No. ab7481, Abcam (Cambridge, UK)) in PBS) for 1 hour at room temperature. The blocking solution was aspirated, and cells were stained with primary antibodies overnight at 4⁰C, diluted to 1:200 and specific for HMB45 (cat. No. ab787, Abcam) and MelanA (cat. No. ab51061, Abcam). Cells were then rinsed three times with PBS, and secondary antibody Alexa Fluor 488 (Invitrogen) diluted to 1:500 was added for 1 hour at room temperature. After three washes, cells were stained with DAPI and examined using a Leica DMI600 B inverted fluorescence microscope.
Quantitative real-time Reverse-Transcription PCR. RT-qPCR reactions were performed as previously described [22] using TaqMan Gene Expression Assays: MITF (Hs01117294_m1), TYR (Hs00165976_m1), and PMEL (Hs00173854_m1). Transcript expression levels were normalized to a housekeeping gene, beta-glucuronidase (GUSB; Hs99999908_m1). Expression of endogenous pluripotency markers was assessed using the TaqMan iPSC Sendai Detection Kit (cat. No. A13640, Life Technologies) according to the manufacturer’s protocol.
Incucyte growth rate assessment: For cell growth determination, melanocytes were seeded into 96-well plates at a density of 10,000 cells per well. Cell proliferation was assessed using a confluence assay with the IncuCyteTM FLR (Essen Instruments, Ann Arbor, MI) live-cell imaging system. Cell growth rate was determined by normalizing cell confluence at a given time to the corresponding initial time point. Data are presented as the mean ± SE from three independent experiments.
Viability assay. For viability assays 10,000 melanocytes and 5,000 melanoma cells were seeded into 96-well plates a day prior to nbUVB irradiation. Seventy-two hours after the nbUVB insult cell viability was determined using CellTiterGlo luminescent viability assay (cat. No G7570) purchased from Promega and following manufacturer’s protocol. Luminescence was recorded with Fluoroskan Ascent FL luminometer (Thermo Fisher Scientific).
DNA extraction. 3×105 melanocytes per well were seeded into 6-well plates 24 hours before nbUVB irradiation with 1.5 J/cm2. At different time points after irradiation, genomic DNA was extracted using DNA extraction kit according to manufacturer’s protocol (ref. 740952.250, Macherey-Nagel) and stored at -20⁰C until use.
Quantification of CPDs by ELISA. Genomic DNA (gDNA) was denaturated by incubating sample at 95⁰C for 10 min, followed by immediate cooling on ice for additional 10 min. gDNA was diluted in DNA coating solution (cat. No. 17250, Thermo Scientific™) to yield a final concentration of 2 ng/µL and 50 µL of each sample were loaded per well on a DNA-BIND 96 well plate (ref. 2525, Sigma-Aldrich) in duplicate. Incubation lasted overnight at 37⁰C, then each well was washed 5 times with 150 μl PBST (0.05% Tween-20 (cat. No. P1379, Sigma Aldrich) in 1x PBS). To prevent non-specific antibody binding, 100 μl of 2.5% FBS (cat. No F7524, Sigma) in 1x PBS was loaded into each well for 1 hour at 37⁰C. After 5 washes with 150 μl PBST, 100 μl per well of 1:1000 diluted anti-CPD antibody (clone TDM2, cat. No CAC-NM-DND-001, Cosmo Bio USA) in 2.5% FBS was loaded and incubation lasted for 1 hour at 37⁰C. After 5 washes with 150 μl PBST, plate was incubated with 100 μl per well of 1:1000 diluted secondary HRP conjugated anti-mouse IgG(H+L) antibody (cat. no. W4021, Promega). The plate was washed 5 times with 150 μl PBST and incubated with 100 μl 1-Step™ Turbo TMB-ELISA substrate solution (cat. No. 34022, Thermo Scientific™) for 30 min at room temperature. Reaction was stopped with 100 μl ELISA Stop Solution (cat. No. SS04, Invitrogen™) and absorbance was measured at 450 nm with a reference at 620 nm on the Multiskan FC microplate photometer (Thermo Scientific).
Melanin measurements. A pellet of 106 melanocytes was dissolved in 1 mL Soluene-350 in the presence of 10% dH2O and heated at 60ºC for 3 hours with agitation. Absorbance was measured at 500 nm using Perkin Elmer spectrometer. To convert absorbance value to the total melanin amounts, a standard calibration curve of synthetic melanin (Sigma-Aldrich) was generated. Three independent experiments to evaluate melanin contents were performed per each melanocyte culture.
Generation of stable lines overexpressing MX2 and GFP. Cell cultures were established as described in a publication [22].
Immunoblotting. For immunoblotting analyses, melanocytes were seeded at 3×105 cells per 6-well plates (BD Falcon). 2, 6 and 24 hours after nbUVB treatments cells were harvested and analyzed for protein expression as previously [45]. Primary antibodies used in a study: Cell Signaling (Danvers, MA, USA): PARP (#9532) 1:1000, Caspase 3 (#9662) 1:5000, Cleaved Caspase 3 (#9664) 1:1000, GAPDH (#2188) 1:2000, phospho-ATM s1981 (#13050) 1:1000, ATM (#2873) 1:1000, phospho-CHK1 s345 (#2341) 1:1000, CHK1 (#2345) 1:1000, phospho-CHK2 t68 (#2661) 1:1000, CHK2 (#3440) 1:1000, γH2AX (#9718) 1:1000, phospho-AKT s473 (#4060) 1:2000, AKT (#9272) 1:2000, phospho-H3 s10 (#9701) 1:1000, H3 (#4499) 1:3000, β-actin (#4967), phospho-p38 MAPK thr180/tyr182 (#4631) 1:1000; Santa Cruz Biotechnology (Dallas, TX, USA): GADD45α (sc-6850) 1:1000, p53 (sc-126); Novus Biologicals (Littleton, CO, USA): MX2 (NBP1-81018) 1:2000; Abcam (Cambridge, UK): phospho-CDK1 y15 (ab47594) 1:2000. Secondary antibodies were purchased from Promega (Madison, WI, USA): anti-rabbit (W4011) 1:2000, anti-mouse (W4021) 1:2000. Immunoblots displayed in figures are representative of at least two independent experiments.
3D skin reconstructs. Skin reconstructs were generated as described in [48]. Briefly, each insert of tissue culture trays was coated with 1 mL mixture containing 98 µL 10× EMEM media, 8.4 µL 200 mM L-glutamine, 100 µL FBS, 760 µL collagen I (cat. No. #5005, Advanced Biomatrix) and 20 µL 7.5% sodium bicarbonate and incubated at 37⁰C for 1 hour. Afterward, 7.5×104 fibroblasts in 250 µL DMEM containing 10% FBS were mixed with 274 µL 10× EMEM media, 25 µL 200 mM glutamine, 308 µL FBS, 2320 µL collagen I, and 58 µL 7.5% sodium bicarbonate and pipetted on each insert. After solidification, 2mL of DMEM containing 10% FBS was added inside, and 10 mL was added to the outside of the insert. After four days of incubation at 37 °C in a 5 % CO2 tissue culture incubator, a mixture of iPSC-derived melanocytes and keratinocytes was added on top of the dermal reconstructs (ratio 1:5 melanocytes to keratinocytes) in skin reconstruct medium: keratinocyte serum-free medium (cat. No. 17005042, Gibco) supplemented with 60 µg/mL bovine pituitary extract, 10 ng/mL SCF, 4.5 ng/mL bFGF, 100 nM ET3 and 1 ng/mL epidermal growth factor (EGF). After two days, the EGF concentration was lowered to 0.2 ng/mL, and after two additional days, the surface of the reconstructed tissue was exposed to air for 1.5 weeks, while high-calcium (2.4 mM) medium was added only to the outside of the insert. For the UV irradiation experiment, media outside of the insert were replaced with PBS prior to the exposure, and skin reconstructs were harvested 48 hours after irradiation, fixed in 10% neutral buffered formalin for 3 hours, and assessed for activated caspase 3 by immunohistochemistry.
RPPA. The samples were prepared as previously described [49]. RPPA was performed by the MD Anderson Center RPPA core facility (Houston, TX) as previously described [50]. Unsupervised or supervised hierarchical clustering was performed on normalized log2 median-centered protein values using the Cluster 3.0 software, with centered correlation and complete linkage. Results were visualized using Java TreeView 3.0 software. For UV time-course analysis, normalized Log2 values were median-centered relative to the untreated controls. K-means clustering using Euclidean distance measure on 20 clusters (identified in unsupervised hierarchical clustering), run for 100 iterations, was performed using Cluster 3.0 and visualized with Java TreeView. Clusters with variance greater than 0.10 across time points were selected for biological process analysis using Ingenuity Pathway Analysis (Qiagen).
nbUVB irradiation. Cell plates with a culture dish lid were irradiated at room temperature from above with the narrowband UVB (nbUVB) source consisting of four 20-watt nbUVB lamps with an output peak at 311 nm within the range 305-315 nm and a fluence rate of 0.83 mW/cm2. (TL20W/01, Philips, Holland). UV output was measured before each irradiation session with a calibrated thermopile detector coupled to an optic power meter Gentec-EO SOLO2 (Gentec EO, Quebec, Canada). IR interference was removed by IR filter (XLP12 model). Prior to irradiation, the cell medium was removed from the wells and substituted with DPBS (cat. No. D8662, Sigma-Aldrich) supplemented with 0.1% glucose. After exposure, DPBS was discarded and cells in culturing media were returned to the incubator and harvested at different time points for corresponding assays.
Statistical analyses. Statistical analysis was performed applying SPSS package Version 18, (SPSS inc., Chicago, IL). A two-tailed paired Student’s t-test was used for evaluation of in vitro results. A P-value of less than 0.05 was considered statistically significant.
5. Conclusions
This study demonstrates that iPSC-derived melanocytes generated from patient fibroblasts carrying distinct MC1R pigmentation variants constitute a robust, genetically defined human model for studying UV-induced cellular responses. Our results establish that UV sensitivity is a donor-encoded phenotype, faithfully recapitulated in iPSC-derived melanocytes through genotype-specific differences in DNA-damage signaling, checkpoint activation, and repair kinetics.
Mechanistically, we identify MX2 as a physiological enhancer of UV-induced p53/p38 signaling and apoptosis, acting independently of AKT, thereby expanding its role beyond interferon-mediated functions previously described in melanoma. These findings reveal MX2 as a critical component of the melanocyte UV stress response and underscore the value of iPSC-derived cells for mechanistic and translational research in pigmentation biology, DNA repair, and photoprotection.
Collectively, our work provides a human-relevant and reproducible platform for investigating the molecular determinants of UV-induced disease susceptibility, aligning with emerging New Approach Methodologies (NAMs) for mechanistic toxicology and personalized photoprotection strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.G.R.-S., A.S., M.H.; formal analysis, E.G.R.-S., A.S.; visualization, E.G.R.-S., A.S.; writing original draft preparation, E.G.R.-S., A.S., M.H.; methodology, A.S., M.J., L.L., M.H; investigation, A.S., M.J., L.L., M.H., S.O.; data curation, A.S., M.J., L.L., M.H., S.O.; writing, review and editing, E.G.R.-S., A.S., M.J., L.L., M.H., S.O., J.N.-B., M.H.; supervision, J.N.-B., M.H.; funding acquisition, J.N.-B., M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by NIH grants U54 CA224070, SPORE P50 CA174523, R01 CA258113, R01 CA259295, P30 CA010815, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and Cancer Research UK C588/A19167, C8216/A6129, and C588/A10721.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of The Wistar Institute (2802240, approved on July 16, 2019).” for studies involving humans.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data supporting the findings of this study are included within the article and its Supplementary Materials. Additional datasets generated and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
Acknowledgments
We gratefully acknowledge the Wistar Institute Imaging Facility for providing access to advanced imaging platforms and for their expert technical support. We also thank the Penn iPSC/ES Cell Core (University of Pennsylvania) for their assistance with iPSC generation and characterization.
Conflicts of Interest
Author A.S. is currently employed by One Carbon Therapeutics. This affiliation did not influence the design, execution, interpretation, or reporting of the results presented in this manuscript. All other authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Abbreviation | Definition |
| α-MSH | Alpha-melanocyte-stimulating hormone |
| AKT | Protein kinase B |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related |
| bFGF | Basic fibroblast growth factor |
| CDK1 | Cyclin-dependent kinase 1 |
| cAMP | Cyclic adenosine monophosphate |
| CHK1 | Checkpoint kinase 1 |
| CHK2 | Checkpoint kinase 2 |
| CPD | Cyclobutane pyrimidine dimer |
| DDR | DNA damage response |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DPBS | Dulbecco’s phosphate-buffered saline |
| EB | Embryoid body |
| EGF | Epidermal growth factor |
| FACS | Fluorescence-activated cell sorting |
| FBS | Fetal bovine serum |
| GADD45α | Growth arrest and DNA damage-inducible protein alpha |
| GFP | Green fluorescent protein |
| GUSB | Beta-glucuronidase |
| γH2AX | Phosphorylated histone H2AX |
| HRP | Horseradish peroxidase |
| iPSC | Induced pluripotent stem cell |
| IRDS | Interferon-related DNA damage resistance signature |
| MAPK | Mitogen-activated protein kinase |
| MC1R | Melanocortin 1 receptor |
| MITF | Microphthalmia-associated transcription factor |
| MX2 | Myxovirus resistance protein 2 |
| NAMs | New Approach Methodologies |
| nbUVB | Narrowband ultraviolet B |
| NER | Nucleotide excision repair |
| NHM | Normal human melanocytes |
| OSKM | OCT4, SOX2, KLF4, and c-MYC |
| PARP | Poly(ADP-ribose) polymerase |
| PBS | Phosphate-buffered saline |
| PKA | Protein kinase A |
| PMA | Phorbol 12-myristate 13-acetate |
| PMEL | Premelanosome protein |
| QC | Quality control |
| ROS | Reactive oxygen species |
| RPPA | Reverse phase protein array |
| RT-qPCR | Quantitative reverse-transcription polymerase chain reaction |
| SCF | Stem cell factor |
| SSEA-4 | Stage-specific embryonic antigen 4 |
| STAT1 | Signal transducer and activator of transcription 1 |
| TG | Test guideline |
| TYR | Tyrosinase |
| UV | Ultraviolet |
| XAF1 | XIAP-associated factor 1 |
References
- Brash, D.E. UV signature mutations. Photochem Photobiol 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T. Oxidatively generated damage to DNA by UVA radiation in cells and human skin. J Invest Dermatol 2011, 131, 1005–7. [Google Scholar] [CrossRef]
- Bellei, B.; Picardo, M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res Rev 2020, 57, 100981. [Google Scholar] [CrossRef]
- Jeayeng, S.; Wongkajornsilp, A.; Slominski, A.T.; Jirawatnotai, S.; Sampattavanich, S.; Panich, U. Nrf2 in keratinocytes modulates UVB-induced DNA damage and apoptosis in melanocytes through MAPK signaling. Free Radic Biol Med 2017, 108, 918–928. [Google Scholar] [CrossRef]
- Schafer, M.; Werner, S. Nrf2--A regulator of keratinocyte redox signaling. Free Radic Biol Med 2015, 88 Pt B, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Borron, J.C.; Abdel-Malek, Z.; Jimenez-Cervantes, C. MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation. Pigment Cell Melanoma Res 2014, 27, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.L. The genetics of sun sensitivity in humans. Am J Hum Genet 2004, 75, 739–51. [Google Scholar] [CrossRef]
- Fargnoli, M.C.; Pike, K.; Pfeiffer, R.M.; Tsang, S.; Rozenblum, E.; Munroe, D.J.; Golubeva, Y.; Calista, D.; Seidenari, S.; Massi, D.; Carli, P.; Bauer, J.; Elder, D.E.; Bastian, B.C.; Peris, K.; Landi, M.T. MC1R variants increase risk of melanomas harboring BRAF mutations. J Invest Dermatol 2008, 128, 2485–90. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Leachman, S.; Kavanagh, R.J.; Swope, V.; Cassidy, P.; Supp, D.; Sartor, M.; Schwemberger, S.; Babcock, G.; Wakamatsu, K.; Ito, S.; Koshoffer, A.; Boissy, R.E.; Manga, P.; Sturm, R.A.; Abdel-Malek, Z.A. Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J 2010, 24, 3850–60. [Google Scholar] [CrossRef]
- Cui, D.; Qu, R.; Liu, D.; Xiong, X.; Liang, T.; Zhao, Y. The Cross Talk Between p53 and mTOR Pathways in Response to Physiological and Genotoxic Stresses. Front Cell Dev Biol 2021, 9, 775507. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; An, J.; Tai, J.; Tanaka, L.; Sun, X.; Hermanson, P.; Baum, R.; Kawasumi, M.; Green, R.; Gale, M., Jr.; Kalus, A.; Werth, V.P.; Elkon, K.B. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci Rep 2020, 10, 7908. [Google Scholar] [CrossRef]
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res 2019, 32, 623–633. [Google Scholar] [CrossRef]
- Ohta, S.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Kuwahara, R.; Ohyama, M.; Amagai, M.; Matsuzaki, Y.; Yamanaka, S.; Okano, H.; Kawakami, Y. Generation of human melanocytes from induced pluripotent stem cells. PLoS One 2011, 6, e16182. [Google Scholar] [CrossRef] [PubMed]
- Coutant, K.; Magne, B.; Ferland, K.; Fuentes-Rodriguez, A.; Chancy, O.; Mitchell, A.; Germain, L.; Landreville, S. Melanocytes in regenerative medicine applications and disease modeling. J Transl Med 2024, 22, 336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Choi, B.; Tang, M.S. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc Natl Acad Sci U S A 2010, 107, 12180–5. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.L.; Yager, J.W.; Hudson, L.G. Melanocytes and keratinocytes have distinct and shared responses to ultraviolet radiation and arsenic. Toxicol Lett 2014, 224, 407–15. [Google Scholar] [CrossRef]
- Sun, X.; Kim, A.; Nakatani, M.; Shen, Y.; Liu, L. Distinctive molecular responses to ultraviolet radiation between keratinocytes and melanocytes. Exp Dermatol 2016, 25, 708–13. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Roberts, M.J.; Cervantes-Laurean, D.; Jacobson, M.K.; Jacobson, E.L. Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J Invest Dermatol 2003, 121, 578–86. [Google Scholar] [CrossRef]
- Betancor, G. You Shall Not Pass: MX2 Proteins Are Versatile Viral Inhibitors. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–62. [Google Scholar] [CrossRef]
- Juraleviciute, M.; Nsengimana, J.; Newton-Bishop, J.; Hendriks, G.J.; Slipicevic, A. MX2 mediates establishment of interferon response profile, regulates XAF1, and can sensitize melanoma cells to targeted therapy. Cancer Med 2021, 10, 2840–2854. [Google Scholar] [CrossRef]
- Juraleviciute, M.; Pozniak, J.; Nsengimana, J.; Harland, M.; Randerson-Moor, J.; Wernhoff, P.; Bassarova, A.; Oy, G.F.; Troen, G.; Florenes, V.A.; Bishop, D.T.; Herlyn, M.; Newton-Bishop, J.; Slipicevic, A. MX 2 is a novel regulator of cell cycle in melanoma cells. Pigment Cell Melanoma Res 2020, 33, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.K.; Song, F.; Barrett, J.H.; Kumar, R.; Easton, D.F.; Pharoah, P.D.P.; Swerdlow, A.J.; Kypreou, K.P.; Taylor, J.C.; Harland, M.; Randerson-Moor, J.; Akslen, L.A.; Andresen, P.A.; Avril, M.F.; Azizi, E.; Scarra, G.B.; Brown, K.M.; Debniak, T.; Duffy, D.L.; Elder, D.E.; Fang, S.; Friedman, E.; Galan, P.; Ghiorzo, P.; Gillanders, E.M.; Goldstein, A.M.; Gruis, N.A.; Hansson, J.; Helsing, P.; Hocevar, M.; Hoiom, V.; Ingvar, C.; Kanetsky, P.A.; Chen, W.V.; Geno, M.E.L.C.; Essen-Heidelberg, I.; Group, S.D.H.S.; Q, M.; Investigators, Q.; Investigators, A.; Group, A.M.S.; Landi, M.T.; Lang, J.; Lathrop, G.M.; Lubinski, J.; Mackie, R.M.; Mann, G.J.; Molven, A.; Montgomery, G.W.; Novakovic, S.; Olsson, H.; Puig, S.; Puig-Butille, J.A.; Qureshi, A.A.; Radford-Smith, G.L.; van der Stoep, N.; van Doorn, R.; Whiteman, D.C.; Craig, J.E.; Schadendorf, D.; Simms, L.A.; Burdon, K.P.; Nyholt, D.R.; Pooley, K.A.; Orr, N.; Stratigos, A.J.; Cust, A.E.; Ward, S.V.; Hayward, N.K.; Han, J.; Schulze, H.J.; Dunning, A.M.; Bishop, J.A.N.; Demenais, F.; Amos, C.I.; MacGregor, S.; Iles, M.M. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet 2015, 47, 987–995. [Google Scholar] [CrossRef]
- Padariya, M.; Sznarkowska, A.; Kote, S.; Gomez-Herranz, M.; Mikac, S.; Pilch, M.; Alfaro, J.; Fahraeus, R.; Hupp, T.; Kalathiya, U. Functional Interfaces, Biological Pathways, and Regulations of Interferon-Related DNA Damage Resistance Signature (IRDS) Genes. Biomolecules 2021, 11(5). [Google Scholar] [CrossRef]
- Punt, A.; Bouwmeester, H.; Blaauboer, B.J.; Coecke, S.; Hakkert, B.; Hendriks, D.F.G.; Jennings, P.; Kramer, N.I.; Neuhoff, S.; Masereeuw, R.; Paini, A.; Peijnenburg, A.; Rooseboom, M.; Shuler, M.L.; Sorrell, I.; Spee, B.; Strikwold, M.; Van der Meer, A.D.; Van der Zande, M.; Vinken, M.; Yang, H.; Bos, P.M.J.; Heringa, M.B. New approach methodologies (NAMs) for human-relevant biokinetics predictions. Meeting the paradigm shift in toxicology towards an animal-free chemical risk assessment. ALTEX 2020, 37, 607–622. [Google Scholar]
- Svendsen, C.N. Adopting novel alternative methods (NAMs) for biomedical research-What is the right approach? Cell Stem Cell 2025, 32, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–72. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009, 85, 348–62. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key regulatory role of dermal fibroblasts in pigmentation as demonstrated using a reconstructed skin model: impact of photo-aging. PLoS One 2014, 9, e114182. [Google Scholar] [CrossRef]
- Cohen, C.; Flouret, V.; Herlyn, M.; Fukunaga-Kalabis, M.; Li, L.; Bernerd, F. Induced pluripotent stem cells reprogramming overcomes technical limitations for highly pigmented adult melanocyte amplification and integration in 3D skin model. Pigment Cell Melanoma Res 2023, 36, 232–245. [Google Scholar] [CrossRef]
- Zhao, N.; Kabotyanski, E.B.; Saltzman, A.B.; Malovannaya, A.; Yuan, X.; Reineke, L.C.; Lieu, N.; Gao, Y.; Pedroza, D.A.; Calderon, S.J.; Smith, A.J.; Hamor, C.; Safari, K.; Savage, S.; Zhang, B.; Zhou, J.; Solis, L.M.; Hilsenbeck, S.G.; Fan, C.; Perou, C.M.; Rosen, J.M. Targeting eIF4A triggers an interferon response to synergize with chemotherapy and suppress triple-negative breast cancer. J Clin Invest 2023, 133(24). [Google Scholar] [CrossRef]
- Abdel-Malek, Z.A.; Ruwe, A.; Kavanagh-Starner, R.; Kadekaro, A.L.; Swope, V.; Haskell-Luevano, C.; Koikov, L.; Knittel, J.J. alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell Melanoma Res 2009, 22, 635–44. [Google Scholar] [CrossRef] [PubMed]
- Castejon-Grinan, M.; Cerdido, S.; Sanchez-Beltran, J.; Lambertos, A.; Abrisqueta, M.; Herraiz, C.; Jimenez-Cervantes, C.; Garcia-Borron, J.C. Melanoma-associated melanocortin 1 receptor variants confer redox signaling-dependent protection against oxidative DNA damage. Redox Biol 2024, 72, 103135. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Das, K.C. Differential roles of ATR and ATM in p53, Chk1, and histone H2AX phosphorylation in response to hyperoxia: ATR-dependent ATM activation. Am J Physiol Lung Cell Mol Physiol 2008, 294, L998–L1006. [Google Scholar] [CrossRef] [PubMed]
- Mucha, M.; Skrzydlewska, E.; Gegotek, A. Natural protection against oxidative stress in human skin melanocytes. Commun Biol 2025, 8, 1283. [Google Scholar] [CrossRef]
- Ma, T.; He, J.; Long, Q.; Wang, Y.; Chen, F.; Chen, S.; Xu, K.; Cao, Y. Orientin attenuates UVB-induced skin photodamage by inhibiting ROS generation via the AMPK/Nrf2 axis. Int Immunopharmacol 2025, 155, 114655. [Google Scholar] [CrossRef]
- Manrique-Silva, E.; David, M.E.; Maider, A.M.; Garcia-Casado, Z.; Moro, R.; Requena, C.; Traves, V.; Viros, A.; Kumar, R.; Nagore, E. Clinical, histological, and molecular differences in melanoma due to different TERT promoter mutations subtypes. A retrospective cross-sectional study in 684 melanoma patients. Pigment Cell Melanoma Res 2024, 37, 343–351. [Google Scholar] [CrossRef]
- Layish, B.; Goli, R.; Flick, H.; Huang, S.W.; Zhang, R.Z.; Kvaratskhelia, M.; Kane, M. Virus specificity and nucleoporin requirements for MX2 activity are affected by GTPase function and capsid-CypA interactions. PLoS Pathog 2024, 20, e1011830. [Google Scholar] [CrossRef]
- Meng, X.W.; Cheng, Z.L.; Lu, Z.Y.; Tan, Y.N.; Jia, X.Y.; Zhang, M. MX2: Identification and systematic mechanistic analysis of a novel immune-related biomarker for systemic lupus erythematosus. Front Immunol 2022, 13, 978851. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Coomans de Brachene, A.; Alvelos, M.I.; Szymczak, F.; Zimath, P.L.; Castela, A.; Marmontel de Souza, B.; Roca Rivada, A.; Marin-Canas, S.; Yi, X.; Op de Beeck, A.; Morgan, N.G.; Sonntag, S.; Jawurek, S.; Title, A.C.; Yesildag, B.; Pattou, F.; Kerr-Conte, J.; Montanya, E.; Nacher, M.; Marselli, L.; Marchetti, P.; Richardson, S.J.; Eizirik, D.L. Interferons are key cytokines acting on pancreatic islets in type 1 diabetes. Diabetologia 2024, 67, 908–927. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Chatterjee, N.; Carvalho, A.; Alfaro-Moreno, E. Particulate matter-induced oxidative stress - Mechanistic insights and antioxidant approaches reported in in vitro studies. Environ Toxicol Pharmacol 2024, 110, 104529. [Google Scholar] [CrossRef]
- Stucki, A.O.; Barton-Maclaren, T.S.; Bhuller, Y.; Henriquez, J.E.; Henry, T.R.; Hirn, C.; Miller-Holt, J.; Nagy, E.G.; Perron, M.M.; Ratzlaff, D.E.; Stedeford, T.J.; Clippinger, A.J. Use of new approach methodologies (NAMs) to meet regulatory requirements for the assessment of industrial chemicals and pesticides for effects on human health. Front Toxicol 2022, 4, 964553. [Google Scholar] [CrossRef]
- Singhal, A.; Zhao, X.; Wall, P.; So, E.; Calderini, G.; Partin, A.; Koussa, N.; Vasanthakumari, P.; Narykov, O.; Zhu, Y.; Jones, S.E.; Abbas-Aghababazadeh, F.; Kadambat Nair, S.; Belisle-Pipon, J.C.; Jayaram, A.; Parker, B.A.; Yeung, K.T.; Griffiths, J.I.; Weil, R.; Nath, A.; Haibe-Kains, B.; Ideker, T. The Hallmarks of Predictive Oncology. Cancer Discov 2025, 15, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, G.I.; Holm, R.; Emilsen, E.; Rosnes, A.K.; Slipicevic, A.; Florenes, V.A. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PLoS One 2012, 7, e38254. [Google Scholar] [CrossRef]
- Vangipuram, M.; Ting, D.; Kim, S.; Diaz, R.; Schule, B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp 2013, e3779. [Google Scholar]
- Li, L.; Fukunaga-Kalabis, M.; Yu, H.; Xu, X.; Kong, J.; Lee, J.T.; Herlyn, M. Human dermal stem cells differentiate into functional epidermal melanocytes. J Cell Sci 2010, 123 Pt 6, 853–60. [Google Scholar] [CrossRef]
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The three-dimensional human skin reconstruct model: a tool to study normal skin and melanoma progression. J Vis Exp 2011, (54). [Google Scholar]
- Krepler, C.; Xiao, M.; Sproesser, K.; Brafford, P.A.; Shannan, B.; Beqiri, M.; Liu, Q.; Xu, W.; Garman, B.; Nathanson, K.L.; Xu, X.; Karakousis, G.C.; Mills, G.B.; Lu, Y.; Ahmed, T.A.; Poulikakos, P.I.; Caponigro, G.; Boehm, M.; Peters, M.; Schuchter, L.M.; Weeraratna, A.T.; Herlyn, M. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res 2016, 22, 1592–602. [Google Scholar] [CrossRef]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 2006, 5, 2512–21. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular, morphological, and phenotypic characterization of patient-derived melanocyte lines with distinct sun sensitivity profiles. (A) Schematic representation of the experimental workflow. Skin biopsies from patients were reprogrammed into induced pluripotent stem cells (iPSCs) using OCT3/4, SOX2, KLF4, and c-MYC. iPSCs were subsequently differentiated into melanocytes through exposure to SCF, EDN3, and bFGF. (B) Representative phase-contrast micrographs showing the morphology of embryoid bodies (EBs) and differentiated melanocytes derived from iPSCs. Differentiated melanocytes exhibit characteristic dendritic morphology. Immunofluorescence staining of melanocyte-specific markers MelanA and HMB45 in SF3, SF4, H1m, and NHM160 cell lines. Nuclei were counterstained with DAPI. Scale bar: 50 μm. (C) Relative mRNA expression levels of melanocyte lineage genes MITF, PMEL, and TYR in SF3, SF4, H1m, and NHM160, as determined by quantitative RT-PCR. Expression values are normalized to housekeeping genes. (D) Quantification of intracellular melanin content (pg/cell) across the same cell lines, revealing differences in pigment production capacity. (E) Summary of MC1R gene variants identified in each cell line, along with corresponding sun sensitivity scores (adapted from Newton-Bishop et al., Eur. J. Cancer, 2011) and phenotypic classification. Lines SF3, SF4, SF5, SF7, SF8, and SF9 exhibit distinct MC1R polymorphisms and sun sensitivity profiles, which correlate with pigmentation phenotypes.
Figure 1.
Molecular, morphological, and phenotypic characterization of patient-derived melanocyte lines with distinct sun sensitivity profiles. (A) Schematic representation of the experimental workflow. Skin biopsies from patients were reprogrammed into induced pluripotent stem cells (iPSCs) using OCT3/4, SOX2, KLF4, and c-MYC. iPSCs were subsequently differentiated into melanocytes through exposure to SCF, EDN3, and bFGF. (B) Representative phase-contrast micrographs showing the morphology of embryoid bodies (EBs) and differentiated melanocytes derived from iPSCs. Differentiated melanocytes exhibit characteristic dendritic morphology. Immunofluorescence staining of melanocyte-specific markers MelanA and HMB45 in SF3, SF4, H1m, and NHM160 cell lines. Nuclei were counterstained with DAPI. Scale bar: 50 μm. (C) Relative mRNA expression levels of melanocyte lineage genes MITF, PMEL, and TYR in SF3, SF4, H1m, and NHM160, as determined by quantitative RT-PCR. Expression values are normalized to housekeeping genes. (D) Quantification of intracellular melanin content (pg/cell) across the same cell lines, revealing differences in pigment production capacity. (E) Summary of MC1R gene variants identified in each cell line, along with corresponding sun sensitivity scores (adapted from Newton-Bishop et al., Eur. J. Cancer, 2011) and phenotypic classification. Lines SF3, SF4, SF5, SF7, SF8, and SF9 exhibit distinct MC1R polymorphisms and sun sensitivity profiles, which correlate with pigmentation phenotypes.
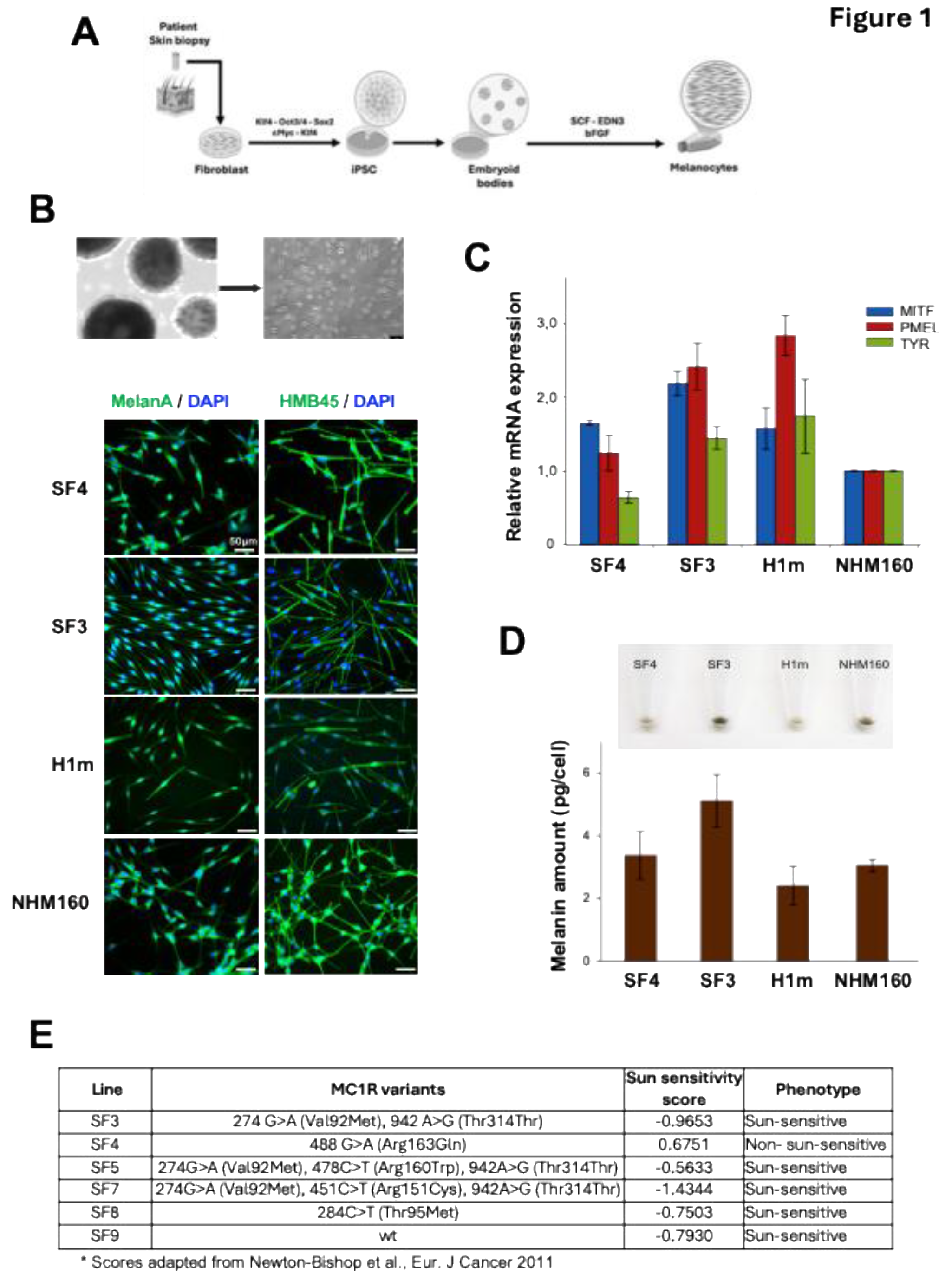
Figure 2.
Donor-specific UVB sensitivity drives checkpoint activation and apoptosis in iPSC-derived melanocytes. (A) Viability of iPSC-derived and normal human melanocytes 72 hours after exposure to increasing doses of narrowband UVB (nbUVB; 0–1.5 J/cm²). Data represent mean ± SE of three independent experiments. (B) Comparison between in vitro viability of melanocytes 72 hours after 1.5 J/cm² nbUVB exposure and the corresponding patients’ sun-sensitivity scores derived from self-reported pigmentation traits. A negative correlation is observed between sun sensitivity and UVB-induced cytotoxicity. (C) Growth rate assessment of melanocyte lines using a confluence-based assay with the IncuCyte™ FLR live-cell imaging system. Cell confluence was normalized to the initial time point. Data represent mean ± SE of n=3. (D) Cell cycle distribution of melanocytes 24 hours post-UVB exposure (1.5 J/cm²), evaluated by flow cytometry using propidium iodide staining. Bar graphs show the percentage of cells in G1, S, and G2/M phases (mean ± SE, n=3). (E) Immunoblot analysis of G2/M phase-specific markers phospho-Histone H3 (Ser10) and phospho-CDK1 (Tyr15) at 2, 6, and 24 hours post-UVB exposure. Total Histone H3 was used as a loading control. (F) Western blot analysis of apoptosis markers cleaved caspase-3 and cleaved PARP at 2, 6, and 24 hours post-UVB exposure. GAPDH was used as a loading control. (G) SF3 and SF4 melanocytes were incorporated into 3D skin reconstructs, irradiated with 300 mJ/cm² UV, and analyzed 48 hours later by immunohistochemistry for cleaved caspase-3 to assess apoptosis in a tissue context.
Figure 2.
Donor-specific UVB sensitivity drives checkpoint activation and apoptosis in iPSC-derived melanocytes. (A) Viability of iPSC-derived and normal human melanocytes 72 hours after exposure to increasing doses of narrowband UVB (nbUVB; 0–1.5 J/cm²). Data represent mean ± SE of three independent experiments. (B) Comparison between in vitro viability of melanocytes 72 hours after 1.5 J/cm² nbUVB exposure and the corresponding patients’ sun-sensitivity scores derived from self-reported pigmentation traits. A negative correlation is observed between sun sensitivity and UVB-induced cytotoxicity. (C) Growth rate assessment of melanocyte lines using a confluence-based assay with the IncuCyte™ FLR live-cell imaging system. Cell confluence was normalized to the initial time point. Data represent mean ± SE of n=3. (D) Cell cycle distribution of melanocytes 24 hours post-UVB exposure (1.5 J/cm²), evaluated by flow cytometry using propidium iodide staining. Bar graphs show the percentage of cells in G1, S, and G2/M phases (mean ± SE, n=3). (E) Immunoblot analysis of G2/M phase-specific markers phospho-Histone H3 (Ser10) and phospho-CDK1 (Tyr15) at 2, 6, and 24 hours post-UVB exposure. Total Histone H3 was used as a loading control. (F) Western blot analysis of apoptosis markers cleaved caspase-3 and cleaved PARP at 2, 6, and 24 hours post-UVB exposure. GAPDH was used as a loading control. (G) SF3 and SF4 melanocytes were incorporated into 3D skin reconstructs, irradiated with 300 mJ/cm² UV, and analyzed 48 hours later by immunohistochemistry for cleaved caspase-3 to assess apoptosis in a tissue context.
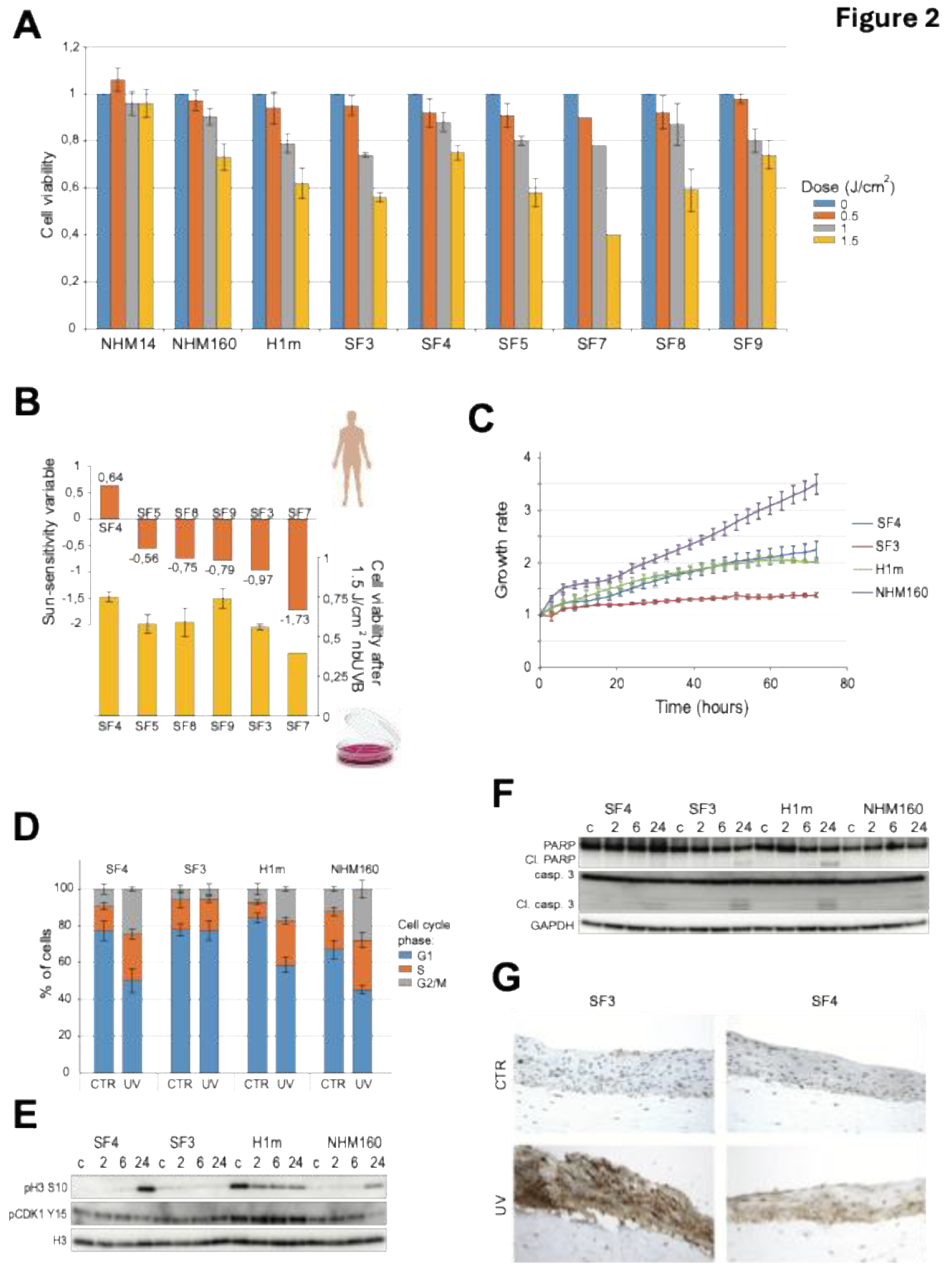
Figure 3.
UV-sensitive iPSC-derived melanocytes exhibit robust DNA damage signaling but impaired CPD repair. (A) Heatmap illustrating differentially activated signaling pathways in SF3 and SF4 melanocytes at 2, 6, and 24 hours after 1.5 J/cm² narrowband UVB (nbUVB) exposure, assessed by reverse phase protein array (RPPA), highlighting pathways involved in DNA damage response, apoptosis, and cell cycle regulation. (B) Validation of RPPA results by immunoblotting for key DNA damage response proteins, including phosphorylated ATM (Ser1981), CHK1 (Ser345), CHK2 (Thr68), γH2AX, GADD45α, and p53, with GAPDH as loading control. (C) Quantification of cyclobutane pyrimidine dimers (CPDs) immediately after nbUVB exposure in SF3, SF4, H1m, and NHM160 melanocytes using ELISA (mean ± SE, n=3). (D) Time-course analysis of CPD repair at 2, 4, 6, and 24 hours post-irradiation, expressed as percentage of unrepaired lesions relative to initial damage. Statistical significance was determined by one-sample t-test (*p < 0.05, ***p < 0.001).
Figure 3.
UV-sensitive iPSC-derived melanocytes exhibit robust DNA damage signaling but impaired CPD repair. (A) Heatmap illustrating differentially activated signaling pathways in SF3 and SF4 melanocytes at 2, 6, and 24 hours after 1.5 J/cm² narrowband UVB (nbUVB) exposure, assessed by reverse phase protein array (RPPA), highlighting pathways involved in DNA damage response, apoptosis, and cell cycle regulation. (B) Validation of RPPA results by immunoblotting for key DNA damage response proteins, including phosphorylated ATM (Ser1981), CHK1 (Ser345), CHK2 (Thr68), γH2AX, GADD45α, and p53, with GAPDH as loading control. (C) Quantification of cyclobutane pyrimidine dimers (CPDs) immediately after nbUVB exposure in SF3, SF4, H1m, and NHM160 melanocytes using ELISA (mean ± SE, n=3). (D) Time-course analysis of CPD repair at 2, 4, 6, and 24 hours post-irradiation, expressed as percentage of unrepaired lesions relative to initial damage. Statistical significance was determined by one-sample t-test (*p < 0.05, ***p < 0.001).
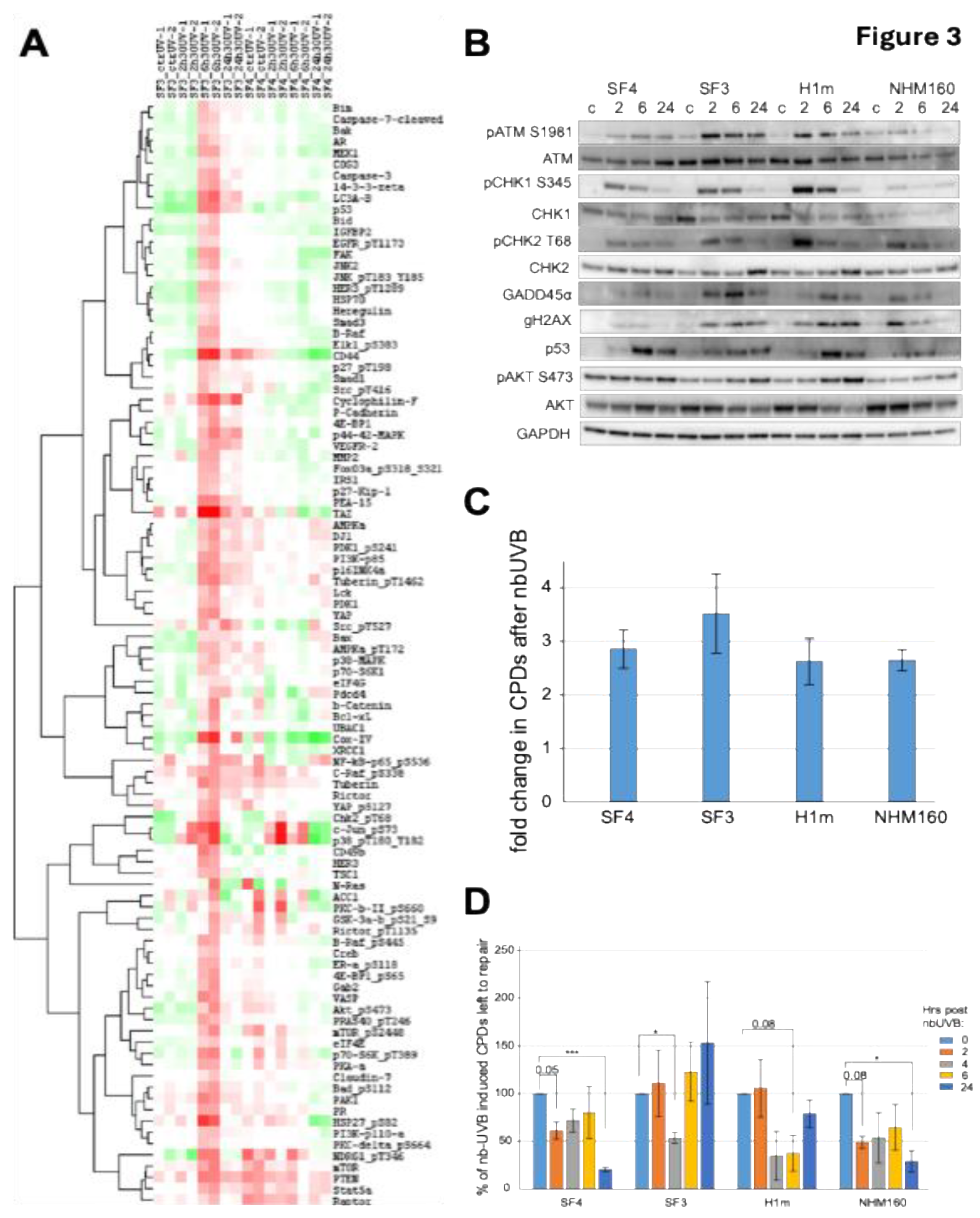
Figure 4.
MX2 amplifies UV-induced stress signaling and reduces melanocyte survival via an AKT-independent mechanism. (A) Immunoblot analysis of MX2 expression in iPSC-derived melanocytes (SF3, SF4, H1m) at 2, 6, and 24 hours after exposure to 1.5 J/cm² narrowband UVB (nbUVB), with GAPDH as loading control. (B) MX2 protein levels in normal human melanocytes NHM9 and NHM160 (left panel) and corresponding cell viability 72 hours after 1.5 J/cm² nbUVB irradiation (right panel). Data represent mean ± SE of n=3. (C) NHM134 melanocytes were treated with 500 ng/mL doxycycline to induce GFP or MX2 expression 24 hours prior to nbUVB exposure (1.5 J/cm²), and cell viability was assessed 72 hours post-irradiation. Data represent mean ± SE of n=3. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05). (D) Immunoblot analysis of NHM134 melanocytes treated as in (C), harvested 24 hours after nbUVB exposure, and evaluated for MX2, p53, phospho-p38, XAF1, and phospho-AKT (Ser473) expression, with β-actin as loading control.
Figure 4.
MX2 amplifies UV-induced stress signaling and reduces melanocyte survival via an AKT-independent mechanism. (A) Immunoblot analysis of MX2 expression in iPSC-derived melanocytes (SF3, SF4, H1m) at 2, 6, and 24 hours after exposure to 1.5 J/cm² narrowband UVB (nbUVB), with GAPDH as loading control. (B) MX2 protein levels in normal human melanocytes NHM9 and NHM160 (left panel) and corresponding cell viability 72 hours after 1.5 J/cm² nbUVB irradiation (right panel). Data represent mean ± SE of n=3. (C) NHM134 melanocytes were treated with 500 ng/mL doxycycline to induce GFP or MX2 expression 24 hours prior to nbUVB exposure (1.5 J/cm²), and cell viability was assessed 72 hours post-irradiation. Data represent mean ± SE of n=3. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05). (D) Immunoblot analysis of NHM134 melanocytes treated as in (C), harvested 24 hours after nbUVB exposure, and evaluated for MX2, p53, phospho-p38, XAF1, and phospho-AKT (Ser473) expression, with β-actin as loading control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



