
Submitted:
22 January 2026
Posted:
23 January 2026
You are already at the latest version
Abstract
Chimeric antigen receptor (CAR) T-cell therapy is an effective treatment for hematologic malignancies. However, it is limited by high costs, risk of severe toxicities such as cytokine release syndrome and neurotoxicity, and heterogeneous patient responses. Current therapy monitoring depends largely on subjective symptom assessment, routine laboratory tests, and basic vital signs, without real-time, quantitative evaluation of CAR T-cell expansion or activation in clinical practice. This lack of timely immune monitoring hampers individualized care and contributes to increased treatment costs. To address this need, we present a proof-of-concept, label-free Rapid Optical Imaging (ROI) biosensor with automated machine learning analysis for direct quantification of functional CAR T-cells from whole blood. This microfluidic platform integrates leukocyte separation, capture, and detection on a single chip, thereby eliminating centrifugation, staining, and operator-dependent interpretation. For validation, 50 μL whole blood samples spiked with Jurkat cells expressing a CD19 CAR underwent red blood cell depletion by agglutination and microfiltration. The leukocyte-enriched fraction was then incubated on a sensor chip functionalized with recombinant CD19 protein. Captured CAR T-cells were imaged by bright-field microscopy and automatically enumerated using a machine learning algorithm. A calibration curve was established across clinically relevant concentrations (1–1,000 cells/mL), with results validated against fluorescence microscopy and flow cytometry. This ROI biosensor enables rapid, quantitative, and label-free CAR T-cell detection from whole blood without specialized equipment or infrastructure. With further development, it could provide a cost-effective point-of-care tool for real-time immune monitoring and improved clinical management of patients receiving CAR T-cell therapy.
Keywords:
chimeric antigen receptor T-cells
; CAR T
; optical imaging
; microfluidic chip
; digital counting
; whole blood
; agglutination
; machine learning
1. Introduction
Chimeric antigen receptor (CAR) T-cell therapy has demonstrated remarkable clinical efficacy in patients with hematologic malignancies, including non-Hodgkin lymphoma, multiple myeloma, and acute lymphoblastic leukemia, leading to the first U.S. Food and Drug Administration (FDA) approval in 2017 [1,2,3]. Despite these advances, several limitations remain: the therapy is costly, approximately half of patients fail to achieve durable responses, and treatment-related toxicities are common and can be severe, particularly among older adults and individuals with comorbidities [2,4,5,6]. Clinical outcomes and adverse events correlate strongly with CAR T-cell expansion and the release of immune activation–related cytokines, which mediate both therapeutic efficacy and toxicities such as cytokine release syndrome (CRS) and neurotoxicity [4,5].
Real-time access to objective, quantitative measures of CAR T-cell expansion and cytokine dynamics could markedly enhance patient management by enabling early detection of CRS and neurotoxicity, supporting pre-emptive interventions prior to progression to severe toxicity, and providing prognostic markers of treatment failure to guide timely initiation of additional diagnostics or salvage therapies.
Current assays for monitoring CAR T-cell expansion and cytokine production include flow cytometry and immunoassays. While suitable for research and clinical trial contexts, these approaches are impractical for routine clinical monitoring given their reliance on costly instruments, specialized expertise, batched testing, and processing times of hours to days [2,5,6,7]. As a result, patient care continues to rely on indirect measures such as subjective symptoms, routine laboratory results, and vital signs, particularly fever, while direct quantification of CAR T-cell activity remains limited to correlative trial studies.
This reliance on delayed and non-specific markers creates a critical “blind spot” in patient monitoring, leading to missed opportunities for early detection of CRS, neurotoxicity, or treatment failure, and limiting precision in clinical decision-making. The paradox is evident: the most advanced and expensive immunotherapy available today is routinely monitored using subjective and indirect clinical parameters that may provide late or misleading information. Beyond clinical challenges, these monitoring gaps contribute significantly to the overall cost of CAR T-cell treatment care, currently estimated at $800,000 to more than $1 million per patient in the United States, driven largely by extended inpatient monitoring and hospitalization [8].
To address these challenges and improve safety, efficacy, and affordability, there is an urgent need for rapid, cost-effective, point-of-care (POC) assays capable of real-time quantification of CAR T-cell expansion and CRS or neurotoxicity-associated cytokine biomarkers in peripheral blood. Access to such objective metrics would enable earlier intervention, support outpatient management, optimize care delivery, and reduce costs while improving patient outcomes.
Recent advances in microfluidic and lab-on-chip technologies highlight the potential of miniaturized blood analysis platforms [9,10,11]; however, their complexity, reliance on specialized materials, and high production costs have limited widespread clinical adoption [12]. Thus, a practical, affordable POC method for monitoring CAR T-cell activity in whole blood remains an unmet need.
Here, we describe the initial development of a simple, POC-compatible diagnostic platform for monitoring CAR T-cell expansion. The approach integrates rapid red blood cell depletion with label-free bright-field optical imaging and automated machine-learning–based cell identification to directly quantify CAR T-cell concentrations from small volumes of whole blood samples. Validation against fluorescence imaging and flow cytometry confirmed the specificity and accuracy of CAR T-cell detection. This proof-of-concept study establishes the feasibility of a low-cost, scalable technology for real-time monitoring of CAR T-cell therapy with the potential to enable earlier detection of treatment failure or toxicities, guide timely interventions, decrease dependence on prolonged hospitalizations, and facilitate safer, more economical outpatient care.
2. Materials and Methods
2.1. Overview of the CAR T-Cell Detection Workflow
Figure 1 summarizes the streamlined workflow for CAR T-cell detection. The protocol consists of four critical stages: red blood cell (RBC) depletion, CAR T-cell capture, imaging, and data analysis. First, we remove RBCs from whole blood samples without centrifugation or RBC disruption procedures. Instead, whole blood is mixed with RBC agglutination antibody solution for 30 minutes to allow RBCs to aggregate, and a membrane filter with a pore size larger than white blood cells filters out the aggregated RBCs. Next, the filtered blood is loaded onto a microfluidic sensor chip functionalized with recombinant CD19 protein to facilitate specific capture of CD19 CAR T-cells through receptor interaction. After 15 minutes of incubation, non-specifically attached cells are removed through washing cycles. The remaining cells are recorded through digital capture of brightfield microscopy images and processed for automated cell identification and counting.
2.2. Sample Delivery and Detection System
As depicted in Figure S1, the sample delivery and detection system consists of four parts. 1) Flow control: Syringe pump (Chemyx Fusion 100, Catalog #: 07100) provides constant flow rate (10 µL/mins); 6-port injection valve (VICI Valco Instruments Co. Inc., #08N-0232H) for switching between washing buffer and the fluorescent protein; 20 mL Sterile syringe for loading washing buffer; and 1 mL sterile syringe for loading cells samples (VWR # 53548-008 & 53548-001). 2) Sensor chip: Homemade microfluidic chips with a straight channel and CD19 functionalized surface for CAR T-cell capture and imaging. 3) Image acquisition: An inverted microscope (Olympus IX81, 40X/ 0.75 NA objective) with CMOS camera for recording bright field and fluorescence image sequences. 4) Data processing: Digital cellular crowding was reduced through a custom Python script; CAR T-cells were identified and quantified with ilastik, a machine-learning based image processing toolkit.
2.3. Microfluidic Chip Fabrication
The microfluidic chip included 4 components (Figure S2). From the bottom to the top, there is a 25 × 50 mm coverslip (Mercedes Medical Glass Coverslips, Fisher Scientific, #15-184-03), a microfluidic channel defined by two layers of double-sided tapes (4 Mil/101.6 μm thick for each layer; #3M 444), top cover glass (Cole-Parmer, 22 x 40 mm, #UX-48511-10) with two 1 mm diameter pre-drilled holes as the inlet and outlet, and two polydimethylsiloxane (PDMS, Momentive Specialty Chemicals;#RTV-615-044) channel connector holders attached to the top cover slides via oxygen plasma treatment. The microfluidic channel defined by the double-sided tape was designed in Adobe Illustrator (2020) and fabricated with a laser cutter (Universal Laser System, VLS 4.15). The channel is linear, with 203 μm in height, 1.0 mm in width, 35 mm in length, and a total volume of 7.1 μL. The PDMS channel connector holders were fabricated with a classic protocol [13,14,15]. 30 mL mixed polydimethylsiloxane was poured into a clean plastic petri dish with a flat surface, degassed in a vacuum desiccator (for 30 mins), and baked in an oven at 60 °C for 60 minutes. Next, the hardened PDMS was peeled off from the dish and cut into small cubes. A hole is punched through each PDMS cube with a hole puncher (Robbins # RBP-075) to hold the stainless-steel flow channel connectors. The chip was assembled bottom up, starting with a functionalized bottom glass, followed by attaching the double-sided tape-defined channel. Then the top cover slide with drilled holes is aligned with the channel end and attached to the tape. Finally, two PDMS holders were aligned with the top glass holes and attached.
2.4. Surface Chemistry
The surface modification process of the sensor chip is depicted in Figure S3. CD19 recombinant protein was immobilized onto the sensing surface (the top surface of the bottom glass) using either (3-glycidyloxypropyl) trimethoxylsilane (GPTMS) or 3-(2,3-Epoxypropoxy) propyltrimethoxysilane (GLYMO) (Sigma-Aldrich #440167) as a coupling agent [16,17,18,19]. The oxirane ring in the coupling agent undergoes nucleophilic addition reaction with the amino group in the target protein, leading to covalent binding of the protein to the glass surface [18,20]. The coverslips (22 x 50 mm) were cleaned with 99% isopropanol (VWR #: 67-63-0) and deionized water (15 MΩ/cm, 0.22 μm filtration) and air-dried using a nitrogen stream. The coverslips were further cleaned using oxygen plasma (Harrick Plasma, # PDC001051102 High frequency, 2.96 W, 3mins). 1% (v/v) GPTMS in anhydrous isopropanol (IPA) was freshly prepared one day before starting the surface modification. The plasma-treated glass coverslips were soaked in the GPTMS solution overnight (24 hours) at room temperature in a glass staining jar, followed by cleaning with anhydrous IPA and distilled water, and drying using a nitrogen stream. The top coverslip and PDMS connector holders were also cleaned using the same washing steps. After the sensor chip was assembled, 0.1 mg/mL human recombinant CD19 protein solution (Sino Biological; #11880-H08H), diluted with 50 mM sodium carbonate buffer (pH = 9.6), was injected into the channel through the inlet and incubated for 30 minutes. The chip was gently washed with 1 mL 1x live cell imaging solution (Thermo Fisher Scientific #A14291DJ) and incubated with 1x blocking reagent (10%, v/v, Roche #11096176001) statically for 30 minutes at room temperature to block nonspecific binding sites. The as-prepared chip was used for CAR T-cell measurement within 6 hours.
2.5. Sample Preparation
To determine the total count of blood cells, fresh whole blood samples were diluted 1000-fold with live cell imaging solution to ensure that the maximum reading threshold of the cell counter was not exceeded. To prepare the Jurkat T-cell samples, 2 mL each of wild-type (WT) Jurkat T-cells and Jurkat CD19 CAR T-cells were centrifuged at 300 g for 5 minutes. The supernatant was discarded, and the cells were re-diluted with live cell imaging solution to eliminate potential interference from FBS and other culture medium components. The concentration of all cell samples was adjusted to ~2 x 103 cells/µL as a standard cell sample before any experiment. This standard sample was then spiked into live cell imaging solution or human whole blood to reach final concentrations of 103, 102, 101, and 100 Jurkat CD19 CAR T-cells/µL. Two types of blood samples were used: Reagent RBCs purchased from Immucor (Catalog #: IG2338), and fresh human whole blood collected from healthy volunteers using K2 EDTA tubes (BD, #23-021-015) (approved by IRB STUDY00008255). The whole-blood spiked samples were designed to mimic clinical patient samples.
To remove RBCs in the blood samples, 25 µL of anti-blood type solution (Ortho Clinical Diagnostic Inc; # 6901934) was gently mixed with 50 µL of whole blood or reagent RBCs spiked samples at room temperature for 30 mins. The agglutinated samples were then transferred to a simple filter system to remove any aggregated RBCs, (Figure 2A). A hydrophilic membrane filter (Sterlitech #PCT25013100) with a 25 µm pore size was inserted between two reusable silicone sample wells (FlexiPERM micro12, # 94.6011.436). A capillary tube (VitroCom #8270) located below the filter provided the capillary force to extract the liquid portion of the agglutinated sample containing non-agglutinated cells for measurement. 50 ±5 µL of filtered samples can be collected after the agglutination and filtering process.
2.6. Sample Loading and Processing
The filtered sample was introduced into the sensor chip through a standard PDMS inlet connection, which is assembled with a 21-gauge blunt needle (Flow-Tips, G.21/0.8mm, PBN-GRN-200) plus a 1 mL syringe (BD, 309628). The CD19 protein immobilized on the chip surface selectively captured the target Jurkat CD19 CAR T-cells. After 15 minutes of incubation, a washing buffer (live cell imaging solution; 20mL syringe(BD,302830)) was perfused through the microfluidic system at a flow rate of 10 µL/min for 1 minute to remove the non-target cells from the region of interest. Two sets of images were acquired for each experiment, one prior to the washing step and the other after the washing step. In total, each set consisted of 30 images covering the entire length of the linear channel.
2.7. Optical Image Acquisition
An inverted microscope (Olympus IX81, 40X/ 0.75 NA objective) with CMOS camera (Hamamatsu ORCA-Flash 4.0 V3; Pixel size: 6.5 μm; View size: 2048 × 2048 pixels) was used to obtain bright field and fluorescent images. A mercury light (Olympus U-LH100HG) and a filter set (Excitation wavelength: 543 ± 10, Emission wavelength: 593 ± 40) were used for fluorescence imaging. The bright field images were recorded with a 0.1 s exposure time, while the exposure time for fluorescence images was 4 s. Optical images were captured in 16-bit monochrome format.
2.8. Cell Culture
This study used two cell lines, Jurkat CD19 CAR T-cells and corresponding wild-type (WT) Jurkat T-cells. Jurkat CD19 CAR T-cells were engineered to express the anti-human CD19 protein, containing a single-chain variable fragment (scFv) derived from the FMC63 monoclonal antibody, which mimics the function of human CAR-19+ T-cells. On the other hand, the WT cells do not express the CAR-CD19 molecule. Both cell lines were cultured and prepared in accordance with the previously published protocol [21]. Specifically, the cells were cultured in Nunc EasYFlask 75 cm2 (Thermo Fisher Scientific, 156472) using RPMI 1640 medium (Gibco, A1049101), supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco, A31606-02) and 1% (v/v) penicillin-streptomycin (Corning, 30-002-CI). Cell density was maintained at 1 × 106 cells/mL.
2.9. Cell Counting with a Cell Counter
An automated cell counter (Logos Biosystems, LUNA-IITM, L40001) was used as a reference method to count cell density in the samples. For proper counting using the cell counter, Jurkat CD19 CAR T-Cells were rendered permeable with 0.1% Triton X-100, and nuclei were visualized using 0.4% trypan blue as a DNA indicator [22,23].
2.10. Immunostaining
Immunostaining was employed to verify the identity of Jurkat CD19 CAR T-cells. The immunostaining protocol was adapted from the manufacturer's instructions and standard cell immunostaining protocols [24]. Briefly, 1 mL of cells (concentration: 1 × 106 cells/mL) in a culture medium were transferred into a 15 mL centrifuge tube and centrifuged at 300 g for 5 minutes. The supernatant was discarded and replaced with the same amount of living cell imaging solution and re-centrifuged. The cells were resuspended in 1 mL 1x blocking reagent (10% v/v) for 30 minutes of blocking. The cells were centrifuged again, and the supernatant was aspirated. The cells were then incubated with Human CD19-phycoerythrin (PE) conjugated protein (1:50 dilution; Acro Biosystems #CD9-HP2H3) for 30 minutes at room temperature in the dark. The centrifugation process was repeated twice with 1 mL of living cell imaging solution to wash off unbound CD19-PE. Finally, 50 μL of stained cell samples were pipetted onto a clean microscope slide attached with a reusable silicone sample well (FlexiPERM micro12 # 94.6011.436) and covered with a microscope coverslip. The sample was observed under a fluorescent microscope with focus adjusted to the central level of cells to minimize the background (Figure S5).
To validate the identity of Jurkat CD19 CAR T-cells captured in a microfluidic chip, Human CD19-PE conjugated protein was injected into the chip system right after the flushing process and incubated for 30 minutes. Then, washing buffer was flowed for 60 seconds at a flow rate of 10 µL/min to wash away the excess fluorescent dye, and the chip was ready for imaging.
2.11. Flow Cytometry
The flow cytometry process was conducted according to a standard protocol [25,26]. Briefly, 1×106 cells were centrifuged at 300 g for 5 mins, washed three times with live cell imaging solution, and counted by the Logos LUNA-II cell counter to verify the cell concentration. The buffer-diluted Jurkat WT/CD19 CAR T-cells were then incubated with 1000 μL 0.5% BSA (Sigma Aldrich # 05470) for 30 mins for blocking, followed by triplicate centrifuging and washing steps. Subsequently, the cells were incubated with 100 μL human CD19-PE conjugated protein (1:50) for another 30 minutes. The cells were then centrifuged and washed three times to remove excessive fluorescent protein and analyzed in an Attune NxT Flow Cytometer.
2.12. Image Processing
Each trial consisted of 30 images of independent regions in the capture zone along the PDMS channel. They were used for Jurkat CD19 CAR T-cells counting and final quantification. These regions were imaged before flushing, after flushing, and after fluorescent staining of each trial to compare the captured cell positivity ratio. As shown in Figure 3A-F and Figure S5, the recorded images were analyzed using Fiji ImageJ [27]. The locations of the CAR T-cells in the bright field imaging were identified based on the morphology of the cells. The region of interest data was then applied to fluorescent images to extract both fluorescence intensity and spatial information. The fluorescent intensity distributions of the stained cells were analyzed and plotted using Python (Figure 3H).
To reduce implicit bias and account for digital crowding, an automated Python pre-filtering algorithm paired with a machine learning (ML) based (bio)image segmentation and classification software, ilastik [28], [29], was used on spiked whole blood trials (Figure 4). The prefiltering algorithm consists of: (1) Images subjected to a localized standard deviation filter; (2) a modified match filter; (3) negative value thresholding; (4) local maxima identification; and (5) radial segmentation of the original brightfield image. This unique method works to eliminate issues caused by cellular crowding and enhances segmentation algorithms by separating each potential target from its neighbors. This method showed great improvement in cell identification accuracy by allowing each cell to be segmented regardless of touching other cells, a difficult common problem in brightfield automated cell segmentation. The machine-learning based cell segmentation and identification program, ilastik, was trained using approximately 100 types of 4 commonly occurring objects in the brightfield image datasets: Car T-cells verified by fluorescent tagging; platelets; red blood cells; and cellular debris/anomalous/broken cells. This software creates profiles of a variety of statistical parameters for later classification on desired datasets. The pre-filtered images were then subjected to this trained model, and cells were automatically segmented, classified, and quantified.
3. Results
Centrifuge-Free, Agglutination-Based RBCs Removal
The ratio of red blood cells (RBCs) to white blood cells (WBCs) in whole blood is approximately 600:1–8, making RBC removal a critical step for effective capture and quantification of CAR T-cells [30]. Classic centrifugation-based RBC removal methods [31,32,33,34] are not POC compatible. Ammonium Chloride Potassium (ACK) lysing buffer can lyse RBCs [35,36,37], but the efficiency is low (~60%) and affects white blood cell viability [38]. Microfluidic RBC removal methods have been explored, including filtration [39,40,41,42,43,44], dielectrophoresis [45,46,47,48,49], or geometrical channels [50,51,52,53]. However, these designs often increase the complexity and cost of the assay [12].
To accomplish RBC removal in a cost-effective POC-compatible manner, we implemented a simple agglutination and filtration method for centrifuge-free RBCs' removal from whole blood samples. By inducing antibody-mediated RBC agglutination followed by filtration through a large-pore membrane, aggregated RBCs were eliminated while preserving WBCs and other nucleated cells. For proof-of-concept testing, 50 µL of whole blood was incubated with 25 µL of the corresponding anti-blood type serum for 30 minutes and then passed through a simple filtration system containing a polycarbonate track-etched (PCTE) filter (Figure 2A). Additional details are mentioned in the Sample Preparation section. Agglutinated RBCs were retained on the filter, and the remaining blood components were collected via capillary force into a test tube. Given the average size of lymphocytes (7 to 15 µm) [54], PCTE filters with pore sizes of 20 µm and 25 µm were evaluated for recovery efficiency using buffer-diluted Jurkat CD19 CAR T-cells and Jurkat wild-type cells. Concentrations of T cells were measured pre- and post-filtration (Figure 2B). Both membranes achieved >95% recovery, with the 25 µm filter showing slightly superior performance; hence, this pore size was selected for subsequent experiments to minimize loss of target cells.
The efficiency of the filtration method in whole blood samples spiked with Jurkat CD19 CAR T-cells was further assessed using hemocytometer counting as the reference standard (Figure S4). WBC counts before and after filtration showed no significant difference compared with undiluted whole blood (P = 0.6353), confirming that this POC-compatible method effectively removes RBCs while maintaining a high WBC recovery rate (Figure 2C).
CAR T-Cell Capture, Detection, and Validation
The surface chemistry used to functionalize the sensor surface for Jurkat CD19 CAR T-cell capture is shown in Figure S3, with details provided in the Methods section. Samples were introduced to the CD19 functionalized sensor surface and incubated for 15 minutes to allow Jurkat CD19 CAR T-cell capture, followed by washing with live cell imaging solution at a flow rate of 10 µL/min to remove non-specifically bound cells. Two primary factors can influence the precision and accuracy of CAR T-cell detection on the chip [55,56]: (1) heterogeneous CAR expression in transfected Jurkat CD19 CAR T-cells, which may result in absent or low receptor levels and weak binding to the CD19-coated surface, and (2) non-specific interactions leading to incorrect identification of non-target blood cells.
To evaluate capture efficiency and specificity, Jurkat CD19 CAR T-cells (positive for CAR expression) and Jurkat wild-type (WT) T-cells (negative control) were tested (Figure 3). Flow cytometry confirmed CD19 CAR expression levels, and capture efficiency on the CD19-coated surface was validated by fluorescence staining of captured cells [57,58]. Following the washing step, 97% of Jurkat CD19 CAR T-cells remained bound to the sensor surface (Figure 3A, B, J), with strong fluorescent signals observed in 88% of captured cells (Figure 3C, G, H, I). This fraction was slightly higher than the 77% CD19 CAR population measured by flow cytometry (Figure 3I, S6), consistent with preferential retention of strongly expressing cells and loss of weak expressers during washing. In contrast, only 8.9% of Jurkat WT T-cells remained attached after washing (Figure 3D, E, J), indicating a low-level of non-specifically bound cells post-washing (Figure 3F, I, S6).
Statistical analysis of ~1,500 cells from each line, tested independently in duplicate assays, was performed by paired evaluation of brightfield and fluorescence images. Although brightfield morphology appeared similar across cells, fluorescence imaging revealed CAR expression on the Jurkat CD19 CAR T-cell surface, with clustered CD19 signals apparent as bright dots (Figure 3G). Quantitative analysis of fluorescence intensity, normalized by dividing pixel intensity standard deviation by the median for each cell, revealed two distinct Gaussian distributions separating Jurkat CD19 CAR T-cells and WT T-cells (Figure 3H; details in Table S1). Notably, approximately 20% of Jurkat CD19 CAR-T cells exhibited absent or low CAR expression, phenotypically resembling WT T-cells and therefore not contributing to the CD19 CAR T-cell population.
Overall, these results demonstrate that the CD19-functionalized surface preferentially captures Jurkat CD19 CAR T-cells, achieving higher detection specificity than bulk flow cytometry, as evidenced by the higher proportion of fluorescently positive cells among captured Jurkat CD19 CAR T-cells (88% vs. 77%). These findings confirm the robustness and specificity of the CD19 capture surface for accurate Jurkat CD19 CAR T-cell detection.
CAR T-Cell Quantification in Whole Blood
As the ultimate goal of this method is point-of-care (POC) diagnosis, experiments were conducted using CAR T-cell–spiked whole blood. These samples produced substantial nonspecific adherence of non-CAR T-cells to the sensor surface, primarily platelets, unfiltered RBCs, and other leukocytes or debris. While the washing step effectively removed CAR-19-negative T cells and RBCs (Figure 3A–F, S7), it was less effective against platelets and other blood components. This resulted in cell crowding and contamination that impaired data processing, generating false negatives and false positives that required manual adjustment of parameters, creating implicit bias and not meeting POC automation requirements.
To address this limitation, we developed a label-free, bright-field imaging-based data processing algorithm for automated Jurkat CD19 CAR T-cell identification and quantification. The algorithm, implemented in Python and integrated with the machine-learning platform ilastik, operates in two stages: (1) broad identification of cells within a relevant size range (Figure 4A–E) and (2) automated classification and counting of identified cells to yield CAR T-cell quantification (Figure 4F). The classifier was trained on four categories using 100 manually curated examples per class: Fluorescence-validated Jurkat CD19 CAR T-cells; platelets; RBCs;, and cell debris. The software extracts and learns multiple optical pixel-based morphological features—including size, granularity, and membrane contour—for high-throughput classification of new datasets [28,59].
System performance was evaluated by spiking Jurkat CD19 CAR T-cells at four different concentrations into undiluted human whole blood from healthy donors. After washing, captured cells across the full channel (30 regions of interest) were enumerated by the algorithm, with whole blood lacking CAR T-cells analyzed as baseline controls. Each concentration was tested in triplicate for reproducibility, with representative images shown in Figure S8. A standard calibration curve was then generated by plotting the number of automated algorithm-identified Jurkat CD19 CAR T-cells against their known input concentrations in spiked whole blood (Figure 4G).
4. Discussion
This proof-of-concept study establishes the feasibility of a low-cost, scalable, POC platform for real-time monitoring of CAR T-cell therapy, directly addressing critical gaps in current patient management. As previously discussed, clinical outcomes and toxicities are closely linked to CAR T-cell expansion and the dynamics of immune activation–associated cytokines, both of which govern therapeutic response and the development of serious, and sometimes life-threatening, complications such as cytokine release syndrome (CRS) and neurotoxicity. Despite their clinical relevance, these parameters are not routinely measured due to the high cost, procedural complexity, and slow turnaround times inherent to conventional assays such as flow cytometry and multiplex cytokine immunoassays (Table S2) [2,5,6,7].
POC devices quantifying CAR T-cell kinetics and cytokine profiles in real time could provide clinicians with objective information that is not captured by routine vital signs or intermittent laboratory testing. Recent advances [60] in microfluidic and lab-on-chip technologies have demonstrated the feasibility of CAR T-cell functional assessment [61], cytokine analysis [62], and low–sample-volume assays [63], and their adoption in both research and early translational settings continues to expand. However, most existing platforms remain optimized for discrete, laboratory-based measurements rather than longitudinal clinical monitoring, and their integration into routine care pathways is still limited. There remains a need for point-of-care approaches that can support timely clinical decision-making, particularly for early precise detection of functional cell level quantification or impending treatment failure. Considering that prolonged hospitalization and monitoring can account for a significant portion of the >$800,000 total per-patient cost of CAR T-cell therapy, reducing reliance on inpatient care has the potential for substantial cost savings across the healthcare system.
The described platform, which integrates centrifuge-free red blood cell removal, label-free optical imaging, and automated machine-learning–based CAR T-cell detection, represents a significant advance toward practical POC patient monitoring. The system demonstrated reliable identification of CAR T-cells at clinically relevant concentrations in whole blood within one hour, and at a material cost of approximately $5 per test (Table S3). This work provides foundational evidence supporting future clinical translation of rapid, deployable biosensors for CAR T-cell monitoring. Ongoing development will extend the platform to multi-analyte cytokine profiling, portable imaging system adaptation, and validation using finger-prick capillary samples in real-world patient cohorts. Collectively, these innovations may redefine monitoring strategies for cellular immunotherapies, improve clinical safety, drive down the cost of care, and expand access to these life-saving treatments.
6. Patents
The technology described in this article is protected by a US patent application (18/778,278) filed by the Arizona Board of Regents on behalf of Arizona State University.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1. Figure S1: Illustration of the flow-driven detection setup; Figure S2: Schematic figure of the fluidic chip structure; Figure S3: Sensor surface modification protocol Figure S4: Standard cell counting images with a hemacytometer; Figure S5: Image processing workflow utilizing Fiji software; Figure S6. Flow cytometry matching results; Figure S7: Post-flush images of Jurkat CD19 CAR T-cells and wild-type Jurkat cells in varying conditions; Figure S8: Representative images of five concentrations used for the calibration experiment; Table S1. Statistical fitting details; Table S2: Comparison of the current representative direct CAR-T cell counting approach; Table S3: Unit price for one chip summary.
Author Contributions
Conceptualization, J.C. and S.W.; methodology, N.Y., R.P., X.Z., W.J., F.Z., E.M., E.F., J.C., and S.W.; software, N.Y., R.P., X.Z., and S.W.; validation, N.Y., E.M., P.L., J.F., E.F., J.C. and S.W.; formal analysis, N.Y., R.P. and S.W.; investigation, N.Y. and W.J.; resources, E.F., S.W., J.C.; data curation, N.Y., and R.P.; writing—original draft preparation, N.Y., R.P., and S.W.; writing—review and editing, N.Y., R.P., E.M., S.W., and J.C.; project supervision and administration, S.W. and J.C.; funding acquisition, S.W. and J.C. All authors have read and agreed to the published version of the manuscript.
Funding
SW and JEC acknowledge financial support from their faculty startup fund. The authors acknowledge resources and support from the KE cores facilities at Arizona State University. ASU-Mayo Clinic Collaborative grant and the Riney Family Foundation grant.
Institutional Review Board Statement
Fresh human whole blood collected from healthy volunteers following approved IRB (STUDY00008255) by Arizona State University.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Source image data are available upon reasonable request from the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol 2017, 35, 1803–1813. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther 2017, 25, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Ruella, M.; Schuster, S.J.; Lymphoma Program Investigators at the University of; P. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med 2021, 384, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri, S.; Timmerman, J.; Larson, S.; Mead, M.D. Axicabtagene Ciloleucel CAR T-cell therapy for relapsed/refractory secondary CNS non-Hodgkin lymphoma: comparable outcomes and toxicities, but shorter remissions may warrant alternative consolidative strategies? Bone Marrow Transplant 2020. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nature Reviews Clinical Oncology 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf, G.F., 2nd; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959 e953. [Google Scholar] [CrossRef] [PubMed]
- Jena, B.; Maiti, S.; Huls, H.; Singh, H.; Lee, D.A.; Champlin, R.E.; Cooper, L.J. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One 2013, 8, e57838. [Google Scholar] [CrossRef]
- Cavallo, M.C.; Cavazza, M.; Bonifazi, F.; Casadei, B.; Cutini, I.; Tonietti, B.; Saccardi, R.; Zinzani, P.; Jommi, C. Cost of implementing CAR-T activity and managing CAR-T patients: an exploratory study. BMC Health Serv Res 2024, 24, 121. [Google Scholar] [CrossRef]
- Kim, H.; Zhbanov, A.; Yang, S. Microfluidic Systems for Blood and Blood Cell Characterization. Biosensors (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Tay, H.M.; Petchakup, C.; He, L.; Gong, L.; Maw, K.K.; Leong, S.Y.; Lok, W.W.; Ong, H.B.; Guo, R.; et al. Label-free microfluidic cell sorting and detection for rapid blood analysis. Lab Chip 2023, 23, 1226–1257. [Google Scholar] [CrossRef]
- Yuan, H.; Miao, Z.; Wan, C.; Wang, J.; Liu, J.; Li, Y.; Xiao, Y.; Chen, P.; Liu, B.F. Recent advances in centrifugal microfluidics for point-of-care testing. Lab Chip 2025, 25, 1015–1046. [Google Scholar] [CrossRef]
- Kant, K.; Shahbazi, M.A.; Dave, V.P.; Ngo, T.A.; Chidambara, V.A.; Than, L.Q.; Bang, D.D.; Wolff, A. Microfluidic devices for sample preparation and rapid detection of foodborne pathogens. Biotechnol Adv 2018, 36, 1003–1024. [Google Scholar] [CrossRef]
- Aran, K.; Sasso, L.A.; Kamdar, N.; Zahn, J.D. Irreversible, direct bonding of nanoporous polymer membranes to PDMS or glass microdevices. Lab on a Chip 2010, 10, 548. [Google Scholar] [CrossRef]
- Wu, D.; Zhao, B.; Dai, Z.; Qin, J.; Lin, B. Grafting epoxy-modified hydrophilic polymers onto poly(dimethylsiloxane) microfluidic chip to resist nonspecific protein adsorption. Lab Chip 2006, 6, 942–947. [Google Scholar] [CrossRef]
- Friend, J.; Yeo, L. Fabrication of microfluidic devices using polydimethylsiloxane. Biomicrofluidics 2010, 4, 026502. [Google Scholar] [CrossRef]
- Ghanbari, A.; Attar, M.M. A study on the anticorrosion performance of epoxy nanocomposite coatings containing epoxy-silane treated nano-silica on mild steel substrate. Journal of Industrial and Engineering Chemistry 2015, 23, 145–153. [Google Scholar] [CrossRef]
- Mrad, M.; Montemor, M.F.; Dhouibi, L.; Triki, E. Deposition of hybrid 3-GPTMS's film on AA2024-T3: Dependence of film morphology and protectiveness performance on coating conditions. Progress in Organic Coatings 2012, 73, 264–271. [Google Scholar] [CrossRef]
- Ojea-Jiménez, I.; Urbán, P.; Barahona, F.; Pedroni, M.; Capomaccio, R.; Ceccone, G.; Kinsner-Ovaskainen, A.; Rossi, F.; Gilliland, D. Highly Flexible Platform for Tuning Surface Properties of Silica Nanoparticles and Monitoring Their Biological Interaction. ACS Applied Materials & Interfaces 2016, 8, 4838–4850. [Google Scholar] [CrossRef]
- Jing, W.; Wang, Y.; Chen, C.; Zhang, F.; Yang, Y.; Ma, G.; Yang, E.H.; Snozek, C.L.N.; Tao, N.; Wang, S. Gradient-Based Rapid Digital Immunoassay for High-Sensitivity Cardiac Troponin T (hs-cTnT) Detection in 1 muL Plasma. ACS Sens 2020. [Google Scholar] [CrossRef]
- Connell, L.S.; Romer, F.; Suarez, M.; Valliant, E.M.; Zhang, Z.; Lee, P.D.; Smith, M.E.; Hanna, J.V.; Jones, J.R. Chemical characterisation and fabrication of chitosan-silica hybrid scaffolds with 3-glycidoxypropyl trimethoxysilane. J Mater Chem B 2014, 2, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Lu, J.; Guo, Y.; Li, D.; Zhang, Z.M.; Tsai, Y.H.; Pi, W.C.; Ahn, J.H.; Gong, W.; Xiang, Y.; et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat Genet 2020, 52, 1384–1396. [Google Scholar] [CrossRef]
- Cheng, R.; Zhang, F.; Li, M.; Wo, X.; Su, Y.-W.; Wang, W. Influence of Fixation and Permeabilization on the Mass Density of Single Cells: A Surface Plasmon Resonance Imaging Study. Frontiers in Chemistry 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Dyes, trypanosomiasis and DNA: a historical and critical review. Biotechnic & Histochemistry 2010, 85, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Burry, R.W. Controls for immunocytochemistry: an update. [CrossRef] [PubMed]
- Finak, G.; Langweiler, M.; Jaimes, M.; Malek, M.; Taghiyar, J.; Korin, Y.; Raddassi, K.; Devine, L.; Obermoser, G.; Pekalski, M.L.; et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci Rep 2016, 6, 20686. [Google Scholar] [CrossRef]
- McKinnon, K.M. Flow Cytometry: An Overview. Curr Protoc Immunol 2018, 120, 5 1 1–5 1 11. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. ilastik: interactive machine learning for (bio)image analysis. Nature Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]
- Zhang, C.; Huber, F.; Knop, M.; Hamprecht, F.A. Yeast cell detection and segmentation in bright field microscopy. Proceedings of the 2014 IEEE 11th International Symposium on Biomedical Imaging (ISBI) 2014, 2014, 1267–1270. [Google Scholar]
- Laura, D. Blood and the cells it contains. In Blood Groups and Red Cell Antigens [Internet]; National Center for Biotechnology Information (US), 2005. [Google Scholar]
- Tatsumi, N.; Tsuda, I; Fau - Fukumori, T.; Fukumori, T; Fau - Hino, M.; Hino, M; Fau - Takubo, T.; Fau - Kondo, Takubo T; Kondo, H.H. A centrifuge-less plasma separation method from whole blood anticoagulated with EDTA-2K for the use of clinical laboratory tests.
- Böyum, A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968, 97, 77–89. [Google Scholar]
- Jia, Y.; Xu, H.; Li, Y.; Wei, C.; Guo, R.; Wang, F.; Wu, Y.; Liu, J.; Jia, J.; Yan, J.; et al. A Modified Ficoll-Paque Gradient Method for Isolating Mononuclear Cells from the Peripheral and Umbilical Cord Blood of Humans for Biobanks and Clinical Laboratories. Biopreservation and Biobanking 2017, 16, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Jaatinen, T.; Laine, J. Isolation of mononuclear cells from human cord blood by Ficoll-Paque density gradient. In Curr Protoc Stem Cell Biol 2007, Chapter 2; Unit 2A.1. [CrossRef]
- Bossuyt, X.; Marti, G.E.; Fleisher, T.A. Comparative analysis of whole blood lysis methods for flow cytometry. Cytometry 1997, 30, 124–133. [Google Scholar] [CrossRef]
- Horn, P.; Bork, S.; Wagner, W. Standardized isolation of human mesenchymal stromal cells with red blood cell lysis. Methods Mol Biol 2011, 698, 23–35. [Google Scholar] [CrossRef]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. Isolation of carbohydrate-specific CD4+ T cell clones from mice after stimulation by two model glycoconjugate vaccines. Nature Protocols 2012, 7, 2180–2192. [Google Scholar] [CrossRef]
- Brown, W.E.; Hu, J.C.; Athanasiou, K.A. Ammonium-Chloride-Potassium Lysing Buffer Treatment of Fully Differentiated Cells Increases Cell Purity and Resulting Neotissue Functional Properties. Tissue Eng Part C Methods 2016, 22, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Chang, Y.; Deng, Q.; You, H. A simple and rapid method for blood plasma separation driven by capillary force with an application in protein detection. Anal Methods 2020, 12, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mauk, M.; Gross, R.; Bushman, F.D.; Edelstein, P.H.; Collman, R.G.; Bau, H.H. Membrane-based, sedimentation-assisted plasma separator for point-of-care applications. Anal Chem 2013, 85, 10463–10470. [Google Scholar] [CrossRef]
- Nabatiyan, A.; Parpia, Z.A.; Elghanian, R.; Kelso, D.M. Membrane-based plasma collection device for point-of-care diagnosis of HIV. J Virol Methods 2011, 173, 37–42. [Google Scholar] [CrossRef]
- Hou, H.W. Microfluidic Devices for Blood Fractionation. Micromachines 2011, 2. [Google Scholar] [CrossRef]
- Hauser, J.; Lenk, G.; Hansson, J.; Beck, O.; Stemme, G.; Roxhed, N. High-Yield Passive Plasma Filtration from Human Finger Prick Blood. Anal Chem 2018, 90, 13393–13399. [Google Scholar] [CrossRef] [PubMed]
- Ulum, M.F.; Maylina, L.; Noviana, D.; Wicaksono, D.H.B. EDTA-treated cotton-thread microfluidic device used for one-step whole blood plasma separation and assay. Lab on a Chip 16 2016, 1492–1504. [Google Scholar] [CrossRef]
- Gascoyne, P.R.; Noshari, J.; Anderson, T.J.; Becker, F.F. Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis 2009, 30, 1388–1398. [Google Scholar] [CrossRef]
- Kim, B.; Oh, S.; You, D.; Choi, S. Microfluidic Pipette Tip for High-Purity and High-Throughput Blood Plasma Separation from Whole Blood. Anal Chem 2017, 89, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Kuan, D.-H.; Wu, C.-C.; Su, W.-Y.; Huang, N.-T. A Microfluidic Device for Simultaneous Extraction of Plasma, Red Blood Cells, and On-Chip White Blood Cell Trapping. Scientific Reports 2018, 8, 15345. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Hata, S.; Yasuda, T. Blood plasma separation and extraction from a minute amount of blood using dielectrophoretic and capillary forces. Sensors and Actuators B: Chemical 2010, 145, 561–569. [Google Scholar] [CrossRef]
- Li, H.; Steckl, A.J. Paper Microfluidics for Point-of-Care Blood-Based Analysis and Diagnostics. Anal Chem 2019, 91, 352–371. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, E.L.; Kelly, P.; Church, C.C.; Brayman, A.A.; Child, S.Z.; Raeman, C.H.; Schery, L. Lysis of erythrocytes by exposure to CW ultrasound. Ultrasound Med Biol 1993, 19, 147–165. [Google Scholar] [CrossRef]
- Davis, J.A. Deterministic hydrodynamics: Taking blood apart. Proceedings of the National Academy of Sciences 2006, 103. [Google Scholar] [CrossRef]
- Petchakup, C.; Tay, H.M.; Li, K.H.H.; Hou, H.W. integrated inertial-impedance cytometry for rapid label-free leukocyte isolation and profiling of neutrophil extracellular traps(NETs). Lab Chip 2019, 19, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Xiang, N.; Wang, J.; Li, Q.; Han, Y.; Huang, D.; Ni, Z. Precise Size-Based Cell Separation via the Coupling of Inertial Microfluidics and Deterministic Lateral Displacement. Anal Chem 2019, 91, 10328–10334. [Google Scholar] [CrossRef]
- Wheater, P.R.; Burkitt, H.G.; Daniels, V.G.; Deakin, P.J. Functional Histology: A Text and Colour Atlas; Churchill Livingstone, 1979. [Google Scholar]
- Mayer, T.; Jagla B Fau - Wyler, M.R.; Wyler Mr Fau - Kelly, P.D.; Kelly Pd Fau - Aulner, N.; Aulner N Fau - Beard, M.; Beard M Fau - Barger, G.; Barger G Fau - Többen, U.; Többen U Fau - Smith, D.H.; Smith Dh Fau - Brandén, L.; Brandén L Fau - Rothman, J.E.; et al. Cell-based assays using primary endothelial cells to study multiple steps in inflammation.
- Ashcroft, R.G.; Lopez, P.A. Commercial high speed machines open new opportunities in high throughput flow cytometry (HTFC). [CrossRef]
- Maino, V.C.; Picker, L.J. Identification of functional subsets by flow cytometry: intracellular detection of cytokine expression. [CrossRef]
- Kummrow, A.; Frankowski, M; Fau - Bock, N.; Bock, N; Fau - Werner, C.; Werner C; Fau - Dziekan, T.; Dziekan, T; Fau - Neukammer, J.; Neukammer, J. Quantitative assessment of cell viability based on flow cytometry and microscopy. [CrossRef]
- Christiansen, E.M.; Yang, S.J.; Ando, D.M.; Javaherian, A.; Skibinski, G.; Lipnick, S.; Mount, E.; O’Neil, A.; Shah, K.; Lee, A.K.; Goyal, P.; Fedus, W.; Poplin, R.; Esteva, A.; Berndl, M.; Rubin, L.L.; Nelson, P.; Finkbeiner, S. In Silico Labeling: Predicting Fluorescent Labels in Unlabeled Images. Cell 2018, Cell, 792–803 e719. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Lim, H.; Chung, A.J. Expanding CAR-T cell immunotherapy horizons through microfluidics. Lab on a Chip 2024, 24, 1088–1120. [Google Scholar] [CrossRef]
- Zeming, K.K.; Quek, K.Y.; Sin, W.-X.; Teo, D.B.L.; Cheung, K.-W.; Goh, C.R.; Kairi, F.; Lee, E.; Lim, F.L.W.I.; Seng, M.S.-F.; et al. Cell trajectory modulation: rapid microfluidic biophysical profiling of CAR T cell functional phenotypes. Nature Communications 2025, 16, 4775. [Google Scholar] [CrossRef]
- Ma, C.; Wang, H.; Liu, L.; Chen, R.; Mukherjee, N.; Tong, J.; Kazmi, S.; Fang, X.; Witkowski, M.T.; Aifantis, I.; et al. Bioengineered immunocompetent preclinical trial-on-chip tool enables screening of CAR T cell therapy for leukaemia. Nature Biomedical Engineering 2025. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a Droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Analytical Chemistry 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
Figure 1.
Workflow of the centrifuge-free rapid optical imaging-based CAR T-cell detection method. A) Sample preparation: 1) A small whole blood sample of 50 µL is collected and then spiked with predetermined concentration of Jurkat CD19 CAR T-cells for calibration curve generation and testing purpose; 2) The whole blood sample is mixed with 25 µL anti-blood type antibody and left at room temperature to agglutinate for 30 mins; 3) The agglutinated sample is then passed through a filter and the fluid is collected through a capillary tube to remove agglutinated RBCs. B) CAR T-cells Capture: 4) The filtered sample is loaded into the functionalized sensor chip and incubated for 15 minutes to capture CAR T-cells to the sensor surface; 5) Unbound cells are washed away with buffer. C) Data collection and analysis: 6) Optical microscopy images of the captured cells on the sensor chip surfaces are collected; 7) These images are processed to count Jurkat CD19 CAR T-cells; 8) Jurkat CD19 CAR T-cell concentration in the blood samples is determined using a calibration curve.
Figure 1.
Workflow of the centrifuge-free rapid optical imaging-based CAR T-cell detection method. A) Sample preparation: 1) A small whole blood sample of 50 µL is collected and then spiked with predetermined concentration of Jurkat CD19 CAR T-cells for calibration curve generation and testing purpose; 2) The whole blood sample is mixed with 25 µL anti-blood type antibody and left at room temperature to agglutinate for 30 mins; 3) The agglutinated sample is then passed through a filter and the fluid is collected through a capillary tube to remove agglutinated RBCs. B) CAR T-cells Capture: 4) The filtered sample is loaded into the functionalized sensor chip and incubated for 15 minutes to capture CAR T-cells to the sensor surface; 5) Unbound cells are washed away with buffer. C) Data collection and analysis: 6) Optical microscopy images of the captured cells on the sensor chip surfaces are collected; 7) These images are processed to count Jurkat CD19 CAR T-cells; 8) Jurkat CD19 CAR T-cell concentration in the blood samples is determined using a calibration curve.
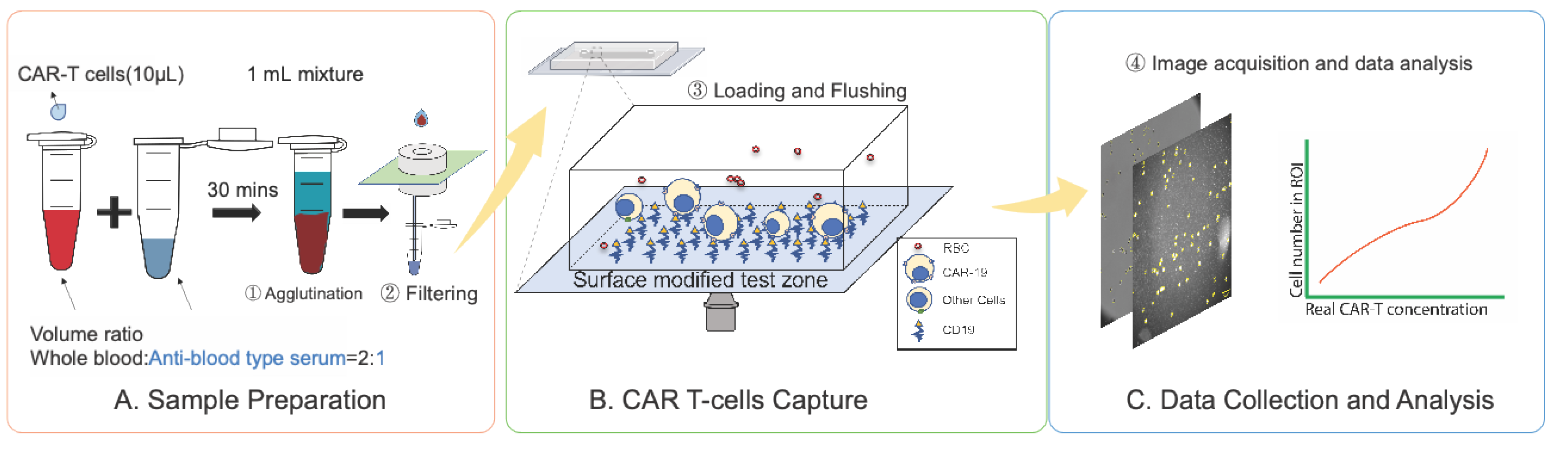
Figure 2.
Principle and result of centrifuge-free RBCs removal from whole blood. A) Schematic of agglutinated sample filtration for removing RBCs from the whole blood sample. B) Comparison of the membrane filter efficiency between 20 µm and 25 µm membrane filters. The filtering efficiency is the ratio of the white blood cell (WBC) concentration after passing through the membrane filter and the concentration before passing through the membrane filter (Efficiency = WBCafter/WBCbefore). Data was generated using LUNA II cell counter C) comparing of the WBCs’ concentration in undiluted whole blood samples (before filtering) and agglutinated and filtered (25 µm filter) samples. The Hemacytometer assay was applied. The standard deviation of triplicate tests determines the error bars (mean±SD).
Figure 2.
Principle and result of centrifuge-free RBCs removal from whole blood. A) Schematic of agglutinated sample filtration for removing RBCs from the whole blood sample. B) Comparison of the membrane filter efficiency between 20 µm and 25 µm membrane filters. The filtering efficiency is the ratio of the white blood cell (WBC) concentration after passing through the membrane filter and the concentration before passing through the membrane filter (Efficiency = WBCafter/WBCbefore). Data was generated using LUNA II cell counter C) comparing of the WBCs’ concentration in undiluted whole blood samples (before filtering) and agglutinated and filtered (25 µm filter) samples. The Hemacytometer assay was applied. The standard deviation of triplicate tests determines the error bars (mean±SD).
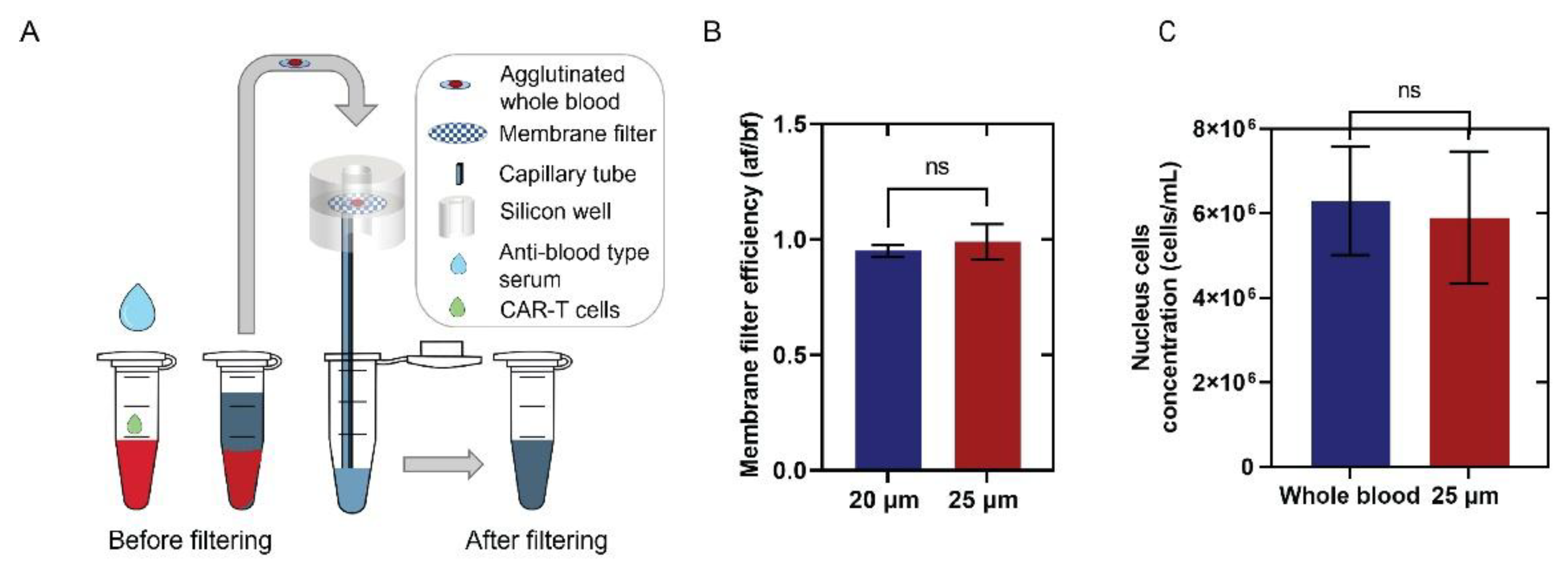
Figure 3.
Validation of CD-19 captured Jurkat CD19 CAR T-cells with fluorescent staining and flow cytometry. Panel A-F are representative brightfield images of Jurkat CD19 CAR T-cells (A, B) and Jurkat wild-type T cells (D,E) before (A, D) and after (B, E) flushing. Panels C and F are fluorescence images of CD19-PE-stained cells after the flushing step. Yellow circles mark the locations of attached cells before flushing. Panel G is bright field (Left) and corresponding fluorescence images (right) of three CAR T-cells with low, medium, and high CD-19 expression levels, where clustered fluorescence signals are clearly visible. Graph H is the histogram showing the statistical distribution of the fluorescence spectrum. Fluorescence spot statistics are represented by the standard deviation of the fluorescence image divided by the median of the image to normalize across different experiments slight background brightness variations. The histogram contains analysis of over 3000 fluorescently stained Jurkat CD19 CAR T-cells and Jurkat wild-type T-cells. Panel I is a comparison of the percentage of identified CAR T-cells in the cultured Jurkat CD19 CAR T-cells line by flow cytometry (77.0±1.7%) and fluorescent images of attached cells (87.7±5.7%) with error bars representing the standard deviation of three or more replicates. No significant difference was observed between the two methods. Panel J is the fraction of Jurkat CD19 CAR T-cells attached to the surface, calculated as the ratio of the number of cells attached after and before flushing. Error bars represent the standard deviation of three or more replicates, and a significant difference was observed between the two groups (P<0.0001).
Figure 3.
Validation of CD-19 captured Jurkat CD19 CAR T-cells with fluorescent staining and flow cytometry. Panel A-F are representative brightfield images of Jurkat CD19 CAR T-cells (A, B) and Jurkat wild-type T cells (D,E) before (A, D) and after (B, E) flushing. Panels C and F are fluorescence images of CD19-PE-stained cells after the flushing step. Yellow circles mark the locations of attached cells before flushing. Panel G is bright field (Left) and corresponding fluorescence images (right) of three CAR T-cells with low, medium, and high CD-19 expression levels, where clustered fluorescence signals are clearly visible. Graph H is the histogram showing the statistical distribution of the fluorescence spectrum. Fluorescence spot statistics are represented by the standard deviation of the fluorescence image divided by the median of the image to normalize across different experiments slight background brightness variations. The histogram contains analysis of over 3000 fluorescently stained Jurkat CD19 CAR T-cells and Jurkat wild-type T-cells. Panel I is a comparison of the percentage of identified CAR T-cells in the cultured Jurkat CD19 CAR T-cells line by flow cytometry (77.0±1.7%) and fluorescent images of attached cells (87.7±5.7%) with error bars representing the standard deviation of three or more replicates. No significant difference was observed between the two methods. Panel J is the fraction of Jurkat CD19 CAR T-cells attached to the surface, calculated as the ratio of the number of cells attached after and before flushing. Error bars represent the standard deviation of three or more replicates, and a significant difference was observed between the two groups (P<0.0001).
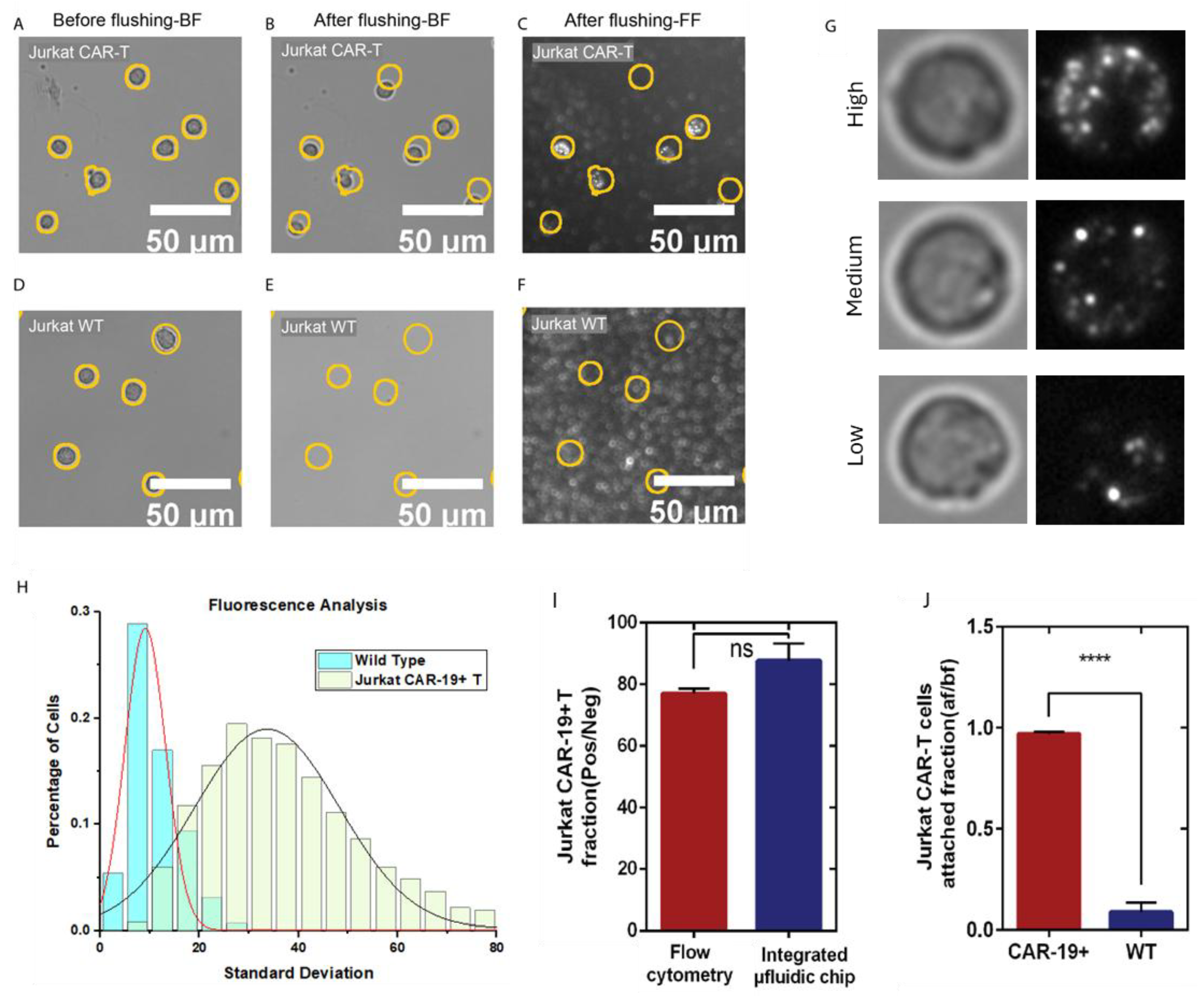
Figure 4.
Automated algorithm overview and results of algorithm output using spiked whole blood at varying CAR T-cell concentrations. (A) Section of a brightfield image representing ~0.1% of the total captured image area. (B) Localized standard deviation filter. (C) Modified match filtering. (D) Negative value thresholding. (E) Identification of local maxima and radial segmentation of the original image. (F) Identification of CAR T-cells using ilastik ML-based software. (G) Output of automated algorithm analyzing whole blood spiked with 4 different concentrations of CAR T-cells with logarithmic fitting with R2 value of 0.99991. Fitting details are shown in Table S1.
Figure 4.
Automated algorithm overview and results of algorithm output using spiked whole blood at varying CAR T-cell concentrations. (A) Section of a brightfield image representing ~0.1% of the total captured image area. (B) Localized standard deviation filter. (C) Modified match filtering. (D) Negative value thresholding. (E) Identification of local maxima and radial segmentation of the original image. (F) Identification of CAR T-cells using ilastik ML-based software. (G) Output of automated algorithm analyzing whole blood spiked with 4 different concentrations of CAR T-cells with logarithmic fitting with R2 value of 0.99991. Fitting details are shown in Table S1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



