Submitted:
18 January 2026
Posted:
21 January 2026
You are already at the latest version
Abstract
Trypanosoma brucei is a protist parasite that causes African trypanosomiasis, or sleeping sickness, in humans and animals. The disease consists of two phases, a hemolymphatic phase and a meningoencephalic phase. However, the parasite could colonize other organs, remain as a reservoir, and evade drug treatment, but the mechanisms it uses are unclear. The purpose of this review is to compile information from different sources to provide an in-depth explanation of the pathophysiology of trypanosomiasis, focusing on its virulence factors, host-parasite interaction, and extravascular migration capacity. All information was collected from various articles, research studies, and experiments that were consistent with the objective presented. In conclusion, trypanosomiasis progresses according to the parasite's ability to modulate and evade the immune response, which generates chronic inflammation and contributes to the progression of the disease. Likewise, thanks to growing evidence of the ability of T. brucei to colonize extravascular tissues, it can be said that trypanosomiasis is not exclusively hematological and that several organs can be affected. Finally, the neuropathogenesis of T. brucei is related to the entry of the parasite into brain tissue (the route of which is still unclear) and different immunomodulators that contribute to neuroinflammation and can aggravate the disease.
Keywords:
veterinary
; public health
; parasites
; trypanosomiasis
1. Introduction
African trypanosomiasis, caused by Trypanosoma brucei, is a parasitic disease with a significant impact on human and veterinary health, especially in endemic regions of Africa, where it affects humans, domestic animals, and wildlife. The pathogenicity of this extracellular hemoparasite is based on a complex host-parasite interaction, characterized by its virulence mechanisms, evasion of the immune system, and ability to persist in tissues, which determine a progressive clinical course that, in the absence of treatment, is potentially fatal [1,4]. In recent years, the field has seen significant advances, particularly in the understanding of antigenic variation mediated by variant surface glycoproteins (VSGs), induced immune dysfunction, and the existence of previously underestimated extravascular reservoirs, such as skin and adipose tissue [2,3,5]. However, discussions persist regarding the relative contribution of host inflammation versus direct parasite damage, as well as the precise mechanisms involved in the transition to neurological involvement [20,21].
This review article aims to integrate current knowledge on the pathophysiology of Trypanosoma brucei, systematically addressing its main virulence factors, immune evasion strategies, host inflammatory response, host-parasite interactions, extravascular invasion and residence in various tissues, and the mechanisms involved in neuropathogenesis. Based on evidence from experimental models and clinically relevant studies, it is highlighted that the persistence of the parasite results from a dynamic balance between immune control and immunopathology, in which antigenic variation, immune modulation, and tissue tropism play a significant role [4,6]. This synthesis seeks to provide a comprehensive overview of the pathophysiological process of infection.
2. Materials and Methods
Information was gathered from various experimental animal studies, research papers, and other literature reviews. PubMed was used to find these sources, and filters were applied to exclude older studies, with a few exceptions where the information was valuable. In addition, articles in English were sought, and the information collected focused on the pathophysiology of trypanosomiasis in animals.
3. Results
3.1. Virulence Factors
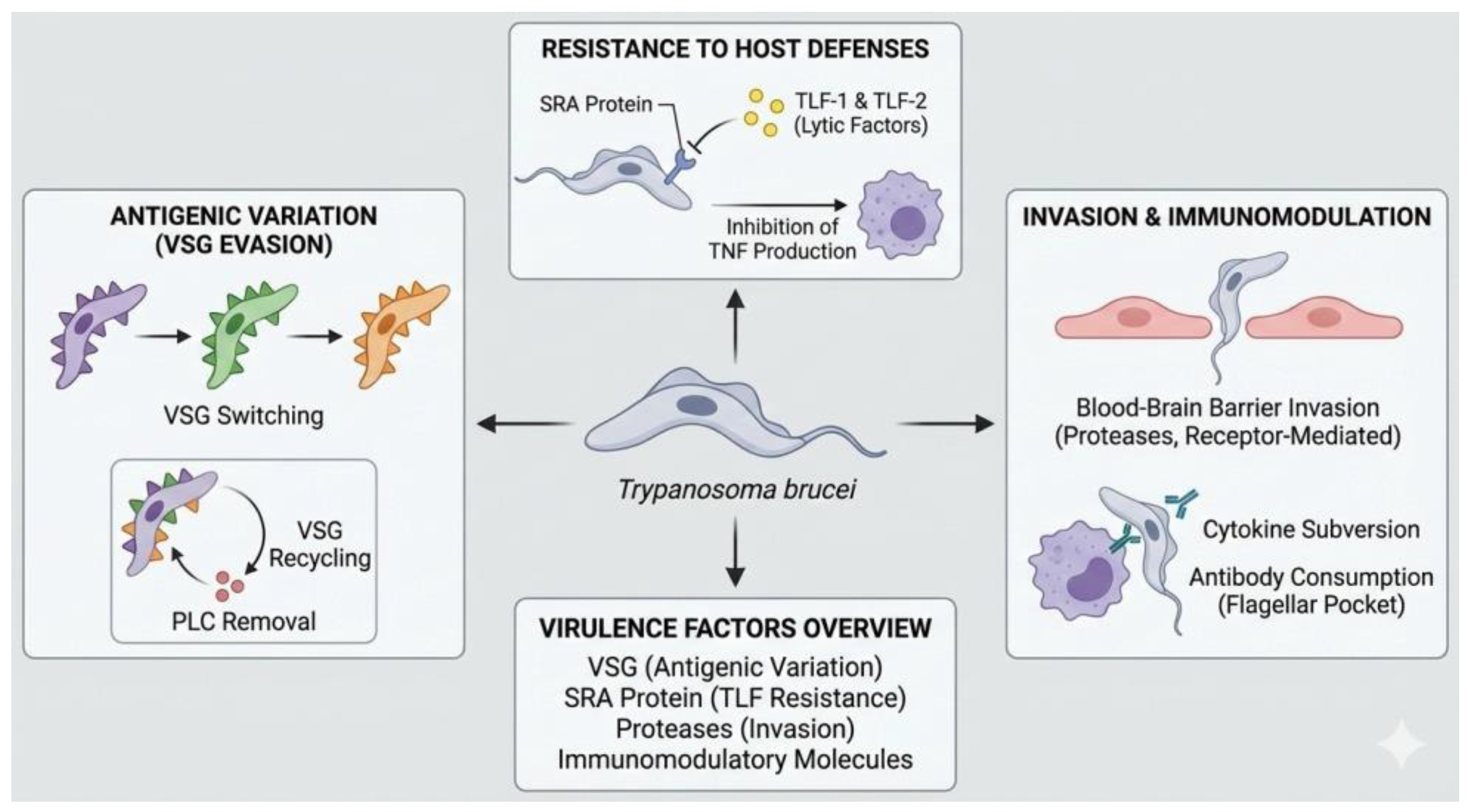
Trypanosoma brucei is an extracellular blood parasite that causes African trypanosomiasis in humans and animals. Its pathogenicity is based on a set of virulence factors that allow it to persist in the host and trigger progressive pathophysiological alterations. The central mechanism of its virulence is the antigenic variation of the variant surface glycoprotein (VSG), a protein that densely coats the parasite’s membrane and constitutes its main interface with the immune system. Although VSG is highly immunogenic, T. brucei has the ability to periodically modify the expressed variant through genetic recombination and alternating activation of telomeric expression sites, allowing the emergence of parasitic subpopulations not recognized by previously generated antibodies. This process underpins the persistence of parasitemia and the chronicity of infection [1].
VSG accounts for approximately 90% of the parasite’s surface proteins and performs an additional protective function by acting as a physical barrier that limits the access of complement and antibodies to invariant membrane antigens. This surface organization reduces complement-mediated lysis and opsonization, favoring the survival of the parasite in the bloodstream. Recent evidence indicates that antigenic diversification does not occur exclusively in blood, but also in extravascular tissue compartments, such as adipose tissue and other interstitial spaces, where T. brucei can persist, diversify its antigenic repertoire, and subsequently re-enter the circulation. These tissue reservoirs play a key role in both immune evasion and the development of organ damage, chronic anemia, and endothelial dysfunction, associated with sustained local inflammation and microvascular alterations [2,3].
Another important component of the virulence of T. brucei is the production of extracellular vesicles derived mainly from the flagellar membrane. These vesicles transport proteins and bioactive factors capable of modifying the host environment, including molecules that alter the integrity and half-life of erythrocytes, promoting their premature elimination by the reticuloendothelial system and contributing to the development of anemia. These vesicles also contain proteins involved in resistance to trypanolytic factors and in the modulation of local immune responses, which increases the parasitic load and prolongs the infection. These processes are associated with persistent systemic inflammation, mediated by proinflammatory cytokines such as TNF-alpha and IL-6, with clinical manifestations relevant to veterinary medicine and public health [1,4].
3.2. Immune Evasion
Immune evasion is one of the most sophisticated features of Trypanosoma brucei biology and is fundamental to its success as a pathogen. This process is mainly based on the antigenic variation of the VSG, which forces the host’s immune system to generate successive humoral responses to constantly changing antigens. Each time an effective response is established against a specific VSG variant, parasites expressing different variants emerge, allowing recurrent parasitemias to be maintained for long periods without complete elimination of the agent [1].
Recent studies have shown that antibodies generated against T. brucei tend to target specific immunodominant epitopes of each VSG, producing highly stereotypical antibody repertoires. This characteristic facilitates immune evasion, as minimal structural modifications in the VSG are sufficient to avoid recognition by pre-existing antibodies. In addition, the high density of VSG contributes to the concealment of invariant antigens that could serve as alternative targets for the immune system, reinforcing evasion of both humoral immunity and innate mechanisms [1,2].
Furthermore, T. brucei reduces the effectiveness of the humoral response through rapid endocytosis of antigen-antibody complexes. Antibodies bound to VSG are internalized and degraded before they can efficiently activate complement or phagocytosis. In the long term, infection also compromises the generation of effective immune memory by affecting the formation and functionality of memory B cells, reducing the host’s ability to respond quickly and effectively to subsequent exposures. This alteration represents a significant obstacle to vaccine development and explains the persistence of chronic T. brucei infections [3].
Figure 1.
Schematic representation of the main virulence mechanisms and immune evasion strategies of Trypanosoma brucei.
Figure 1.
Schematic representation of the main virulence mechanisms and immune evasion strategies of Trypanosoma brucei.
3.3. Host Inflammatory Response to Trypanosoma brucei
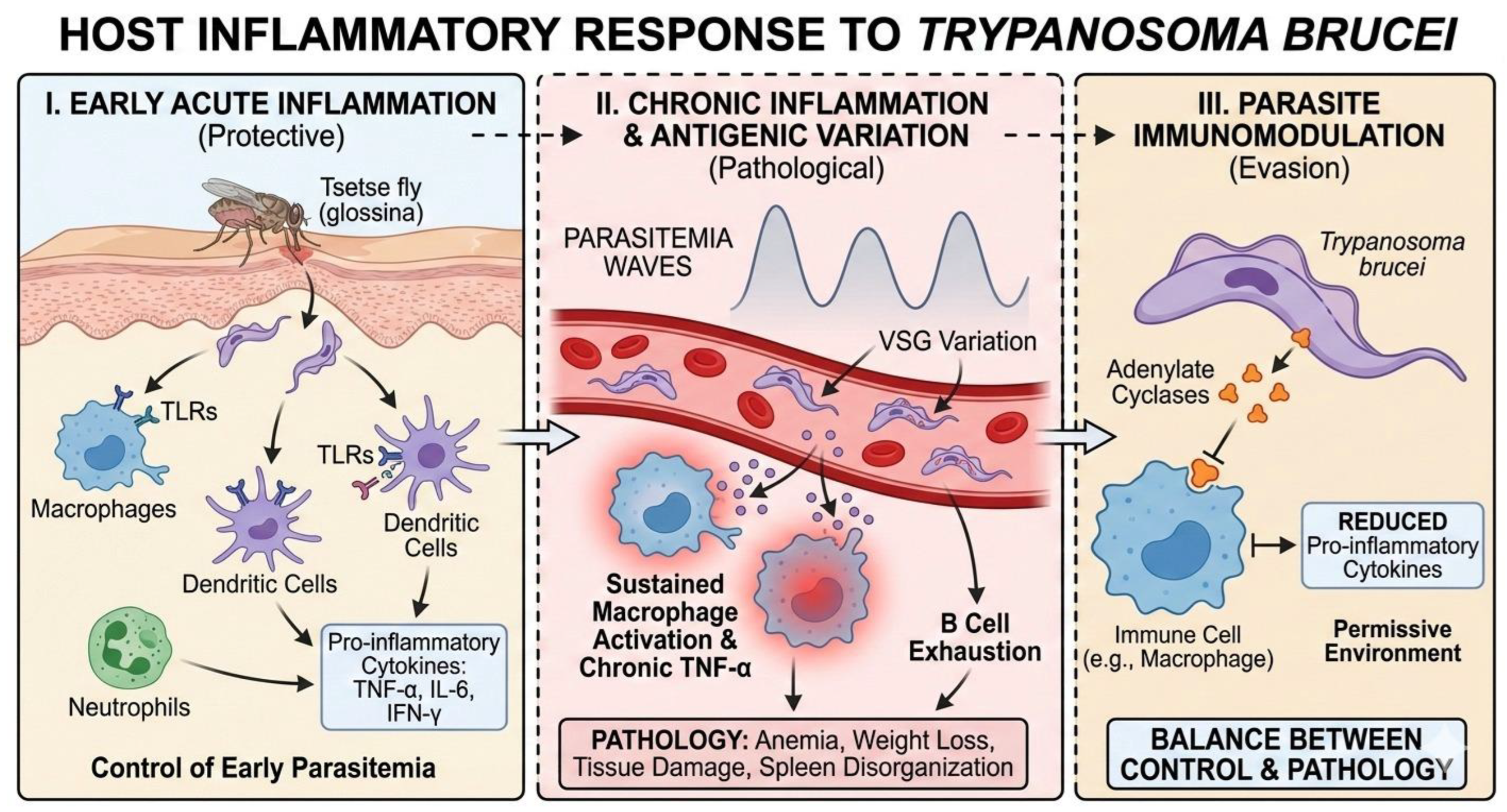
Infection with Trypanosoma brucei triggers a complex and highly dynamic inflammatory response in the mammalian host, which plays a dual role: initially it contributes to parasite control, but subsequently it promotes pathogenesis and disease progression. Following inoculation of the parasite by the tsetse fly, innate immunity is activated, particularly at the skin level, where macrophages, dendritic cells, and neutrophils recognize the parasite through pattern recognition receptors, such as Toll-like receptors (TLRs), inducing the early production of proinflammatory cytokines such as TNF-α, IL-6, and IFN-γ [5].
This initial inflammatory response is crucial for limiting early parasitemia; however, T. brucei has developed effective mechanisms to resist and modulate this response. Studies have shown that sustained macrophage activation leads to chronic TNF-α production, which, while contributing to parasite control, is also associated with anemia, weight loss, and tissue damage, clinical signs characteristic of African trypanosomiasis [6]. In this context, inflammation ceases to be protective and becomes a pathological factor.
At the systemic level, the inflammatory response is strongly influenced by the antigenic variation of the parasite, mediated by variable surface glycoproteins (VSGs). Each peak of parasitemia generates a new inflammatory surge, repeatedly stimulating the immune system and promoting a state of chronic inflammation. This constant stimulation leads to B-cell depletion and disorganization of the spleen architecture, compromising the host’s ability to mount effective immune responses against secondary infections [7,8].
In addition, T. brucei is capable of actively attenuating the host’s inflammatory response by releasing immunomodulatory molecules. Among these are parasitic adenylate cyclases, which interfere with the intracellular signaling of immune cells, reducing the production of proinflammatory cytokines and promoting a more permissive environment for parasite survival [8]. Thus, the host’s inflammatory response to T. brucei represents a delicate balance between immune control and pathological damage, a direct result of an evolutionarily refined interaction between parasite and host.
Figure 2.
Host inflammatory response to Trypanosoma brucei. This panel illustrates the progression of the immune response, starting with the early activation of innate immunity mediated by TLRs and cytokines To control initial infection. It highlights the transition to a chronic phase driven by VSG-mediated antigenic variation, where sustained inflammation leads to pathology (e.g., B cell exhaustion, tissue damage). Finally, the figure depicts the active immunomodulation by the parasite via the release of adenylate cyclases, which attenuate the inflammatory response to favor parasite persistence.
Figure 2.
Host inflammatory response to Trypanosoma brucei. This panel illustrates the progression of the immune response, starting with the early activation of innate immunity mediated by TLRs and cytokines To control initial infection. It highlights the transition to a chronic phase driven by VSG-mediated antigenic variation, where sustained inflammation leads to pathology (e.g., B cell exhaustion, tissue damage). Finally, the figure depicts the active immunomodulation by the parasite via the release of adenylate cyclases, which attenuate the inflammatory response to favor parasite persistence.
3.4. Host–Parasite Interactions in Trypanosoma brucei Infection
Host–parasite interactions in Trypanosoma brucei infection are highly specialized and reflect coevolution aimed at the parasite’s persistence within the mammalian host. From the moment of transmission, T. brucei interacts closely with the host’s immune system, modulating both the innate and adaptive responses to ensure its survival and dissemination [7].
One of the central mechanisms of this interaction is VSG-mediated antigenic variation, which allows the parasite to continuously evade the specific antibodies generated by the host. Each time the humoral response manages to control an antigenic variant, a parasitic subpopulation expresses a different VSG, restarting the cycle of infection and inflammation. This process not only ensures the persistence of the parasite, but also profoundly disrupts the host’s immunological memory [7,9].
Additionally, T. brucei maintains sophisticated molecular interactions with immune system cells. The release and constant turnover of VSGs not only serve to evade antibodies, but also induce polyclonal activation of B lymphocytes, contributing to progressive immunosuppression of the host [10]. This phenomenon limits the effectiveness of specific adaptive responses and promotes susceptibility to concomitant infections, a finding of particular relevance in veterinary medicine and public health.
On the other hand, recent studies have shown that certain essential parasitic receptors remain protected from immune attack thanks to their strategic location and the dense layer of VSG that covers the surface of the parasite. This structural organization prevents antibodies from accessing vulnerable targets, reinforcing the efficiency of immune evasion and highlighting the complexity of host-parasite interactions at the molecular level [10].
Finally, early interactions at the site of inoculation, particularly in the skin, play a decisive role in the course of infection. The local inflammatory response, modulated by components of the parasite and the vector, conditions the systemic dissemination of T. brucei and the subsequent immunological dynamics. These initial interactions represent a critical point where the biology of the parasite and the host response converge, defining the balance between control and disease progression [5].
3.5. Tissue Invasion and Extravascular Residence
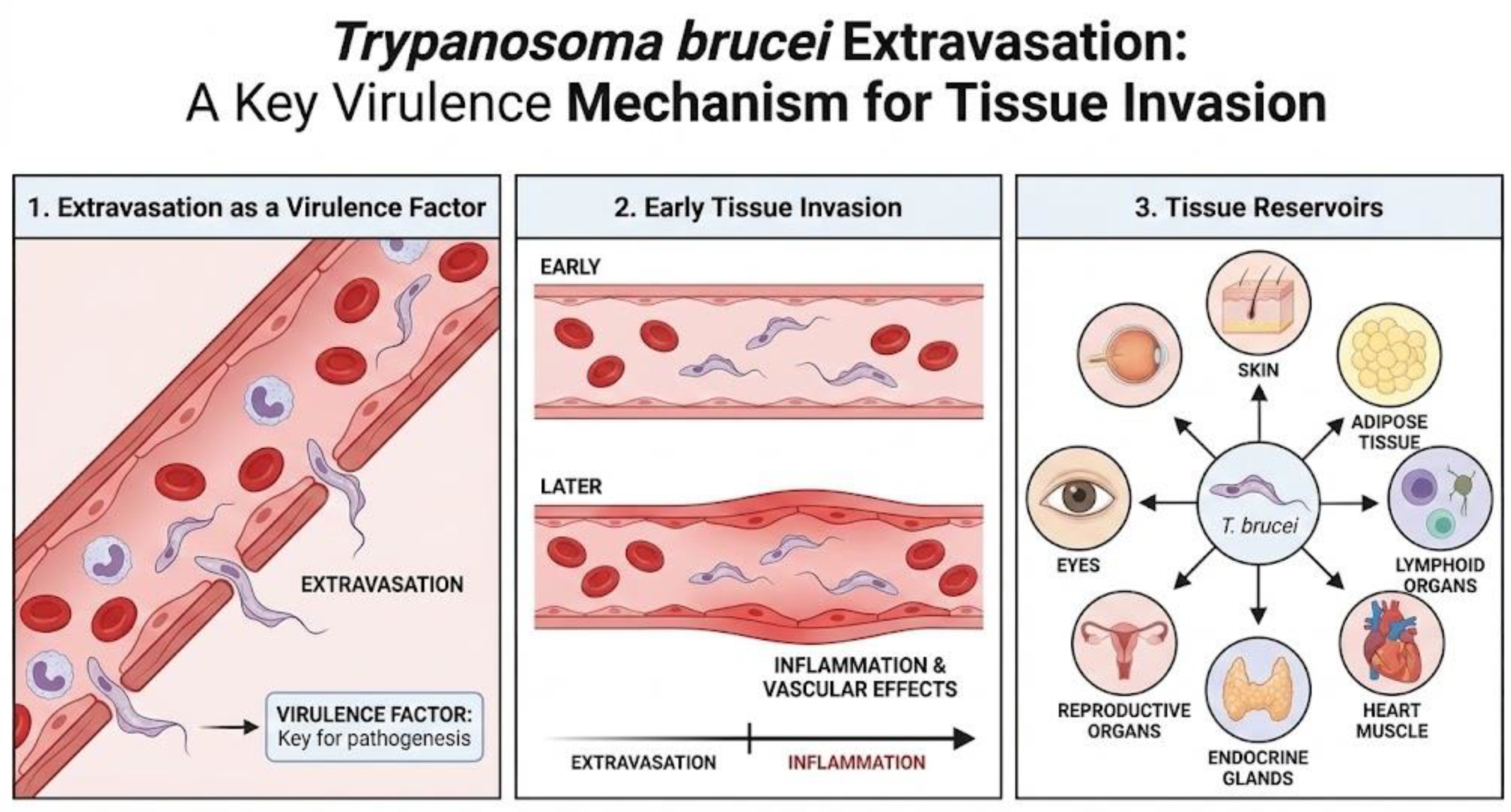
The symptoms produced by Trypanosoma brucei stem from its ability to extravasate and enter extravascular tissues, as has been seen in experimental models, where extravasation occurs before the vascular effects produced by inflammation, demonstrating that it is a key virulence factor [4]. Although there is currently abundant information on the effect of the parasite on the blood and central nervous system, what is known about its effect on other tissues is not entirely clear. For this reason, studies on this subject have increased, allowing us to understand how Trypanosoma brucei reaches different tissues and uses them as reservoirs, such as the skin, adipose tissue, lymphoid organs, heart muscle, endocrine glands, reproductive organs, and even the eyes [2].
Firstly, there is the proliferation of the parasite in the skin, a tissue considered to be an important reservoir, as there have been documented cases of individuals affected by pruritus, skin rashes, localized edema, and dermatitis. In the latter, it is suggested that the presence of trypanosomes affects the immune status of the skin, making it sensitive and vulnerable to other pathogens [11]. On the other hand, according to one study, after simulating the vector transmission of T. brucei in human skin models, it was observed that the infectious metacyclic forms (MCF) of the parasite were activated and established a proliferative population of trypanosomes, and then slowed down their growth, metabolism, DNA synthesis, and protein translation, suggesting that they act as “resting cells,” all of which contributes to the persistence of the parasite in the skin [12]. Similarly, another study demonstrated the presence of live, mobile parasites in the dermis and hypodermis and suggested that these trypanosomes maintain the disease foci despite screening and treatment, representing a new target for diagnosis, as skin tests should be performed to identify the presence of the parasite and prevent reinfestation [13].
Another tissue of importance for the disease is adipose tissue. Initially considered only a lipid store, we now know that it is an immunologically active endocrine organ, as adipocytes secrete various immunomodulators. In the context of trypanosomiasis, a common sign is weight loss, which is sometimes associated with neurological disease. However, it has been suggested that the parasite consumes the host’s reserves, producing this particular sign, in addition to tissue inflammation [14]. One study showed that adipose tissue acts as one of the main reservoirs of the parasite, reaching levels similar to those in the blood. Furthermore, the parasites present in the tissue show genetic variations compared to the blood forms [3]. Research on trypanosomes in adipose tissue has been limited to subcutaneous fat, so there is still a lack of information on gonadal, mesenteric, and perirenal fat [2].
Trypanosomiasis causes a variety of symptoms, including hemolytic anemia, hepatomegaly, splenomegaly, endocrine disorders, pericarditis, myocarditis, tachycardia, and congestive heart failure [15]. In animal hosts, such as cattle, dogs, and sheep, the parasite has been observed in all layers and structures of the heart; however, there is a particular tropism for the atrial myocardium rather than the ventricular myocardium, but this cannot be generalized for all mammalian species. This could be due to the presence of collagen in the atria and the tropism that T. brucei has for it [16]. On the other hand, damage to the heart can occur through direct parasitic action or through the host’s inflammatory response [2].
With regard to the reproductive organs, in humans, loss of libido, impotence, menstrual and fertility problems, miscarriages, stillbirths, premature births, hair loss, gynecomastia, orchitis, and testicular atrophy have been reported [15]. This information coincides with observations in experimental animal models, since trypanosomes were found in the tunica albuginea, pampiniform plexus, and tissue associated with the testicles in experimentally infected sheep, cattle, rabbits, and dogs. Likewise, lesions in the ovaries and uterine wall have been described in infected female dogs [16,17,18]. On the other hand, a study was conducted in a murine model, where the presence of the parasite was detected in the ovaries and uterus, even when there was no active parasitemia. This suggests that the reproductive organs can act as reservoirs for trypanosomes, causing horizontal transmission of the disease and also causing relapses in patients by maintaining the infection in a latent form [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19].
Symptoms often include eye conditions such as optic neuritis, diplopia, iritis, keratitis, conjunctivitis, choroidal atrophy, and iridocyclitis [15 and 20]. These signs have been studied in various animal species and have shown that the parasite enters the eye through the ciliary body and remains latent in the aqueous humor, evading pharmacological treatment, and could therefore be an anatomical reservoir. Unfortunately, no rodent models have been developed in this regard, despite the clinical importance of trypanosomes residing in the eyes [2].
Finally, it is important to mention the presence of T. brucei in the lymphatic system. The information suggests that in this tissue, there is no true extravascular population and that the enlargement of the lymph nodes and spleen observed in affected individuals is due to the body’s immune response and not due to the direct presence of the parasite in these tissues, but it is a route by which the parasite can spread and cause relapses of the disease [2].
3.6. Neuropathogenesis
The course of trypanosomiasis has two distinct phases: a hemolymphatic phase, in which the parasite travels through the blood and lymph, and a meningoencephalitic phase, characterized by the presence of trypanosomes in the central nervous system, causing a wide range of symptoms that vary between animals and humans. These include severe sleep disorders, neurological disorders, coma, and paralysis of the extremities as a result of myelopathy, myelitis, or peripheral motor neuron involvement. Muscle fasciculation may also be a symptom. It can also cause painful hyperesthesia in the extremities, which, when severe, is called Kerandel’s sign in advanced stages. Likewise, abnormal reflexes may occur that indicate frontal lobe involvement, such as the Pout reflex and palmo-mental reflexes [15,16,17,18,19,20,21]. Despite the information on the symptoms of the disease, the mechanism by which the parasite enters the nervous system is still not known with certainty. However, possible ways have been proposed. One is that the trypanosomes enter through the fenestrated vessels of the choroid plexus, accessing the stroma before entering the blood-brain barrier and spreading with the cerebrospinal fluid through the ventricles and subarachnoid space, finally colonizing the pia mater. Another way is through the fenestrated vessels of the circumventricular organs, i.e., those with an incomplete blood-brain barrier. Finally, the parasites could also enter the neuropil directly from the blood vessels. The mechanism of entry remains a mystery, but it has been suggested that it could be a combination of the three routes [15 and 20]. On the other hand, an important factor in neuropathogenesis is the presence of immunomodulators. It has been shown that there is a proportional association between the concentration of IFN-y and TNF-⍺ and a more severe neuroinflammatory response, while higher levels of interleukin 10 and 6 are associated with milder neuroinflammation [22 and 23].
Figure 3.
Mechanism for tissue invasion and affected tissues in trypanosomiasis.
4. Discussion
Infection with Trypanosoma brucei is a typical model of highly dynamic host–parasite interaction, in which the virulence mechanisms of the parasite and the immune response of the host are closely intertwined. The findings reviewed show that the pathogenesis of African trypanosomiasis does not depend exclusively on the parasite load, but rather on the parasite’s ability to modulate, evade, and redirect the host’s immune responses, generating a state of chronic inflammation that contributes decisively to tissue damage and the clinical progression of the disease [1,4,7].
One of the central axes of this interaction is antigenic variation mediated by variable surface glycoproteins (VSG), considered the main determinant of parasitic persistence. This mechanism not only allows T. brucei to effectively evade humoral immunity, but also imposes cyclical immune stimulation that leads to progressive exhaustion of the host’s immune system. The repeated generation of inflammatory responses to each wave of parasitemia promotes the disorganization of secondary lymphoid organs, such as the spleen, compromising the formation of effective immune memory and explaining the susceptibility to secondary infections observed in both infected humans and animals [7,8].
The host inflammatory response emerges as a varied component of the infection. In initial stages, the activation of innate immunity, characterized by the production of proinflammatory cytokines such as TNF-α, IFN-γ, and IL-6, is essential for the initial control of parasitemia [5,6]. However, the persistence of the parasite and sustained immune stimulation transform this response into a pathogenic factor, strongly associated with the development of anemia, cachexia, organ dysfunction, and progressive tissue damage. This phenomenon illustrates how T. brucei exploits the host’s defensive mechanisms to promote its own survival, reinforcing the concept of immunopathology as the central axis of the disease [6,8].
Growing evidence of the ability of T. brucei to colonize extravascular tissue compartments significantly broadens the traditional understanding of trypanosomiasis as an exclusively blood-borne infection. The identification of reservoirs in skin, adipose tissue, reproductive organs, heart, and eye demonstrates that parasite persistence is supported by a strategic anatomical distribution that allows the parasite to evade both the immune response and pharmacological action [2,11,12,13,14]. From a veterinary perspective, these findings are particularly relevant, as they explain clinical recurrence, silent transmission, and the difficulty of achieving complete eradication of the parasite in animal hosts.
Furthermore, tissue residence is associated with metabolic and phenotypic adaptations of the parasite, such as those observed in adipose tissue, where T. brucei adjusts its lipid metabolism and exhibits genetic variations compared to blood forms [3,14]. These adaptations not only enhance parasite survival, but also contribute to local and systemic inflammation, exacerbating the characteristic clinical signs of the disease. The existence of these extravascular niches poses significant diagnostic and therapeutic challenges, especially in veterinary settings where subclinical detection and persistent transmission represent a considerable health risk [2,13].
Finally, the neuropathogenesis associated with T. brucei reflects the culmination of the host–parasite interaction, where the combination of parasitic invasion and neuroinflammation determines clinical severity. The association between proinflammatory cytokines and neurological damage, together with the modulatory role of IL-10 and IL-6 in protecting against neuroinflammation, underscores the complexity of the immune mechanisms involved and reinforces the need for therapeutic approaches that not only target parasite elimination but also modulate the host immune response [20,22,23].
Overall, the evidence analyzed reveals that African trypanosomiasis is the result of a complex interaction, resulting from a progressive adaptation between Trypanosoma brucei and its host, in which immune evasion, chronic inflammation, and tissue invasion converge to sustain the infection and determine its clinical impact. Understanding these processes from an integrated perspective is essential for the development of new diagnostic, therapeutic, and control strategies, especially in the fields of veterinary medicine and public health.
Supplementary Materials
The following supporting information can be downloaded at: https://www.dropbox.com/scl/fo/0mp0hjc6rxfr40c386fjk/AIHOJIk0Dm7kLsUXcD1JCs?rlkey=5bn60twy6ygzjd23ze51tuj3u&st=dz60qpy1&dl=0.
Funding
This research received no external funding.
Acknowledgments
During the preparation of this manuscript, the authors used Gemini for the purposes of creation of illustrations. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- de Castro Neto, A.L.; da Silveira, J.F.; Mortara, R.A. Comparative Analysis of Virulence Mechanisms of Trypanosomatids Pathogenic to Humans. Front Cell Infect Microbiol 2021, 11.
- Crilly, N.P.; Mugnier, M.R. Thinking Outside the Blood: Perspectives on Tissue-Resident Trypanosoma Brucei. PLoS Pathog 2021, 17.
- Machado, H.; Bizarra-Rebelo, T.; Costa-Sequeira, M.; Trindade, S.; Carvalho, T.; Rijo-Ferreira, F.; Pacheco, B.R.; Serre, K.; Figueiredo, L.M. Trypanosoma Brucei Triggers a Broad Immune Response in the Adipose Tissue. PLoS Pathog 2021, 17, doi: 10.1371/journal.ppat.1009933. [CrossRef]
- Morrison, L.J.; Steketee, P.C.; Tettey, M.D.; Matthews, K.R. Pathogenicity and Virulence of African Trypanosomes: From Laboratory Models to Clinically Relevant Hosts. Virulence 2023, 14.
- Alfituri, O.A.; Quintana, J.F.; MacLeod, A.; Garside, P.; Benson, R.A.; Brewer, J.M.; Mabbott, N.A.; Morrison, L.J.; Capewell, P. To the Skin and Beyond: The Immune Response to African Trypanosomes as They Enter and Exit the Vertebrate Host. Front Immunol 2020, 11, doi:10.3389/fimmu.2020.01250. [CrossRef]
- Antoine-Moussiaux, N.; Büscher, P.; Desmecht, D. Host-Parasite Interactions in Trypanosomiasis: On the Way to an Antidisease Strategy. Infect Immun 2009, 77, 1276–1284, doi:10.1128/IAI.01185-08. [CrossRef]
- Ponte-Sucre, A. An Overview of Trypanosoma Brucei Infections: An Intense Host–Parasite Interaction. Front Microbiol 2016, 7, doi:10.3389/fmicb.2016.02126. [CrossRef]
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune Evasion Strategies of Trypanosoma Brucei within the Mammalian Host: Progression to Pathogenicity. Front Immunol 2016, 7, doi:10.3389/fimmu.2016.00233. [CrossRef]
- Pays, E.; Vanhamme, L.; Pérez-Morga, D. Antigenic Variation in Trypanosoma Brucei: Facts, Challenges and Mysteries. Curr Opin Microbiol 2004, 7, 369–374, doi: 10.1016/j.mib.2004.05.001. [CrossRef]
- Moreno, C.J.G.; Temporão, A.; Torres, T.; Sousa Silva, M. Trypanosoma Brucei Interaction with Host: Mechanism of VSG Release as Target for Drug Discovery for African Trypanosomiasis. Int J Mol Sci 2019, 20, 1484, doi:10.3390/ijms20061484. [CrossRef]
- Camara, M.; Soumah, A.M.; Ilboudo, H.; Travaillé, C.; Clucas, C.; Cooper, A.; Kuispond Swar, N.-R.; Camara, O.; Sadissou, I.; Calvo Alvarez, E.; et al. Extravascular Dermal Trypanosomes in Suspected and Confirmed Cases of Gambiense Human African Trypanosomiasis. Clinical Infectious Diseases 2021, 73, 12–20, doi:10.1093/cid/ciaa897. [CrossRef]
- Reuter, C.; Hauf, L.; Imdahl, F.; Sen, R.; Vafadarnejad, E.; Fey, P.; Finger, T.; Jones, N.G.; Walles, H.; Barquist, L.; et al. Vector-Borne Trypanosoma Brucei Parasites Develop in Artificial Human Skin and Persist as Skin Tissue Forms. Nat Commun 2023, 14, 7660, doi:10.1038/s41467-023-43437-2. [CrossRef]
- Capewell, P.; Cren-Travaillé, C.; Marchesi, F.; Johnston, P.; Clucas, C.; Benson, R.A.; Gorman, T.-A.; Calvo-Alvarez, E.; Crouzols, A.; Jouvion, G.; et al. The Skin Is a Significant but Overlooked Anatomical Reservoir for Vector-Borne African Trypanosomes. Elife 2016, 5, doi:10.7554/eLife.17716. [CrossRef]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; Van Den Abbeele, J.; et al. Trypanosoma Brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 2016, 19, 837–848, doi: 10.1016/j.chom.2016.05.002. [CrossRef]
- Kennedy, P.G.E. The Continuing Problem of Human African Trypanosomiasis (Sleeping Sickness). Ann Neurol 2008, 64, 116–126, doi:10.1002/ana.21429. [CrossRef]
- Ikede, B.O.; Losos, G.J. Pathological Changes in Cattle Infected with Trypanosoma Brucei. Vet Pathol 1972, 9, 272–277, doi:10.1177/030098587200900407. [CrossRef]
- Morrison, W.I.; Murray, M.; Sayer, P.D.; Preston, J.M. The Pathogenesis of Experimentally Induced Trypanosoma Brucei Infection in the Dog. I. Tissue and Organ Damage. Am J Pathol 1981, 102, 168–181.
- Ikede, B.O.; Losos, G.J. Pathology of the Disease in Sheep Produced Experimentally by Trypanosoma Brucei. Vet Pathol 1972, 9, 278–289, doi:10.1177/030098587200900408. [CrossRef]
- Biteau, N.; Asencio, C.; Izotte, J.; Rousseau, B.; Fèvre, M.; Pillay, D.; Baltz, T. Trypanosoma Brucei Gambiense Infections in Mice Lead to Tropism to the Reproductive Organs, and Horizontal and Vertical Transmission. PLoS Negl Trop Dis 2016, 10, e0004350, doi: 10.1371/journal.pntd.0004350. [CrossRef]
- Kennedy, P.G.E.; Rodgers, J. Clinical and Neuropathogenetic Aspects of Human African Trypanosomiasis. Front Immunol 2019, 10, doi:10.3389/fimmu.2019.00039. [CrossRef]
- Figarella, K. Neuropathogenesis Caused by Trypanosoma Brucei, Still an Enigma to Be Unveiled. Microbial Cell 2021, 8, 73–76, doi:10.15698/mic2021.04.745. [CrossRef]
- MacLean, L.; Reiber, H.; Kennedy, P.G.E.; Sternberg, J.M. Stage Progression and Neurological Symptoms in Trypanosoma Brucei Rhodesiense Sleeping Sickness: Role of the CNS Inflammatory Response. PLoS Negl Trop Dis 2012, 6, e1857, doi: 10.1371/journal.pntd.0001857. [CrossRef]
- Sternberg, J.M.; Rodgers, J.; Bradley, B.; MacLean, L.; Murray, M.; Kennedy, P.G.E. Meningoencephalitic African Trypanosomiasis: Brain IL-10 and IL-6 Are Associated with Protection from Neuro-Inflammatory Pathology. J Neuroimmunol 2005, 167, 81–89, doi: 10.1016/j.jneuroim.2005.06.017. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



