
Submitted:
20 January 2026
Posted:
21 January 2026
You are already at the latest version
Abstract
The dystrophin gene encodes multiple dystrophin isoforms with tissue-specific functions, including several shorter isoforms expressed in the central nervous system and retina. While Duchenne muscular dystrophy (DMD) has historically been charac-terized as a primary myopathy resulting from loss of the full-length dystrophin Dp427, increasing clinical evidence indicates that dysfunction of shorter dystrophin isoforms contributes to significant extramuscular pathology, including retinal disease. In par-ticular, loss of the Dp71 isoform has been implicated in retinal inflammation, blood–retinal barrier breakdown, and pathological angiogenesis. In this study, we investigated whether low-level residual expression of Dp71 is sufficient to mitigate retinal inflammation in the mdx3Cv mouse model, which displays reduced—but not absent—expression of multiple dystrophin isoforms. Western blot analysis revealed that mdx3Cv retinas express approximately 4% of wild-type Dp71 protein levels. Despite this marked reduction, mdx3Cv mice did not exhibit the in-flammatory phenotype previously observed in Dp71-null mice. Retinal VEGF protein levels and VEGF receptor (FLT-1 and KDR) mRNA expression were preserved, while VEGF mRNA levels were modestly reduced. Furthermore, expression of inflammatory markers ICAM-1 and ALOX5AP, leukocyte adhesion to retinal vasculature, Aqua-porin-4 expression, and BRB permeability to albumin were all comparable to wild-type littermates. Together, these findings demonstrate that minimal residual expression of Dp71 is sufficient to preserve retinal vascular homeostasis and prevent inflammatory and permeability defects in the mdx3Cv retina. These results further suggest that partial dystrophin restoration—at levels achievable with current exon-skipping or gene-based therapies—may be adequate to prevent or attenuate retinal pathology in DMD, providing a realistic and clinically relevant therapeutic target.
Keywords:
Duchenne muscular dystrophy
; dystrophin
; Dp71
; retina
; Müller glial cell
; Blood–Retinal Barrier (BRB)
; VEGF signaling
; retinal inflammation
; mdx3Cv mouse
; vascular permeability
1. Introduction
The dystrophin gene (dmd), with 79 exons and over 2.3 million base pairs, is the largest in the human genome, and is highly conserved across the animal kingdom [1,2]. The use of various internal promoters allows for the synthesis of five different isoforms of the dystrophin protein [1]; the longest, Dp427, measures 427 kDa and is ubiquitous in muscle cells, while the shorter dystrophin isoforms (Dp260, Dp140, Dp116, Dp71, Dp40) are expressed in a wide range of cell types throughout the gastrointestinal, genitourinary, and central nervous systems [3,4]. Mutations in this gene resulting in dysfunction of Dp427 lead to Duchenne Muscular Dystrophy (DMD) [1,2,3,5], a fatal X-linked recessive disease affecting approximately 1 in 3,600 live male births [6,7]. The disease was first described in 1868 and has primarily been characterized by its muscular phenotype, marked by progressive myocyte destruction and ultimate cardiomyopathy [8].
As emerging therapies prolong life and slow the progression of muscular degeneration in these patients [9], it becomes increasingly apparent that dysfunction of shorter dystrophin isoforms in extramuscular tissues also provokes clinically relevant disease. Investigation of the role of these smaller isoforms in the central nervous system has led to the recognition of the neuropsychologic phenotype of this disease [4,10,11,12,13], with DMD patients having a greater prevalence of intellectual disability, specific learning disorders, autism spectrum disorder, ADHD (Attention-Deficit/Hyperactivity Disorder), and epilepsy than the general population [4,11,12,13].
Additionally, some patients with DMD develop a proliferative retinopathy that presents clinically with a profound decrease in visual acuity [14,15,16,17], similar to proliferative diabetic retinopathy (DR) or wet age-related macular degeneration (AMD). Colorblindness is also more prevalent in DMD patients than the general population, and up to ⅔ of the cohort of patients deficient in the shorter dystrophins may be red-green colorblind [18]. Moreover, electroretinography (ERG) shows abnormalities even in DMD patients with clinically normal vision [19,20,21]. Under scotopic (low light) conditions, these patients exhibit decreased b-wave amplitude and increased implicit time (latency from stimulus), attributed to an impairment in the neurotransmission between cone / rod cells and second order neurons like bipolar cells in the retina [19,22,23].
Isoforms Dp427, Dp260, and Dp71 have all been identified in the retina, with the two-former localizing in the outer plexiform layer, and Dp71 in the inner limiting membrane [24]. Alongside utrophin, β-dystroglycan, δ- and γ-sarcoglycans, and α1-syntrophin, Dp71 and Dp260 form part of the dystrophin associated protein complex (DAPC) in the retina, which anchors to the cytoskeleton of Müller glial cells and allows for the assembly of postsynaptic ion channels (e.g. Kir4.1K+, AQP4) and lipid rafts [23,25]. Under normal conditions, Müller glial cells regulate the integrity of the blood-retinal barrier (BRB) and prevent pathogenic angiogenesis by secreting antiproliferative factors such as pigment epithelium-derived factor (PEDF) [26,27,28]. Conversely, injury to this glia promotes BRB breakdown and neovascularization, the mechanism of which is understood to involve K+ ion transport and the secretion of proinflammatory mediators as vascular endothelial growth factor (VEGF) and intracellular adhesion molecule 1 (ICAM-1) [26,29,30,31].
In contrast to the Dp71-null model, the mdx3Cv mouse used in the present study exhibits modest expression of several dystrophin isoforms, including Dp71 [32]. Specifically, this low level Dp71 expression is sufficient to support the retention of DAPC component β-dystroglycan in the internal limiting membrane and surrounding retinal vasculature of mdx3Cv mice, despite its absence in the outer plexiform layer [32,33]. In this study, we evaluate whether the mdx3Cv mouse model exhibits a form of retinal inflammation similar to that described in Dp71-null mice. We address this question by first quantifying Dp71 expression in mdx3Cv compared with wild-type (WT) littermates, and then examining how this modest protein expression influence the retinal expression of VEGF, FLT-1, KDR, ICAM-1, ALOX5AP, and AQP4. We additionally explore how this limited Dp71 expression affects vascular permeability using a range of quantitative metrics. Finally, we discuss how these findings may inform future research directions and therapeutic strategies.
2. Materials and Methods
2.1. Animal Model
This study was conducted using 15-week-old mdx3Cv mice. This animal model carries a point mutation in intron 65 introduced using chemical mutagen N-ethyl-N-nitrosurea (ENU), and exhibits reduced expression of both the full-length Dp427 isoform and shorter dystrophin isoforms, including Dp71 [34]. Littermates lacking this mutation were used as wild-type controls for all analyses.
2.2. mRNA Quantification using RT PCR
RNA was extracted from the retinas of mdx3Cv and wildtype littermate mice using the RNeasy Mini Kits (Qiagen), and isolated using the QIAcube automated purification system (Qiagen). The Agilent 2100 Bioanalyzer (Agilent Technologies) was used to measure the concentration and integrity of the extracted RNA. Next, 5 ug of total RNA was primed with oligo-dT and cDNA was synthesized using reverse transcriptase (Super Script II) at 42°C for one hour. Real time quantitative polymerase chain reaction (RT-qPCR) was performed using TaqMan probe-based assays (Applied Biosystems), with the following probes: VEGFA (Mm01281449_m1), KDR (Mm01222421_m1), FLT-1 (Mm00438980_m1), ALOX5AP (Mm00802100_m1), AQP4 (Mm00802131_m1), ICAM-1 (Mm00516023_m1) and ACTB (Mm00607939_s1). Ct values of RT-qPCR were analyzed per manufacturer’s instructions using the ΔΔCt method. Each sample was measured in triplicate, outliers were eliminated, and values were normalized with respect to β-actin quantities.
2.3. Protein Quantification and Analysis
Protein was extracted from the retinas homogenized in RIPA lysis buffer (Sigma), and total protein concentration in the lysates was measured with the BCA protein assay kit (Thermo Scientific). A specific ELISA kit (R&D Systems) was used to determine the quantity of VEGF-A protein in the lysate, per manufacturer’s instructions. Each sample was measured in triplicate and outliers were removed. The Infinite M200 spectrophotometer (TECAN) was used to read the plates.
A standard Western Blot was performed as previously described. Protein extracts from the retinas were deposited in Tris-Acetate NuPAGE 4-12% gels (Life technologies, France) and electrotransferred to polyvinylidine difluoride (PVDF) membranes per manufacturer protocols. The PVDF membranes were blocked in phosphate-buffered saline (PBS) with 0.1% Tween 20, 1% bovine serum albumin (BSA), and 5% dry milk (Bio-Rad, Hertz, United Kingdom) for 1 hour at room temperature, then incubated overnight with primary anti-dystrophin H4 antibody (pan-specific for all dystrophin isoforms) in the same blocking solution. The PVDF membranes were washed and incubated with goat-anti-Mouse horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch Laboratories). The ECL Western PLUS system was used to read the blots (GE Healthcare, Germany).
2.4. Quantifying Leukocytes Adherent to Retinal Vasculature
The mice were first perfused with PBS via intracardiac route (through the left ventricle) to remove non-adherent leukocytes. Next, perfusion with fluorescein isothiocyanate (FITC) conjugated with concanavalin A lectin (ConA; Vector laboratory) was used to label the retinal vasculature and adherent leukocytes. PBS was then reperfused to rinse out excess ConA. The retinas were mounted on slides and photographed with a fluorescent microscope (Zeiss Axiovert 200M), and adherent leukocytes were counted. Secondary immunolabeling with an anti-CD45 antibody (BD PharMingen) was performed to confirm that counted cells were leukocytes.
2.5. Measuring Retinal Vascular Permeability
Anesthetized mice received 45 mg/kg of Evans Blue dye (Sigma-Aldrich, Germany) via intrapenile injection. Blood samples were collected 3 hours after dye injection, then mice were perfused with 10 mL citrate buffer (0.05M, pH 3.5) at 37°C through left ventricle intracardiac access. Both eyes were then enucleated, and the retinas were carefully dissected. Retinas were dried in a Speed-Vac for 5h, weighed, and Evans Blue was extracted by incubation in 100 µL of formamide at 70°C for 18h. The supernatant of each retina was filtered using 30k omega membrane nanosep tubes at 14,000 g and 4°C for 2 hours. Each blood sample was centrifuged at 18,000 g for 15 minutes. Absorbance at 620nm (maximal for Evans Blue) and 740 nm (minimal for Evans Blue) was measured for both types of supernatants using the TECAN infinite M1000 spectrophotometer. The difference in absorbances A620-A740 was used for calculations, and concentrations of Evans Blue were calculated using a standard curve. BRB permeability was expressed in µL of Evans Blue per g of dry retina per hour according to the following equation:
2.6. Statistical Analysis
All data are expressed as mean + standard error of the mean. Statistical analysis was performed using the Prism 4 program (GraphPad, San Diego, CA) and the Mann-Whitney test. Statistical significance was defined as p values < 0.05.
3. Results
3.1. Dp71 expression in mdx3Cv mouse model
Using Western blot–based protein quantification, we observed a marked reduction in Dp71 expression in the retinae of mdx3Cv mice compared with wild-type (WT) littermates. Densitometric analysis, normalized to β-actin, revealed that mdx3Cv mice expressed approximately 4% of WT Dp71 protein levels, representing a statistically significant decrease (Figure 1). Despite this substantial reduction, Dp71 protein remained detectable in all mdx3Cv retinal samples, indicating residual expression rather than complete loss of the isoform. This low-level persistence of Dp71 is consistent with previous characterizations of the mdx3Cv model and distinguishes it from Dp71-null mice, in which the isoform is entirely absent. The presence of residual Dp71 expression in mdx3Cv retinas provides a biologically relevant context for assessing whether minimal dystrophin restoration is sufficient to preserve retinal homeostasis in subsequent analyses.
3.2. VEGF Pathway Gene Expression Remains Intact
Having quantified Dp71 expression in mdx3Cv mice, we next investigated the impact of this partial dystrophin expression on retinal inflammatory signaling. Whereas we previously demonstrated that complete loss of Dp71 in the Dp71-null mouse model induces retinal inflammation through increased activation of the VEGF pathway [35], the mdx3Cv model exhibited a distinct profile. Specifically, mdx3Cv mice showed a significant reduction in VEGF mRNA expression compared with wild-type (WT) littermates (Figure 2A). Despite this transcriptional decrease, VEGF protein levels were preserved in mdx3Cv retinas, with no significant difference relative to WT controls (Figure 2B).
We then assessed the expression of VEGF receptors using real-time quantitative PCR. Consistent with the preserved VEGF protein levels, the mRNA expression of both FLT-1 (VEGFR-1) and KDR (VEGFR-2) did not differ significantly between mdx3Cv mice and WT littermates (Figure 2C and D).
3.3. Cell-mediated inflammation is unchanged from WT in mdx3Cv
We previously reported that complete loss of Dp71 in the Dp71-null mouse model leads to a significant increase in ICAM-1 mRNA and protein expression, accompanied by a marked rise in leukocytes adherent to the retinal vasculature [35]. In contrast, the present study found no significant difference in ICAM-1 mRNA levels between mdx3Cv mice and wild-type (WT) littermates (Figure 3A). Consistent with this finding, visualization of leukocytes adherent to the retinal vasculature following intracardiac perfusion with fluorescein isothiocyanate (FITC)–conjugated concanavalin A showed no significant difference in leukocyte adhesion between mdx3Cv mice and WT controls (Figure 3B).
3.4. Preserved Permeability of the Blood-Retinal Barrier to Inflammatory Markers
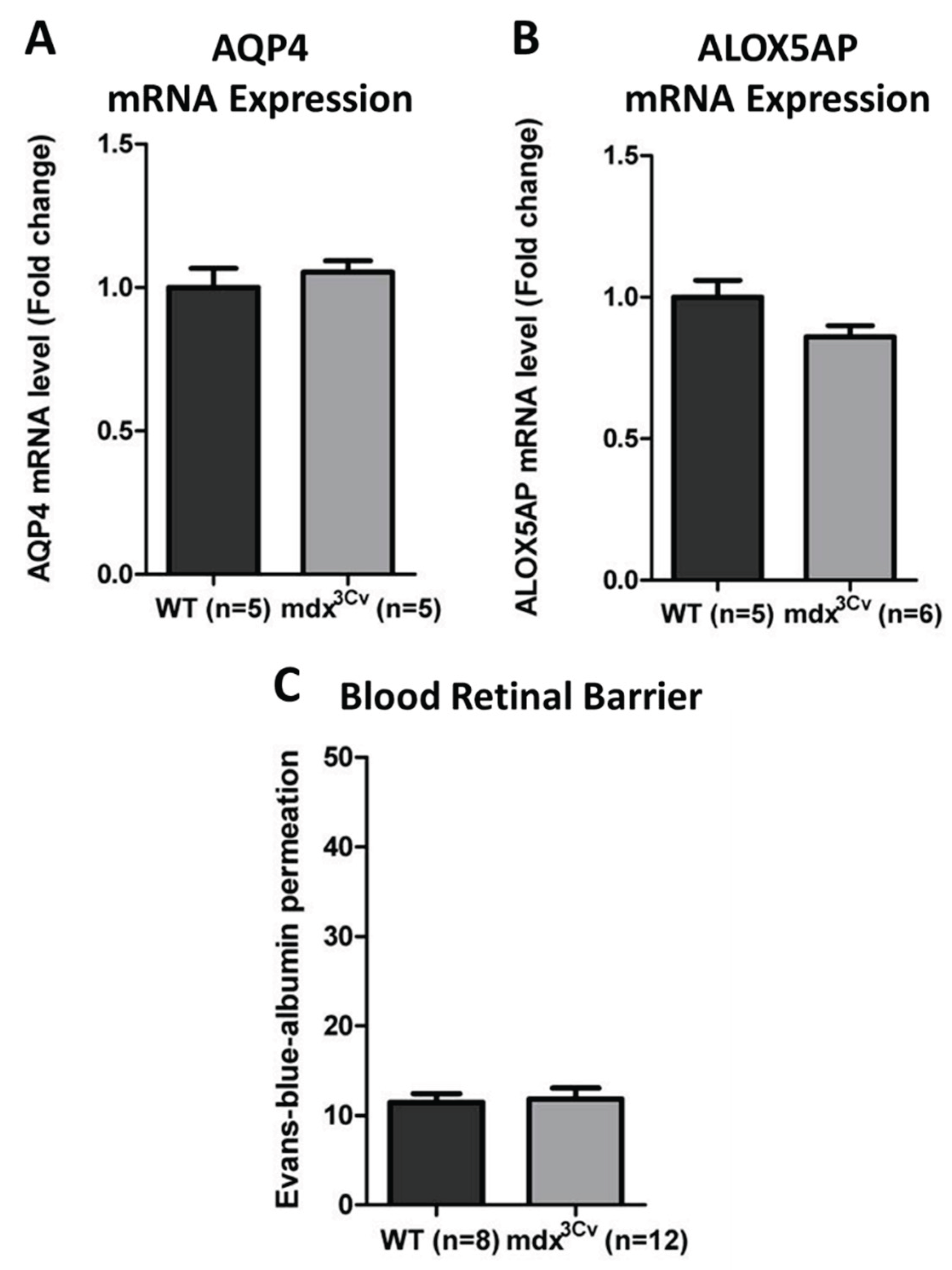
In contrast with the increased permeability of the BRB previously reported in the Dp71-null mouse model [35], this study found no significant difference in BRB permeability between mdx3Cv mice and WT littermates, across three different metrics. Firstly, the mRNA expression levels of the transmembrane channel Aquaporin 4 (AQP4) were unchanged between mdx3Cv and WT littermate (Figure 4A). Additionally, there was no significant difference in the mRNA expression of arachidonate 5-lipoxygenase activating protein (ALOX5AP) between cohorts (Figure 4B). Finally, the BRB permeability to albumin was equivalent in both experimental groups (Figure 4C).
4. Discussion
The Dp71-null mouse carries a complete knock-out of the Dp71 dystrophin isoform, which is associated with increased VEGF secretion by Müller glial cells, elevated AQP4 mRNA expression despite reduced protein abundance, increased ICAM-1 expression, increased adherent leukocytes, and increased retinal capillary permeability [35]. Dysregulated expression of these inflammatory markers is associated with retinal disease in both DMD patients, and other patient populations. Analyzing the expression of these same markers in the mdx3Cv mouse enables us to evaluate the extent to which a modest, 4% level of Dp71 expression is sufficient to reduce retinal inflammation.
Vascular endothelial growth factors (VEGF) are a family of growth factors that promote the proliferation, differentiation, survival, and increased permeability of vascular endothelial cells, with VEGF-A being the principal mediator of these effects [36]. VEGF receptors are class V tyrosine kinases expressed predominantly in vascular and lymphatic endothelial cells [37]. These receptors dimerize upon ligand binding and activate a complex signal transduction pathway that promotes inflammatory cell migration (primarily VEGFR-1 or FLT-1), vascular endothelial proliferation (primarily VEGFR-2 or KDR), and lymphangiogenesis in inflammatory and malignant states (primarily VEGFR-3) [37,38]. This phospholipase C gamma pathway directly activates the transcription of genes that result in inflammation and angiogenesis, while the indirect branch of this pathway serves to deactivate pro-apoptotic factors [38]. The VEGFR-2 mediated response is also responsible for mediating capillary permeability, and is considered the most relevant receptor for both physiologic and pathologic vascular proliferation. Increased signaling through the VEGF pathway is a major component of the understood mechanism behind DR, wet AMD, and the similarly-appearing retinopathy of Duchenne muscular dystrophy [35,36,37,39].
Intracellular adhesion molecule 1 (ICAM-1) is an immunoglobulin superfamily transmembrane protein with basal expression in vascular, epithelial, and immune tissues, and plays a key role in mediating leukocyte adhesion and extravasation in response to proinflammatory stimuli [40]. ICAM-1 intersects with the VEGF pathway because activation of VEGFRs generates the intermediate messenger diacylglycerol (DAG), which subsequently activates the NF-kB pathway, resulting in increased expression of several proinflammatory genes, including ICAM-1 [38]. The dysregulation, and particularly the amplification of ICAM-1 expression is associated with a plethora of disease states, including inflammatory myopathies, atherosclerosis, and cancer [41,42]. Specifically within the retina, ICAM-1 overexpression has been implicated in the pathogenesis of other proliferative retinopathies, and this upregulation may have a dose-dependent effect on the severity of phenotype and the risk of developing severe complications [36,39,43,44,45,46]. At the tissue level, increased ICAM-1 expression leads to enhanced leukocyte infiltration and greater BRB permeability to Evans blue dye and horseradish peroxidase [41,47].
AQP4 is the predominant water channel in the retina, where it is selectively expressed by Müller glial cells and astrocytes, with strong enrichment at perivascular endfeet and the inner limiting membrane (ILM) [48]. Through its strategic localization, AQP4 plays a critical role in retinal water homeostasis, potassium buffering, and maintenance of BRB integrity. Dysregulation of AQP4 expression or localization has been implicated in several retinal pathologies, most notably diabetic retinopathy (DR), where increased oxidative stress and inflammatory signaling induce AQP4 upregulation and contribute to retinal edema and vascular leakage [49]. In the Dp71-null mouse, loss of the dystrophin-associated protein complex (DAPC) from Müller glial endfeet leads to mislocalization and dysregulation of AQP4, despite paradoxically increased mRNA levels, ultimately resulting in impaired water handling and increased BRB permeability [35]. In contrast, our data show that AQP4 mRNA expression remains unchanged in the mdx3Cv retina relative to wild-type controls. This preservation of AQP4 expression is consistent with the retention of key DAPC components, including β-dystroglycan, at the ILM in this model [32,50], and correlates with the absence of detectable BRB leakage. These findings suggest that even minimal residual expression of Dp71 is sufficient to stabilize Müller glial polarity and preserve normal AQP4 regulation. Importantly, these findings support a threshold model in which complete loss of Dp71 is required to trigger the AQP4-mediated permeability phenotype, whereas low-level dystrophin expression is sufficient to maintain retinal fluid homeostasis.
ALOX5AP is an essential cofactor for 5-lipoxygenase (5-LOX) and plays a central role in leukotriene biosynthesis, generating potent lipid mediators that promote leukocyte recruitment, vascular permeability, and amplification of inflammatory cascades [51]. Elevated leukotriene signaling has been implicated in a variety of inflammatory and vascular diseases, including atherosclerosis, asthma, and proliferative retinopathies [51]. Within the retina, leukotriene pathway activation contributes to endothelial dysfunction and BRB breakdown, acting in concert with VEGF and ICAM-1–mediated mechanisms to promote leukostasis and vascular leakage [52,53,54]. Increased ALOX5AP expression has been reported in experimental models of retinal inflammation and correlates with disease severity in DR and ischemic retinopathies [51]. In the present study, ALOX5AP mRNA expression was not significantly altered in mdx3Cv mice compared with wild-type littermates. This finding further supports the absence of an active inflammatory milieu in the mdx3Cv retina and aligns with the preserved ICAM-1 expression, reduced leukocyte adhesion, and intact BRB permeability observed in this model. Collectively, these results indicate that low-level Dp71 expression prevents activation of both protein-based (VEGF/ICAM-1) and lipid-mediated (leukotriene) inflammatory pathways, underscoring the central role of Müller glial dystrophin in coordinating retinal immune homeostasis.
An intriguing observation in this study is the reduction of VEGF mRNA expression in mdx3Cv retinas despite preservation of VEGF protein levels comparable to wild-type controls. Such discordance between transcript and protein abundance has been reported in multiple biological systems and may reflect post-transcriptional, translational, or protein stability–based regulatory mechanisms [55,56]. VEGF expression is tightly controlled at multiple levels, including mRNA stability, alternative splicing, microRNA-mediated repression, and protein sequestration within the extracellular matrix [55,57]. In the retina, VEGF is a key mediator of pathological angiogenesis and vascular permeability in diseases such as diabetic retinopathy and age-related macular degeneration, where its dysregulated upregulation leads to BRB breakdown [58]. Notably, VEGF protein can be stored in association with heparan sulfate proteoglycans and released in response to physiological cues, allowing protein levels to remain stable even when transcriptional output fluctuates [56]. Additionally, Müller glial cells and endothelial cells may differentially regulate VEGF transcription and secretion, resulting in cell-type–specific buffering of protein availability. One possible explanation for our findings is that partial Dp71 expression restores sufficient DAPC integrity to normalize VEGF protein trafficking, stabilization, or secretion, thereby preventing pathological VEGF accumulation despite reduced transcription. Alternatively, compensatory translational upregulation or reduced protein turnover may contribute to maintained VEGF protein levels. Importantly, the preservation of VEGF receptor expression (FLT-1 and KDR) and the absence of downstream inflammatory or permeability changes suggest that VEGF signaling remains functionally regulated in mdx3Cv mice, rather than pathologically activated. Further investigation will be required to elucidate the precise mechanisms underlying this transcription–translation uncoupling, including assessment of VEGF isoform distribution, microRNA regulation, and cell-specific expression patterns within the retina.
Our data demonstrates that the mdx3Cv mouse experiences regularization in proinflammatory markers to wild type levels, despite exhibiting only low expression of Dp71. These findings suggest that even partial dystrophin restoration may be sufficient to prevent to prevent retinal pathology in DMD patients, thereby helping to define a realistic therapeutic target. While dystrophin protein restoration remains a challenge, recent preclinical work shows steps in the right direction. Dp71ab-containing plasmid transfection successfully promoted human myocyte proliferation, and shows promise in terms of application to other cell lineages [59]. Exon skipping using tricycloDNA-small antisense oligonucleotides has achieved anywhere from 5-15% restoration of Dp427 (a different isoform of dystrophin) throughout brain tissues in animal models, resulting in improvement in the neurologic phenotype observed in these animals [60]. A similar approach achieved a dose-dependent restoration across all dystrophin isoforms in the brain, between 10-30% of wild type levels [61]. Most relevantly, the usage of viral vector and exon skipping techniques has restored 2-14% of Dp71 in various brain tissues [62]. Further study is needed to evaluate the efficacy of these protein restoration strategies within the retina.
While these strategies have yet to reach practical clinical applicability, our growing understanding of the pathophysiology of Duchenne’s retinopathy (and its similarity to diabetic and age-related retinopathies) opens the door to promising alternative therapeutic approaches. Firstly, as VEGF pathway overactivation is unequivocally implicated in this disease process, an extensive class of anti-VEGF therapies becomes relevant, including pegaptanib (VEGF antagonist), ranibizumab (anti-VEGF antibody fragment), aflibercept (VEGF decoy receptor), and bevacizumab (anti-VEGF monoclonal antibody) [36]. These anti-VEGF intravitreal injections represent the currently standard of care for DR and wet AMD, but response varies significantly from patient to patient and the need for frequent reinjection limits practicality and patient comfort [37]. Inhibition of HMGB1, an upstream modulator to VEGF, has shown therapeutic efficacy in early DR, and several herbal derivatives are known HMGB1 inhibitors with anti-inflammatory properties that have demonstrated improved outcomes in other animal models of inflammatory disease [36,63,64]. Moreover, anti-inflammatory drugs of various mechanisms (e.g. intravitreal glucocorticoids, topical nonsteroidal anti-inflammatory drugs (NSAIDs), antioxidants) have been similarly effective for DR and AMD, and may be more practical and comfortable for patients than VEGF inhibition [65]. Finally, laser coagulation remains a destructive but highly effective intervention for controlling retinal neovascularization [38]. Perhaps the most relevant recommendation is to add routine ophthalmologic evaluation into the standard of care for DMD patients.
5. Conclusions
In summary, this study demonstrates that minimal residual expression of the dystrophin isoform Dp71—approximately 4% of wild-type levels—is sufficient to preserve retinal vascular integrity and prevent inflammatory pathology in the mdx3Cv mouse model. Unlike the Dp71-null phenotype, mdx3Cv mice exhibit preserved VEGF signaling balance, normal ICAM-1 and ALOX5AP expression, absence of leukostasis, intact Aquaporin-4 regulation, and maintained BRB integrity.
These findings provide important mechanistic insight into the role of dystrophin in Müller glial cells and support a threshold model of dystrophin function in the retina. Critically, they suggest that complete dystrophin restoration may not be necessary to achieve meaningful therapeutic benefit, and that partial correction—within the range currently attainable by exon-skipping, viral gene delivery, or antisense approaches—may be sufficient to prevent retinal disease in Duchenne muscular dystrophy.
Beyond DMD, these results further reinforce the conceptual parallels between Duchenne retinopathy, diabetic retinopathy, and age-related macular degeneration, highlighting shared inflammatory and vascular mechanisms. Routine ophthalmologic screening should therefore be considered an integral component of DMD clinical care, particularly as life expectancy increases. Future studies should focus on functional retinal outcomes, including electroretinography, and on evaluating whether targeted dystrophin restoration strategies can reverse established retinal pathology.
Author Contributions
Conceptualization and Methodology, O.V. and R.T. and A.R.; Funding acquisition and resources, B.E.M and A.G. and R.T. and A.R.; Design of experiments, A.R. and O.V.; Experimental work: B.E.M; Data analysis, B.E.M. and O.V.; Original draft preparation, J.K. and O.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Association Française contre les Myopathies (AFM) through a research project grant (no. 14853) awarded to A.R.; by Sanofi, which supported the PhD training of Brahim El Mathari through a two-year secondment while maintaining his salary; by the Institut National de la Santé et de la Recherche Médicale (INSERM); by the French State program “Investissements d’Avenir” managed by the Agence Nationale de la Recherche (LIFESENSES: ANR-10-LABX-65); and by the ECOS-Nord program (no. M11S02).
Institutional Review Board Statement
All experiments received authorization from the French Research Ministry and followed the guidelines of our mouse facility in compliance with the European Directive 2010/63/EU, in agreement with the ARRIVE guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
The primary data for this study are available from the authors upon request.
Acknowledgments
The authors wish to thank Stéphane Fouquet from the Plateforme d’imagerie of the Institut de la Vision and the skillful technical assistance of Marie Darche. We dedicate this work to the memory of Pr Ramin Tadayoni, whose scientific insight and dedication significantly advanced research in ophthalmology.
Conflicts of Interest
AFM, ECOS Nord, Labex, ANR DysTher are public or non-commercial organizations. Sanofi supported this work by allowing the secondment of Brahim El Mathari to the laboratory for a two-year period to carry out his PhD while continuing to pay his salary. Sanofi had no role in the study design, data collection, analysis, interpretation, or decision to publish. Authors also disclose the following conflict of interests with commercial entities: Ramin Tadayoni is a board member of and consultant for Alcon, Switzerland; Novartis, Switzerland; Allergan, USA; Bausch and Lomb, USA; Alimera, USA; Bayer, Germany; FCIZeiss, France; Thrombogenics, Belgium; Roche, Switzerland; Genentech, USA; Zeiss, Germany. He has received lecture fees from Alcon, USA; Bausch and Lomb, USA; Novartis, Switzerland; Allergan, USA; Bayer, Germany; Alimera, USA, Zeiss, Germany, and meeting expenses from Novartis, Switzerland; Alcon, Switzerland; Allergan, USA; Bausch and Lomb, USA; Bayer, Germany; Alimera, USA.
Abbreviations
The following abbreviations are used in this manuscript:
| ALOX5AP | Arachidonate 5-Lipoxygenase Activating Protein |
| AQP4 | Aquaporin-4 |
| AMD | Age-related Macular Degeneration |
| BRB | Blood–Retinal Barrier |
| DAPC | Dystrophin-Associated Protein Complex |
| DMD | Duchenne Muscular Dystrophy |
| Dp | Dystrophin Protein Isoform |
| DR | Diabetic Retinopathy |
| ERG | Electroretinography |
| FLT-1 | Fms-Like Tyrosine kinase 1 (VEGFR-1) |
| ICAM-1 | Intracellular Adhesion Molecule 1 |
| ILM | Inner Limiting Membrane |
| KDR | Kinase Insert Domain Receptor (VEGFR-2) |
| RT-qPCR | Reverse Transcription quantitative Polymerase Chain Reaction |
| SEM | Standard Error of the Mean |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| WT | Wild Type |
References
- Ehmsen, J.; Poon, E.; Davies, K. The Dystrophin-Associated Protein Complex. J Cell Sci 2002, 115, 2801–2803. [Google Scholar] [CrossRef]
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The Human Dystrophin Gene Requires 16 Hours to Be Transcribed and Is Cotranscriptionally Spliced. Nat Genet 1995, 9, 184–190. [Google Scholar] [CrossRef]
- Tokarz, S.A.; Duncan, N.M.; Rash, S.M.; Sadeghi, A.; Dewan, A.K.; Pillers, D.A. Redefinition of Dystrophin Isoform Distribution in Mouse Tissue by RT-PCR Implies Role in Nonmuscle Manifestations of Duchenne Muscular Dystrophy. Mol Genet Metab 1998, 65, 272–281. [Google Scholar] [CrossRef]
- Vaillend, C.; Aoki, Y.; Mercuri, E.; Hendriksen, J.; Tetorou, K.; Goyenvalle, A.; Muntoni, F. Duchenne Muscular Dystrophy: Recent Insights in Brain Related Comorbidities. Nat Commun 2025, 16, 1298. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M. Dystrophin, Its Interactions with Other Proteins, and Implications for Muscular Dystrophy. Biochim Biophys Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, V.; Pavlakis, S. Duchenne Muscular Dystrophy. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J Rare Dis 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- GB, D.D.B.; Duchenne De Boulogne GB De La Paralysie Musculaire Pseudo-Hypertrophique Ou Paralysie Myo-Sclérosique : Extrait Des Archives Générales de Médecine; Editeur des archives générales de médecine, 1868.
- Markati, T.; Oskoui, M.; Farrar, M.A.; Duong, T.; Goemans, N.; Servais, L. Emerging Therapies for Duchenne Muscular Dystrophy. Lancet Neurol 2022, 21, 814–829. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Jiménez-López, E.; Saz-Lara, A.; Martínez-García, I.; Martínez-Vizcaíno, V. Global Prevalence of Intellectual Developmental Disorder in Dystrophinopathies: A Systematic Review and Meta-Analysis. Dev Med Child Neurol 2023, 65, 734–744. [Google Scholar] [CrossRef]
- Perumal, A.R.; Rajeswaran, J.; Nalini, A. Neuropsychological Profile of Duchenne Muscular Dystrophy. Appl Neuropsychol Child 2015, 4, 49–57. [Google Scholar] [CrossRef]
- Ramani, P.K.; Fawcett, K.; Guntrum, D.; Samuel, H.; Ciafaloni, E.; Veerapandiyan, A. Epilepsy Characteristics in Duchenne and Becker Muscular Dystrophies. Child Neurol Open 2023, 10, 2329048X231159484. [Google Scholar] [CrossRef]
- Parravicini, S.; Quaranta, C.A.; Dainesi, M.I.; Berardinelli, A. The Hidden Face of Duchenne (Neuro)Muscular Dystrophy. Preliminary Evidence of Social Cognition Impairment as a Feature of the Neuropsychological Phenotype of DMD. Front Psychol 2024, 15, 1504174. [Google Scholar] [CrossRef]
- Fagan, X.J.; Levy, J.; Al-Qureshi, S.; Harper, C.A. Proliferative Retinopathy in Duchenne Muscular Dystrophy and Its Response to Bevacizumab. Clin Exp Ophthalmol 2012, 40, 906–907. [Google Scholar] [CrossRef]
- Park, S.H.; Jo, Y.J.; Lee, J.J.; Park, S.W.; Lee, J.E. Proliferative Retinopathy Developed in a Duchenne Muscular Dystrophy Patient with Normal Cardiac Function. J Retin 2019, 4, 36–39. [Google Scholar] [CrossRef]
- Louie, K.; Apte, R.S.; Mori, K.; Gehlbach, P. Severe Proliferative Retinopathy in a Patient with Advanced Muscular Dystrophy. Br J Ophthalmol 2004, 88, 1604–1605. [Google Scholar] [CrossRef]
- Hahn, P.; Lin, P.; Fekrat, S. Ultra-Widefield Imaging of Duchenne Muscular Dystrophy-Associated Proliferative Retinal Vasculopathy Improved with Panretinal Laser Photocoagulation Alone. Ophthalmic Surg Lasers Imaging Retina 2013, 44, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.F.; Oliveira, A.G.F.; Feitosa-Santana, C.; Zatz, M.; Ventura, D.F. Red-Green Color Vision Impairment in Duchenne Muscular Dystrophy. Am J Hum Genet 2007, 80, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Kröger, S. The Neurobiology of Duchenne Muscular Dystrophy: Learning Lessons from Muscle? Trends Neurosci 2000, 23, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Jägle, H.; Theodorou, M.; Moore, A.T.; Muntoni, F.; Thompson, D.A. Ocular and Neurodevelopmental Features of Duchenne Muscular Dystrophy: A Signature of Dystrophin Function in the Central Nervous System. Eur J Hum Genet 2016, 24, 562–568. [Google Scholar] [CrossRef]
- Pillers, D.A.; Bulman, D.E.; Weleber, R.G.; Sigesmund, D.A.; Musarella, M.A.; Powell, B.R.; Murphey, W.H.; Westall, C.; Panton, C.; Becker, L.E. Dystrophin Expression in the Human Retina Is Required for Normal Function as Defined by Electroretinography. Nat Genet 1993, 4, 82–86. [Google Scholar] [CrossRef]
- Jiang, X.; Mahroo, O.A. Negative Electroretinograms: Genetic and Acquired Causes, Diagnostic Approaches and Physiological Insights. Eye (Lond) 2021, 35, 2419–2437. [Google Scholar] [CrossRef]
- Barboni, M.T.S.; Joachimsthaler, A.; Roux, M.J.; Nagy, Z.Z.; Ventura, D.F.; Rendon, A.; Kremers, J.; Vaillend, C. Retinal Dystrophins and the Retinopathy of Duchenne Muscular Dystrophy. Prog Retin Eye Res 2023, 95, 101137. [Google Scholar] [CrossRef]
- Howard, P.L.; Dally, G.Y.; Wong, M.H.; Ho, A.; Weleber, R.G.; Pillers, D.A.; Ray, P.N. Localization of Dystrophin Isoform Dp71 to the Inner Limiting Membrane of the Retina Suggests a Unique Functional Contribution of Dp71 in the Retina. Hum Mol Genet 1998, 7, 1385–1391. [Google Scholar] [CrossRef]
- Satz, J.S.; Philp, A.R.; Nguyen, H.; Kusano, H.; Lee, J.; Turk, R.; Riker, M.J.; Hernández, J.; Weiss, R.M.; Anderson, M.G.; et al. Visual Impairment in the Absence of Dystroglycan. J Neurosci 2009, 29, 13136–13146. [Google Scholar] [CrossRef]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Müllercell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J Neurosci 2012, 32, 15715–15727. [Google Scholar] [CrossRef]
- Tout, S.; Chan-Ling, T.; Holländer, H.; Stone, J. The Role of Müller Cells in the Formation of the Blood-Retinal Barrier. Neuroscience 1993, 55, 291–301. [Google Scholar] [CrossRef]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller Cell Changes in Human Diabetic Retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Paulson, O.B.; Newman, E.A. Does the Release of Potassium from Astrocyte Endfeet Regulate Cerebral Blood Flow? Science 1987, 237, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of Astrocyte-Vascular Coupling and the Blood-Brain Barrier by Invading Glioma Cells. Nat Commun 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.D.; Distler, A.M.; Kern, T.S.; Mieyal, J.J. Glutaredoxin Regulates Autocrine and Paracrine Proinflammatory Responses in Retinal Glial (Muller) Cells. J Biol Chem 2009, 284, 4760–4766. [Google Scholar] [CrossRef]
- Dalloz, C.; Claudepierre, T.; Rodius, F.; Mornet, D.; Sahel, J.; Rendon, A. Differential Distribution of the Members of the Dystrophin Glycoprotein Complex in Mouse Retina: Effect of the Mdx(3Cv) Mutation. Mol Cell Neurosci 2001, 17, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Koulen, P.; Blake, D.J.; Kröger, S. Dystrophin and Beta-Dystroglycan in Photoreceptor Terminals from Normal and mdx3Cv Mouse Retinae. Eur J Neurosci 1999, 11, 2121–2133. [Google Scholar] [CrossRef]
- Cox, G.A.; Phelps, S.F.; Chapman, V.M.; Chamberlain, J.S. New Mdx Mutation Disrupts Expression of Muscle and Nonmuscle Isoforms of Dystrophin. Nat Genet 1993, 4, 87–93. [Google Scholar] [CrossRef]
- El Mathari, B.; Sene, A.; Charles-Messance, H.; Vacca, O.; Guillonneau, X.; Grepin, C.; Sennlaub, F.; Sahel, J.-A.; Rendon, A.; Tadayoni, R. Dystrophin Dp71 Gene Deletion Induces Retinal Vascular Inflammation and Capillary Degeneration. Hum Mol Genet 2015, 24, 3939–3947. [Google Scholar] [CrossRef]
- Wei, L.; Sun, X.; Fan, C.; Li, R.; Zhou, S.; Yu, H. The Pathophysiological Mechanisms Underlying Diabetic Retinopathy. Front Cell Dev Biol 2022, 10, 963615. [Google Scholar] [CrossRef] [PubMed]
- Callan, A.; Heckman, J.; Tah, G.; Lopez, S.; Valdez, L.; Tsin, A. VEGF in Diabetic Retinopathy and Age-Related Macular Degeneration. International Journal of Molecular Sciences 2025, 26, 4992. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kotwani, A. Exploring the Various Aspects of the Pathological Role of Vascular Endothelial Growth Factor (VEGF) in Diabetic Retinopathy. Pharmacol Res 2015, 99, 137–148. [Google Scholar] [CrossRef]
- Khalfaoui, T.; Lizard, G.; Beltaief, O.; Colin, D.; Ben Hamida, J.; Errais, K.; Ammous, I.; Zbiba, W.; Tounsi, L.; Zhioua, R.; et al. Immunohistochemical Analysis of Cellular Adhesion Molecules (ICAM-1, VCAM-1) and VEGF in Fibrovascular Membranes of Patients with Proliferative Diabetic Retinopathy: Preliminary Study. Pathol Biol (Paris) 2009, 57, 513–517. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A Master Regulator of Cellular Responses in Inflammation, Injury Resolution, and Tumorigenesis. J Leukoc Biol 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Shimizu, J.; Kawai, M.; Kanazawa, I. [논문]Sarcolemmal Coexpression of Intercellular Adhesion Molecule-1 (ICAM-1) and HLA-DR in Inflammatory Myopathy. Neuropathology : official journal of the Japanese Society of Neuropathology 1994, 14, 149–157. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in Inflammation and in the Regulation of Vascular Permeability. Am J Physiol Heart Circ Physiol 2008, 295, H926–H927. [Google Scholar] [CrossRef]
- Yao, Y.; Du, J.; Li, R.; Zhao, L.; Luo, N.; Zhai, J.Y.; Long, L. Association between ICAM-1 Level and Diabetic Retinopathy: A Review and Meta-Analysis. Postgrad Med J 2019, 95, 162–168. [Google Scholar] [CrossRef]
- Barile, G.R.; Chang, S.S.; Park, L.S.; Reppucci, V.S.; Schiff, W.M.; Schmidt, A.M. Soluble Cellular Adhesion Molecules in Proliferative Vitreoretinopathy and Proliferative Diabetic Retinopathy. Curr Eye Res 1999, 19, 219–227. [Google Scholar] [CrossRef]
- Limb, G.; Chignell, A. Vitreous Levels of Intercellular Adhesion Molecule 1 (ICAM-1) as a Risk Indicator of Proliferative Vitreoretinopathy. Br J Ophthalmol 1999, 83, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Limb, G.A.; Webster, L.; Soomro, H.; Janikoun, S.; Shilling, J. Platelet Expression of Tumour Necrosis Factor-Alpha (TNF-α), TNF Receptors and Intercellular Adhesion Molecule-1 (ICAM-1) in Patients with Proliferative Diabetic Retinopathy. Clin Exp Immunol 1999, 118, 213–218. [Google Scholar] [CrossRef]
- Ma, N.; Hunt, N.H.; Madigan, M.C.; Chan-Ling, T. Correlation between Enhanced Vascular Permeability, up-Regulation of Cellular Adhesion Molecules and Monocyte Adhesion to the Endothelium in the Retina during the Development of Fatal Murine Cerebral Malaria. Am J Pathol 1996, 149, 1745–1762. [Google Scholar]
- Nagelhus, E.A.; Ottersen, O.P. Physiological Roles of Aquaporin-4 in Brain. Physiol Rev 2013, 93, 1543–1562. [Google Scholar] [CrossRef]
- Li, J.; Patil, R.V.; Verkman, A.S. Mildly Abnormal Retinal Function in Transgenic Mice without Müller Cell Aquaporin-4 Water Channels. Invest Ophthalmol Vis Sci 2002, 43, 573–579. [Google Scholar]
- Claudepierre, T.; Dalloz, C.; Mornet, D.; Matsumura, K.; Sahel, J.; Rendon, A. Characterization of the Intermolecular Associations of the Dystrophin-Associated Glycoprotein Complex in Retinal Müller Glial Cells. J Cell Sci 2000, 113 Pt 19, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Henderson, W.R. Leukotrienes. N Engl J Med 2007, 357, 1841–1854. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog Retin Eye Res 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic Retinopathy. N Engl J Med 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.F.; Schlingemann, R.O. Molecular Basis of the Inner Blood-Retinal Barrier and Its Breakdown in Diabetic Macular Edema and Other Pathological Conditions. Prog Retin Eye Res 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat Rev Drug Discov 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF Receptor Signalling - in Control of Vascular Function. Nat Rev Mol Cell Biol 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Ocular Neovascularization. J Mol Med (Berl) 2013, 91, 311–321. [Google Scholar] [CrossRef]
- Farea, M.; Rani, A.Q.M.; Maeta, K.; Nishio, H.; Matsuo, M. Dystrophin Dp71ab Is Monoclonally Expressed in Human Satellite Cells and Enhances Proliferation of Myoblast Cells. Sci Rep 2020, 10, 17123. [Google Scholar] [CrossRef]
- Karuppasamy, M.; Alexander, M.S. Restoration of Brain Dystrophin Using Tricyclo-DNA ASOs Restores Neurobehavioral Deficits in DMD Mice. Mol Ther Nucleic Acids 2023, 32, 841–842. [Google Scholar] [CrossRef]
- Zarrouki, F.; Relizani, K.; Bizot, F.; Tensorer, T.; Garcia, L.; Vaillend, C.; Goyenvalle, A. Partial Restoration of Brain Dystrophin and Behavioral Deficits by Exon Skipping in the Muscular Dystrophy X-Linked (Mdx) Mouse. Ann Neurol 2022, 92, 213–229. [Google Scholar] [CrossRef]
- Vacca, O.; Zarrouki, F.; Izabelle, C.; Belmaati Cherkaoui, M.; Rendon, A.; Dalkara, D.; Vaillend, C. AAV-Mediated Restoration of Dystrophin-Dp71 in the Brain of Dp71-Null Mice: Molecular, Cellular and Behavioral Outcomes. Cells 2024, 13, 718. [Google Scholar] [CrossRef]
- Feng, L.; Liang, L.; Zhang, S.; Yang, J.; Yue, Y.; Zhang, X. HMGB1 Downregulation in Retinal Pigment Epithelial Cells Protects against Diabetic Retinopathy through the Autophagy-Lysosome Pathway. Autophagy 2022, 18, 320–339. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; He, L.; Long, W.; Zhou, Q.; Zhu, S.; Wang, P.; Fan, S.; Wang, H. Novel Mechanisms of Herbal Therapies for Inhibiting HMGB1 Secretion or Action. Evid Based Complement Alternat Med 2015, 2015, 456305. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Morescalchi, F.; Cancarini, A.; Russo, A.; Rezzola, S.; Costagliola, C. Diabetic Retinopathy, a Vascular and Inflammatory Disease: Therapeutic Implications. Diabetes Metab 2019, 45, 517–527. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Reduced but detectable Dp71 expression in the retina of mdx3Cv mice. (A) Representative Western blot showing Dp71 and β-actin expression in retinal extracts from mdx3Cv and WT littermate mice. Approximate molecular weights (kDa) are indicated on the left; (B) Quantification of Dp71 protein levels normalized to β-actin, expressed relative to WT controls. Protein abundance was quantified by densitometric analysis using Fiji ImageJ software. Data are presented as mean ± SEM (n = 4 per group). Statistical significance is assessed relative to WT littermate controls (* p < 0.05).
Figure 1.
Reduced but detectable Dp71 expression in the retina of mdx3Cv mice. (A) Representative Western blot showing Dp71 and β-actin expression in retinal extracts from mdx3Cv and WT littermate mice. Approximate molecular weights (kDa) are indicated on the left; (B) Quantification of Dp71 protein levels normalized to β-actin, expressed relative to WT controls. Protein abundance was quantified by densitometric analysis using Fiji ImageJ software. Data are presented as mean ± SEM (n = 4 per group). Statistical significance is assessed relative to WT littermate controls (* p < 0.05).
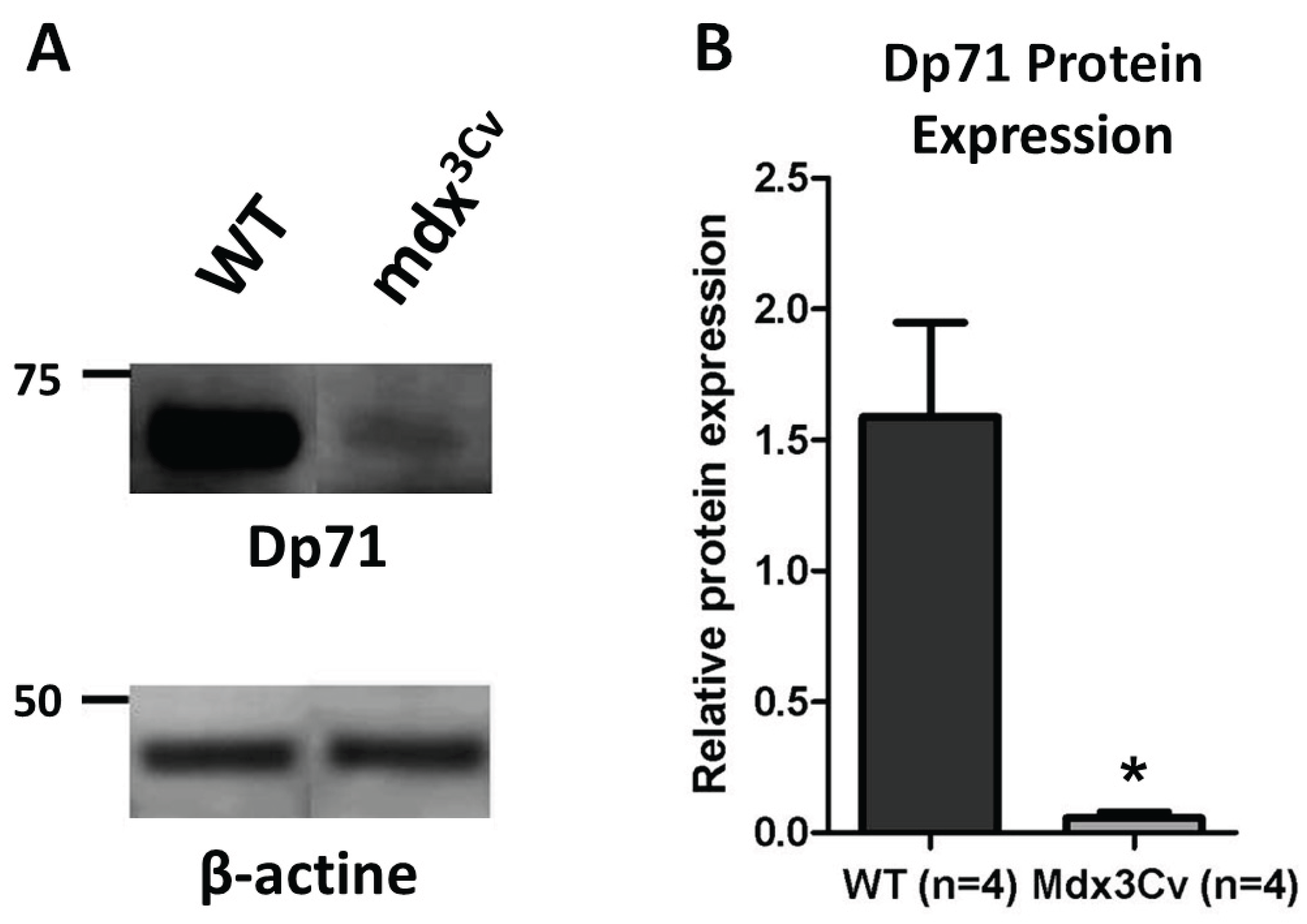
Figure 2.
Preservation of VEGF signaling components in the retina of mdx3Cv mice. (A) VEGF mRNA expression in mdx3Cv versus WT littermates, showing a significant reduction in mdx3Cv mice; (B) VEGF protein expression in mdx3Cv and WT littermates, demonstrating preserved protein levels; (C) FLT-1 (VEGFR-1) mRNA expression in mdx3Cv versus WT littermates; (D) KDR (VEGFR-2) mRNA expression in mdx3Cv versus WT littermates. mRNA expression was quantified by RT-qPCR and normalized to endogenous β-actin. VEGF protein levels were quantified by ELISA and normalized to total protein. Data are presented as mean ± SEM (n = 6 per group). Statistical significance is assessed relative to WT littermate controls (** p < 0.01).
Figure 2.
Preservation of VEGF signaling components in the retina of mdx3Cv mice. (A) VEGF mRNA expression in mdx3Cv versus WT littermates, showing a significant reduction in mdx3Cv mice; (B) VEGF protein expression in mdx3Cv and WT littermates, demonstrating preserved protein levels; (C) FLT-1 (VEGFR-1) mRNA expression in mdx3Cv versus WT littermates; (D) KDR (VEGFR-2) mRNA expression in mdx3Cv versus WT littermates. mRNA expression was quantified by RT-qPCR and normalized to endogenous β-actin. VEGF protein levels were quantified by ELISA and normalized to total protein. Data are presented as mean ± SEM (n = 6 per group). Statistical significance is assessed relative to WT littermate controls (** p < 0.01).
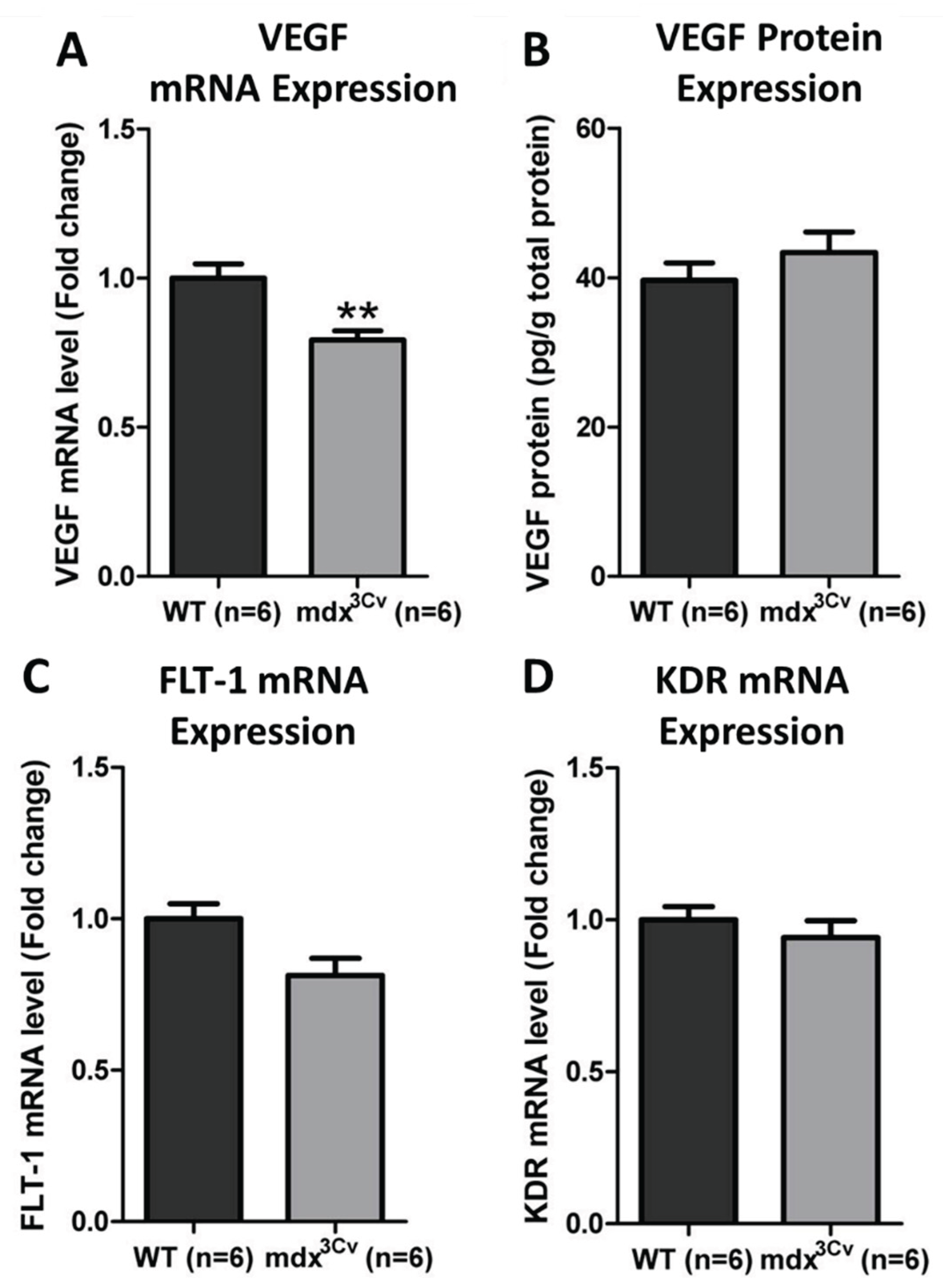
Figure 3.
Inflammatory markers in the retina of mdx3Cv versus WT littermate mice. (A) ICAM-1 mRNA expression in mdx3Cv (n = 5) and WT (n = 6) littermates, quantified by RT-qPCR and normalized to endogenous β-actin. (B) Number of leukocytes adherent to the retinal vascular lumen in mdx3Cv (n = 8) and WT (n = 9) littermates, quantified by direct visualization following FITC–conjugated concanavalin A perfusion. Data are presented as mean ± SEM.
Figure 3.
Inflammatory markers in the retina of mdx3Cv versus WT littermate mice. (A) ICAM-1 mRNA expression in mdx3Cv (n = 5) and WT (n = 6) littermates, quantified by RT-qPCR and normalized to endogenous β-actin. (B) Number of leukocytes adherent to the retinal vascular lumen in mdx3Cv (n = 8) and WT (n = 9) littermates, quantified by direct visualization following FITC–conjugated concanavalin A perfusion. Data are presented as mean ± SEM.
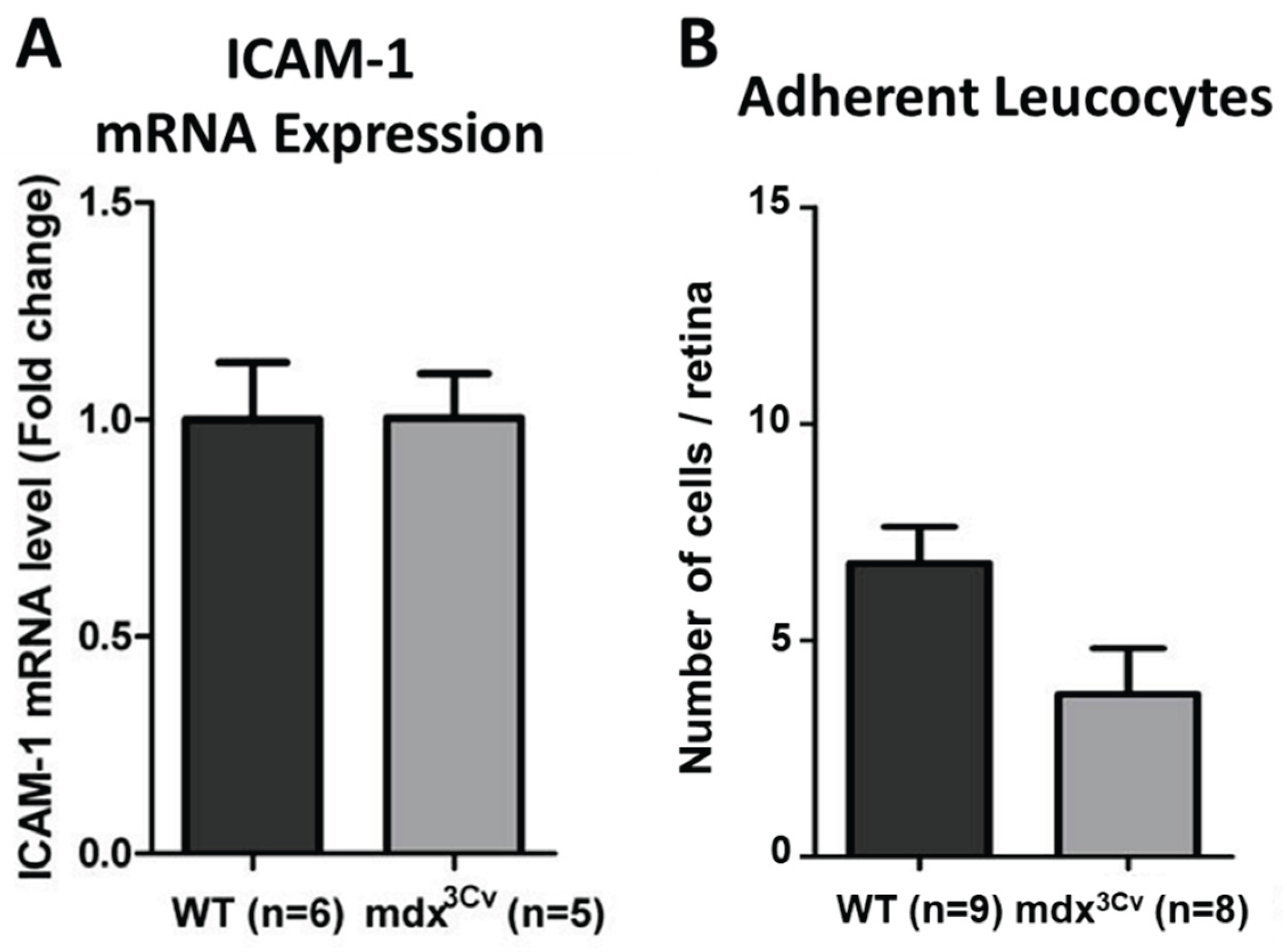
Figure 4.
Preservation of BRB integrity and inflammatory markers in mdx3Cv mice. (A) Quantity of AQP4 mRNA in mdx3Cv (n = 5) versus wild-type (WT) littermates (n = 5); (B) Quantity of ALOX5AP mRNA in mdx3Cv (n = 6) versus WT littermates (n = 5); (C) BRB permeability to albumin in mdx3Cv (n = 12) versus WT littermates (n = 8), quantified using Evans Blue–bound albumin extravasation. mRNA expression was quantified by RT-qPCR and normalized to endogenous β-actin. Data are presented as mean ± SEM. Statistical significance is assessed relative to WT littermate controls (* p < 0.05, ** p < 0.01).
Figure 4.
Preservation of BRB integrity and inflammatory markers in mdx3Cv mice. (A) Quantity of AQP4 mRNA in mdx3Cv (n = 5) versus wild-type (WT) littermates (n = 5); (B) Quantity of ALOX5AP mRNA in mdx3Cv (n = 6) versus WT littermates (n = 5); (C) BRB permeability to albumin in mdx3Cv (n = 12) versus WT littermates (n = 8), quantified using Evans Blue–bound albumin extravasation. mRNA expression was quantified by RT-qPCR and normalized to endogenous β-actin. Data are presented as mean ± SEM. Statistical significance is assessed relative to WT littermate controls (* p < 0.05, ** p < 0.01).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



