
Submitted:
14 January 2026
Posted:
20 January 2026
You are already at the latest version
Abstract
Staphylococcus aureus remains to be one of the leading causes of global mortality. The most common class of antibiotics used to treat S. aureus infections are Next-Generation β-lactams (NGBs), as they are highly efficacious and have low adverse effects. NGB resistance in S. aureus is classically attributed to Penicillin-Binding Protein-2a (PBP2a), but previous studies from our group have also implicated altered expression of Penicillin-Binding Protein-4 (PBP4) with high-level NGB resistance. PBP4 is the only low-molecular mass (LMM) PBP present in S. aureus; it is also the only known LMM PBP with transpeptidase activity, giving it the unique ability to bring about peptidoglycan cross-linking. In this article, we review some of the recent findings from our group, which reveal that mutations associated with PBP4 lead to altered protein expression and NGB resistance in both MSSA and MRSA backgrounds. We discuss the clinical relevance of PBP4-associated mutations, particularly in Methicillin Resistant Lacking mec (MRLM) isolates, as well as the combined effect of altered expression of PBP4 and GdpP. Finally, this review summarizes the potential role played by PBP4 in S. aureus virulence. Together, we highlight the increasing relevance of PBP4 as a mediator of NGB resistance and discuss its potential to be an important factor during infection diagnosis and therapy.
Keywords:
Staphylococcus aureus
1. Introduction
Staphylococcus aureus, the Gram- positive opportunistic pathogen, can cause skin and soft tissue infections, bacteremia, osteomyelitis and sepsis (among other infections) in humans. The bacterium is also well-known for causing persistent or relapsing infections due to its ability to evade the action of antibiotic treatment through various mechanisms of drug resistance or tolerance and plays a significant impact on global health and economy. In 2019 alone, S. aureus infections resulted in over a million deaths in individuals older than 15 years. It was also the leading bacterial cause of death in 135 countries and resulted in a YLL burden (years of life lost due to mortality) of 34.3 million. S. aureus was also the leading cause of mortality associated with bloodstream infections, resulting in 299,000 global deaths in 2019 alone [1,2]. Treatment of S. aureus infections is largely dependent on antibiotics that target vital cellular processes and inhibit bacterial growth and survival. However, the bacterium is infamous for developing resistance towards virtually all available antibiotics and rendering them ineffective [3]. Development of vaccines targeting S. aureus has been consistently unsuccessful due to a plethora of virulence factors, high genetic variability among strains and a range of diseases caused by the bacteria [4]. The highly pathogenic nature of the bacteria, combined with its ability to evade antibiotic treatment and the lack of an efficient vaccine makes S. aureus a successful modern pathogen.
Today, the most common classes of antibiotic drugs used to treat S. aureus infections include β-lactams, glycopeptides, cyclic lipopeptides and oxazolidones [5]. β-lactams, in particular, are deemed largely successful due to their high efficacy, superior tissue distribution and low adverse effects compared to the other drug classes and account for 65% of all prescribed injectable antibiotics in the United States [6]. They inhibit the formation of the bacterial cell wall by targeting penicillin-binding proteins (PBPs), enzymes that perform peptidoglycan cross-linking, thereby causing cell lysis and death [7]. The bacterial cell wall, a continuous, outer layer of the cell, is an important and essential feature of the cell as it provides shape, structure and protection from external stressors [8] (Figure 1). Almost all bacterial species possess a cell wall, with the exception of mycoplasma and L-form bacteria [9]. The cell wall not only serves as a physical barrier from the environment, but also plays important roles in cell metabolism, cell division, pathogenesis and antibiotic resistance, which makes it an attractive anti-bacterial target [10]. It is a complex structure mainly consisting of sugars, lipids and protein molecules, the composition of which varies among Gram-positive and Gram-negative bacteria. Gram-positive cell wall is comprised of a thick layer of peptidoglycan along with teichoic acid polymers and surface proteins [11] (Figure 1). Gram-negative bacteria also possess a peptidoglycan, but it is relatively thin and is present in between the inner and the outer cell membrane, the latter of which has a lipopolysaccharide layer (LPS) anchored to it.
2. Peptidoglycan
The structure, shape and integrity of the cell is attributed to the peptidoglycan (PG), a continuous polymer spanning the cell surface. The PG is a scaffolding structure composed of glycan polymers interconnected through peptide chains [11]. In Gram-positive bacteria, the PG is present in multiple layers and is approximately 20-40 nm thick. Gram-negative bacteria have one to few layers of PG and range from 1.5 to 10 nm in thickness [12]. The PG is dynamic is nature; the thickness of the structure varies based on the stage of cell division, cell growth and pathogenesis [13]. The single repeating unit of the PG, a monomer, is comprised of a disaccharide, a stem peptide and a bridge peptide. The disaccharide is composed of N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) and the stem peptide is associated with NAM at the D-lactyl site [14,15]. This stem peptide consists of five amino acids: L-alanine-D-iso-glutamine-L-lysine-D-alanine-D-alanine [8] (Figure 2). The NAM-NAG disaccharide and L-alanine, the first amino acid of the stem peptide, are largely conserved in most bacterial species. In certain Gram-positive bacteria such as Bacillus subtilis [16] and Listeria monocytogenes and a majority of Gram-negative species, the 3rd position of the stem peptide consists of meso-diaminopimelic acid (m-DAP) instead of L-lysine [17] (Figure 3). Notably, the D-alanine-D-alanine at the stem peptide terminus are highly conserved, a feature exploited by PG targeting antibiotics such as glycopeptides [18] (Figure 4). The 3rd position of the stem peptide comprises of a bridge peptide, the composition of which varies among bacterial species [15]. In S. aureus, the bridge peptide comprises of five units of glycine and is referred to as the penta-glycine bridge [8]. The bridge peptide in Streptococcus pneumoniae [19] and Enterococcus faecalis [20] are relatively shorter and are composed of L-alanine-L-serine and L-alanine-L-alanine, respectively (Figure 3). It is unknown whether the length of the bridge peptide has an effect on PG integrity and elasticity. Linear polymerization of NAM-NAG units is formed via a glycosidic bond among adjacent disaccharides, whereas cross-linking occurs via formation of an amide bond between the terminal amino acid of the bridge peptide and D-alanine, the 4th amino acid of the stem peptide of adjacent disaccharides [8]. The average length of the glycan chains and the extent of PG cross-linking vary among species. In S. aureus, the average PG chain length is typically shorter, comprised of around six monomers [21]. However, 80-90% of the stem peptides in S. aureus are cross-linked [22]. Cross-linking of stem peptides has been demonstrated to be essential for peptidoglycan integrity and cell survival in S. aureus [23]. B. subtilis have longer PG chains of around 50-250 monomers but only 40% of the stem peptides are cross-linked [16]. E. coli strains have a large variety in PG chain lengths, ranging from 1-30 monomers, but only 30% of them are typically cross-linked [24]. Length of PG chains or the degree of cross-linking, however, are likely to not have an effect on PG thickness or cell shape [17]. Linear polymerization and cross-linking of adjacent PG disaccharides are both performed by a group of enzymes collectively called Penicillin-Binding Proteins (PBPs) [25].
Peptidoglycan Synthesis
Synthesis of the PG begins intracellularly with the assembly of a membrane-anchored monomer, which is subsequently flipped across the membrane and incorporated into the growing PG chain in the extracellular space [26] (Figure 4). In S. aureus, NAG is first synthesized from glucosamine-6-phosphate by the enzymes GlmM and GlmU in the cytosol, which is then converted to NAM by the enzymes MurA and MurB [27]. The Mur ligases, MurC, MurD, MurE and MurF further catalyze the sequential addition of L-alanine, D-iso-glutamic acid, L-lysine and D-alanyl-D-alanine to NAM respectively, completing the formation of the stem peptide [28]. The terminal D-alanyl-D-alanine dipeptide is synthesized from two molecules of L-alanine via the activity of the enzymes encoded by alr1 and ddl. Alr1 (alanine racemase) converts L-alanine to D-alanine, while Ddl (alanine ligase) forms the D-alanyl-D-alanine dipeptide [29]. The assembly of the PG monomer is an attractive antibiotic target, as causing its inhibition leads to stalling of the PG synthesis process at the cytosolic stage. The antibiotic fosfomycin [30] inhibits the activity of MurA by selectively binding to the active site of the enzyme, causing the enzyme to be inactivated. Similarly, D-cycloserine inhibits Alr1 and Ddl, preventing the formation of the D-alanyl-D-alanine dipeptide [29]. The assembled precursor is linked to the lipid carrier undecaprenyl-diphosphate by the membrane translocase MraY, yielding lipid I. The formation of lipid I is inhibited by the antibiotic tunicamycin that inhibits MraY [31]. MurG catalyzes the generation of lipid II by adding NAG to lipid I precursor. The addition of the penta-glycine bridge to lipid II is catalyzed by the enzymes FemX/A/B, completing the synthesis of a PG monomer [23]. Finally, D-iso-glutamic acid, the 2nd amino acid of the stem peptide is converted to D-iso-glutamine by amidation performed by MurT/GatD [32,33]. The lipid II assembly is then flipped towards the outer leaflet of the membrane via the flippase, MurJ [34]. In the extracellular region, lipid-II linked monomers are polymerized and cross-linked by the transglycosylation and transpeptidation activities of PBPs [35]. The nascent, lipid-linked PG chains are released from the lipid carrier by the SagB-SpdC complex and are subsequently incorporated into the pre-existing PG [36], while the lipid carrier is recycled to continue to transport more PG monomers [37]. The processing of lipid II is an important target of various antibiotics. β-lactams such as nafcillin or oxacillin inhibit the transpeptidation activity of the proteins, while moenomycin [38] or flavomycin inhibit transglycosylation. Glycopeptides such as vancomycin or teicoplanin specifically bind to the D-alanyl-D-alanine stem peptide of lipid II-linked PG to inhibit PBP-mediated transpeptidation [18]. Lysostaphin, a metalloprotease bacteriocin synthesized by S. simulans specifically cleaves the bridge peptide between the third and fourth glycine residues [39]. Bacitracin, a bacteriocin produced by Bacillus licheniformis, blocks the recycling of lipid carriers [40].
The PG synthesis and cross-linking machinery is concentrated at the septum, the division site of replicating bacteria while the mature, pre-existing PG is dispersed towards the peripheral region of the cell. The process of PG synthesis is highly regulated, and works in co-ordination with enzymes of the cell division machinery such as FtsZ, GpsB or DivIC and cell wall hydrolases such as Atl, SagB, LytN or LytM [41]. PBPs and cell wall hydrolases work in concert to maintain a balance in PG synthesis and hydrolysis, ensuring cell viability.
3. Penicillin-Binding Proteins (PBPs)
The substrates of penicillin were found to be a group of enzymes that perform cross-linking of bacterial PG. These enzymes, due to their affinity towards penicillin, were termed as Penicillin-Binding Proteins (PBPs) [42]. PBPs are membrane-anchored proteins found in all bacterial species. They are multi-domain structures, each with an associated function [43]. They have the ability to polymerize and/or cross-link the PG monomers through transglycosylation [38] (formation of a glycosidic bond between adjacent NAM-NAG disaccharides) and transpeptidation [7] (formation of the cross-bridge between adjacent PG stem peptides) activities, respectively (Figure 5). Certain PBPs also have carboxypeptidation (hydrolysis of terminal D-alanine of stem peptide) or endopeptidation (hydrolysis of bridge peptide) activities. PBPs, in addition to maintaining the shape and thickness of the PG, also control the extent of PG cross-linking. Each PBP has one or two of the above-mentioned functions due to which bacteria often possess multiple PBPs.
3.1. Classification of PBPs
PBPs are present in both Gram-positive and Gram-negative bacteria and are primarily categorized based on their molecular mass. The molecular mass of each PBP also dictates its nomenclature, as they are annotated based on their pattern of separation following SDS-PAGE [35]. PBPs are broadly categorized into three classes based on their domains and associated functions.
Class A PBPs are high molecular-mass (HMM) PBPs and contain both, the glycosyltransferase domain and the transpeptidase domain. They are bi-functional and bring about polymerization of the glycan chain, as well as cross-linking of stem peptide. The glycosyltransferase domain associated with class A PBPs is present at the N-terminal region (Figure 6). The general structure of this domain is conserved among bacterial species, with five highly conserved amino acid motifs surrounding two catalytically active glutamic acid residues [38]. It is separated from the transpeptidase domain by a spacer to allow both transglycosylation and transpeptidation activities simultaneously. Transglycosylation activity is exclusive to class A PBPs, making them essential PBPs.
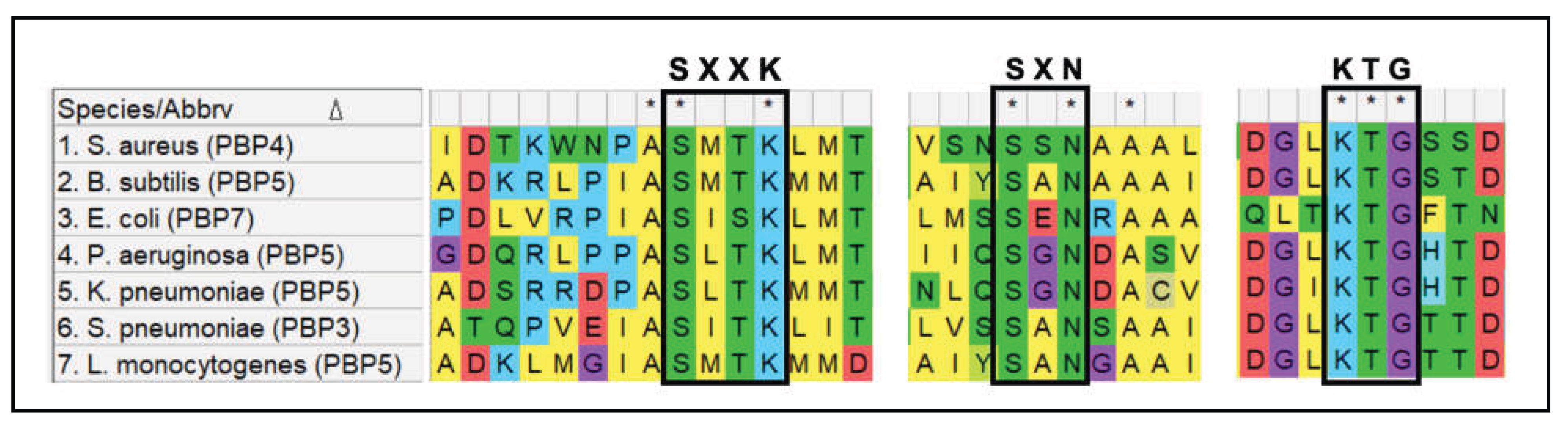
Class B PBPs are mono-functional HMM proteins, with only transpeptidation activity. The transpeptidase domain of class A and class B PBPs are structurally similar, with the consistent characteristic of a penicillin-binding (PB) domain. The PB domain includes the transpeptidation active site (SxxK motif) and its associated conserved motifs (SxN and the KTG motifs) [44,45]. Transpeptidation begins with the binding of PBPs to the PG stem peptide, where the active site serine of the SxxK motif attacks the terminal D-alanyl-D-alanine peptide bond, forms an acyl-enzyme complex with the stem peptide and concomitantly releases the terminal D-alanine. The C-terminal peptidyl moiety is subsequently transferred onto the terminal glycine of the bridge peptide of the adjacent PG monomer, completing the bridge formation [43]. The SxN motif is associated with substrate binding and deacetylation following transpeptidation, whereas lysine of the KTG motif has been associated with activation of the SxN motif [46]. β-lactams, being structurally analogous to the D-alanyl-D-alanine dipeptide, also bind to the SxxK motif. The antibiotics first form a non-covalent Michaelis complex with the PB domain region, following which the active site serine launches a nucleophile attack on the β-lactam ring and forms an acyl-enzyme intermediate, inhibiting further transpeptidase activity [47].
Class C PBPs are low molecular-mass (LMM) proteins that have PG hydrolase activity, either in the form of carboxypeptidation or endopeptidation. Carboxypeptidation and endopeptidation activities bring about a decrease in the extent of PG cross-linking due to which they play vital roles in PG remodeling and recycling [43]. LMM PBPs also have a PB-domain associated with them, including the transpeptidase domain with the conserved SxxK, SxN and KTG motifs. This allows LMM PBPs to recognize and bind the D-alanyl-D-alanine stem peptide. However, while they can attack the stem peptide and form an acyl-enzyme complex, they are unable to accept a recipient amino acid to transfer the peptidyl moiety onto, thus resulting in a shorter stem peptide and a released D-alanine. PBP4 of S. aureus is an exception, as it is the only known LMM PBP with transpeptidation activity.
3.2. PBPs of S. aureus
S. aureus naturally contains four PBPs- PBP1 through PBP4. The PBP with the highest molecular mass, PBP1, is a class B, mono-functional transpeptidase. PBP1 deletion leads to abnormal septal formation, suggesting that PBP1, in addition to cross-linking the PG, is also part of the cell division machinery [48,49]. In addition to the transpeptidase domain, the C-terminal of PBP1 contains two PASTA (penicillin-binding protein and serine/threonine kinase-associated) domains. The PASTA domains of PBP1 are essential for cell growth and protein functionality, and are shown to bind to uncross-linked PG [49]. PBP2 belongs to class A, and thus has the ability to perform both transglycosylation and transpeptidation activities. It is the only class A PBP in S. aureus, due to which it is essential for bacterial growth and survival [50]. PBP3, a class B PBP, is associated with cross-linking of the peripheral PG, in addition to its role in cell elongation during cell division via its association with proteins involved in cell division, such as RodA [51,52]. PBP4 is the only low molecular mass (LMM) PBP of S. aureus. LMM PBPs belong to class C and are characterized by their carboxypeptidase and/or endopeptidase activities. However, PBP4 of S. aureus has transpeptidation activity in addition to carboxypeptidation, making it the only known LMM PBP to be able to perform PG cross-linking [53,54,55].
3.3. Penicillin Binding Protein-2A (PBP2a)
Resistance to penicillin, the first β-lactam used for treatment of bacterial infections was detected soon after the drug was introduced clinically to treat infections [56]. Penicillin resistance is attributed to a penicillinase, now known as β-lactamase. β-lactamase, encoded by the blaZ gene, cleaves and inactivates the β-lactam ring in penicillin and renders it inactive [57]. Subsequent years witnessed the development of methicillin, a β-lactam used to treat infections caused by strains resistant to penicillin. In 1961, strains resistant to methicillin were detected, marking the surge of methicillin resistant S. aureus (MRSA). β-lactam resistance in MRSA is attributed to PBP2a, a horizontally-acquired PBP with low affinity towards the drug that results in decreased drug binding and resistance to methicillin [58]. PBP2a is encoded by mecA, a gene part of the mobile genetic element Staphylococcal Cassette Chromosome mec (SCC-mec) and confers broad-spectrum resistance to subsequently introduced types of β-lactams such as cephalosporins and carbapenems. While the three transpeptidase motifs of PBP2a remain conserved, kinetic studies revealed that the acyl-enzyme complex formation between PBP2a and β-lactams occurs insufficiently, allowing β-lactam resistance to occur [59]. In 2019, MRSA led to over 100,000 deaths associated with drug resistance, demonstrating its significance as a pathogen responsible for a high health and economic burden, over 50 years after it was initially identified [2].
Strains lacking mecA (or, mecC [60], a homolog of mecA) are classified as MSSA (methicillin susceptible S. aureus), with narrow-spectrum resistance to early generation β-lactams such as penicillin. MSSA strains are treated with new generation β-lactams (NGBs) such as nafcillin or oxacillin [61]. Infections caused by MRSA, identified by the presence of mecA are treated with advanced generation cephalosporins such as ceftaroline (or ceftabiprole) that are designed to specifically bind and inhibit PBP2a [62]. Recent years have seen the detection of ceftaroline-resistant MRSA owing to structural alterations in PBP2a that lead to decreased affinity towards ceftaroline [63]. Ceftaroline resistance warrants the use of antibiotics belonging to other drug classes such as vancomyin, or daptomycin. However, these alternative classes of antibiotics, albeit effective, have significant adverse effects such as nephrotoxicity and pose the risk of infection relapse [64].
3.4. PBP4
PBP4 of S. aureus has transpeptidation activity in addition to carboxypeptidation, making it the only known LMM PBP to be able to perform PG cross-linking [53,54,55]. Studies revealed the ability of PBP4 to add amino acids onto not only PG polymers, but also Lipid I and Lipid II, suggesting its propensity towards transpeptidation over carboxypeptidation [65]. Multiple sequence alignment of PBP4 from S. aureus and LMM PBPs from other bacterial species such as B. subtilis (PBP5), E. coli (PBP7), Listeria monocytogenes (PBP5), P. aeruginosa (PBP5), K. pneumoniae (PBP5) and S. pneumoniae (PBP3) suggest that the active site and associated motifs were highly conserved in PBP4 (Figure 7). This indicated that the transpeptidation activity of PBP4 was not attributed to any potential alterations in the active site. Thus, the exact mechanism by which PBP4 of S. aureus is able to perform PG cross-linking remains unknown. The conditions that dictate S. aureus PBP4 to perform either, carboxypeptidase or transpeptidase activity are also unknown. However, deletion or inactivation of PBP4 from S. aureus led to significant decrease in cell wall cross-linking, suggesting its propensity towards transpeptidation [66].
4. Non-Classical Mechanisms of New Generation β-Lactams (NGB) Resistance in S. aureus
4.1. Pbp4-Associated Mutations Lead to β-Lactam Resistance in S. aureus
In addition to MSSA or MRSA isolates, recent years have seen a rise in detection of mecA-negative, β-lactam resistant isolates of S. aureus, potentially suggesting the presence of certain non-classical NGB resistance mechanisms [67,68,69]. This increasing detection of mecA-negative, β-lactam resistant S. aureus, combined with the ability of S. aureus to rapidly acquire novel resistance mechanisms prompted studies that explored potentially novel, non-classical mechanisms of β-lactam resistance that were independent of mecA. To that end, our early studies involved passaging of S. aureus in increasing concentrations of the advanced generation β-lactam, ceftobiprole until the bacteria gained the ability to survive in presence of the drug [70,71]. The parental, susceptible strain used for the study was COLnex, the mecA, blaZ devoid variant of COL, a prominent, archaic MRSA strain. Serial passaging of COLnex in sub-inhibitory concentrations of ceftobiprole for 21 days resulted in the generation of CRB, a resistant variant, with an MIC of 128 mg/L. The MIC for CRB was 512-fold more than that for COLnex, which has an MIC of less than 0.25 mg/L for ceftobiprole. In order to identify the potential genetic alterations that led to the resistant phenotype in CRB, whole genome sequencing was carried out with COLnex and CRB. CRB contained mutations associated with two specific genes- gdpP and pbp4. gdpP contained a missense mutation in its coding region. pbp4 had two missense mutations (E183A and F241R) associated with it, in addition to a 36 bp duplication upstream of the start codon, in the regulatory region [72]. Of the two candidate genes, PBP4 is one of the targets of β-lactam binding and has been previously associated with low-level β-lactam resistance [73]. More importantly, the deletion of pbp4 in CRB led to significant decrease in MIC to ceftobiprole, such that it was comparable with the parent strain, COLnex, suggesting that PBP4 was likely responsible for the resistant phenotype seen in CRB [74].
Following the indication that alterations associated with PBP4 expression potentially led to resistance to β-lactams, studies were carried out to determine the specific phenotypes associated with the pbp4 mutations in CRB. The regulatory-site mutation and the missense mutations were each individually introduced into COLnex, resulting in COLnex Ppbp4*(CRB) and COLnex pbp4**(CRB), respectively. Since the Ppbp4*(CRB) mutation was located in the regulatory region, its effect on protein expression was first analyzed. Bocillin-Fl binding assay revealed significantly increased levels of PBP4 in COLnex Ppbp4*(CRB) compared to COLnex wild-type, suggesting that the mutation led to alterations in regulation of PBP4 expression [74]. Since PBP4 is involved in PG cross-linking, the PG from COLnex wild-type and the COLnex Ppbp4*(CRB) mutant were purified and the muropeptide composition was analyzed by HPLC. COLnex Ppbp4*(CRB) had significantly enhanced PG cross-linking compared to COLnex wild-type, as witnessed by an increase in the amount of long-chained oligomeric units, or the “hump” and a corresponding decrease in short-chained muropeptides such as monomers and dimers. COLnex Ppbp4*(CRB) was also highly resistant to β-lactams, with a reported 16-fold increase in MIC for nafcillin, 128-fold increase in MIC for ceftaroline, and 4-fold increase in MIC for ceftobiprole when compared to COLnex [74]. Taken together, this study revealed that the pbp4 regulatory-site mutation in CRB resulted in increased expression of PBP4, subsequently leading to increased PG cross-linking that resulted in β-lactam resistance. This mechanism of β-lactam resistance was novel, as hitherto known resistance mechanisms were dependent either on drug inactivation (as displayed by the β-lactamase PC1, mediated by blaZ), or on reduced drug affinity (as displayed by the expression of PBP2a, mediated by mecA) [3], and thus warranted further investigation.
In order to assess the effect of pbp4 missense mutations, the pbp4 gene from either wild-type bacteria (pbp4 wt), or from CRB (pbp4**) was introduced into an isogenic, heterologous host and challenged with β-lactam treatment. Variants of pbp4 were introduced into COLnex Δpbp4 via the constitutively expressing plasmid, pTXΔ and a population assay was performed with nafcillin. It was seen that complementation with pbp4** led to significantly higher survival in presence of nafcillin, when compared to complementation with pbp4 wild-type [75]. Introduction of both, the regulatory-site and missense mutations into COLnex (COLnex Ppbp4*(CRB) pbp4** (CRB)) also led to an increased level of β-lactam resistance (16-fold increase in MIC for nafcillin) and was higher than that seen by either COLnex Ppbp4*(CRB) or COLnex pbp4** (CRB) [76]. Subsequent studies were performed by serially passaging NGB-susceptible strains in other antibiotics such as nafcillin or ceftaroline. Similar to that seen in CRB, the resultant variants were significantly more resistant to NGBs compared to their respective parent strains, and had accumulated mutations associated with pbp4 [75]. Taken together, these studies demonstrated that PBP4 played an important role in non-classical β-lactam resistance, through altered expression and/or function.
4.2. Relevance of Pbp4-Associated Mutations in Clinically Isolated Strains
On determining that pbp4 regulatory site and missense mutations played an important role in β-lactam resistance in laboratory-passaged strains, our group conducted studies to determine if the mutations and their associated phenotypes were also relevant in clinically isolated strains of S. aureus. Findings of a retrospective study conducted in Belgium revealed the presence of clinical isolates of S. aureus that were phenotypically resistant to β-lactams, but were devoid of mec genes. These strains, termed either as MRLM (Methicillin resistant lacking mec) or BORSA (Borderline-Oxacillin Resistant S. aureus), contained mutations associated with pbp1, pbp2, pbp3, pbp4 (including the regulatory region) yjbH and gdpP [67]. To determine if the mutations detected in clinical strains had similar phenotypic effects as those displayed by the laboratory-generated strains, the effects of representative pbp4 associated mutations were assessed by introducing them into isogenic, heterologous host bacterial strains. Assessment of 5 representative regulatory-site associated mutations (Ppbp4*) via a luciferase reporter assay revealed that 3 of the 5 mutation types had significantly increased promoter activity when compared to the wild-type promoter, suggesting that these mutations potentially led to increased PBP4 expression [77]. Indeed, introduction of one of these mutations (T to A substitution 266 bps upstream the pbp4 start codon) into the genome of COLnex [Colnex Ppbp4* (Strain 1)] revealed increased PBP4 expression when compared to COLnex wild-type. Moreover, Colnex Ppbp4* (Strain 1) also had increased survival when exposed nafcillin and cefoxitin when compared to COLnex wild-type, suggesting its ability to be resistant to next generation β-lactams (NGBs).
Similarly, the effect of missense mutations associated with the pbp4 gene were assessed by introducing a representative mutation, R200L, into the genome of COLnex, generating COLnex pbp4** (R200L). When challenged with β-lactams, COLnex pbp4** (R200L) had significantly increased survival when compared to COLnex wild-type, suggesting that the R200L mutation played an important role in β-lactam resistance. Interestingly, the R200L missense mutation was able to mediate increased β-lactam resistance compared to other missense mutations, including those seen in CRB, suggesting that each missense mutation potentially had unique mechanisms of resistance [77]. These studies confirmed that pbp4-associated mutations are not only prominent in clinically isolated, β-lactam resistant strains of S. aureus, but that they also play a significant role in contributing towards the resistant phenotype. pbp4-associated mutations have since been detected in two other surveillance studies [68,69]. An analysis to determine the relevance of these mutations revealed that of a total of 194 MRLM isolates detected, 84% (163 isolates) contained at least one type of pbp4-associated mutation [78], signifying the global prevalence of these mutations, and the potential impact of PBP4 mediated NGB resistance.
The findings of the above discussed studies indicated that S. aureus strains with regulatory-site associated mutations are significantly more resistant to NGBs, when compared to strains consisting of missense mutations, suggesting that PBP4 overexpression and increased PG cross-linking were vital for high-level β-lactam resistance. Further studies were therefore focused on the regulatory-site associated mutations. While the phenotypes associated with the regulatory-site associated mutations were well described in MSSA background [75], the effect of these mutations in MRSA strains was unknown. In order to determine this, three different types of representative regulatory-site associated mutations were introduced into SF8300, a derivative of USA300, one of the most prominent community-associated MRSA (CA-MRSA) strains detected in the U.S [79]. Growth studies indicated that SF8300 containing regulatory-site associated mutations had increased survival in presence of NGBs, when compared to SF8300 wild-type [80]. Moreover, the increase in resistance to NGBs corresponded to the levels of PBP4 overexpression displayed by the mutants, further reiterating that PBP4 overexpression was the cause of NGB resistance. These findings suggested that PBP4 overexpression supplemented PBP2a-associated resistance seen in MRSA strains, and that MRSA strains containing PBP4-associated mutations were potentially more challenging to treat.
pbp4 shares its regulatory region (Ppbp4) with abcA, a gene that encodes for an ATP-binding cassette transporter protein. ABC transporters are notorious for enabling resistance to chemotherapeutic agents in both prokaryotes and eukaryotes via export activity [81,82]. Since AbcA has previously been associated with antibiotic resistance, it was necessary to determine whether the regulatory site-associated mutations altered abcA expression that potentially led to AbcA-mediated NGB resistance. qRT-PCR to determine the effect of regulatory-site associated mutations indicated that while the presence of mutations in Ppbp4 led to increased levels of pbp4, they also led to a corresponding decrease in levels of abcA transcripts. These findings suggested that the regulatory-site associated mutations potentially led to a decrease in AbcA expression, and that the resistance phenotype was likely attributed to PBP4 overexpression.
5. Auxiliary Effects of Pbp4-Associated Mutations
5.1. Synergistic Effect of Pbp4-Associated Mutations and GdpP Alterations
While pbp4-associated mutations led to high-level β-lactam resistance, the increase in resistance was not as substantial as that reported by CRB, as CRB was 512-fold more resistant than COLnex to nafcillin, whereas COLnex Ppbp4* (CRB) pbp4** (CRB) was only 16-fold more resistant than COLnex [76]. While CRB did not contain mutations associated with any other PBPs, it did contain mutations associated with GdpP, a phosphodiesterase that cleaves the second messenger cyclic-di-AMP (CDA) [71,83]. This GdpP associated mutation, N182K, located upstream the DHH/DHHA1 functional domain of the gene, leads to premature truncation of the protein, rendering it functionally inactive. Loss of GdpP activity has been associated with NGB tolerance, a phenomenon where bacteria can survive NGB challenges without resulting in a change in MIC [84]. However, altered functioning of neither PBP4, nor GdpP, on their own, accounted for the high-level, broad-spectrum resistance to β-lactams seen in CRB. It was thus necessary to determine if the high MIC seen in CRB was potentially attributed to altered functioning of both PBP4 and GdpP [78]. Alterations in PBP4 and GdpP were introduced in SF8300ex (Wtex), to generate the triple mutant, Wtex Ppbp4* pbp4** ΔgdpP. Interestingly, MIC assay for the triple mutant revealed a drastic increase in resistance to nafcillin and oxacillin (64- fold), along with ceftaroline (8- fold) when compared to the parent strain. This increase in MIC values seen in the triple mutant was significantly higher than Wtex ΔgdpP as well as Wtex Ppbp4* pbp4**, suggesting a synergistic role of altered functions of PBP4 and GdpP. Validation of this synergism was performed either by including the pbp4 non-functional mutation, S75A in the triple mutant, or, by complementing it with a functional GdpP, both of which resulted in susceptibility to the selected NGBs. Additionally, the increase in MIC for the triple mutant was comparable to that displayed by SF8300, an MRSA background strain. Moreover, in case of ceftaroline, the MIC was 4-fold higher than that of SF8300. These findings were substantiated with the help of growth assays, which demonstrated that the triple mutants had increased survival in presence of NGBs. These studies demonstrated that alterations in PBP4 and GdpP could synergistically result in MRSA-like, high-level NGB resistance.
While the above study substantiated that both PBP4 and GdpP alterations were required for MRSA-like, broad spectrum NGB resistance, the mechanism by which it occurs remains elusive. GdpP alterations have been postulated to cause increased PBP4 expression; however, bocillin assay revealed that PBP4 overexpession levels remained unchanged, without or with ΔgdpP. Similarly, the loss of GdpP did not have any effect on the PG cross-linking, as both the double mutant (Wtex Ppbp4* pbp4**) and the triple mutant had similar PG profiles [78].
5.2. PBP4’s Role in Virulence
In addition to resistance, studies have suggested that PBP4 potentially has a role in virulence. While PBP4 was identified as a critical factor for bone invasion during osteomyelitis in mice [85], other studies have suggested that strains lacking PBP4 were more virulent than strains containing PBP4 [13]. Findings from our group suggested that PBP4 overexpression led to decreased virulence in a C. elegans infection model [80]. Co-infection of C. elegans with SF8300 and SF8300 Ppbp4* demonstrated significantly decreased bacterial colonization by SF8300 Ppbp4* compared to its wild-type counterpart, suggesting that the mutant was potentially deficient in its ability to sufficiently cause virulence. Taken together, the exact role played by PBP4 in bacterial pathogenesis remains unknown and requires further investigation.
6. Conclusion and Future Outlooks
The findings discussed in this review put forth the argument to consider PBP4 as a factor during clinical diagnosis and treatment of S. aureus infections. Identification of MRSA is done by genomic techniques such as PCR for the mecA gene, a gold standard test attributed to it being rapid, accurate and cost effective [86]. However, PCR for mecA is not always sufficient, as the presence of mecC instead of mecA yields a false-negative result for PCR tests specific to mecA. PCR for other elements of the SCC-mec cassette are also included during testing [60]. Phenotypic tests such as MIC determination by broth micro-dilution and disk tests with cefoxitin or oxacillin have proven to be rapid and accurate, due to which they are often performed in combination with PCR to differentiate MRSA and MSSA [87]. However, these tests may not be sufficient for accurate detection of MRLM, or other non-classical NGB resistant strains. MRLM strains, due to lack of mec genes, are likely to be diagnosed and consequently treated as MSSA, resulting in their exposure to sub-inhibitory doses of β-lactams. Not only would this treatment be ineffective, but it could also lead to further accumulation of resistance mutations. Infections caused by MRLM strains with increased expression of PBP4 likely require to be treated with increased doses of β-lactam when compared to infections caused by MSSA strains, or require to be treated with other classes of antibiotics. The presence of pbp4-associated mutations in MRSA background strains introduces further complications to pathogen diagnosis and treatment. Thus, detection PBP4 expression levels during infection diagnosis could potentially help lead to more specific and effective treatment options.
While this review summarizes the current understandings of PBP4-mediated NGB resistance, there is much that remains unknown. The exact alterations brought about by the regulatory site-associated mutations need to be identified. While it is likely that the mutations lead to alterations in binding of regulatory factors, identification of those proteins could be beneficial for designing treatment options. Further, identification of the mechanism(s) of synergy between alterations in PBP4 and GdpP is necessary. The effect of PBP4-overexpression on other cell wall targeting antibiotics is unknown. Lastly, the effect of PBP4-overexpression on antibiotic resistance and virulence in suitable host systems should also be determined by performing ex vivo and in vivo experiments.
References
- Ikuta, K.S.; Swetschinski, L.R.; Robles Aguilar, G.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Davis Weaver, N.; Wool, E.E.; Han, C.; Gershberg Hayoon, A.; et al. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. The Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.A. Challenges for a universal Staphylococcus aureus vaccine. Clin Infect Dis 2012, 54, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Lade, H.; Kim, J.S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus. Antibiotics (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb Perspect Med 2016, 6. [Google Scholar] [CrossRef]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci U S A 1965, 54, 1133–1141. [Google Scholar] [CrossRef]
- Kim, S.J.; Chang, J.; Singh, M. Peptidoglycan architecture of Gram-positive bacteria by solid-state NMR. Biochimica et Biophysica Acta (BBA) - Biomembranes 2015, 1848, 350–362. [Google Scholar] [CrossRef]
- Salton, M.R.J.; Kim, K.S. Chapter 2-Structure. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston Copyright © 1996, The University of Texas Medical Branch at Galveston.: Galveston (TX), 1996. [Google Scholar]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiology Reviews 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Garde, S.; Chodisetti, P.K.; Reddy, M. Peptidoglycan: Structure, Synthesis, and Regulation. EcoSal Plus 2021, 9. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Clauson, M.; Hong, J.; Murphy, A.B. Gram positive and Gram negative bacteria differ in their sensitivity to cold plasma. Scientific Reports 2016, 6, 38610. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.A.F.; Carnell, O.T.; Lafage, L.; Gray, J.; Biboy, J.; Gibson, J.F.; Pollitt, E.J.G.; Tazoll, S.C.; Turnbull, W.; Hajdamowicz, N.H.; et al. Staphylococcus aureus cell wall structure and dynamics during host-pathogen interaction. PLoS Pathog 2021, 17, e1009468. [Google Scholar] [CrossRef] [PubMed]
- Ghuysen, J.M.; Strominger, J.L. STRUCTURE OF THE CELL WALL OF STAPHYLOCOCCUS AUREUS, STRAIN COPENHAGEN. I. PREPARATION OF FRAGMENTS BY ENZYMATIC HYDROLYSIS. Biochemistry 1963, 2, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 1972, 36, 407–477. [Google Scholar] [CrossRef]
- Forrest, T.M.; Wilson, G.E.; Pan, Y.; Schaefer, J. Characterization of cross-linking of cell walls of Bacillus subtilis by a combination of magic-angle spinning NMR and gas chromatography-mass spectrometry of both intact and hydrolyzed 13C- and 15N-labeled cell-wall peptidoglycan. J Biol Chem 1991, 266, 24485–24491. [Google Scholar] [CrossRef]
- Apostolos, A.J.; Pires, M.M. Impact of crossbridge structure on peptidoglycan crosslinking: A synthetic stem peptide approach. Methods Enzymol 2022, 665, 259–279. [Google Scholar] [CrossRef]
- Kim, S.J.; Matsuoka, S.; Patti, G.J.; Schaefer, J. Vancomycin Derivative with Damaged d-Ala-d-Ala Binding Cleft Binds to Cross-linked Peptidoglycan in the Cell Wall of Staphylococcus aureus. Biochemistry 2008, 47, 3822–3831. [Google Scholar] [CrossRef]
- Severin, A.; Tomasz, A. Naturally occurring peptidoglycan variants of Streptococcus pneumoniae. J Bacteriol 1996, 178, 168–174. [Google Scholar] [CrossRef]
- Rajagopal, M.; Walker, S. Envelope Structures of Gram-Positive Bacteria. Curr Top Microbiol Immunol 2017, 404, 1–44. [Google Scholar] [CrossRef]
- Boneca, I.G.; Huang, Z.H.; Gage, D.A.; Tomasz, A. Characterization of Staphylococcus aureus cell wall glycan strands, evidence for a new beta-N-acetylglucosaminidase activity. J Biol Chem 2000, 275, 9910–9918. [Google Scholar] [CrossRef]
- Gally, D.; Archibald, A.R. Cell wall assembly in Staphylococcus aureus: proposed absence of secondary crosslinking reactions. J Gen Microbiol 1993, 139, 1907–1913. [Google Scholar] [CrossRef]
- Monteiro, J.M.; Covas, G.; Rausch, D.; Filipe, S.R.; Schneider, T.; Sahl, H.-G.; Pinho, M.G. The pentaglycine bridges of Staphylococcus aureus peptidoglycan are essential for cell integrity. Scientific Reports 2019, 9, 5010. [Google Scholar] [CrossRef]
- Glauner, B.; Höltje, J.V.; Schwarz, U. The composition of the murein of Escherichia coli. J Biol Chem 1988, 263, 10088–10095. [Google Scholar] [CrossRef]
- Ghuysen, J.M. Serine beta-lactamases and penicillin-binding proteins. Annu Rev Microbiol 1991, 45, 37–67. [Google Scholar] [CrossRef]
- Jarick, M.; Bertsche, U.; Stahl, M.; Schultz, D.; Methling, K.; Lalk, M.; Stigloher, C.; Steger, M.; Schlosser, A.; Ohlsen, K. The serine/threonine kinase Stk and the phosphatase Stp regulate cell wall synthesis in Staphylococcus aureus. Scientific Reports 2018, 8, 13693. [Google Scholar] [CrossRef] [PubMed]
- Komatsuzawa, H.; Fujiwara, T.; Nishi, H.; Yamada, S.; Ohara, M.; McCallum, N.; Berger-Bächi, B.; Sugai, M. The gate controlling cell wall synthesis in Staphylococcus aureus. Mol Microbiol 2004, 53, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Patin, D.; Boniface, A.; Kovač, A.; Hervé, M.; Dementin, S.; Barreteau, H.; Mengin-Lecreulx, D.; Blanot, D. Purification and biochemical characterization of Mur ligases from Staphylococcus aureus. Biochimie 2010, 92, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Neuhaus, F.C. Mechanism of D-cycloserine action: alanine racemase from Escherichia coli W. J Bacteriol 1972, 110, 978–987. [Google Scholar] [CrossRef]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann N Y Acad Sci 1974, 235, 364–386. [Google Scholar] [CrossRef]
- Yoo, J.; Mashalidis, E.H.; Kuk, A.C.Y.; Yamamoto, K.; Kaeser, B.; Ichikawa, S.; Lee, S.-Y. GlcNAc-1-P-transferase–tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nature Structural & Molecular Biology 2018, 25, 217–224. [Google Scholar] [CrossRef]
- Figueiredo, T.A.; Sobral, R.G.; Ludovice, A.M.; de Almeida, J.M.F.; Bui, N.K.; Vollmer, W.; de Lencastre, H.; Tomasz, A. Identification of Genetic Determinants and Enzymes Involved with the Amidation of Glutamic Acid Residues in the Peptidoglycan of Staphylococcus aureus. PLOS Pathogens 2012, 8, e1002508. [Google Scholar] [CrossRef]
- Münch, D.; Roemer, T.; Lee, S.H.; Engeser, M.; Sahl, H.G.; Schneider, T. Identification and in vitro Analysis of the GatD/MurT Enzyme-Complex Catalyzing Lipid II Amidation in Staphylococcus aureus. PLOS Pathogens 2012, 8, e1002509. [Google Scholar] [CrossRef]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiology Reviews 2008, 32, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, K.; Owens, T.W.; Page, J.E.; Santiago, M.; Kahne, D.; Walker, S. Structure and reconstitution of a hydrolase complex that may release peptidoglycan from the membrane after polymerization. Nat Microbiol 2021, 6, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mollo, A.; Kahne, D.; Ruiz, N. The Bacterial Cell Wall: From Lipid II Flipping to Polymerization. Chem Rev 2022, 122, 8884–8910. [Google Scholar] [CrossRef]
- Lovering, A.L.; de Castro, L.H.; Lim, D.; Strynadka, N.C. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 2007, 315, 1402–1405. [Google Scholar] [CrossRef]
- Schneewind, O.; Fowler, A.; Faull, K.F. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 1995, 268, 103–106. [Google Scholar] [CrossRef]
- Siewert, G.; Strominger, J.L. Bacitracin: an inhibitor of the dephosphorylation of lipid pyrophosphate, an intermediate in the biosynthesis of the peptidoglycan of bacterial cell walls. Proc Natl Acad Sci U S A 1967, 57, 767–773. [Google Scholar] [CrossRef]
- Wang, M.; Buist, G.; van Dijl, J.M. Staphylococcus aureus cell wall maintenance – the multifaceted roles of peptidoglycan hydrolases in bacterial growth, fitness, and virulence. FEMS Microbiology Reviews 2022, 46. [Google Scholar] [CrossRef]
- Blumberg, P.M.; Strominger, J.L. Interaction of penicillin with the bacterial cell: penicillin-binding proteins and penicillin-sensitive enzymes. Bacteriol Rev 1974, 38, 291–335. [Google Scholar] [CrossRef]
- Goffin, C.; Ghuysen, J.M. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev 1998, 62, 1079–1093. [Google Scholar] [CrossRef]
- Macheboeuf, P.; Contreras-Martel, C.; Job, V.; Dideberg, O.; Dessen, A. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev 2006, 30, 673–691. [Google Scholar] [CrossRef]
- Lovering, A.L.; Safadi, S.S.; Strynadka, N.C. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 2012, 81, 451–478. [Google Scholar] [CrossRef]
- Alexander, J.A.N.; Chatterjee, S.S.; Hamilton, S.M.; Eltis, L.D.; Chambers, H.F.; Strynadka, N.C.J. Structural and kinetic analyses of penicillin-binding protein 4 (PBP4)-mediated antibiotic resistance in Staphylococcus aureus. J Biol Chem 2018, 293, 19854–19865. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Terrak, M. Glycosyltransferases and Transpeptidases/Penicillin-Binding Proteins: Valuable Targets for New Antibacterials. Antibiotics (Basel) 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.F.; Henriques, A.O.; Pinho, M.G.; de Lencastre, H.; Tomasz, A. Role of PBP1 in cell division of Staphylococcus aureus. J Bacteriol 2007, 189, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Caballero, S.; Mahasenan, K.V.; Kim, C.; Molina, R.; Feltzer, R.; Lee, M.; Bouley, R.; Hesek, D.; Fisher, J.F.; Muñoz, I.G.; et al. Integrative structural biology of the penicillin-binding protein-1 from Staphylococcus aureus, an essential component of the divisome machinery. Comput Struct Biotechnol J 2021, 19, 5392–5405. [Google Scholar] [CrossRef]
- Łeski, T.A.; Tomasz, A. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol 2005, 187, 1815–1824. [Google Scholar] [CrossRef]
- Pinho, M.G.; de Lencastre, H.; Tomasz, A. Cloning, characterization, and inactivation of the gene pbpC, encoding penicillin-binding protein 3 of Staphylococcus aureus. J Bacteriol 2000, 182, 1074–1079. [Google Scholar] [CrossRef]
- Reichmann, N.T.; Tavares, A.C.; Saraiva, B.M.; Jousselin, A.; Reed, P.; Pereira, A.R.; Monteiro, J.M.; Sobral, R.G.; VanNieuwenhze, M.S.; Fernandes, F.; et al. SEDS-bPBP pairs direct lateral and septal peptidoglycan synthesis in Staphylococcus aureus. Nat Microbiol 2019, 4, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Wyke, A.W.; Ward, J.B.; Hayes, M.V.; Curtis, N.A. A role in vivo for penicillin-binding protein-4 of Staphylococcus aureus. Eur J Biochem 1981, 119, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Loskill, P.; Pereira, P.M.; Jung, P.; Bischoff, M.; Herrmann, M.; Pinho, M.G.; Jacobs, K. Reduction of the peptidoglycan crosslinking causes a decrease in stiffness of the Staphylococcus aureus cell envelope. Biophys J 2014, 107, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Srisuknimit, V.; Qiao, Y.; Schaefer, K.; Kahne, D.; Walker, S. Peptidoglycan Cross-Linking Preferences of Staphylococcus aureus Penicillin-Binding Proteins Have Implications for Treating MRSA Infections. J Am Chem Soc 2017, 139, 9791–9794. [Google Scholar] [CrossRef]
- Dyke, K.G.; Jevons, M.P.; Parker, M.T. Penicillinase production and intrinsic resistance to penicillins in Staphylococcus aures. Lancet 1966, 1, 835–838. [Google Scholar] [CrossRef]
- Pantosti, A.; Sanchini, A.; Monaco, M. Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol 2007, 2, 323–334. [Google Scholar] [CrossRef]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef]
- Lim, D.; Strynadka, N.C. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat Struct Biol 2002, 9, 870–876. [Google Scholar] [CrossRef]
- Paterson, G.K.; Harrison, E.M.; Holmes, M.A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol 2014, 22, 42–47. [Google Scholar] [CrossRef]
- Abraham, L.; Bamberger, D.M. Staphylococcus aureus Bacteremia: Contemporary Management. Mo Med 2020, 117, 341–345. [Google Scholar]
- Parish, D.; Scheinfeld, N. Ceftaroline fosamil, a cephalosporin derivative for the potential treatment of MRSA infection. Curr Opin Investig Drugs 2008, 9, 201–209. [Google Scholar] [PubMed]
- Long, S.W.; Olsen, R.J.; Mehta, S.C.; Palzkill, T.; Cernoch, P.L.; Perez, K.K.; Musick, W.L.; Rosato, A.E.; Musser, J.M. PBP2a mutations causing high-level Ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 2014, 58, 6668–6674. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Muraoka, A.; Bedenbaugh, M.; Childress, B.; Pernot, L.; Wiencek, M.; Peterson, Y.K. The Chemical Relationship Among Beta-Lactam Antibiotics and Potential Impacts on Reactivity and Decomposition. Front Microbiol 2022, 13, 807955. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Lebar, M.D.; Schirner, K.; Schaefer, K.; Tsukamoto, H.; Kahne, D.; Walker, S. Detection of Lipid-Linked Peptidoglycan Precursors by Exploiting an Unexpected Transpeptidase Reaction. Journal of the American Chemical Society 2014, 136, 14678–14681. [Google Scholar] [CrossRef]
- Memmi, G.; Filipe, S.R.; Pinho, M.G.; Fu, Z.; Cheung, A. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob Agents Chemother 2008, 52, 3955–3966. [Google Scholar] [CrossRef]
- Argudin, M.A.; Roisin, S.; Nienhaus, L.; Dodemont, M.; de Mendonca, R.; Nonhoff, C.; Deplano, A.; Denis, O. Genetic diversity amongStaphylococcus aureusisolates showing oxacillin and/or cefoxitin resistance not linked to the presence of mec genes. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef]
- Ba, X.; Kalmar, L.; Hadjirin, N.F.; Kerschner, H.; Apfalter, P.; Morgan, F.J.; Paterson, G.K.; Girvan, S.L.; Zhou, R.; Harrison, E.M.; et al. Truncation of GdpP mediates β-lactam resistance in clinical isolates of Staphylococcus aureus. J Antimicrob Chemother 2019, 74, 1182–1191. [Google Scholar] [CrossRef]
- Sommer, A.; Fuchs, S.; Layer, F.; Schaudinn, C.; Weber, R.E.; Richard, H.; Erdmann, M.B.; Laue, M.; Schuster, C.F.; Werner, G.; et al. Mutations in the gdpP gene are a clinically relevant mechanism for β-lactam resistance in meticillin-resistant Staphylococcus aureus lacking mec determinants. Microbial Genomics 2021, 7. [Google Scholar] [CrossRef]
- Banerjee, R.; Gretes, M.; Basuino, L.; Strynadka, N.; Chambers, H.F. In Vitro Selection and Characterization of Ceftobiprole-Resistant Methicillin-Resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2008, 52, 2089–2096. [Google Scholar] [CrossRef]
- Banerjee, R.; Gretes, M.; Harlem, C.; Basuino, L.; Chambers, H.F. A mecA-negative strain of methicillin-resistant Staphylococcus aureus with high-level β-lactam resistance contains mutations in three genes. Antimicrob Agents Chemother 2010, 54, 4900–4902. [Google Scholar] [CrossRef]
- Greninger, A.L.; Chatterjee, S.S.; Chan, L.C.; Hamilton, S.M.; Chambers, H.F.; Chiu, C.Y. Whole-Genome Sequencing of Methicillin-Resistant Staphylococcus aureus Resistant to Fifth-Generation Cephalosporins Reveals Potential Non-mecA Mechanisms of Resistance. PLoS One 2016, 11, e0149541. [Google Scholar] [CrossRef]
- Henze, U.U.; Berger-Bächi, B. Staphylococcus aureus penicillin-binding protein 4 and intrinsic beta-lactam resistance. Antimicrob Agents Chemother 1995, 39, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.M.; Alexander, J.A.N.; Choo, E.J.; Basuino, L.; da Costa, T.M.; Severin, A.; Chung, M.; Aedo, S.; Strynadka, N.C.J.; Tomasz, A.; et al. High-Level Resistance of Staphylococcus aureus to β-Lactam Antibiotics Mediated by Penicillin-Binding Protein 4 (PBP4). Antimicrob Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.S.; Chen, L.; Gilbert, A.; da Costa, T.M.; Nair, V.; Datta, S.K.; Kreiswirth, B.N.; Chambers, H.F. PBP4 mediates β-lactam resistance by altered function. Antimicrob Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Basuino, L.; Jousselin, A.; Alexander, J.A.N.; Strynadka, N.C.J.; Pinho, M.G.; Chambers, H.F.; Chatterjee, S.S. PBP4 activity and its overexpression are necessary for PBP4-mediated high-level β-lactam resistance. J Antimicrob Chemother 2018, 73, 1177–1180. [Google Scholar] [CrossRef]
- Satishkumar, N.; Alexander, J.A.N.; Poon, R.; Buggeln, E.; Argudín, M.A.; Strynadka, N.C.J.; Chatterjee, S.S. PBP4-mediated β-lactam resistance among clinical strains of Staphylococcus aureus. J Antimicrob Chemother 2021, 76, 2268–2272. [Google Scholar] [CrossRef]
- Lai, L.Y.; Satishkumar, N.; Cardozo, S.; Hemmadi, V.; Marques, L.B.; Huang, L.; Filipe, S.R.; Pinho, M.G.; Chambers, H.F.; Chatterjee, S.S. Altered PBP4 and GdpP functions synergistically mediate MRSA-like high-level, broad-spectrum β-lactam resistance in Staphylococcus aureus. mBio 2024, 15, e0288923. [Google Scholar] [CrossRef]
- Carrel, M.; Perencevich, E.N.; David, M.Z. USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000-2013. Emerg Infect Dis 2015, 21, 1973–1980. [Google Scholar] [CrossRef]
- Satishkumar, N.; Lai, L.Y.; Mukkayyan, N.; Vogel, B.E.; Chatterjee, S.S. A Nonclassical Mechanism of β-Lactam Resistance in Methicillin-Resistant Staphylococcus aureus and Its Effect on Virulence. Microbiol Spectr 2022, 10, e0228422. [Google Scholar] [CrossRef]
- Villet, R.A.; Truong-Bolduc, Q.C.; Wang, Y.; Estabrooks, Z.; Medeiros, H.; Hooper, D.C. Regulation of expression of abcA and its response to environmental conditions. J Bacteriol 2014, 196, 1532–1539. [Google Scholar] [CrossRef]
- Yoshikai, H.; Kizaki, H.; Saito, Y.; Omae, Y.; Sekimizu, K.; Kaito, C. Multidrug-Resistance Transporter AbcA Secretes Staphylococcus aureus Cytolytic Toxins. The Journal of Infectious Diseases 2015, 213, 295–304. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Abbott, J.C.; Burhenne, H.; Kaever, V.; Grundling, A. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 2011, 7, e1002217. [Google Scholar] [CrossRef]
- Poon, R.; Basuino, L.; Satishkumar, N.; Chatterjee, A.; Mukkayyan, N.; Buggeln, E.; Huang, L.; Nair, V.; Argudín, M.A.; Datta, S.K.; et al. Loss of GdpP Function in Staphylococcus aureus Leads to β-Lactam Tolerance and Enhanced Evolution of β-Lactam Resistance. Antimicrobial Agents and Chemotherapy 2022, 66, e01431-01421. [Google Scholar] [CrossRef]
- Masters, E.A.; de Mesy Bentley, K.L.; Gill, A.L.; Hao, S.P.; Galloway, C.A.; Salminen, A.T.; Guy, D.R.; McGrath, J.L.; Awad, H.A.; Gill, S.R.; et al. Identification of Penicillin Binding Protein 4 (PBP4) as a critical factor for Staphylococcus aureus bone invasion during osteomyelitis in mice. PLoS Pathog 2020, 16, e1008988. [Google Scholar] [CrossRef]
- Pillai, M.M.; Latha, R.; Sarkar, G. Detection of methicillin resistance in Staphylococcus aureus by polymerase chain reaction and conventional methods: a comparative study. J Lab Physicians 2012, 4, 83–88. [Google Scholar] [CrossRef]
- Farahani, A.; Mohajeri, P.; Gholamine, B.; Rezaei, M.; Abbasi, H. Comparison of different phenotypic and genotypic methods for the detection of methicillin-resistant Staphylococcus aureus. N Am J Med Sci 2013, 5, 637–640. [Google Scholar] [CrossRef]
Figure 1.
Structure and composition of the S. aureus cell wall. The cell wall is a continuous, outermost layer of the cell and is composed of polymers including the peptidoglycan and teichoic acids, in addition to surface anchored proteins. Figure created with BioRender.com.
Figure 1.
Structure and composition of the S. aureus cell wall. The cell wall is a continuous, outermost layer of the cell and is composed of polymers including the peptidoglycan and teichoic acids, in addition to surface anchored proteins. Figure created with BioRender.com.
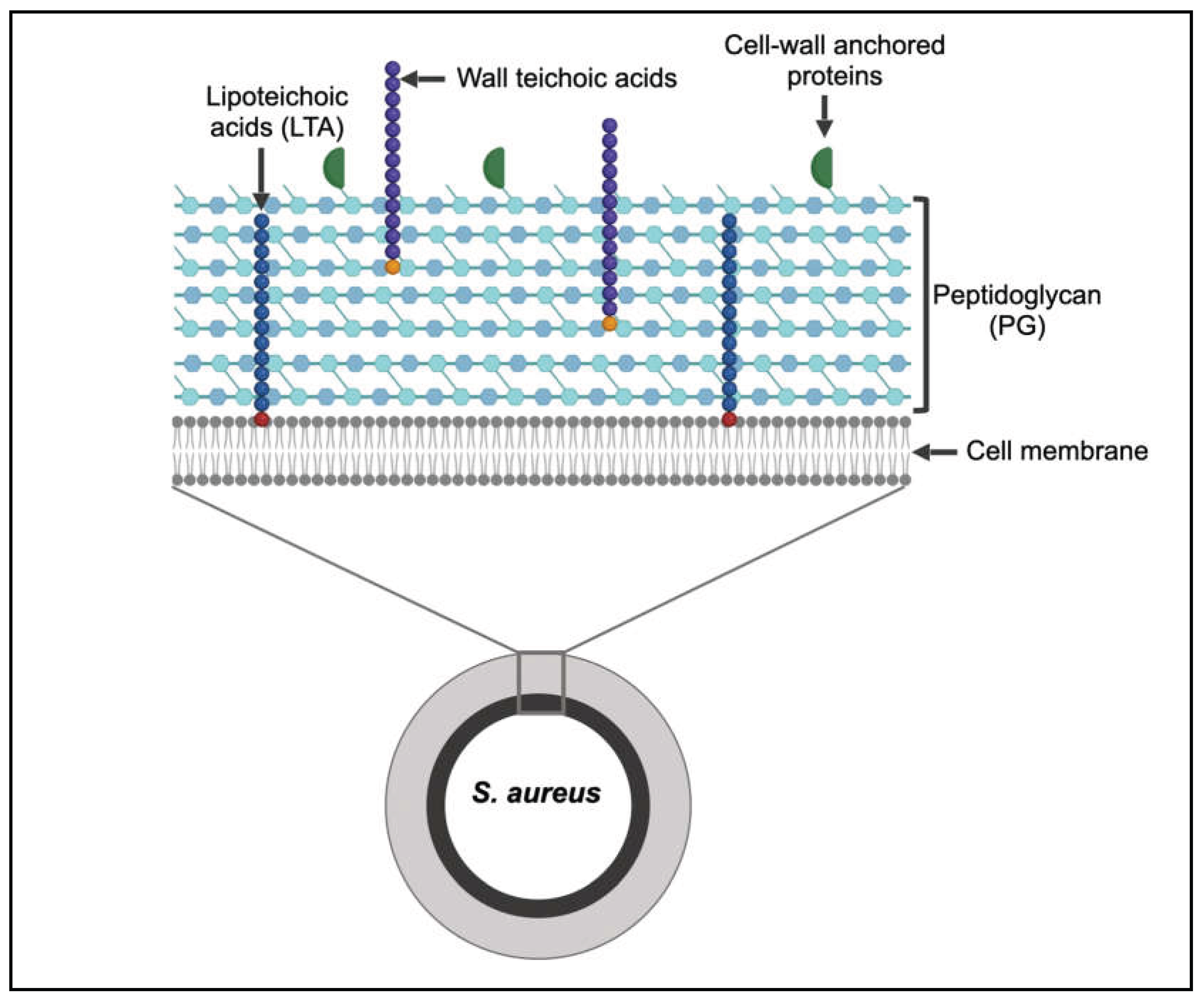
Figure 2.
Components of the peptidoglycan monomer in S. aureus.
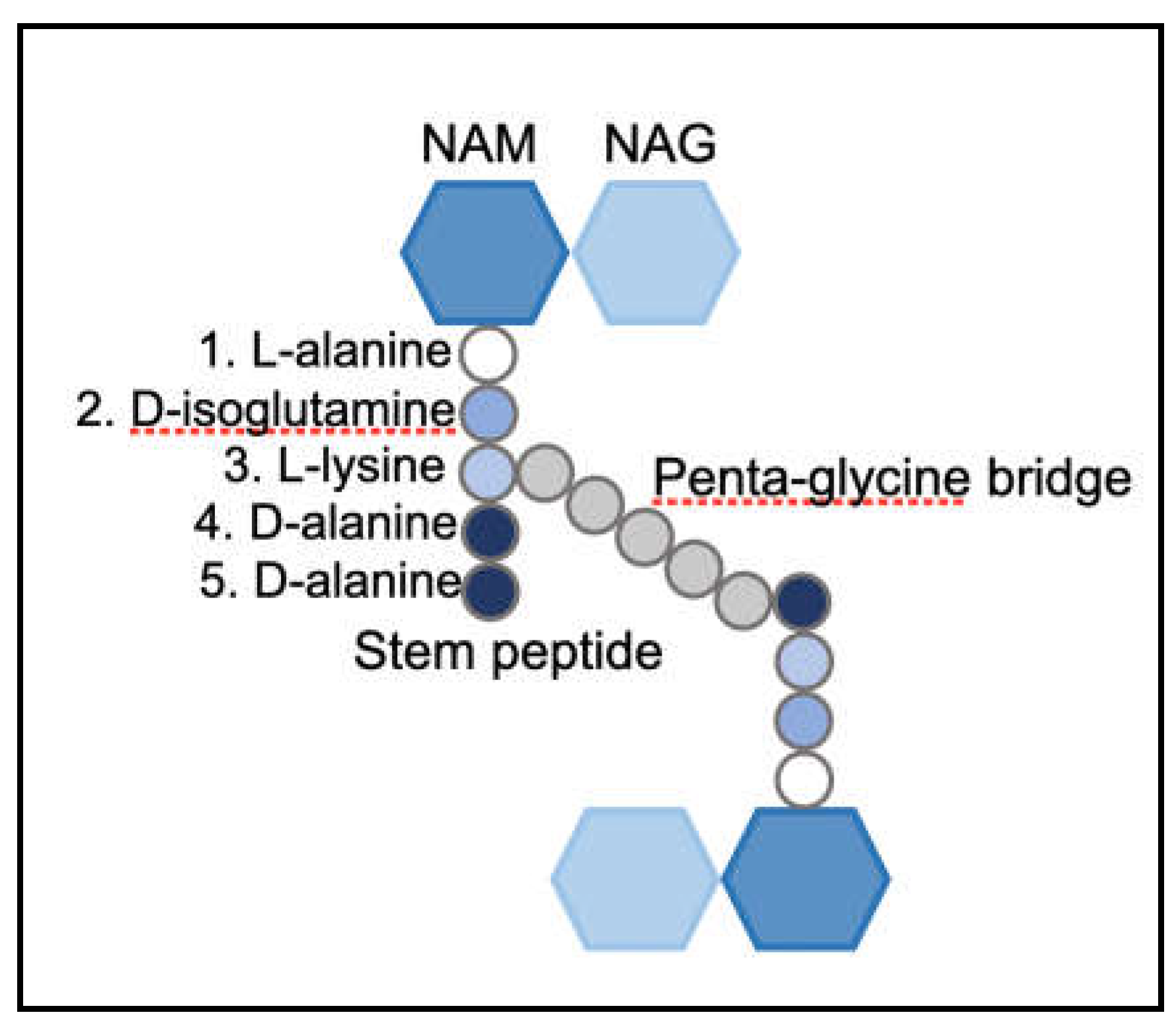
Figure 3.
Variations in composition of peptidoglycan monomers among bacterial species.
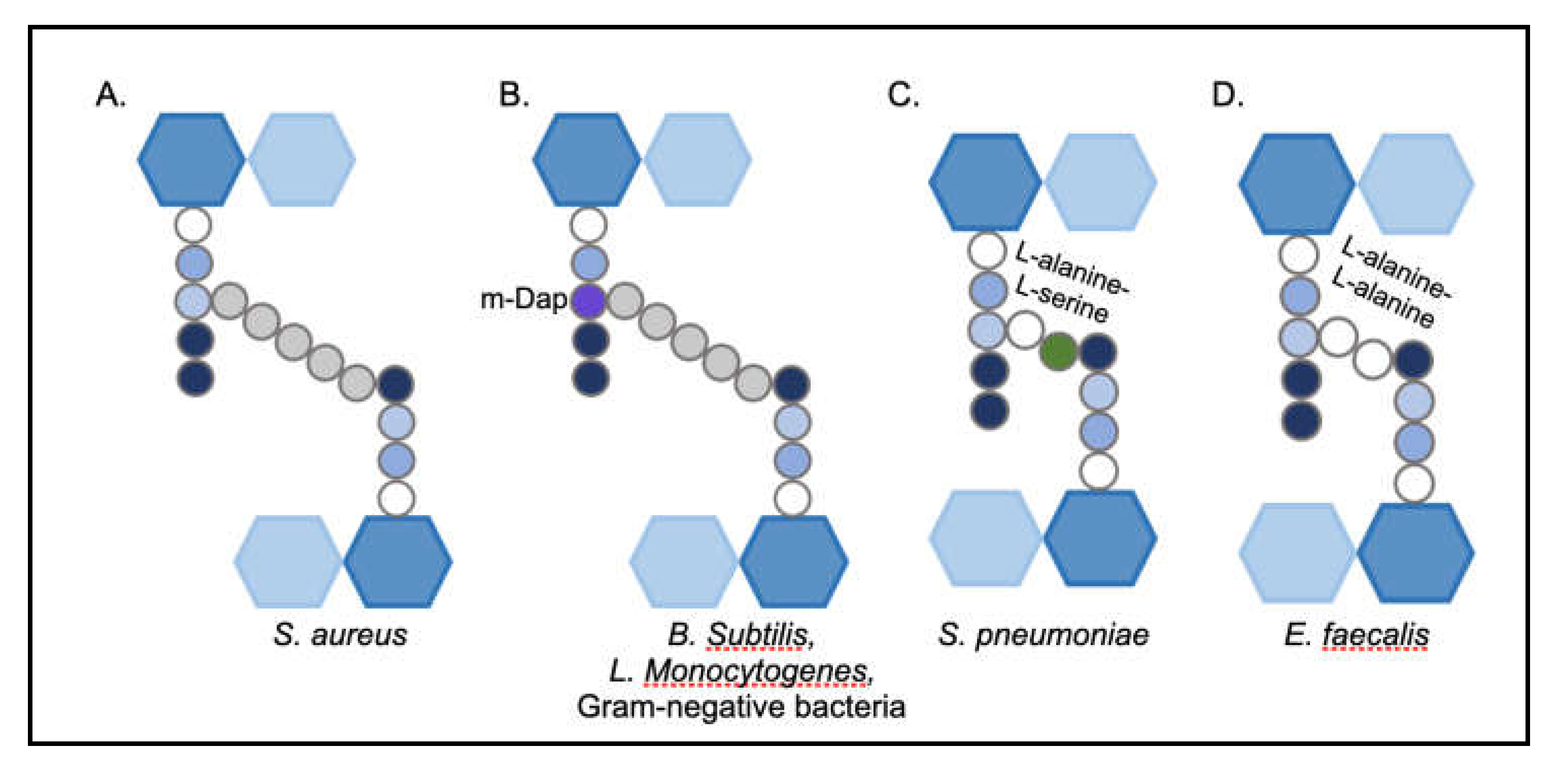
Figure 4.
S. aureus PG synthesis begins in the cell cytosol, following which the monomer is assembled and anchored onto the cell membrane (Lipid II). Lipid II is flipped to the extra-cellular space, where it is cross-linked by PBPs. Antibiotics targeting the PG synthesis process are indicated.
Figure 4.
S. aureus PG synthesis begins in the cell cytosol, following which the monomer is assembled and anchored onto the cell membrane (Lipid II). Lipid II is flipped to the extra-cellular space, where it is cross-linked by PBPs. Antibiotics targeting the PG synthesis process are indicated.
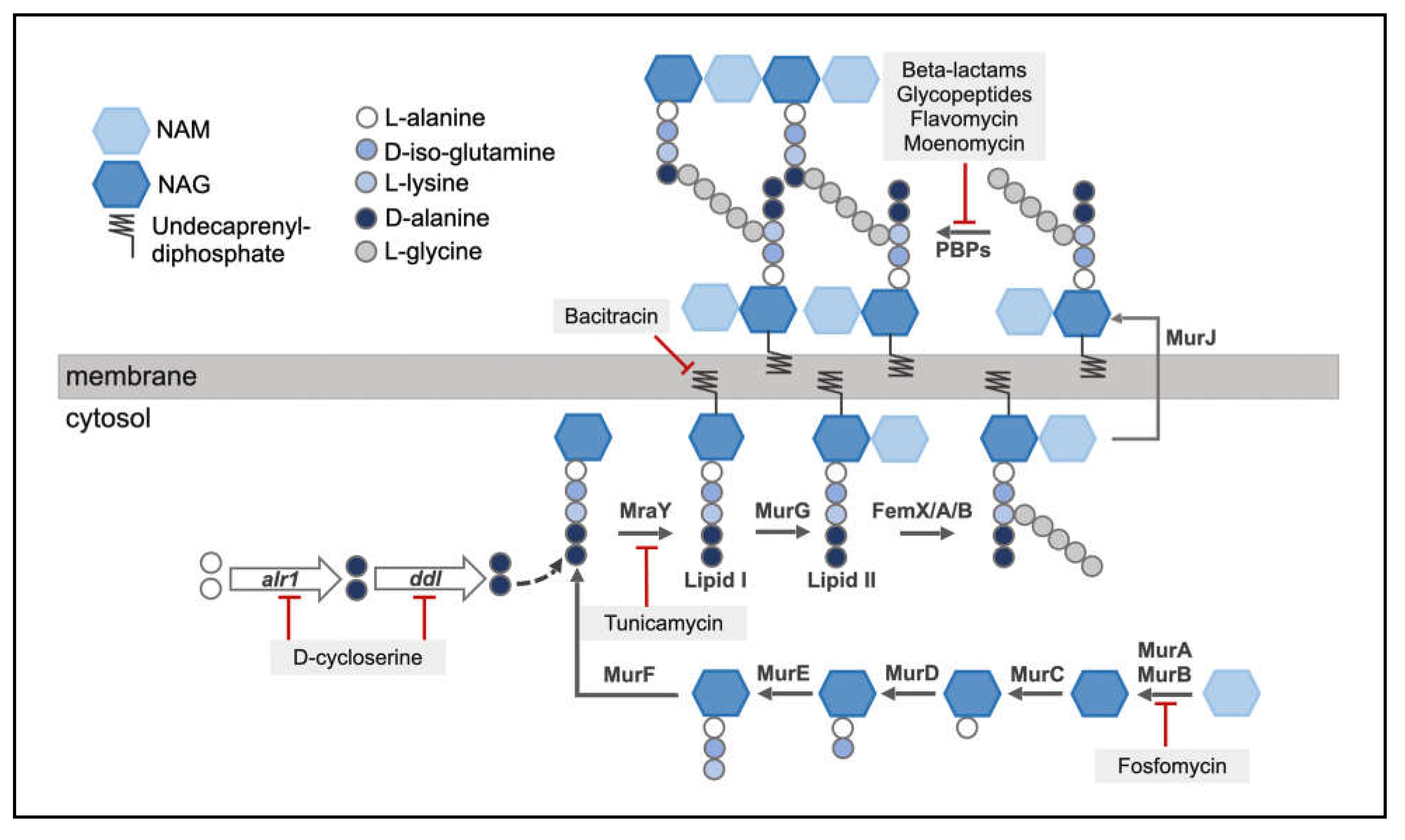
Figure 5.
Functions performed by PBPs in S. aureus.
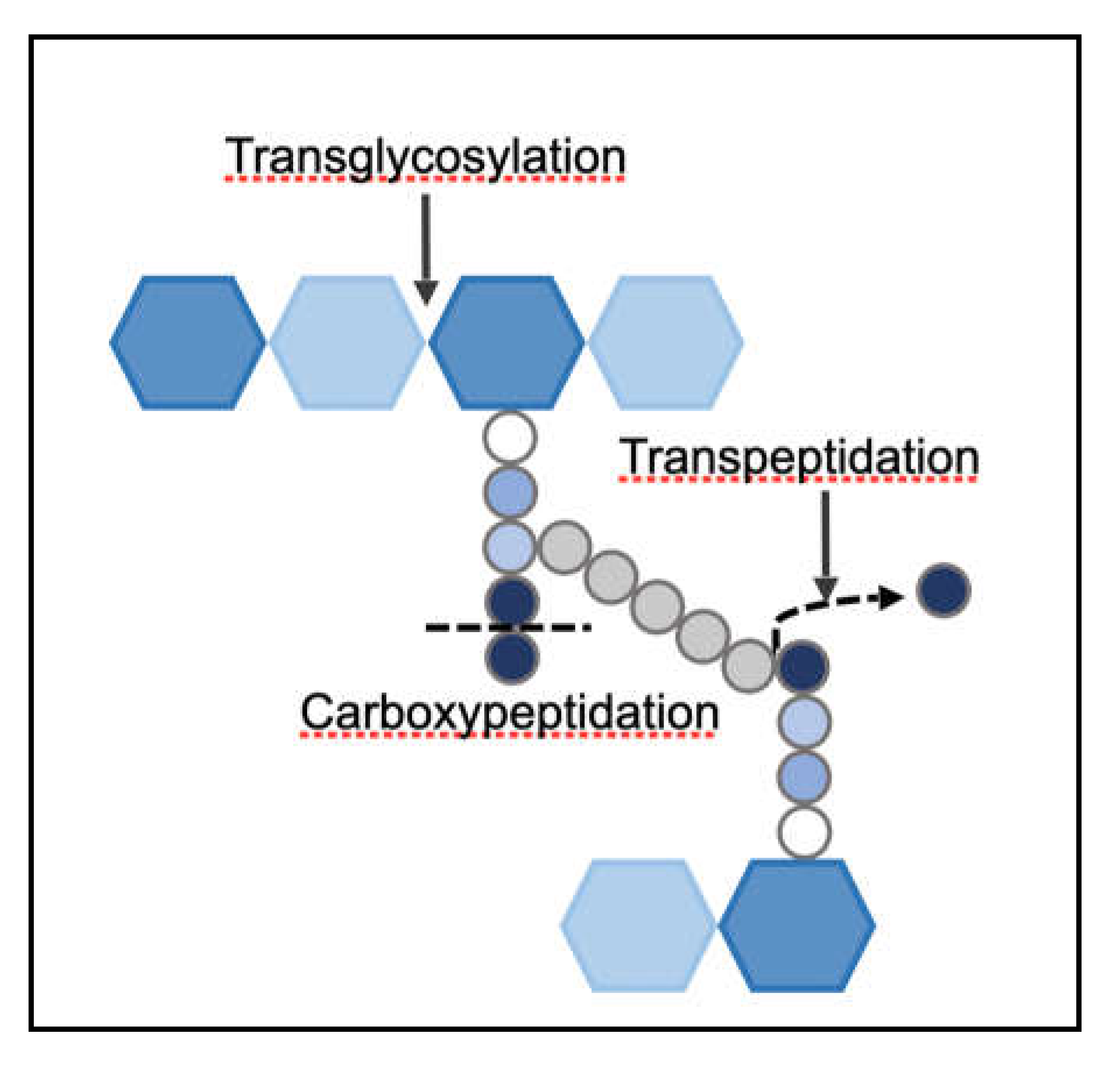
Figure 6.
PBPs are composed of multiple domains with different functions.
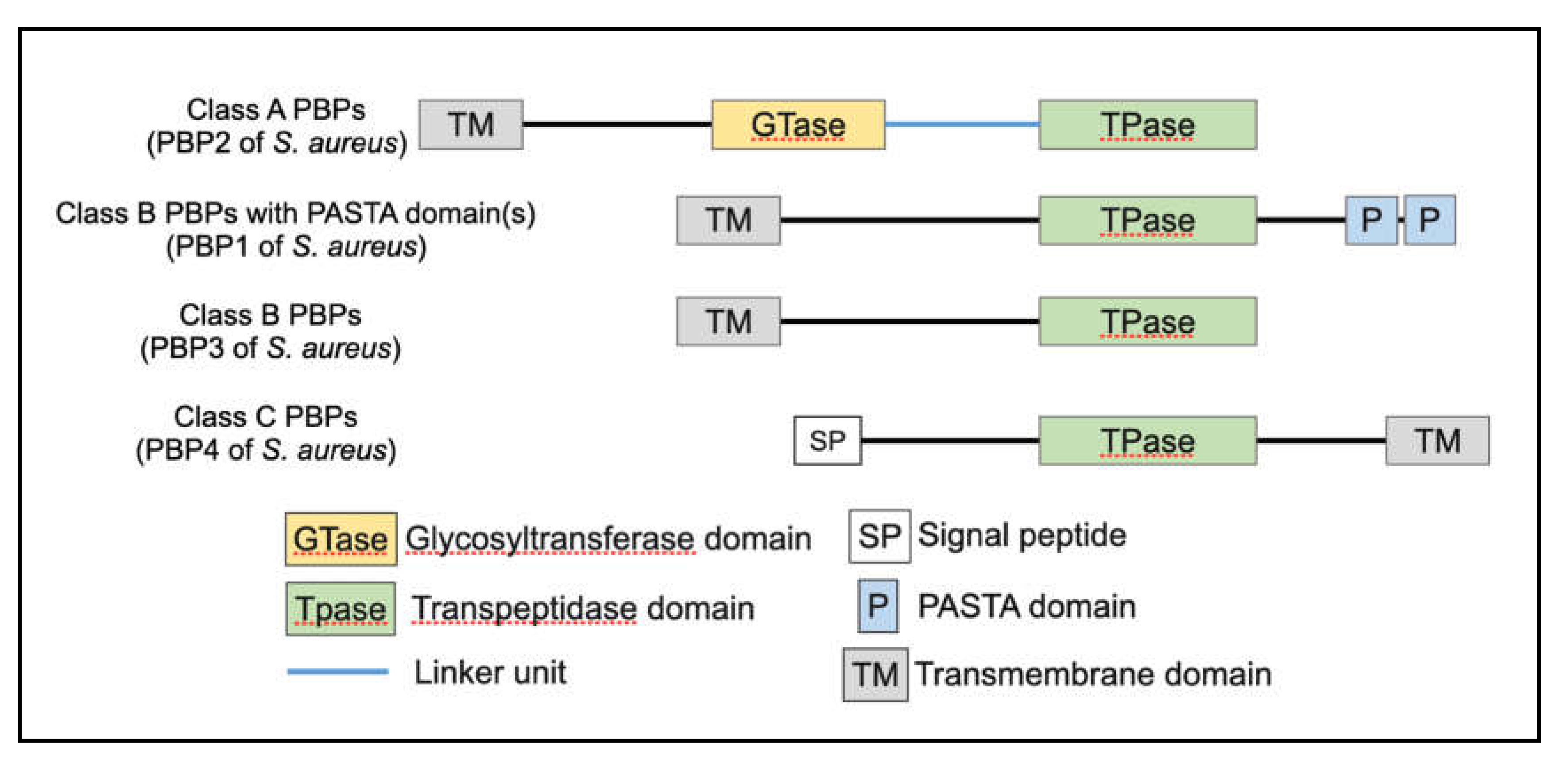
Figure 7.
Multiple sequence alignment of LMM-PBPs in various bacterial species.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



