
Submitted:
19 January 2026
Posted:
19 January 2026
You are already at the latest version
Abstract
Background and Clinical Significance: Plexiform fibromyxoma (PFM) is a rare benign gastric mesenchymal neoplasm characterized by multinodular plexiform growth of bland spindle cells in a myxoid or fibromyxoid stroma. We report a case of the cellular form of PFM with limited myxoid stroma and aberrant KIT expression, resulting in diagnostic difficulty by biopsy. Case presentation: A 59-year-old woman presented with a slowly enlarging 15-mm gastric antral submucosal tumor. A resected specimen by laparoscopic and endoscopic cooperative surgery revealed spindle cell proliferation forming plexiform nodules with a myxoid background in limited areas. Positive immunoreactivity of a subset of spindle cells for KIT suggested a diagnosis of gastrointestinal stromal tumor (GIST), although DOG1 was negative. In addition, diffuse staining for CD10, smooth muscle actin, and D2-40 were confusing. MALAT1::GLI1 fusion was detected by next-generation sequencing analysis. Consequently, a diagnosis of PFM was established. Conclusions: This case expands the morphologic and immunophenotypic spectrum of PFM and indicates the possible diagnostic utility and biological significance of D2-40 expression. Although molecular confirmation of MALAT1::GLI1 fusion is definitive for diagnosis of PFM, the findings of present case may aid diagnosis in challenging cases that mimic GIST.
Keywords:
plexiform fibromyxoma
; stomach
; D2-40
; MALAT1-GLI1 fusion
; variant morphology
1. Introduction
Plexiform fibromyxoma (PFM) is a rare benign gastric mesenchymal neoplasm first described by Takahashi et al. in 2007 as “plexiform angiomyxoid myofibroblastic tumor” [1]. The term “plexiform fibromyoma” was proposed by Miettinen et al. in 2009 [2] and then PFM was officially listed as a distinct type of gastric mesenchymal neoplasm by the World Health Organization (WHO) in 2010 [3]. In the English literature, more than 130 cases have been reported [4]. PFM typically presents a multinodular plexiform growth of bland spindle cells embedded in myxoid or fibromyxoid stroma with increased vascularity [3]. Molecular studies have identified fusions of recurrent metastasis associated lung adenocarcinoma transcript 1 (MALAT1):: glioma-associated oncogene homologue 1 (GLI1) that drive GLI1 overexpression in a subset of PFM [5,6]. Here, we present the case of a cellular form of PFM with limited myxoid matrix, showing diffuse D2-40 expression, resulting in a diagnostic challenge.
2. Case Presentation
A 59-year-old woman was referred to our hospital for evaluation of a gastric submucosal tumor at the antrum that was detected at a medical checkup, and which has been under surveillance for several years. She has no remarkable past medical history or family history. Serial endoscopy revealed an enlargement of the submucosal tumor from 10 mm to 15 mm over the preceding 12 months. The laboratory data were unremarkable. Contrast-enhanced computed tomography demonstrated a well-circumscribed exophytic mass along the greater curvature and no evidence of metastatic lesion was noted. Microscopic examination of an endoscopic biopsy specimen suggested a benign or low-grade spindle cell neoplasm, i.e., gastrointestinal stromal tumor (GIST) or myopericytomatous tumor, but failed to establish a definitive diagnosis. Local resection of the stomach was performed using laparoscopic and endoscopic cooperative surgery. Postoperative course was uneventful and the patient was discharged on postoperative day 7. No evidence of recurrent tumor was identified at the time of writing (8 months after surgery).
2.1 Pathological Findings
Gross examination revealed a lobulated, solid submucosal tumor, measuring 15 × 11 × 10 mm, presenting as a protruding lesion (Figure 1). The cut surface was homogeneous without hemorrhage or necrosis.
Histologically, the tumor had a plexiform appearance and tongue-like extension into the muscularis propria, forming a lobulated mass, which was composed of bland-looking spindle cells without significant nuclear atypia, arranged in fascicular or streaming patterns with only limited fibromyxoid or myxoid stroma (Figure 2). The mitotic count was 11 per 50 high-power fields (HPF). No necrosis or lymphovascular invasion was identified, and all surgical margins were negative.
Immunohistochemical analysis indicated the tumor cells were focally positive for KIT (Figure 3a), but negative for DOG1, making a diagnosis of GIST unlikely. Succinate dehydrogenase (SDH)-deficient GIST was also excluded because SDHB expression was retained. Other differential diagnoses, including schwannoma, leiomyoma, glomus tumor, gastroblastoma, inflammatory myofibroblastic tumor (IMT), inflammatory fibroid polyp, and perivascular epithelioid cell tumor (PEComa), were also excluded based on a combination of morphology and results of extensive immunohistochemical workup (Table 1). Of note, the tumor cells stained positive for smooth muscle actin (SMA), CD10, and D2-40 (Figure 3b-e). However, cytokeratin (AE1/AE3), CD34, desmin, h-caldesmon, synaptophysin, S100 protein, HMB45, Melan A, and anaplastic lymphoma kinase (ALK) were negative. Diffuse D2-40 expression made mesothelioma and follicular dendritic cell sarcoma diagnostic concerns, but mesothelial markers (calretinin and WT-1), and follicular dendritic cell markers (CD21, CD23, and ICOS) were negative. The Ki-67 labeling index was 5.2% (91/1736) at the hot spot (Figure 3f).
Finally, because the diagnosis of PMF remained a serious diagnostic consideration, the analysis of a next-generation sequencing panel (Archer FusionPlex Sarcoma Panel) supported by the consultation system of the National Cancer Center (Tokyo, Japan) was requested. This revealed a MALAT1::GLI1 fusion, indicating a diagnosis of PFM. Although the mitotic activity appeared higher compared with that in a previous series of PFM cases, our case was considered not to fit the morphological characteristics of a malignant epithelioid tumor with GLI1 rearrangement [4,7,8].
3. Discussion
PFM is characterized by a distinctive plexiform growth pattern composed of bland spindle to stellate cells embedded within a myxoid or fibromyxoid stroma, often accompanied by vascularity [3]. This morphological appearance frequently overlaps with other gastrointestinal stromal tumors, such as myxoid GIST, nerve sheath tumor, smooth muscle tumor, glomus tumor, gastroblastoma, IMT, and inflammatory fibroid polyp [2,4,5]. PFM presents a significant diagnostic challenge because of its rarity and a variety of mimics, particularly when using small biopsy samples. In the current case, cellular morphology with only limited myxoid stroma and focal KIT expression were also a source of diagnostic confusion.
The molecular features of PFM have been investigated [4,5]. PFM lacks KIT, platelet-derived growth factor-A, and SDH gene mutations that are definitional alterations of GISTs. In 2016, Spans et al. [6] reported that a minor subset of PFM (3/16 cases, 18%) harbored MALAT1::GLI1 gene fusions. Banerjee et al. [10] reported PTCH1 inactivation via gene or chromosomal deletion in 2 of 8 cases of PFM (5 cases used for NGS analysis).
It should be noted that MALAT1::GLI1 fusion has been demonstrated in other tumors, including gastroblastoma and malignant epithelioid tumor with GLI1 rearrangement, which is a recently described epithelioid neoplasm of the soft tissue, typically with S100 protein expression and the variable expression of cytokeratin [7]. In the gastrointestinal tract, only one case involving the jejunum has been reported [8]. Although the mitotic activity and Ki-67 labeling index in our case were relatively higher (11/50 HPF and 5.2%, respectively) compared with those in a previous series of PFM cases (median: 1/50 HPF and less than 2%, respectively) [2,11], the immunophenotype, i.e., negative staining for S100 and cytokeratin, and absence of epithelioid morphology, distinct nuclear atypia, necrosis, and lymphovascular space invasion justified the diagnosis of PFM. Although the follow-up period was limited, there was no evidence of recurrent disease 3 months after resection.
A diagnosis of PFM depends on morphology and immunophenotyping to exclude other mesenchymal tumors, especially GIST. To date, no specific IHC marker for the diagnosis of PFM has been established. The utility of GLI1 IHC has been questioned [5], because MALAT1::GLI1 fusion has been identified in a minor subset of PFM. Hu et al. [11] investigated the immunohistochemical profile of 10 cases of PFM and demonstrated that all cases were positive for vimentin and SMA, and some cases were positive for CD10 (5/10), desmin (5/10), h-caldesmon (6/10), and progesterone receptor (6/10), whereas CD34, S100 protein, estrogen receptor, anaplastic lymphoma kinase (ALK), KIT, and DOG1 were all negative.
The current case highlights the diffuse D2-40 expression in PFM. D2-40 is a monoclonal antibody directed against podoplanin, a transmembrane glycoprotein involved in lymphangiogenesis, cell migration, and epithelial–mesenchymal transition [12]. Our literature search revealed only three single case reports of PFM referring to D2-40 expression. Of these three cases, one showed diffuse expression [13], whereas the remaining two cases were negative [14,15], although, unfortunately, these three case reports did not investigate MALAT1::GLI1 fusion. D2-40 IHC is a commonly used diagnostic tool among pathologists to identify lymphatic vessels, and tumors that could be differential diagnosis of PFM were usually negative for this marker (Table 1); therefore, it might be useful for the diagnosis of PFM. Future studies should examine the utility of D2-40 involving a series of PFM cases with confirmed MALAT1::GLI1 fusion.
4. Conclusions
PFM can be a diagnostic challenge because of the limited or absence of a myxoid background and focal non-specific KIT expression. Diffuse D2-40 expression might be a surrogate for MALAT1::GLI1 fusion when establishing a diagnosis of PFM in an appropriate clinicopathologic context. In this regard, the spectrum of morphology, as represented by the current case, is an essential component for avoiding diagnostic misinterpretations. Further investigations are expected to reveal the diagnostic utility and biological significance of D2-40 expression in PFM.
Author Contributions
KW, KT, KO and KK drafted the manuscript. KE, SI, KY, YK, and MI were involved in patient care and clinical data acquisition. KK, KO, HY and YM performed the pathological diagnosis and supervised the work. YT contributed to clinical management. All authors read and approved the final manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The requirement for ethics committee approval was waived for this single-patient case report in accordance with institutional policy.
Informed Consent Statement
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
The genomic results were provided by the Section of the Promotion and Support for Pathology Diagnosis, Division of Cancer Information Services, NCC Institute for Cancer Control. We thank J. Ludovic Croxford, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PFM | Plexiform fibromyxoma |
| IHC | Immunohistochemistry |
| DOG1 | Discovered on gastrointestinal stromal tumor 1 |
| SMA | Smooth muscle actin |
| SDHB | Succinate dehydrogenase subunit B |
| MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| GLI1 | Glioma-associated oncogene homologue 1 |
| GIST | Gastrointestinal stromal tumor |
| HE | Hematoxylin-eosin |
| SDH | Succinate dehydrogenase |
| HPF | High-power fields |
| IMT | Inflammatory myofibroblastic tumor |
| PEComa | Perivascular epithelioid cell tumor |
| CD | Cluster of differentiation |
| ICOS | Inducible T-cell co-stimulator |
| NGS | Next-generation sequencing |
| WT-1 | Wilms tumor 1 |
References
- Takahashi, Y.; Shimizu, S.; Ishida, T.; Aita, K.; Toida, S.; Fukusato, T.; Mori, S. Plexiform Angiomyxoid Myofibroblastic Tumor of the Stomach. Am. J. Surg. Pathol. 2007, 31, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M; Makhlouf, HR; Sobin, LH; Lasota, J. Plexiform fibromyxoma: a distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am J Surg Pathol 2009, 33, 1624–32. [Google Scholar] [CrossRef]
- Miettinen, M; Fletcher, CD; Kindblom, LG; Tsui, WM. Mesenchymal tumors of the stomach. In WHO Classification of Tumours of the Digestive System, 4th edn; Bosman, FT, Carneiro, F, Hruban, R, Teise, ND, Eds.; IARC: Lyon, 2010; pp. 74–9. [Google Scholar]
- Arslan, M.E.; Li, H.; Fu, Z.; A Jennings, T.; Lee, H. Plexiform fibromyxoma: Review of rare mesenchymal gastric neoplasm and its differential diagnosis. World J. Gastrointest. Oncol. 2021, 13, 409–423. [Google Scholar] [CrossRef]
- Szczepanski, J.; Westerhoff, M.; Schechter, S. Plexiform Fibromyxoma: A Review and Discussion of the Differential Diagnosis of Gastrointestinal Mesenchymal Tumors. Arch. Pathol. Lab. Med. 2025, 149, e298–e304. [Google Scholar] [CrossRef]
- Spans, L.; Fletcher, C.D.; Antonescu, C.R.; Rouquette, A.; Coindre, J.; Sciot, R.; Debiec-Rychter, M. Recurrent MALAT1–GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J. Pathol. 2016, 239, 335–343. [Google Scholar] [CrossRef]
- Antonescu, CR; Agaram, NP; Sung, YS; Zhang, L; Swanson, D; Dickson, BC. A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol 2018, 42, 553–60. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.J.; McEvoy, C.R.E.; Byrne, D.J.; Iravani, A.; Browning, J.; Choong, D.Y.-H.; Yellapu, B.; O’hAire, S.; Smith, K.; Luen, S.J.; et al. A Malignant Neoplasm From the Jejunum With a MALAT1-GLI1 Fusion and 26-Year Survival History. Int. J. Surg. Pathol. 2020, 28, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M; Wang, ZF; Lasota, J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol 2009, 33, 1401–8. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S; Corless, CL; Miettinen, MM; Noh, S; Ustoy, R; Davis, JL; et al. Loss of the PTCH1 subset of plexiform fibromyxoma. J Transl Med 2019, 17, 246. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Chen, H.; Liu, Q.; Wei, J.; Feng, Y.; Fu, W.; Zhang, M.; Wu, H.; Gu, B.; Ren, J. Plexiform fibromyxoma of the stomach: a clinicopathological study of 10 cases. 2017, 10, 10926–10933. [Google Scholar] [PubMed]
- Ordóñez, N.G.; G., N. Podoplanin: A Novel Diagnostic Immunohistochemical Marker. Adv. Anat. Pathol. 2006, 13, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ebi, M.; Nagao, K.; Sugiyama, T.; Yamamoto, K.; Saito, T.; Kurahashi, S.; Yamaguchi, Y.; Adachi, K.; Tamura, Y.; Izawa, S.; et al. Gastric Plexiform Fibromyxoma Resected Using Nonexposed Endoscopic Wall-Inversion Surgery: A Case Report. Case Rep. Gastroenterol. 2022, 16, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Kane, JR; Lewis, N; Lin, R; Villa, C; Larson, A; Wayne, JD; et al. Plexiform fibromyxoma with cotyledon-like serosal growth: a case report of a rare gastric tumor and review of the literature. Oncol Lett 2016, 11, 2189–94. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, F. Plexiform Fibromyxoma: A Rare Mesenchymal Tumor Found in the Esophagus. Am. J. Gastroenterol. 2019, 115, 648–648. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
a) Endoscopic appearance. The submucosal tumor covered by non-neoplastic mucosa is shown. b) Gross appearance of a formalin-fixed specimen.
Figure 1.
a) Endoscopic appearance. The submucosal tumor covered by non-neoplastic mucosa is shown. b) Gross appearance of a formalin-fixed specimen.
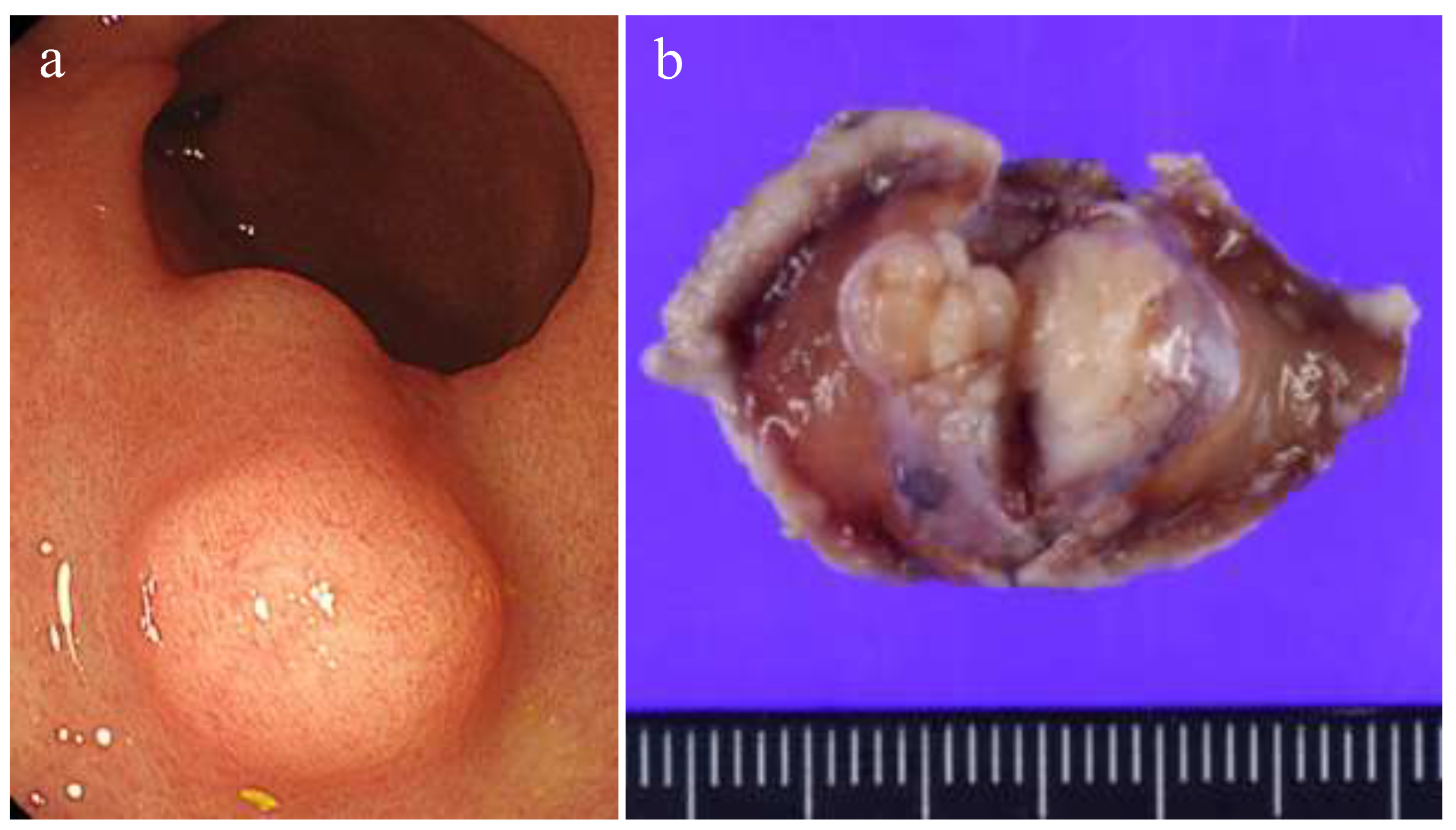
Figure 2.
Histological findings of the tumor. a) Roupe image. The tumor proliferated with a plexiform pattern and infiltrated into the muscularis propria. b) The area of high-cellularity without mucinous stroma (original magnification: × 200). Inset shows a mitotic area (× 400). c) An area of intermediate-cellularity with a small amount of fibromyxoid stoma (× 200). d) Area showing focal myxoid stroma (× 200).
Figure 2.
Histological findings of the tumor. a) Roupe image. The tumor proliferated with a plexiform pattern and infiltrated into the muscularis propria. b) The area of high-cellularity without mucinous stroma (original magnification: × 200). Inset shows a mitotic area (× 400). c) An area of intermediate-cellularity with a small amount of fibromyxoid stoma (× 200). d) Area showing focal myxoid stroma (× 200).
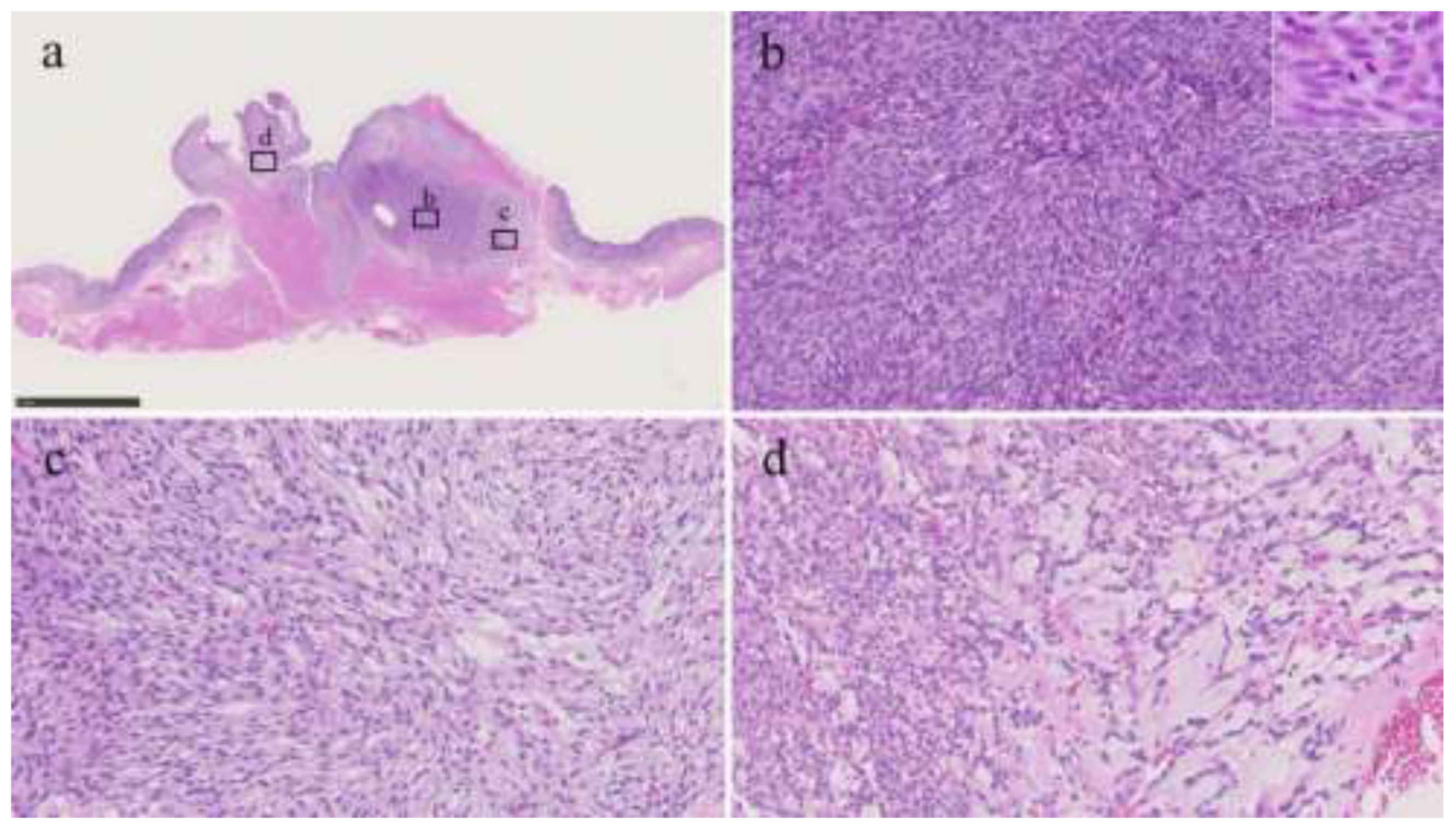
Figure 3.
Immunohistochemical analysis. a) Tumor cells focally express KIT (original magnification: × 200). Tumor cells diffusely positive for b) SMA (× 200) and c) CD10 (× 200). d) Roupe image of D2-40 immunohistochemistry. The entire tumor expressed D2-40. e) Tumor cells are diffusely positive for D2-40 (o× 200). f) The Ki-67 labelling index was approximately 5% (× 200).
Figure 3.
Immunohistochemical analysis. a) Tumor cells focally express KIT (original magnification: × 200). Tumor cells diffusely positive for b) SMA (× 200) and c) CD10 (× 200). d) Roupe image of D2-40 immunohistochemistry. The entire tumor expressed D2-40. e) Tumor cells are diffusely positive for D2-40 (o× 200). f) The Ki-67 labelling index was approximately 5% (× 200).
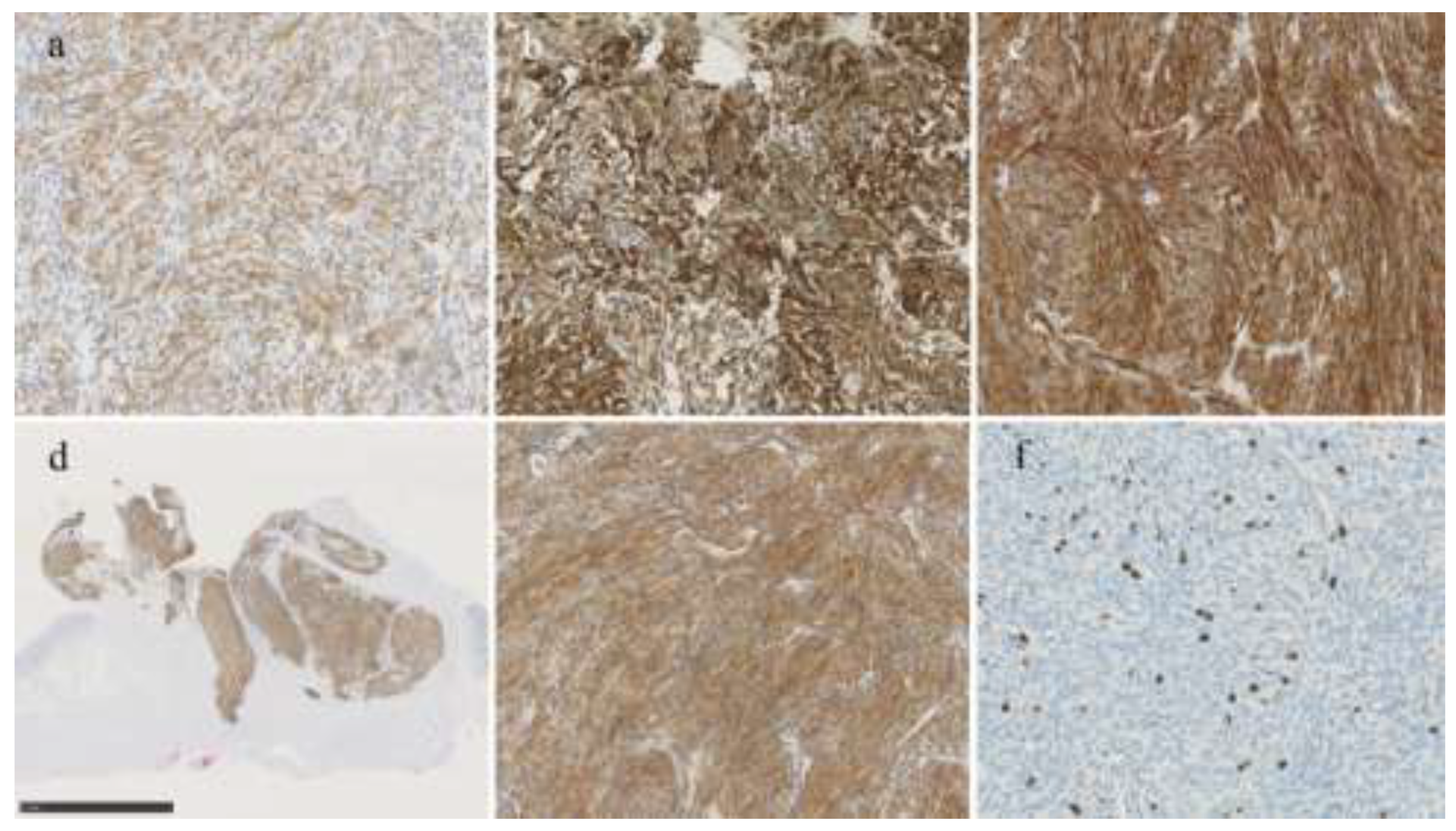

Table 1.
Possible differential diagnoses for plexiform fibromyxoma.
| Tumors | Typical morphology | Useful immunohistochemistry | Molecular alteration |
| Plexiform fibromyxoma | Multinodular and plexiform low-power architecture; uniform spindle cells in a myxoid, fibromyxoid, or collagenous stroma | None (usually positive for SMA, occasionally positive for CD10, desmin, caldesmon, D2-40) | MALAT1::GLI1, PTCH1 inactivation |
| GIST | Intramural, submucosal, or subserosal mass; spindle cell, epithelial, or mixed morphology | Positive for KIT, DOG-1 | KIT, PDGFRA |
| SDH-deficient GIST | Characteristic epithelioid morphology and typically multinodular with plexiform mural involvement | Positive for KIT, DOG-1, loss of SDHB | SDHB, No KIT or PDGFRA mutation |
| Leiomyoma | Fascicles of spindle cells with eosinophilic cytoplasm | Positive for desmin, SMA | |
| Schwannoma | Well-circumscribed mass; areas of cellular and hypocellular spindle cells | Positive for S100 | NF1, NF2 |
| Perineurioma | Uniform spindle cells, most are small colonic polyps | Positive for EMA, Negative for S100 | NF2, BRAF (associated serrated polyp) |
| Glomus tumor | Round glomoid cells; sometimes plexiform growth | Positive for SMA, collagen IV | NOTCH, BRAF |
| PEComa | Epithelioid and/or spindle cells with granular eosinophilic-to-clear cytoplasm | Positive for smooth muscle (desmin, SMA) and melanocytic (HMB45, melan-A, PNL2, MITF, tyrosinase) markers | TSC1/2, TFE3 (minority) |
| Inflammatory fibroid polyp | Hypocellular, with short spindled-to-stellate cells, infiltration of eosinophils and lymphocytes. | Positive for CD34, PDGFRA | PDGFRA |
| Inflammatory myofibroblastic tumor | Loose fascicles of spindle cells without pleomorphism, infiltration of lymphocytes and plasma cells | Positive for SMA, ALK | ALK, ROS1 |
| Solitary fibrous tumor | Fascicles of uniform ovoid-to-spindle cells, staghorn vessels | Positive for CD34, STAT6 | NAB2::STAT6 |
| Synovial sarcoma | Spindle cell (monophasic) or spindle cell with epithelioid-to-glandular (biphasic) | None (focal positivity for keratin and EMA) | SS18::SSX1/2/4 |
| Gastroblastoma | Biphasic with spindle cells and epithelial cells | Epithelial cells are positive for CK, and spindle and epithelial cells are focally positive for CD10 and CD56 | MALAT1::GLI1 |
| Malignant epithelioid tumor with GLI1 rearrangement | Epithelioid, ovoid, round to spindle | Occasionally positive for S100, a subset case focally positive for CK | MALAT1::GLI1 |
SMA, smooth muscle actin; CD, cluster of differentiation; MALAT1, metastasis associated lung adenocarcinoma transcript 1; GLI1, glioma-associated oncogene homologue 1; PTCH1, patched 1; GIST, gastrointestinal stromal tumor; DOG-1, discovered on GIST-1; PDGFRA, platelet-derived growth factor receptor alpha; SDH, succinate dehydrogenase; SDHB, succinate dehydrogenase complex iron sulfur subunit B; NF, neurofibromin; EMA, epithelial membrane antigen; PEComa, perivascular epithelioid cell tumor; HMB45, human melanoma black 45; melan-A, melanoma antigen recognized by T cells 1; MITF, microphthalmia-associated transcription factor; TFE3, transcription factor binding to IGHM enhancer 3; TSC, tuberous sclerosis complex; ALK, anaplastic lymphoma kinase; STAT6, signal transducer and activator of transcription 6; NAB2, NGFI-A binding protein 2; CK, cytokeratin
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



