
Submitted:
15 January 2026
Posted:
19 January 2026
You are already at the latest version
Abstract
Pituitary neuroendocrine tumors (PitNETs) constitute a significant proportion of primary intracranial neoplasms and were historically differentiated based on clinical hormone excess syndromes and tinctorial properties. The 5th edition of the WHO classification introduces a paradigm shift towards the lineage-based taxonomy based on the cell-specific expression of transcription factors (TFs). This overview focuses on the biological justifications and diagnostic value of the core TFs of PIT1, TPIT and SF1 which signify the somatotroph, lactotroph, thyrotroph, corticotroph and gonadotroph lineages respectively. By focusing on TF expressions instead of hormone immunoreactivity, pathologists can better subtype clinically non-functioning tumors, effectively relegating the previously overutilized, null cell category, to about 1% of cases. The TF-based classification is also essential in discriminating high-risk histotypes of silent corticotroph tumors, sparsely granulated somatotrophs, and immature PIT1-lineage PitNETs, which are linked to a higher invasiveness and recurrence. We suggest a practical, stepwise immunohistochemical diagnostic algorithm with the integration of ancillary markers (e.g. GATA3 and ERα) to refine lineage assignment. New molecular correlates such as GNAS and USP8 mutations also add to this framework and guide the use of individualized treatment involving somatostatin analogs or dopamine agonists. And lastly, we discuss the ongoing issues of diagnosis of triple-negative and multilineage tumors and the growing importance of DNA methylation profiling and artificial intelligence in standardized reporting and improving precision management.
Keywords:
pituitary neuroendocrine tumors
; transcription factor–based classification
; immunohistochemistry
; PitNETs
; PIT1
; TPIT
; WHO 5th edition classification
1. Introduction
Pituitary neuroendocrine tumors (PitNETs) are epithelial neuroendocrine neoplasms (NENs) which arise from adenohypophyseal cells [1]. Pituitary tumors make up about 10 to 15 percent of all intracranial neoplasms making them the second most common primary intracranial neoplasm and most common neuroendocrine neoplasm in the human body [2,3]. It is estimated that clinically relevant PitNETs occur at a prevalence of 78-116 cases per 100,000 people [1,2,3]. Patients typically present either with sellar mass effects, such as visual impairment, headache, or hypopituitarism, or with syndromes due to hormonal hypersecretion, such as acromegaly or Cushing disease [3,4,5]. Although the majority of PitNETs are non-cancerous and clinically indolent, there are subsets that demonstrate significant variability and aggressiveness with invasive growth, high proliferation, and resistance to standard treatments [5,6,7].
In the past, adenohypophyseal neoplasms were referred to as pituitary adenomas (PAs) [6]. Initial taxonomies were simple as they were mostly based on tinctorial properties of acidophil, basophil, and chromophobe [4,8]. Further developments in pathology led to the shift towards a morpho-functional paradigm, which combined clinical status (functional or non-functional), hormonal profiles (immunohistochemistry), and ultrastructure (electron microscopy) [4,8,9]. On this basis, the 2004 WHO classification defined seven main types - prolactin (PRL), growth hormone (GH), adrenocorticotrophic hormone (ACTH), thyroid-stimulating hormone (TSH), and gonadotropin (LH/FSH) secreting lesions - and subdivided them into 13 ultrastructural types [4]. Despite this, the hormonal classification had significant limitations. The common use of the term ‘pituitary adenoma’ was problematic because it implied a purely benign behavior which contradicted the existence of tumors that are locally invasive, clinically aggressive, or, in rare cases, metastatic [4,6,7,8]. The WHO 2004 classification introduced the category ‘atypical adenoma’, defined by proliferation markers such as Ki-67 index of >3% or diffuse p53 staining. However, this category has proven unreliable due to inconsistent prognosis [4,5,6,10]. It also included the poorly defined category of null cell adenoma (non-secretory adenoma) [4,9]. The weaknesses of hormone immunohistochemistry (IHC) suggested that the use of secretory products alone cannot be enough to capture the complexity of tumor cell lineage.
As a response to these limitations, in 2017, the International Pituitary Pathology Club suggested the new term “Pituitary Neuroendocrine Tumor (PitNET)” as more reflective of the epithelial neuroendocrine etiology of such tumors and their spectrum of behavior ranging from indolent to locally invasive [1,3,4,5,8,11,12]. This nomenclature has since been preferred in the 5th edition of the WHO Classification of Endocrine and Neuroendocrine Tumors (2022), although the term “pituitary adenoma” is acceptable [3,5,11]. This modern classification identifies the cell lineage of a tumor by transcription factor (TF) IHC. Three major TF families are currently in common use in diagnostic IHC: PIT1 (for somatotroph, lactotroph or thyrotroph lineage), TPIT (for corticotroph lineage), and SF-1 (for gonadotroph lineage), along with supporting markers such as GATA3, ERα [4,10,11,12]. The implementation of TF-based IHC has enabled more accurate subtyping of PitNETs. It allows reclassification of many tumors previously labeled as ‘null-cell tumors’ as gonadotroph PitNETs and helps define high-risk subtypes such as sparsely granulated somatotroph, silent corticotroph, Crooke cell, and immature PIT1-lineage tumors, with important implications for prognosis and therapy [2,4,5,9,11,12,13].
However, there are still practical issues, including limitations in IHC standardization (e.g. fixation protocols, cut-off levels) and the unreliability of antibody supply (e.g., SF-1 and TPIT). TF-negative tumors (null-cell tumors) and rare multilineage tumors (e.g., PIT1 -SF1 co-expressions) still pose a challenge to the existing classification system, so they require further molecular research [4,9,11,12,13].
This review provides a practical overview of TF-based classification of PitNETs in the 5th WHO era, focusing on the biology of lineage-defining transcription factors, stepwise immunohistochemical algorithms, molecular and clinicopathologic correlates, and common diagnostic challenges. Emphasis is placed on pragmatic guidance for routine surgical pathology and multidisciplinary decision-making.
2. Historical and Conceptual Background
2.1. Evolution of Pituitary Tumor Classification
The classification of adenohypophyseal neoplasms has undergone a significant change, evolving from systems based largely on clinical and morphological criteria to frameworks incorporating immunohistochemical and molecular stratification [5,7,9]. Initial classification systems were largely established based on the clinical presentation of one of the hormone excess syndromes, such as acromegaly, hyperthyroidism, or Cushing disease [5]. Early classification schemes also relied on tinctorial properties of the neoplastic cells, using conventional stains such as hematoxylin–eosin to separate tumors into acidophilic, basophilic, or chromophobic types. This early classification, however, was subjective and poorly reproducible [14].
The classification system progressed with the establishment of IHC, which provided the possibility to directly detect the pituitary hormones in the tumor cells (e.g., GH, PRL, ACTH, TSH, FSH, LH) [3,10,14]. IHC enabled a more precise matching of tumor histology with their clinical endocrine syndromes and made it possible to classify tumors by simply the hormone that they synthesize (e.g. a GH-producing adenoma) [3,10].
Despite these developments, the hormone-based classification had some strong clinicopathological contradictions [1]. Many clinically non-functioning PitNETs (NF-PitNETs), without overt clinical manifestations of hormonal hypersecretion, were found to be positive for at least one pituitary hormone on IHC. Such tumors were referred to as silent neoplasms or silent adenomas [15]. An example of such a tumor is silent corticotroph PitNET (historically ‘silent corticotroph adenoma’) which produces ACTH but does not induce hypercortisolism [12,15]. Besides, the tumors which contain no identifiable anterior pituitary hormones using IHC were classified to the ambiguous group of null cell adenomas [14,15]. These gaps in the earlier classification highlight the necessity of a paradigm of classification based on the fundamental biology of pituitary cell differentiation and not necessarily based on hormone detection only. The major changes in the WHO classification of pituitary tumors over time are summarized in Table 1.
2.2. Rationale for Lineage-Based, TF-Centered Classification
The limitations of hormone-based classification systems prompted a shift toward cell lineage–based classification using pituitary-specific transcription factors (TFs). TFs are regulatory proteins important for embryogenesis and differentiation of adenohypophyseal cells and regulate the expression of the hormone-encoding genes. This classification system recognizes three major cell lineages, each of which is controlled by a specific core TF [10,12,15]:
- PIT1-lineage specifies somatotrophs (GH-secreting), lactotrophs (PRL-secreting), and thyrotrophs (TSH-secreting).
- TPIT-lineage specifies corticotrophs (ACTH-secreting).
- SF1-lineage specifies gonadotrophs (FSH/LH-secreting).
This lineage-based approach was adopted in the 2017 WHO classification of pituitary tumors and further refined in the 2022 WHO Classification of Endocrine and Neuroendocrine Tumors. In the 2017 WHO classification, the term ‘pituitary neuroendocrine tumor’ (PitNET) was introduced to align pituitary tumors with the broader classification of neuroendocrine neoplasms [1,3,5,15]. The incorporation of TFs has revolutionized diagnosis of PitNETs, especially the non-functioning tumors. The systematic use of TF IHC has shown that a large proportion of tumors previously diagnosed as null cell adenomas actually represent silent lineage-specific tumors [1,9,12]. This observation explains why TF-based classification can be used to represent cellular origin more accurately than hormone expression only [3].
TF-based classification is important for the recognition and classification of the high-risk subtypes regardless of clinical hormonal activity. The WHO classification associates several histological subtypes with potentially aggressive behavior, and a TF-based classification is essential for their identification [5]:
- PIT1-lineage aggressive tumors include sparsely granulated somatotroph tumors (more aggressive and often resistant to somatostatin analogue (SSA) therapy than their densely granulated counterparts), acidophil stem cell tumors, and immature PIT1-lineage tumors (formerly classified as silent subtype 3 adenomas with poor differentiation and increased potential for recurrence and invasiveness).
- TPIT-lineage aggressive tumors include silent corticotroph tumors (that are more likely to exhibit enhanced proliferative and invasive capacity compared to silent gonadotroph tumors), and the rare but highly aggressive Crooke cell tumors.
TF-based classification provides a basis for predicting tumor behavior, responsiveness to medical treatment, and overall prognosis by assigning each PitNET to a specific cell lineage (PIT1, TPIT, or SF-1) [1].
3. Biology of Pituitary Lineage-Defining Transcription Factors
3.1. PIT1 (POU1F1) and PIT1-Lineage Tumors
The Pituitary-specific positive transcription factor (PIT1) or POU1F1 is a nuclear transcription factor of the POU-domain family, which is necessary in the differentiation, proliferation, and survival of the somatotroph, lactotroph and thyrotroph cells [12,17]. During pituitary embryogenesis, PIT1 expression is induced downstream of PROP1 TF and continues to be expressed in the adult pituitary, regulating the production of GH, PRL, and TSH [12].
PIT1-lineage PitNETs are divided into subgroups based on granulation pattern, hormone expression, and cytoskeletal features [5,14]:
- Somatotroph PitNETs are classified into densely granulated and sparsely granulated tumors. Densely granulated tumors include GH positive acidophilic cells with characteristic perinuclear keratin pattern. Sparsely granulated tumors often have chromophobic cytology and contain fibrous bodies (cytoplasmic keratin aggregates) and are associated with more aggressive behavior and poorer clinical response following treatment with first-generation somatostatin analogues.
- Lactotroph PitNETs are mostly sparsely granulated and show a typical juxtanuclear “dot-like” immunostaining of PRL. The rare densely granulated types show diffuse cytoplasmic PRL expression.
- Thyrotroph PitNETs are rare and often have fibrotic or spindle-cell morphology, secrete TSH and are positive to GATA3 and PIT1 TF.
- Among plurihormonal PIT1-lineage tumors, immature PIT1-lineage tumors and acidophil stem cell tumors are often large and clinically aggressive, whereas mature plurihormonal PIT1-lineage tumors show more variable behavior. These entities exhibit lineage infidelity, with co-expression of several PIT1-lineage hormones without full terminal differentiation.
The main anterior pituitary cell lineages and their associated hormones are illustrated in Figure 1.
3.2. TPIT (TBX19) and Corticotroph Tumors
T-box pituitary transcription factor (TPIT), encoded by TBX19 gene, is the master regulator of the corticotroph lineage and controls the final differentiation of cells that produce pro-opiomelanocortin (POMC) [18,19]. TPIT binds to the POMC promoter together with other transcription factors including Pitx1 to drive the production of ACTH [12,18].
TPIT-lineage PitNETs are typically divided into two clinico-pathological categories:
- Sparsely granulated or silent corticotroph tumors (SCTs) are clinically non-functional but immunoreactive to TPIT and ACTH [5,20]. These tumors present as macroadenomas at the time of diagnosis and portray more aggressive clinical behaviors such as greater recurrence and invasion rates as compared to gonadotroph tumors [19].
Within this lineage, a distinctive variant termed Crooke cell PitNET is recognized. It is a highly aggressive corticotroph tumor characterized by extensive perinuclear hyaline (keratin) ring-like cytoplasmic change in the majority of tumor cells. The hyaline changes indicate a paradoxical state of cells, where the cells exhibit characteristics of the hormone-mediated feedback inhibition and at the same time show elevated proliferative indices and invasive potential [5].
3.3. SF1 (NR5A1) and Gonadotroph Tumors
Steroidogenic Factor 1 (SF1), encoded by NR5A1 gene, is a nuclear receptor TF which plays a crucial role in the differentiation and functional regulation of the gonadotroph lineage, which produces FSH and LH [12,21]. Unlike the PIT1 and TPIT lineages, which are often characterized by the clinical endocrine syndromes, SF1-lineage PitNETs are, in general, clinically non-functional and are only revealed through the presence of mass effect symptoms [12,22].
SF1-lineage PitNETs constitute the majority of clinically non-functioning pituitary adenomas [5]. These tumors usually show patchy or weakly stained FSH, LH, and the alpha -subunit on IHC. But diffuse nuclear SF1expression is the most sensitive and specific marker of identifying these tumors. Even though these tumors tend to be quite large, they tend to exhibit a more indolent behavior with reduced recurrence rates after complete resection compared to silent corticotroph or immature PIT1-lineage tumors [14,20].
3.4. Supporting and Additional Markers: GATA3, ERα, Others
The diagnostic accuracy is further enhanced by the assessment of ancillary markers such as GATA3 and ERα that complement the lineage assignment [5]:
- GATA binding protein 3 (GATA3) is a nuclear protein expressed in a subset of PIT1-lineage tumors, particularly thyrotroph PitNETs, and can also be detected in some gonadotroph tumors [17]. It is a useful adjunct marker for confirming thyrotroph differentiation (co-localizing with PIT1) and can aid in lineage assignment when SF-1 immunostaining is inconclusive [12].
- Estrogen receptor alpha (ERα ) is a TF that plays a crucial role alongside PIT1 in the differentiation and maintenance of lactotrophs and mammosomatotrophs [17]. Its expression has been associated with higher DRD2 expression and may contribute to the good clinical responsiveness of many lactotroph tumors to dopamine agonist therapy [20].
Some other important markers are PROP1, PITX1, PITX2. PROP1, an early developmental TF which precedes PIT1, is upregulated in certain immature PitNETs. It is considered to be a marker of the progenitor state rather than a terminal lineage. PITX1 and PITX2 complement TPIT in corticotroph differentiation, especially activating POMC gene expression [12].
3.5. Link with Germline and Somatic Driver Events
It is becoming well known that molecular alterations in PitNETs align with TF-defined cell lineages. In the PIT1 lineage, GNAS mutations (gain-of-function) have been detected in up to 40 percent of somatotroph tumors, particularly in densely granulated subtypes, and are associated with smaller tumor size and increased responsiveness to somatostatin analog therapy. Germline alterations such as AIP mutations (often associated with young-onset, large somatotroph PitNETs) and MEN1 mutations (frequently associated with lactotroph and other PitNETs) are enriched in tumors with more aggressive clinical behavior [5,22,23].
In the TPIT-lineage, gain-of-function somatic USP8 mutations are detected in 30 to 50 percent of functioning corticotroph tumors [20,22]. This alteration results in defects of epidermal growth factor receptor (EGFR) degradation, which stimulates transcription of POMC and, consequently, hypersecretion of ACTH. Other molecular alterations seen in the USP8 wild-type cases include USP48 and BRAF mutations that result in increased stimulation of the MAPK pathway [19,22].
By contrast, SF-1-lineage PitNETs have not been linked to a single dominant recurrent driver event; instead, they appear as distinct clusters in methylation and transcriptomic studies, while their somatic mutational landscape remains relatively heterogeneous and less well characterized [22].
4. Practical TF-Based Immunohistochemical Classification
4.1. Recommended IHC Panels and Technical Issues
In the 5th edition of the WHO Classification, accurate diagnosis of PitNETs requires a shift from hormone-only panels to a lineage-based diagnostic approach using transcription factors (TFs). The minimal recommended panel of any pituitary neoplasms should include the three fundamental TFs (PIT1, TPIT and SF1) as well as pan-neuroendocrine markers such as synaptophysin and chromogranin A [5,24,25]. Ki-67 can be used to assess proliferative activity, although PitNETs are not graded into G1-G3 categories as in other NENs [5,26]. For a comprehensive evaluation, an extended immunohistochemical panel is recommended that includes GATA3, ERα, and low molecular weight cytokeratin (LMWCK) such as CAM5.2 clone, to the core TF and neuroendocrine markers [5,14,25].
There are significant challenges in technical execution and interpretation. Pre-analytic variables such as tissue fixation and antigen retrieval are critical. Prolonged formalin fixation can cause extensive cross-linking and epitope masking, leading to reduced antigen detectability and false-negative staining [10,27]. Pitfalls include patchy or weak nuclear staining that may be observed in gonadotroph tumors with SF1 expression. In addition, a universal cut-off point of TF positivity does not exist; different studies have used various cut-offs ranging from 5 to 80 percent, while some studies have used a proportion system [9].

Table 2.
Recommended minimal and extended TF-IHC panel.
| Markers | Primary Function |
|---|---|
| Minimal Panel | |
| PIT1 | Lineage marker for somatotroph, lactotroph, and thyrotroph cells |
| TPIT | Lineage marker for corticotroph cells |
| SF1 | Lineage marker for gonadotroph cells |
| Synaptophysin | Pan-neuroendocrine marker to confirm neuroendocrine differentiation; highly sensitive |
| Chromogranin A | Pan-neuroendocrine marker; less sensitive than synaptophysin |
| Ki-67 (MIB1) | Proliferation marker to assess tumor growth potential |
| Extended Panel | |
| GATA3 | Marker for thyrotroph and gonadotroph |
| ERα | Marker for lactotroph and mammosomatotroph |
| CAM5.2 | Distinguishes high-risk subtypes; detects fibrous bodies of sparsely granulated somatotroph PitNETs |
4.2. Stepwise Algorithm for Work-Up of a Pituitary Region Neoplasm
The diagnostic work-up starts with the H&E-stains to verify the presence of a monomorphic population of cells with a “salt and pepper” like chromatin appearance of neuroendocrine differentiation. The first line IHC includes synaptophysin and chromogranin A [25]. After the neuroendocrine lineage has been confirmed, then the TF panel is utilized to place the tumor into one of the three major lineages. In case of PIT1+, it is further categorized into the somatotroph, lactotroph, thyrotroph, or plurihormonal PIT1-lineage tumors. TPIT+ and SF1+ define corticotroph and gonadotroph tumors respectively [14,25]. Lineage assignment can be refined using hormone IHC, GATA3, ERα, and LMWCK like CAM5.2. If the tumor is negative for all core TFs, pathologists should first exclude posterior pituitary tumors (PPTs) and other sellar lesions; a diagnosis of true null cell PitNET should be reserved for rare cases in which no lineage-defining TF or alternative origin can be demonstrated. Pathologists should test for markers like TTF1 (pituicyte-derived tumors), GFAP (glial lesion), and neuronal markers like neurofilament (gangliocytomas or neurocytomas) [5,14,28].
4.3. Mapping TF/Hormone Patterns to WHO 5th PitNET Subtypes
The WHO 5th edition PitNETs classification incorporates the expression of TFs, hormonal profiles, and clinical status to define specific tumor subtypes. This framework offers a robust basis for diagnosis, prognosis, and treatment decisions. Table 3 provides an overview of the main characteristics of the major PitNET subtypes.
4.4. Distinguishing PitNETs from TTF-1–Positive Posterior Pituitary Tumors
There is a critical diagnostic distinction between PitNETs and PPTs. Pituicytes give rise to non-neuroendocrine neoplasms or PPTs that include pituicytoma, spindle cell oncocytoma (SCO), granular cell tumor (GCT), and ependymal pituicytoma. These tumors share strong nuclear expression of TTF-1, which is highly characteristic of posterior pituitary (pituicyte) tumors in the sellar region [14,28].
PPTs consistently express nuclear TTF-1 and are negative for pituitary hormones and lineage-defining TFs (PIT1, TPIT, SF-1), in contrast to PitNETs. Neuroendocrine markers such as synaptophysin and chromogranin A may be weak or variable, reflecting their pituicyte/glial-like nature rather than true adenohypophyseal origin [14,28,29]. On the other hand, PitNETs are almost invariably TTF1 negative. Demonstration of TTF-1 positivity is especially important in spindle cell or oncocytic sellar tumors, in which SCO could otherwise be mistaken for an oncocytic null cell PitNET or thyrotroph PitNET [14,28]. Recognizing PPTs as TTF-1–positive, non-adenohypophyseal tumors is crucial for accurate classification and prognostication, and prevents mislabeling them as PitNETs.
5. Molecular and Clinicopathologic Correlates
5.1. Genetic and Epigenetic Features by Lineage
PIT1-lineage PitNETs consist of somatotroph, lactotroph, and thyrotroph tumors with different but closely related molecular signature characteristics [22]. The PIT1-lineage has a distinctive pattern of diffuse DNA hypomethylation and chromosomal instability in comparison to other groups [30]. 30 to 40 percent of somatotroph PitNETs have heterozygous gain-of-function mutations in the GNAS gene that increases the cAMP production leading to increase in cell proliferation activity [20,31]. Some studies suggest that GNAS-mutated somatotroph PitNETs may show higher expression of DRD2 and somatostatin receptors, which could influence responsiveness to medical therapy. Most lactotroph PitNETs are sporadic. A subset of prolactinomas harbor somatic variants, including mutations in the splicing factor gene SF3B1, which have been associated with more aggressive clinical behavior and reduced responsiveness to standard treatment. These tumors also express higher levels of DRD2. Thyrotroph PitNETs have increased Wnt4 gene expression and subsets of SSTR [20]. On the other hand, germline mutations in the Aryl hydrocarbon receptor Interacting Protein (AIP) gene in the PIT1-lineage have been linked to the aggressive and large, sparsely granulated somatotroph tumors in the young patients who in most cases are resistant to standard medical treatment [20,23,31].
TPIT-lineage PitNETs have recurring somatic mutations within the USP8 gene found in around 20 to 60 percent of corticotrophic tumors [19,20,22]. These gain-of-function mutations avoid the breakdown of epidermal growth factor receptor (EGFR), which increases the transcription of POMC and leads to ACTH hyper-secretion [19,20,22,31]. In the USP8 wild-type, regular mutations of USP48 and BRAF (V600E) have been observed, which also increase POMC promoter activity [19,22,31]. In the case of aggressive or metastatic TPIT-lineage tumors, mutations in ATRX and TP53 are becoming important indicators of poor clinical behavior [6].
To date, SF-1-lineage PitNETs have not been linked to a single dominant recurrent driver mutation; their somatic mutational landscape appears more heterogeneous [22]. Nevertheless, these tumors do not share methylation clusters with other PitNETs and are often hypermethylated to silence tumor suppressor genes such as MEG3 [20,22]. But, some somatotroph PitNETs and even corticotroph PitNETs can have gonadotroph signatures [30]. Although it is usually indolent, fast-growing gonadotroph tumors exhibit selective mRNA expression of genes associated with epithelial-mesenchymal transition (EMT) [15,22].
5.2. Proliferation, High-Risk Histotypes, and Aggressiveness
The WHO has recognized that certain histopathological subtypes of PitNETs have a high risk of invasion and recurrence, many of which are defined in the PIT1- and TPIT-lineages [6,20,27]. In the PIT1-lineage, sparsely granulated somatotroph PitNETs are considered high-risk, as they are more likely to be larger in size and more invasive in comparison with their densely granulated counterparts [27]. In the TPIT-lineage, silent corticotroph tumors are clinically aggressive. These tumors are characterized by a high rate of cavernous sinus invasion, and development of post-surgery recurrence as opposed to other clinically non-functional PitNETs, especially silent gonadotroph tumors [15,19,20]. Crooke cell tumor is a rare but aggressive type of TPIT-lineage tumor, which is marked by extensive Crooke hyaline change, and shows high invasive growth and therapeutic resistance [5,19,20,27]. High-risk histotypes also include plurihormonal PIT1-lineage tumors, particularly the immature PIT1-lineage tumor (previously referred to as silent subtype 3). These tumors are often very large and invasive, and they usually have high recurrence rates, especially in young patients [5,12,20].
Aggressive behavior in these high-risk subtypes is frequently associated with elevated proliferative indices (e.g., Ki-67 ≥3% and increased mitotic activity), although clinically aggressive behavior can also be observed in tumors with relatively low proliferation [15].
5.3. Treatment Implications
Determination of the lineage forms a critical framework to classify therapeutic interventions for PitNETs. PIT1-lineage tumors express different subtypes of SSTR. Densely granulated somatotroph tumors typically have high SSTR2 expression and usually show the best response to first-generation somatostatin analogs (SSAs), such as octreotide and lanreotide [10,22,31]. In contrast, sparsely granulated somatotroph tumors have less SSTR2 expression and are therefore less sensitive to first-generation SSAs. In these circumstances, they have a better response to second-generation analogs like pasireotide or GH receptor antagonists like pegvisomant [22,31]. Lactotroph PitNETs are uniquely characterized by strong expression of ERα in the cell, which is associated with high DRD2 expression, which explains their high sensitivity to dopamine receptor agonists like cabergoline [22,32].
In the TPIT-lineage, corticotroph tumors express SSTR5 and relatively low levels of SSTR2, as chronic hypercortisolism downregulates SSTR2 expression [31,33]. This receptor expression justifies the existing state of pasireotide being the only pituitary-targeting therapeutic agent approved in Cushing disease [22,33]. EGFR-targeted therapy such as gefitinib in tumors with USP8 mutations, has shown potential in attenuating ACTH secretion and cell proliferation in preclinical studies [19,22,31].
In all the lineages, temozolomide remains the first-line systemic therapy to aggressive and metastatic PitNETs, and therapy outcome has a reverse relationship with a profile of O6-methylguanine DNA methyltransferase (MGMT) expression [10,27,32]. Key molecular drivers, high-risk histotypes and their therapeutic correlates across PIT1-, TPIT-, and SF-1-lineage PitNETs are summarized in Table 4.
6. Diagnostic Challenges and Pitfalls
6.1. TF-Negative or “Triple Negative” Neoplasms
TF based classification has significantly reduced tumor types which were previously referred to as null-cell adenomas [5]. In the current 5th WHO era, true null-cell PitNETs, characterized by total absence of PIT1, TPIT, and SF1, are very rare, approximately one percent or less of pituitary neoplasms [34]. Most of the tumors that were once classified as null-cells adenomas have been reclassified as silent gonadotroph PitNETs with the help of SF1 IHC [10]. When a tumor has a triple-negative phenotype, technical factors should be ruled out first because poor fixation or failure to retrieve antigens may result in false negative nuclear staining [9]. After technical variables are excluded, non-adenohypophyseal sellar lesions that mimic PitNET architecture, such as PPTs and metastatic NENs, should be included in the differential diagnosis [5,17]. Rarely, true TF negative PitNETs can be highly undifferentiated or primitive neoplasms with no markers of terminal differentiation [9]. In these situations, re-staining using optimized protocols and an extended IHC panel (that includes GATA3, NeuroD1) can clarify a diagnosis [12]. In ambiguous triple-negative cases, DNA methylation profiling has demonstrated a powerful supplementary method, being able to put the tumors under discrete classes of molecules even in the absence of lineage-specific protein expression [6,35].
6.2. Multilineage/Mixed-TF Tumors
One major area of diagnostic challenge in the 5th WHO era is PitNETs that express more than a single lineage-defining TFs in a monomorphous population of tumor cells. This is sometimes referred to as lineage infidelity. Among multilineage PitNETs, co-expression of PIT1 and SF-1 is one of the patterns most frequently described, particularly within PIT1-lineage tumors [35]. While some cases may have double adenomas or collision lesions where two different tumors co-exist, other cases show diffuse co-expression in every tumor cell [12]. These unusual tumors are usually classified within the spectrum of mixed or plurihormonal PitNETs, although their optimal taxonomic position is still under discussion [5].
Most recent epigenetic results, especially relating to DNA methylation profiling, suggest that PIT1/SF1 co-expressing tumors (recently proposed as somatogonadotroph PitNETs) are molecular clusters, which are dissimilar to the conventional somatotroph PitNETs [26,35]. Clinically, these multi-lineage PitNETs, particularly those that co-express PIT1/SF1, are linked to the more aggressive phenotype, such as higher cavernous sinus invasion rates (45 to 82 percent) and a higher probability of postoperative recurrence [35]. Nevertheless, the existing data is still scarce, and more longitudinal studies are needed to give a clear picture on the real prognostic value of multilineage PitNETs.
6.3. Discordant Hormone and TF Profiles
Discordance between hormone IHC and TF expression is a common diagnostic situation that requires careful combination of morphologic, immunophenotypic and clinical evidence. Common discordant patterns include hormone negative tumors that express lineage-defining TFs [15]. In such cases, TF expression indicates transcriptional lineage commitment, and the absence or low levels of hormone staining may be caused by an inefficient production of hormones, a high rate of intracellular degradation, or a failure to form secretory granules [1,5]. On the other hand, tumors that are hormone positive, yet lack the anticipated TF, are more likely to be due to technical issues, including, but not limited to, poor fixation, low sensitivity of antibodies, or non-specific staining of entrapped non-neoplastic adenohypophyseal cells [9]. The 5th WHO classification establishes a hierarchical method of diagnosis in which TF expression is more important than hormone immunoreactivity in assigning the lineage [1,5]. The diagnosis of diffuse nuclear TPIT expression is therefore adequate to make a diagnosis of silent corticotroph PitNETs in the absence of ACTH immunoreactivity or with a focal localization of ACTH immunoreactivity. However, the results of hormone and TF should be considered within the broader clinical and biochemical context to differentiate the truly silent tumors from lesions related to subtle or intermittent hormonal hypersecretion.
6.4. Differentiating Null Cell PitNETs from Non-PitNET Sellar Tumors
Diagnosis of a true PitNET as opposed to non-adenohypophyseal sellar neoplasms is a difficult task. Since the definition of null-cell PitNETs is based on exclusion, histopathologists have to carefully remove mimickers, such as pituicytoma, spindle -cell oncocytoma, and granular-cell tumor [5,17,28]. These posterior pituitary neoplasms consistently show nuclear TTF-1 expression and lack pituitary hormone and PitNET TF expression; neuroendocrine markers such as synaptophysin may be weak or variable. TTF-1 is therefore a highly useful discriminatory marker in the differential diagnosis with PitNETs. A broad IHC panel is required in the comprehensive diagnostic work up of a TF/hormone negative sellar mass. TTF1 is critical for identifying posterior pituitary lineage, GFAP for glial or pituicyte-derived lesions, EMA for ependymal pituicytomas or meningiomas [14,28]. Metastatic NENs should also be systematically ruled out, as they tend to express synaptophysin, chromogranin like PitNETs but lack pituitary specific TFs. But they express site-specific markers like CDX2 (intestinal), TTF1/Napsin A (lung) [36,37]. An additional diagnostic pitfall arises when a non-pituitary neoplasm entraps normal adenohypophyseal tissue and the tissue may appear as multilineage differentiation. As a result, it becomes important to assess the reticulin or collagen IV framework, which is usually disrupted by PitNETs, but is preserved by non-neoplastic pituitary tissue entrapped within the acinar space [5]. Combination of morphology, IHC, and structural evaluations is crucial to avoid misclassification and provide correct diagnosis in these cases which are difficult to diagnose.
7. Implementation in Reporting and Future Directions
7.1. How to Report: Minimum Dataset and Suggested Wording
The 5th edition of the WHO Classification of Endocrine and Neuroendocrine Tumors requires the inclusion of morphological, immunophenotypic, and appropriate clinical data in pathology reporting. At least, pituitary tumor reports should include the description of morphological characteristics of the adenohypophyseal cells population, as well as the findings of a comprehensive IHC panel [5,9].
The lineage assignment is to be primarily performed based on the analysis of lineage-determining TFs, such as PIT1, TPIT, and SF1, supported with ERα and GATA3 in required cases. This TF panel should be conjugated with six anterior pituitary hormones and the a-subunit [9]. LMWCK staining, especially with CAM5.2 clones, is important in the identification of high-risk subtypes such as sparsely granulated somatotroph PitNETs, which display perinuclear fibrous bodies [5,27]. The level of proliferative activity should be reported with the help of the Ki-67 labelling index, which is measured in hot spots and reported in percentage or number of positive cells per mm2 [26]. Although the independent prognostic value of p53 IHC is controversial, it should be added where possible to identify the underlying genomic instability in aggressive tumors [12,27].
The final diagnosis should follow the WHO 5th edition nomenclature and designate the lesion as a PitNET with a specific histological subtype (e.g., ‘immature PIT1-lineage PitNET’). A diagnosis of null cell PitNET should be reserved as a diagnosis of exclusion for tumors that are negative for all lineage-defining TFs and pituitary hormones, after confirming adequate technical quality and excluding posterior pituitary tumors and other non-adenohypophyseal sellar lesions [5].
7.2. Impact on Multidisciplinary Management
The correct TF-based lineage and subtype assignment has a direct implication on the postoperative management and long-term monitoring, which enforces the cooperation between pathologists, endocrinologists, neurosurgeons and oncologists [26,27]. In the PIT1 lineage, subtyping can be refined especially in relation to making therapeutic decisions. Densely granulated somatotroph PitNET, as indicated by a strong expression of SSTR2, respond strongly to first-generation SSAs, but sparsely granulated with lower levels of SSTR2 and relative predominance of SSTR5 may need second-generation agents, like pasireotide or GH receptor antagonists like pegvisomant [5,27]. Similarly, in lactotroph PitNET, ERα expression correlates with high DRD2 and reliably predicts sensitivity for dopamine agonists [32]. The lineage assignment also helps to classify high-risk subtypes such as silent corticotroph PitNETs, Crooke cell tumors and immature PIT1-lineage PitNETs. They have a notably high proportion of cavernous sinus invasion, early recurrence and diminished response to standard medical treatment. These patients often warrant closer postoperative monitoring with more frequent MRI follow-up and earlier consideration of adjuvant radiotherapy or systemic treatment such as temozolomide [34,38].
Moreover, certain IHC and morphological patterns suggest the review of underlying genetic syndromes. The presence of sparsely granulated somatotroph PitNETs in young patients should make physicians consider AIP mutations whereas multifocal PIT1-lineage tumors might indicate MEN1 syndrome [5,26]. Thus, accurate pathological diagnosis does not only inform immediate management, but also informs genetic counseling, management follow up strategies and clinical trial eligibility.
7.3. Future Perspectives
The future of pituitary pathology lies in the adoption of TF-based histopathology combined with pangenomic methods, particularly, DNA methylation [20]. Methylation profiling has emerged as a molecular gold standard and can solve the diagnostically challenging cases, such as TF-negative, multilineage, or poorly differentiated PitNETs whose diagnosis is uncertain with conventional IHC [12,35].
Routine diagnosis is expected to further transform along with the advancement in digital pathology and artificial intelligence. TF expression, Ki-67 labeling indices, and mitotic activity can be objectively measured with automated image analysis systems and decreases the extensive inter-observer variability introduced with manual scoring [1,26]. Furthermore, the recent machine-learning models that combine histopathology with radiologic characteristics have shown a promise and capability in predicting specific subtypes such as densely versus sparsely granulated somatotroph PitNETs, and even TF-defined lineage patterns pre-operatively, which may inform the surgical planning and initial therapeutic choices.
Despite such advances, the extensive clinical application is dependent on standardization. TF antibody clone harmonization, consensus IHC positivity thresholds, and robust inter-laboratory quality control initiatives will be necessary to provide diagnostic reproducibility [13,26]. With such technical and logistical obstacles resolved, combined morphologic, immunophenotypic, and molecular hierarchy will redesign precision diagnostics and customized therapy.
8. Conclusions
The conversion to a TF-based classification system has become the essential foundation of modern diagnostic PitNET. Routine assessment of core TFs; PIT1, TPIT, and SF1 as well as auxiliary markers, GATA3 and ERα, have made pathologists to transition away from the subjective morphological criteria and have reduced the ambiguous null cell category dramatically, reclassifying the vast majority as silent gonadotroph tumor or other lineage-specific neoplasms. Lineage-level subtyping can be used to recognize high-risk variants early, whose unique biological features demand heightened surveillance and multidisciplinary interventions. Despite these progresses, there are still diagnostic holes, including handling of highly rare cases of so-called triple-negative neoplasms and tumors with multilineage infidelity, which today push the limits of classical classifications, and potentially have a more ominous prognosis. Future developments will necessitate a smooth incorporation of pangenomic signatures such as DNA methylation and transcriptomic profiling with the new digital pathology and AI based algorithms to normalize IHC readings and negate inter-observer bias. These sophisticated lineage-based and molecular characterizations will eventually be the springboard to end the age of one-size-fits-all model to an age of actually accurate and personalized management of patients with PitNETs.
Author Contributions
“Conceptualization, Y.W. and O.I.; methodology, Y.W.; writing—original draft preparation, N.P.; writing—review and editing, Y.W. and O.I.; supervision, D.K.; project administration, D.K. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACTH | Adrenocorticotropic Hormone |
| AIP | Aryl Hydrocarbon Receptor–Interacting Protein |
| ATRX | Alpha Thalassemia/Mental Retardation Syndrome X-Linked |
| BRAF | B-Raf Proto-Oncogene |
| cAMP | Cyclic Adenosine Monophosphate |
| CDX2 | Caudal Type Homeobox 2 |
| DRD2 | Dopamine Receptor D2 |
| EGFR | Epidermal Growth Factor Receptor |
| EMA | Epithelial Membrane Antigen |
| EMT | Epithelial–Mesenchymal Transition |
| ERα | Estrogen Receptor Alpha |
| FSH | Follicle-Stimulating Hormone |
| GATA3 | GATA Binding Protein 3 |
| GCT | Granular Cell Tumor |
| GFAP | Glial Fibrillary Acidic Protein |
| GH | Growth Hormone |
| GNAS | Guanine Nucleotide-binding Protein alpha stimulating |
| H&E | Hematoxylin and Eosin |
| IHC | Immunohistochemistry |
| LH | Luteinizing Hormone |
| LMWCK | Low Molecular Weight Cytokeratin |
| MAPK | Mitogen-Activated Protein Kinase |
| MEG3 | Maternally Expressed Gene 3 |
| MEN1 | Multiple Endocrine Neoplasia Type 1 |
| MGMT | O6-Methylguanine-DNA Methyltransferase |
| MRI | Magnetic Resonance Imaging |
| NEN | Neuroendocrine Neoplasm |
| NF | Non-Functioning |
| NF-PitNET | Non-Functioning Pituitary Neuroendocrine Tumor |
| NR5A1 | Nuclear Receptor Subfamily 5 Group A Member 1 |
| PA | Pituitary Adenoma |
| PAS | Periodic Acid–Schiff |
| PIT1 | Pituitary-Specific Positive Transcription Factor 1 |
| POU1F1 | POU Class 1 Homeobox 1 |
| PitNET | Pituitary Neuroendocrine Tumor |
| PITX1 | Pituitary Homeobox 1 |
| PITX2 | Pituitary Homeobox 2 |
| POMC | Pro-opiomelanocortin |
| PROP1 | Prophet of PIT1 |
| PRL | Prolactin |
| PRLR | Prolactin Receptor |
| PPT | Posterior Pituitary Tumor |
| SCT | Silent Corticotroph Tumor |
| SCO | Spindle Cell Oncocytoma |
| SF1 | Steroidogenic Factor 1 |
| SF3B1 | Splicing Factor 3B Subunit 1 |
| SSTR | Somatostatin Receptor |
| SSTR2 | Somatostatin Receptor Type 2 |
| SSTR5 | Somatostatin Receptor Type 5 |
| SSA | Somatostatin Analogue |
| TBX19 | T-Box Transcription Factor 19 |
| TF | Transcription Factor |
| TP53 | Tumor Protein p53 |
| TPIT | T-Box Pituitary Transcription Factor |
| TSH | Thyroid-Stimulating Hormone |
| TTF-1 | Thyroid Transcription Factor 1 |
| USP8 | Ubiquitin-Specific Protease 8 |
| USP48 | Ubiquitin-Specific Protease 48 |
| V600E | Valine-to-Glutamic Acid Substitution at Codon 600 |
| WHO | World Health Organization |
References
- Asa, S.L.; Mete, O.; Cusimano, M.D.; McCutcheon, I.E.; Perry, A.; Yamada, S.; Nishioka, H.; Casar-Borota, O.; Uccella, S.; La Rosa, S.; et al. Pituitary neuroendocrine tumors: a model for neuroendocrine tumor classification. Mod. Pathol. 2021, 34, 1634–1650. [Google Scholar] [CrossRef]
- Villa, C.; Birtolo, M.F.; Perez-Rivas, L.G.; Righi, A.; Assie, G.; Baussart, B.; Asioli, S. Grading and staging for pituitary neuroendocrine tumors. Brain Pathol. 2024, 35, e13299. [Google Scholar] [CrossRef]
- Dai, C.; Kang, J.; Liu, X.; Yao, Y.; Wang, H.; Wang, R. How to Classify and Define Pituitary Tumors: Recent Advances and Current Controversies. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Trouillas, J.; Jaffrain-Rea, M.-L.; Vasiljevic, A.; Raverot, G.; Roncaroli, F.; Villa, C. How to Classify Pituitary Neuroendocrine Tumors (PitNET)s in 2020. Cancers 2020, 12, 514. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Casar-Borota, O.; Burman, P.; Lopes, M.B. The 2022 WHO classification of tumors of the pituitary gland: An update on aggressive and metastatic pituitary neuroendocrine tumors. Brain Pathol. 2024, 35, e13302. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; Melmed, S. Pituitary adenomas: biology, nomenclature and clinical classification. Rev. Endocr. Metab. Disord. 2025, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, S.; De Marinis, L. From Pituitary Adenoma to Pituitary Neuroendocrine Tumors: How Molecular Pathways may Impact the Therapeutic Management? Endocrine, Metab. Immune Disord. - Drug Targets 2021, 21, 1744–1759. [Google Scholar] [CrossRef]
- Goyal-Honavar, A.; Chacko, G. Practical approaches to diagnosing PitNETs/adenomas based on cell lineage. Brain Pathol. 2024, 35, e13298. [Google Scholar] [CrossRef]
- Kontogeorgos, G. Update on pituitary adenomas in the 2017 World Health Organization classification: innovations and perspectives. Hormones 2021, 20, 287–291. [Google Scholar] [CrossRef]
- Wan, X.-Y.; Chen, J.; Wang, J.-W.; Liu, Y.-C.; Shu, K.; Lei, T. Overview of the 2022 WHO Classification of Pituitary Adenomas/Pituitary Neuroendocrine Tumors: Clinical Practices, Controversies, and Perspectives. Curr. Med Sci. 2022, 42, 1111–1118. [Google Scholar] [CrossRef]
- Burcea, I.F.; Năstase, V.-N.; Poiană, C. Pituitary transcription factors in the immunohistochemical and molecular diagnosis of pituitary tumours — a systematic review. Endokrynol. Polska 2021, 72, 53–63. [Google Scholar] [CrossRef]
- Inomoto, C.; Tahara, S.; Oyama, K.; Kimura, M.; Matsuno, A.; Teramoto, A.; Osamura, R.Y. Molecular, functional, and histopathological classification of the pituitary neuroendocrine neoplasms. Brain Tumor Pathol. 2021, 38, 183–188. [Google Scholar] [CrossRef]
- Kobalka, P.J.; Huntoon, K.; Becker, A.P. Neuropathology of Pituitary Adenomas and Sellar Lesions. Neurosurgery 2021, 88, 900–918. [Google Scholar] [CrossRef]
- Portovedo, S.; Neto, L.V.; Soares, P.; de Carvalho, D.P.; Takiya, C.M.; Miranda-Alves, L. Aggressive nonfunctioning pituitary neuroendocrine tumors. Brain Tumor Pathol. 2022, 39, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Lenders, N.F.; E Earls, P.; Inder, W.J.; I McCormack, A. The evolution in pituitary tumour classification: a clinical perspective. Endocr. Oncol. 2023, 3, e220079. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Ezzat, S. Genomics and Epigenomics of Pituitary Tumors: What Do Pathologists Need to Know? Endocr. Pathol. 2021, 32, 3–16. [Google Scholar] [CrossRef]
- Drouin, J. The corticotroph cells from early development to tumorigenesis. J. Neuroendocr. 2022, 34, e13147. [Google Scholar] [CrossRef] [PubMed]
- Hinojosa-Amaya, J.M.; Lam-Chung, C.E.; Cuevas-Ramos, D. Recent Understanding and Future Directions of Recurrent Corticotroph Tumors. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Mouchtouris, N.; Smit, R.D.; Piper, K.; Prashant, G.; Evans, J.J.; Karsy, M. A review of multiomics platforms in pituitary adenoma pathogenesis. Front. Biosci. 2022, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Relav, L.; Doghman-Bouguerra, M.; Ruggiero, C.; Muzzi, J.C.D.; Figueiredo, B.C.; Lalli, E. Steroidogenic Factor 1, a Goldilocks Transcription Factor from Adrenocortical Organogenesis to Malignancy. Int. J. Mol. Sci. 2023, 24, 3585. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.C.; Wang, E.J.; Aghi, M.K. Recent advancements in the molecular biology of pituitary adenomas. Expert Rev. Endocrinol. Metab. 2022, 17, 293–304. [Google Scholar] [CrossRef]
- Chang, M.; Yang, C.; Bao, X.; Wang, R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 2021, 11. [Google Scholar] [CrossRef]
- Möller, K.; Uhlig, R.; Gorbokon, N.; Dum, D.; Menz, A.; Büscheck, F.; Luebke, A.M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; et al. Comparison of INSM1 immunostaining with established neuroendocrine markers synaptophysin and chromogranin A in over 14,000 neuroendocrine and non-neuroendocrine tumors. Mol. Cell. Endocrinol. 2023, 581, 112106. [Google Scholar] [CrossRef]
- Gheorghișan-Gălățeanu, A.-A.; Ilieșiu, A.; Lambrescu, I.M.; Țăpoi, D.A. The Complex Histopathological and Immunohistochemical Spectrum of Neuroendocrine Tumors—An Overview of the Latest Classifications. Int. J. Mol. Sci. 2023, 24, 1418. [Google Scholar] [CrossRef]
- Guaraldi, F.; Ambrosi, F.; Ricci, C.; Di Sciascio, L.; Asioli, S. Histopathology of growth hormone-secreting pituitary tumors: State of the art and new perspectives. Best Pr. Res. Clin. Endocrinol. Metab. 2024, 38, 101894. [Google Scholar] [CrossRef]
- Kontogeorgos, G.; Thodou, E.; Osamura, R.Y.; Lloyd, R.V. High-risk pituitary adenomas and strategies for predicting response to treatment. Hormones 2022, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P. Histopathological Types, Clinical Presentation, Imaging Studies, Treatment Strategies, and Prognosis of Posterior Pituitary Tumors: An Updated Review. J. Clin. Med. 2025, 14, 4553. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, R.; Dum, D.; Gorbokon, N.; Menz, A.; Büscheck, F.; Luebke, A.M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; Fraune, C.; et al. Synaptophysin and chromogranin A expression analysis in human tumors. Mol. Cell. Endocrinol. 2022, 555, 111726. [Google Scholar] [CrossRef]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.-L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e5. [Google Scholar] [CrossRef]
- Serioli, S.; Agostini, L.; Pietrantoni, A.; Valeri, F.; Costanza, F.; Chiloiro, S.; Buffoli, B.; Piazza, A.; Poliani, P.L.; Peris-Celda, M.; et al. Aggressive PitNETs and Potential Target Therapies: A Systematic Review of Molecular and Genetic Pathways. Int. J. Mol. Sci. 2023, 24, 15719. [Google Scholar] [CrossRef]
- Fleseriu, M.; Varlamov, E.V.; Akirov, A.; Langlois, F.; Petersenn, S.; Melmed, S. Prolactin-secreting adenomas: pathogenesis, diagnosis, and management. Lancet Diabetes Endocrinol. 2025. [Google Scholar] [CrossRef]
- Peverelli, E.; Treppiedi, D.; Mantovani, G. Molecular mechanisms involved in somatostatin receptor regulation in corticotroph tumors: the role of cytoskeleton and USP8 mutations. Endocr. Oncol. 2022, 2, R24–R30. [Google Scholar] [CrossRef]
- Lenders, N.F.; Inder, W.J.; McCormack, A.I. Towards precision medicine for clinically non-functioning pituitary tumours. Clin. Endocrinol. 2021, 95, 398–409. [Google Scholar] [CrossRef]
- Faraj, C.A.; McCutcheon, I.E.; Gubbiotti, M.A. PIT-1/SF-1 co-expression in pituitary neuroendocrine tumors (PitNETs) with comprehensive review of the literature: How should we best characterize these neoplasms? Ann. Diagn. Pathol. 2024, 74, 152398. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Wenig, B.M. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Overview of the 2022 WHO Classification of Head and Neck Neuroendocrine Neoplasms. Head Neck Pathol. 2022, 16, 123–142. [Google Scholar] [CrossRef]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Nf, L.; AI, M. Refractory Pit1 plurihormonal tumours and thyrotroph adenomas. Pituitary 2023, 26, 182–186. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Anterior pituitary cell lineages and their hormones.
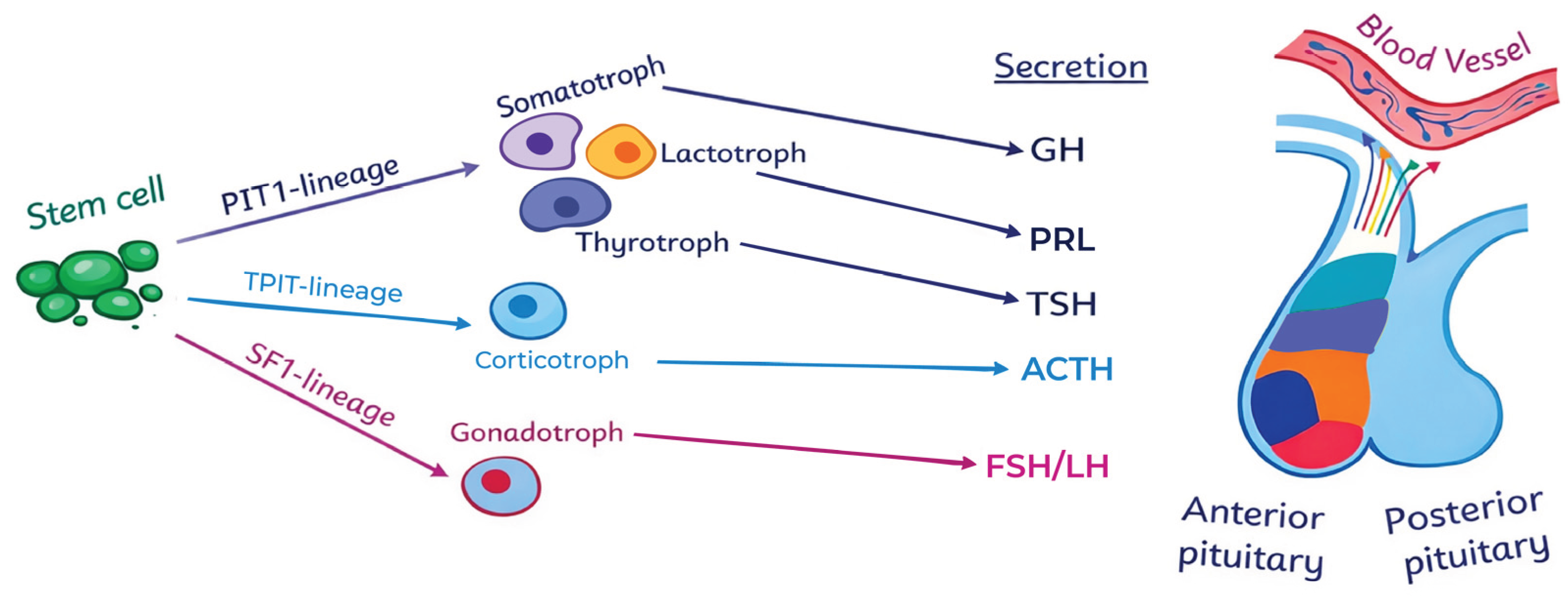
Table 1.
Evolution of WHO classification of pituitary tumors [16].
Table 1.
Evolution of WHO classification of pituitary tumors [16].
| CATEGORY | 2004 WHO | 2017 WHO | 2022 WHO (5TH ED.) |
|---|---|---|---|
| Terminology | Adenoma | Adenoma vs tumor vs PitNET | Pituitary Neuroendocrine Tumor (PitNET) |
| Ihc Basis | Hormonal | Transcription factors (TF) & hormonal | Transcription factors (TF) & hormonal |
| Type / Lineage | ‘Typical’ | SF1 lineage Gonadotroph |
SF1 lineage Gonadotroph |
| ‘Atypical’ | TPIT lineage Corticotroph PIT1 lineage Lactotroph (sparsely granulated, densely granulated, ASC) Somatotroph (sparsely granulated, densely granulated, mammosomatotroph, mixed somatotroph–lactotroph) Thyrotroph Plurihormonal (PIT1-positive plurihormonalᵃ, unusual combinations) |
TPIT lineage Corticotroph PIT1 lineage Lactotroph (sparsely granulated, densely granulated) Somatotroph (sparsely granulated, densely granulated) Mammosomatotrophᵇ Mixed somatotroph and lactotrophᵇ Thyrotroph Mature plurihormonal PIT1 lineageᶜ Immature PIT1 lineageᶜ Acidophil stem cellᵇ |
|
| No Distinct Cell Lineage | Null cell | Null cell | Null cell Plurihormonal |
| Proliferative Markers | Ki-67 > 3% Elevated mitotic index p53 ↑ |
Ki-67 > 3% Elevated mitotic index — |
— — — |
| Carcinoma / Metastasis | Craniospinal or distant metastases | Craniospinal or distant metastases | Term omitted; replaced with “Metastatic PitNET”ᶜ |
a-Newly defined in 2017 WHO classification; b-Newly described as separate ‘type’ rather than ‘subtype’ in 2022 WHO classification; c-Newly defined in 2022 WHO classification. IHC, immunohistochemistry; TF, transcription factor.
Table 3.
TF- based classification of PitNETs according to WHO 5th edition. Adapted from WHO and recent studies [5,14,25].
| PitNET Subtype | Transcription Factor(s) | Hormone IHC Pattern | Clinical / Functional Status | Key Comments |
|---|---|---|---|---|
| Densely Granulated Somatotroph PitNET | PIT1+ | GH+, α-subunit+ | Acromegaly | Acidophilic cells; diffuse GH staining; perinuclear LMWCK; usually responsive to SSA |
| Sparsely Granulated Somatotroph PitNET | PIT1+ | GH weak/focal | Acromegaly or clinically NF | High-risk subtype; chromophobic cells; fibrous bodies (CAM5.2+); aggressive behavior and reduced SSA response |
| Lactotroph PitNET | PIT1+, ERα+ | PRL+ | Hyperprolactinemia | Most are sparsely granulated with juxtanuclear “dot-like” PRL; excellent dopamine agonist response |
| Thyrotroph PitNET | PIT1+, GATA3+ | TSH+, α-subunit+ | Hyperthyroidism (rare) | Often fibrotic or spindle-celled; rare but diagnostically challenging |
| Corticotroph PitNET | TPIT+ | ACTH+ | Cushing disease | Basophilic cells; strong PAS positivity; typically, microadenomas |
| Crooke Cell Tumor | TPIT+ | ACTH+ (often weak) | Cushing disease or NF | High-risk variant; perinuclear hyaline keratin rings; aggressive and invasive |
| Silent Corticotroph PitNET | TPIT+ | ACTH+ | Clinically NF | High-risk; larger size, invasive growth, higher recurrence rate than gonadotroph tumors |
| Gonadotroph PitNET | SF1+, GATA3+ (± ERα) | FSH/LH/α-subunit (often weak or negative) | Clinically NF | Most common PitNET subtype; diffuse nuclear SF-1 is most reliable marker |
| Mature Plurihormonal PIT1-Lineage PitNET | PIT1+ | Multiple PIT1-lineage hormones | Variable (often functional) | Well-differentiated cells; distinct from aggressive immature subtype |
| Immature PIT1-Lineage PitNET | PIT1+ (ERα/GATA3 variable) | Multihormonal (focal/patchy) | Clinically NF or mixed | High-risk; lineage infidelity; large, invasive tumors; formerly “silent subtype 3” |
| Acidophil Stem Cell PitNET | PIT1+ | Usually GH+, PRL+ | Acromegaly, Hyperprolactinemia | High-risk; scattered fibrous bodies |
| Null Cell PitNET | PIT1− / TPIT− / SF-1− | No hormone expression | Clinically NF | Diagnosis of exclusion; very rare with modern TF IHC |
| Mixed/ plurihormonal PitNET | Multiple combination | Multiple combination | Variable | Variable |
Table 4.
Molecular Drivers, High-Risk Histotypes, and Therapeutic Correlates of TF-defined PitNETs. Adapted from WHO classification and recent studies [17,22,23].
| Tf-Defined Group | Typical Molecular Drivers / Alterations | High‐Risk Histotypes (Who 5th) | General Therapeutic Considerations |
|---|---|---|---|
| PIT1 Lineage | Somatic (GNAS, SF3B1, PRLR); Germline (AIP, MEN1) | Sparsely granulated somatotroph; Immature PIT1-lineage PitNET; Acidophil stem cell PitNET | SSR–directed therapy (octreotide, pasireotide); GH receptor antagonists (pegvisomant) in resistant acromegaly; dopamine agonists for lactotroph tumors |
| TPit Lineage | USP8, USP48, BRAF, ATRX; additional signaling pathway alterations reported | Silent corticotroph PitNET; Crooke cell tumor | Pasireotide preferred due to SSTR5 expression; emerging molecularly targeted approaches under investigation in selected cases |
| SF-1 Lineage | No recurrent somatic driver mutations identified; epigenetic alterations (MEG3) | Rare aggressive gonadotroph PitNETs; most clinically non-functioning | Surgery remains primary therapy; limited and variable response to medical therapies |
| TF-Negative/Mixed PITNets | Copy number alterations; TP53 abnormalities reported in aggressive disease | Metastatic PitNET | Temozolomide as first-line systemic therapy; treatment response associated with low MGMT expression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



