
Submitted:
15 January 2026
Posted:
16 January 2026
You are already at the latest version
Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that play central roles in post-transcriptional gene regulation and cellular homeostasis maintenance. Dysregula-tion of miRNA expression is increasingly recognized as a key contributor to tissue injury during the acute phase and to disease progression in the chronic phase. Chronic kidney disease (CKD) commonly progresses and ultimately leads to kidney failure through interstitial fibrosis, which is the final common pathway of CKD progression. Interstitial fibrosis is driven not only by fibroblast activation but also by a phenotypic transition of injured tubular epithelial cells, infiltrating macrophages, and peritubular capillary cells. These multifaceted cellular pathways induce and exacerbate interstitial fibrosis, and several miRNAs have been identified as important regulators of these pathways. In addition to fibrotic pathophysiological features, disease-specific dysregulation of miRNAs has been increasingly detected in various causes of CKD, including diabetic kidney disease, chronic glomerulonephritis, and nephrosclerosis. In this review, we provide an integrated overview of miRNA-mediated regulation in CKD, with particular emphasis on cell lineage functions within fibrotic pathways and disease-specific roles. Finally, we discuss the emerging potential of miRNAs as biomarkers and therapeutic targets for CKD and highlight future research directions.

Keywords:
MicroRNA
; renal interstitial fibrosis
; chronic kidney disease
1. Introduction
Non-coding RNAs (ncRNAs) are RNA molecules transcribed from DNA that are not translated into proteins. They contribute to several biological processes, including protein synthesis and intracellular transport, RNA maturation, DNA replication, and most prominently, the regulation of gene expression. They are commonly classified according to their length into small ncRNAs (<200 nucleotides) and long ncRNAs (>200 nucleotides). Among the small ncRNAs, microRNAs (miRNAs) are 18–22 nucleotides in length and are single-stranded transcripts that mediate gene silencing through RNA interference. Long ncRNAs include linear lncRNAs and circRNAs [1].
MiRNAs regulate gene expression by binding to partially complementary sites, typically within 3’ untranslated regions (3’UTR) of the target mRNAs [2,3,4]. This interaction represses translation and promotes mRNA decay via an RNA-induced silencing complex. A single miRNA can target multiple mRNAs, while individual mRNAs can be regulated by several miRNAs [5,6]. Through these mechanisms, miRNAs influence several biological processes, including development, differentiation, cell proliferation, apoptosis, cancer metastasis, inflammation, and fibrosis [7]. MicroRNAs were first described in 1993, and since then, over 2,000 human microRNAs have been identified in miRBase [8]. Recent renal research has identified various dysregulated miRNAs in acute kidney injury (AKI) and chronic kidney disease (CKD) [9,10,11,12,13,14,15]. Otherwise, lncRNAs are transcribed from intergenic, exonic, or distal protein-coding regions of the genome by RNA polymerase, followed by 3’-polyadenylation and 5’-end capping. LncRNAs are functional molecules that play various roles by interacting with mRNAs, miRNAs, DNAs, proteins, and small molecules. Another lncRNA promotes the miRNA-induced silencing complex (mi-RISC) to repress mRNA translation. Additionally, circRNAs are generated from precursor mRNAs by back-splicing on a 5’ splice site to a 3’ splice site. Backsplicing originates from lariat-driven circularization, RNA-binding proteins (RBPs), intron pairing-driven circularization, and intron circularization. By covalent joining on the 5’ and 3’ ends, the circular structure contributes to the stability against RNA degradation and de-adenylation, which gives a longer half-life than linear RNAs. Accumulating evidence has demonstrated the mechanisms of action of these circRNAs. Some circRNAs act as miRNA sponges and inhibit the activity of one or multiple miRNAs [1]. Recently, many researchers have reported that lncRNAs and circRNAs directly or indirectly regulate alternative splicing of downstream target genes associated with kidney injury [14,16,17].
CKD affects approximately 10% of the global population [18]. Importantly, delaying early-stage CKD progression provides economic benefits and prevents the development of end-stage kidney disease (ESKD) and cardiovascular complications [19]. Although CKD arises from many heterogeneous diseases that irreversibly alter the function and structure of the kidney, such as diabetes, hypertension, and nephritis, it is characterized by progressive and irreversible nephron loss, microvascular damage, decreased regenerative capacity, inflammation, oxidative stress, and metabolic changes, ultimately leading to renal failure [20,21,22]. In CKD progression, tubulointerstitial fibrosis is the key mechanism of renal dysfunction, marked by excessive ECM deposition and loss of epithelial cells. This process is largely driven by activated myofibroblasts, which primarily derive from interstitial fibroblasts, with contributions from pericytes, circulating fibrocytes, and transitional processes involving tubular and endothelial cells. [21,22,23,24]. Numerous studies link altered miRNA expression to renal interstitial fibrosis initiation and progression. We summarize recent advances in the roles of dysregulated miRNAs, focusing on intercellular communication, mesenchymal transition, and fibroblast activation. We also highlight potential miRNAs as novel biomarkers and antifibrotic therapeutic targets in various renal diseases.
2. Mechanisms of Renal Interstitial Fibrosis
Renal fibrosis can develop in both the glomerular and tubulointerstitial compartments as a maladaptive wound-healing response, resulting in excessive deposition of ECM proteins [25,26]. Renal interstitial fibrosis involves myofibroblast activation, excessive ECM accumulation, and the loss or dysfunction of renal tubular epithelial cells. Understanding the mechanisms behind interstitial fibrosis and developing effective interventions are crucial.
Resident interstitial fibroblasts and perivascular mesenchymal cells, located between the tubular basement membrane and peritubular capillaries, help maintain ECM homeostasis. Following injury, these cells proliferate and differentiate into ECM-producing myofibroblasts. Their activation is influenced by interactions among injured tubular epithelial cells, endothelial cells, pericytes, and infiltrating immune cells. The exact contributions of these cellular sources to the myofibroblast population are still debated in the literature [27,28,29,30,31].
In injured kidneys, such as those with ischemic, drug, toxic, or obstructive injuries, the tubular epithelial cells may fail to complete adaptive repair. Instead, they can become “stuck” in the cell cycle and begin to release inflammatory and pro-fibrotic mediators [32]. These sustained signals activate nearby fibroblasts and attract immune cells, thereby promoting interstitial fibrosis. These processes propagate interstitial fibrosis through sustained myofibroblast activation, capillary rarefaction, and inflammatory signaling. In a murine unilateral ureteral obstruction (UUO) model, lineage-tracing studies suggested that the majority of myofibroblasts arise from resident interstitial mesenchymal cells, including fibroblasts and pericyte-related cells [27,33]. EMT [34,35], EndMT [36], and macrophage-to-mesenchymal transition (MMT) [37] have been implicated in renal interstitial fibrogenesis. The relative contributions of interstitial myofibroblasts to fibrosis has not been clearly defined, but their transition processes are important for reshaping cell states and promoting pro-fibrotic signaling in injured tissues. Renal interstitial fibrosis results from the interaction of kidney cells, immune cells, and the extracellular matrix. We will discuss the miRNAs that regulate fibroblast-to-myofibroblast activation as well as MMT, EMT, and EndMT, emphasizing their roles in fibrotic remodeling.
3. MiRNAs in Renal Interstitial Fibrosis
Although there are currently no effective treatments that reverse established renal interstitial fibrosis, miRNAs have emerged as promising mechanistic regulators and potential therapeutic targets [11,38]. Over the past decade, various studies on miRNAs in renal interstitial fibrosis have expanded rapidly, and numerous miRNAs related to the mechanisms and progression of renal interstitial fibrosis have been identified in diverse animal models, as well as in human plasma or urine [10,39,40]. MiRNAs typically bind seed sequences in the 3’ UTR of target mRNAs and reduce protein output by repressing translation and/or promoting mRNA decay. Accordingly, many studies have examined specific miRNA–mRNA axes in renal interstitial fibrosis. Most investigations have been conducted using the following approaches: 1) depletion of specific miRNAs from specific cell types in kidneys using conditional knockout models; 2) analyses of differential miRNA expression in renal diseases to identify potential pathogenetic and/or therapeutic miRNA species; and 3) investigation of miRNA regulation of specific mRNA that play pathogenic roles in renal disease. Many miRNAs were initially identified using differential expression analysis, indicating an association, not causality. Gain- and loss-of-function studies are required to confirm a causal role. Therefore, we preferentially selected studies that provided evidence not limited to associations and supported a causal role.
In the following section, we highlight studies that elucidate the role of cell–type–specific miRNAs in generating or promoting renal interstitial fibrosis. We also focused on the miRNA-mediated processes that influence cellular plasticity and the emergence or activation of myofibroblasts in renal interstitial fibrosis.
4. Regulation of Myofibroblast Activation Derived from Resident Renal Fibroblasts
Myofibroblast activation and subsequent ECM accumulation are central events in renal interstitial fibrosis [21,22,25]. Activated myofibroblasts are the principal effector cells of fibrosis because they produce large amounts of ECM [23,26]. The acquisition of α-smooth muscle actin (α-SMA) expression is a hallmark of fibroblast-to-myofibroblast activation [21,23]. Although myofibroblasts are scarce in the normal renal interstitium, their numbers markedly increase in fibrotic kidneys. Therefore, elucidation of the mechanisms underlying myofibroblast activation remains a key issue in renal fibrosis [21,22,24]. Following renal injury, injured tubular epithelial cells and infiltrating immune cells release a variety of pro-fibrotic mediators through autocrine and paracrine signaling, thereby promoting myofibroblast activation within the fibrotic microenvironment [21,22,24,26]. In the following section, we summarize the recent studies on miRNAs that regulate the activation of myofibroblasts derived from resident interstitial fibroblasts.
MiR-21 is one of the most extensively studied miRNAs involved in the relationship between renal fibrosis. Several studies have revealed that miR-21 expression is upregulated and is mainly prevalent in activated fibrotic regions, including tubulointerstitial fibroblasts, using in situ hybridization [41,42]. Glowacki et al. reported that miR-21 is strongly upregulated in the kidneys of mice with UUO and in human kidneys with advanced fibrosis. Moreover, circulating miR-21 levels were higher in renal transplant recipients with severe interstitial fibrosis/tubular atrophy (IF/TA) and were independently associated with IF/TA scores in multivariate linear regression analysis. In vitro, primary mouse kidney fibroblasts stimulated with transforming growth factor-β (TGF-β) revealed the upregulation of miR-21 and actin alpha 2 (acta2), known as a myofibroblast marker [41]. Sun et al. demonstrated that miR-21 is robustly upregulated in UUO kidneys and in TGF-β1–stimulated normal rat kidney-49F (NRK-49F) cells. MiR-21 mimics enhance the expression of collagen type I alpha 1 chain (Col1a1), fibronectin, and α-SMA, whereas antagonist or genetic deletion of miR-21 attenuates fibroblast activation and ameliorates UUO-induced interstitial fibrosis. Mechanistically, miR-21 directly targets and suppresses Smad7 and enhances TGF-β/Smad3 signaling. Additionally, miR-21, programmed cell death protein 4 (PDCD4), and activation protein-1 (AP-1) form an autoregulatory loop that sustains AP-1–driven miR-21 expression and fibroblast activation [42]. PDCD4 is a tumor suppressor that negatively regulates AP-1 activity. AP-1 is a transcription factor complex, which promotes pro-fibrotic gene expression [43]. Li et al. showed that melatonin attenuates TGF-β1–induced fibroblast-to-myofibroblast transition in NRK-49F cells and UUO-induced renal fibrosis. Melatonin reduces the expression of α-SMA, collagen I, and fibronectin; inhibits phosphorylation of signal transducers and activators of transcription3 (STAT3); downregulates miR-21-5p level; and upregulates its antifibrotic targets of Sprouty1 (Spry1) and phosphatase and tensin homolog (PTEN). MiR-21-5p mimics or knockdown of Spry1 or PTEN partially reverses these protective effects, indicating that regulation of the miR-21-5p/PTEN and/or miR-21-5p/Spry1 axes plays a central role in fibrosis [44]. Zhao et al. identified tubular epithelial cell–derived exosomes as a source of miR-21 fibrosis in UUO kidneys. TGF-β1–stimulated NRK-52E cells release miR-21–rich exosomes that were taken up by NRK-49F cells, leading to an increase in the α-SMA, collagen I, and fibronectin levels. Pharmacological inhibition of exosome release with GW4869 or genetic deletion of Rab27a reduces fibroblast activation and UUO-induced fibrosis. Moreover, miR-21–deficient exosomes preserve PTEN expression, suppress Akt activation, and ameliorate renal interstitial fibrosis in vivo [45]. In a murine model of chronic renal allograft dysfunction, renal miR-21a-5p expression was markedly upregulated, and therapeutic silencing of miR-21a-5p with a locked nucleic acid (LNA) inhibitor attenuated interstitial fibrosis, infiltration of inflammatory cells, Banff lesion scores, and improved graft function. MiR-21a-5p expression in renal fibroblasts is induced by macrophage-derived interleukin-6 (Il-6) via STAT3 activation. In addition, LPS-activated macrophages released small extracellular vesicles that are enriched in mature miR-21a-5p. These vesicles are taken up by renal fibroblasts that induce a pro-fibrotic phenotype, with increased expression of Il-6, connective tissue growth factor (CTGF), α-SMA, and collagens. Schauerte et al. have identified Notch2 as a novel direct target of miR-21a-5p. Notch2 is a membrane-bound receptor that plays a critical role in kidney development. Consistent with previous reports showing that Notch2 signaling restrains the fibroblast-to-myofibroblast transition, downregulation of Notch2 via miR-21a-5p upregulation promotes myofibroblast differentiation and interstitial fibrosis [46].
MiR-26a-5p was markedly downregulated in human kidneys with interstitial nephritis and in UUO-injured mouse kidneys. Systemic infusion of miR-26a-5p mimic after UUO surgery ameliorates renal interstitial inflammation and fibrosis, lowers serum creatinine, and reduces TGF-β1 expression. In TGF-β1–stimulated NRK-49F cells, miR-26a-5p overexpression attenuates fibroblast activation by decreasing TGF-β receptor I and II, phosphorylation of Smad3, expression of NF-κB, matrix metalloproteinase-9, and mesenchymal markers such as vimentin, fibronectin, and α-SMA, thereby limiting fibroblast-to-myofibroblast transition [47].
Both miR-29 and miR-30 families are widely recognized as anti-fibrotic regulators of renal interstitial fibrosis. Their expression is consistently reduced in fibrotic human kidneys and murine models of UUO, angiotensin II (Ang II) infusion, and ischemic reperfusion injury (IRI). In these models, overexpression of miR-29 or miR-30 through mimic- or vector-mediated restoration suppressed fibroblast activation and expression of myofibroblast markers, limited extracellular matrix accumulation, and attenuated interstitial fibrosis [48,49,50,51,52].
In mice with UUO, renal expression of p53 and miR-34a was upregulated. Transfection of a miR-34a mimic into NRK-49F cells increases the expression of Col1a1, Col1a2, and Tgf-β1 and robustly induces α-SMA. Inhibition of miR-34a reduces the expression of TGF-β1-induced Acta2. Importantly, miR-34a does not activate the TGF-β/Smad pathway, and TGF-β1 does not increase miR-34a expression. Thus, miR-34a-induced α-SMA upregulation occurs independently of canonical TGF-β/Smad signaling, indicating that the p53/miR-34a axis promotes fibroblast-to-myofibroblast differentiation and contributes to renal interstitial fibrosis [53].
Sakuma et al. generated mice with Dicer deletion in platelet-derived growth factor receptor (Pdgfr)-β–positive mesenchymal cells and showed that loss of Dicer exacerbates UUO- and folic acid-induced interstitial fibrosis, accompanied by Pdgfr-β overexpression and miR-9-5p downregulation. In primary renal fibroblasts, inhibition of miR-9-5p further increased Pdgfrb and Acta2 under TGF-β1 stimulation. These findings indicate that miR-9-5p restrains Pdgfr-β–driven myofibroblast differentiation and may function as an antifibrotic regulator in renal interstitial fibrosis [54]. Table 1 summarizes other recent studies supporting the role of miRNAs in fibroblast activation.
5. Regulation of Epithelial-Mesenchymal Transition (EMT)
EMT is a phenotypic process in which epithelial cells lose their epithelial characteristics and acquire mesenchymal features [61]. Tubular epithelial cells have been proposed to undergo EMT-related changes during renal fibrosis, although the extent to which they are fully converted into interstitial myofibroblasts remains controversial [62]. Current concepts emphasize partial EMT, in which tubular cells acquire mesenchymal traits while remaining epithelial, and promote fibrosis through sustained pro-fibrotic signaling [35,62]. Consistent with this, injured tubular cells in fibrotic kidneys often show increased mesenchymal marker expression (e.g., vimentin, α-SMA, collagen I) together with reduced epithelial marker expression [34,35]. Collectively, EMT-associated epithelial plasticity is considered an important mechanistic component of renal fibrogenesis within the broader framework of kidney fibrosis pathways [34,61]. In this section, we discuss recent studies on miRNAs related to EMT processes in renal interstitial fibrosis.
The expression of miR-17 was upregulated in the serum of patients with diabetic nephropathy (DN) and in both the serum and kidney tissues of db/db mice. Systemic administration of an LNA–miR-17 inhibitor attenuated renal fibrotic changes in db/db mice, as determined by the histological assessment of PAS and Masson staining. In vitro, TGF-β1–induced HK-2 cells (and human mesangial cells) increased the expression of miR-17, whereas miR-17 inhibition increased the levels of E-cadherin and decreased those of vimentin, fibronectin, and collagen I. Mechanistically, Smad7 was identified as a direct target of miR-17 by dual-luciferase reporter assays, and the inhibition of miR-17 resulted in upregulated Smad7 expression, suggesting that miR-17 promotes TGF-β1–driven fibrotic/EMT-like responses through Smad7 suppression [63].
MiR-19 is also associated with renal tubular EMT. The expression of miR-19 is upregulated in peripheral blood from patients with renal fibrosis and in UUO kidneys and is also induced by TGF-β1 in NRK-52E cells. MiR-19 suppresses PTEN by binding its 3’UTR, followed by activating Akt signaling, decreasing E-cadherin expression, and increasing α-SMA/fibronectin expression. miR-19 inhibition restores PTEN and blunts TGF-β1–induced EMT. In mice with UUO, tail vein miR-19 agomir increased collagen deposition in Masson’s and Sirius Red staining, whereas miR-19 antagomir alleviated interstitial fibrosis [64]. Beyond miR-19, multiple EMT-linked miRNAs converge on the PTEN/AKT axis, including miR-21 in UUO kidneys treated with pure total flavonoids from Smilax glabra (PTFS) and miR-382 in aristolochic acid (AA)–induced renal injury model [65,66]. The miR-21 family is also an important factor in the EMT during renal interstitial fibrosis. In rats with UUO, PTFS suppressed EMT and mitigated renal interstitial fibrosis, as assessed by the evaluation of reduction of expression of α-SMA and restoration of E-cadherin in real-time polymerase chain reaction (PCR) and western blot. PTFS also modulated the miR-21/PTEN/PI3K–Akt axis in TGF-β1–stimulated HK-2 cells. Additionally, overexpression of miR-21 reduced the expression of PTEN and E-cadherin and promoted α-SMA and phosphorylation of PI3K/Akt, whereas inhibition of miR-21 produced the opposite phenotype, indicating that blockade of miR-21–mediated PTEN suppression is a key anti-EMT mechanism in UUO-induced fibrosis [65]. In aristolochic acid (AA)-induced nephropathy, the renal expression of miR-382 is upregulated during the AKI-to-CKD transition, concomitant with progressive tubulointerstitial fibrosis. Genetic deletion or pharmacological inhibition of miR-382 partially reversed inflammatory and fibrotic responses and mitigated EMT-like changes. Mechanistically, miR-382 directly targeted the 3’UTR of PTEN by luciferase assays, leading to PTEN loss and downstream activation of AKT signaling. In tubular epithelial cells, inhibition of PTEN and/or overexpression of miR-382 aggravated the loss of epithelial markers and induction of mesenchymal markers, supporting the PTEN/AKT-dependent EMT program. NF-κB acted upstream as NF-κB siRNA attenuated AA-induced upregulation of miR-382, supporting an NF-κB–dependent miR-382/PTEN/AKT cascade in tubular injury [66].
The expression of miR-23a is upregulated in renal tissues of patients with DN and in HK-2 cells exposed to HG, whereas the Ski-related novel protein N (SnoN), known as a critical negative regulator of the TGF-β/Smad signal pathway, is downregulated in diabetic kidneys among patients and HG-exposed HK-2 cells. Silencing miR-23a restores SnoN expression and attenuates HG-induced EMT and fibrogenic responses. Conversely, miR-23a overexpression suppresses SnoN expression and aggravates EMT and ECM production. Additionally, overexpression of SnoN inhibits miR-23a, whereas SnoN knockdown partially reverses this protection, supporting that the miR-23a/SnoN axis drives tubular EMT and renal fibrogenesis in vitro [67]. Similarly, miR-130a-3p has also been reported to promote TGF-β1–induced EMT and fibrotic responses in tubular epithelial cells by directly targeting SnoN. Inhibition of miR-130a-3p restores SnoN, reduces p-Smad2/3, increases Smad7, and attenuates EMT markers in HK-2 and primary human renal proximal tubular epithelial cells [68].
Members of the miR-27 family exert both pro- and anti-fibrotic effects. In db/db mice and HG-treated HK-2 cells, the expression of miR-27a-3p was upregulated, whereas that of prohibitin and transmembrane BAX inhibitor motif-containing 6 (TMBIM6), which are direct targets of miR-27a-3p, was downregulated. In a previous study, prohibitin, a multifunctional protein, was ubiquitously present in multiple cellular compartments. TMBIM6 ameliorates IRI-induced AKI by regulating mitochondrial homeostasis [69]. Silencing of miR-27a-3p attenuates renal interstitial fibrosis, accompanied by a reduction in pro-fibrotic factors and restoration of E-cadherin expression. Anti–miR-27a-3p also alleviates mitochondrial dysfunction and endoplasmic reticulum (ER) stress signaling, thereby limiting apoptosis and matrix accumulation. In HG-treated HK-2 cells, restoring prohibitin or TMBIM6 recapitulated the protective effects of miR-27a-3p inhibition, supporting miR-27a-3p/prohibitin or TMBIM6 axis in diabetic tubulointerstitial injury [70]. In STZ-induced DN of rats and HG-stimulated NRK-52E cells, miR-27a is increased in parallel with renal fibrosis and activation of Wnt/β-catenin signaling, including β-catenin nuclear translocation. Mechanistically, miR-27a directly targets the 3’UTR of secreted frizzled-related protein 1 (Sfrp1), known as a Wnt antagonist by luciferase assays. Functionally, inhibition of miR-27a decreases collagen IV and α-SMA levels and restores E-cadherin. Conversely, Sfrp1 knockdown aggravated EMT-like changes, whereas miR-27a inhibition partially reversed these effects. Collectively, these findings support a pro-fibrotic axis of miR-27a/Sfrp1/Wnt–β-catenin in diabetic renal injury [71]. By contrast, miR-27b-3p expression was downregulated in UUO kidneys and in TGF-β1–treated HK-2 cells. Restoration of miR-27b-3p suppresses TGF-β1–induced EMT and apoptosis, concomitant with reduction of α-SMA, collagen III, fibronectin, and vimentin. STAT1 was identified as a direct target of miR-27b-3p using dual-luciferase assays, and its overexpression counteracted the anti-apoptotic effects of miR-27b-3p. In vivo, overexpression of miR-27b-3p alleviated UUO-associated tubulointerstitial injury and fibrosis, supporting that miR-27b-3p is an anti-fibrotic EMT-limiting regulator [72].
Recently, several studies have demonstrated that the modulation of lncRNA activity in the kidney is an important mechanism in EMT. The upregulation of lcnRNA H19 alleviated interstitial fibrosis and EMT, accompanied by a reduction in lipid deposition and inflammatory responses. lncRNA H19 is capable of increasing ACSL1 levels by sponging miR-130a-3p, leading to the suppression of EMT, inflammatory response, and prevention of interstitial fibrosis [73].
Other recent studies supporting the role of miRNAs in the EMT are summarized in Table 2.
6. Regulation of Endothelial-Mesenchymal Transition (EndMT)
EndMT is a phenotypic program in which endothelial cells lose endothelial features and acquire mesenchymal characteristics and has been implicated in kidney injury and fibrosis [36]. In injured renal capillaries, endothelial dysfunction enhances local inflammation, ECM deposition, and capillary rarefaction, thereby promoting tubulointerstitial fibrosis [109]. During EndMT, endothelial cells pass through an intermediate state with co-expression of endothelial and mesenchymal markers, accompanied by increased mesenchymal markers (e.g., α-SMA, collagen I, fibronectin, vimentin) and reduced endothelial markers (e.g., von Willebrand Factor (vWF), CD31, Vascular Endothelial (VE)-cadherin) [36]. TGF-β signaling drives EndMT through canonical Smad-dependent pathways and non-canonical pathways, such as MAPK and PI3K/Akt [36]. Noncoding RNAs, including miRNAs, are recognized as regulators of endothelial dysfunction [110]. Here, we summarize the relationship between EndMT and miRNAs.
In an in vitro study, TGF-β1 stimulation of human umbilical vein endothelial cells (HUVECs) reduced VE-cadherin expression and increased the mesenchymal marker SM22α. During this process, HUVECs displayed cellular hypertrophy, lost sprouting capacity, and downregulated miR-20a. MiR-20a targeted TGF-β receptors (ALK5, TGF-βR2) and SARA, which are crucial for TGF-β signaling. Additionally, FGF2 increased miR-20a expression and inhibited EndMT in TGFβ1-stimulated endothelial cells. HUVECs transfected with miR-20a mimics blunted TGF-β1 stimulation, while anti-miR-20a abrogated FGF2's protective effect against TGF-β1-induced EndMT. These results indicate that FGF2 inhibits TGF-β signaling via miR-20a upregulation during EndMT [111].
Huang et al. found that the expression of miR-29a-3p in fibrotic mouse kidneys was reduced after unilateral IRI. Intravenous delivery of human umbilical cord mesenchymal stem cell-derived exosomes containing high levels of miR-29a-3p improved microvascular integrity, reduced vascular rarefaction, and attenuated interstitial fibrosis. This benefit was supported by miR-29a-3p gain and loss of experiments and was linked to the direct targeting of COL1A1 in fibroblasts and TNFR1 in endothelial cells [49].
Wang et al. found that elevated parathyroid hormone (PTH) levels drive EndMT and calcific remodeling in CKD rats induced by 5/6 nephrectomy and a high-phosphate diet. In vitro and in vivo studies showed that endothelial miR-29a-5p is significantly reduced and targets γ-secretase-activating protein (GSAP). Overexpression of miR-29a-5p inhibits PTH-induced EndMT, and the use of miR-29a-5p mimics or γ-secretase inhibitors blocks the Notch1 pathway, preventing EndMT. PTH induces valvular EndMT through the miR-29a-5p/GSAP/Notch1 pathway. [112].
miR-126-3p was detected in renal endothelial cells by in situ hybridization and was reduced in fibrotic kidneys after UUO. In HUVECs, EndMT induced by TGF-β2 plus Il-1β was associated with a reduction in miR-126-3p levels. Transfection with a miR-126-3p mimic preserved CD31 expression and reduced fibronectin levels, supporting partial protection against EndMT, although it did not restore vWF or VE-cadherin levels. These findings suggest that the loss of miR-126-3p accompanies endothelial phenotypic instability and that replenishment may help maintain endothelial features [113].
Qian et al. found that lncRNA taurine-upregulated gene 1 (TUG1) is upregulated in UUO kidneys and hypoxia-injured HUVECs. Silencing of TUG1 reduced renal fibrosis, restored vascular endothelial growth factor (VEGF), and lowered the α-SMA, TGF-β1, and HIF-1α expression. It also preserved CD31-positive endothelium, reduced CD31 and αSMA-double positive EndMT-phenotypic cells, and improved peritubular capillary perfusion on fluorescence microangiography. Mechanistically, cytoplasmic TUG1 directly bound miR-542-3p and miR-542-3p also bound HIF-1α using luciferase assays and bioinformatics analysis. TUG1 knockdown increased miR-542-3p and inhibition of miR-542-3p reversed the protection, supporting a miR-542-3p/HIF-1α/VEGF axis [114].
7. Regulation of Macrophage-Mesenchymal Transition
Macrophages adopt different phenotypes and play crucial roles in tissue homeostasis in both normal and fibrotic kidneys. They are often classified as pro-inflammatory (M1) or anti-inflammatory (M2) macrophages [115]. M1 macrophages contribute to the host defense and aggravate kidney injury, whereas M2 macrophages are associated with injury resolution and repair. However, in later stages of renal injury, M2 macrophages can also produce pro-fibrotic mediators, including Il-10 and TGF-β1, thereby contributing to renal interstitial fibrosis [115,116]. Prolonged pro-fibrotic activation has been linked to macrophage-to-myofibroblast transitions (MMT), in which macrophages acquire myofibroblast-like features and contribute as matrix-producing cells during renal fibrosis [116,117]. Because MMT is considered an important mechanism and a potential therapeutic target, we discuss the miRNA regulation of MMT in renal interstitial fibrosis in the following section[116,118].
Biopsy specimens from patients with chronic active renal graft rejection showed CD68/α-SMA double-positive cells consistent with MMT, and mice after IRI likewise exhibited numerous F4/80 and α-SMA double-positive cells in the renal interstitium at day 28. After IRI, the mouse kidneys displayed tubular senescence markers (p21/p16). In addition, paclitaxel-treated senescent HK-2 cells released extracellular vesicles enriched with miR-20a-5p and miR-21-5p. These miRNAs drive M2-like polarization and expression of fibrotic markers in macrophages. These miRNAs mechanistically suppress Smad7, increase phosphorylation of Smad3, and form a positive feedback loop with TGF-β1. Mimic transfection in macrophages recapitulated the EV-driven phenotype by increasing M2-associated features, elevating TGF-β1, and upregulating myofibroblast markers, whereas miR inhibitors attenuated these pro-fibrotic markers and partially blunted the effects induced by senescent HK-2 cells-derived EVs [119].
In LPS-stimulated RAW264.7 cells, miR-92a-3p overexpression augmented M1-associated inflammatory responses with increases in iNOS, Il-6, and TNF/TNF-α, whereas miR-92a-3p inhibition blunted these changes. Mechanistically, LIN28A was validated as a direct target of miR-92a-3p, and LIN28A overexpression rescued the pro-inflammatory effects of miR-92a-3p. LIN28 is known for its selective suppression of miRNA expression and influences cell proliferation, differentiation, and metabolic processes across various cell types [120]. In vivo, systemic administration of a miR-92a-3p inhibitor attenuated UUO-induced renal inflammation and interstitial fibrosis, accompanied by restored expression of LIN28A and reduced expression of iNOS and α-SMA. While MMT was not directly examined, miR-92a-3p was shown to amplify the M1-type inflammatory activation of macrophages by targeting LIN28A and worsen renal inflammation and fibrosis in vivo [121].
In a mouse model of AA-induced nephropathy, miR-382 was found to be enriched in kidney macrophages isolated by flow cytometry. Systemic and macrophage-specific genetic deletion of miR-382 reduced CD206+M2-like polarization and attenuated interstitial fibrosis, whereas miR-382 overexpression in bone marrow-derived macrophages aggravated renal injury and fibrosis. Mechanistically, miR-382 directly targeted signal regulatory protein-alpha (SIRP-α), thereby enhancing phosphorylation of STAT3 and promoting a pro-fibrotic macrophage program. These findings highlight a miRNA-controlled macrophage axis that can amplify pro-fibrotic signaling and tubular injury in renal fibrosis [122].
Renal miR-150 levels increased in the FA–induced AKI-to-CKD model and in HK-2 cells co-cultured with macrophages. This upregulation was accompanied by reduced suppressor of cytokine signal 1 (SOCS1) expression and increased levels of α-SMA, fibronectin, and collagen I. SOCS1 is an antifibrotic effector protein [123]. Systemic administration of LNA–anti–miR-150 restored SOCS1 expression, reduced renal IFN-γ/Il-6/TNF-α expression, diminished macrophage accumulation, and attenuated tubulointerstitial fibrosis. In vitro co-culture with macrophages increased miR-150 expression in HK-2 cells. This was accompanied by reduced SOCS1, activation of the JAK/STAT pathway, and upregulation of pro-fibrotic markers. These changes were partially reversed upon treatment with a miR-150 antagonist. Although MMT was not directly assessed, the data highlighted miRNA-driven macrophage–tubular crosstalk that may create a pro-fibrotic milieu permissive to MMT [124].
8. Regulation of Pericyte-To-Myofibroblast Transition
Pericytes are perivascular cells located on the abluminal surface of capillary endothelial cells. Under physiological conditions, they stabilize the microvascular network and maintain vascular homeostasis through angiogenic growth factors [125]. During kidney injury, endothelial cell–derived signals, including VEGF, TGF-β1, and PDGF, can activate neighboring pericytes [37,126]. Activated pericytes detach from endothelial cells, lose their vessel-stabilizing functions, migrate into the interstitial space, and transition into myofibroblasts [126]. This pericyte-to-myofibroblast transition contributes to peritubular capillary rarefaction and promotes interstitial fibrosis [37,126].
A recent study explored the miRNA-dependent regulation of pericyte-to-mesenchymal transition in renal interstitial fibrosis using primary kidney pericytes. Hu et al. found that bone marrow mesenchymal stem cell–derived exosomes delivered miR-34c-5p to TGF-β1–stimulated primary renal pericytes, suppressing α1,6-fucosyltransferase (FUT8) and reducing core fucosylation of pro-fibrotic receptors. This inhibition decreased pro-fibrotic signaling and extracellular matrix production in vitro and improved renal interstitial fibrosis in vivo. The transfer of miR-34c-5p was driven by the CD81–EGFR complex, with similar effects observed in renal fibroblasts and macrophages [127].
9. Clinical Utilities of miRNAs in Acute Kidney Injury and Chronic Kidney Disease
9.1. IgA Nephropathy (IgAN)
IgAN is the most prevalent form of primary glomerulonephritis and predominantly affects children and young adults. The most striking feature of IgAN is the mesangial deposition of IgA or IgA-containing immune complexes [128,129]. Pathological risk stratification is commonly guided by the Oxford classification (MEST-C), in which the T-score reflects the extent of tubular atrophy/interstitial fibrosis (TA/IF), T0 (0–25%), T1 (26–50%), and T2 (>50%). [130]. Notably, TA/IF represents one of the strongest predictors of long-term renal outcome across cohorts, underscoring the clinical importance of interstitial fibrotic remodeling in IgAN [131,132]. However, the mechanisms driving tubulointerstitial fibrosis and optimal therapeutic strategies remain unclear. Recent studies have revealed that ncRNAs are regulators of inflammation-fibrotic programs; therefore, we summarize recent evidence on ncRNA-associated pathways relevant to interstitial fibrosis in IgAN.
Fang et al. reported that miR-382 promotes renal tubulointerstitial fibrosis by targeting heat shock protein 60 (HSPD1), which is accompanied by decreased thioredoxin (Trx) expression and increased oxidative stress (e.g., elevated 3-nitrotyrosine [3-NT]). In kidney biopsy specimens from IgA nephropathy patients with moderate-to-severe tubulointerstitial fibrosis, HSPD1 and Trx were reduced whereas 3-NT was increased, supporting the clinical relevance of this pathway [133].
Duan et al. showed that miR-185-5p directly targets tight junction protein 1 (TJP1) in tubular epithelial cells. miR-185-5p–mediated TJP1 suppression promoted a profibrotic phenotype in HK-2 cells, with increased α-SMA, fibronectin, and collagens. Clinically, urinary miR-185-5p levels were significantly higher in IgAN patients with Oxford T1–T2 lesions than in those with T0, supporting its potential utility as a noninvasive marker of tubulointerstitial injury [134].
Szeto et al. reported that intrarenal miR-21 expression was significantly increased in IgAN compared with hypertensive nephrosclerosis and showed a modest correlation with the severity of tubulointerstitial fibrosis. Higher intrarenal miR-21 levels were associated with poorer kidney event–free survival in univariate analysis, although the association became borderline after adjustment for histologic scarring. These findings suggested that intrarenal miR-21 expression was increased in patients with IgAN, modestly correlated with the severity of histologic damage, and predictive of subsequent kidney function loss [135].
9.2. Diabetic Nephropathy (DN)
DN is the leading cause of CKD and ESKD [136,137]. Recent data show that approximately 529 million people worldwide will be affected by diabetes in 2021, and it is predicted that the number will increase to 1.3 billion by 2050 [138,139]. Parallel to the increasing prevalence of diabetes, the global incidence of DN has markedly increased. In fact, around 30-40% of people with type 1 or type 2 diabetes develop DN [140].
Hyperglycemic conditions induce the glycation of proteins and lipids, the formation of advanced glycation end products, and activated polyol pathway activity, leading to glomerular and tubular injury [141]. Moreover, hyperglycemia triggers proinflammatory mediators like TNF-α, IL-1, and IL-6, leading to endothelial dysfunction and tubulointerstitial fibrosis. DN involves a complex interplay of metabolic changes, hemodynamic alterations, and inflammation. Pathological features of DN include mesangial expansion, glomerular hypertrophy, tubulointerstitial fibrosis from ECM protein accumulation, podocyte dysfunction, and basement membrane thickening [141].
Activation of TGF-β/Smad signaling is a key mechanism in renal fibrosis, influencing inflammation, oxidative stress, endothelial dysfunction, and podocyte injury, which leads to proteinuria in DN [14,142]. In addition, hyperglycemic condition-mediated reactive oxygen species (ROS) lead to cellular injury, endothelial dysfunction, and activation of pro-inflammatory signaling pathways, such as NF-κB and JAK/STAT, all of which contribute to mesangial expansion, glomerulosclerosis, and tubulointerstitial fibrosis [142].
Recently, several studies have demonstrated that modulation of ncRNA activity in the kidney is an important mechanism underlying DN pathogenesis.
Ding et al. reported that miR-10a and -10b are downregulated in the kidneys of STZ-treated DN mice and patients with DN, as well as in HG-treated human glomerular podocytes and tubular epithelial cells [143]. In another experiment [144], miR-10a/10b overexpression reduces renal fibrosis in STZ-treated diabetic mice by inhibiting TGF-β/Smad signaling, a process reversed by anti-miR-10a and -10b. Luciferase reporter analysis in human glomerular podocytes and tubular epithelial cells revealed that miR-10a and -10b target the 3’UTR of TGFBR1, which regulates fibronectin and α-SMA. In summary, miR-10a/b modulates renal fibrosis by directly targeting TGFBR1 and influencing TGF-β/Smad signaling.
Fu et al. found that miR-17 was significantly upregulated in the serum of patients with DN and in the serum and kidneys of db/db mice. In addition, the miR-17 inhibitor attenuated renal fibrosis by suppressing TGF-β1/Smad7 signaling. Luciferase reporter experiment demonstrated that miR-17 bound to the 3’UTR of human Smad7 using human renal mesangial cells (HRMCs) and HK-2 cells. In conclusion, miR-17 regulates renal fibrotic genes such as E-cadherin, vimentin, fibronectin, and collagen I via TGF-β1/Smad7 signaling [63].
In a study by Kim et al., it was found that renal miR-144-3p regulates TGF-β1-induced oxidative stress and fibrosis. This miRNA is upregulated in the urine and kidneys of spontaneously hypertensive rats (SHRs) and STZ-treated SHRs. A luciferase reporter assay showed that miR-144-3p binds to the 3’UTR of NRF2, a negative regulator of oxidative stress linked to metabolism and inflammation. TGF-β1 suppresses NRF2 and increases ROS, resulting in renal fibrosis via miR-144-3p upregulation. These findings suggest that targeting the miR-144-3p/NRF2 pathway may offer therapeutic potential for CKD in the context of hypertension and diabetes [145].
Another type of ncRNA, circRNA, has been reported to play an important role in the regulation of renal fibrosis in DN. Jiang et al. demonstrated that circ_0054633 was upregulated more in the serum of patients with DN than in those without DN, which was positively correlated with the renal fibrotic area. Attenuating upregulation of circ_005463 protected HG-treated HRMCs from cell proliferation and ECM accumulation via downregulation of TGF-β1/SMAD3 signaling. To investigate the underlying mechanism, they co-transfected with luciferase reporter vectors containing the circ_0054633 or circ_0054633 mutant and miR-NC or miR-136-5p mimic. These results have demonstrated the role of circ_0054633 as a sponge for miR-136-5p. In addition, the experiment showed that miR-136-5p bound to the 3’UTR of SMAD3 in HRMCs. Furthermore, silencing of circ_0054633 in db/db mice attenuated collagen Ⅳ and fibronectin activation from TGF-β1/SMAD3 signaling via regulation of miR-136-5p/SMAD3 signaling. In summary, this study suggests that circ_0054633 plays a clinically important role in the regulation of renal fibrosis via miR-136-5p/SMAD3 [146].
LncRNAs also have functional roles in interactions with small molecules, such as mRNAs, miRNAs, or proteins. Wang et al. identified the role of lncRNA small nucleolar RNA host gene 14 (SNHG14) in DN. SNHG14 expression was elevated in the kidneys of mice with STZ-induced DN and in HG-treated HRMCs. Next, they confirmed that SNHG14 silencing inhibited cell proliferation and fibrosis in HG-treated HRMCs and kidneys of DN mice. Luciferase reporter experiments demonstrated that SNHG14 bound to miR-30e-5p in HRMCs and miR-30e-5p also bound to the 3’UTR of SRY-box transcription factor 4 (SOX4) expression in HRMCs. SOX4 is a critical factor involved in tubular epithelial cell (TEC) dedifferentiation and fibroblast activation [147]. In summary, SNHG14 reduces cell proliferation and fibrotic phenotype in DN via the miR-30e-5p/SOX4 axis [148].
Recent studies on the association between ncRNAs and fibrosis activation in DN are summarized in Table 3.
9.3. Hypertensive Nephropathy
Hypertensive nephropathy refers to chronic kidney damage caused by prolonged high blood pressure. It is the leading cause of CKD and ESKD [160]. Pathologically, hypertensive nephropathy is characterized by arterial and arteriolar sclerosis, glomerulosclerosis, and tubulointerstitial fibrosis, which together result in the progressive loss of renal function. In recent years, non-coding RNAs, including miRNAs and lncRNAs, have emerged as important regulators of gene expression in renal disease. Given their central roles in gene regulation, miRNAs and lncRNAs have been intensively studied for their contribution to the pathogenesis of hypertensive nephropathy and their potential as diagnostic biomarkers or therapeutic targets.
Several miRNAs are associated with renal injury in patients with chronic hypertension. Broad miRNA expression profiles in hypertensive models and patient samples suggest a signature of dysregulated miRNAs that correlate with kidney tissue damage and functional decline. In a biopsy-based study of patients with hypertensive nephrosclerosis, Wang et al. reported increased intrarenal expression of several miRNAs, including members of the miR-200 family (miR-200a, miR-200b, miR-141, and miR-429) as well as miR-205 and miR-192, compared to controls [161]. In that study, higher intrarenal levels of these miRNAs correlated with the degree of proteinuria, and miR-200a and miR-205 showed inverse correlations with the estimated glomerular filtration rate. The miR-200 family regulates epithelial–mesenchymal transition (EMT) by targeting the transcription factor ZEB1/2. Consistent with this regulatory axis, ZEB1 expression was inversely correlated with miR-429, and ZEB2 expression was inversely correlated with miR-200a, miR-200b, and miR-429 in biopsy specimens. Taken together, these observations support an association between intrarenal miRNA dysregulation and clinical/pathological severity in hypertensive nephrosclerosis; however, the cross-sectional nature of the data limits causal inference regarding whether these miRNAs are protective responses or contributors to injury [161].
Another miRNA linked to renal fibrosis in hypertension is miR-21. miR-21 is a well-known pro-fibrotic miRNA found in many organs. In a mouse model of deoxycorticosterone acetate (DOCA)-salt hypertension, which induces high blood pressure and kidney injury, miR-21 was among the most upregulated miRNAs in injured kidneys [162]. Notably, urinary miR-21 (normalized to creatinine) levels increased early after DOCA-salt treatment (detectable by day 4 in their analysis) and were proposed to increase earlier than albuminuria, suggesting its potential utility as an early indicator of hypertensive kidney injury in that model [70]. The same study also reported an increased expression of additional miRNAs (including miR-146b, miR-155, and miR-132) in the kidney during DOCA-salt exposure [162].
Conversely, some miRNAs appear to play protective roles by blunting the impact of hypertension on the kidneys. One striking example is miR-204-5p, which is highly enriched in the renal tissue [163]. In patients with hypertension or hypertensive nephrosclerosis, levels of miR-204-5p in the kidneys are significantly lower than in normotensive controls. This downregulation is mirrored in animal models, such as hypertensive Dahl salt-sensitive rats and Ang II-induced hypertensive mice, which show reduced renal miR-204 levels. Knocking out the mir-204 gene in mice exacerbates albuminuria, interstitial fibrosis, and arteriolar thickening. Additionally, inhibiting miR-204 in hypertensive rats worsens renal artery sclerosis and fibrosis. MiR-204 normally targets SHP2 mRNA; its loss results in SHP2 upregulation and overactivation of the STAT3 pathway, leading to inflammation and fibrosis. Thus, miR-204 serves as a protective mechanism against hypertension-related renal injury by regulating SHP2/STAT3 signaling.
One of the best-characterized lncRNAs is TUG1, which has emerged as a promoter of Ang II-mediated renal injury. A recent study showed that TUG1 expression is significantly increased in renal tubular epithelial cells following Ang II treatment [164]. In vivo, mouse models of Ang II-induced hypertension and fibrosis exhibit upregulation of TUG1 in the kidneys, particularly in areas of tubulointerstitial fibrosis. Tug1 expression is higher in human kidney biopsies with fibrotic lesions than in non-fibrotic samples. It interacts with the mineralocorticoid receptor (MR) and acts as a competing endogenous RNA, sequestering miR-29b-3p, an anti-fibrotic miRNA. This interaction promotes fibrotic responses by relieving the repression of extracellular matrix-related genes. Reducing TUG1 expression lessened angiotensin II-induced pro-fibrotic gene expression and renal fibrosis markers, while increased TUG1 expression worsened fibrosis. These findings highlight TUG1's role in linking renin–angiotensin–aldosterone system signaling to pro-fibrotic pathways in hypertensive kidney disease [164].
10. Conclusions
Recent studies indicate shared miRNA regulators across multiple fibrotic pathways, indicating that these miRNAs are potential novel therapeutic targets for renal interstitial fibrosis (Figure 1). For example, miR-21, miR-29, and miR-214 play key roles in resident fibroblast-to-myofibroblast mesenchymal transition, and miR-21, miR-27, miR-30, miR-124, and miR-214 regulate EMT.
Recent advancements in molecular diagnosis have improved our understanding of CKD with interstitial fibrosis. However, the causes of CKD remain unclear for many patients. miRNAs are promising targets for early diagnosis and therapy due to their roles in renal function and the progression of interstitial fibrosis. Despite this potential, most preclinical findings have not translated to clinical applications. Mechanistic dissection of miRNA regulatory networks will be essential for translating experimental findings into clinical interventions.
Author Contributions
Conceptualization, H.S. and S.K.; writing—original draft preparation, H.S, S.K., Y.K., A.K.; writing—review and editing, H.S. and S.K.; visualization, H.S. and S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Suhara Memorial Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
During the preparation of this manuscript, the authors used Editage (http//www.editage.com) for the purposes of editing and reviewing the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| AA | Aristolochic acid |
| ACSL1 | Acyl-CoA synthetase long-chain family member 1 |
| AKI | Acute kidney injury |
| α-SMA | Alpha-smooth muscle actin |
| Ang II | Angiotensin II |
| AP-1 | Activator protein 1 |
| BAT | Brown adipose tissue |
| BAX | BCL2-associated X protein |
| BUMPT | Boston University mouse proximal tubule cells |
| CBM | Coalbed methane |
| CD31 | Cluster of differentiation 31 |
| CD68 | Cluster of differentiation 68 |
| CKD | Chronic kidney disease |
| COL1A1 | Collagen type I alpha 1 chain |
| CREBBP | CREB-binding protein |
| CTGF | Connective tissue growth factor |
| circRNA | Circular RNA |
| DN | Diabetic nephropathy |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| EndMT | Endothelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| ESKD | End-stage kidney disease |
| EV | Extracellular vesicle |
| FA | Folic acid |
| FGF2 | Fibroblast growth factor 2 |
| FUT8 | α1,6-fucosyltransferase |
| GAB1 | GRB2-associated binding protein 1 |
| GSAP | γ-secretase–activating protein |
| HG | High glucose |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| HK-2 | Human kidney proximal tubular epithelial cell line |
| HUVEC | Human umbilical vein endothelial cell |
| IF/TA | Interstitial fibrosis/tubular atrophy |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| IRI | Ischemia–reperfusion injury |
| JAK | Janus kinase |
| LNA | Locked nucleic acid |
| lncRNA | Long non-coding RNA |
| MAPK | Mitogen-activated protein kinase |
| METTL7A | Methyltransferase-like 7A |
| miRNA / miR | MicroRNA |
| miRISC | MicroRNA-induced silencing complex |
| MMT | Macrophage–mesenchymal transition |
| MMP | Matrix metalloproteinase |
| NF-κB | Nuclear factor kappa B |
| NRK-49F | Normal rat kidney fibroblast cell line |
| NRK-52E | Normal rat kidney tubular epithelial cell line |
| PAS | Periodic acid–Schiff |
| PDCD4 | Programmed cell death protein 4 |
| PDGFR-β | Platelet-derived growth factor receptor beta |
| PI3K | Phosphoinositide 3-kinase |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PTEN | Phosphatase and tensin homolog |
| PTFS | Pure total flavonoids from Smilax glabra |
| rAAV | Recombinant adeno-associated virus |
| RECK | Reversion-inducing cysteine-rich protein with kazal motifs |
| RBP | RNA-binding protein |
| RFA | Radiofrequency ablation |
| RISC | RNA-induced silencing complex |
| RUNX1 | Runt-related transcription factor 1 |
| SARA | Smad anchor for receptor activation |
| Sfrp1 | Secreted frizzled-related protein 1 |
| SIRP-α | Signal regulatory protein alpha |
| SMAD | Mothers against decapentaplegic homolog |
| SnoN | Ski-related novel protein N |
| SOCS1 | Suppressor of cytokine signaling 1 |
| SPRY1 | Sprouty homolog 1 |
| STAT | Signal transducer and activator of transcription |
| STZ | Streptozotocin |
| TGF-β | Transforming growth factor beta |
| TGF-βRI/II | Transforming growth factor beta receptor I/II |
| TLR4 | Toll-like receptor 4 |
| TNF | Tumor necrosis factor |
| TPM1 | Tropomyosin 1 |
| TUG1 | Taurine upregulated gene 1 |
| UUO | Unilateral ureteral obstruction |
| VE-cadherin | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| vWF | von Willebrand factor |
References
- Kawaguchi, S. Noncoding RNAs as Key Regulators for Cardiac Development and Cardiovascular Diseases. J Cardiovasc Dev Dis 2023, 10. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–5. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–97. [Google Scholar] [CrossRef]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit Rev Biochem Mol Biol 2014, 49, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–33. [Google Scholar] [CrossRef]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of miRNA-Mediated Gene Regulation from Common Downregulation to mRNA-Specific Upregulation. Int J Genomics 2014, 2014, 970607. [Google Scholar] [PubMed]
- O'Brien, J. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Gomez, I.G.; Nakagawa, N.; Duffield, J.S. MicroRNAs as novel therapeutic targets to treat kidney injury and fibrosis. Am J Physiol Renal Physiol 2016, 310, F931–44. [Google Scholar] [CrossRef]
- Peters, L.J.F. MicroRNAs in Chronic Kidney Disease: Four Candidates for Clinical Application. Int J Mol Sci 2020, 21. [Google Scholar]
- Cerqueira, D.M.; Tayeb, M.; Ho, J. MicroRNAs in kidney development and disease. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Mahtal, N. MicroRNAs in kidney injury and disease. Nat Rev Nephrol 2022, 18, 643–662. [Google Scholar]
- Tsuji, K. MicroRNAs as Biomarkers and Therapeutic Targets for Acute Kidney Injury. Diagnostics (Basel) 2023, 13. [Google Scholar] [CrossRef]
- Bravo-Vázquez, L.A. Exploring the Therapeutic Significance of microRNAs and lncRNAs in Kidney Diseases. Genes (Basel) 2024, 15. [Google Scholar]
- Liu, Z. microRNAs in kidney diseases: Regulation, therapeutics, and biomarker potential. Pharmacol Ther 2024, 262, 108709. [Google Scholar] [PubMed]
- Brandenburger, T. Noncoding RNAs in acute kidney injury. Kidney Int 2018, 94, 870–881. [Google Scholar] [CrossRef]
- Liu, Z. Non-coding RNAs in kidney injury and repair. Am J Physiol Cell Physiol 2019, 317, C177–C188. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, G.B.D.C.K.D., Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733.
- Kalantar-Zadeh, K. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu Rev Physiol 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther 2023, 8, 129. [Google Scholar] [CrossRef]
- Nakagawa, N.; Duffield, J.S. Myofibroblasts in Fibrotic Kidneys. Curr Pathobiol Rep 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Kramann, R. Mechanisms of kidney fibrosis and routes towards therapy. Trends Endocrinol Metab 2024, 35, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int 2006, 69, 213–7. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012, 18, 1028–40. [Google Scholar] [CrossRef]
- Chen, Y.T. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 2011, 80, 1170–81. [Google Scholar] [CrossRef]
- Feng, Y.L. Small molecules against the origin and activation of myofibroblast for renal interstitial fibrosis therapy. Biomed Pharmacother 2021, 139, 111386. [Google Scholar] [CrossRef] [PubMed]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol 2011, 92, 158–67. [Google Scholar] [CrossRef]
- Lin, S.L. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 2008, 173, 1617–27. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 2011, 7, 684–96. [Google Scholar]
- Yang, L. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010, 16, 535–43, 1p following 143. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends Endocrinol Metab 2016, 27, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Zhuang, S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front Physiol 2020, 11, 569322. [Google Scholar] [PubMed]
- Jacobs, M.E. Endothelial to mesenchymal transition in kidney fibrosis. Nephrol Dial Transplant 2024, 39, 752–760. [Google Scholar]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 2014, 124, 2299–306. [Google Scholar]
- Lv, W. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol Genomics 2018, 50, 20–34. [Google Scholar]
- Chung, A.C.; Lan, H.Y. MicroRNAs in renal fibrosis. Front Physiol 2015, 6, 50. [Google Scholar]
- Fan, Y. Emerging role of miRNAs in renal fibrosis. RNA Biol 2020, 17, 1–12. [Google Scholar]
- Glowacki, F. Increased circulating miR-21 levels are associated with kidney fibrosis. PLoS One 2013, 8, e58014. [Google Scholar] [CrossRef]
- Sun, Q. The feedback loop between miR-21, PDCD4 and AP-1 functions as a driving force for renal fibrogenesis. J Cell Sci 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z. The autoregulatory feedback loop of microRNA-21/programmed cell death protein 4/activation protein-1 (MiR-21/PDCD4/AP-1) as a driving force for hepatic fibrosis development. J Biol Chem 2013, 288, 37082–93. [Google Scholar] [CrossRef] [PubMed]
- Li, N. Melatonin ameliorates renal fibroblast-myofibroblast transdifferentiation and renal fibrosis through miR-21-5p regulation. J Cell Mol Med 2020, 24, 5615–5628. [Google Scholar] [CrossRef]
- Zhao, S. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting PTEN in obstructed kidneys. Theranostics 2021, 11, 8660–8673. [Google Scholar] [CrossRef] [PubMed]
- Schauerte, C. Antagonism of profibrotic microRNA-21 improves outcome of murine chronic renal allograft dysfunction. Kidney Int 2017, 92, 646–656. [Google Scholar] [CrossRef]
- Chung, Y.H. MicroRNA-26a-5p Restoration Ameliorates Unilateral Ureteral Obstruction-Induced Renal Fibrosis in Mice Through Modulating TGF-beta Signaling. Lab Invest 2023, 103, 100131. [Google Scholar] [CrossRef]
- Huang, H. The MicroRNA MiR-29c Alleviates Renal Fibrosis via TPM1-Mediated Suppression of the Wnt/beta-Catenin Pathway. Front Physiol 2020, 11, 331. [Google Scholar] [CrossRef]
- Huang, J. Mesenchymal cell-derived exosomes and miR-29a-3p mitigate renal fibrosis and vascular rarefaction after renal ischemia reperfusion injury. Stem Cell Res Ther 2025, 16, 135. [Google Scholar] [CrossRef]
- Li, H. TGF-beta-mediated upregulation of Sox9 in fibroblast promotes renal fibrosis. Biochim Biophys Acta Mol Basis Dis 2018, 1864, 520–532. [Google Scholar] [CrossRef]
- Sun, C.M. Fer exacerbates renal fibrosis and can be targeted by miR-29c-3p. Open Med (Wars) 2021, 16, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H. rAAV9-mediated supplementation of miR-29b improve angiotensin-II induced renal fibrosis in mice. Mol Med 2021, 27, 89. [Google Scholar] [CrossRef]
- Saito, S. MiR-34a induces myofibroblast differentiation from renal fibroblasts. Clin Exp Nephrol 2023, 27, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, H. Inducible deletion of microRNA activity in kidney mesenchymal cells exacerbates renal fibrosis. Sci Rep 2024, 14, 10963. [Google Scholar] [CrossRef]
- Yang, J. Exosomal miR-122-5p from tubular cells ameliorates renal interstitial fibrosis by regulating fibroblasts via HIF-1alpha. Cell Death Discov 2025, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. Tubular cell-derived exosomal miR-150-5p contributes to renal fibrosis following unilateral ischemia-reperfusion injury by activating fibroblast in vitro and in vivo. Int J Biol Sci 2021, 17, 4021–4033. [Google Scholar] [CrossRef]
- Zhang, W. miR-155-5p Implicates in the Pathogenesis of Renal Fibrosis via Targeting SOCS1 and SOCS6. Oxid Med Cell Longev 2020, 2020, 6263921. [Google Scholar] [CrossRef]
- Zhang, X. MicroRNA-181 exerts an inhibitory role during renal fibrosis by targeting early growth response factor-1 and attenuating the expression of profibrotic markers. Mol Med Rep 2019, 19, 3305–3313. [Google Scholar] [CrossRef]
- Chen, B. The miRNA-184 drives renal fibrosis by targeting HIF1AN in vitro and in vivo. Int Urol Nephrol 2019, 51, 543–550. [Google Scholar] [CrossRef]
- Dai, R. Renal tubular epithelial cell-derived Exosomal miR-330-3p plays a key role in fibroblast activation and renal fibrosis by regulating CREBBP. Stem Cell Res Ther 2025, 16, 203. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–90. [Google Scholar] [CrossRef]
- Menon, M.C.; Ross, M.J. Epithelial-to-mesenchymal transition of tubular epithelial cells in renal fibrosis: a new twist on an old tale. Kidney Int 2016, 89, 263–6. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chu, D.; Geng, X. Downregulation of miR-17 suppresses TGF-beta1-mediated renal fibrosis through targeting Smad7. Mol Cell Biochem 2021, 476, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miR-19 promotes development of renal fibrosis by targeting PTEN-mediated epithelial-mesenchymal transition. Int J Clin Exp Pathol 2020, 13, 642–654. [Google Scholar]
- Luo, Q. Total flavonoids from Smilax glabra Roxb blocks epithelial-mesenchymal transition and inhibits renal interstitial fibrosis by targeting miR-21/PTEN signaling. Journal of Cellular Biochemistry 2018, 120, 3861–3873. [Google Scholar] [CrossRef]
- Wang, X. Upregulation of miR-382 contributes to renal fibrosis secondary to aristolochic acid-induced kidney injury via PTEN signaling pathway. Cell Death & Disease 2020, 11. [Google Scholar]
- Xu, H. Down-regulation of miR-23a inhibits high glucose-induced EMT and renal fibrogenesis by up-regulation of SnoN. Hum Cell 2018, 31, 22–32. [Google Scholar] [CrossRef]
- Ai, K. miR-130a-3p inhibition protects against renal fibrosis in vitro via the TGF-β1/Smad pathway by targeting SnoN. Experimental and Molecular Pathology 2020, 112. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics 2020, 10, 384–397. [Google Scholar] [CrossRef]
- Wu, L. Involvement of miR-27a-3p in diabetic nephropathy via affecting renal fibrosis, mitochondrial dysfunction, and endoplasmic reticulum stress. J Cell Physiol 2021, 236, 1454–1468. [Google Scholar] [CrossRef]
- Shi, M. MicroRNA-27a targets Sfrp1 to induce renal fibrosis in diabetic nephropathy by activating Wnt/beta-Catenin signalling. Biosci Rep 2020, 40. [Google Scholar] [CrossRef]
- Bai, L. MiR-27b-3p inhibits the progression of renal fibrosis via suppressing STAT1. Hum Cell 2021, 34, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y. Up-regulation of long non-coding RNA H19 ameliorates renal tubulointerstitial fibrosis by reducing lipid deposition and inflammatory response through regulation of the microRNA-130a-3p/long-chain acyl-CoA synthetase 1 axis. Non-coding RNA Research 2024, 9, 1120–1132. [Google Scholar] [CrossRef]
- Chung, K.W. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J Am Soc Nephrol 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Kim, J. TLR7 activation by miR-21 promotes renal fibrosis by activating the pro-inflammatory signaling pathway in tubule epithelial cells. Cell Commun Signal 2023, 21, 215. [Google Scholar] [CrossRef]
- Liu, E. METTL3/N6-methyladenosine/ miR-21-5p promotes obstructive renal fibrosis by regulating inflammation through SPRY1/ERK/NF-kappaB pathway activation. J Cell Mol Med 2021, 25, 7660–7674. [Google Scholar] [CrossRef]
- Zhang, Y. Brown adipose tissue transplantation ameliorates diabetic nephropathy through the miR-30b pathway by targeting Runx1. Metabolism 2021, 125, 154916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. miR-30b-5p modulate renal epithelial-mesenchymal transition in diabetic nephropathy by directly targeting SNAI1. Biochem Biophys Res Commun 2021, 535, 12–18. [Google Scholar] [CrossRef]
- Zhao, Y. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell 2017, 16, 387–400. [Google Scholar] [CrossRef]
- Wang, H.J. MiR-32-5p knockdown inhibits epithelial to mesenchymal transition and renal fibrosis by targeting SMAD7 in diabetic nephropathy. Hum Exp Toxicol 2021, 40, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. MicroRNA-34a Promotes Renal Fibrosis by Downregulation of Klotho in Tubular Epithelial Cells. Molecular Therapy 2019, 27, 1051–1065. [Google Scholar] [CrossRef]
- Xu, M. MiR-92a-3p Knockdown Attenuates Transforming Growth Factor-β1-induced Tubulointerstitial Fibrosis by Targeting LIN28A-mediated EMT Pathway. Journal of Physiological Investigation 2024, 67, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. MiR-92d-3p suppresses the progression of diabetic nephropathy renal fibrosis by inhibiting the C3/HMGB1/TGF-beta1 pathway. Biosci Rep 2021, 41. [Google Scholar] [CrossRef]
- Ma, J. Up-regulation of microRNA-93 inhibits TGF-β1-induced EMT and renal fibrogenesis by down-regulation of Orai1. Journal of Pharmacological Sciences 2018, 136, 218–227. [Google Scholar] [CrossRef]
- Wang, Q. MicroRNA-101 inhibits renal tubular epithelial-to-mesenchymal transition by targeting TGF-beta1 type I receptor. Int J Mol Med 2021, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Baicalin suppresses renal fibrosis through microRNA-124/TLR4/NF-κB axis in streptozotocin-induced diabetic nephropathy mice and high glucose-treated human proximal tubule epithelial cells. Journal of Physiology and Biochemistry 2020, 76, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Bai, X. MicroRNA-130b improves renal tubulointerstitial fibrosis via repression of Snail-induced epithelial-mesenchymal transition in diabetic nephropathy. Scientific Reports 2016, 6. [Google Scholar] [CrossRef]
- Liu, L. miR-136 improves renal fibrosis in diabetic rats by targeting down-regulation of tyrosine kinase SYK and inhibition of TGF-β1/Smad3 signaling pathway. Renal Failure 2020, 42, 513–522. [Google Scholar] [CrossRef]
- Liao, W. MicroRNA-140-5p Mediates Renal Fibrosis Through TGF-β1/Smad Signaling Pathway by Directly Targeting TGFBR1. Frontiers in Physiology 2020, 11. [Google Scholar] [CrossRef]
- Yi, Z. METTL3 aggravates renal fibrogenesis in obstructive nephropathy via the miR-199a-3p/PAR4 axis. European Journal of Pharmacology 2024, 982. [Google Scholar]
- Liu, X. Upregulation of miR-200a improves ureteral obstruction-induced renal fibrosis via GAB1/Wnt/β-catenin signaling. Nefrología (English Edition) 2023, 43, 21–31. [Google Scholar] [CrossRef]
- Chen, S.-J. miR-204 regulates epithelial-mesenchymal transition by targeting SP1 in the tubular epithelial cells after acute kidney injury induced by ischemia-reperfusion. Oncology Reports 2017, 37, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Jin, J. Exosomes as nanostructures deliver miR-204 in alleviation of mitochondrial dysfunction in diabetic nephropathy through suppressing methyltransferase-like 7A-mediated CIDEC N6-methyladenosine methylation. Aging (Albany NY) 2024, 16, 3302–3331. [Google Scholar] [CrossRef] [PubMed]
- Dong, J. MicroRNA-204-5p Ameliorates Renal Injury via Regulating Keap1/Nrf2 Pathway in Diabetic Kidney Disease. Diabetes, Metabolic Syndrome and Obesity 2024, 17, 75–92. [Google Scholar] [CrossRef]
- Ma, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest 2020, 130, 5011–5026. [Google Scholar] [CrossRef]
- Bai, M. MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney International 2019, 95, 1389–1404. [Google Scholar] [CrossRef]
- Sun, M. MicroRNA-302b mitigates renal fibrosis via inhibiting TGF-beta/Smad pathway activation. Braz J Med Biol Res 2021, 54, e9206. [Google Scholar] [CrossRef]
- Dong, X. MicroRNA-363-3p Inhibits the Expression of Renal Fibrosis Markers in TGF-β1-Treated HK-2 Cells by Targeting TGF-β2. Biochemical Genetics 2021, 59, 1033–1048. [Google Scholar] [CrossRef]
- Liang, M. Exosomes from miR-374a-5p-modified mesenchymal stem cells inhibit the progression of renal fibrosis by regulating MAPK6/MK5/YAP axis. Bioengineered 2022, 13, 4517–4527. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. MiR-455-3p suppresses renal fibrosis through repression of ROCK2 expression in diabetic nephropathy. Biochem Biophys Res Commun 2018, 503, 977–983. [Google Scholar] [CrossRef]
- Wang, L. Emodin ameliorates renal injury and fibrosis via regulating the miR-490-3p/HMGA2 axis. Front Pharmacol 2023, 14, 1042093. [Google Scholar] [CrossRef]
- Xue, X. MicroRNA-494-3p Exacerbates Renal Epithelial Cell Dysfunction by Targeting SOCS6 under High Glucose Treatment. Kidney Blood Press Res 2022, 47, 247–255. [Google Scholar]
- Wu, Q.-S. MicroRNA-630 alleviates inflammatory reactions in rats with diabetic kidney disease by targeting toll-like receptor 4. World Journal of Diabetes 2024, 15, 488–501. [Google Scholar] [PubMed]
- Zhang, C. Mechanistic study on lncRNA XIST/miR-124-3p/ITGB1 axis in renal fibrosis in obstructive nephropathy. Experimental Cell Research 2024, 442. [Google Scholar] [CrossRef]
- Xia, W. Knockdown of lncRNA MALAT1 attenuates renal interstitial fibrosis through miR-124-3p/ITGB1 axis. Sci Rep 2023, 13, 18076. [Google Scholar] [CrossRef] [PubMed]
- Hao, J. Silencing of LncRNA KCNQ1OT1 confers an inhibitory effect on renal fibrosis through repressing miR-124-3p activity. Bioengineered 2022, 13, 10399–10411. [Google Scholar] [CrossRef]
- Liu, L. Exploration of the mechanism of NORAD activation of TGF-β1/Smad3 through miR-136-5p and promotion of tacrolimus-induced renal fibrosis. Renal Failure 2023, 45. [Google Scholar] [CrossRef]
- Zhang, L.-C. Knockdown of the Long Noncoding RNA LUCAT1 Inhibits High-Glucose-Induced Epithelial-Mesenchymal Transition through the miR-199a-5p–ZEB1 Axis in Human Renal Tubular Epithelial Cells. In BioMed Research International; 2020. [Google Scholar]
- Lipphardt, M. The third path of tubulointerstitial fibrosis: aberrant endothelial secretome. Kidney Int 2017, 92, 558–568. [Google Scholar] [PubMed]
- Piao, X. Noncoding RNAs: Versatile regulators of endothelial dysfunction. Life Sci 2023, 334, 122246. [Google Scholar] [CrossRef]
- Correia, A.C. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-beta signaling. J Cell Sci 2016, 129, 569–79. [Google Scholar] [CrossRef]
- Wang, L. PTH-induced EndMT via miR-29a-5p/GSAP/Notch1 pathway contributed to valvular calcification in rats with CKD. Cell Prolif 2021, 54, e13018. [Google Scholar] [CrossRef]
- Jordan, N.P. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Qian, L. LncRNA TUG1 mitigates chronic kidney disease through miR-542-3p/HIF-1alpha/VEGF axis. Heliyon 2025, 11, e40891. [Google Scholar] [PubMed]
- Jiang, Y. Macrophages in organ fibrosis: from pathogenesis to therapeutic targets. Cell Death Discov 2024, 10, 487. [Google Scholar] [PubMed]
- Wei, J.; Xu, Z.; Yan, X. The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front Immunol 2022, 13, 934377. [Google Scholar]
- Meng, X.M. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis 2016, 7, e2495. [Google Scholar] [CrossRef]
- Zhang, J. Regulatory mechanisms of macrophage-myofibroblast transdifferentiation: A potential therapeutic strategy for fibrosis. Biochem Biophys Res Commun 2024, 737, 150915. [Google Scholar] [CrossRef]
- Zhong, Q. Senescent renal tubular cells derived extracellular vesicles transported miR-20a and miR-21 induced macrophage-to-myofibroblast transition in renal fibrosis after ischemia reperfusion injury. Int J Biol Sci 2025, 21, 940–954. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, K.; Huang, Y. Lin28 modulates cell growth and associates with a subset of cell cycle regulator mRNAs in mouse embryonic stem cells. RNA 2009, 15, 357–61. [Google Scholar] [CrossRef] [PubMed]
- Xu, M. MiR-92a-3p Promotes Renal Injury and Fibrosis Through Facilitating M1 Macrophage Polarization via Targeting LIN28A. Physiol Res 2024, 73, 755–767. [Google Scholar] [CrossRef]
- Wang, X. MicroRNA-382 Promotes M2-Like Macrophage via the SIRP-alpha/STAT3 Signaling Pathway in Aristolochic Acid-Induced Renal Fibrosis. Front Immunol 2022, 13, 864984. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J Am Soc Nephrol 2013, 24, 1073–87. [Google Scholar] [CrossRef]
- Luan, J. miR-150-Based RNA Interference Attenuates Tubulointerstitial Fibrosis through the SOCS1/JAK/STAT Pathway In Vivo and In Vitro. Mol Ther Nucleic Acids 2020, 22, 871–884. [Google Scholar] [CrossRef]
- Chou, Y.H. Update of pericytes function and their roles in kidney diseases. J Formos Med Assoc 2024, 123, 307–317. [Google Scholar] [CrossRef]
- Chang, F.C. Novel insights into pericyte-myofibroblast transition and therapeutic targets in renal fibrosis. J Formos Med Assoc 2012, 111, 589–98. [Google Scholar] [CrossRef]
- Hu, X. Bone marrow mesenchymal stem cell-derived exosomal miR-34c-5p ameliorates RIF by inhibiting the core fucosylation of multiple proteins. Mol Ther 2022, 30, 763–781. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.J. Updating the International IgA Nephropathy Prediction Tool for use in children. Kidney Int 2021, 99, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Liu, C. Astragaloside IV Inhibits Galactose-Deficient IgA1 Secretion via miR-98-5p in Pediatric IgA Nephropathy. Front Pharmacol 2021, 12, 658236. [Google Scholar] [CrossRef]
- El Karoui, K.; Fervenza, F.C.; De Vriese, A.S. Treatment of IgA Nephropathy: A Rapidly Evolving Field. J Am Soc Nephrol 2024, 35, 103–116. [Google Scholar] [CrossRef]
- Haas, M. A Multicenter Study of the Predictive Value of Crescents in IgA Nephropathy. Journal of the American Society of Nephrology 2017, 28, 691–701. [Google Scholar] [CrossRef]
- Edstrom Halling, S.; Soderberg, M.P.; Berg, U.B. Predictors of outcome in paediatric IgA nephropathy with regard to clinical and histopathological variables (Oxford classification). Nephrol Dial Transplant 2012, 27, 715–22. [Google Scholar]
- Fang, Y. miR-382 Contributes to Renal Tubulointerstitial Fibrosis by Downregulating HSPD1. Oxid Med Cell Longev 2017, 2017, 4708516. [Google Scholar] [CrossRef]
- Duan, Z.Y. Urinary miR-185-5p is a biomarker of renal tubulointerstitial fibrosis in IgA nephropathy. Front Immunol 2024, 15, 1326026. [Google Scholar]
- Szeto, C.C. Kidney microRNA-21 Expression and Kidney Function in IgA Nephropathy. Kidney Med 2021, 3, 76–82 e1. [Google Scholar]
- Chen, Z.; Malek, V.; Natarajan, R. Update: the role of epigenetics in the metabolic memory of diabetic complications. Am J Physiol Renal Physiol 2024, 327, F327–f339. [Google Scholar] [PubMed]
- Natesan, V.; Kim, S.J. Diabetic Nephropathy - a Review of Risk Factors, Progression, Mechanism, and Dietary Management. Biomol Ther (Seoul) 2021, 29, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, K.; Garg, S.S.; Gupta, J. Targeting epigenetic regulators for treating diabetic nephropathy. Biochimie 2022, 202, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Advances in the Epigenetic Mechanisms of Diabetic Nephropathy Pathogenesis. Diabetes Metab Syndr Obes 2025, 18, 2629–2639. [Google Scholar]
- Gembillo, G. Planetary Health Diet and Body Mass Distribution in Relation to Kidney Health: Evidence from NHANES 2003-2018. Nutrients 2025, 17. [Google Scholar]
- Garcia-Fernandez, N. Matrix Metalloproteinases in Diabetic Kidney Disease. J Clin Med 2020, 9. [Google Scholar]
- Yaribeygi, H. The Emerging role of lipoprotein(a) in diabetic kidney disease: possible pathophysiological links and unresolved mechanisms. Diabetes Res Clin Pract 2025, 231, 113040. [Google Scholar]
- Ding, H. MicroRNA-10 negatively regulates inflammation in diabetic kidney via targeting activation of the NLRP3 inflammasome. Mol Ther 2021, 29, 2308–2320. [Google Scholar]
- Li, J. MicroRNA-10a/b inhibit TGF-β/Smad-induced renal fibrosis by targeting TGF-β receptor 1 in diabetic kidney disease. Mol Ther Nucleic Acids 2022, 28, 488–499. [Google Scholar]
- Kim, S.K. Renal microRNA-144-3p is associated with transforming growth factor-β1-induced oxidative stress and fibrosis by suppressing the NRF2 pathway in hypertensive diabetic kidney disease. Free Radic Biol Med 2024, 225, 546–559. [Google Scholar] [CrossRef]
- Jiang, Q. Circ_0054633 silencing suppresses hyperglycemia-induced extracellular matrix accumulation in renal mesangial cells by regulating MiR-136-5p/SMAD3 signaling. Noncoding RNA Res 2025, 13, 84–93. [Google Scholar]
- Du, H. Critical role of transcription factor SOX4 in tubular epithelial cell dedifferentiation and fibroblast activation during kidney fibrosis. Kidney Int 2025. [Google Scholar]
- Wang, Y. LncRNA SNHG14 silencing attenuates the progression of diabetic nephropathy via the miR-30e-5p/SOX4 axis. J Diabetes 2024, 16, e13565. [Google Scholar]
- Hao, J. The axis of miR-30a/AIF-1/TRPC6/calcineurin A/NFAT2 regulated the death modalities and inflammation of renal tubular epithelial cells in diabetic kidney disease via exosome. Life Sci 2025, 377, 123760. [Google Scholar]
- Zhuang, L. miR-543 regulates high glucose-induced fibrosis and autophagy in diabetic nephropathy by targeting TSPAN8. BMC Nephrol 2022, 23, 89. [Google Scholar]
- Sheng, S. miR-23a-3p regulates the inflammatory response and fibrosis in diabetic kidney disease by targeting early growth response 1. In Vitro Cell Dev Biol Anim 2021, 57, 763–774. [Google Scholar] [PubMed]
- Zhang, J. miR-1225-3p regulates fibrosis in mesangial cells via SMURF2-mediated ubiquitination of ChREBP in diabetic kidney disease. Ren Fail 2025, 47, 2484632. [Google Scholar]
- Hagiwara, S. miR-214 and Its Primary Transcript Dnm3os Regulate Fibrosis and Inflammation Through RAGE Signaling in Diabetic Kidney Disease. Diabetes 2025, 74, 1205–1219. [Google Scholar]
- Zhao, P. Mechanism of miR-365 in regulating BDNF-TrkB signal axis of HFD/STZ induced diabetic nephropathy fibrosis and renal function. Int Urol Nephrol 2021, 53, 2177–2187. [Google Scholar]
- Chen, S. circ_000166/miR-296 Aggravates the Process of Diabetic Renal Fibrosis by Regulating the SGLT2 Signaling Pathway in Renal Tubular Epithelial Cells. Dis Markers 2022, 2022, 6103086. [Google Scholar] [PubMed]
- Liu, J. CircRNA circ-ITCH improves renal inflammation and fibrosis in streptozotocin-induced diabetic mice by regulating the miR-33a-5p/SIRT6 axis. Inflamm Res 2021, 70, 835–846. [Google Scholar] [PubMed]
- Shi, S. Knockdown of LncRNA-H19 Ameliorates Kidney Fibrosis in Diabetic Mice by Suppressing miR-29a-Mediated EndMT. Front Pharmacol 2020, 11, 586895. [Google Scholar] [PubMed]
- Wang, T. LncTUG1 ameliorates renal tubular fibrosis in experimental diabetic nephropathy through the miR-145-5p/dual-specificity phosphatase 6 axis. Ren Fail 2023, 45, 2173950. [Google Scholar]
- Yin, Q.; Guo, N.; Liao, R. LncRNA GAS5 reduces blood glucose levels and alleviates renal fibrosis in diabetic nephropathy by regulating the miR-542-3p/ERBB4 axis. Diabetol Metab Syndr 2025, 17, 30. [Google Scholar]
- Zhang, Y. Epigenetics of Hypertensive Nephropathy. Biomedicines 2024, 12. [Google Scholar] [CrossRef]
- Wang, G. Intrarenal expression of miRNAs in patients with hypertensive nephrosclerosis. Am J Hypertens 2010, 23, 78–84. [Google Scholar]
- Chen, C. Urinary miR-21 as a potential biomarker of hypertensive kidney injury and fibrosis. Sci Rep 2017, 7, 17737. [Google Scholar] [PubMed]
- Cheng, Y. Endogenous miR-204 Protects the Kidney against Chronic Injury in Hypertension and Diabetes. J Am Soc Nephrol 2020, 31, 1539–1554. [Google Scholar] [PubMed]
- Zhang, J. Long Noncoding RNA Tug1 Promotes Angiotensin II-Induced Renal Fibrosis by Binding to Mineralocorticoid Receptor and Negatively Regulating MicroR-29b-3p. Hypertension 2021, 78, 693–705. [Google Scholar] [PubMed]
Figure 1.
Myofibroblast activation in renal interstitial fibrosis. Resident interstitial fibroblasts, pericytes, injured renal tubular epithelial cells undergoing epithelial-to-mesenchymal transition (EMT), endothelial cells undergoing endothelial-to-mesenchymal transition (EndMT), and activated macrophages derived from circulating monocytes undergoing macrophage-to-mesenchymal transition (MMT) are all known sources of myofibroblasts in fibrotic processes. Various miRNAs have been implicated in these processes. Some miRNAs exert protective effects (shown in blue), whereas others directly promote renal intersitital fibrosis (shown in red).
Figure 1.
Myofibroblast activation in renal interstitial fibrosis. Resident interstitial fibroblasts, pericytes, injured renal tubular epithelial cells undergoing epithelial-to-mesenchymal transition (EMT), endothelial cells undergoing endothelial-to-mesenchymal transition (EndMT), and activated macrophages derived from circulating monocytes undergoing macrophage-to-mesenchymal transition (MMT) are all known sources of myofibroblasts in fibrotic processes. Various miRNAs have been implicated in these processes. Some miRNAs exert protective effects (shown in blue), whereas others directly promote renal intersitital fibrosis (shown in red).
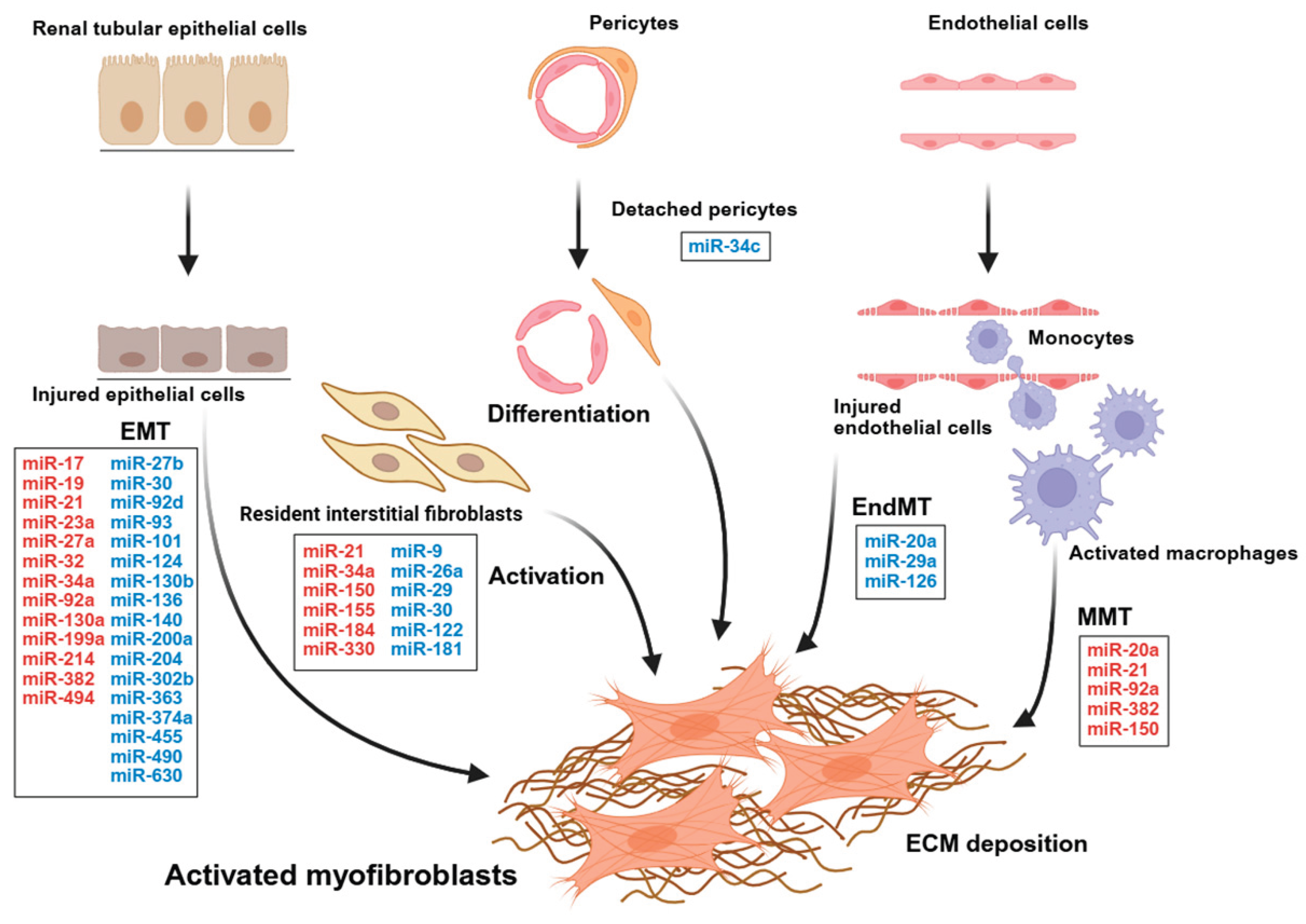

| NcRNA | Expression | Experimental models | Targets of ncRNAs | Ref |
|---|---|---|---|---|
| miR-9-5p | Down | UUO- and FA-induced mouse Mouse primary renal fibroblast |
miR-9-5p/Pdgfrβ | [54] |
| miR-21 | Up | Renal transplant recipients with severe renal interstitial fibrosis UUO-induced mouse TGF-β1 induced primary renal fibroblasts |
Not assessed | [41] |
| miR-21 | Up | UUO-induced mouse TGF-β1-induced NRK-49F |
miR-21/PDCD4/AP-1 | [42] |
| miR-21 | Up | UUO-induced mouse Exosomes derived from TGF-β1-induced NRK-52E transfer to NRK-49F |
miR-21/PTEN | [45] |
| miR-21a-5p | Up | Murine chronic renal allograft dysfunction LPS-activated macrophage co-culture and macrophage-derived small vesicles uptake by NRK-49F |
miR-21a-5p/Notch2 | [46] |
| miR-21-5p | Up | UUO-induced mouse TGF-β1-induced NRK-49F |
miR-21-5p/PTEN/Spry1 | [44] |
| miR-26a-5p | Down | Human kidney tissues with interstitial nephritis UUO-induced mouse TGF-β1-induced NRK-49F |
miR-26a-5p/TGF-β receptor I and II | [47] |
| miR-29a-3p | Down | Human CKD kidney biopsy Unilateral IRI-induced mouse HucMSC-derived exosomes and TGF-β1 induced NRK-49F and HUVEC |
miR-29a-3p/col1a1 (fibroblasts) | [49] |
| miR-29b | Down | Ang II-infused mouse with rAAV9-miR-29b delivery | Collagens (Predicted targets) |
[52] |
| miR-29c | Down | UUO-induced mouse with intrarenal pelvic injection of rAAV6-FSP1 TGF-β1-induced NRK-49F |
miR-29c/TPM1 | [48] |
| miR-29c-3p | Down | UUO-induced mouse TGF-β1-induced NRK-49F |
miR-29c-3p/Fer | [51] |
| miR-30 | Down | Human kidney tissues with renal fibrosis UUO- and FA-induced mouse TGF-β1 induced NRK-49F |
miR-30/SOX9 | [50] |
| miR-34a | Up | UUO-induced mouse TGF-β1-induced NRK-49F |
Not identified (p53/miR-34a axis) |
[53] |
| miR-122-5p | Down | UUO-induced mouse injected with exosomes isolated from UUO-kidneys and/or TGF-β1-induced HK-2 NRK-49F treated with exosomes derived from TGF-β1-induced -HK-2 cells |
miR-122-5p/HIF-1α | [55] |
| miR-150-5p | Up | Unilateral IRI-induced mouse injected with exosomes isolated from hypoxia-treated NRK-52E NRK-49F treated with exosomes isolated from hypoxia-treated NRK-52E |
miR-150-5p/SOCS1 | [56] |
| miR-155-5p | Up | Human kidney tissues with renal fibrosis UUO-induced rat HK-2 and NRK-49F |
miR-155-5p/SOCS1, SOCS6 | [57] |
| miR-181 | Down | Serum samples from patients with renal fibrosis UUO-induced mouse Ang II-stimulated NRK-49F |
miR-181/Egr-1 | [58] |
| miR-184 | Up | Serum samples from patients with renal fibrosis UUO-induced mouse Ang II-stimulated NRK-49F |
miR-184/HIF1AN | [59] |
| miR-330-3p | Up | Urine samples from patients with CKD Adenine-induced mouse with exosomes isolated from UA-treated NRK-52E NRK-49F treated with exosomes isolated from UA-stimulated NRK-52E |
miR-330-3p/CREBBP | [60] |
Ang II, Angiotensin II; AP-1, activation protein-1; CKD, Chronic kidney disease; Col1a1, Collagen type I alpha 1 chain; CREBBP, CREB-binding protein; ECM, Extracellular matrix; Egr-1, early growth response factor-1; FA, Folic acid; HG, High glucose; HIF-1α, Hypoxia-inducible factor 1 alpha; HK-2, Human kidney proximal tubular epithelial cell; HucMSC, Human umbilical cord mesenchymal cell; HUVEC, Human umbilical vein endothelial cell; IRI, Ischemia–reperfusion injury; LPS, Lipopolysaccharide; Notch2, neurogenic locus notch homolog protein 2; NRK-49F, Normal rat kidney fibroblast cell; NRK-52E, Normal rat kidney tubular epithelial cell; PDCD4, programmed cell death protein 4; PDGFR-β, Platelet-derived growth factor receptor beta; PTEN, Phosphatase and tensin homolog; SOX9, Sex-determining region Y-box containing gene 9; rAAV, Recombinant adeno-associated virus; SOCS, Suppressor of cytokine signaling; TGF-β, Transforming growth factor beta; UA, Uric acid; UUO, Unilateral ureteral obstruction.
| NcRNA | Expression | Experimental models | Targets of ncRNAs | Ref |
|---|---|---|---|---|
| miR-17 | Up | Serum samples from patients with DN db/db mouse TGF-β1 induced HK-2 |
miR-17/Smad7 | [63] |
| miR-19 | Up | Serum samples from patients with renal fibrosis UUO-induced mouse TGF-β1 induced NRK-52E |
miR-19/PTEN | [64] |
| miR-21 | Up | Aging rat kidney PPARα-deficient mouse TGF-β1 induced NRK-52E |
miR-21/PPARα | [74] |
| miR-21 | Up | UUO-induced rat TGF-β1 induced HK-2 |
miR-21/PTEN | [65] |
| miR-21 | Up | Adenine diet induced mouse LPS-stimulated NRK-52E |
Not assessed (miR-21 activates TLR7 signaling) |
[75] |
| miR-21-5p | Up | UUO-induced mouse HK-2 |
Not assessed (SPRY1/ERK/NF-κB axis) |
[76] |
| miR-23a | Up | Kidney tissues from patients with DN HG-treated with HK-2 |
miR-23a/SnoN | [67] |
| miR-27a | Up | STZ-induced rat HG-treated with NRK-52E |
miR-27a/Sfrp1 | [71] |
| miR-27a-3p | Up | db/db mouse HG-treated with HK-2 |
miR-27a-3p/Prohibitin, TMBIM6 | [70] |
| miR-27b-3p | Down | UUO-induced mouse TGF-β1 induced HK-2 |
miR-27b-3p/STAT1 | [72] |
| miR-30b | Down | HFD and STZ-induced mouse BAT transplantation TGF-β1 induced HK-2 |
miR-30b/Runx1, Snail1 | [77] |
| miR-30b-5p | Down | db/db mouse HG-treated with HK-2 |
miR-30b-5p/Snail1 | [78] |
| miR-30c | Down | db/db mouse HG-treated with HK-2 |
miR-30c/Snail1 | [79] |
| miR-32-5p | Up | STZ-induced rat HG-treated with HK-2 |
miR-32-5p/Smad7 | [80] |
| miR-34a-5p | Up | Kidney tissues from patients with renal fibrosis UUO-induced mouse TGF-β1 induced HK-2 |
miR-34a-5p/Klotho | [81] |
| miR-92a-3p | Up | UUO-induced mouse TGF-β1 induced HK-2 |
miR-92a-3p/LIN28A | [82] |
| miR-92d-3p | Down | Kidney tissues from patients with renal fibrosis UUO-induced mouse TGF-β1 induced HK-2 |
Not assessed (C3/HMGB1/ TGF-β1 pathway) |
[83] |
| miR-93 | Down | Kidney tissues from patients with renal fibrosis TGF-β1 induced HK-2 |
miR-93/Orail1 | [84] |
| miR-101 | Down | Mercury chloride (HgCl₂)-induced rat TGF-β1 induced HK-2 |
miR-101/TGF-βRI, Smad3 | [85] |
| miR-124 | Down | STZ-induced mouse HG-treated with HK-2 |
miR-124/TLR4 | [86] |
| miR-130a-3p | Up | TGF-β1 induced renal tubular epithelial cells | miR-130a-3p/SnoN | [68] |
| miR-130b | Down | Plasma samples and kidney tissues from patients with DN STZ-induced rat HG-treated with NRK-52E |
miR-130b/Snail | [87] |
| miR-136 | Down | STZ-induced rat HG-treated with NRK-52E |
miR-136/SYK | [88] |
| miR-140-5p | Down | UUO-induced mouse TGF-β1 induced HK-2 |
miR-140-5p/TGF-βRI | [89] |
| miR-199a-3p | Up (METTL3-dependent) |
Kidney tissues from patients with hydronephrosis UUO-induced mouse TGF-β1 induced HK-2 |
miR-199a-3p/Par4 | [90] |
| miR-200a | Down | UUO-induced rat TGF-β1 induced HK-2 |
miR-200a-3p/GAB1 | [91] |
| miR-204 | Down | Unilateral IRI-induced mouse Hypoxia-treated with HK-2 |
miR-204/SP-1 | [92] |
| miR-204 | Down | HFD and STZ-induced rat with ADSC-derived exosome delivery HG-treated with HK-2 and NRK-52E |
miR-204/METTL7A | [93] |
| miR-204-5p | Down | db/db mouse HG-treated with HK-2 |
miR-204-5p/Keap1 | [94] |
| miR-214 | Up | Kidney tissues from patients with DN Akita mouse and STZ-induced mouse HG-treated with RPTC and BUMPT |
miR-214/ULK1 | [95] |
| miR-214 | Up | Kidney tissues and urine sample from patients with CKD IRI-, UUO-, albumin overload-induced mouse PTC |
miR-214/mt-Nd6, mt-Nd4l | [96] |
| miR-302b | Down | UUO-induced mouse TGF-β1 induced HK-2 |
miR-302b/TGF-βRII | [97] |
| miR-363-3p | Down | TGF-β1 induced HK-2 | miR-363-3p/TGF-β2 | [98] |
| miR-374a-5p | Down | UUO-induced mouse MSC-derived exosome delivery TGF-β1 induced HK-2 |
miR-374a-5p/MAPK6 | [99] |
| miR-382 | Up | Kidney tissues from patients with IgAN AA-induced mouse AA-treated with mouse TEC |
miR-382/PTEN | [66] |
| miR-455-3p | Down | HFD and STZ-induced rat HG- or TGF-β1-treated with HK-2 or HMC |
miR-455-3p/ROCK2 | [100] |
| miR-490-3p | Down | UUO-induced mouse TGF-β1 induced NRK-52E |
miR-490-3p/HMGA2 | [101] |
| miR-494-3p | Up | HG-treated with HK-2 | miR-494-3p/SOCS6 | [102] |
| miR-630 | Down | STZ-induced rat HG-treated with NRK-52E |
miR-630/TLR4 | [103] |
| lncRNA H19 | Down | UUO-induced mouse TGF-β1 induced HK-2 |
lncRNA H19/miR-130a-3p/ACSL1 | [73] |
| lncRNA XIST | Up | Kidney tissues from patients with ON UUO-induced mouse TGF-β1 induced HK-2 |
lncRNA XIST/miR-124-3p/ITGB1 | [104] |
| lncRNA MALAT1 |
Up | Kidney tissues from patients with ON UUO-induced mouse TGF-β1 induced HK-2 and NRK-49F |
lncRNA MALAT1/ miR-124-3p/ITGB1 |
[105] |
| lncRNA KCNQ1OT1 | Up | UUO-induced mouse TGF-β1 induced HK-2 |
lncRNA KCNQ1OT1/miR-124-3p | [106] |
| lncRNA NORAD |
Up | Tacrolimus induced rat Tacrolimus induced NRK-52E |
lncRNA NORAD/miR-136-5p/SYK | [107] |
| lncRNA LUCAT1 |
Up | HG-treated with HK-2 | lncRNA LUCAT1/ miR-199a-5p/ZEB1 |
[108] |
AA, Aristolochic acid; ACSL1, long-chain acyl-CoA synthetase 1; ADSC, Adipose-derived stem cells; BAT, Brown adipose tissue; BUMPT, Boston University mouse proximal tubule cells; CKD, Chronic kidney disease; DN, Diabetic nephropathy; ERK, Extracellular signal-regulated kinase; GAB1, GRB2 associated binding protein 1; HG, High glucose; HFD, High-fat diet; HK-2, Human kidney proximal tubular epithelial cell; HMGA2, High migration protein A2; IgAN, IgA nephropathy; IRI, Ischemia–reperfusion injury; Keap1, Kelch-like ECH-associated protein 1; LIN28A, Lin-28 homolog A; LPS, Lipopolysaccharide, MAPK, Mitogen-activated protein kinase; METTL3, Methyltransferase-like 3; METTL7A, Methyltransferase-like 7A; MSC, Mesenchymal stem cells; NF-κB, Nuclear factor kappa B; NRK-49F, Normal rat kidney fibroblast cell; NRK-52E, Normal rat kidney tubular epithelial cell; ON, Obstructive nephropathy; Par4, Prostate apoptotic response 4; PPARα, Peroxisome proliferator-activated receptor α; PTC, Proximal tubular cells; PTEN, Phosphatase and tensin homolog; RECK, Reversion Inducing Cysteine Rich Protein with Kazal Motifs; ROCK2, Rho-associated coiled coil-containing protein kinase 2; RPTC, Renal proximal tubular cells; ROS, Reactive oxygen species; Runx 1, Runt-related transcription factor 1; Snail 1, Snail family zinc finger 1; Sfrp1, Secreted frizzled-related protein 1; SnoN, Ski-related novel protein N; SOCS, Suppressor of cytokine signaling; STAT1, Signal transducers and activators of transcription 1; STZ, Streptozotocin; SYK, Spleen tyrosine kinase; TGF-β, Transforming growth factor beta; TGF-βRI/II, Transforming growth factor beta receptor I/II; TMBIM6, transmembrane BAX inhibitor motif containing 6; TLR, Toll-like receptor; ULK1, Unc-51-like autophagy-activating kinase 1; UUO, Unilateral ureteral obstruction.
| NcRNA | Expression | Experimental models | Targets of ncRNAs | Ref |
|---|---|---|---|---|
| miR-30a | Down | db/db mouse HG-treated mouse renal tubular epithelial cells |
miR-30a/AIF-1/TRPC6/ calcineurin A/NFAT2 |
[149] |
| miR-543 | Down | db/db mouse HG-treated with HK-2 |
miR-543/TSPAN8 | [150] |
| miR-23a-3p | Down | HFD- or STZ- induced mouse BSA-stimulated HK-2 |
miR-23a-3p/Egr1 | [151] |
| miR-17 | Up | Serum samples from patients with DN db/db mouse kidney TGF-β1 induced HK-2 |
miR-17/Smad7 | [63] |
| miR-92d-3p | Down | Kidney tissues from patients with DN HFD and STZ-induced mouse TGF-β1 and C3 stimulated HK-2 |
Not assessed (C3/HMGB1/ TGF-β1 pathway) |
[83] |
| miR-1225-3p | Up | Kidney tissues from patients with DN STZ-induced mouse HG-treated with MCs |
miR-1225-3p/ARHGAP5/SMURF2 | [152] |
| miR-214 | Up (in mouse) |
Kidney tissues from patients with DN STZ-induced mouse and KK-Ay/Ta mouse MCs |
miR-214/ DIAPH1 | [153] |
| miR-10a/10b | Down | Kidney tissues from patients with DN STZ-induced mouse HG-treated with human podocytes and HK-2 |
miR-10a/b/ TGF-β1 | [144] |
| miR-144-3p | Up | STZ-induced SHR and WKY rat TGF-β1 induced NRK-52E |
miR-144-3p/NRF2 | [145] |
| miR-365 | Up | STZ-treated mouse HG-treated with HK-2 |
miR-365/BDNF | [154] |
| Circ_000166 | Up | STZ-treated mouse HG-treated with HK-2 |
Circ_000166/miR-296/SGLT2 | [155] |
| Circ_0054633 | Up | Serum and urine samples from patients with DN db/db mouse HG-treated with HRMCs |
Circ_0054633/MiR-136-5p/SMAD3 | [146] |
| Circ_ITCH | Down | STZ-treated mouse HG-treated with rat MCs |
Circ_ITCH/miR-33a-5p/SIRT6 | [156] |
| LncRNA H19 | Up | STZ-induced CD-1 mouse TGF-β2 treated with HMVECs |
LncRNA_H19/miR-29a/ TGF-βR |
[157] |
| LncRNA SNHG14 |
Up | STZ-treated mouse HG-treated with human MCs |
LncRNA SNHG14 /miR-30e-5p/SOX4 |
[148] |
| LncRNA TUG1 | Up | Uni-nephrectomy and STZ-treated mouse HG-treated with HK-2 |
TUG1 /miR-145-5p/DUSP6 | [158] |
| LncRNA GAS5 | Down | db/db mouse | LncRNA GAS5/miR-542-3p/ERBB4 | [159] |
AIF-1, allograft inflammatory factor 1; ARHGAP5, Rho GTPase-activating protein 5; BDNF, brain-derived neurotrophic factor; BSA, bovine serum albumin; DIAPH1, diaphanous related formin 1; DN, diabetic nephropathy; DUSP6, dual-specificity phosphatase 6; Egr1, early growth response 1; ERBB4, erb-b2 receptor tyrosine kinase 4; HFD, high-fat diet; HG, high glucose; HK-2, human kidney proximal tubular epithelial cell; HMVECs, human microvascular endothelial cells; HRMCs, human renal mesangial cells; MCs, mesangial cells; NFAT2, nuclear factor of activated T-cells 2; NRF2, nuclear factor erythroid 2–related factor 2; SHR, spontaneously hypertensive rat; SGLT2, sodium–glucose cotransporter 2; SIRT6, sirtuin 6; SMAD3, SMAD family member 3; SMURF2, SMAD specific E3 ubiquitin protein ligase 2; SOX4, SRY-box transcription factor 4; STZ, streptozotocin; TGF-β, transforming growth factor beta; TSPAN8, tetraspanin 8; TRPC6, transient receptor potential cation channel subfamily C member 6; WKY, Wistar–Kyoto rat.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



